 Kohei Ito
Kohei Ito Ryo Niwa
Ryo Niwa Ken Kobayashi1
Ken Kobayashi1- 1BIOTA Inc., Tokyo, Japan
- 2Graduate School of Medicine, Kyoto University, Kyoto, Japan
- 3Faculty of Applied Biological Sciences, Gifu University, Gifu, Japan
- 4Tsuchida Sake Brewing Company, Gunma, Japan
Introduction: In Kimoto-style fermentation, a fermentation starter is produced before the primary brewing process to stabilize fermentation. Nitrate-reducing bacteria, mainly derived from brewing water, produce nitrite, and lactic acid bacteria such as Leuconostoc can proliferate because of their tolerance toward low temperature and their low nutritional requirements. Later, Lactobacillus becomes the dominant genus, leading to weakly acidic conditions that contribute to control yeasts and undesired bacterial contaminants. However, the sources of these microorganisms that play a pivotal role in Sake brewing have not yet been revealed. Thus, comprehensive elucidation of the microbiome is necessary.
Methods: In this study, we performed 16S rRNA amplicon sequencing analysis after sampling from floor, equipment surfaces, and raw materials for making fermentation starters, including koji, and water in Tsuchida Sake brewery, Gunma, Japan.
Results: Amplicon sequence variants (ASVs) between the external environments and the fermentation starter were compared, and it was verified that the microorganisms in the external environments, such as built environments, equipment surfaces, and raw materials in the sake brewery, were introduced into the fermentation starter. Furthermore, various adventitious microbes present in the fermentation starter of early days and from the external environments were detected in a nonnegligible proportion in the starter, which may impact the taste and flavor.
Discussion: These findings illuminate the uncharacterized microbial dark matter of sake brewing, the sources of microbes in Kimoto-style fermentation.
1. Introduction
1.1. Description of microbial transition of a fermentation starter of Kimoto-style Sake
Fermentation is an ancient biotechnology that has been used to improve the safety, storage capacity, flavor, and nutrition of food. The most modern fermented foods are manufactured by adding fermentation starters for improving stability and reproducibility. In contrast, the fermentation process has traditionally been carried out by utilizing microorganisms from raw materials, starter cultures, and the environment around the fermentation batches. Among fermented foods, Sake is the traditional alcoholic beverage of Japan. Sake is made from rice through starch saccharification by Aspergillus oryzae and alcoholic fermentation by Saccharomyces cerevisiae. The beverage is traditionally brewed using a fermentation starter called Moto.
Moto is a yeast mash made from a nutritious mixture of rice, Koji, and water. In the preliminary stage of the primary brewing process, Moto is manufactured to prevent the proliferation of undesired contaminants and to promote smooth alcoholic fermentation. Based on the manufacturing methods of Moto, Sake is divided into three types, Sokujo, Kimoto, and Yamahai. Sokujo-style Moto is a modern method to add food-grade quality lactic acid and decrease pH before alcoholic fermentation by yeast. Lactic acid inhibits contaminations of unintended yeasts and bacteria from external environments into the fermentation starter. Since Sokujo relies on lactic acid to control the pH of Moto, a desirable condition can be created relatively quickly, leading to stable production of the starter. Meanwhile, the Kimoto-style Moto is a traditional preparation method for the starter culture and is manufactured by inducing the growth of lactic acid bacteria properly. Lactic acid bacteria produce lactic acid, which contributes to creating an environment conducive to the growth of yeast. The Yamahai style is similar to the Kimoto style but made without grinding rice. Koji, mold (Aspergillus oryzae) on rice, degrades solid rice and initiates alcoholic fermentation by yeast. Repeatedly, in both methods, indigenous lactic acid bacteria, which are indispensable in the production of Sake and Moto, grow without being added to the culture. In other words, these lactic acid bacteria are originating from the external environments.
1.2. Impacts of Kuratsuki bacteria and adventitious microbes from the external environments
The microorganisms that inhibit surrounding the fermentation reactor, or more precisely, in the breweries, are described as Kuratsuki in Japanese. Massive efforts have been made to investigate Kuratsuki microbes. One of the significant findings is the study isolating the genus Kocuria from sake in multiple batches from the same Sake manufacturer in Japan (Terasaki and Nishida, 2020). The study also found that Kocuria was the unique genus common to all the studied breweries and concluded that the isolated Kocuria were Kuratsuki microbes. In addition, several Kuratsuki microbes have also been isolated, including Staphylococcus sp. and Bacillus sp., as well as lactic acid bacteria considered necessary for Sake production (Terasaki and Nishida, 2020; Kanamoto et al., 2021; Takahashi et al., 2021). As an alternative method to identify Kuratsuki microbes, a study using amplicon sequencing was reported in 2014 (Bokulich et al., 2014). A comparison between Kimoto-style Moto and microbes on the surfaces of the brewery was conducted by 16S rRNA and ITS amplicon sequencing. These authors showed that some genera present on the surfaces of fermentation equipment were also detected in Kimoto-style fermentation.
1.3. Tracking the microbiomes from the external environment of Moto and their dynamics during the fermentation process
To characterize the source of Kuratsuki microbes more deeply, we conducted 16S rRNA amplicon sequencing at Tsuchida Sake Brewery in Gunma Prefecture, Japan. We collected the samples from floors, raw materials, and equipment surfaces such as a fermentation tank and a wood stand for growing Koji. This brewery produces yeast-free and bacteria-free Moto, which means external microbiomes initiate the preparation of Moto. We reported their unique microbial transition that Lactobacillus did not become dominant in the fermentation starter (Ito et al., 2022). We utilized this sequence data of Moto and compared microbial communities between the built environments and the starter culture by counting shared amplicon sequencing variants (ASVs), showing identical sequences in the V3-V4 region of 16S rRNA. Our results suggest that adventitious microbes from the built environment create the initial microbial status, and Kuratsuki microbes giving successive fermentation, are screened during competitions in the fermentation.
2. Materials and methods
2.1. Sample collection
Samples were collected from 10 locations, including floor, raw materials, and equipment surfaces in the brewery, by a swabbing method (Figure 1). All samples were collected at Tsuchida Sake Brewing Company (Gunma, Japan) between October and November 2021. Sterile cotton-tipped swabs (Isohelix Swab: SK-3, Cell Projects Ltd., Harrietsham, UK) were moistened with lysis buffers (BuccalFix Stabilization and Lysis Buffers: BFX, Cell Projects Ltd., Harrietsham, UK) and streaked across an area of the target surface (A square of 20 cm in length and width) for 3 min. Samples were immediately frozen and stored until DNA extraction. We collected two samples from different locations on the same sampling targets except for water and used them for library preparation, respectively. We performed this project in a manner of duplicates because we set a hypothesis that microbes on equipment or floor are not rich and distributed heterogeneously. All equipment surfaces were swabbed before fermentation. A map and pictures of each floor and equipment are shown in Supplementary Figure 1. The description of the roles of each equipment is listed in Table 1.
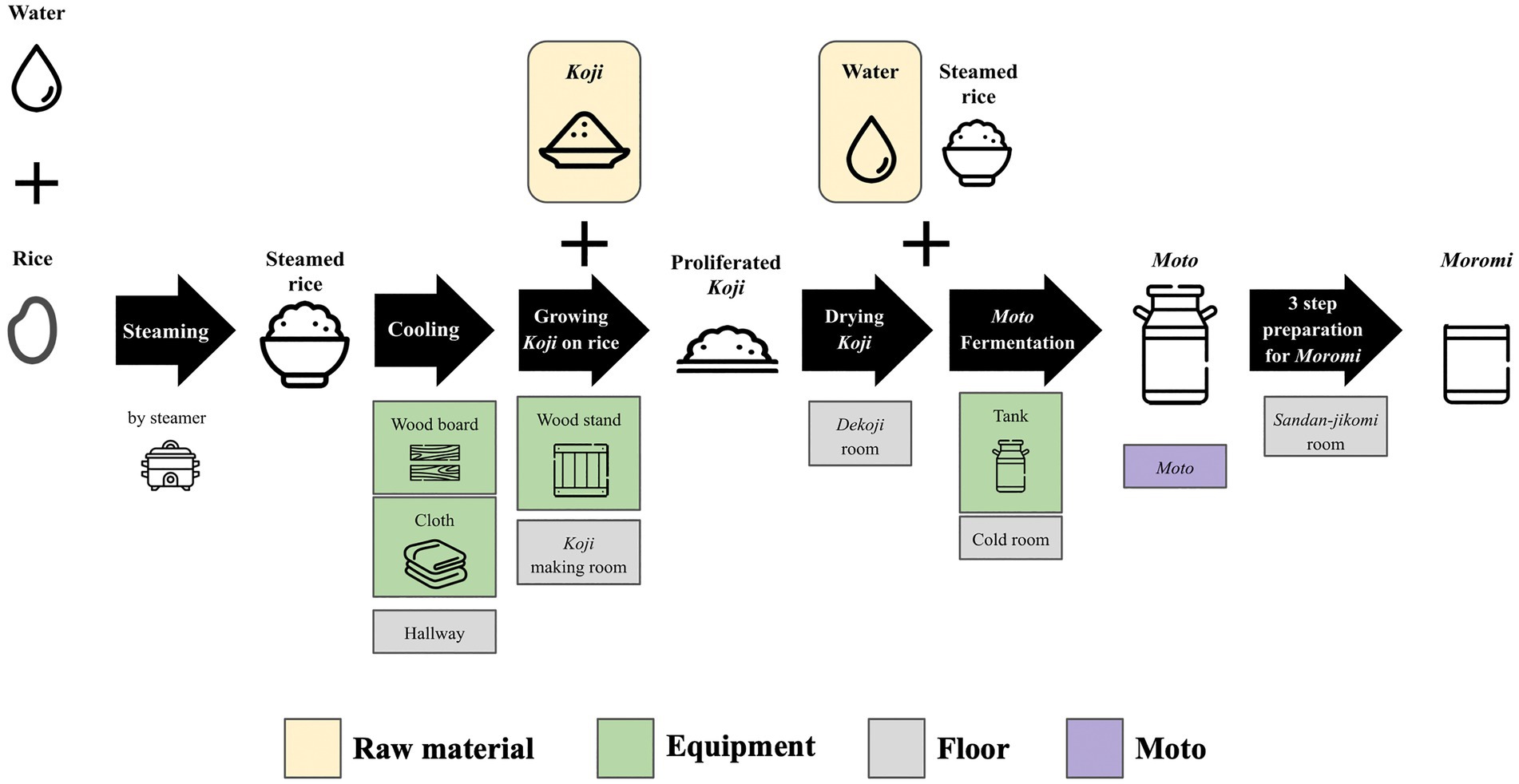
Figure 1. An overview of multiple processes for Sake brewing with sampling targets in the study. The group of Floor includes the brewery’s floor samples. The group of Raw materials includes water and Koji. The group of Equipments includes the wood board, wood stand, cloth, and tank for Moto Fermentation. 10 samples with grid lines were collected in this study. Sample collection was performed in duplicate. A fungus called Aspergillus oryzae, which is capable of secreting amylases, is cultivated on steamed rice to create Koji. This proliferated Koji, along with steamed rice and water, is then placed in an open-top tank to initiate the fermentation process and produce Moto. The starter is subsequently mixed with additional Koji, steamed rice, and water, and fermentation for a period of 3–5 weeks is done to make Moromi through three-step preparation (Sandan-jikomi). Moromi is separated into Sake and spent rice by a filter press.
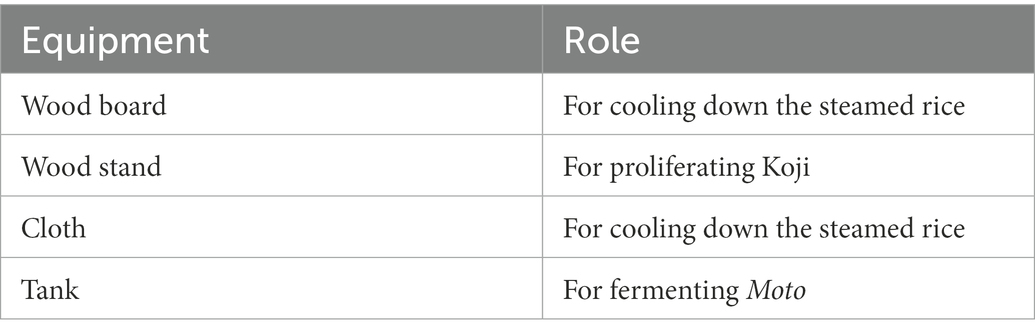
Table 1. The roles of each piece of equipment surveyed in this study.
2.2. Total DNA extraction and high-throughput sequencing
Samples were subjected to 750 μl of lysis buffer supplied with the GenFind V2 DNA extraction kit (Beckman Coulter, Indianapolis, U.S.). The suspension was vortexed for 10 min, heat-treated at 100°C for 10 min, and centrifuged for 5 min at 15,000 rpm. The supernatant was mixed with E.Z. beads (AMR, Tokyo, Japan). DNA fragmentation was performed on an MM-400 (Retsch, Haan, Germany) at a maximum speed of 3 min. The rest of the DNA purification process was performed using a GenFind v2 DNA extraction kit (Beckman Coulter, Indianapolis, U.S.) following the manufacturer’s protocol. DNA was eluted with 80 μl of water. Primers 341F (5′ -TCGTCGGCAGCGTCAGATGTGTATAAGAGAGACACCTACGGGNGGCWGCAG-3′) and 806R (5′-GTCTCGTGGGCTCGGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′) were utilized to amplify the V3-V4 region of the 16S rRNA gene, and adapter sequences were added by KAPA HiFi HotStart ReadyMix (Roche, Basel, Switzerland) Thermal conditions for PCR were 95°C for 3 min; 32 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s; and a final extension at 72°C for 5 min. Secondary PCR was performed with forward and reverse primer sequences designated by Illumina. The thermal condition was set to be the same as the first PCR but for 8 cycles. After AMpure XP purification (Beckman Coulter, Indianapolis, U.S.), the libraries were sequenced on the Illumina MiSeq platform (Illumina, Inc., San Diego, USA) by paired-end sequencing of 300 bp. Preparation of DNA samples and libraries and amplicon sequencing were performed at GenomeRead Inc. in Kagawa, Japan.
2.3. Microbiome analysis
Microbiome analysis was performed as previously reported (Ito et al., 2022). Briefly, raw FASTQ files were imported into the QIIME2 platform as qza files. Denoising and quality control of reads was performed by qiime dada2 denoise-paired, and reads were classified into amplicon sequencing variants (ASVs) (Callahan et al., 2016). The SILVA database SSU 138 was utilized with qiime feature-classifier classify-sklearn for taxonomic assignment (Quast et al., 2013; Bokulich et al., 2018). ASVs classified as chloroplast, mitochondria, and unassigned were excluded from statistical analysis. One sample from the cold room floor did not have enough sequencing depth and was excluded from the downstream analysis. The depth of sequence reads differed among samples, and we subsampled from each sample to 4,724 reads each to normalize the data. Subsampling is an approach for inferring microbiome differences between samples and has been reported to be a suitable analytical method when analyzing new datasets (Hughes and Hellmann, 2005). To evaluate the effect of sequence read counts on microbiome diversity assessment, we examined changes in the value of the number of ASVs over a range of reading counts from 0 to 8,000 by rarefaction curves. The rarefaction curves of the number of ASVs were leveled off when the number of reads reached approximately 4,000 (Supplementary Figure 2).
2.4. Calculation of shared ASVs
Sequence data obtained in this study were confronted to those previously obtained (PRJDB13924) to detect ASVs present in both datasets (Ito et al., 2022). Shared ASVs in this study were defined as ASVs present at more than 1% were considered to detect those shared by different types of data sets. The calculation was conducted by R version 4.2.1 and Phyloseq (McMurdie and Holmes, 2013) version 1.40.0.
2.5. Statistical analysis
Kruskal–Wallis tests were used to compare the relative abundances among sample types. Mann–Whitney U tests were used to compare all combinations of elements within the sample types. All multiple testing corrections were performed by computing FDRs using the Benjamini-Hochberg method, and adjusted p values <0.05 were considered statistically significant. Differentially abundant genera between groups were identified using linear discriminant analysis (LDA) effect size (LEfSe) analysis (Segata et al., 2011). Only bacterial genera reaching the LDA score threshold of 3.0 and with a p value lower than 0.01 are shown. Data visualization was performed by R version 4.2.1, the ggplot2 package and ggprism (Dawson, 2022).
3. Results
3.1. The phylum-level bacterial profiles in the Sake brewery
The number of reads after DADA2-based denoising is described in Supplementary Table 1. The relative abundances of the top 7 phyla are shown as a heatmap (Figure 2A). The bacterial profiles of floor surfaces showed relatively high abundances of Bacteroidota, Proteobacteria, Firmicutes, and Actinobacteria. Equipment surfaces (wood board, wood stand, cloth, tank) showed similar trends as floor surfaces, but cloth and tank showed lower relative abundance in Bacteroidota and higher relative abundance in Deinococcota. Raw materials (Koji and water) are rich with Firmicutes and Proteobacteria. The relative abundances of the four phyla were significantly different among the three sample types (Figures 2B–E). Firmicutes had the highest median of over 30% for raw materials, and Mann–Whitney U tests also showed a significant difference in raw materials against floor surfaces (value of p = 0.05) and equipment surfaces (value of p = 0.016) (Figure 2B). Otherwise, floor surfaces and equipment surfaces showed higher relative abundances in Bacteroidota, Deinococcota, and Actinobacteria (Figures 2C–E).
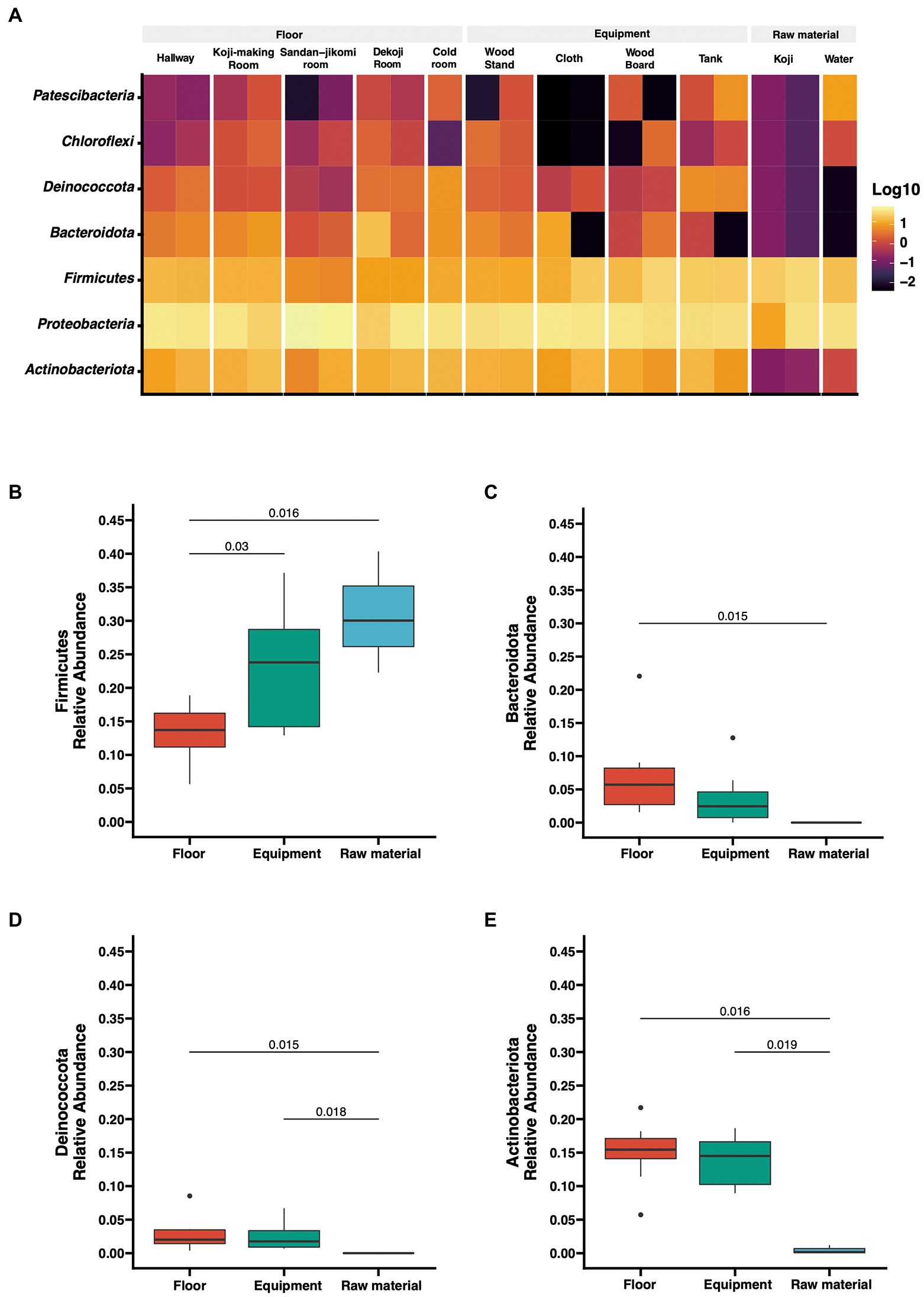
Figure 2. (A) Taxon abundance heatmap at the phylum level. Heatmap depicts the relative abundances (log10 scale) of the top 7 phyla across all surfaces. (B–E) Box plot representation for phylum-level comparison among each group. Four phyla (Firmicutes, Bacteroidota, Deinococcota, Actinobacteriota) were compared across groups in the brewery. Statistical analysis was computed using Mann-U tests.
3.2. Distribution patterns of bacterial genera associated with Kimoto-style fermentation
Latilactobacillus sakei has been described a dominant bacterial species in Moto (Tsuji et al., 2018). However, our analysis is based on SILVA database SSU 138, which curates at the genus-level. Therefore, we could not assess that the Lactobacillus reads fit with this species. The top 15 genera are shown in a stacked bar graph (Figure 3); the rest are noted as the remainder. Sphingomonas was widely detected with a high abundance ratio within samples grouped in floor surfaces (4.2 ~ 33.5%). 28.3 and 27.5% Halomonas were detected from the Sandan-jikomi (three-step preparation for Moromi, fermentation mash) room floor. Koji had high abundances in Anaerobacillus (30 and 8.7%, respectively) and Staphylococcus (20.1 and 4.7%, respectively). The heatmaps sorted by Firmicutes, Bacteroidota, Deinococcota, and Actinobacteria are shown in Supplementary Figure 3A–D to identify the unique genera within each sample. The relative abundance of the genus Lactobacillus, considered to be necessary for brewing, was found in more than 1% of only one sample of the cold room floor (3.6%) and the wood board (2.8%) (Supplementary Figure 3B). Both surface types Leuconostoc, the dominant species of Moto in the brewery, was found in the tank, Koji making room floor, Dekoji room floor, and cold room floor (Supplementary Figure 3B). Bacillus was widely detected in water (8.7 and 1.1%, respectively), wood board (8.3 and 0%, respectively), and cloth (8.8, and 1.7%, respectively) (Supplementary Figure 3B). LFfSe analysis identified Anaerobacillus, Methylobacterium, Escherichia, and Delftia as differentially abundant in raw materials compared with other sample types (Supplementary Figure 4).
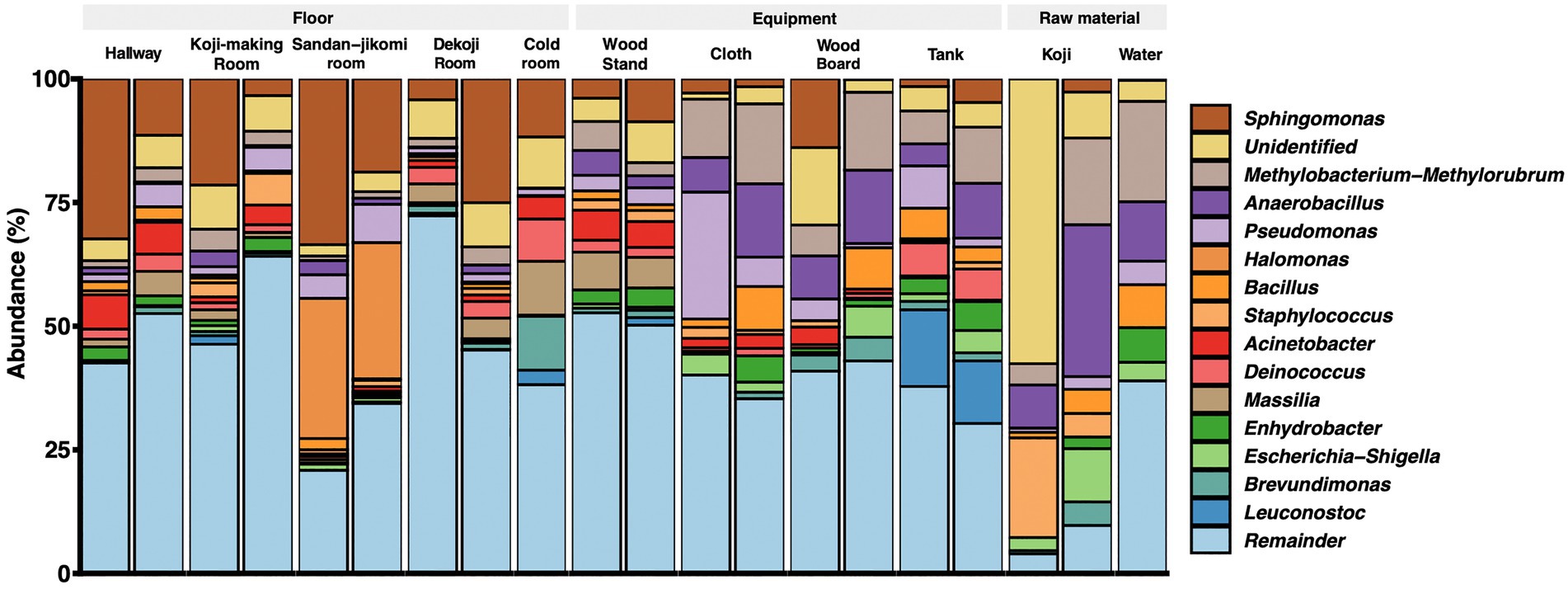
Figure 3. Genus-level taxonomic composition across the environmental samples.
3.3. Changes in the relative abundances of microbes from the external environment during Moto fermentation
Principal coordinates analysis (PCoA) based on unweighted UniFrac distances was performed (Figure 4A). The dots were divided into three major groups according to their sample types. Raw materials and equipment surfaces are located between Moto and the group of floor surfaces. Floor surface samples differ substantially from Moto.
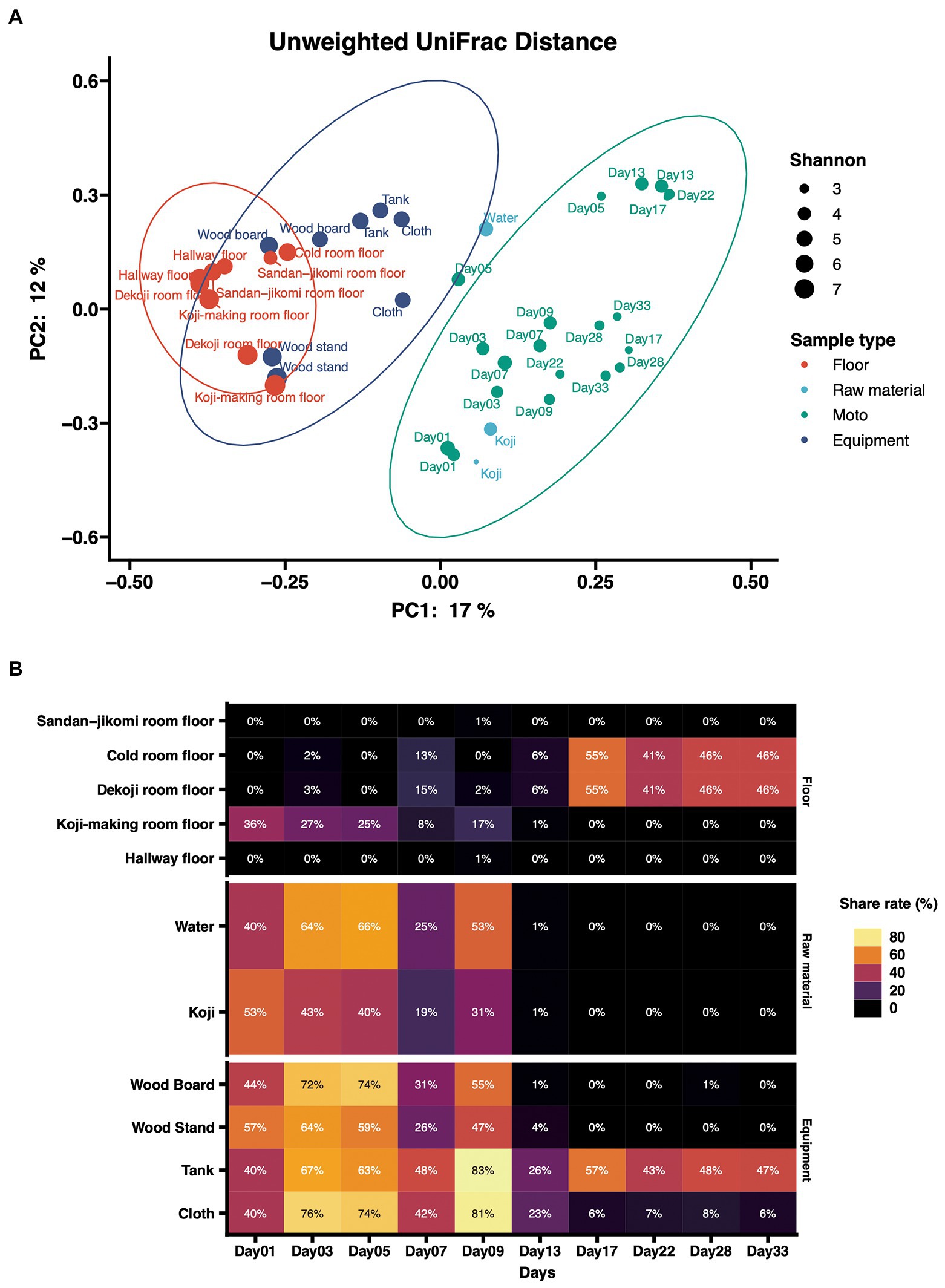
Figure 4. (A) Principal-coordinate analysis (PCoA) by Weighted UniFrac distance of environmental samples and Moto samples. The dot size shows the values of Shannon diversity of each sample. (B) Time-series changes in the relative abundance of shared ASVs in Moto during the Kimoto-style fermentation period. More than 40% of the microbiomes in Moto on Day 33 shared the ASVs with the tank surface, cold room floor, and Koji-making room floor samples.
The results of the shared ASV analysis are shown as a heatmap (Figure 4B). Bacteria confirmed on the surface of all raw materials and equipment surfaces showed shared ASVs with Moto samples throughout Day 1 to Day 9. The Koji making room also showed them from Day 1 to Day 5. The ratio of shared ASVs increased on Day 3 and Day 5. The tank and cloth reached 83 and 81%, respectively, on Day 9. Day 13 showed a decrease in the ratio of the tank (26%) and cloth (23%) and an increase in the cold room (6%) and Dekoji room (6%). From Day 17 to Day 33, only the tank, Dekoji room floor, and cold room floor showed shared ASVs. The tank, cold room, and Dekoji room showed more than 40% in the later days. Further details of ASV-assigned genera shared by Moto on Day 33 are shown in Table 2. ASVs assigned to Pseudomonas in Moto were shared between the tank and cloth, and Serratia and Hafnia-Obesumbacterium were shared by cloth. Tank, cold room, and Dekoji room showed Leuconostoc sharing with Moto. The shared ASV of Leuconostoc consisted of a single ASV unit.
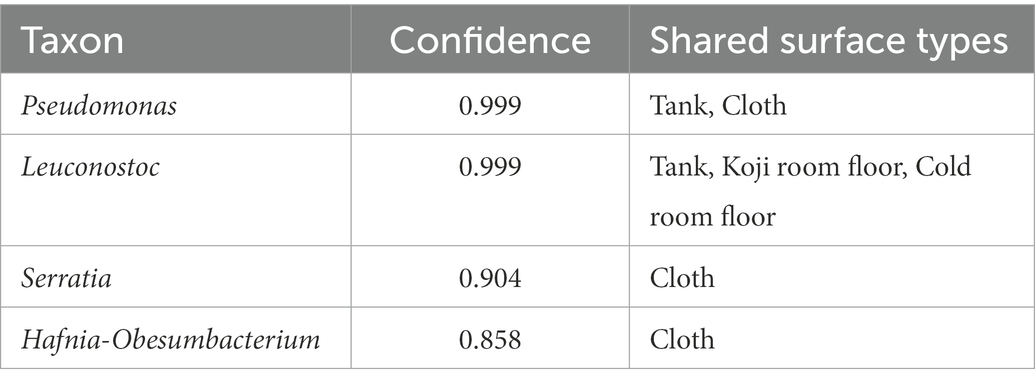
Table 2. The taxon of shared ASV (Genus-level) between Moto on Day 33 and other environments.
4. Discussion
4.1. Bacterial profiles differed by sample types
We found different phyla profiles between the raw materials and environmental samples (floor surfaces and equipment surfaces). The microbial composition of floor surface samples was diverse compared with the rest of the two sample types (Figures 3, 4A). Previous studies indicate that floors in indoor environments are known to be rich in microbes (Hospodsky et al., 2012; Ruiz-Calderon et al., 2016). For this reason, we included floor samples rather than walls and ceilings. The built environments are also complex structures, and microbial communities differ by conditions such as surface materials and the possibility of human interaction. In fermentation starter production, the manufacturer inputs multiple ingredients and uses equipment surfaces. Also, Moto is produced in multiple rooms based on the steps of production. These direct interactions with sources of microbes can make microbiomes in fermentation starters of early days exceedingly divergent. Firmicutes are detected in high abundance on dry (e.g., floors, cabinets, microwaves) and cold (e.g., inside of the refrigerators) surfaces in the kitchen (Flores et al., 2013) and refrigerator surfaces (Einson et al., 2018), but in this study, they were most abundant in raw materials (Figure 2B).
Genus-level composition (Figure 3A) shows that Koji samples have Bacillus, Anaerobacillus, Staphylococcus, and Sphingomonas. These genera were confirmed in Sokujo-style Moto by Sanger sequencing of their 16S rRNA sequences in the previous report (Terasaki et al., 2017). In this study, these genera were especially detected in the architectural samples. As other features, Koji is made by bare hands; therefore, it is considered that Staphylococcus, a genus encompassing species known to be present on skin (Cogen et al., 2008), may have been contaminated in the Koji and the floors.
We reported that Leuconostoc was dominant in the fermentation of Moto, and the switch of the predominant lactic acid bacteria from Leuconostoc to Lactobacillus was not observed in the fermentation process of this brewery (Ito et al., 2022). Several studies reported that Lactobacillus was detected in tanks (Bokulich et al., 2014, 2015) that can be directly in contact with raw materials and Moto. However, Lactobacillus was detected only on the cold room floor and the wood board. In contrast, Moto did not contact these sites. However, Leuconostoc was detected in the tank of Moto. This result suggests that appreciable concentrations of Leuconostoc are introduced from the tank to the fermentation reaction of Moto. This consideration is consistent with findings in a previous report (Bokulich et al., 2014) that surface contact is the most significant predictor of bacterial composition in the Sake brewery. We also concluded that the sources of Kuratsuki microbes are possibly surfaces of equipment surfaces and raw materials that have high chances of direct contact with Moto. Therefore, the Moto in Tsuchida Sake Brewery may not have shown the lactic acid bacteria switch from Leuconostoc to Lactobacillus in the fermentation process due to the lack of the sources of Lactobacillus.
4.2. Shared ASV analysis revealed the interaction between environmental samples and Moto
In the PCoA of unweighted UniFrac distances (Figure 4A), raw materials are located near the fermentation starter because they are an input of Moto. Next are the equipment surfaces that are in direct contact with Moto. The floor is not in direct contact with the fermentation starter and thus differs substantially from the group structure of Moto. In this study, the floor was employed as the floor surfaces group. This is based on our expectation that microbes may fall on the floor during manufacture. However, experimental data indicate that surfaces that can be in direct contact may have a more significant impact on Moto. The bacterial profile of brewing equipment surfaces has yet to be fully revealed, and further research is needed.
In this study, we investigated how bacteria are transferred to Moto as a source. Bacteria on the equipment surfaces and floor surfaces were confirmed in the early stage of Moto (Figure 4B). Thus, the shared ASVs results indicate that Leuconostoc, which plays an essential role in Moto, was screened within adventitious microbes, and eventually became the dominant species.
4.3. Technical limitations in 16 rRNA amplicon sequencing
Because of the limited classification resolution of 16S rRNA amplicon sequencing, it is challenging to rigorously predict microbial sharing between fermentation starters and the environment. Calculating shared ASVs or statistical methods is a convenient way to track microbial sources. However, high-resolution sequencing, such as metagenome sequencing, must be performed to clarify precisely which places or sources in bacteria affect fermentation. Lineages of bacteria between two groups can be compared by metagenome-assembled genomes (MAGs) and show more precision in tracking at the level of whole chromosomal sequences, not just the restricted regions of 16S rRNA.
We reported that the ethanol concentration increased as fermentation progressed (Ito et al., 2022). According to ethanol production from yeast, bacteria should decrease gradually. However, 16S rRNA amplicon sequencing is a DNA-based detection method, and it is impossible to discern between viable and nonviable bacteria. In fermented foods, viable bacteria can be considered to have a significant influence, and conventional microbiology experiments or flow cytometry with staining is required to verify the viable cells. In addition, fungi also play a pivotal role in Sake fermentation. Therefore, metagenome sequencing is necessary to unveil the interaction between bacteria and fungi, especially for fermentation utilizing Koji, Aspergillus oryzae, and Saccharomyces cerevisiae.
Also, microbes in the built environments and equipment are not as abundant as in fermented food. Kuratsuki microbes inhabit breweries in a heterogeneous manner. In our samples, we confirmed the variety of relative abundances in some genera between duplicated experiments (Figure 3). We sampled each duplicate set from different locations. This can explain by biases in library preparation, including PCR or sampling bias in each experiment. Multiple sampling points in the exact locations can give higher resolutions in rare ASVs, and shared ASVs analysis can show precision results.
4.4. Kuratsuki microbiota as a community structure and their impact on sake brewing
Tracing the dynamic network of microbial communities further contributes to understanding this traditional liquor. According to the previous study, microbial diversity decreases with the number of fermentation days of Moto (Bokulich et al., 2014). Another study performing microbiome analysis of Moto from five different Sake breweries reported that the bacterial communities of Moto were very diverse, but that diversity decreased over time through multiple microbial transition patterns (Takahashi et al., 2021). It was also reported that ethanol produced by yeast inhibits the growth of adventitious microbes (Terasaki and Nishida, 2020). Microbes produce metabolites that influence microbial dynamics over time. Both metabolome and microbiota interact mutually. Profiling the metabolome of Sake breweries can deepen the understanding of the roles of Kuratsuki microbes.
Some strains have been isolated from Moto and Sake. Latilactobacillus sakei was isolated from yamahai-moto (Tsuji et al., 2018). Lactiplantibacillus plantarum was also isolated from different yamahai-moto. These lactic acid bacteria were not confirmed in our study, but they can also be Kuratsuki microbes and further study in different breweries should be conducted.
Furthermore, some adventitious microbes have been found to possess activity interfering with the growth of bacteria that spoil Sake, potentially benefiting Sake brewing (Taniguchi et al., 2010) or producing beneficial chemical components that contribute to the flavor, taste, and quality of Sake (Akaike et al., 2020).
Mapping the microbiomes in Sake breweries may also be important in terms of quality and safety in Sake production (De Filippis et al., 2021). In the past, studies have yet comprehensively examined changes in microbes sharing between different sample types (floor surfaces and equipment surfaces) and Moto in a brewery during the fermentation process with a lens of amplicon sequencing. Previous studies have isolated Kocuria and Bacillus as viable microbes (Kanamoto et al., 2021; Terasaki et al., 2021), indicating that certain microbes can live in Moto, such as lactic acid bacteria and yeast, and may be involved in Sake brewing. The possibility of horizontal gene transfer among microorganisms in Sake breweries (Terasaki et al., 2021) has also been demonstrated; instead of targeting only specific bacteria, such as lactic acid bacteria, the community-level structure should be targeted as Kuratsuki microbiota. Isolation of viable microbes and whole genome sequencing can give a deep insight into gene transfer.
Finally, understanding microbial diversity and dynamics are about preserving traditional food culture and food production safety. Sake requires attention to spoilage, such as microbial contamination and spoilage, which can cause spoilage and may be helpful for safety and quality control. This research can be an essential milestone in unraveling the mysteries of fermentation phenomena. Because each brewery produces different flavors of sake with different ingredients and equipment surfaces, it is necessary to further research to profile microbiomes in sake breweries. Additionally, these contributions can lead to process control, spoilage prevention, food safety, and the protection of traditional cultures.
Data availability statement
The data presented in the study are deposited in online repositories, accession number can be found at: NCBI, DRR423705.
Author contributions
KI and RN conceived this study, wrote the original draft, and performed the statistical analysis. KI and KK designed the experiments. RN performed microbiome analysis. YT, GH, and KK collected samples and provided the records required for discussion. TN supervised the study and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by Tsuchida Sake Brewing Company. RN is a graduate student of the Medical Innovation Program at Kyoto University and supported by the JST SPRING program, Grant Number JPMJSP2110. We cited icons for Figure 1 from https://www.flaticon.com/free-icon/.
Acknowledgments
Samples were collected with the help of members of the Tsuchida Sake Brewery. GenomeRead Inc. performed amplicon sequencing. All authors thank Morgenrot Inc. for providing the computational environment for the analysis.
Conflict of interest
KI is a board member of BIOTA Inc., Tokyo, Japan. RN and KK are employed by BIOTA Inc. GH and YT belong to Tsuchida Sake Brewing Company, Gunma, Japan.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Tsuchida Sake Brewing Company. The funder was involved in the study design and collection.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1112638/full#supplementary-material
References
Akaike, M., Miyagawa, H., Kimura, Y., Terasaki, M., Kusaba, Y., Kitagaki, H., et al. (2020). Chemical and bacterial components in sake and sake production process. Curr. Microbiol. 77, 632–637. doi: 10.1007/s00284-019-01718-4
Bokulich, N. A., Bergsveinson, J., Ziola, B., and Mills, D. A. (2015). Mapping microbial ecosystems and spoilage-gene flow in breweries highlights patterns of contamination and resistance. elife 4:e04634. doi: 10.7554/eLife.04634
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bokulich, N. A., Ohta, M., Lee, M., and Mills, D. A. (2014). Indigenous bacteria and fungi drive traditional kimoto sake fermentations. Appl. Environ. Microbiol. 80, 5522–5529. doi: 10.1128/AEM.00663-14
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Cogen, A. L., Nizet, V., and Gallo, R. L. (2008). Skin microbiota: a source of disease or defence? Br. J. Dermatol. 158, 442–455. doi: 10.1111/j.1365-2133.2008.08437.x
Dawson, C. (2022). ggprism: A ggplot2’ Extension Inspired by ‘GraphPad Prism.’ Comprehensive R Archive Network (CRAN). Available at: https://CRAN.R-project.org/package=ggprism (Accessed November 28, 2022).
De Filippis, F., Valentino, V., Alvarez-Ordóñez, A., Cotter, P. D., and Ercolini, D. (2021). Environmental microbiome mapping as a strategy to improve quality and safety in the food industry. Curr. Opin. Food Sci. 38, 168–176. doi: 10.1016/j.cofs.2020.11.012
Einson, J. E., Rani, A., You, X., Rodriguez, A. A., Randell, C. L., Barnaba, T., et al. (2018). A vegetable fermentation facility hosts distinct microbiomes reflecting the production environment. Appl. Environ. Microbiol. 84:e01680-18. doi: 10.1128/AEM.01680-18
Flores, G. E., Bates, S. T., Caporaso, J. G., Lauber, C. L., Leff, J. W., Knight, R., et al. (2013). Diversity, distribution and sources of bacteria in residential kitchens. Environ. Microbiol. 15, 588–596. doi: 10.1111/1462-2920.12036
Hospodsky, D., Qian, J., Nazaroff, W. W., Yamamoto, N., Bibby, K., Rismani-Yazdi, H., et al. (2012). Human occupancy as a source of indoor airborne bacteria. PLoS One 7:e34867. doi: 10.1371/journal.pone.0034867
Hughes, J. B., and Hellmann, J. J. (2005). The application of rarefaction techniques to molecular inventories of microbial diversity. Methods Enzymol 397, 292–308. doi: 10.1016/S0076-6879(05)97017-1
Ito, C., Niwa, R., Yamagishi, Y., Kobayashi, K., Tsuchida, Y., Hoshino, G., et al. (2022). A unique case in which Kimoto-style fermentation was completed with Leuconostoc as the dominant genus without transitioning to Lactobacillus. bioRxiv. doi: 10.1101/2022.09.05.505584v1.full
Kanamoto, E., Terashima, K., Shiraki, Y., and Nishida, H. (2021). Diversity of Bacillus isolates from the sake brewing process at a sake brewery. Microorganisms 9:1760. doi: 10.3390/microorganisms9081760
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ruiz-Calderon, J. F., Cavallin, H., Song, S. J., Novoselac, A., Pericchi, L. R., Hernandez, J. N., et al. (2016). Walls talk: microbial biogeography of homes spanning urbanization. Sci. Adv. 2:e1501061. doi: 10.1126/sciadv.1501061
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Takahashi, M., Morikawa, K., Kita, Y., Shimoda, T., Akao, T., and Goto-Yamamoto, N. (2021). Changes in bacterial and chemical components and growth prediction for Lactobacillus sakei during Kimoto-style fermentation starter preparation in sake brewing: a comprehensive analysis. Appl. Environ. Microbiol. 87. doi: 10.1128/AEM.02546-20
Taniguchi, M., Ishiyama, Y., Takata, T., Nakanishi, T., Kaneoke, M., Watanabe, K. I., et al. (2010). Growth-inhibition of hiochi bacteria in namazake (raw sake) by bacteriocins from lactic acid bacteria. J. Biosci. Bioeng. 109, 570–575. doi: 10.1016/j.jbiosc.2009.11.015
Terasaki, M., Fukuyama, A., Takahashi, Y., Yamada, M., and Nishida, H. (2017). Bacterial DNA detected in Japanese rice wines and the fermentation starters. Curr. Microbiol. 74, 1432–1437. doi: 10.1007/s00284-017-1337-4
Terasaki, M., Kimura, Y., Yamada, M., and Nishida, H. (2021). Genomic information of Kocuria isolates from sake brewing process. AIMS Microbiol. 7, 114–123. doi: 10.3934/microbiol.2021008
Terasaki, M., and Nishida, H. (2020). Bacterial DNA diversity among clear and cloudy sakes, and sake-kasu. Open Bioinformat. J. 13, 74–82. doi: 10.2174/1875036202013010074
Tsuji, A., Kozawa, M., Tokuda, K., Enomoto, T., and Koyanagi, T. (2018). Robust domination of Lactobacillus sakei in microbiota during traditional Japanese sake starter yamahai-moto fermentation and the accompanying changes in metabolites. Curr. Microbiol. 75, 1498–1505. Available at:. doi: 10.1007/s00284-018-1551-8
Keywords: Japanese sake, microbial fermentation, Kuratsuki, microbiome, Kimoto-style
Citation: Ito K, Niwa R, Kobayashi K, Nakagawa T, Hoshino G and Tsuchida Y (2023) A dark matter in sake brewing: Origin of microbes producing a Kimoto-style fermentation starter. Front. Microbiol. 14:1112638. doi: 10.3389/fmicb.2023.1112638
Edited by:
Pasquale Russo, University of Milan, ItalyReviewed by:
Giuseppe Spano, University of Foggia, ItalyMonique Zagorec, INRA Centre Angers-Nantes Pays de la Loire, France
Copyright © 2023 Ito, Niwa, Kobayashi, Nakagawa, Hoshino and Tsuchida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kohei Ito, ✉ a29oZWlAYmlvdGEubmUuanA=
†These authors have contributed equally to this work