Abstract
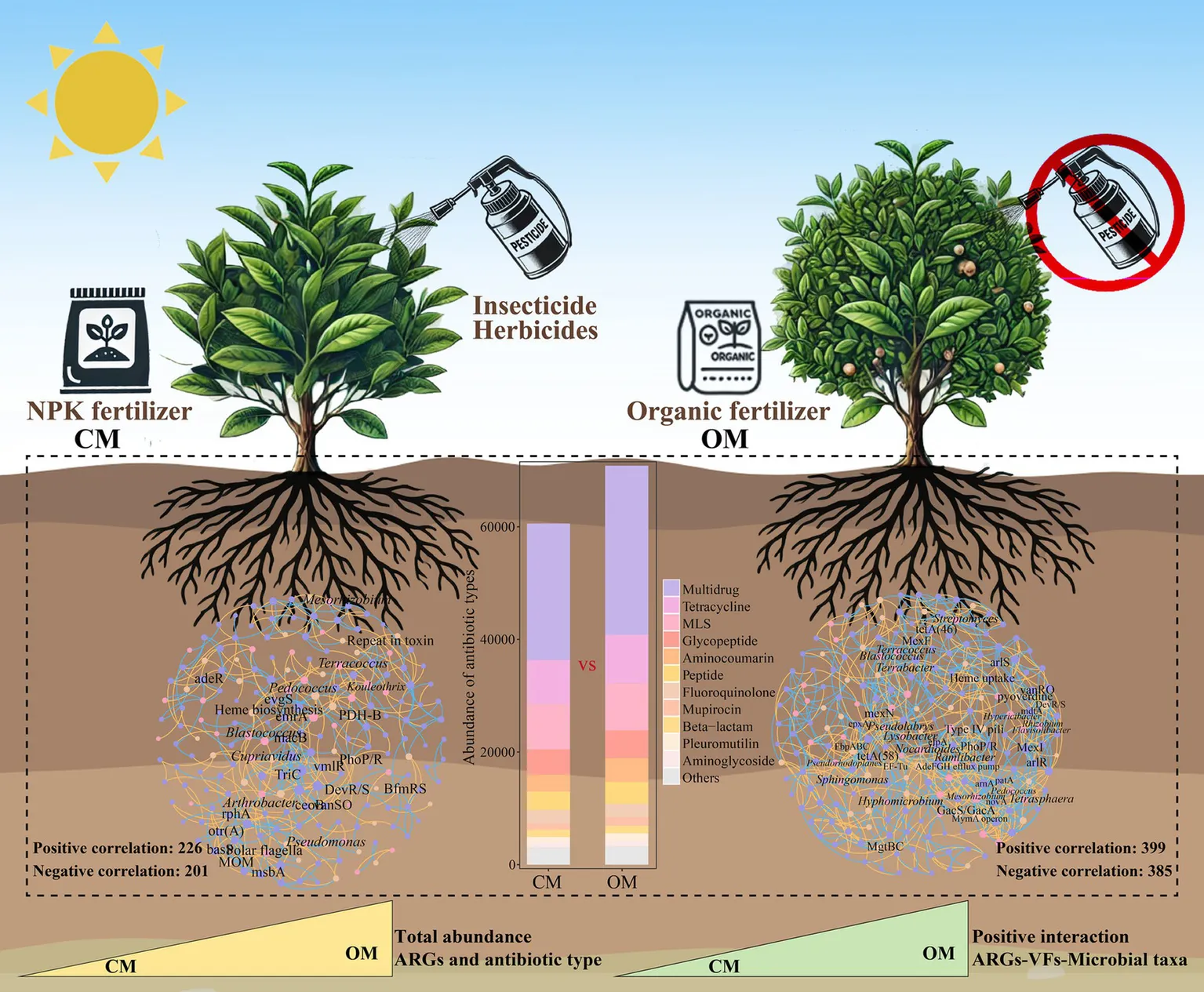
Sustainable organic management practices have gained significant attentions for its potential health and environmental benefits. However, the spread of antibiotic resistance genes (ARGs) and virulence factors (VFs) in soils, plants, and agricultural products has severely limited the development of organic managements on agriculture. At present, the distribution and assembly of ARGs and VFs in organic managed tea plantation systems remains largely unknown. Here, we used metagenomic analysis to explore soil microbial taxa, ARGs and VFs in 20 years of conventional managed (CM) and organic managed (OM) tea plantation soils. Results showed that total abundance of ARGs in OM was 16.9% (p < 0.001) higher than that in CM, and the increased ARGs were rpoB2, evgS, MuxB, TaeA, and efrA. As for VFs, OM significantly increased the abundance of adherence, stress protein and actin-based motility compared to CM. Moreover, OM increased the relative abundance of soil microbial taxa harboring ARGs and VFs, which were Streptomyces, Pseudomonas, and Terrabacter, compared to CM. Network analysis suggested that OM increased the positive interactions of microbial taxa-ARGs, microbial taxa-VFs and ARGs-VFs compared to CM. Impact of stochastic process on the assembly of soil microbial taxa, ARGs and VFs in OM was stronger than that in CM. Overall, these findings provide a basis for integrating ARGs, VFs and pathogen hosts to assess the ecological and health risks in long-term organic managed soils, and increased efforts need to be done in reducing ARGs, VFs and bacterial pathogens in fertilizers for organic managements on agriculture.
1 Introduction
In last decades, the widespread use and misuse of antibiotics in medical has led to the prevalence of antibiotic resistance genes (ARGs) in microbial communities (Kuppusamy et al., 2018; Wang J. H. et al., 2022). With the increase of ARGs and antibiotic resistant bacteria (ARBs) in the environment, antibiotic resistance poses a serious threat to ecological security and public health (Tiedje et al., 2019; Zhang et al., 2022). Soils are probably the most important hosts of ARGs, and a large number and diversity of ARGs have been found in soils around the world (Xiao et al., 2016; Braga et al., 2017). The ARGs in farmland soils can enter to plant stems, leaves and agricultural products, and further spread to humans along the food chain, posing a major threat to human health (Zhang et al., 2019; Song et al., 2023). Besides, the virulence factors (VFs) enable pathogenic bacteria to colonize a host, establish infection and confer virulence, thereby enabling its bacterial hosts to invade humans or animals and cause disease (Wu H. J. et al., 2008; Liang et al., 2020). When the ARGs coexist with VFs in the genome, the risk of the genome to humans or animals will increase (Liang et al., 2020). Many studies have reported that anthropogenic activities (such as irrigation, landfilling of waste, fertilization and cropping practices, especially organic fertilization) significantly enriched the abundance of ARGs in soils (Wang et al., 2014; Wu et al., 2017; Wang et al., 2018). Hence, it is urgent to assess to distribution and interaction of ARGs and VFs in organic managed farmland systems.
Tea (Camellia sinensis L.) plantations are widely distributed in tropical and subtropical acidic soils (Wang et al., 2016). Normally, high nitrogen fertilization rate was applied to obtain high yield in tea plantations (Yang et al., 2023), but its misuse may trigger negative environmental impacts (Wang L. L. et al., 2020). Organic management, such as using livestock manure, can reduce chemical fertilizer application, maintain soil fertility and improve soil biodiversity (Guo et al., 2017; Ekman et al., 2020). However, inputs of livestock-derived organic fertilizers may introduce ARGs and ARBs into farmland soils (Han et al., 2018; Sanz et al., 2022). Therefore, antibiotic resistance may spread from organically amended soils to crops, products and ultimately to consumers (Yang et al., 2018). Recent findings shown that the coexistence and convergence of ARGs and VFs in pathogenic bacteria significantly increased the risk of microbial contaminants in the environment (Yang et al., 2018; Liang et al., 2020; Li et al., 2023). Therefore, it is necessary to understand the characteristics of ARGs and VFs in long-term organic managed soils.
Studies have demonstrated that soil properties and microbial diversity are the main drivers that influence ARGs and VFs distribution (Nõlvak et al., 2016; Wei et al., 2022; Wu et al., 2023a; Wang L. et al., 2024). It is found that abundance and diversity of ARGs in soil are related to soil type and nutrient content in organic farming systems (Wang L. et al., 2022). Besides, soil physicochemical variables, such as soil organic carbon (SOC), total nitrogen (TN), soil TN:TP ratio and microbiomass-P, are strongly associated with the distribution and prevalence of ARGs or VFs in soils (Guo et al., 2020; Wang L. et al., 2020; Kang et al., 2023). Furthermore, soil microorganisms carrying ARGs can influence plant-associated microbiota through direct contact between the plant rhizosphere and bulk soil environment, and ultimately accelerate the evolution of ARGs in plant compartments (Chen et al., 2018). Environmental heterogeneity has been shown to determine the diversity and distribution of bacterial communities, and soil property variables induced by fertilization may indirectly influence ARGs distribution by shaping soil bacterial communities (Li H. et al., 2022; Wu et al., 2023b). Therefore, it is essential to elucidate the complexity and correlation of microbial communities and soil properties impacts on ARGs and VFs distribution in long-term organic managed soils.
Here, we used macrogenomic sequencing to analyze soil microbial communities, antibiotic resistance genes (ARGs) and Virulence factors (VFs) in conventional and organic managed tea plantation soils after 20 years. The aims of this study were (I) to investigate the effects of conventional and organic managements on soil microbial communities, ARGs and VFs; (II) to explore the biotic and abiotic factors that affect the composition and distribution of ARGs and VFs.
2 Materials and methods
2.1 Study site and soil sampling
Soil samples of the 0–20 cm layer were collected in July 2023 from a 20-year managed tea plantation (22.48°N, 100.58°E) in Pu’er City, Yunnan Province, China. The study site has a typical subtropical monsoon climate, with an annual mean precipitation of 1,311 mm and annual mean temperature of 21.5°C. Two treatments: conventional managed (CM, only NPK fertilizer) or organic managed (OM, livestock organic fertilizer) tea plantation soils with 20 years were selected in this study. The tea variety in the experimental area is Yunkang 10. The long-term experimental field was managed according to local practices, which usually received NPK fertilizer or sheep manure compost for the past two decades. The organic management fertilization method includes basal fertilizer (November to December every year) and topdressing (May of the following year). The basal fertilizer was 12,000 kg ha−1 and the topdressing was 3,000 kg ha−1. The organic matter content, total nutrient content and pH of the organic fertilizer were 60, 5% and 7.5, respectively. In conventional management, the compound fertilizer (1,050 kg ha−1; N-P-K: 22-5-5) was applied in June each year, and Glyphosate and Diafenthiuron were used for weeding and pest extermination, respectively. In order to ensure the representativeness of the soil samples, we established 6 plots (20 m × 5 m) for each fertilization treatment to collect soil. Each plot used a five-point sampling method to collect soil, and five individual samples were mixed to obtain a duplicate sample. In total, 12 soil samples (2 treatments × 6 replicates) were obtained, then the soil samples were stored at 4°C and −80°C, respectively.
2.2 Analysis of soil properties and enzyme activities
Soil pH was determined using a pH meter (1:2.5, w/v). Soil organic carbon (SOC) and total nitrogen (TN) were determined by the K2Cr2O7 oxidation–reduction titration and Kjeldahl digestion methods (Bao, 2000), respectively. Soil ammonium nitrogen (NH4+-N) and nitrate nitrogen (NO3−-N) were determined using a microplate spectrophotometer (Thermo1510, Multiskan Go; Thermo Scientific Inc., Waltham, MA, United States). Activity of β-1, 4-glucosidase (BG), β-cellobiohydrolase (CE), β-xylosidase (BX), β-1,4-N-acetylglucosaminidase (NAG) and L-leucine aminopeptidase (LAP) were determined by a microplate spectrophotometer (Ex. 360 nm; Em. 450 nm; Thermo Scientific Inc., Waltham, MA, United States) using 4-methylumbelliferone (MUF) and 7-amino-4-methylcoumarin (AMC) coupled substrates (Marx et al., 2001).
2.3 DNA extraction, metagenomics sequencing and data analysis
Total microbial genomic DNA was extracted from 0.5 g soil using the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, United States), and the quality of extracted DNA was measured using NanoDrop® ND-2000 spectrophotometer (Thermo Scientific Inc., Waltham, MA, United States). The shotgunmetagenomic sequencing were performed using Novaseq6000 (Shanghai Majorbio Bio-pharm Technology Co., Ltd., Shanghai, China).
Raw sequences were trimmed and filtered using fastp version 0.20.0 software. Reads with average quality score lower than 20, containing more than three “N,” with length shorter than 50 bp and those reads matching the Illumina background sequences (artifact, spike-ins or phiX) were all removed. CD-HIT version 4.6.1 software was used for clustering, and the longest gene was selected as the representative sequence to construct a non-redundant gene set. Use BLASTP version 2.3.0 software to compare the non-redundant gene set with the NR database version 20,200,604, and obtain the species annotation results through the taxonomic information database corresponding to the NR database (Altschul et al., 1997).
2.4 ARGs and VFs analysis
We use the Comprehensive Antibiotic Resistance Database (CARD version 3.0.9) with Antibiotic Resistance Ontology (ARO) as its core for annotation of antibiotic resistance genes (ARGs) (Yang et al., 2022). The non-redundant gene sets were compared to the CARD database using BLASTP version 2.3.0 software, and the annotation of E to 1e−5 was selected. The setting parameters for ARGs annotation were ≥90% of sequence identity and ≥25 amino acids of alignment length. The ARGs obtained were classified by type (antibiotics to which the genes are resistant) and subtype (antibiotic resistance genes). To identify virulence factors (VFs) sequences in our data, open reading frames (ORFs) were compared against the virulence factor database (VFDB version 2020.07.03) using blastx with the E-value to 1e−5. The ORF with identity ≥90% and coverage ≥ 90% was annotated as a VFs (Liu B. et al., 2022; Liu W. B. et al., 2022). In addition, we annotated the species of ARGs or VFs to identify host bacteria.
2.5 Statistical analysis
Unpaired t tests were performed for significance analysis of two groups, and p values were adjusted by the false discovery rate test. Heatmap, boxplot and stacked chart were created using the OmicStudio.1 Principal coordinate analysis (PCoA) and redundancy analysis (RDA) were performed using the “vegan” package in R version 4.2.2 (Oksanen et al., 2013). Procrustes analysis was performed to examine the correlations between soil microbial communities, ARGs and VFs, and the sum of squares (M2) and p-value were used to determine the consistency of two datasets. The neutral community model (NCM) was performed using the “hmisc” and “minpack.lm” packages in R version 4.2.2 to evaluate the impact of stochastic dispersal on the assembly of soil microbial communities, ARGs and VFs (Ning et al., 2019). We selected soil microbial taxa, ARGs and VFs with relative abundance greater than 0.1% to construct the co-occurrence network. The correlations were computed using the “Hmisc” package in R version 4.2.2, with a strict absolute value threshold set to 0.9. To increase the credibility of the network analysis, only correlations with adjusted p values less than 0.01 were retained. Network visualization was performed using the Gephi version 0.9.2 software (Bastian et al., 2009; Yu et al., 2023).
3 Results
3.1 Soil properties and enzyme activities
Soil properties and enzyme activities varied greatly in OM and CM soils (Supplementary Table S1). The soil pH, TN content and SOC content in OM was 29.9, 9.3 and 6.6% (p < 0.05) higher than that in CM. The soil activities of NAG and LAP in OM was 26.8 and 184.1% (p < 0.05) higher than that in CM. However, no difference was detected in content of NO3−-N and NH4+-N, and activity of BG and CE between OM and CM (Supplementary Table S1).
3.2 Microbial diversity and community composition
A total of 6.1–7.6 Gb high quality clean reads was obtained after quality control for each sample (Supplementary Table S2). The filtered sequences were assembled de novo and 233,226 to 550,838 sequences were obtained, with a 481–598 bp for N50 and 332–345 bp for N90, for each sample. Each sample has 264,897–678,746 ORFs with a mean 373–419 bp per sample (Supplementary Table S3). Furthermore, bacteria and archaea dominated in total sequences with proportion of 65.3 and 19.5%, respectively (Supplementary Table S4).
The Shannon index of soil microbial communities in OM was 3.6% higher than that in CM (Figure 1a, P < 0.001). Similarly, OM significantly altered soil microbial community composition compared to CM (Figure 1). Actinobacteria, Proteobacteria, Acidobacteria, and Chloroflexi dominanted in abundance at the phylum level, with relative abundance of 31.9–40.7%, 27.7–36.5%, 10.8–14.9%, and 6.8–10.7%, respectively (Figure 1b). OM increased relative abundance of Proteobacteria, Gemmatimonadetes and Bacteroidota by 24.1, 56.6 and 37.4%, while reduced relative abundance of Actinobacteria and Chloroflexi by 12.6 and 34.2% compared to CM (Figure 1b, P < 0.001). Further analysis showed OM significantly increased relative abundance of the genera Nocardioides, Terrabacter, Rhodococcus, Arthrobacter, Streptomyces, Sphingomonas, Pseudolabrys, Lysobacter, and Pseudomonas within the changed phyla compared to CM (Figure 1c; Supplementary Table S5, p < 0.001).
Figure 1
3.3 Abundance and composition of ARGs and VFs
PCoA demonstrated that composition of ARGs were significantly separated in OM and CM (Figure 2a, P = 0.004). The total abundance of ARGs in OM was 16.9% higher than that in CM (Figure 2b, P < 0.001). A total of 21 ARGs and 945 subtypes were detected in OM and CM. Multidrug, Tetracycline, MLS and Glycopeptide were the major ARGs components in all samples, with abundance of 23788.9–30589.7, 7709.7–8851.8, 7681.3–8466.5 and 4324.6–5001.8, respectively (Figure 2c). Among them, OM significantly enriched the abundance of 13 antibiotics (i.e., Multidrug, Tetracycline, MLS, Glycopeptide, Aminocoumarin, Peptide, Mupirocin, Beta-lactam, Pleuromutilin, Fosfomycin, Triclosan, Diaminopyrimidine, and Bicyclomycin), while decreased the abundance of 5 antibiotics (Fluoroquinolone, Aminoglycoside, Rifamycin, Elfamycin, and Nucleoside), compared to CM (Supplementary Table S6). Subsequently, differential analysis on the top abundant 30 subtypes showed that OM enriched relative abundance of 10 subtypes (rpoB2, evgS, TaeA, MuxB, efrA, otr(A), tetB(P), mdtC, efrB, and vanRM), while decreased relative abundance of 16 subtypes (macB, tetA(58), oleC, bcrA, mtrA, msbA, efpA, arlR, kdpE, tlrC, baeS, facT, patA, evgA, patB, and lmrC), compared to CM (Figure 2d; Supplementary Table S7). Furthermore, the potential host test of ARGs showed that Mycobacterium (Unclassified), Bradyrhizobium (B. sp._35–63-5), Streptomyces (S. sp._CEV_2–1, S. sp._ADI95-17, S. chartreusis, S. sp._LAM7114 and S. rishiriensis), Saccharopolyspora (S. shandongensis and S. hirsuta) and Actinomadura (A. amylolytica and A. hibisca) were shared hosts for both CM and OM. A total of 23 unique hosts were detected in CM, such as Bacillus (B. cereus), Dictyobacter (D. kobayashii and D. aurantiacus), Bradyrhizobium (B. sp._35–63-5) and Amycolatopsis (A. vastitatis and A. kentuckyensis), while 10 unique hosts were detected in OM, such as Terrabacter (Terrabacter. sp._3264), Pseudomonas (P. aeruginosa), Pseudonocardia (P. hierapolitana), Tetrasphaera (T. sp._HKS02) and Microbispora (M. sp._GKU_823) (Supplementary Table S8).
Figure 2
PCoA demonstrated that composition of VFs were significantly separated between OM and CM (Figure 3a, P = 0.003). Iron uptake system, adherence, secretion systems, regulation, toxin and antiphagocytosis were the dominant encoded functions for both OM and CM, with relative abundance of 23.61–26.55, 16.44–18.67, 12.16–12.71, 11.32–12.26, 9.82–11.21, and 9.53–9.94, respectively (Figure 3b). Among them, OM enriched the relative abundance of adherence, stress protein, serum resistance, phase variation, complement protease, exoenzyme and actin-based motility, while decreased the relative abundance of iron uptake system, secretion system, regulation, toxin and magnesium uptake system, compared to CM (Supplementary Table S9). Further analysis of the top abundant 40 VFs showed that OM significantly increased VFs related to the putative hosts of Acinetobacter baumannii (AdeFGH), Francisella tularensis subsp. (repeat in toxin and EF-Tu), Pseudomonas aeruginosa (HSI-I and alginate), Legionella pneumophila subsp. (Hsp60), Pseudomonas stutzeri (pyridine-2,6-dithiocarboxylic acid), Pseudomonas syringae pv (GacS/GacA), Mycobacterium smegmatis str (proteasome-associated proteins) (Figure 3c; Supplementary Table S10). However, OM significantly decreased VFs associated with Aeromonas hydrophila subsp. (Polar flagella and repeat in toxin), Mycobacterium sp. (MymA operon), Mycobacterium tuberculosis (PDIM, PhoP/R and PhoP), Mycobacterium ulcerans (GPL locus) (Figure 3c; Supplementary Table S10).
Figure 3
3.4 Assembly processes and environmental drivers of microbial taxa, ARGs and VFs
The neutral community model fitting results showed that the explained variance of soil microbial communities (R2OM = 0.906, R2CM = 0.871), ARGs (R2OM = 0.925, R2CM = 0.775) and VFs (R2OM = 0.936, R2CM = 0.706) in OM was higher than that in CM (Figure 4). These results underscore the important role played of stochastic processes in shaping assembly of soil microbial communities, ARGs and VFs, particularly in OM. We found that OM decreased the m value (the migration rate of community) of soil microbial communities, ARGs and VFs, indicating that the species and gene dispersal was lower, compared to CM (Figure 4).
Figure 4
Soil properties and enzyme activities correlated significantly with composition of soil microbial communities (F = 5.33, p = 0.004), ARGs (F = 15.01, p = 0.003) and VFs (F = 21.30, p = 0.003). RDA result illustrated that RDA1 and RDA2 explained 48.2% of the microbial variations (Figure 5a), 83.2% of the ARGs variations (Figure 5b) and 79.9% of the VFs variations (Figure 5c). Furthermore, soil pH, TN, SOC, NAG and LAP were the main environmental factors driving the composition of soil microbial communities, ARGs and VFs (Supplementary Table S11).
Figure 5
3.5 Relationships between microbial taxa, ARGs and VFs
The Procrustes analysis showed that ARGs and VFs of soil exhibited goodness-of-fit based on the Bray–Curtis dissimilarity metrics (M2 = 0.116, p < 0.001, permutations = 999), indicating significant correlations between ARGs and VFs (Figure 6a). Similarly, Procrustes analysis showed that ARGs (M2 = 0.182, p < 0.002, permutations = 999; Figure 6b) and VFs (M2 = 0.157, p < 0.001, permutations = 999; Figure 6c) correlated significantly with microbial communities, respectively. Networks showed that OM increased the number of node, edge, correlation, average degree, graph density, modularity and average clustering coefficient of the network, while reduced the average path length, compared to CM (Figures 6d,e; Supplementary Table S12). These results suggested that organic management leads to tighter relationships between microbial taxa, ARGs and VFs. Furthermore, OM increased the positive interaction of microbial taxa-ARGs (116 vs. 35), microbial taxa-VFs (40 vs. 21), and ARGs-VFs (119 vs. 57) compared to CM (Supplementary Table S13). Within the network of CM, Mesorhizobium (6 subtypes), Kouleothrix (5 subtypes) and Blastococcus (4 subtypes) were highly correlated with ARGs, and Cupriavidus (3 VFs), Blastococcus (2 VFs) and Kouleothrix (2 VFs) were highly correlated with VFs. Within the network of OM, Streptomyces (11 subtypes), Flavisolibacter (9 subtypes), Rhizobium (8 subtypes) Blastococcus (8 subtypes), Terrabacter (8 subtypes) and Nocardioides (7 subtypes) were highly correlated with ARGs, and Pseudomonas (5 VFs), Hypericibacter (4 VFs), Ramlibacter (4 VFs) and Hyphomicrobium (3 VFs) were highly correlated with VFs (Figures 6d,e).
Figure 6
4 Discussion
The composition of microbial communities and antibiotic resistance in agricultural soils was closely related to soil health, food production safety and human welfare (Tshikantwa et al., 2018; Bertola et al., 2021). Organic management was reported to increase soil microbial community diversity and change its community structure (Schmidt et al., 2019; Li J. et al., 2022; Shu et al., 2022). This may be attributed to organic matter addition increased organic carbon and available nitrogen contents in the soils (Wu T. et al., 2008), which provided a favorable nutritional environment for microorganisms. On the other hand, organic fertilizer can effectively regulate soil acidification and improve the living environment for soil microorganisms (Chepkorir et al., 2018; Ye et al., 2022), especially in tea plantations. Although organic fertilizer input brings many benefits to the soils, the application of livestock derived organic fertilizers also has the risk of contamination with antibiotics, virulence factors and pathogenic bacteria (Bloem et al., 2017).
In this study, OM significantly changed the ARGs composition and increased its abundance, which was consistent with previous findings (Chen et al., 2016; Sun et al., 2019). Livestock manure contains high levels of antibiotics, ARBs and ARGs (Fang et al., 2014; Wang X. R. et al., 2024), which can potentially spread into the environment when applied to agricultural fields (Joy et al., 2013). We found OM enriched the abundant of Multidrug, Tetracycline, MLS and Glycopeptide antibiotic types compared to CM. Previous studies have confirmed that application of manure introduced extra antibiotics into the agricultural ecosystems (Wang L. et al., 2020; Zhu et al., 2022; Xiao et al., 2023). Further, we explore the differences of main subtypes of the microbial risk genes and OM significantly enriched resistance genes affiliated to multidrug (rpoB2, evgS, MuxB and efrA) and Pleuromutilin (TaeA). These genes may increase soil resistance to multidrug and Pleuromutilin by encoding efflux pump or antibiotic target alteration (Alcock et al., 2020). In contrast, resistance genes significantly enriched in CM included MLS (macB and oleC), Tetracycline (tetA(58)), Aminocoumarin (novA) and Peptide (bcrA), and these genes were closely related to antibiotic efflux (Alcock et al., 2020). Virulence factors related to adherence, stress protein, serum resistance, phase variation, complement protease, exoenzyme and actin-based motility were significantly enriched in OM. Bacterial pathogens are able to adhere to host cells by produce a protein or polysaccharide surface layer, and specific enzymes participate in the invasion of host cells and tissues after adhesion (Huang et al., 2016). For intracellular survival, stress proteins affected their persistence and survival (Hingley-Wilson et al., 2010). The significant enrichment of these virulence factors in organically managed agricultural systems suggests that bacterial pathogens may have an enhanced ability to colonize and persist in these environments. Taken together, these results indicated that organic management increased some of ARGs and VFs, which may pose a serious threat to public health (Carmeli et al., 2016; Coll et al., 2018).
Microbial risk depends not only on diversity and abundance of microbial communities, ARGs and VFs, but also on their patterns of coexistence in the same niche (Martínez et al., 2015; Liang et al., 2020). Procrustes analysis and network analysis found significant correlations between microbial communities, ARGs and VFs (Che et al., 2019; Li et al., 2023), and OM increased network complexity (Xie et al., 2018), which may promote the coexistence of microbial taxa, ARGs and VFs thereby increase the risk of microbial contamination (Liang et al., 2020; Zhu et al., 2022). We found that OM increased more positive interactions of microbial taxa-ARGs, microbial taxa-VFs and ARGs-VFs which further supported this conclusion. Within the network, species in CM (such as Kouleothrix and Blastococcus) and OM (such as Streptomyces, Blastococcus, Flavisolibacter, Terrabacter, Pseudomonas) were significantly associated with many ARGs and VFs, suggesting that microbial taxa may carry various ARG subtypes and VFs (Yin et al., 2022). It is worth noting that Streptomyces (S. rishiriensis), Pseudomonas (P. aeruginosa) and Terrabacter (T. sp._3264) with significantly higher relative abundance in OM were identified as potential hosts carrying ARGs, which was consistent with previous findings (Ye et al., 2018; Sheam et al., 2020; Song et al., 2022). Meanwhile, several species affiliated to Pseudomonas were potential hosts of VFs in OM, including P. aeruginosa, P. stutzeri and P. syringae pv. The P. aeruginosa was a significant pathogen which increased risks of mortality and zoonotic diffusion among patients with sepsis (Dulger, 2020). These results indicate that close links existed between microbial taxa, ARGs and VFs in organic managed tea plantation soils, indicating the significance roles of microbial community succession in growth and spread of ARGs and VFs in soils.
Deterministic and stochastic processes play important roles in assembly of soil microbial communities, ARGs and VFs (Evans et al., 2017; Wang L. et al., 2024; Wang M. M. et al., 2024). Our results support the prominent role of stochastic processes in shaping the assembly of soil microbial communities, ARGs and VFs, particularly in organic managed systems. The higher stochastic assembly from ARGs and VFs in organic managed soils resulted in a more stable antibiotic resistome and virulence factor than that from conventional managed soils (Hou et al., 2021). For soil microbial communities, long-term organic management increased resource availability to reduce resource competitiveness, which resulted in the dominance of stochasticity in soil microbial community assembly process (Badri et al., 2013; Chaparro et al., 2013). To some extent, the regulation principle of ARGs and VFs assembly by environmental stress is similar to regulation of soil microbial community assembly by resources (Liu B. et al., 2022; Liu W. B. et al., 2022). The importance of soil properties on soil microbial communities, ARGs and VFs varies under different management strategies (Cycon et al., 2019; Tang et al., 2023; Wu et al., 2023a). In this study, soil microbial communities, ARGs and VFs were strongly influenced by soil pH, TN, SOC, NAG, and LAP, as proved by previous findings (Zhu et al., 2022; Wu et al., 2023c; Shen et al., 2024). It has been reported that soil pH strongly affected the adsorption and desorption behavior of ARGs (Liu et al., 2010) and organic carbon, total nitrogen, and available potassium altered the distribution of ARGs in soils (Zhu et al., 2022). Furthermore, considering that ARGs and VFs are existed in microbial potential hosts, and the strong correlation between soil properties, enzyme activities and the resistance group may be mediated by soil microbial communities (Li T. T. et al., 2022).
5 Conclusion
Our study found that ARGs and VFs could be transferred into soils by agricultural managements in tea plantation. Organic management significantly increased diversity and abundance of ARGs and VFs, and increased relative abundance of microbial hosts harboring ARGs and VFs have significant impacts on soil and human health compared to conventional management. The assembly of microbial communities, ARGs and VFs in organic managed soils was more driven by stochastic processes than that in conventional managed soils. Furthermore, organic management increased the coexistence of microbial taxa-ARGs, microbial taxa-VFs and ARGs-VFs. Taken together, these findings provide more comprehensive insights into the spread, ecological processes and coexistence patterns of ARGs and VFs in tea plantation soils under long-term organic management.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, PRJNA1139650.
Author contributions
TY: Conceptualization, Data curation, Formal analysis, Writing – original draft, Writing – review & editing. LC: Data curation, Formal analysis, Writing – review & editing. QZ: Writing – review & editing. JY: Writing – review & editing. HZ: Writing – review & editing. ZZ: Writing – review & editing. YY: Conceptualization, Data curation, Funding acquisition, Project administration, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Major Science and Technology Program in Yunnan Province (202202AE090029) and the National Natural Science Foundation of China (31901470).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1580450/full#supplementary-material
Footnotes
References
1
AlcockB. P.RaphenyaA. R.LauT. T.TsangK. K.BouchardM.EdalatmandA.et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res.48, D517–D525. doi: 10.1093/nar/gkz935
2
AltschulS. F.MaddenT. L.SchäfferA. A.ZhangJ. H.ZhangZ.MillerW.et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res.25, 3389–3402. doi: 10.1093/nar/25.17.3389
3
BadriD. V.ChaparroJ. M.ZhangR.ShenQ.VivancoJ. M. (2013). Application of natural blends of phytochemicals derived from the root exudates of Arabidopsis to the soil reveal that phenolic-related compounds predominantly modulate the soil microbiome. J. Biol. Chem.288, 4502–4512. doi: 10.1074/jbc.M112.433300
4
BaoS. D. (2000). Soil agro-chemistrical analysis. 3rdEdn. Beijing: China Agricultural Press.
5
BastianM.HeymannS.JacomyM. (2009). Gephi: an open source software for exploring and manipulating networks. Proceedings of the International AAAI Conference on Web and Social Media3, 361–362.
6
BertolaM.FerrariniA.VisioliG. (2021). Improvement of soil microbial diversity through sustainable agricultural practices and its evaluation by-omics approaches: a perspective for the environment, food quality and human safety. Microorganisms9:1400. doi: 10.3390/microorganisms9071400
7
BloemE.AlbihnA.ElvingJ.HermannL.LehmannL.SarviM.et al. (2017). Contamination of organic nutrient sources with potentially toxic elements, antibiotics and pathogen microorganisms in relation to P fertilizer potential and treatment options for the production of sustainable fertilizers: a review. Sci. Total Environ.607-608, 225–242. doi: 10.1016/j.scitotenv.2017.06.274
8
BragaL. P. P.AlvesR. F.DelliasM. T. F.NavarreteA. A.BassoT. O.TsaiS. M. (2017). Vinasse fertirrigation alters soil resistome dynamics: an analysis based on metagenomic profiles. Biodata Min.10:17. doi: 10.1186/s13040-017-0138-4
9
CarmeliY.ArmstrongJ.LaudP. J.NewellP.StoneG.WardmanA.et al. (2016). Ceftazidime-avibactam or best available therapy in patients with ceftazidimeresistant Enterobacteriaceae and Pseudomonas aeruginosa complicated urinary tract infections or complicated intra-abdominal infections (REPRISE): a randomised, pathogen-directed, phase 3 study. Lancet Infect. Dis.16, 661–673. doi: 10.1016/S1473-3099(16)30004-4
10
ChaparroJ. M.BadriD. V.BakkerM. G.SugiyamaA.ManterD. K.VivancoJ. M. (2013). Root exudation of phytochemicals in Arabidopsis follows specific patterns that are developmentally programmed and correlate with soil microbial functions. PLoS One8:e55731. doi: 10.1371/journal.pone.0055731
11
CheY.XiaY.LiuL.LiA. D.YangY.ZhangT. (2019). Mobile antibiotic resistome in wastewater treatment plants revealed by Nanopore metagenomic sequencing. Microbiome7, 1–13. doi: 10.1186/S40168-019-0663-0/FIGURES/5
12
ChenQ. L.AnX. L.LiH.SuJ. Q.MaY. B.ZhuY. G. (2016). Long-term field application of sewage sludge increases the abundance of antibiotic resistance genes in soil. Environ. Int.92-93, 1–10. doi: 10.1016/j.envint.2016.03.026
13
ChenQ. L.AnX. L.ZhengB. X.MaY. B.SuJ. Q. (2018). Long-term organic fertilization increased antibiotic resistome in phyllosphere of maize. Sci. Total Environ.645, 1230–1237. doi: 10.1016/j.scitotenv.2018.07.260
14
ChepkorirB. M.AnnS.MbiraK. G. (2018). Effect of enriched sheep manure rates on physico-chemical parameters of tea soil in timbilil tea estate, Kericho, Kenya. Int. J. Plant Soil Sci.25, 1–7. doi: 10.9734/IJPSS/2018/44866
15
CollF.PhelanJ.Hill-CawthorneG. A.NairM. B.MallardK.AliS.et al. (2018). Genome-wide analysis of multi-and extensively drug-resistant Mycobacterium tuberculosis. Nat. Genet.50:764. doi: 10.1038/s41588-017-0029-0
16
CyconM.MrozikA.Piotrowska-SegetZ. (2019). Antibiotics in the soil environmentdegradation and their impact on microbial activity and diversity. Front. Microbiol.10:412419. doi: 10.3389/fmicb.2019.00338
17
DulgerD. (2020). Statistical analysis of the relationship between mortality and nosocomial factors in patients with septicemia and the importance of Pseudomonas aeruginosa. Turk. J. Vet. Anim. Sci.44, 573–580. doi: 10.3906/vet-1912-13
18
EkmanJ.GoldwaterA.BradburyM.MatthewsJ.RogersG. (2020). Persistence of human pathogens in manure-amended Australian soils used for production of leafy vegetables. Agriculture11:14. doi: 10.3390/agriculture11010014
19
EvansS.MartinyJ. B.AllisonS. D. (2017). Effects of dispersal and selection on stochastic assembly in microbial communities. ISME J.11, 176–185. doi: 10.1038/ismej.2016.96
20
FangH.WangH. F.CaiL.YuY. L. (2014). Prevalence of antibiotic resistance genes and bacterial pathogens in long-term manured greenhouse soils as revealed by metagenomic survey. Environ. Sci. Technol.49, 1095–1104. doi: 10.1021/es504157v
21
GuoJ.LiuW.ZhuC.LuoG.KongY.LingN.et al. (2017). Bacterial rather than fungal community composition is associated with microbial activities and nutrient-use efficiencies in a paddy soil with short-term organic amendments. Plant Soil424, 335–349. doi: 10.1007/s11104-017-3547-8
22
GuoX. P.ZhaoS.ChenY. R.YangJ.HouL. J.LiuM.et al. (2020). Antibiotic resistance genes in sediments of the Yangtze estuary: from 2007 to 2019. Sci. Total Environ.744:140713. doi: 10.1016/j.scitotenv.2020.140713
23
HanX. M.HuH. W.ChenQ. L.YangL. Y.LiH. L.ZhuY. G.et al. (2018). Antibiotic resistance genes and associated bacterial communities in agricultural soils amended with different sources of animal manures. Soil Biol. Biochem.126, 91–102. doi: 10.1016/j.soilbio.2018.08.018
24
Hingley-WilsonS. M.LougheedK. E. A.FergusonK.LeivaS.WilliamsH. D. (2010). Individual Mycobacterium tuberculosis universal stress protein homologues are dispensable in vitro. Tuberculosis90, 236–244. doi: 10.1016/j.tube.2010.03.013
25
HouL. Y.WangH. J.ChenQ. F.SuJ. Q.GadM.LiJ. W.et al. (2021). Fecal pollution mediates the dominance of stochastic assembly of antibiotic resistome in an urban lagoon (Yundang lagoon), China. J. Hazard. Mater.417:126083. doi: 10.1016/j.jhazmat.2021.126083
26
HuangY.WangQ. L.ChengD. D.XuW. T.LuN. H. (2016). Adhesion and invasion of gastric mucosa epithelial cells by Helicobacter pylori. Front. Cell. Infect. Microbiol.6:159. doi: 10.3389/fcimb.2016.00159
27
JoyS. R.Bartelt-HuntS. L.SnowD. D.GilleyJ. E.WoodburyB. L.ParkerD. B.et al. (2013). Fate and transport of antimicrobials and antimicrobial resistance genes in soil and runoff following land application of swine manure slurry. Environ. Sci. Technol.47, 12081–12088. doi: 10.1021/es4026358
28
KangJ.QiuW.ZhangW.LiuJ.YangZ.WuZ.et al. (2023). Understanding how various forms of phosphorus stress affect microbiome functions and boost plant disease resistance: insights from metagenomic analysis. Sci. Total Environ.904:166899. doi: 10.1016/j.scitotenv.2023.166899
29
KuppusamyS.KakarlaD.VenkateswarluK.MegharajM.YoonY. E.LeeY. B. (2018). Veterinary antibiotics (VAs) contamination as a global agro-ecological issue: a critical view. Agric. Ecosyst. Environ.257, 47–59. doi: 10.1016/j.agee.2018.01.026
30
LiY.KongF. G.LiS.WangJ.HuJ. R.ChenS.et al. (2023). Insights into the driving factors of vertical distribution of antibiotic resistance genes in long-term fertilized soils. J. Hazard. Mater.456:131706. doi: 10.1016/j.jhazmat.2023.131706
31
LiT. T.LiR. C.CaoY. F.TaoC. Y.DengX. H.OuY. N.et al. (2022). Soil antibiotic abatement associates with the manipulation of soil microbiome via long-term fertilizer application. J. Hazard. Mater.439:129704. doi: 10.1016/j.jhazmat.2022.129704
32
LiJ.YangY.WenJ.MoF.LiuY. (2022). Continuous manure application strengthens the associations between soil microbial function and crop production: evidence from a 7-year multisite field experiment on the Guanzhong plain. Agric. Ecosyst. Environ.338:108082. doi: 10.1016/j.agee.2022.108082
33
LiH.ZhengX.TanL.ShaoZ.CaoH.XuY. (2022). The vertical migration of antibiotic-resistant genes and pathogens in soil and vegetables after the application of different fertilizers. Environ. Res.203:111884. doi: 10.1016/j.envres.2021.111884
34
LiangJ. S.MaoG. N.YinX. L.MaL. P.LiuL.BaiY. H.et al. (2020). Identification and quantification of bacterial genomes carrying antibiotic resistance genes and virulence factor genes for aquatic microbiological risk assessment. Water Res.168:115160. doi: 10.1016/j.watres.2019.115160
35
LiuW. B.ChengY. F.GuoJ. J.DuanY. H.WangS.XuQ. C.et al. (2022). Long-term manure inputs induce a deep selection on agroecosystem soil antibiotic resistome. J. Hazard. Mater.436:129163. doi: 10.1016/j.jhazmat.2022.129163
36
LiuY.XuZ.WuX.GuiW.ZhuG. (2010). Adsorption and desorption behavior of herbicide diuron on various Chinese cultivated soils. J. Hazard. Mater.178, 462–468. doi: 10.1016/j.jhazmat.2010.01.105
37
LiuB.ZhengD.ZhouS.ChenL.YangJ. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res.50, D912–D917. doi: 10.1093/nar/gkab1107
38
MartínezJ. L.CoqueT. M.BaqueroF. (2015). What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol.13, 116–123. doi: 10.1038/nrmicro3399
39
MarxM. C.WoodM.JarvisS. C. (2001). A microplate fluorimetric assay for the study of enzyme diversity in soils. Soil Biol. Biochem.33, 1633–1640. doi: 10.1016/S0038-0717(01)00079-7
40
NingD. L.DengY.TiedjeJ. M.ZhouJ. Z. (2019). A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. USA116, 16892–16898. doi: 10.1073/pnas.1904623116
41
NõlvakH.TruuM.KangerK.TampereM.EspenbergM.LoitE.et al. (2016). Inorganic and organic fertilizers impact the abundance and proportion of antibiotic resistance and integron-integrase genes in agricultural grassland soil. Sci. Total Environ.562, 678–689. doi: 10.1016/j.scitotenv.2016.04.035
42
OksanenJ.BlanchetF. G.KindtR.LegendreP.MinchinP.O’HaraR. B.et al. (2013). Vegan: community ecology package. R Package Version 2.0–10.
43
SanzC.CasadoM.Navarro-MartinL.CañamerasN.CarazoN.MatamorosV.et al. (2022). Implications of the use of organic fertilizers for antibiotic resistance gene distribution in agricultural soils and fresh food products. A plot-scale study. Sci. Total Environ.815:151973. doi: 10.1016/j.scitotenv.2021.151973
44
SchmidtJ. E.VannetteR. L.IgweA.BlundellR.CasteelC. L.GaudinA. C. (2019). Effects of agricultural management on rhizosphere microbial structure and function in processing tomato plants. Appl. Environ. Microbiol.85, e01064–e01019. doi: 10.1128/AEM.01064-19
45
SheamM.HaqueZ.NainZ. (2020). Towards the antimicrobial, therapeutic and invasive properties of Mikania micrantha Knuth: a brief overview. J. Adv. Biotechnol. Exp. Ther.3, 92–101. doi: 10.5455/jabet.2020.d112
46
ShenC.HeM.ZhangJ.LiuJ.WangY. (2024). Response of soil antibiotic resistance genes and bacterial communities to fresh cattle manure and organic fertilizer application. J. Environ. Manag.349:119453. doi: 10.1016/j.jenvman.2023.119453
47
ShuX. Y.HeJ.ZhouZ. H.XiaL. L.HuY. F.ZhangY. L.et al. (2022). Organic amendments enhance soil microbial diversity, microbial functionality and crop yields: a meta-analysis. Sci. Total Environ.829:154627. doi: 10.1016/j.scitotenv.2022.154627
48
SongR.SunY.LiX.DingC.HuangY.DuX.et al. (2022). Biodegradable microplastics induced the dissemination of antibiotic resistance genes and virulence factors in soil: a metagenomic perspective. Sci. Total Environ.828:154596. doi: 10.1016/j.scitotenv.2022.154596
49
SongD.TangX.TariqA.PanK.LiD. (2023). Regional distribution and migration potential of antibiotic resistance genes in croplands of Qinghai Tibet plateau. Environ. Res.231:116233. doi: 10.1016/j.envres.2023.116233
50
SunY. M.QiuT. L.GaoM.ShiM. M.ZhangH. F.WangX. M. (2019). Inorganic and organic fertilizers application enhanced antibiotic resistome in greenhouse soils growing vegetables. Ecotoxicol. Environ. Saf.179, 24–30. doi: 10.1016/j.ecoenv.2019.04.039
51
TangS.MaQ.MarsdenK. A.ChadwickD. R.LuoY.KuzyakovY.et al. (2023). Microbial community succession in soil is mainly driven by carbon and nitrogen contents rather than phosphorus and Sulphur contents. Soil Biol. Biochem.180:109019. doi: 10.1016/j.soilbio.2023.109019
52
TiedjeJ. M.FangW.ManaiaC. M.VirtaM.ShengH.LipingM.et al. (2019). Antibiotic resistance genes in the human-impacted environment: a one health perspective. Pedosphere29, 273–282. doi: 10.1016/S1002-0160(18)60062-1
53
TshikantwaT. S.UllahM. W.HeF.YangG. (2018). Current trends and potential applications of microbial interactions for human welfare. Front. Microbiol.9:1156. doi: 10.3389/fmicb.2018.01156
54
WangY. C.HaoX. Y.WangL.XiaoB.WangX. C.YangY. J. (2016). Diverse Colletotrichum species cause anthracnose of tea plants (Camellia sinensis (L.) O. Kuntze) in China. Sci. Rep.6:35287. doi: 10.1038/srep35287
55
WangL. L.LiQ.CoulterJ. A.XieJ. H.LuoZ. Z.ZhangR. Z.et al. (2020). Winter wheat yield and water use efficiency response to organic fertilization in northern China: a meta-analysis. Agric. Water Manag.229:105934. doi: 10.1016/j.agwat.2019.105934
56
WangL.LiY.ZhaoZ.ZhuM.HuT. (2022). Tidal flat aquaculture pollution governs sedimentary antibiotic resistance gene profiles but not bacterial community based on metagenomic data. Sci. Total Environ.833:155206. doi: 10.1016/j.scitotenv.2022.155206
57
WangF. H.QiaoM.SuJ. Q.ChenZ.ZhouX.ZhuY. G. (2014). High throughput profiling of antibiotic resistance genes in urban park soils with reclaimed water irrigation. Environ. Sci. Technol.48, 9079–9085. doi: 10.1021/es502615e
58
WangL.WangJ. H.WangJ.ZhuL.ConkleJ. L.YangR. (2020). Soil types influence the characteristic of antibiotic resistance genes in greenhouse soil with long-term manure application. J. Hazard. Mater.392:122334. doi: 10.1016/j.jhazmat.2020.122334
59
WangJ. H.WangL.ZhuL.WangJ.XingB. (2022). Antibiotic resistance in agricultural soils: source, fate, mechanism and attenuation strategy. Crit. Rev. Environ. Sci. Technol.52, 847–889. doi: 10.1080/10643389.2020.1835438
60
WangF.XuM.StedtfeldR. D.ShengH.FanJ.LiuM.et al. (2018). Long-term effect of different fertilization and cropping systems on the soil antibiotic resistome. Environ. Sci. Technol.52, 13037–13046. doi: 10.1021/acs.est.8b04330
61
WangX. R.ZhangX.LiN.YangZ. Z.LiB. X.ZhangX. L.et al. (2024). Prioritized regional management for antibiotics and heavy metals in animal manure across China. J. Hazard. Mater.461:132706. doi: 10.1016/j.jhazmat.2023.132706
62
WangL.ZhangT. L.XiangQ.FuC. X.QiaoM.DingL. J.et al. (2024). Selective enrichment of virulence factor genes in the plastisphere under antibiotic and heavy metal pressures. J. Hazard. Mater.465:133319. doi: 10.1016/j.jhazmat.2023.133319
63
WangM. M.ZhaoJ. Y.LiuY.HuangS. J.ZhaoC. Y.JiangZ. K.et al. (2024). Decipher soil resistance and virulence gene risks in conventional and organic farming systems. J. Hazard. Mater.468:133788. doi: 10.1016/j.jhazmat.2024.133788
64
WeiZ.ShenW.FengK.FengY.HeZ.LiY.et al. (2022). Organic fertilizer potentiates the transfer of typical antibiotic resistance gene among special bacterial species. J. Hazard. Mater.435:128985. doi: 10.1016/j.jhazmat.2022.128985
65
WuT.ChellemiD. O.GrahamJ. H.MartinK. J.RosskopfE. N. (2008). Comparison of soil bacterial communities under diverse agricultural land management and crop production practices. Microb. Ecol.55, 293–310. doi: 10.1007/s00248-007-9276-4
66
WuJ.GuoS.LiK.LiZ.XuP.JonesD. L.et al. (2023b). Effect of fertilizer type on antibiotic resistance genes by reshaping the bacterial community and soil properties. Chemosphere336:139272. doi: 10.1016/j.chemosphere.2023.139272
67
WuJ.GuoS.LinH. Y.LiK. J.LiZ. T.WangJ. Y.et al. (2023c). Uncovering the prevalence and drivers of antibiotic resistance genes in soils across different land-use types. J. Environ. Manag.344:118920. doi: 10.1016/j.jenvman.2023.118920
68
WuD.HuangX. H.SunJ. Z.GrahamD. W.XieB. (2017). Antibiotic resistance genes and associated microbial community conditions in aging landfill systems. Environ. Sci. Technol.51, 12859–12867. doi: 10.1021/acs.est.7b03797
69
WuH. J.WangA. H.JenningsM. P. (2008). Discovery of virulence factors of pathogenic bacteria. Curr. Opin. Chem. Biol.12, 93–101. doi: 10.1016/j.cbpa.2008.01.023
70
WuJ.WangJ.LiZ.GuoS.LiK.XuP.et al. (2023a). Antibiotics and antibiotic resistance genes in agricultural soils: a systematic analysis. Crit. Rev. Environ. Sci. Technol.53, 847–864. doi: 10.1080/10643389.2022.2094693
71
XiaoR. H.HuangD. L.DuL.SongB.YinL. S.ChenY. S.et al. (2023). Antibiotic resistance in soil-plant systems: a review of the source, dissemination, influence factors, and potential exposure risks. Sci. Total Environ.869:161855. doi: 10.1016/j.scitotenv.2023.161855
72
XiaoK. Q.LiB.MaL. P.BaoP.ZhouX.ZhangT.et al. (2016). Metagenomic profiles of antibiotic resistance genes in paddy soils from South China. FEMS Microbiol. Ecol.92:fiw023. doi: 10.1093/femsec/fiw023
73
XieW. Y.YuanS. T.XuM. G.YangX. P.ShenQ. R.ZhangW. W.et al. (2018). Long-term effects of manure and chemical fertilizers on soil antibiotic resistome. Soil Biol. Biochem.122, 111–119. doi: 10.1016/j.soilbio.2018.04.009
74
YangL.LiuW.ZhuD.HouJ.MaT.WuL.et al. (2018). Application of biosolids drives the diversity of antibiotic resistance genes in soil and lettuce at harvest. Soil Biol. Biochem.122, 131–140. doi: 10.1016/j.soilbio.2018.04.017
75
YangX. D.NiK.ShiY. Z.YiX. Y.JiL. F.WeiS. R.et al. (2023). Metagenomics reveals N-induced changes in carbon-degrading genes and microbial communities of tea (Camellia sinensis L.) plantation soil under long-term fertilization. Sci. Total Environ.856:159231. doi: 10.1016/j.scitotenv.2022.159231
76
YangC.ZhaoY.CaoW.XingM.XuX.WangZ.et al. (2022). Metagenomic analysis reveals antibiotic resistance genes and virulence factors in the saline-alkali soils from the Yellow River Delta, China. Environ. Res.214:113823. doi: 10.1016/j.envres.2022.113823
77
YeM.SunM.ZhaoY.JiaoW.XiaB.LiuM.et al. (2018). Targeted inactivation of antibiotic-resistant Escherichia coli and Pseudomonas aeruginosa in a soil-lettuce system by combined polyvalent bacteriophage and biochar treatment. Environ. Pollut.241, 978–987. doi: 10.1016/j.envpol.2018.04.070
78
YeJ.WangY.JiaX.WuZ.WangH. (2022). Improvement of soil acidification in tea plantations by long-term use of organic fertilizers and its effect on tea yield and quality. Front. Plant Sci.13:1055900. doi: 10.3389/fpls.2022.1055900
79
YinY.ZhuD.YangG.SuJ.DuanG. (2022). Diverse antibiotic resistance genes and potential pathogens inhabit in the phyllosphere of fresh vegetables. Sci. Total Environ.815:152851. doi: 10.1016/j.scitotenv.2021.152851
80
YuT. B.FangX. Y.LiuY.ZangH. D.ZengZ. H.YangY. D. (2023). Irrigation rather than fertilization drives the abundance, community structure and assembly process of soil denitrifiers. Agric. Ecosyst. Environ.357:108688. doi: 10.1016/j.agee.2023.108688
81
ZhangY. J.HuH. W.ChenQ. L.SinghB. K.YanH.ChenD.et al. (2019). Transfer of antibiotic resistance from manure-amended soils to vegetable microbiomes. Environ. Int.130:104912. doi: 10.1016/j.envint.2019.104912
82
ZhangZ.ZhangQ.WangT.XuN.LuT.HongW.et al. (2022). Assessment of global health risk of antibiotic resistance genes. Nat. Commun.13:1553. doi: 10.1038/s41467-022-29283-8
83
ZhuL.LianY.LinD.HuangD.YaoY.JuF.et al. (2022). Insights into microbial contamination in multi-type manure-amended soils: the profile of human bacterial pathogens, virulence factor genes and antibiotic resistance genes. J. Hazard. Mater.437:129356. doi: 10.1016/j.jhazmat.2022.129356
Summary
Keywords
organic management, antibiotic resistance genes, virulence factors, assembly process, soil microorganisms
Citation
Yu T, Cheng L, Zhang Q, Yang J, Zang H, Zeng Z and Yang Y (2025) Characterization of antibiotic resistance genes and virulence factors in organic managed tea plantation soils in southwestern China by metagenomics. Front. Microbiol. 16:1580450. doi: 10.3389/fmicb.2025.1580450
Received
20 February 2025
Accepted
14 April 2025
Published
01 May 2025
Volume
16 - 2025
Edited by
Rosa Alduina, University of Palermo, Italy
Reviewed by
Jun Zhao, Nanjing Normal University, China
Thi Thuy Do, Department of Agriculture Food and the Marine, Ireland
Updates
Copyright
© 2025 Yu, Cheng, Zhang, Yang, Zang, Zeng and Yang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yadong Yang, yadong_tracy@cau.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.