 Fei Wu
Fei Wu Tingting Zhang1,2†
Tingting Zhang1,2† Yiquan Zhang
Yiquan Zhang Renfei Lu
Renfei Lu- 1Department of Clinical Laboratory, Nantong Third People’s Hospital, Affiliated Nantong Hospital 3 of Nantong University, Nantong, China
- 2School of Medicine, Nantong University, Nantong, Jiangsu, China
Vibrio vulnificus is an opportunistic, global pathogen that naturally inhabits sea water and is responsible for most vibriosis-related deaths. We investigated the genetic characteristics of V. vulnificus isolated from the clinical blood culture specimen of a patient with hepatitis B virus cirrhosis in 2018 (named as V. vulnificus VV2018) by whole genome sequencing (WGS). VV2018 belonged to a novel sequencing type 620 (ST620) and comprised two circular chromosomes, containing 4,389 potential coding sequences (CDSs) and 152 RNA genes. The phylogenetic tree of single nucleotide polymorphisms (SNPs) using 26 representative genomes revealed that VV2108 grouped with two other V. vulnificus strains isolated from humans. The pan-genome of V. vulnificus was constructed using 26 representative genomes to elucidate their genetic diversity, evolutionary characteristics, and virulence and antibiotic resistance profiles. The pan-genome analysis revealed that VV2018 shared a total of 3,016 core genes (≥99% presence), including 115 core virulence factors (VFs) and 5 core antibiotic resistance-related genes, and 309 soft core genes (≥95 and <99% presence) with 25 other V. vulnificus strains. The varG gene might account for the cefazolin resistance, and comparative analysis of the genetic context of varG revealed that two genes upstream and downstream of varG were conserved. The glycosylation (pgl) like genes were found in VV2018 compared with Pgl-related proteins in Neisseria that might affect the adherence of the strain in hosts. The comparative analysis of VV2018 would contribute to a better understanding of the virulence and antibiotic resistance profiles of V. vulnificus. Meanwhile much work remains to be done to better understand the function of pgl-like genes in V. vulnificus.
Introduction
Vibrio vulnificus is a gram-negative, rod-shaped bacterium that is widely distributed throughout marine and brackish environments (Gulig et al., 2005). V. vulnificus is found in association with zooplankton, crabs, and various filter feeders such as oysters (Jones et al., 2014). V. vulnificus is also known as an opportunistic pathogen transmitted through the consumption of raw/undercooked seafood or by direct contact causing serious wound infections and sepsis (Baker-Austin and Oliver, 2020). Several underlying medical conditions have been identified as risk factors for V. vulnificus infection, including chronic liver disease, diabetes mellitus, kidney disease, autoimmune disease, hematological disorders and malignancy (Menon et al., 2014). V. vulnificus is responsible for more than 95% of seafood-related deaths in the United States (Haftel and Sharman, 2023). Several other risk factors contribute to the high pathogenicity of V. vulnificus in humans, such as the presence of a capsule, the availability of iron and possession of the vcg gene (Jones and Oliver, 2009). Recent studies indicate that global climate change, resulting in increased surface water temperatures, enables the global distribution and spread of V. vulnificus (Paz et al., 2007; Heng et al., 2017).
The pan-genome refers to the pool of genetic material that is present in a group of bacteria (Tettelin et al., 2005). It is made up of the core genome (genes shared by all strains) and the accessory genome (genes shared by some strains and not all) (Iranzadeh and Mulder, 2019), including soft core genes (≥95 and <99% presence), shell genes (≥15 and <95% presence) and cloud genes (≥0 and <15% presence). The boundaries of the core genome can be extrapolated from highly-conserved genes. Pan-genome analysis has provided new insights into interspecies differentiation and whole sets of genes shared among a group of bacteria (Medini et al., 2005; Lapierre and Gogarten, 2009). Meanwhile, a large range of genomic diversity is observed for pathogenic V. vulnificus strains (López-Pérez et al., 2019). Although multiple virulence factors (VFs) and antibiotic resistance profiles have been identified independently (Horseman and Surani, 2011), the diversity of VFs and resistance genes among V. vulnificus strains remains unknown. Despite the frequent occurrence of the pathogen, the number of cases reported are relatively low, indicating that not all strains of V. vulnificus are equally virulent (Strom and Paranjpye, 2000; Rosche et al., 2010).
In this work, we report the complete genome sequence of V. vulnificus isolated from the blood culture specimen of a clinical patient with hepatitis B virus cirrhosis in 2018 (named as V. vulnificus VV2018), in Nantong, Jiangsu Province, China (Wu et al., 2023), and show that this strain belonged to a novel sequence type (ST620). We characterized the genomic features of this strain to reveal the putative molecular mechanisms underlying its virulence and antibiotic resistance profiles. Furthermore, pan-genome analysis revealed the distribution of VFs and resistance-related genes among V. vulnificus strains. Comparative analysis revealed that the genetic context of varG was conserved with a sequence of approximately 3 kbp encoding ompV-varG-nodD. Meanwhile, using comparative analysis, we first identified putative pgl-like genes in VV2018, that might affect the adherence of the strain in hosts; however, much work still needs to be done to confirm this putative effect.
Materials and methods
Bacterial strain and genomic DNA extraction
VV2018 was isolated from the blood culture specimen of a clinical patient with hepatitis B virus cirrhosis in 2018, in Nantong, Jiangsu Province, China (Wu et al., 2023). The clinical blood sample was used with the approval of the Ethics Committee of Affiliated Nantong Hospital 3 of Nantong University. The strain was identified using the bioMérieux VITEK 2 compact instrument (bioMérieux, Marcy-l’Étoile, France) and average nucleotide identity (ANI) analysis. The genomic DNA of VV2018 was extracted using a TIANamp Bacteria DNA Kit (Tiangen Biotech Company Ltd., Beijing, China), according to the manufacturer’s protocol.
Assessment of antibiotic resistances
The antibiotic resistance profiles were assessed through minimal inhibitory concentration (MIC) assays (Liu and Crosa, 2012). Briefly, a final suspension of 10 cfu/mL in broth supplemented with 2% NaCl and 1 mM CaCl2-H2O were distributed in triplicate throughout a 96-well microtiter plate. Escherichia coli ATCC 25922 was used as the susceptible-control reference bacterial strain for MIC assays. Cells were challenged with 0.25–1,024 g/mL antibiotics. MICs were determined by detection of cell pellet formation in the bottom of the wells of the 96-well plate by turbidometry at 600 nm using Multiskan GO (Thermo Fisher Scientific, USA). Drug susceptibility was determined according to the Clinical and Laboratory Standards Institute (CLSI) drug susceptibility test standard from 2018.
Genomic DNA sequencing, assembly and annotation
Whole genome sequencing (WGS) and assembly were conducted at Azenta Life Sciences (Suzhou, China). Sequences of VV2018 were obtained using PacBio Sequel platform (Pacific Biosciences, Menlo Park, CA, USA) and Illumina HiSeq X Ten platform (Illumina, San Diego, CA, USA). The PacBio reads were assembled by Hifiasm v0.13-r308 and Canu v2.2 (Koren et al., 2017; Cheng et al., 2021), and then the Illumina reads were mapped onto the assembled contigs to correct the primary assembly and control assembly quality using Pilon 1.22 and Quiver (Chin et al., 2013; Walker et al., 2014). The genome completeness and contamination of all V. vulnificus strains we used were further evaluated by checkM with default settings (Parks et al., 2015). Prokka 1.14.6 was used to predict potential CDSs (Seemann, 2014). The functional annotation of these CDSs was performed by DIAMOND (Buchfink et al., 2021) against the non-redundant protein sequence (NR) database of the National Center for Biotechnology Information (NCBI). Kyoto Encyclopedia of Genes and Genomes (KEGG), Cluster of Orthologous Groups (COG), UniProt/Swiss-Prot, Pfam, CAZymes, virulence factors of pathogenic bacteria (VFDB) and Antibiotic Resistance Genes Database (ARDB) were also used to annotate the functions of CDSs (Liu and Pop, 2009; Levasseur et al., 2013; Kanehisa et al., 2017; Galperin et al., 2021; Mistry et al., 2021; Liu et al., 2022; UniProt Consortium, 2023). The rRNA and tRNA sequences were annotated by RNAmmer (Lagesen et al., 2007) and tRNAscan-SE (Lowe and Chan, 2016), respectively. The mobile genetic elements (MGEs) were annotated using ISfinder (Siguier et al., 2006). Genomic islands (GIs), prophages, and CRISPR-Cas systems were identified using online tools IslandViewer 4, PHAge Search Tool (PHAST), and CRISPRCasFinder software, respectively (Arndt et al., 2016; Bertelli et al., 2017; Couvin et al., 2018). Multilocus sequence typing (MLST) was performed by analyzing the housekeeping genes on the MLST website.1 The basic characteristics of the chromosomes were visualized by the CGView Comparison Tool (Petkau et al., 2010).
Comparative sequence analysis
All the available V. vulnificus complete genome sequences (n = 25) were downloaded from the NCBI database with checkM values (Supplementary Table 1). The genome sequences were re-annotated with Prokka 1.14.6 and pan-genome analysis was conducted based on the output of Prokka using Roary with a BLASTP identity cutoff of 90% (Page et al., 2015). For genome similarity assessment, digital DNA-DNA hybridization (dDDH) values were computed using web tool GGDC 3.0 (formula 2, identities/HSP length) (Meier-Kolthoff et al., 2022). Whole genome ANI between pairwise V. vulnificus strains was calculated with Pyani software available at https://github.com/widdowquinn/pyani. The core genome of these strains was produced by Harvest software v1.1.2 (Treangen et al., 2014) using the V. vulnificus CMCP6 genome as a reference. Recombination events were removed from the core-genome alignment using Gubbins v2.2.0 (Croucher et al., 2015). Single nucleotide polymorphisms (SNPs) were then extracted from the recombination-free core genome alignment using the script available at https://github.com/sanger-pathogens/snp-sites. The multi-alignments were aligned with the ClustalW in MEGA 11.0 and analyzed using GeneDoc 2.7.0 (Nicholas and Nicholas, 1997; Tamura et al., 2021). The maximum likelihood (ML) phylogenetic tree of SNPs was constructed using RAxML in the GTRGAMMA model (1,000 bootstrap) (Stamatakis, 2014) and was visualized using Figtree v1.4.4.2 The neighbor-joining phylogenetic trees of PglC and PglD performed by MEGA 11.0. CD-HIT used to cluster the retained sequences using the genome sequence of VV2018 as the reference with identity of 80% and coverage of 90% (Li and Godzik, 2006).
Results and discussion
Genome characteristics of VV2018
The complete genome of VV2018 comprised two chromosomes, Chr I and Chr II. The genome completeness of VV2018 was 100%, and the contamination was 0.05% accessed by checkM. Chr I consisted of 3,264,146 bp with a GC content of 46.60% containing 2,874 predicted CDSs, 106 tRNA genes and 31 rRNA genes. Chr II consisted of 1,816,653 bp with a GC content of 47.19% containing 1,515 predicted CDSs, 13 tRNA genes and 3 rRNA genes (Table 1 and Figure 1). MLST revealed that VV2018 belonged to a novel ST620 and was very close to ST387, with eight loci, isolated from humans in China.
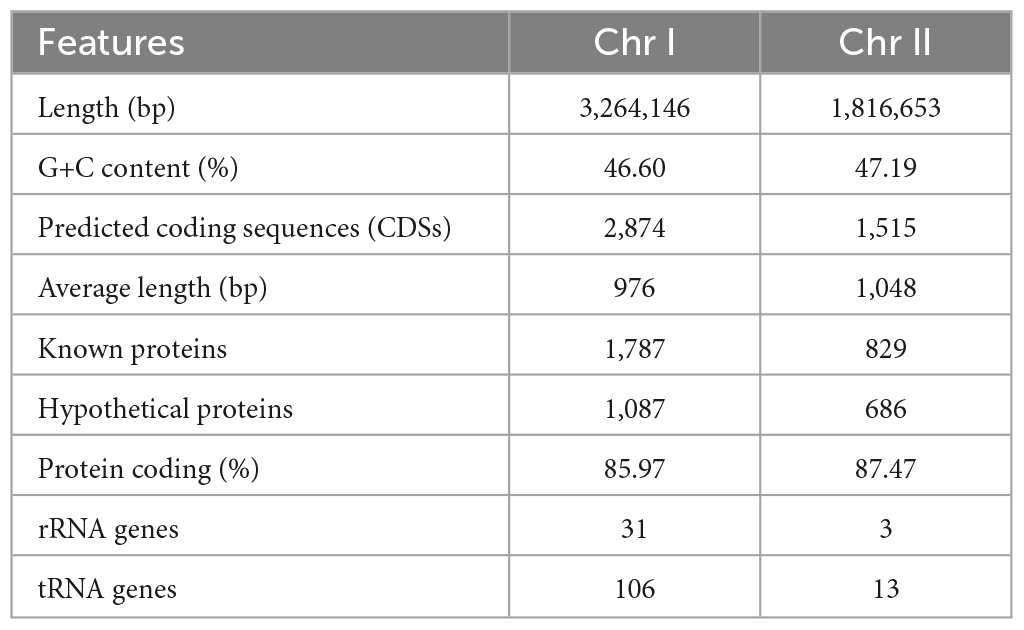
Table 1. General features of VV2018.
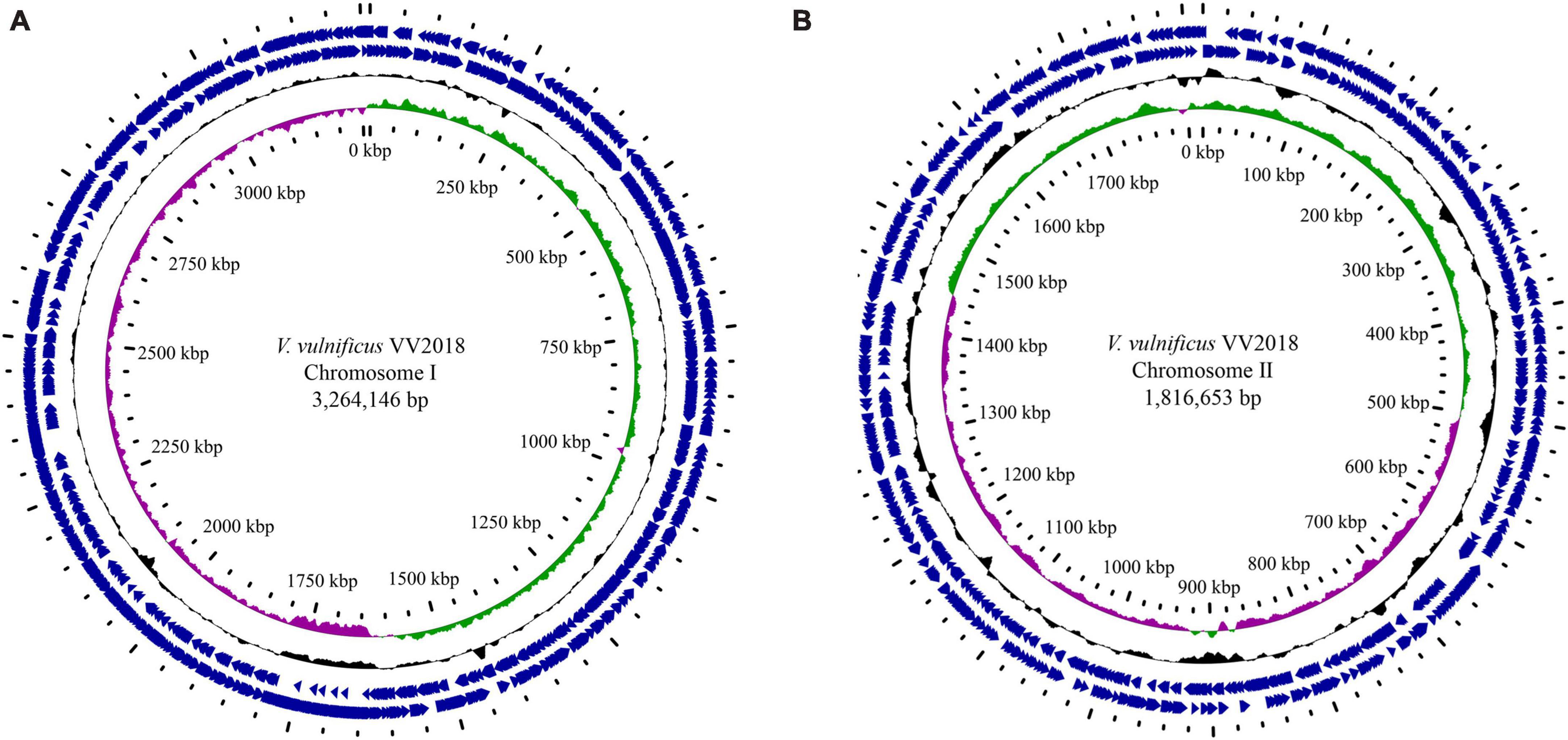
Figure 1. Circular genome maps of VV2018. (A) Chromosome I. (B) Chromosome I. Counting from the center toward the outside: (1) the innermost circle shows the position in kbp. (2) GC skew (G-C/G+C), with a positive GC skew toward the outside and a negative GC skew toward the inside. (3) GC content, with an average of 50%, whereby a G+C content of more than 50% is shown toward the outside, otherwise, inward. (4) Genes encoded in the leading strands (outward) or lagging strands (inward).
The distribution of VV2018 CDSs into COG functional categories is shown in Supplementary Figure 1. Except for genes with unknown functions (8.57%), most genes were related to signal transduction mechanisms, amino acid transport and metabolism, transcription, and carbohydrate transport and metabolism. The annotation of genes of VV2018 in KEGG pathway analysis showed that the most genes were involved in metabolism, including carbohydrate metabolism, amino acid metabolism, metabolism of cofactors and vitamins, and energy metabolism (Supplementary Figure 2).
Virulence factors and resistance-related genes
A total 151 putative VFs were predicted among Chr I (106, 70.20%) and Chr II (45, 29.80%). These genes were mainly associated with motility (polar flagellar proteins), immune evasion (capsular polysaccharide and iron uptake), secretion system (type II secretion system proteins), adherence (type IV pilus, lipooligosaccharide and OmpU) and toxin (RTX toxin) (Table 2). Iron uptake from host cells plays a key role in the survival of V. vulnificus (Dittmann et al., 2019). RTX toxin (rtxABCD) and OmpU have been shown to play important roles in the infection and pathogenesis, respectively, of V. vulnificus (Goo et al., 2006; Liu and Crosa, 2012). The vvhA and tlh were another two toxin genes, encoding cytolysin-hemolysin and thermolabile hemolysin, which induced acute cell death and were important in the pathogenesis and dissemination of these bacteria (Wang et al., 2015; Song et al., 2016). In this case, the patient’s temperature was 40.2°C after infection, which was accompanied by chills, unbearable low back pain, and forced position. Thus, the serious infection of this case might have a strong relationship with the mixing effect of multiple VFs (Wu et al., 2023).
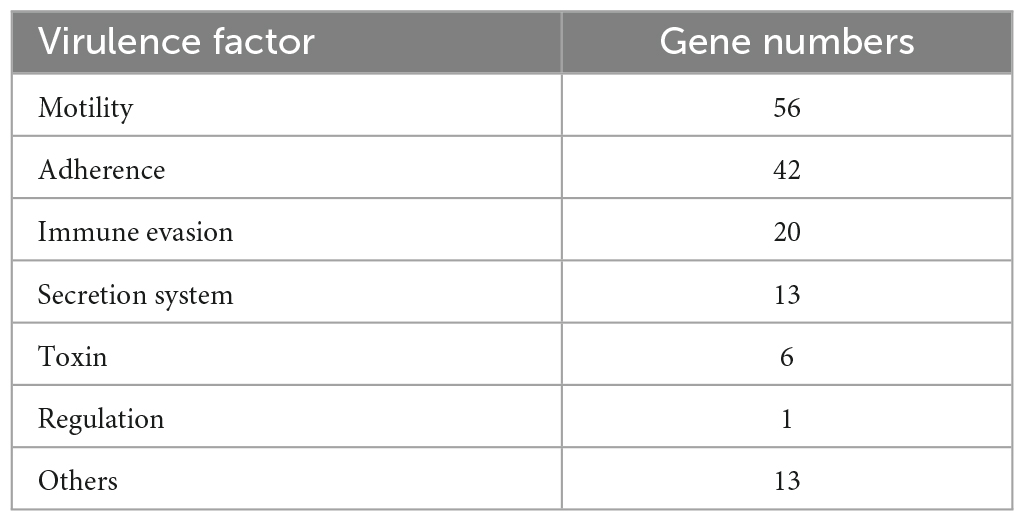
Table 2. The annotation of VFs of the VV2018 in VFDB databases.
Six antibiotic resistance-related genes were identified in the genome of VV2018, including dfrA3 (encoding a dihydrofolate reductase), qnrVC1 (encoding a pentapeptide repeat protein), catB9 (encoding a type B-5 chloramphenicol O-acetyltransferase), tet (Bertelli et al., 2017) (encoding a tetracycline efflux pump), crp (encoding a cAMP-receptor protein) and varG (showing resistance to penicillin, carbapenems and cephalosporins in vitro). CRP is a global regulator that not only regulates the expression of the multidrug efflux pump but also impacts the expression of multiple VFs (Nishino et al., 2008; Zhan et al., 2008). The antibiotic resistance pattern of VV2018 is shown in Table 3. This isolate was susceptible to most tested antibiotics, including tetracycline and chloramphenicol, with the exception of cefazolin. The fact that VV2018 showed susceptibility to tetracycline may be the result of acetylation-mediated down-regulation of tetA gene (Pang et al., 2020). The study also reported that all of Vibrio cholerae strains harboring catB9 gene were susceptible to chloramphenicol (Lepuschitz et al., 2019). Further work needs to be done to study catB9 in Vibrio. V. vulnificus has been reported to show complete resistance against cefazolin (Pan et al., 2013). The varG gene has been shown to have beta-lactamase activity against penicillin, carbapenems, and cephalosporins in vitro (Lin et al., 2017), which might account for the cefazolin resistance of VV2018.
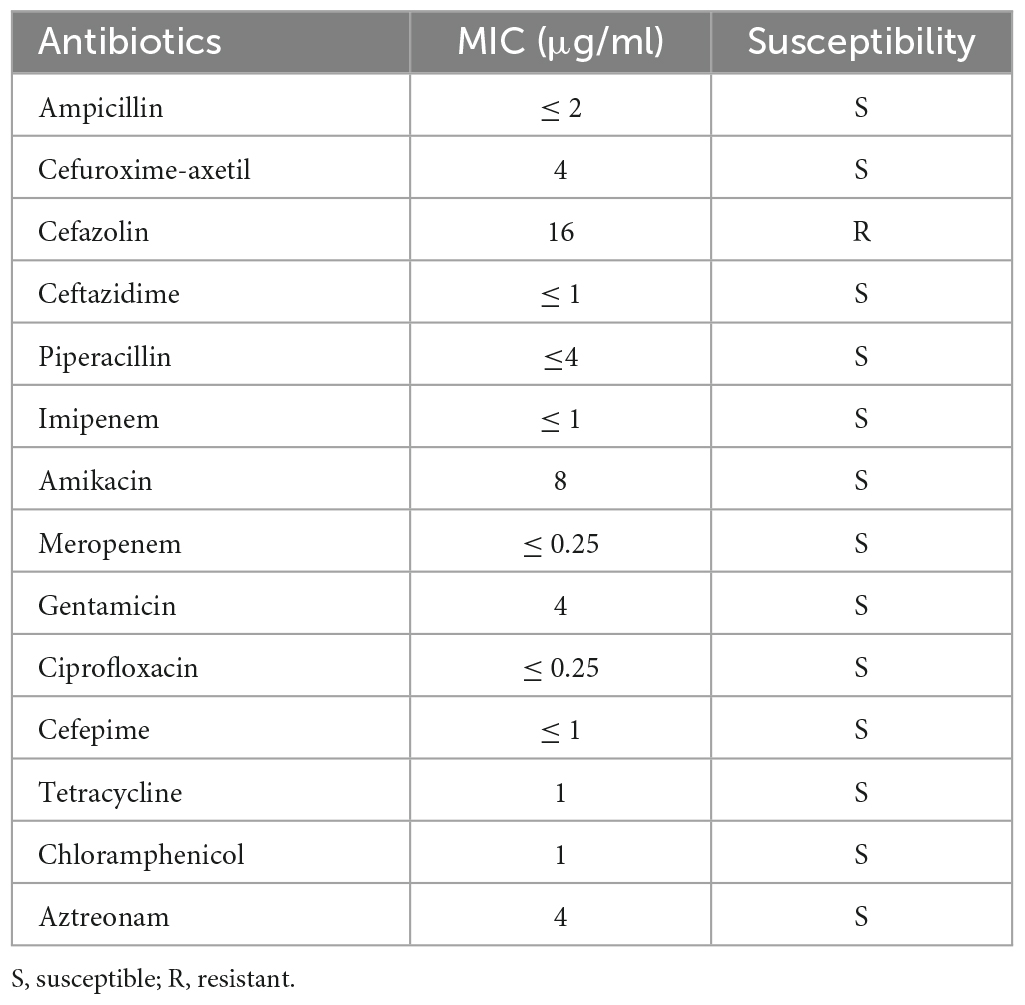
Table 3. The antibiotic resistance profile of VV2018.
Genomic islands, prophages and CRISPR-Cas systems
Large parts of the genome designated as genomic islands (GIs) and phages were transferred from one bacterium to another (Canchaya et al., 2003; Dobrindt et al., 2004). Twenty GIs and nine GIs were detected on Chr I and Chr II in VV2018, respectively (Supplementary Tables 2, 3). The length of GIs on Chr I ranged from 4 kbp to 163 kbp. In the GIs of Ch I, a total of 6 transposase genes were predicted, all of which were classified into the IS4, IS481 and IS5 families. Meanwhile, one integrases (intS) and two tyrosine recombinases (xerC and xerD) were encoded. Two genes were predicted to encode type I restriction enzyme proteins. The length of GIs on Chr II ranged from 4 kbp to 86 kbp, containing one tyrosine recombinase-encoding genes (xerC). Meanwhile, sulfate permease genes (cysTWA) which allowed the bacteria survive in selenite environment by decreasing the expression, were found on Chr II_GI2 (Tempel et al., 2022). Only one incomplete prophage sequence was predicted on Chr I with a length of 9.7 kbp encoding genes with unknown functions (Supplementary Table 4). Thus, further work needs to be done to investigate the functions of these genes.
One CRISPR locus was predicted without Cas genes on Chr I (Supplementary Table 5), the phenomenon that CRISPR locus without Cas genes was also found in other Vibrio strains, Listeria monocytogenes and Staphylococcus, indicating that it was unable to effectively exert adaptive immunity (Mandin et al., 2007; Zhang et al., 2019, 2021). There were four direct repeats with a length of 32 bp and three spacers. The sequences of spacers closely matched other V. vulnificus strains in the NCBI database.
Comparative genome analysis of VV2018
The ANI and dDDH values of 26 V. vulnificus strains are summarized in Supplementary Table 6. The ANI value between VV2018 and other V. vulnificus strains was 97.14% (range 95.41 to 98.45%) (Figure 2A). The most similar strain compared to VV2018 was V. vulnificus FORC_017 (98.45% identity) isolated from human in South Korea. The heatmap showed that all 26 V. vulnificus strains were divided into two clusters, most strains including VV2018 in cluster 1 were isolated from human, and most strains in cluster 2 were isolated from seafood or unknown places. Meanwhile, the phylogenetic tree of all 26 V. vulnificus strains constructed on the basis of the core SNPs showed that VV2018 was grouped with two other V. vulnificus strains isolated from human (V. vulnificus FORC_009 and V. vulnificus FORC_016) (Figure 2B). The dDDH values among 26 V. vulnificus strains were more than 60%. An ANI cut-off of around 95% did not correspond to an absolute dDDH value (70% cut-off for dDDH). A previous study reported that the value of 70% dDDH could not be used as absolute boundary, but still a gap between 60 and 70% similarity seemed to embrace clear-cut clusters of organisms, given the large extent of diversity among prokaryotes (Richter and Rosselló-Móra, 2009).
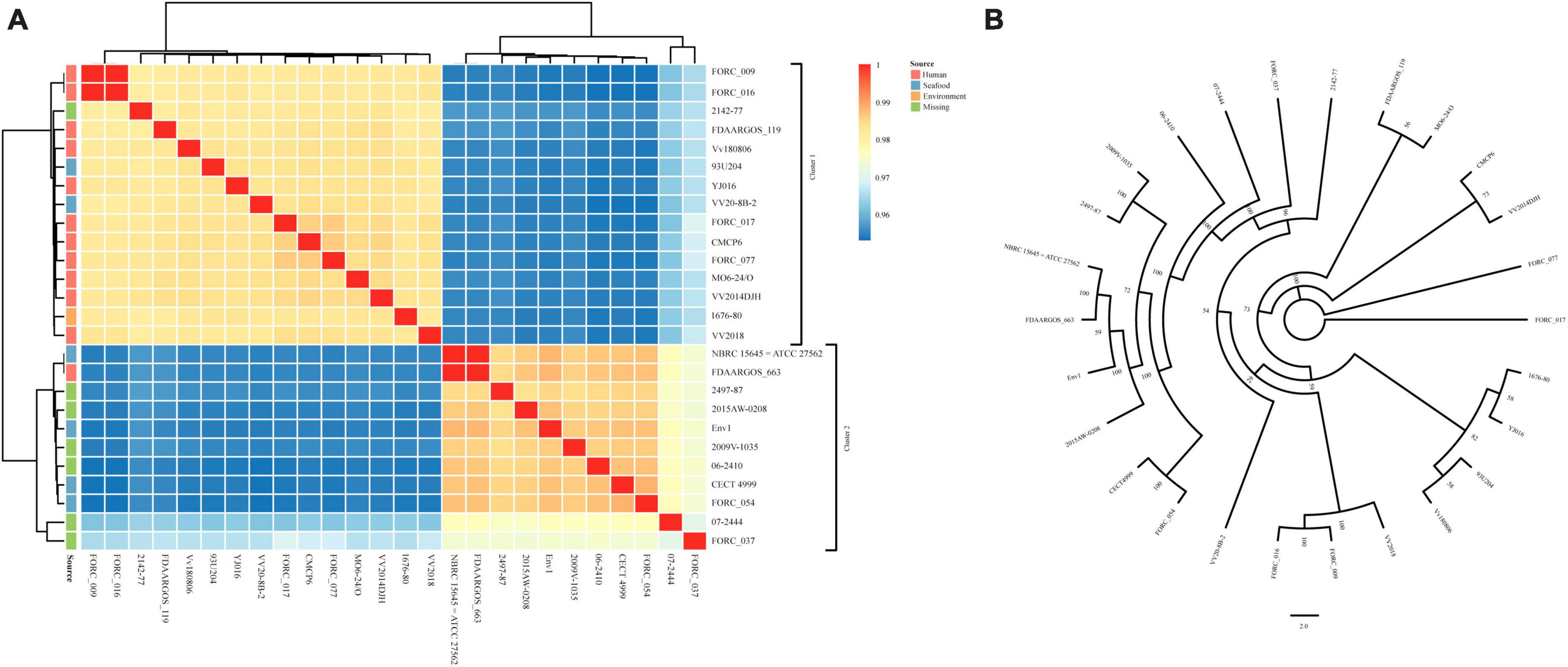
Figure 2. Comparative analysis between VV2018 and 25 other V. vulnificus strains. (A) Heatmap of ANI of 26 V. vulnificus strains. Blue color represents low identity and red color represents high identity. (B) An unrooted maximum-likelihood phylogeny tree of VV2018 with 25 other V. vulnificus strains based on core genome SNPs.
VV2018 shared a total of 3,016 core genes and 314 soft core genes with other 25 V. vulnificus strains according to the pan-genome analysis. A total of 138 strain-specific genes, accounting for 3.14%, were identified in VV2018. The functions of the majority of VV2018 specific genes (78.26%) were unknown, the other specific genes were involved in functional categories of replication/recombination/repair (7.97%) and cell wall/membrane/envelope biogenesis (4.36%) (Supplementary Table 7).
A total of 180 specific VFs were identified in 26 V. vulnificus strains and 115 VFs were included in core genes. The heatmap based on the presence and absence of all VFs clearly showed that the distribution of virulence genes differed between V. vulnificus strains (Figure 3A), and the VFs in VV2018 were similar to those in V. vulnificus 07-2444. Meanwhile, the differences in VFs between strains were among adherence and the immune system. In addition, some V. vulnificus strains isolated from humans were closely clustered with those isolated from seafood, indicating that these strains may cause foodborne infection.
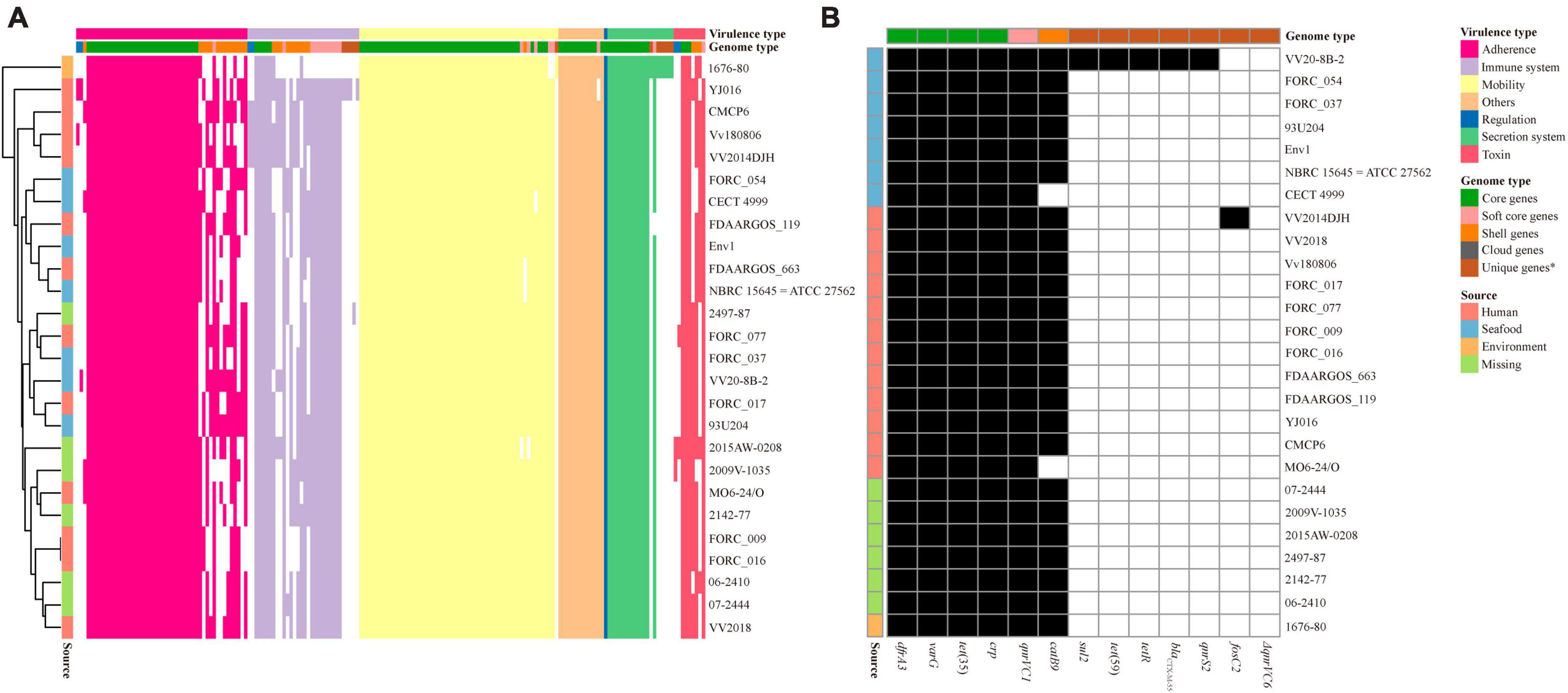
Figure 3. Presence/absence pattern of VFs and resistance-related genes in each V. vulnificus genome. (A) Presence/absence pattern of VFs in each V. vulnificus genome. (B) Presence/absence pattern of resistance-related genes in each V. vulnificus genome. *Unique genes were in only one strain included in cloud genes.
Due to the misuse of antibiotics, V. vulnificus in seafood and aquatic environments are exhibiting resistance to multiple antibiotics (Elmahdi et al., 2016). V. vulnificus resistance toward common antibiotics has reached alarming levels in many countries which has serious implications for the treatment methods for bacterial infections (Heng et al., 2017). The distribution of antibiotic resistance genes in the V. vulnificus strains was also investigated. All 26 V. vulnificus strains possessed the resistance-related genes dfrA3, varG, tet (Bertelli et al., 2017), qnrVC1 and crp (Figure 3B). The prevalence of dfrA3, varG, tet (Bertelli et al., 2017), qnrVC1, and crp genes in these strains suggested that these genes may increase the resistance of these strains to trimethoprim, penicillin, tetracycline, quinolone and oxacillin. The phenicol resistance gene catB9 were also in most genomes of V. vulnificus strains, except V. vulnificus CECT 4999 and V. vulnificus MO6-24/O. In addition, among these strains, V. vulnificus VV2014DJH carried the fosfomycin resistance gene fosC2. Moreover, V. vulnificus VV20-8B-2 isolated from seafood possessed the most antibiotic resistance genes than other V. vulnificus strains, indicating that its antibiotic resistance may be more extensive.
The genetic context of the varG gene
The genetic context of the resistance-related genes of VV2018 was almost the same compared with other 25 V. vulnificus strains, except the varG gene. The gene varG might account for the cefazolin resistance of VV2018, and showed resistance to penicillin, carbapenems, and cephalosporins in vitro (Lin et al., 2017), however, the genetic context of varG was unknown in V. vulnificus strains. The varG gene was present in all 26 V. vulnificus strains, and the sequences containing varG were clustered into eight clusters with coverage of 90% and identity of 80% (Supplementary Table 8). The largest cluster was cluster 3 containing 12 sequences, and most of them (8/12, 75.0%) were isolated from humans including VV2018. Eight representative sequences were chosen for further analysis (Figure 4). The results of this gene neighborhood analysis of representative sequences revealed that a few transposase genes (IS30, IS5 and IS110) were upstream and downstream of varG. The genetic context of varG was conserved and the genes upstream and down of varG were ompV and nodD that were present in 96.15% of the sequences. These observations indicated that sequence rearrangement rarely occurs in the varG-encoding region, and genetic commonalities of ompV-varG-nodD across sources strongly suggested the structure of ompV-varG-nodD was conserved in V. vulnificus strains.
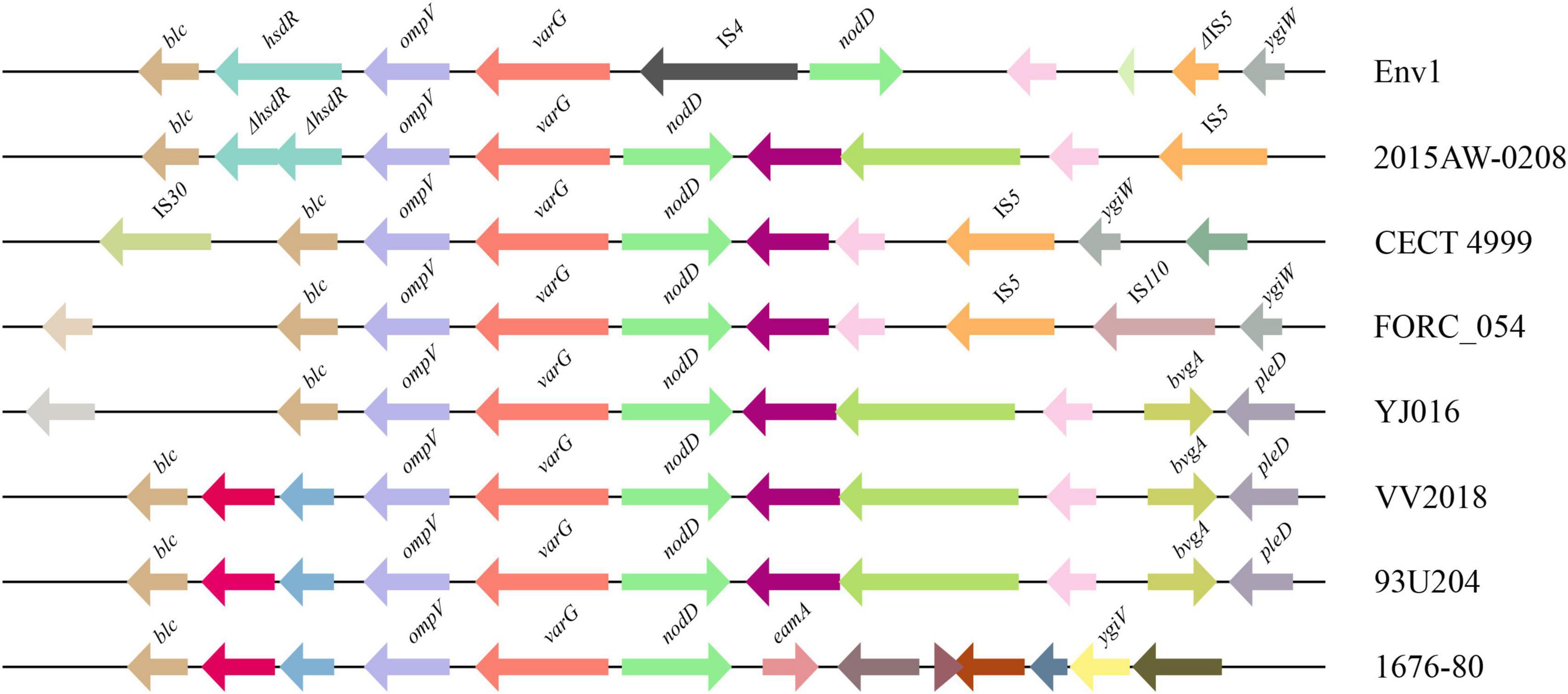
Figure 4. Comparative analysis of the varG-related regions of eight representatives from 26 sequences. The direction of genes is indicated by an arrow. Homologous genes are shown in the same colors.
Molecular characterization and comparative analysis of pgl-like genes
Two (LNNJENCE_00235 and LNNJENCE_00237) of six genes on Chr I_GI4 were glycosyl transferases, belonging to the GT4 family (Supplementary Table 9). LNNJENCE_00237 was compared with PglA in Neisseria including N. elongata subsp. glycolytica, with 37% identity. Meanwhile, four genes downstream of LNNJENCE_00237 also shared a high identity compared with Pgl-related proteins in Neisseria such as N. elongata subsp. glycolytica ATCC29315 (Table 4 and Figure 5). Broad-spectrum O-linked protein glycosylation (pgl) systems have been defined in Neisseria, such as N. gonorrhoeae, N. meningitidis, and N. elongata subspecies glycolytica (Ku et al., 2009; Vik et al., 2009; Naess et al., 2023). The pglA and pglBCD may be involved in pilin glycosylation (Power et al., 2003).
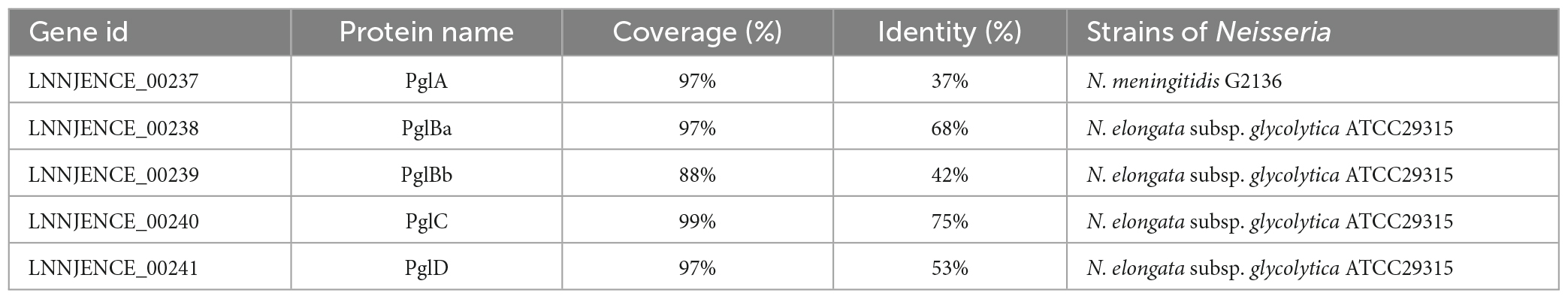
Table 4. The result of BLASTP of Pgl in VV2018 against Pgl in Neisseria.
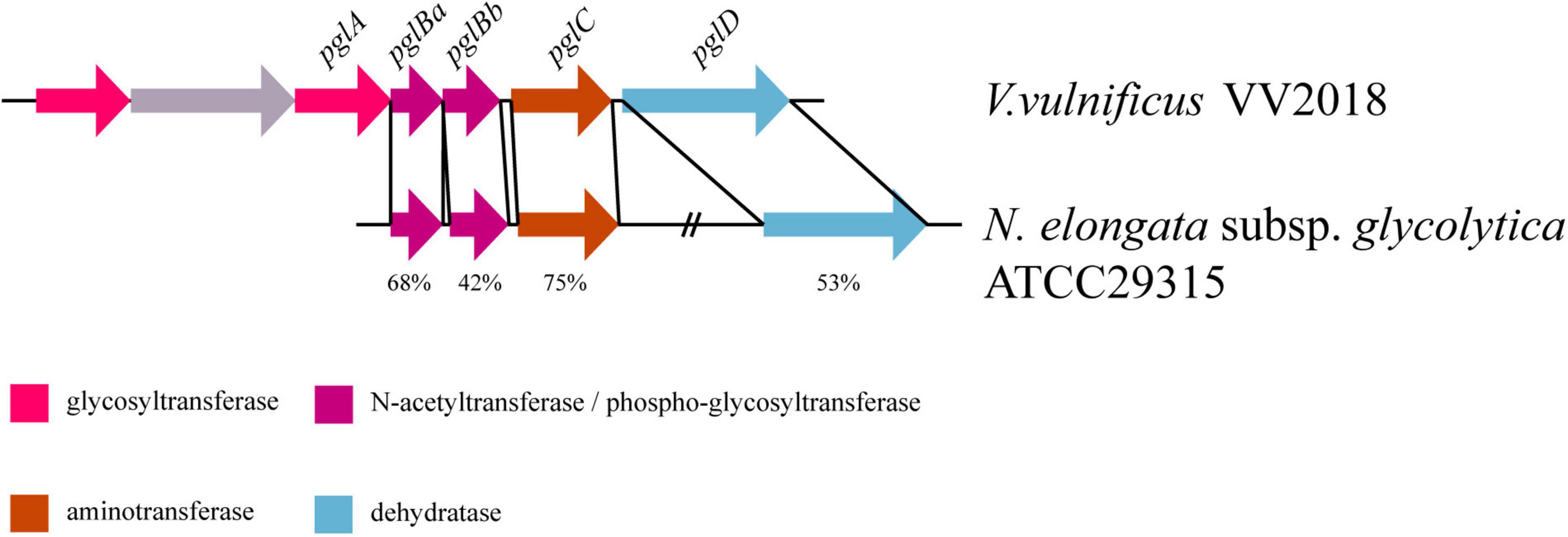
Figure 5. Comparative pgl gene content and synteny between VV2018 and N. elongata subsp. glycolytica strain ATCC29315. The direction of genes is indicated by an arrow. Homologous genes are shown in the same colors.
We selected six sequences of Neisseria for further analysis and the result of the multiple sequence alignment showed that the sequences of PglC and PglD were highly conserved (Figure 6A and Supplementary Figure 3A). The phylogenetic tree of PglC and PglD showed that the PglC of VV2018 was closest to the protein of N. weaveri LMG 5135, and PglD of VV2018 was closest to N. bacilliformis ATCC BAA-1200 and N. elongata subsp. glycolytica ATCC29315 (Figure 6B and Supplementary Figure 3B). Meanwhile, comparative genomic analysis revealed that the sequence of VV2018 containing pgl-like genes was similar to the sequences of V. vulnificus 07-2444, V. vulnificus YJ016 and V. vulnificus FORC_017 (Figure 7A). The result showed that the pglA gene was only found in V. vulnificus 07-2444 with coverage of 76% and identity of 85%. The other genes pglBa, pglBb, pglC, and pglD also had high homology with identity ranging from 90 to 100%. In addition, in the genome of V. parahaemolyticus, the pglB2 gene is associated with a pglC and pglD homolog, suggesting that a complete glycosylation system might also be present (Chamot-Rooke et al., 2007). In V. cholerae, O-glycosylation via PglLVc and possibly RbmD could represent a fine-tuned feedback mechanism controlling release of type II secretion system (T2SS) effectors by modulation of secretion efficacy (Vorkapic et al., 2019). Therefore, pgl-like genes (pglABCD) may play important roles in Vibrio species.

Figure 6. Comprehensive comparisons of PglC by sequence similarity and phylogenetic analysis. (A) Multisequence alignment of PglC with amino acids. Multisequence alignment was conducted using ClustalW. (B) A neighbor-joining phylogenetic tree of PglC was estimated by MEGA, and the sequence of Morococcus cerebrosus (M. cerebrosus) CIP 81.93 was used as outgroup. *The markers of length such as 10, 30, that are provided by software.
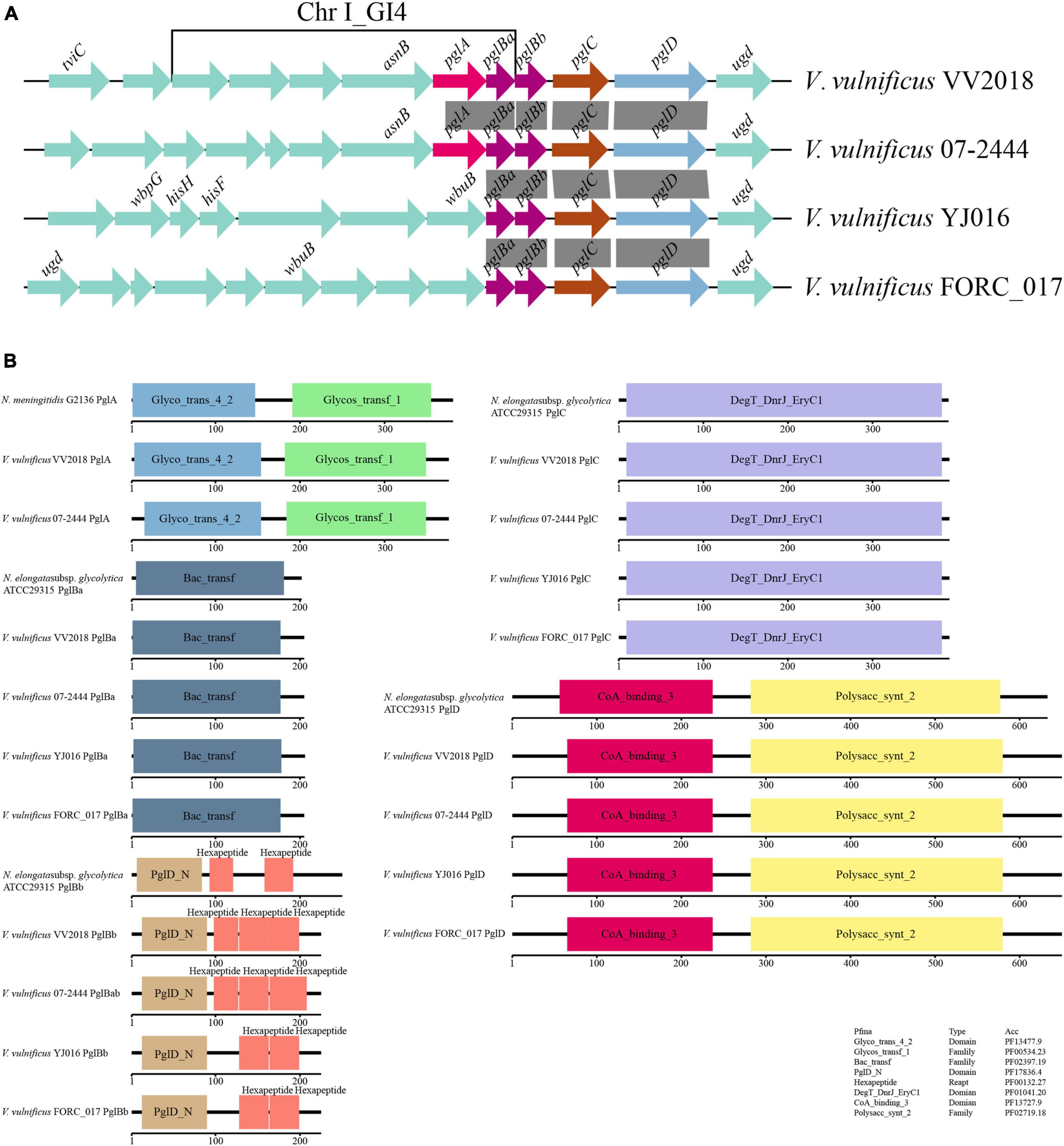
Figure 7. Comparative genomic analysis of pgl-like genes. (A) Comparative genomic analysis of the genetic context of pgl genes in VV2018 with the sequences carrying their homologous genes in three other V. vulnificus strains. The direction of genes is indicated by an arrow. Homologous pgl genes are shown in the same colors, syntenic regions between the sequences are displayed as gray blocks. All other genes are shown in green. (B) Schematic diagram depicting the total domain structure of PglA, PglBa, PglBb, PglC and PglD. Each scaled box denotes a functional module labeled with the term short name.
The total domain structures of PglA, PglBa, PglBb, PglC and PglD were also analyzed and compared with protein sequences in Neisseria. The domains of PglA, PglBa, PglC and PglD were similar, except for PglBb (Figure 7B). The PglD_N domain was found in the N-terminus of all PglBb sequences, besides the number and location of hexapeptides. There were three hexapeptides in PglBb of VV2018 and V. vulnificus 07-2444. The pglBb of V. vulnificus YJ016 and V. vulnificus FORC_017 had two hexapeptides in the second and third position. The pglBb of N. elongata subsp. glycolytica strain ATCC29315 had two hexapeptides in the first and third position. A number of different transferase protein families contain hexapeptide repeats, such as galactoside acetyltransferase-like proteins (Wang et al., 2002). It has been shown that most hexapeptide acyltransferases form catalytic trimers with three symmetrical active sites (Bergfeld et al., 2007). This is the first study to report that pglA and pglBCD genes were found in V. vulnificus strains. The pgl-like genes may affect the adherence of the strain, however, much more work is need to prove this hypothesis.
Conclusion
In this study, we investigated the genomic features of VV2018 with novel strain ST620 that was isolated from the blood culture specimen of a clinical patient with hepatitis B virus cirrhosis in China. Multiple VFs and resistance genes were identified in the genome sequence of VV2018. Pan-genome analysis of 26 V. vulnificus strains revealed their pan-genome characteristics, evolutionary relationships, and virulence and antibiotic resistance profiles. This study provides a snapshot of the genomic diversity and evolution of different strains that contribute to the pathogenic diversity of 26 V. vulnificus strains. We also found that the resistance gene varG was present in all 26 V. vulnificus strains and the genes upstream and downstream were conserved. In addition, it’s the first to report the presence of pgl-like genes in V. vulnificus based on amino acid sequence homologies with genes in Neisseria. The pgl-like genes may affect the adherence of the strain in hosts, and much work still needs to be done to confirm this theory.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in this article/Supplementary material.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Affiliated Nantong Hospital 3 of Nantong University. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
TZ, QW, XuL, and XiL collected the strains and performed the experiments. MZ and TZ analyzed the experimental results. FW performed the bioinformatic analysis. FW, YZ, and RL co-led the writing of the manuscript. YZ and RL designed the work. All authors read and approved the final manuscript.
Funding
This study was supported by the Nantong Commission of Health Science Foundation for Youths (QA2020026).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1240835/full#supplementary-material
Footnotes
References
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Baker-Austin, C., and Oliver, J. D. (2020). Vibrio vulnificus. Trends Microbiol. 28, 81–82. doi: 10.1016/j.tim.2019.08.006
Bergfeld, A. K., Claus, H., Vogel, U., and Mühlenhoff, M. (2007). Biochemical characterization of the polysialic acid-specific O-acetyltransferase NeuO of Escherichia coli K1. J. Biol. Chem. 282, 22217–22227. doi: 10.1074/jbc.M703044200
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Buchfink, B., Reuter, K., and Drost, H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Canchaya, C., Fournous, G., Chibani-Chennoufi, S., Dillmann, M., and Brüssow, H. (2003). Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 6, 417–424. doi: 10.1016/S1369-5274(03)00086-9
Chamot-Rooke, J., Rousseau, B., Lanternier, F., Mikaty, G., Mairey, E., Malosse, C., et al. (2007). Alternative Neisseria spp. type IV pilin glycosylation with a glyceramido acetamido trideoxyhexose residue. Proc. Natl. Acad. Sci. U. S. A. 104, 14783–14788. doi: 10.1073/pnas.0705335104
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15. doi: 10.1093/nar/gku1196
Dittmann, K. K., Porsby, C. H., Goncalves, P., Mateiu, R. V., Sonnenschein, E. C., Bentzon-Tilia, M., et al. (2019). Tropodithietic acid induces oxidative stress response, cell envelope biogenesis and iron uptake in Vibrio vulnificus. Environ. Microbiol. Rep. 11, 581–588. doi: 10.1111/1758-2229.12771
Dobrindt, U., Hochhut, B., Hentschel, U., and Hacker, J. (2004). Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2, 414–424. doi: 10.1038/nrmicro884
Elmahdi, S., DaSilva, L. V., and Parveen, S. (2016). Antibiotic resistance of Vibrio parahaemolyticus and Vibrio vulnificus in various countries: A review. Food Microbiol. 57, 128–134. doi: 10.1016/j.fm.2016.02.008
Galperin, M. Y., Wolf, Y. I., Makarova, K. S., Vera Alvarez, R., Landsman, D., and Koonin, E. V. (2021). COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 49, D274–D281. doi: 10.1093/nar/gkaa1018
Goo, S. Y., Lee, H. J., Kim, W. H., Han, K. L., Park, D. K., Lee, H. J., et al. (2006). Identification of OmpU of Vibrio vulnificus as a fibronectin-binding protein and its role in bacterial pathogenesis. Infect. Immun. 74, 5586–5594. doi: 10.1128/IAI.00171-06
Gulig, P. A., Bourdage, K. L., and Starks, A. M. (2005). Molecular pathogenesis of Vibrio vulnificus. J. Microbiol. 43, 118–131.
Haftel, A., and Sharman, T. (2023). Vibrio Vulnificus. StatPearls. Treasure Island (FL): StatPearls Publishing.
Heng, S. P., Letchumanan, V., Deng, C. Y., Ab Mutalib, N. S., Khan, T. M., Chuah, L. H., et al. (2017). Vibrio vulnificus: An environmental and clinical burden. Front. Microbiol. 8:997. doi: 10.3389/fmicb.2017.00997
Horseman, M. A., and Surani, S. (2011). A comprehensive review of Vibrio vulnificus: an important cause of severe sepsis and skin and soft-tissue infection. Int. J. Infect. Dis. 15, e157–e166. doi: 10.1016/j.ijid.2010.11.003
Iranzadeh, A., and Mulder, N. J. (2019). “Bacterial Pan-Genomics,” in Microbial Genomics in Sustainable Agroecosystems: Volume 1, eds V. Tripathi, P. Kumar, P. Tripathi, and A. Kishore (Singapore: Springer Singapore), 21–38.
Jones, J. L., Lüdeke, C. H., Bowers, J. C., DeRosia-Banick, K., Carey, D. H., and Hastback, W. (2014). Abundance of Vibrio cholerae, V. vulnificus, and V. parahaemolyticus in oysters (Crassostrea virginica) and clams (Mercenaria mercenaria) from Long Island sound. Appl. Environ. Microbiol. 80, 7667–7672. doi: 10.1128/AEM.02820-14
Jones, M. K., and Oliver, J. D. (2009). Vibrio vulnificus: disease and pathogenesis. Infect. Immun. 77, 1723–1733. doi: 10.1128/IAI.01046-08
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Ku, S. C., Schulz, B. L., Power, P. M., and Jennings, M. P. (2009). The pilin O-glycosylation pathway of pathogenic Neisseria is a general system that glycosylates AniA, an outer membrane nitrite reductase. Biochem. Biophys. Res. Commun. 378, 84–89. doi: 10.1016/j.bbrc.2008.11.025
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lapierre, P., and Gogarten, J. P. (2009). Estimating the size of the bacterial pan-genome. Trends Genet. 25, 107–110. doi: 10.1016/j.tig.2008.12.004
Lepuschitz, S., Baron, S., Larvor, E., Granier, S., Pretzer, C., Mach, R., et al. (2019). Phenotypic and genotypic antimicrobial resistance traits of Vibrio cholerae Non-O1/Non-O139 isolated from a large austrian lake frequently associated with cases of human infection. Front. Microbiol. 10:2600. doi: 10.3389/fmicb.2019.02600
Levasseur, A., Drula, E., Lombard, V., Coutinho, P., and Henrissat, B. (2013). Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41. doi: 10.1186/1754-6834-6-41
Li, W., and Godzik, A. (2006). Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Lin, H. V., Massam-Wu, T., Lin, C. P., Wang, Y. A., Shen, Y. C., Lu, W. J., et al. (2017). The Vibrio cholerae var regulon encodes a metallo-β-lactamase and an antibiotic efflux pump, which are regulated by VarR, a LysR-type transcription factor. PLoS One 12:e0184255. doi: 10.1371/journal.pone.0184255
Liu, B., and Pop, M. (2009). ARDB–antibiotic resistance genes database. Nucleic Acids Res. 37, D443–D447. doi: 10.1093/nar/gkn656
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Liu, M., and Crosa, J. H. (2012). The regulator HlyU, the repeat-in-toxin gene rtxA1, and their roles in the pathogenesis of Vibrio vulnificus infections. Microbiologyopen 1, 502–513.
López-Pérez, M., Jayakumar, J. M., Haro-Moreno, J. M., Zaragoza-Solas, A., Reddi, G., Rodriguez-Valera, F., et al. (2019). Evolutionary model of cluster divergence of the emergent marine pathogen vibrio vulnificus: From genotype to ecotype. mBio 10, e2852–e2818. doi: 10.1128/mBio.02852-18
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Mandin, P., Repoila, F., Vergassola, M., Geissmann, T., and Cossart, P. (2007). Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res. 35, 962–974. doi: 10.1093/nar/gkl1096
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594.
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., and Göker, M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Menon, M. P., Yu, P. A., Iwamoto, M., and Painter, J. (2014). Pre-existing medical conditions associated with Vibrio vulnificus septicaemia. Epidemiol. Infect. 142, 878–881.
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G., Sonnhammer, E. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419.
Naess, L. M., Maugesten, I. S., Caugant, D. A., Kassu, A., Aseffa, A., and Børud, B. (2023). Genetic, functional, and immunogenic analyses of the o-linked protein glycosylation system in Neisseria meningitidis serogroup A ST-7 isolates. J. Bacteriol. 205:e0045822. doi: 10.1128/jb.00458-22
Nicholas, K. B., and Nicholas, H. B. (eds) (1997). GeneDoc: a tool for editing and annotating multiple sequence alignments. Berlin: ScienceOpen.
Nishino, K., Senda, Y., and Yamaguchi, A. (2008). CRP regulator modulates multidrug resistance of Escherichia coli by repressing the mdtEF multidrug efflux genes. J. Antibiot. 61, 120–127. doi: 10.1038/ja.2008.120
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pan, J., Zhang, Y., Jin, D., Ding, G., Luo, Y., Zhang, J., et al. (2013). Molecular characterization and antibiotic susceptibility of Vibrio vulnificus in retail shrimps in Hangzhou, People’s Republic of China. J. Food Prot. 76, 2063–2068. doi: 10.4315/0362-028X.JFP-13-161
Pang, R., Li, Y., Liao, K., Guo, P., Li, Y., Yang, X., et al. (2020). Genome- and proteome-wide analysis of lysine acetylation in vibrio vulnificus Vv180806 reveals its regulatory roles in virulence and antibiotic resistance. Front. Microbiol. 11:591287. doi: 10.3389/fmicb.2020.591287
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Paz, S., Bisharat, N., Paz, E., Kidar, O., and Cohen, D. (2007). Climate change and the emergence of Vibrio vulnificus disease in Israel. Environ. Res. 103, 390–396. doi: 10.1016/j.envres.2006.07.002
Petkau, A., Stuart-Edwards, M., Stothard, P., and Van Domselaar, G. (2010). Interactive microbial genome visualization with GView. Bioinformatics 26, 3125–3126. doi: 10.1093/bioinformatics/btq588
Power, P. M., Roddam, L. F., Rutter, K., Fitzpatrick, S. Z., Srikhanta, Y. N., and Jennings, M. P. (2003). Genetic characterization of pilin glycosylation and phase variation in Neisseria meningitidis. Mol. Microbiol. 49, 833–847. doi: 10.1046/j.1365-2958.2003.03602.x
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Rosche, T. M., Binder, E. A., and Oliver, J. D. (2010). Vibrio vulnificus genome suggests two distinct ecotypes. Environ. Microbiol. Rep. 2, 128–132.
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36.
Song, E. J., Lee, S. J., Lim, H. S., Kim, J. S., Jang, K. K., Choi, S. H., et al. (2016). Vibrio vulnificus VvhA induces autophagy-related cell death through the lipid raft-dependent c-Src/NOX signaling pathway. Sci. Rep. 6:27080. doi: 10.1038/srep27080
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Strom, M. S., and Paranjpye, R. N. (2000). Epidemiology and pathogenesis of Vibrio vulnificus. Microbes Infect. 2, 177–188.
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tempel, S., Bedo, J., and Talla, E. (2022). From a large-scale genomic analysis of insertion sequences to insights into their regulatory roles in prokaryotes. BMC Genomics 23:451. doi: 10.1186/s12864-022-08678-3
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U. S. A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
UniProt Consortium (2023). UniProt: The universal protein knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531.
Vik, A., Aas, F. E., Anonsen, J. H., Bilsborough, S., Schneider, A., Egge-Jacobsen, W., et al. (2009). Broad spectrum O-linked protein glycosylation in the human pathogen Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. U. S. A. 106, 4447–4452. doi: 10.1073/pnas.0809504106
Vorkapic, D., Mitterer, F., Pressler, K., Leitner, D. R., Anonsen, J., Liesinger, L., et al. (2019). A broad spectrum protein glycosylation system influences type II protein secretion and associated phenotypes in Vibrio cholerae. Front. Microbiol. 10:2780. doi: 10.3389/fmicb.2019.02780
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, R., Zhong, Y., Gu, X., Yuan, J., Saeed, A., and Wang, S. (2015). The pathogenesis, detection, and prevention of Vibrio parahaemolyticus. Front. Microbiol. 6:144. doi: 10.3389/fmicb.2015.00144
Wang, X. G., Olsen, L. R., and Roderick, S. L. (2002). Structure of the lac operon galactoside acetyltransferase. Structure 10, 581–588.
Wu, Q., Wu, Y., Zhang, T., Wu, F., Zhang, Y., and Lu, R. (2023). Vibrio vulnificus septicemia in a hospitalized patient with hepatitis B virus-associated cirrhosis: A case report. Heliyon 9:e18905. doi: 10.1016/j.heliyon.2023.e18905
Zhan, L., Han, Y., Yang, L., Geng, J., Li, Y., Gao, H., et al. (2008). The cyclic AMP receptor protein, CRP, is required for both virulence and expression of the minimal CRP regulon in Yersinia pestis biovar microtus. Infect. Immun. 76, 5028–5037. doi: 10.1128/IAI.00370-08
Zhang, E., Zhou, W., Zhou, J., He, Z., Zhou, Y., Han, J., et al. (2021). CRISPR-Cas systems are present predominantly on chromosome and its relationship with MEGs in Vibrio species. Arch. Microbiol. 204:76. doi: 10.1007/s00203-021-02656-1
Keywords: V. vulnificus, pan-genome, virulence, resistance, pgl
Citation: Wu F, Zhang T, Wu Q, Li X, Zhang M, Luo X, Zhang Y and Lu R (2023) Complete genome sequence and comparative analysis of a Vibrio vulnificus strain isolated from a clinical patient. Front. Microbiol. 14:1240835. doi: 10.3389/fmicb.2023.1240835
Received: 15 June 2023; Accepted: 17 October 2023;
Published: 31 October 2023.
Edited by:
Ludmila Chistoserdova, University of Washington, United StatesReviewed by:
Fabini Orata, Bio-Conversion Databank Foundation, CanadaSatyabrata Bag, 3B BlackBio Biotech India Ltd., India
Copyright © 2023 Wu, Zhang, Wu, Li, Zhang, Luo, Zhang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yiquan Zhang, emhhbmd5aXF1YW5xQDE2My5jb20=; Renfei Lu, cmFpbm1hbjc4QDE2My5jb20=
†These authors have contributed equally to this work