 Anna Marciniak1*
Anna Marciniak1* Marzena Skrzypczak-Zielińska2
Marzena Skrzypczak-Zielińska2 Oliwia Zakerska-Banaszak2
Oliwia Zakerska-Banaszak2 Elżbieta Nowakowska3Anna Kozaczka4Brunon Zemła5Andrzej Szpak6Dariusz Godlewski1Jadwiga Charzewska7
Elżbieta Nowakowska3Anna Kozaczka4Brunon Zemła5Andrzej Szpak6Dariusz Godlewski1Jadwiga Charzewska7 Dorothy R. Pathak8
Dorothy R. Pathak8- 1Center of Cancer Prevention and Epidemiology OPEN, Poznan, Poland
- 2Institute of Human Genetics, Polish Academy of Sciences, Poznan, Poland
- 3Affidea International Cancer Center Poznań, Poznan, Poland
- 4Department of Nephrology, Transplantation and Internal Medicine, Medical University of Silesia, Katowice, Poland
- 5Department of Nuclear Medicine and Endocrine Oncology, Maria Skłodowska-Curie Memorial Cancer Centre and Institute of Oncology, Gliwice, Poland
- 6Witold Chodźko Institute of Rural Medicine, Lublin, Poland
- 7Department of Nutrition and Nutritional Value of Food, National Institute of Public Health NIH-National Research Institute, Warsaw, Poland
- 8Department of Epidemiology and Biostatistics, Michigan State University, East Lansing, MI, United States
Introduction: The human microbiota can be a critical component in the development and progression of various diseases, including cancer. This study aims to investigate the composition of the urinary and oral microbiota in Polish breast cancer (BC) patients relative to healthy controls (HCs) and to predict relevant metabolic pathways of microbiota in studied groups.
Methods: Urine and oral samples from 48 participants, 24 BC cases and 24 HCs, randomly selected from 417 BC cases and 514 HCs, were analyzed using next-generation sequencing of bacterial 16S rRNA gene (V1-V9) and fungal ITS regions, along with bioinformatics tools to identify and compare microbial communities and predict relevant pathways of microbiota in the studied groups.
Results: BC case urine microbiota contained an increased abundance of Corynebacterium (5.2-fold, but not significant) and Gammaproteobacteria including unknown genus and Pseudomonas (1.7- and 1.8-fold) and decreased abundance of Family XI (0.3-fold) and Bifidobacteriaceae (0.4-fold) compared to HCs. Oral BC microbiota contains higher levels of the bacterial families P5D1–392, Leptotrichiaceae, and Pasteurellaceae (3.3-, 3.3-, and 1.9-fold, respectively), whereas the genera Cellulosimicrobium, Pseudomonas, and Pantoea were significantly less abundant (0.4-, 0.3-, and 0.3-fold, respectively). At the species level, the most differentiating species between BC and HC was uncultured Pseudomonas sp. (1.8-fold) in urine and Pantoea agglomerans (0.2-fold) in oral microbiota. Fungal composition did not show any significant differences between the groups. Functional analysis based on Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt2) predicted, e.g. enhanced hydrogen production and benzoyl-CoA degradation in BC cases, as well as reduced CMP-diacetamido-8-epilegionaminic acid biosynthesis.
Discussion: The study underscores the potential significance of the microbiota in BC pathogenesis. Further research is needed to elucidate the mechanisms underlying microbiota–tumor interactions and to explore the clinical applications.
Graphical Abstract. Skrzypczak-zielinska, M. (2025) (https://BioRender.com/o59f811).
Introduction
Based on the World Health Organization (WHO) data, breast cancer (BC) is the most frequently diagnosed cancer in the world. In 2020, 2.3 million people were diagnosed with BC and 685,000 cases died from this disorder (Sung et al., 2021). It is estimated that by 2040, the number of newly diagnosed BC worldwide will increase by 40% and the number of deaths will escalate by 50% (Arnold et al., 2022). In Poland, the incidence and mortality of BC are a significant health problem. In 2020 represented 23% of all cancer cases, and was the second cause of cancer-related deaths among Polish women (Wojciechowska et al., 2023).
The human body is inhabited by a microbiota consisting mainly of bacteria, fungi, archaea, and viruses (Proctor et al., 2019). The composition and role of the intestinal microbiota is crucial in maintaining the homeostasis of the host organism. It is involved in a variety of physiological processes, including immune system modulation, metabolic homeostasis, and defense against pathogens, with its influence extending to distant organs and potentially impacting cancer development (Álvarez-Mercado et al., 2019, 2023; Vivarelli et al., 2019). In the context of BC, the microbiota may contribute to pathogenesis by regulating steroid hormone levels, energy metabolism, the synthesis of bioactive metabolites (e.g., genotoxins, lipopolysaccharides, and vitamins), and immune response modulation. It has been demonstrated that the bacterial enzyme β-glucuronidase (GUS) participates in estrogen reactivation, which may contribute to BC progression (Al-Ansari et al., 2021). Short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate, may support cellular stability and modulate immune responses, potentially exerting protective effects. In contrast, secondary bile acids exhibit both carcinogenic and anti-carcinogenic properties, depending on the biological context and microbiota composition (Miko et al., 2019). The complex interactions between the microbiota and the human organism suggest that its disturbances, known as dysbiosis, may not only contribute to the development of BC but also influence its progression and treatment efficacy (Viswanathan et al., 2023). Research findings demonstrate that tumor tissues exhibit reduced microbial diversity compared to healthy tissues, along with an increased abundance of bacteria from the Pseudomonadaceae and Enterobacteriaceae families. In contrast, Propionibacterium and Staphylococcus are more frequently found in healthy tissues, suggesting their potential role in maintaining microenvironmental homeostasis (Tzeng et al., 2021).
For BC, several studies evaluated the presence of various bacteria in the human gut, breast tissue, and oral microbiome (Huybrechts et al., 2020; Samkari et al., 2022; Thu et al., 2023) with inconsistent findings. While Wang et al. (2017). and Nearing et al. (2020) found no differences in the distribution of oral microbiome between BC cases and healthy subjects, Wu et al. (2022) observed a lower abundance of Porphyromonas and Fusobacterium genus in BC cases.
The urinary microbiota has been less studied; however, recent studies showed its association with oncological disease (Wolfe and Brubaker, 2015; Perez-Carrasco et al., 2021), including bladder, colon, and prostate cancer. For BC, Wang et al. demonstrated increased amounts of Gram-positive bacteria, including Corynebacterium, Staphylococcus, Actinomyces and Propionibacteriaceae in the urine microbiota of BC cases (Wang et al., 2017).
Moreover, although fungi constitute a minor part of the microbiota community, their association with carcinogenesis has been established. Therefore, their composition and abundance could be important in BC cases. For example, Candida albicans can support cancer progression (Ramirez-Garcia et al., 2013).
The current research investigated the composition of urinary and oral microbiota in Polish BC patients relative to healthy subjects, marking Poland’s first study of its kind. The findings may help identify potential biomarkers and offer new prevention or treatment options for BC through microbiota intervention.
Materials and methods
Study population
This study is based on a subset of samples collected as part of the Polish Women’s Health Study (PWHS) which was conducted between 2000 and 2006 (Wajszczyk et al., 2021). The biological samples were collected between 2004 and 2006 with participants from five Regional Cancer Registries located in three regions in Poland. The study followed the Helsinki Declaration and received approval from the local Bioethics Committee of the Greater Poland Medical Chamber (resolution no 44/2024 issued on 20 March 2024). BC cases were consecutively included in the study, simultaneously from Regional Registries, starting on 1 January 2000, until the assumed number of cases (200 per register) was exhausted, regardless of the type of breast cancer. Control samples were matched by age and residence. For further details, see Wajszczyk et al. (2021) and Łukasiewicz-Śmietańska et al. (2024). Urine and oral wash samples were collected from 514 healthy women and 417 women with BC under the sub-study “Breast Cancer Gene-Diet Interaction in Polish Women.” This pilot project includes samples from 48 participants—randomly selected 24 women diagnosed with BC and 24 healthy controls equally from each of the five study sites (48 urine samples and 47 oral wash samples) using a simple, dependent sampling method. Participants collected midstream morning urine in 50 mL containers with Vitamin C after fasting for 8 h; menstruating participants were asked to collect morning urine samples between the 20th and 25th day of the cycle. Oral wash samples were collected using 50 mL Falcon tubes and Scope mouthwash after fasting for 1 h. Urine samples and oral washes were transported in a carrier on ice to freezers at study sites, then to Warsaw for storage at −80°C, and finally to Poznań in 2018, where they remain at −80°C.
DNA extraction
Microbial genomic DNA was extracted using the Sherlock AX kit (A&A Biotechnology, Gdansk, Poland) according to the manufacturer’s protocol. After defrosting each urine sample, 4 mL of sterile 1 M TRIS-EDTA buffer was added, mixed, and centrifuged at 3,645 x g at 4°C for 10 min. Next, the supernatant was discarded. The urine pellet at the volume of 300 μL was lysed with 300 μL of lysis buffer, 20 μL of proteinase K (20 mg/mL), 20 μL of lysozyme (10 mg/mL), 5 μL of mutanolysin (10 U/μL), and 10 μL of lyticase (10 U/1 μL). Oral rinses were directly lysed without concentration. The isolated microbial material was quantified and characterized using a spectrophotometer NanoDrop™ 2,000 (Thermo fisher scientific, Inc., Waltham, MA, United States).
Next-generation sequencing (NGS)
Bacterial 16S ribosomal RNA (rRNA) gene libraries including all nine hypervariable V1-V9 region and fungal ITS region libraries for metaprofiling analysis were prepared using the QIAseq 16S/ITS Panels Reagent Kit Screening Panel (QIAGEN) according to the manufacturer’s protocol. The obtained amplification products were combined, mixed, and then cleaned using the magnetic QIAseq Beads (QIAGEN) and subjected to indexing with unique oligonucleotide sequences placed on a commercial plate QIAseq 16S/ITS 96-Index I (QIAGEN). Amplicon libraries were purified, and the following were checked using a High Sensitivity DNA Assay on an Agilent 2,100 Bioanalyzer System (Agilent, Santa Clara, CA, USA) and pooled to one collective NGS library with a final concentration of 8 pm. The bacteriophage Φ-X174 (PhiX) control library was added at 3%. Sequencing was performed on an Illumina MiSeq platform using a MiSeq Reagent Kit v3 (600 cycles).
Bioinformatics
Sequencing reads (paired-end) of the 16S rRNA gene and the ITS1-ITS2 region were analyzed using the Detect Amplicon Sequence Variants algorithm, based on DADA2 (Callahan et al., 2016), in CLC Genomics Workbench v. 24.02 with CLC Microbial Genomics Module v. 24.1.1 (QIAGEN Bioinformatics, Aarhus, Denmark). The Detect Amplicon Sequence Variants analysis included the following steps: (1) initial error filtering (maximum expected errors per read: 1.0) and length trimming (first/s read length: 250 bp for both reads), (2) dereplication, (3) denoising, (4) removal of chimeric sequences, and (5) merging of unique read pairs (with a minimum overlap of <12 bases). Taxonomic annotation of amplicon sequence variant (ASV) abundance tables was performed using the Assign Taxonomies to Sequences in Abundance Table tool, with a minimum similarity threshold of 80%. ASVs were mapped against the following reference databases: SILVA SSU 99% (release 138.1) for bacterial classification and UNITE 97% (version 9.0) for fungal classification. Bioinformatics processing made it possible to obtain and export Excel files with a qualitative–quantitative list of microorganisms on different taxonomic levels: phylum, class, order, family, genus, and species. Alpha diversity was calculated using the ASV richness. Beta diversity was specified based on a Bray–Curtis dissimilarity matrix. To predict the functional potential of the microbial community of urine and oral cavity microbiota in the BC patients and healthy control groups, the PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) tool integrated within CLC Genomics Workbench v. 24.02 with CLC Microbial Genomics Module v. 24.1.1 (QIAGEN Bioinformatics, Aarhus, Denmark) was used. The analysis was performed based on ASV abundances from study samples using the default prediction model. Prior to functional prediction, ASVs were filtered based on a minimum relative abundance threshold of 0.01%, and normalization was applied to account for 16S rRNA copy number variation. Metabolic pathways were annotated using the MetaCyc database. The differential abundance of pathways was assessed using the Kruskal–Wallis test. Visualization of statistically significant (p < 0.05) functional profiles and pathway abundances was performed using heatmaps generated in GraphPad Prism 8 (GraphPad Software, Inc., San Diego, United States) software.
Statistical analysis
The normality of microbiota percentage abundance levels at different taxonomic levels was assessed using the Shapiro–Wilk test. When the data did not follow the normal distribution pattern, the non-parametric Mann–Whitney test for median values of cases and controls was applied for the comparison. For the relative abundance analyses, we restricted to taxon present in at least 20% of the population. Calculations were carried out using PQStat 1.8.6 (PQStat Software, Poznan. Poland) and Graph Pad Prism 8 (GraphPad Software, Inc., San Diego, United States) software, and all tests were considered significant at a p-value of ≤0.05. The FDR-adjusted p-values were calculated according to the Benjamini–Hochberg procedure (Benjamini and Hochberg, 1995).
Results
A total of 48 participants were included in this study: 24 women with BC and 24 HC. The characteristics of the study groups are shown in Table 1 and were reported during the interview. BC cases were similar to healthy controls with regard to all presented parameters (p > 0.05).
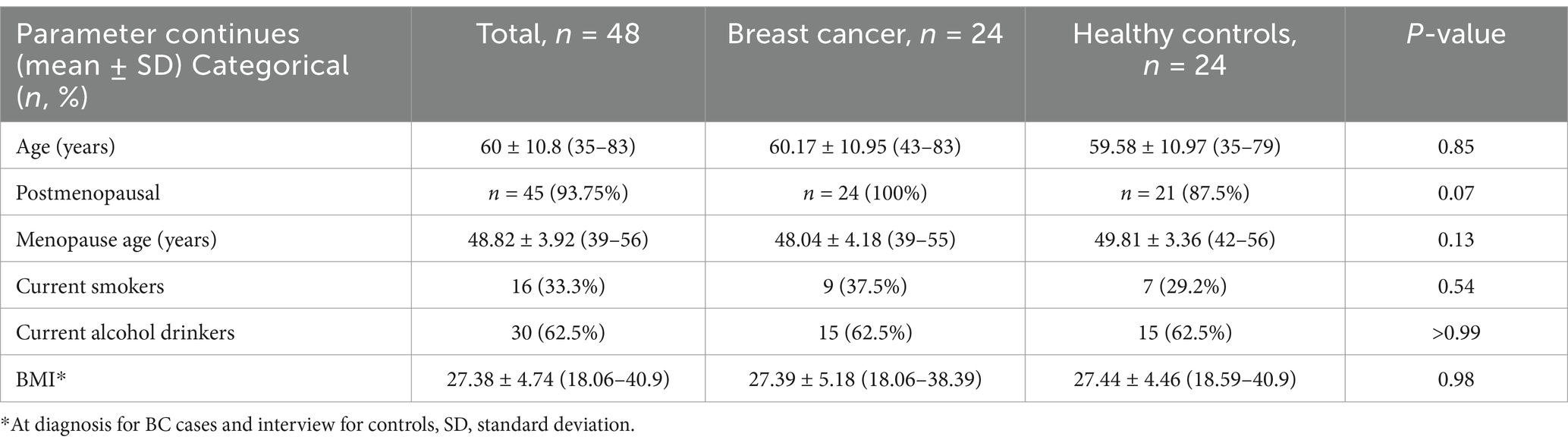
Table 1. Characteristic of the study population.
In the NGS analysis for urine and oral metaprofiling, the obtained parameters were as follows: the flow cell cluster density 1,006 ± 32, the Phred base-calling score GQX ≥ 30, an average of 74.87%, and the passing filter (PF) reads 22,703,315 constituting 92.25% of the total reads 24,597,294. In the bioinformatics analysis, the number of input reads per sample was on average 410,976 [SD 68509.5 (median 402,000, min. 278,462, max. 698,298)], and the number of reads after filtering on length, ambiguity, and expected errors was at the level of 24639.6 (SD 6637, median 24,318, min. 12,764, max. 47,286). Refraction curves of amplicon sequence variant (ASV) richness at read depth are presented in Figure 1A. In our study, both urine and oral rinse samples of BC patients and controls demonstrated similar microbial taxonomic diversity based on the deepest available analysis of all regions V1-V9 of the 16S rRNA gene (Figures 1B,C). Analysis of the beta diversity determined by the Bray–Curtis dissimilarity revealed that the bacterial microbiota of urine samples create a separate cluster from oral microbiota samples (Figure 1D).
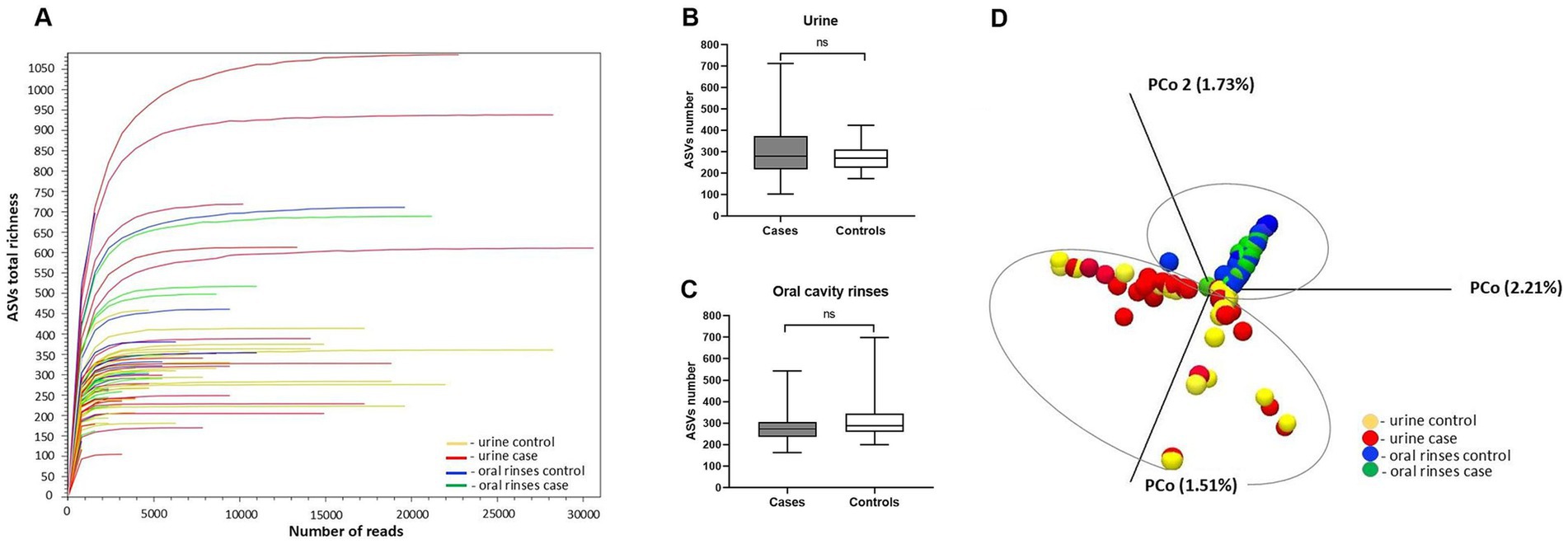
Figure 1. Alpha and beta diversity of the urine and oral cavity rinses microbiota between patients and healthy controls: (A) refraction curves of amplicon sequence variant (ASV) richness at read depth in all analyzed samples; (B) ASV richness in urine samples; (C) ASV richness in oral cavity rinses samples; (D) comparison between sample diversities by principal coordinate analysis (PCoA) with a Bray–Curtis dissimilarity matrix.
Based on a total number of identified ASVs, the urine samples presented a similar richness of microbiota compared to oral rinses, 12,086 vs. 12,035, respectively, of which 492 ASVs are found in both types of samples, giving a total richness of 24,613 ASVs. To narrow down the number of ASVs and to exclude sporadically occurring microorganisms, only ASVs that were observed in at least 20% of the samples within each group were counted (Figure 2).
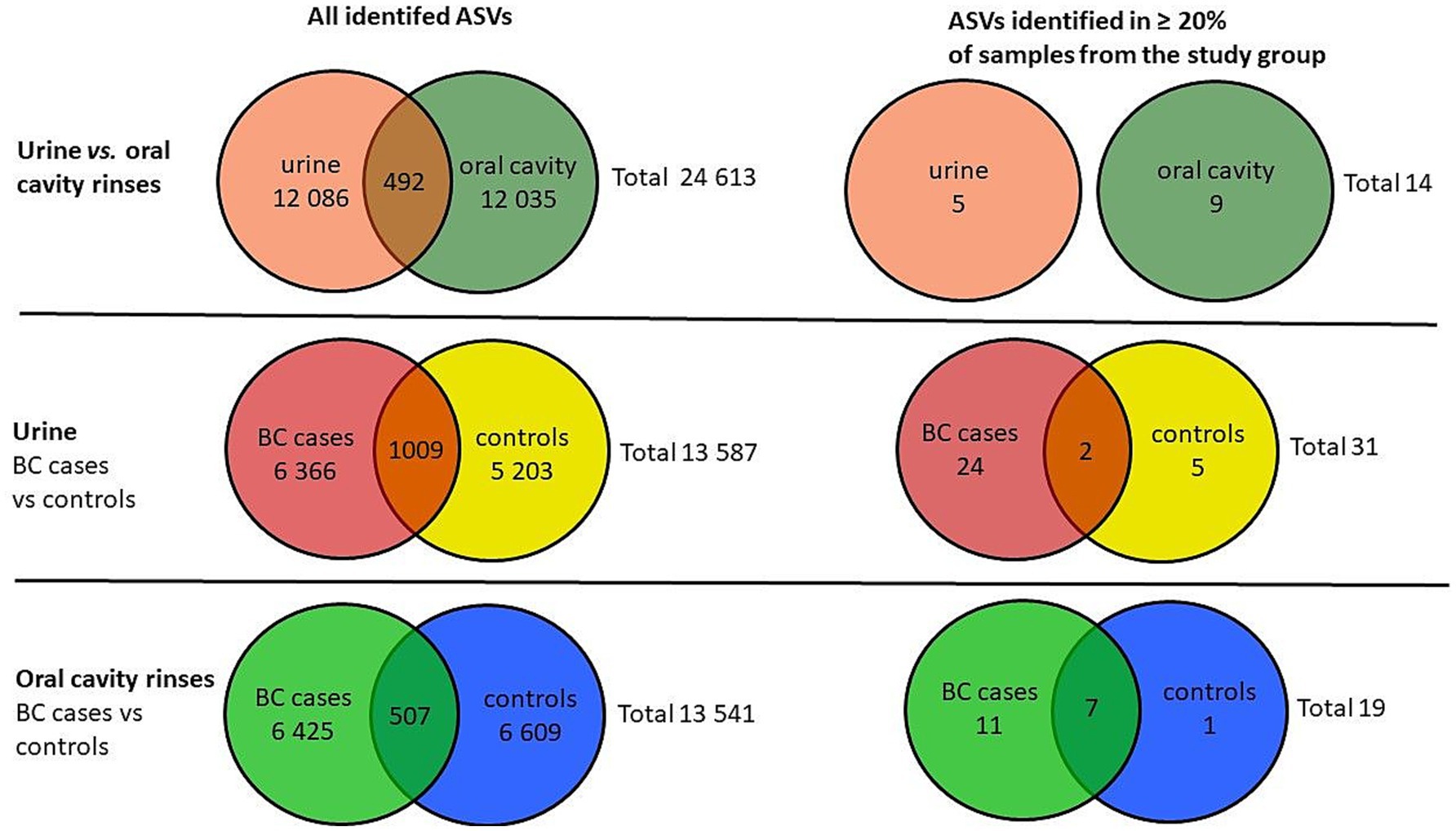
Figure 2. Venn diagrams of the number of identified ASVs in urine and oral cavity samples of BC cases and the control group.
After applying this criterion, urine samples presented 5 unique ASVs and 9 oral rinses, and none were joint. For urine, BC cases showed more unique ASVs than controls (24 vs. 5), with 2 ASVs present in both groups. In oral rinses, BC patients demonstrated also a richer composition of unique ASVs (11) compared to healthy controls (1) (Figure 2).
NGS raw data are available in the SRA database (accession number PRJNA1180501).
Metagenomic profile of the urine samples
We analyzed the bacterial taxonomic composition of urine sample microbiota in both the BC patients and the control group. A total of 13 phyla, 19 classes, 39 orders, 83 families, 159 genera, and 265 species were detected (Supplementary Tables S1–S6). After applying our filter in the biostatistics (selection of taxa occurring in at least 20% of the samples), the number on each taxonomic level was reduced to 5 phyla, 8 classes, 12 orders, 18 families, 17 genera, and 47 species (Figure 3—species not shown). Obtained results are presented in tabular form in the Supplementary Tables S1–S6.

Figure 3. Bacterial taxonomy in the urine samples. Columns show names of taxonomic levels identified in urine samples of BC patients and healthy controls occurring in at least 20% of the samples in the total group. The first column represents the phylum level. The subsequent taxonomy groups are class, order, family, and genus. The green underline indicates greater abundance among healthy controls, and the red underline indicates greater abundance among cases.
The most abundant bacteria for the whole study group at the phylum level in urine were the following: Bacillota (commonly known as Firmicutes, 32.66%, SD 37.78), Pseudomonadota (known as Proteobacteria, 31.16%, SD 29.81), and Actinomycetota (known as Actinobacteriota, 25.77%, SD 25.44). Bacteria classified at these three phyla accounted for 86.22% of cases and 88.83% of controls. However, the composition of urine microbiota at the phylum level did not differ significantly statistically between BC cases and HCs.
Differences in abundance between BC cases and HCs were observed at further taxonomic classification levels (Figures 3, 4). For the results discussed below, we provide p-values (p ≤ 0.05), for the difference in bacterial abundance between BC cases and HCs on the Mann–Whitney test, before applying FDR correction. Both sets of p-values are reported in Supplementary Tables S1–S6.
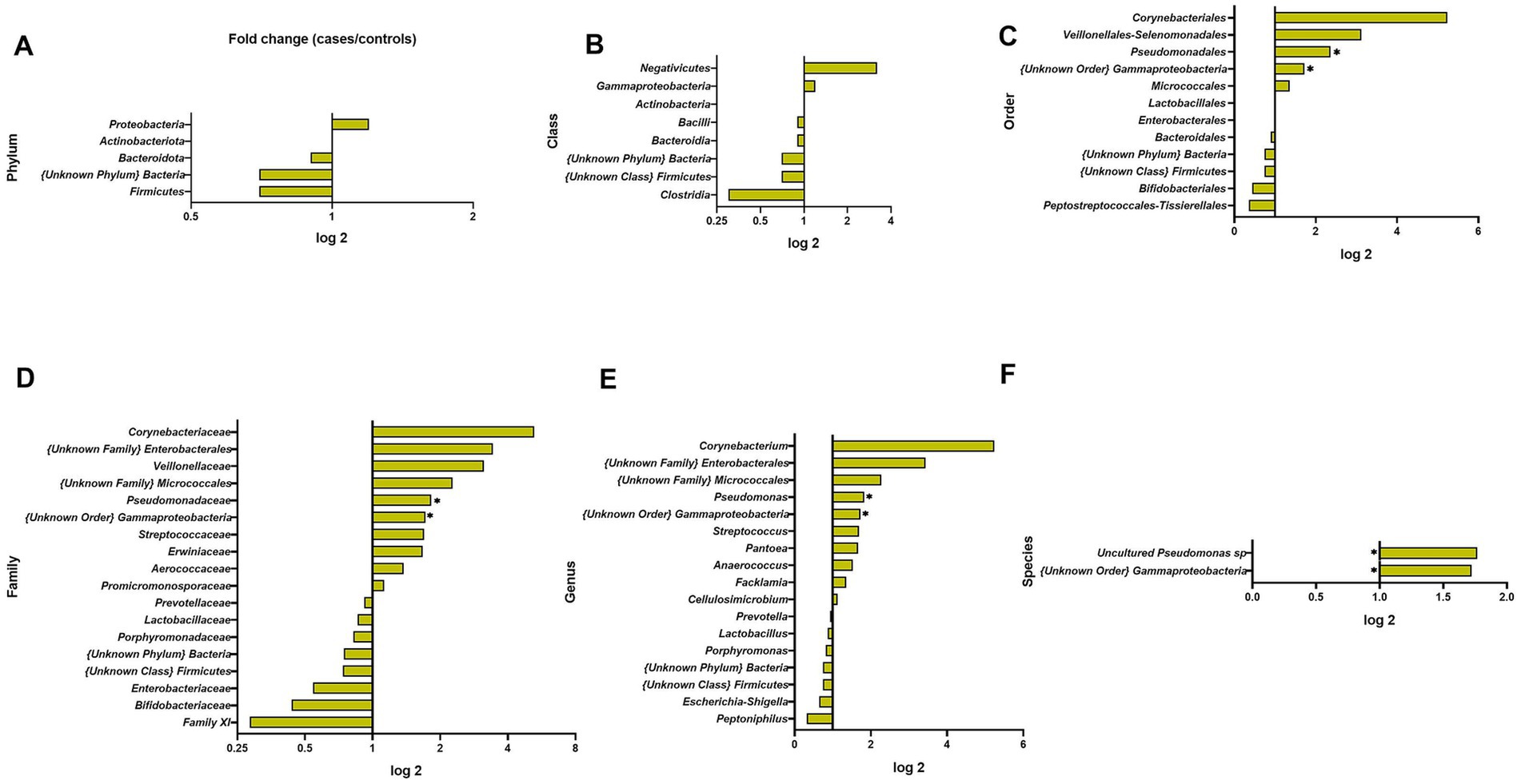
Figure 4. Fold changes of urine bacterial abundance between cases and healthy controls at different taxonomic levels: (A) phylum; (B) class; (C) order; (D) family; (E) genus; (F) species. * indicates statistical significance at p ≤ 0.05, before FDR correction. ** p ≤ 0.01.
At the bacterial class level, the results showed a lower abundance of Clostridia in BC patients compared to HCs (0.3-fold) and a higher level of Negativicutes (3.2-fold); these differences were not statistically significant (Figure 4).
At the bacterial order level, the most abundant in the whole study group were Lactobacillales (26.53%, SD 35.48), Enterobacterales (20.21%, SD 26.21), and Micrococcales (17.33%, SD 20.72) and did not differ significantly between BC cases and HCs. In BC samples, we observed a significantly higher presence of Pseudomonadales (2.4-fold) and {Unknown Order} Gammaproteobacteria (1.7-fold) (p = 0.0077, p = 0.0424, respectively).
At the family level, the urine samples of BC cases and HCs were dominated by Lactobacillaceae (18.56%, SD 32.65), Promicromonosporaceae (13.09%, SD 16.05), and Enterobacteriaceae (11.04%, SD 27.24) and did not differ between BC cases and HCs. BC cases had a 1.8-fold and 1.7-fold significantly higher abundance of Pseudomonadaceae (p = 0.0257) and Unknown Order} Gammaproteobacteria (p = 0.0424) (Figure 4).
At the genus level, we observed 17 types of bacteria, of which Lactobacillus (17.50%, SD 31.28) and Cellulosimicrobium (12.82%, SD 15.99) predominated among urine samples and did not differ between BC cases and HCs. However, the less common genera of the microbiota composition differentiated between BC patients and HCs: Pseudomonas and {Unknown Order} Gammaproteobacteria were significantly more common in BC (1.8-fold, p = 0.0257, 1.7-fold p = 0.0424, respectively, Figure 4).
At the species level, a significant difference revealed the uncultured Pseudomonas sp. (31.8-fold, p = 0.028) and Gammaproteobacteria {unknown order} (1.7-fold, p = 0.042) in urine with greater abundance among BC patients (Figure 4).
Fungal analysis revealed the presence of two phyla, Ascomycota and Basidiomycota in urine material, detected in 16.6% of samples (8 of 48). At lower taxonomic levels, urine samples represented 8 classes, 13 orders, 17 families, 19 genera, and 18 species (detailed in Supplementary Table S13, respectively). The occurrence of these taxa was sporadic, typically limited to individual samples. No differences in urine fungal composition were observed between BC patients and controls. The only notable distinction was a higher abundance of the phylum Basidiomycota (p = 0.0392), class Agaricomycetes (p = 0.0392), order Capnodiales (p = 0.0192), and family Cladosporiaceae (p = 0.0192) in the urine of BC patients compared to controls. However, this difference was no longer significant after adjustment.
Metagenomic profile of oral rinse samples
A total of 9 phyla, 15 classes, 45 orders, 53 families, 80 genera, and 240 species of bacteria were detected, wherein after applying our filter in the biostatistics (selection of taxa occurring in at least 20% of the samples), the number on each taxonomic level was reduced to 6 phyla, 8 classes, 14 orders, 21 families, 24 genera, and 44 species (Figure 5—species not shown). All findings are presented in tabular format within the Supplementary Tables S7–S12.

Figure 5. Bacterial taxonomy in the oral rinse samples. Columns show phyla identified in oral rinse samples of BC patients and healthy controls occurring in at least 20% of the samples in the total group. The first column represents the phylum level. The subsequent taxonomy groups are class, order, family, and genus. The green underline indicates greater abundance among healthy controls, the red underline indicates greater abundance among cases.
At the phylum level, Firmicutes (51.2%, SD 26.4) and Bacteroidota (30.5%, SD 23.6) were most abundant in both BC cases and HCs accounted for 84.04% of all bacteria present in cases and 79.28% in controls. The distribution of bacterial phyla did not reveal statistically significant differences between BC cases and healthy controls (Figure 6).
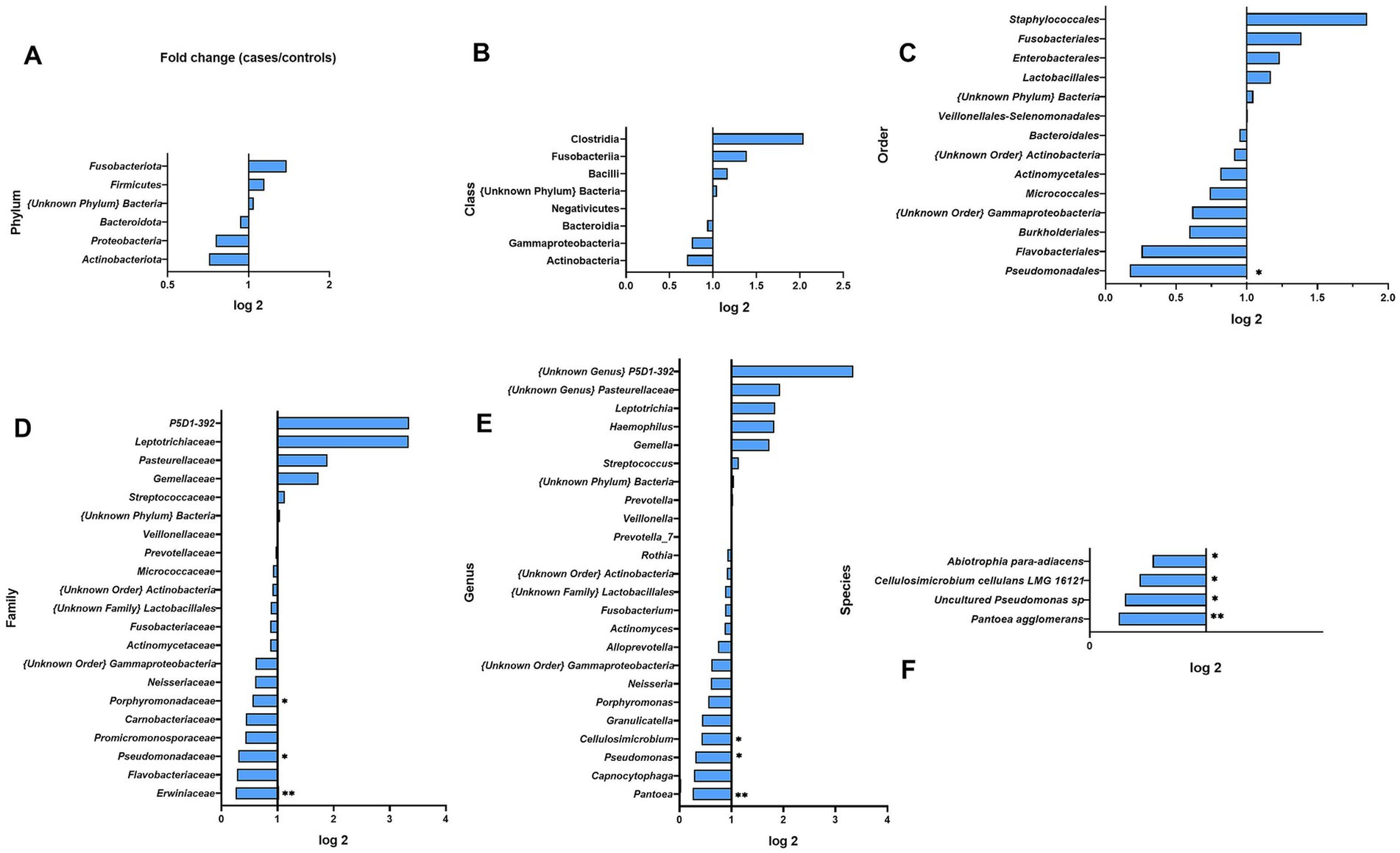
Figure 6. Fold changes of oral wash bacterial abundance between cases and healthy controls at different taxonomic levels: (A) phylum; (B) class; (C) order; (D) family; (E) genus; (F) species. * indicates statistical significance at p ≤ 0.05, before FDR correction. ** p ≤ 0.01.
At the class level, the most abundant ones were Bacilli (46.47%, SD 29.81 in BC and 39.69%, SD 28.12 in HCs) and Bacteroidia (29.39%, SD 23.64 in BC and 31.53%, SD 37.85 in HC); however, significant differences were not observed between groups.
At the order level, the most abundant were Lactobacillales (42.44%, SD 28.34) and Bacteroidales (30.5%, SD 22.59), which accounted for 75.01% in BC patients and 69.95% in controls. Only Pseudomonadales had significantly lower levels in BC cases relative to HCs (0.2-fold, p = 0.0203).
At the family level, the oral rinse samples of both groups were dominated by Streptococcaceae (35.87%, SD 26.66) and Prevotellaceae (29.01%, SD 22.77). We observed a 0.3-fold, 0.4-fold, and 0.3-fold lower abundance of Erwiniaceae, Promicromonosporaceae, and Pseudomonadaceae among BC cases (p = 0.0091, p = 0.0332, and p = 0.0470) (Figure 6). We observed a higher richness among BC cases of Leptotrichiaceae (3.3-fold) and P5D1–392 family (3.3-fold) but not statistically significant.
At the genus level, we observed 24 representatives of bacteria, and the most numerous were Streptococcus (35.66%, SD 26.42) and Prevotella_7 (23.74%, SD 22.04). Genera of Pantoea, Cellulosimicrobium, and Pseudomonas were less abundant in BC patients (0.3-, 0.4-, and 0.3-fold, p = 0.0091, p = 0.0332, and p = 0.0470 respectively).
At the species level, four species differed significantly between BC cases and HCs. As shown in Figure 6 and Supplementary Table S12 all of those species, Abiotrophia para-adiacens, Cellulosimicrobium cellulans LMG 16121, Pseudomonas sp. (uncultured), and Pantoea agglomerans were less abundant among BC cases (0.2-, 0.4-, 0.3-, and 0.5-fold; p = 0.0074, p = 0.0332, p = 0.0470, and p = 0.0476).
Fungal analysis revealed in oral cavity material the presence of two phyla, Ascomycota and Basidiomycota, detected in 19.14% of samples (9 of 47). At lower taxonomic levels, oral cavity samples represented 8 classes, 13 orders, 17 families, 19 genera, and 18 species (detailed in Supplementary Table S14). The occurrence of these taxa was sporadic, typically limited to individual samples. No differences in oral cavity fungal composition were observed between BC patients and controls.
Functional profile analysis of the urine and oral cavity microbiota
Using PICRUSt2 software, based on inferred Enzyme Commission (EC) profiles, we identified 1,352 predicted pathways in urine samples and 1,323 in oral rinses. Among them, three metabolites from urine and seven from oral rinses were identified as most significantly differentiating between BC patients and healthy controls (p < 0.05, Kruskal–Wallis test). These are presented in Figure 7A and Supplementary Table S15 for the urine metabolome, and in Figure 7B and Supplementary Table S16 for the oral metabolome.
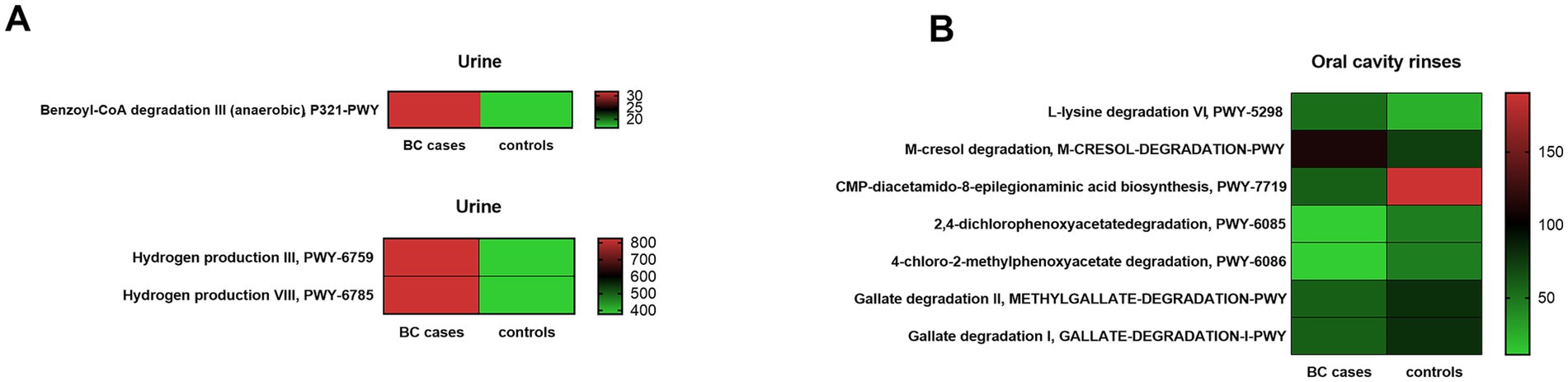
Figure 7. Functional pathways predicted by PICRUSt2 differentiate BC cases and healthy control (HC) groups in urine (A) and oral rinses (B). Differentiating MetaCyc pathways identified by Kruskal–Wallis test (p < 0.01). The color scale (red means higher and green means lower) corresponds to the raw abundance, based on the number of reads. The column on the right side corresponds to the number of sequencing reads.
Prediction analysis in our urine microbiota data found a significantly higher content of enzymes responsible for benzoyl-CoA degradation and hydrogen production in BC cases. A detailed list of predicted urine metabolome enzymes with pathway description information is provided in Supplementary Table S15.
Based on oral microbiota data, we observed a significant predomination of enzymes, such as involved in L-lysine degradation and m-cresol degradation for BC cases, whereas significant reduction in the BC group compared to the control was observed for CMP-diacetamido-8-epilegionaminic acid biosynthesis, 2,4-dichlorophenoxyacetate degradation, 4-chloro-2-methylphenoxyacetate degradation, methylgallate, and gallate degradation. A detailed list of predicted oral rinse metabolome enzymes with pathway description information is provided in Supplementary Table S16.
Discussion
In our study, metagenomic profiling based on next-generation sequencing allowed us to classify bacteria and fungi inhabiting the urine and oral cavity of BC patients and healthy participants.
Despite contamination of urine samples by bacteria originating from the uroepithelium, urethra, or genital tract, analyzing urine microbiota can provide valuable health insights into its role in disease development (Gottschick et al., 2017; Perez-Carrasco et al., 2021). Previous research on the urine microbiota indicated its lower richness compared to the number of bacteria inhabiting other niches of the human body (Pearce et al., 2014), a finding confirmed in our study, where urine samples had fewer ASVs than oral rinse samples. This is probably the result of greater external environmental pressure in the case of oral wash samples compared to urine samples.
We observed a slight increase in abundance (not significant statistically) of Proteobacteria phylum in the group of BC patients (34.5%) compared to healthy controls (27.7%) (Figure 4A). This phylum includes a wide variety of potentially pathogenic genera Escherichia, Klebsiella, and Shigella, whereas Firmicutes were presented decreased in BC cases to healthy controls, with an abundance of 25.8 and 35.3%, respectively. In general, the Proteobacteria increase with Firmicutes depletion is a microbial signature of dysbiosis in gut microbiota (Kumar and Kumar, 2022). Furthermore, in urine BC samples, we detected a significant increase in the genera Pseudomonas (p = 0.0257) and Gammaproteobacteria (p = 0.0424) compared to controls. This was also observed at the species level, although the fold change was not so high (Figures 4E,F). There are reports that some strains of bacteria, including Pseudomonas, produce toxins that can affect host cells, damaging their DNA and causing genetic changes that promote the development of cancer. On the other hand, there is evidence that cancer cells can stimulate the secretion of toxins by bacteria, such as those from the Pseudomonas genus (Choi et al., 2023). When considering the potential link between Gammaproteobacteria and BC, we came across research showing that tumor-adjacent-normal tissue was found to contain higher relative abundances of Gammaproteobacteria (unclassified) in comparison with healthy normal tissue (Giallourou et al., 2021). The exact role of this bacteria in BC development is still unclear; however, it is known that Gammaproteobacteria is associated with a specific lipid profile.
The highest fold change (5.2-fold) with a predominance in BC was presented for Corynebacterium. Although most species of Corynebacterium are symbiotic to the host, some of them may become pathogenic and lead to infections (Burkovski, 2018). Importantly, in urine samples of healthy women, we observed a higher content of probiotic bacteria from the Bifidobacteriaceae and Lactobacillaceae families compared to BC. Moreover, BC patients had an increased abundance of Micrococcales in their urine. Micrococcales belongs to the Actinomycetes class, which in general are commensal organisms and can also be an opportunistic pathogen, particularly in a host with an impaired immune system (Li et al., 2022). The presence of the Micrococcales bacteria has also been reported in a study analyzing microbiota composition in BC and healthy tissue in Slovakian and Chinese women (Hadzega et al., 2021). In that study, a higher level of Micrococcales was observed in the breast tumor tissue of Slovakian women in comparison with healthy breast tissue samples but not for the Chinese women. The observed enrichment of Micrococcales in the breast tissue among the Slovakian BC cases and in urine among Polish BC cases, two similar ethnic groups, makes this bacterium a possible candidate as a biomarker for BC in the Polish population.
At this moment, it is difficult to clearly interpret the role of bacteria that significantly distinguish BC patients from healthy women, calling them radical pathobionts or commensals. As defined by the International Cancer Microbiome Consortium, there is a large and poor-defined space between a “pathogen” coexisting with the disease of the host (e.g., Helicobacter pylori) and the “commensal” which may contribute to disease under certain conditions. Therefore, the term “dynamic symbiont” was introduced and is often used, also in our study, when interpreting the bacterial genera, such as Micrococcales, Pseudomonas, and Gammaproteobacteria which were enriched in the urine of BC women (Scott et al., 2019).
Searching in our study for the microbial signature of BC in oral rinses, we have observed a significant depletion of bacterial species Abiotrophia para-adiacens, Cellulosimicrobium cellulans LMG 16121, Pseudomonas sp. (uncultured), and Pantoea agglomerans in BC cases compared to healthy controls.
To discover the potential functional role of bacteria in the host, we applied metabolomic analysis by PICRUSt2 software to predict relevant pathways of urine and oral cavity microbiota in BC patients and HC. Of the 1,352 identified pathways based on interfered Enzyme Commission (EC) profiles in urine samples and 1,323 in oral rinses, 10 pathways together from both niches differentially enriched between the BC patients and HCs were indicated (Supplementary Tables S15, S16). Based on the urine microbiota composition, we have identified three processes that mostly differentiate the two studied groups, benzoyl-CoA degradation III and hydrogen production III and VIII, which dominate in BC cases. Urinary hydrogen peroxide was postulated to be a biomarker of oxidative stress in general malignancies (Banerjee et al., 2003), whereas increased benzoyl-CoA degradation III (anaerobic) in urine samples of BC patients could be the effect of different factors. Benzoyl-CoA is the most common central intermediate of anaerobic aromatic metabolism, including those aromatic compounds derived from diet, drugs such as chemotherapy, or tumor metabolism (Koch et al., 1993). However, based on the oral rinse microbiota, we have observed that m-cresol and L-lysine degradation was significantly increased in BC cases. There are speculations that cancers alter aromatic compound metabolism leading to an increased degradation process of m-cresol, but there is a lack of evidence. Studies on full microbial metabolites are needed to elucidate these process correlation with BC development.
Fungi were remarkably less present in the tested samples of both urine and oral rinses in our study. In general, we have detected two main fungal phyla, Ascomycota and Basidiomycota, and also Incertae sedis phylum. In the case of oral rinses, we did not find any relevant differences in the composition of mycobiota between patients and healthy women. However, urine samples of BC cases showed a significant reduction in Basidiomycota abundance and enrichment in Agaricomycetes, Capnodiales, Cladosporiaceae, and Cladosporium abundance, whereas global investigations revealed a strong correlation of particular fungi presence with cancer development (Hosseini et al., 2022). Among them, Aspergillus, Candida, Coccidioides, Cunninghamella, Geotrichum, Pleistophora, Rhodotorula, Filobasidiella, Mucor, Trichophyton, Epidermophyton, Fonsecaea, Pseudallescheria, Penicillium, Ajellomyces, Alternaria, Rhizomucor, Piedraia, and Malassezia were found to be linked with breast cancer development (Banerjee et al., 2018). However, these observations concern microbial niches such as intestines or cancer tissue. In our study, we were focused on microbial composition in oral rinses and urine only.
To summarize, the current study has shown bacterial differences in the urinal and oral microbiota composition between BC patients and healthy Polish women. Although the identified differences in bacterial abundance did not maintain statistical significance after FDR adjustment, they suggest trends for further analysis using the full PWHS dataset. We acknowledge the pilot study’s small sample size limits the statistical power. The obtained results will guide us in larger investigations, in which we plan to perform metabolomic analysis parallel to metagenomics profiling in the full sample of 514 HCs and 417 BC cases. Metabolomics data would be a valuable complement to our research, particularly due to estrobolome characterization.
Our study represents the first attempt to compare groups of women that do not differ on age, menopausal status, smoking and drinking habits, and BMI. In previous studies on the diversity of urine and/or oral microbiota in patients with BC conducted by Wu et al. (2022)and Wang et al. (2017), significant differences in the selection of the study and control groups were observed on age, menopausal status, mean BMI, alcohol, tobacco, and antibiotics intake, which authors indicate could have influenced their results. In a research by Nearing et al., although has a fairly extensive description of the study population available, there is a lack of data regarding menopausal status (Nearing et al., 2020). By controlling for age, menopausal status, BMI, smoking, and alcohol consumption, we minimized their impact, allowing us to detect subtle microbiota changes. These findings may have significant implications for the health of women in Poland, suggesting potential new directions for research into BC prevention, diagnostics, and treatment.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found at: http://www.ncbi.nlm.nih.gov/bioproject/1180501.
Ethics statement
The studies involving humans were approved by Bioethics Committee of the Greater Poland Medical Chamber. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
AM: Conceptualization, Formal analysis, Methodology, Resources, Software, Visualization, Writing – original draft, Writing – review & editing. MS-Z: Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. OZ-B: Formal analysis, Methodology, Resources, Software, Visualization, Writing – original draft. EN: Data curation, Writing – review & editing. AK: Data curation, Writing – review & editing. BZ: Data curation, Writing – review & editing. AS: Data curation, Writing – review & editing. DG: Data curation, Funding acquisition, Project administration, Supervision, Writing – review & editing. JC: Data curation, Investigation, Supervision, Writing – review & editing. DP: Data curation, Funding acquisition, Investigation, Methodology, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. DP financed specimen and clinical data collection by grants: R01CA069670-NIH/NCI; R03CA092838; R03CA096436. Additional work with further study after 2006 has been financed in case JC by former Institute of Food and Nutrition and National Institute of Public Health-NIH-National Research Institute, Warsaw, AM, and DG financed study development and realization by Center of Cancer Prevention and Epidemiology OPEN, Poznan.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1538224/full#supplementary-material
References
Al-Ansari, M. M., AlMalki, R. H., Dahabiyeh, L. A., and Abdel Rahman, A. M. (2021). Metabolomics-microbiome crosstalk in the breast cancer microenvironment. Meta 11, 758–770. doi: 10.3390/metabo11110758
Álvarez Mercado, A. I., Del Valle Cano, A., Fernández, M. F., Del Valle Cano, A., Fernández, M. F., and Fontana, L. (2023). Gut microbiota and breast Cancer: the dual role of microbes. Cancers 15:443. doi: 10.3390/cancers15020443
Álvarez-Mercado, A. I., Navarro-Oliveros, M., Robles-Sánchez, C., Plaza-Díaz, J., Sáez-Lara, M. J., Muñoz-Quezada, S., et al. (2019). Microbial population changes and their relationship with human health and disease. Microorganisms 7:68. doi: 10.3390/microorganisms7030068
Arnold, M., Morgan, E., Rumgay, H., Mafra, A., Singh, D., Laversanne, M., et al. (2022). Current and future burden of breast cancer: global statistics for 2020 and 2040. Breast 66, 15–23. doi: 10.1016/j.breast.2022.08.010
Banerjee, D., Madhusoodanan, U. K., Nayak, S., and Jacob, J. (2003). Urinary hydrogen peroxide: a probable marker of oxidative stress in malignancy. Clin. Chim. Acta 334, 205–209. doi: 10.1016/s0009-8981(03)00236-5.12867293
Banerjee, S., Tian, T., Wei, Z., Shih, N., Feldman, M. D., Peck, K. N., et al. (2018). Distinct microbial signatures associated with different breast Cancer types. Front. Microbiol. 9:951. doi: 10.3389/fmicb.2018.00951
Benjamini, Y., and Hochberg, Y. (1995). ‘Controlling the false discovery rate: a practical and powerful approach to multiple testing’, journal of the Royal Statistical Society. Series B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Burkovski, A. (2018). The role of corynomycolic acids in Corynebacterium-host interaction. Antonie Van Leeuwenhoek 111, 717–725. doi: 10.1007/s10482-018-1036-6
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). Dada2: high resolution sample inference from illumina amplicon data repository. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Choi, J. K., Naffouje, S. A., Goto, M., Wang, J., Christov, K., Rademacher, D. J., et al. (2023). Cross-talk between cancer and Pseudomonas aeruginosa mediates tumor suppression. Commun. Biol. 6:16. doi: 10.1038/s42003-022-04395-5
Giallourou, N., Urbaniak, C., Puebla-Barragan, S., Vorkas, P. A., Swann, J. R., and Reid, G. (2021). Characterizing the breast cancer lipidome and its interaction with the tissue microbiota. Commun. Biol. 4:1229. doi: 10.1038/s42003-021-02710-0
Gottschick, C., Deng, Z. L., Vital, M., Masur, C., Abels, C., Pieper, D. H., et al. (2017). The urinary microbiota of men and women and its changes in women during bacterial vaginosis and antibiotic treatment. Microbiome 5:99. doi: 10.1186/s40168-017-0305-3
Hadzega, D., Minarik, G., Karaba, M., Kalavska, K., Benca, J., Ciernikova, S., et al. (2021). Uncovering microbial composition in human breast Cancer primary tumour tissue using transcriptomic RNA-seq. Int. J. Mol. Sci. 22:9058. doi: 10.3390/ijms22169058
Hosseini, K., Ahangari, H., Chapeland-Leclerc, F., Ruprich-Robert, G., Tarhriz, V., and Dilmaghani, A. (2022). Role of fungal infections in carcinogenesis and Cancer development: a literature review. Adv. Pharm. Bull. 12, 747–756. doi: 10.34172/apb.2022.076
Huybrechts, I., Zouiouich, S., Loobuyck, A., Vandenbulcke, Z., Vogtmann, E., Pisanu, S., et al. (2020). The human microbiome in relation to Cancer risk: a systematic review of epidemiologic studies. Cancer Epidemiol. Biomarkers Prev. 29, 1856–1868. doi: 10.1158/1055-9965.EPI-20-0288
Koch, J., Eisenreich, W., Bacher, A., and Fuchs, G. (1993). Products of enzymatic reduction of benzoyl-CoA, a key reaction in anaerobic aromatic metabolism. Eur. J. Biochem. 211, 649–661. doi: 10.1111/j.1432-1033.1993.tb17593.x
Kumar, S., and Kumar, A. (2022). Microbial pathogenesis in inflammatory bowel diseases. Microb. Pathog. 163:105383. doi: 10.1016/j.micpath.2021.105383
Li, X., Liu, Y., Yang, X., Li, C., and Song, Z. (2022). The Oral microbiota: community composition, influencing factors, pathogenesis, and interventions. Front. Microbiol. 13:895537. doi: 10.3389/fmicb.2022.895537
Łukasiewicz-Śmietańska, D., Godlewski, D., Nowakowska, E., Szpak, A., Chabros, E., Juszczyk, G., et al. (2024). Association of the bitter taste genes TAS2R38 and CA6 and breast cancer risk; a case-control study of polish women in Poland and polish immigrants in USA. PLoS One 19:e0300061. doi: 10.1371/journal.pone.0300061
Miko, E., Kovács, T., Sebő, É., Tóth, J., Csonka, T., Ujlaki, G., et al. (2019). Microbiome-microbial metabolome-cancer cell interactions in breast cancer-familiar, but unexplored. Cells 8, 293–326. doi: 10.3390/cells8040293
Nearing, J. T., DeClercq, V., Van Limbergen, J., and Langille, M. G. I. (2020). Assessing the variation within the Oral microbiome of healthy adults. mSphere 5, e00451–e00420. doi: 10.1128/mSphere.00451-20
Pearce, M. M., Hilt, E. E., Rosenfeld, A. B., Zilliox, M. J., Thomas-White, K., Fok, C., et al. (2014). The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. mBio 5, e01283–e01214. doi: 10.1128/mBio.01283-14
Perez-Carrasco, V., Soriano-Lerma, A., Soriano, M., Gutiérrez-Fernández, J., and Garcia-Salcedo, J. A. (2021). Urinary microbiome: yin and Yang of the urinary tract. Front. Cell. Infect. Microbiol. 11:617002. doi: 10.3389/fcimb.2021.617002
Proctor, L. M., Creasy, H. H., Fettweis, J. M., Lloyd-Price, J., Mahurkar, A., Zhou, W., et al. (2019). The integrative human microbiome project. Nature 569, 641–648. doi: 10.1038/s41586-019-1238-8
Ramirez-Garcia, A., Arteta, B., Abad-Diaz-de-Cerio, A., Pellon, A., Antoran, A., Marquez, J., et al. (2013). Candida albicans increases tumor cell adhesion to endothelial cells in vitro: intraspecific differences and importance of the mannose receptor. PLoS One 8:e53584. doi: 10.1371/journal.pone.0053584
Samkari, A. A., Alsulami, M., Bataweel, L., Altaifi, R., Altaifi, A., Saleem, A. M., et al. (2022). Body microbiota and its relationship with benign and malignant breast tumors: a systematic review. Cureus 14:e25473. doi: 10.7759/cureus.25473
Scott, A. J., Alexander, J. L., Merrifield, C. A., Cunningham, D., Jobin, C., Brown, R., et al. (2019). International Cancer microbiome consortium consensus statement on the role of the human microbiome in carcinogenesis. Gut 68, 1624–1632. doi: 10.1136/gutjnl-2019-318556
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. doi: 10.3322/caac.21660
Thu, M. S., Chotirosniramit, K., Nopsopon, T., Hirankarn, N., and Pongpirul, K. (2023). Human gut, breast, and oral microbiome in breast cancer: a systematic review and meta-analysis. Front. Oncol. 13:1144021. doi: 10.3389/fonc.2023.1144021
Tzeng, A., Sangwan, N., Jia, M., Liu, C. C., Keslar, K. S., Downs-Kelly, E., et al. (2021). Human breast microbiome correlates with prognostic features and immunological signatures in breast cancer. Genome Med. 13:60. doi: 10.1186/s13073-021-00874-2
Viswanathan, S., Parida, S., Lingipilli, B. T., Krishnan, R., Podipireddy, D. R., and Muniraj, N. (2023). Role of gut microbiota in breast Cancer and drug resistance. Pathogens 12:468. doi: 10.3390/pathogens12030468
Vivarelli, S., Salemi, R., Candido, S., Falzone, L., Santagati, M., Stefani, S., et al. (2019). Gut microbiota and Cancer: from pathogenesis to therapy. Cancers 11:38. doi: 10.3390/cancers11010038
Wajszczyk, B., Charzewska, J., Godlewski, D., Zemła, B., Nowakowska, E., Kozaczka, M., et al. (2021). Consumption of dairy products and the risk of developing breast Cancer in polish women. Nutrients 13:4420. doi: 10.3390/nu13124420
Wang, H., Altemus, J., Niazi, F., Green, H., Calhoun, B. C., Sturgis, C., et al. (2017). Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget 8, 88122–88138. doi: 10.18632/oncotarget.21490
Wojciechowska, U., Barańska, K., Miklewska, M., and Didkowska, J. A. (2023). Cancer incidence and mortality in Poland in 2020. Biuletyn Polskiego Towarzystwa Onkologicznego Nowotwory 73, 129–145. doi: 10.5603/NJO.2023.0026
Wolfe, A. J., and Brubaker, L. (2015). “Sterile urine” and the presence of Bacteria. Eur. Urol. 68, 173–174. doi: 10.1016/j.eururo.2015.02.041
Wu, Z., Byrd, D. A., Wan, Y., Ansong, D., Clegg-Lamptey, J. N., Wiafe-Addai, B., et al. (2022). The oral microbiome and breast cancer and nonmalignant breast disease, and its relationship with the fecal microbiome in the Ghana breast health study. Int. J. Cancer 151, 1248–1260. doi: 10.1002/ijc.34145
Keywords: breast cancer, next-generation sequencing, oral microbiota, urinary microbiota, functional analysis
Citation: Marciniak A, Skrzypczak-Zielińska M, Zakerska-Banaszak O, Nowakowska E, Kozaczka A, Zemła B, Szpak A, Godlewski D, Charzewska J and Pathak DR (2025) Urinary and oral microbiota in Polish women: a pilot case-control study of breast cancer. Front. Microbiol. 16:1538224. doi: 10.3389/fmicb.2025.1538224
Edited by:
Le Liu, Southern Medical University, ChinaReviewed by:
Madjid Morsli, Centre Hospitalier Universitaire de Nîmes, FranceSandeep Kasaragod, University of Glasgow, United Kingdom
Corinna Lesley Seidel, Ludwig Maximilian University of Munich, Germany
Copyright © 2025 Marciniak, Skrzypczak-Zielińska, Zakerska-Banaszak, Nowakowska, Kozaczka, Zemła, Szpak, Godlewski, Charzewska and Pathak. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Marciniak, bWFyY2luaWFrLmFubmFAb3Blbi5wb3puYW4ucGw=