Abstract
Tick-borne diseases (TBDs) pose a significant public health challenge, as their incidence is increasing due to the effects of climate change and ecological shifts. The interplay between tick-borne pathogens and the host microbiome is an emerging area of research that may elucidate the mechanisms underlying disease susceptibility and severity. To investigate the diversity of microbial communities in ticks infected with vertebrate pathogens, we analyzed the microbiomes of 142 tick specimens. The presence of Rickettsia and Anaplasma pathogens in individual samples was detected through PCR. Our study aimed to elucidate the composition and variation of microbial communities associated with three tick species, which are known vectors for various pathogens affecting both wildlife and humans. We employed high-throughput sequencing techniques to characterize the microbial diversity and conducted statistical analyses to assess the correlation between the presence of specific pathogens and the overall microbial community structure. Pathogen screening revealed an overall positivity rate of 51.9% for Anaplasma and 44.6% for spotted fever group rickettsia (SFGR). Among the three tick species (Dermacentor silvarum, Haemaphysalis concinna, and Haemaphysalis japonica) analyzed, D. silvarum (the predominant species) exhibited the highest pathogen prevalence. The results indicate significant variation in microbial diversity between tick samples, with the presence of Anaplasma and SFGR associated with distinct changes in the microbial community composition. These findings underscore the complex interactions between ticks and their microbial inhabitants, enriching our understanding of tick-borne diseases.
1 Introduction
Ticks are hematophagous arthropods that primarily infest mammals, reptiles, and birds in the wild, causing serious disease in humans who are exposed to tick bites (Adrion et al., 2015; Duron and Gottlieb, 2020; Tokarz and Lipkin, 2021). Ticks play an essential role as vectors in the dynamics of vector-borne diseases (Simo et al., 2017). Hard ticks exhibit stage-specific feeding behavior, requiring a single prolonged blood meal per developmental instar (Wikel, 2013). During their multi-day attachment period, hard ticks engage in telmophagy, a cyclical process characterized by alternating anticoagulant saliva secretion and blood ingestion through the hypostome. During this time, pathogens are transmitted through saliva, which helps initiate infection in the host (Maldonado-Ruiz et al., 2019). Concurrently, symbiotic bacteria enter the host through the tick bite. In mice, it has been demonstrated that ticks replace the majority of the pre-existing commensal bacteria on the host’s skin during blood intake (Boulanger et al., 2023), and this alteration may play an important role in transmitting pathogens. The first attempts to characterize the full tick microbiome (Andreotti et al., 2011) indicated that it is complex and of varied origin. Since then, high-throughput sequencing technologies have been utilized to further study the tick microbiome (Budachetri et al., 2014; Grandi et al., 2023; Zhang et al., 2023). Currently, 16S rRNA genes are frequently utilized as target genes for amplicon sequencing, with the V1, V3, and V4 regions being of particular value (Sperling et al., 2017). The majority of tick-borne pathogens are of significant natural epidemiological importance, including their role in Lyme disease, anaplasmosis, rickettsiosis, and tick-borne encephalitis (Dantas-Torres et al., 2012; Madison-Antenucci et al., 2020). The microbiome plays a pivotal role in regulating various physiological processes in ticks, including immunity, nutrition, and digestion (Duron et al., 2018; Zhong et al., 2024). The environment has a significant impact on tick microbiome abundance. The prevalence of tick-borne pathogens is closely related to geographic location, climate, and other abiotic factors (Van Treuren et al., 2015).
The sampling area for this study is located on the border of China, Russia, and North Korea. This region is known for its rich biodiversity and favorable climate, which fosters the proliferation of tick communities. A substantial number of migratory birds visit the area each year, making it a potential source of pathogen transmission. This phenomenon renders the region a significant public health concern. Consequently, scientists around the world are engaged in efforts to control the spread of disease by studying the microbiomes of disease vectors (Moreira et al., 2009; Wu et al., 2019; Huang et al., 2020). Of particular importance is the elucidation of the relationship between vertebrate pathogenic infections and the arthropod microbiomes of vectors. Through the analysis of a vertical transmission model in Rickettsia raoultii, it has been demonstrated that interactions between rickettsiae and tick microbiome components contribute to the horizontal transmission of pathogenic rickettsiae (Du et al., 2023). In Ixodes scapularis, infection by anamorphs disrupts midgut microbial homeostasis, thereby facilitating the colonization by these anamorphs (Abraham et al., 2017). Conversely, the microbiome of the arthropod vector significantly influences the susceptibility of pathogens to vector-borne transmission. A substantial body of evidence has demonstrated that the gut microbial homeostasis of arthropods has a significant impact on influencing the colonization of the gut by pathogens (Gonzalez-Ceron et al., 2003; Finney et al., 2015; Gall et al., 2016; Pavanelo et al., 2020). Multiple arthropod endosymbionts show varying degrees of correlation with pathogens (Novakova et al., 2017; Budachetri et al., 2018), suggesting that the vector microbiome may have a direct influence on vectorial competence. Recently, a study conducted in the border region of Yunnan, China, discovered a mosquito gut commensal that is effective in blocking mosquito-borne viruses, representing a novel method for controlling mosquito-borne viral transmission (Zhang et al., 2024). The aforementioned evidence suggests the presence of intricate interactions between the host microbiome and pathogens. Therefore, understanding the composition of the microbiome is imperative for its rational application.
The objective of this study is to analyze the bacterial community composition of tick vectors in the region at the border of China, Russia, and North Korea in order to elucidate the distribution of tick-borne pathogens in the area and to collect fundamental data for the prevention and control of tick-borne diseases.
2 Materials and methods
2.1 Tick collection, nucleic acid preparation, and high-throughput sequencing
Ticks were collected from April to May 2022 using standard dragging methods with corduroy cloths in low-lying scrub vegetation and forest-grassland ecotones in the Yanbian Korean Autonomous Prefecture, Jilin Province, China. Collections were conducted once a month at three sampling sites, namely Yanji (43°48′N, 129°23′E), Tumen (42°58′N, 129°50′E), and Longjing (42°46′N, 129°26′E), resulting in a total of two sampling rounds at each site (Figure 1). A total of 442 questing ticks were collected, including 326 Dermacentor silvarum, 54 Haemaphysalis japonica, and 62 Haemaphysalis concinna (Table 1). The collected ticks were placed in 1.5 mL centrifuge tubes based on their morphological characteristics for analysis of mitochondrial 16S ribosomal DNA (16S rDNA) gene sequences and species identification (Black and Piesman, 1994; Jia et al., 2020). The ticks were then cleaned by vortexing for 30 s in a 1% bleach solution (Binetruy et al., 2019b). This was followed by three consecutive 1-min washes using sterilized, DNA-free water. After washing, the samples were dried using sterile filter paper and stored at −80°C. Sequencing was performed on 142 single-tick pools stratified by species and sex (Table 2). Personalbio Co., Ltd. managed the entire workflow, including cryogenic homogenization in lysis buffer, DNA extraction from supernatants using DNeasy Blood & Tissue Kit (Qiagen, Hilden, North Rhine-Westphalia, Germany) amplification of bacterial 16S rRNA V3–V4 regions with primers 338F (5′-ACTCCTACGGGAGGCAGCA-3′)/806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Claesson et al., 2009), library preparation with TruSeq Nano Kit (Illumina, San Diego, California, CA, United States), and paired-end sequencing (2 × 250 bp) on an Illumina NovaSeq 6000 platform (Klindworth et al., 2013; Bonnet et al., 2017). Post-sequencing, DNA aliquots were transported under cold-chain conditions for subsequent pathogen PCR validation. Sequencing datasets were classified into non-infected and infected groups based on PCR validation and species metadata, with detailed stratification criteria provided in Supplementary Table S1. The remaining tick samples were individually placed in disposable zirconia bead grinding tubes and ground using a low-temperature tissue grinding homogenizer. The genomic DNA from ticks was then extracted using a genomic DNA extraction kit (Tiangen, China).
Figure 1
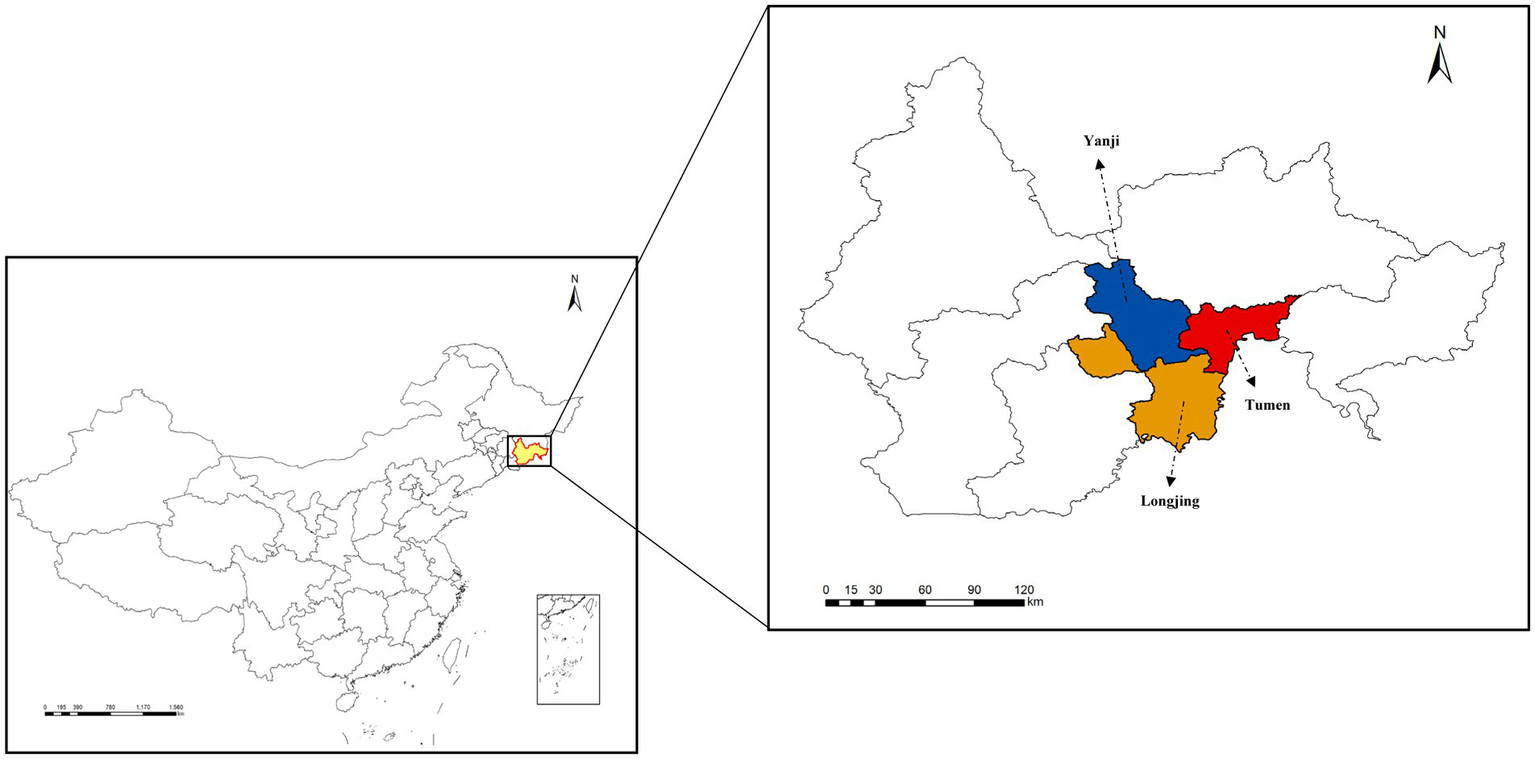
Map of the study area. Yanbian Korean Autonomous Prefecture of Jilin Province, China. Black arrows indicate the sampling locations.
Table 1
| Species | No. of males | No. of females | No. of infected (%) | |||
|---|---|---|---|---|---|---|
| SFG Rickettsia | Anaplasma | |||||
| Male | Female | Male | Female | |||
| D. silvarum | 149 | 177 | 48.3 | 55.9 | 61.7 | 66.1 |
| H. japonica | 17 | 37 | 17.6 | 18.9 | 11.7 | 16.2 |
| H. concinna | 39 | 23 | 33.3 | 17.3 | 23.0 | 17.3 |
| Total | 442 | 44.6 | 51.9 | |||
Number of tick samples and prevalence of tick-borne pathogens in this study.
Table 2
| Samples ID | Number | Species | Sex |
|---|---|---|---|
| DSMt 1–32 | 32 | D. silvarum | Male |
| DSFt 33–65 | 33 | D. silvarum | Female |
| HJMt 66–76 | 11 | H. japonica | Male |
| HJFt 77–99 | 23 | H. japonica | Female |
| HCMt 100–124 | 25 | H. concinna | Male |
| HCFt 125–142 | 18 | H. concinna | Female |
Information on tick samples for high-throughput sequencing.
2.2 Bioinformatics and statistical analyses
Microbiome bioinformatics analysis was performed using QIIME2 (v2019.4) (Caporaso et al., 2010; Rai et al., 2021). Raw sequences were demultiplexed, trimmed of primers using the cutadapt plugin, and processed through the DADA2 plugin (Callahan et al., 2016) for quality filtering, denoising, merging, and chimera removal to generate a non-singleton amplicon sequence variant (ASV) table. Taxonomy was assigned to amplicon sequence variants (ASVs) using the classify-sklearn naïve Bayes taxonomy classifier in the feature-classifier plugin (Bokulich et al., 2018) against the Greengenes 13.8 Database (DeSantis et al., 2006), excluding mitochondrial, chloroplast, and unassigned sequences, along with rare ASVs (relative abundance <0.005%) (Bokulich et al., 2013). Alpha diversity metrics (ACE/Observed/Fisher’s alpha/Shannon/Simpson/Chao1) were calculated on rarefied ASV tables (10,000 reads/sample) and compared across groups using the Wilcoxon rank-sum test with Benjamini–Hochberg false discovery rate correction. Beta diversity analysis based on Bray–Curtis distances revealed significant differences. For group comparisons, we performed Kruskal–Wallis tests with Benjamini–Hochberg correction (Benjamini and Hochberg, 2018) and PERMANOVA (Anderson, 2001). LEfSe analysis (Segata et al., 2011) identified differentially abundant taxa using Kruskal–Wallis screening (p < 0.05), LDA effect size >2.0, and all-against-all validation. Group-specific ASVs, identified via ASV-level Venn diagrams, were quantitatively compared between infected and non-infected groups using the Mann–Whitney U-test. Dominant phyla and genera (>1% mean abundance) were visualized in compositional bar plots (ggplot2), while the microbial community structure was illustrated through PCoA ordinations (QIIME2 View) (Asnicar et al., 2015).
2.3 Pathogen detection, phylogenetics, and prevalence analysis
The prevalence of two pathogens was detected by PCR amplification of the gene fragments of spotted fever group rickettsia (SFGR) (ompA) and Anaplasma spp. (16S rRNA). The sequences of the primers are shown in Supplementary Table S2. Target DNA fragments purified with a Gel Extraction Kit (Omega, Norcross, Georgia, GA, United States) were ligated into the pMD19-T vector (Takara, Japan) via TA cloning and transformed into E. coli DH5α competent cells (Tiangen, Beijing, China). Positive clones selected by antibiotic resistance and colony PCR underwent plasmid extraction using the Omega Plasmid Mini Kit (Omega, Norcross, Georgia, GA, United States), followed by Sanger sequencing at Jilin kumei Biotechnology Co., Ltd. The corrected sequences were then searched for similarity in the GenBank database using the National Center for Biotechnology Information (NCBI) Basic Local Alignment Search Tool (BLAST) search engine. Then, the representative sequences of the pathogens were aligned using MEGA11 software, and a phylogenetic tree was constructed using the neighbor-joining method, with the number of bootstrap replicates set to 1,000 and the Kimura’s two-parameter model. Differences in pathogen positivity rates between tick species were analyzed using Pearson’s chi-squared test, with statistical significance defined as a two-tailed p-value of <0.05.
3 Results
3.1 Infection of ticks with pathogens
PCR-based surveillance revealed an overall Anaplasma positivity rate of 51.9% (Table 1), with D. silvarum exhibiting a significantly higher infection prevalence than other tick species (χ2 = 12.302, df = 1, p < 0.001). For D. silvarum, female ticks demonstrated a marginally elevated positivity rate (66.1%) compared to male ticks (61.7%). Phylogenetic analysis of representative Anaplasma 16S rRNA sequences indicated 99.8–100% similarity to GenBank references, clustering in a monophyletic clade with Anaplasma capra isolates from South Korea (LC432117.1), Qinghai (MG940873.1), and Xian (MG869483.1), China (Figure 2A). Concurrently, SFGR showed an overall positivity rate of 44.6%, with the highest prevalence observed in D. silvarum female ticks (55.9%) and the lowest in H. concinna female ticks (17.3%). Comparative analysis of SFGR ompA gene sequences revealed 97.3–99.37% similarity to GenBank entries, with the closest phylogenetic proximity to Rickettsia raoultii from Yunnan Province, China (MN550901.1). These sequences formed a distinct clade that included R. raoultii strains from Russia (EU036986.1) and Thailand (OM281195.1) (Figure 2B).
Figure 2
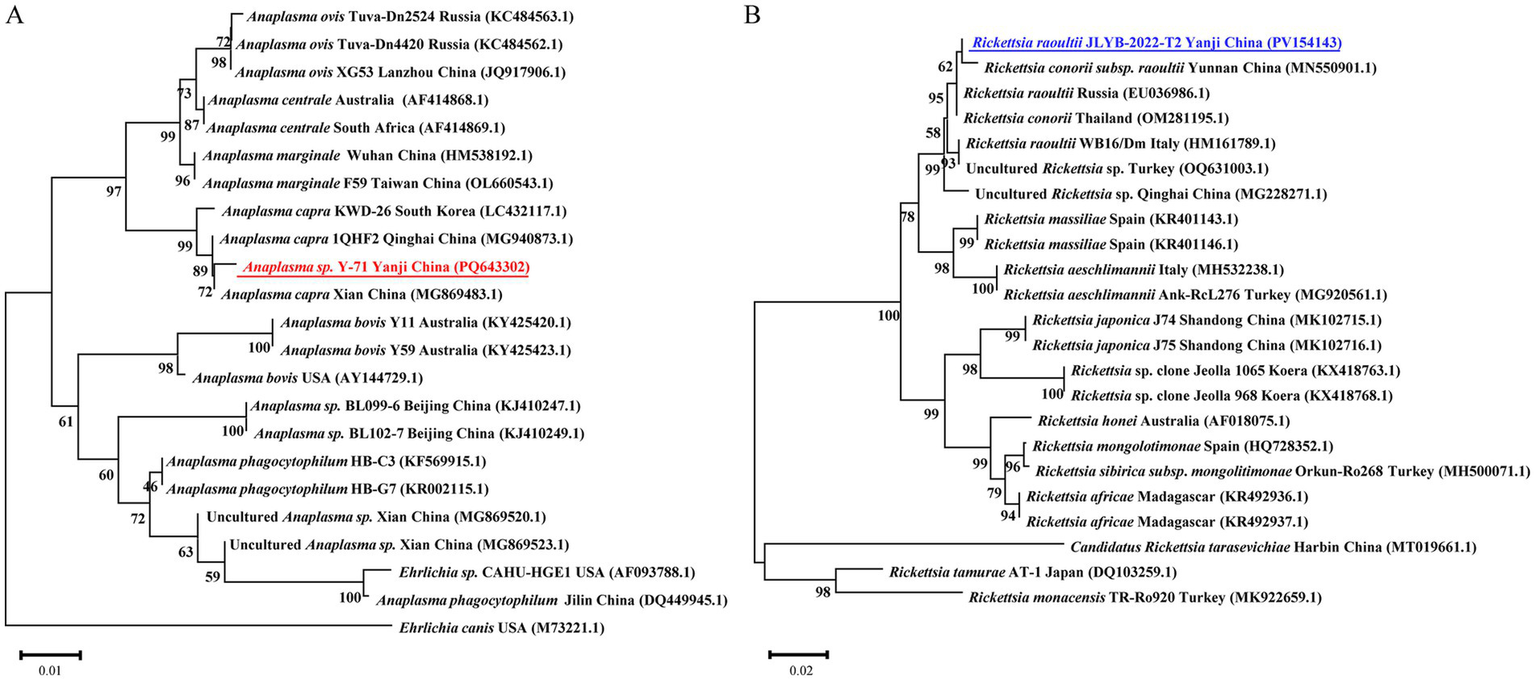
Neighbor-joining phylogenies of (A)Anaplasma spp. 16S rRNA gene sequences and (B) spotted fever group rickettsia ompA. Bootstrap support values from 1,000 replicates are indicated at branch nodes. Sequences from this study are bolded in blue (A; PV154143) and red (B; PQ643302).
3.2 Diversity of microbiota in ticks
To investigate the diversity of microbial communities in ticks infected with vertebrate pathogens, we analyzed the microbiomes of 142 tick specimens. The presence of Rickettsia and Anaplasma pathogens in individual samples was determined using PCR methods. Figure 3A illustrates the distribution of alpha diversity across experimental groups. There were no statistically significant differences in alpha diversity between SFGR-infected and non-infected groups across tick species (Mann–Whitney U-test, p > 0.05). However, Anaplasma-infected groups exhibited significantly higher alpha diversity indices compared to their non-infected counterparts (p < 0.01, Benjamini–Hochberg corrected). Notably, this trend remained consistent across all three tick species investigated. Beta diversity analysis revealed a significant divergence in the microbial community structure among the tick species (PERMANOVA, R2 = 0.32, p < 0.001). However, congeneric Haemaphysalis species (H. japonica vs. H. concinna) exhibited closer clustering in the PCoA ordination based on Bray–Curtis dissimilarity (Figure 3B). Within each tick species, microbial communities showed minimal separation between experimental groups (infected vs. non-infected), indicating that pathogen exposure did not significantly alter β-diversity patterns (PERMANOVA, R2 = 0.02–0.05, p > 0.1).
Figure 3
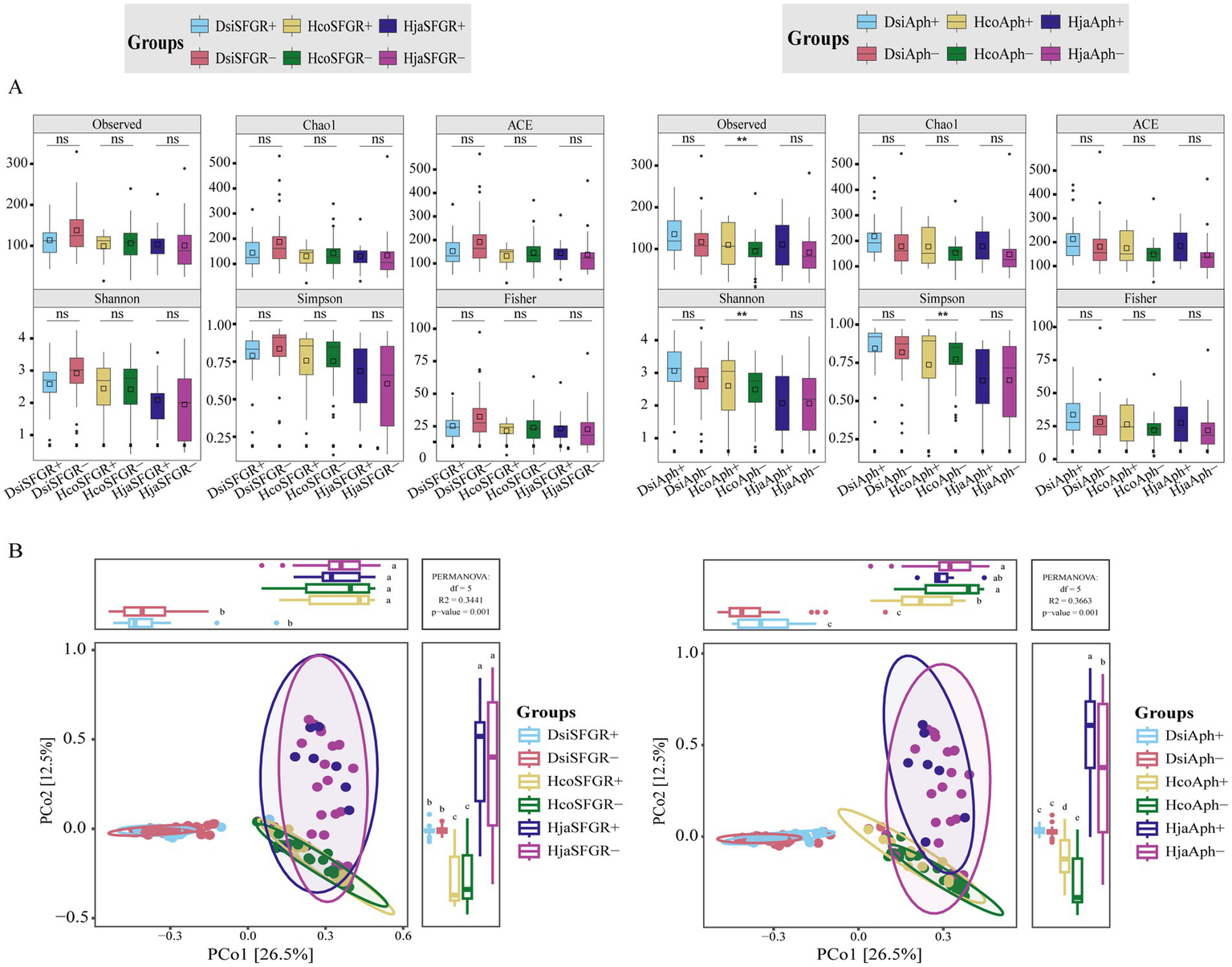
Tick microbiota diversity in pathogen-infected and uninfected groups. (A) α-diversity comparisons (six indices): SFGR-infected vs. SFGR-uninfected, Anaplasma-infected vs. Anaplasma-uninfected. Significance: Mann–Whitney U-test with Benjamini–Hochberg (**p < 0.01, ns: not significant). (B) β-diversity: Bray–Curtis PERMANOVA for each pathogen. Dsi, Dermacentor silvarum; Hco, Haemaphysalis concinna; Hja, Haemaphysalis japonica.
3.3 Composition of microbiota in ticks
Figure 4A presents an amplicon sequence variant (ASV)-level Venn diagram comparing the microbial composition across infection status groups. The analysis revealed that non-infected groups had more unique ASVs than their pathogen-infected counterparts (mean ± SEM: 1111.0 ± 78.5 vs. 410.3 ± 47.7; Mann–Whitney U-test, p < 0.01). This pattern remained consistent across all analyzed tick species, demonstrating enhanced microbial specificity in non-infected arthropods. A total of 5,954 ASVs were classified into 33 phyla, 362 families, and 804 genera. Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes were the dominant phyla. In addition, other phyla, including Planctomycetes, TM7, Thermi, Acidobacteria, Fusobacteria, and Gemmatimonadetes, were observed. At the phylum level, Proteobacteria, Firmicutes, and Actinobacteria dominated the microbial profiles of all three tick species (Figure 4B). Notably, D. silvarum exhibited a significantly higher abundance of Firmicutes (18.3 ± 2.1%) than the Haemaphysalis species. Genus-level profiling identified Pseudomonas, Coxiella, Rhodococcus, and Carnobacterium as core taxa. A notable observation was that Rickettsia-infected ticks exhibited a significantly higher relative abundance of Pseudomonas compared to non-infected controls, whereas Anaplasma-infected ticks showed an inverse trend with reduced Pseudomonas population (Figure 4C).
Figure 4
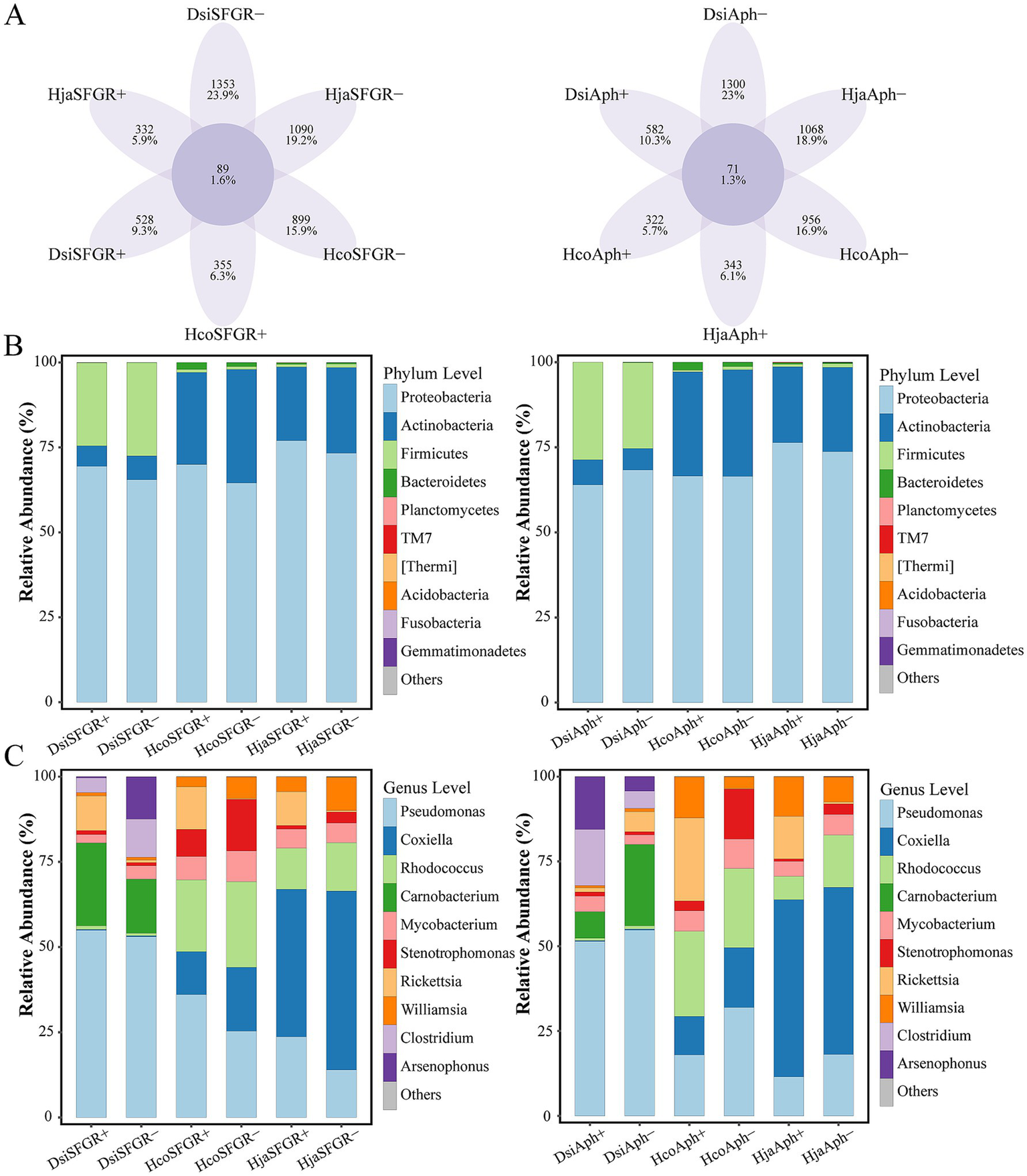
Microbial community divergence across taxonomic ranks in ticks. (A) Comparative analysis of unique and shared amplicon sequence variants (ASVs) between pathogen-infected and non-infected groups. (B) Phylum-level and (C) genus-level differential abundance of microbiota between infected and non-infected groups. The top 10 most abundant phyla (B) and genera (C) are displayed. Dsi, Dermacentor silvarum; Hco, Haemaphysalis concinna; Hja, Haemaphysalis japonica.
3.4 Impact of microbiota on vectors
LEfSe was employed to analyze how the microbiota varied across groups of different species. The results revealed differences in bacterial community composition between each tick species. In H. japonica, LEfSe analysis (LDA—linear discriminant analysis, LDA score >2, p < 0.05) identified five significantly divergent taxonomic units at the genus level, while SFGR-infected samples exhibited specific enrichment of microbial taxa, including Brevundimonas, Clavibacter, Hyphomicrobium, Sphingopyxis, and Pediococcus. The results indicate that these taxa are potential biomarkers of infection status. Non-infected samples were predominantly enriched in Caulobacteraceae, implying their antagonistic role in pathogen suppression or host homeostasis (Figure 5A). Notably, Sphingomonas and Sphingopyxis showed specific enrichment in SFGR-infected D. silvarum and H. concinna, respectively (Figures 5B–C). When grouped by the Anaplasma infection status, the genus of Spirosoma emerged as a conserved potential biomarker in both Haemaphysalis tick species (H. japonica and H. concinna). Notably, no significant enrichment of Spirosoma was detected in infected D. silvarum samples compared to their uninfected counterparts (Figures 5D–F).
Figure 5
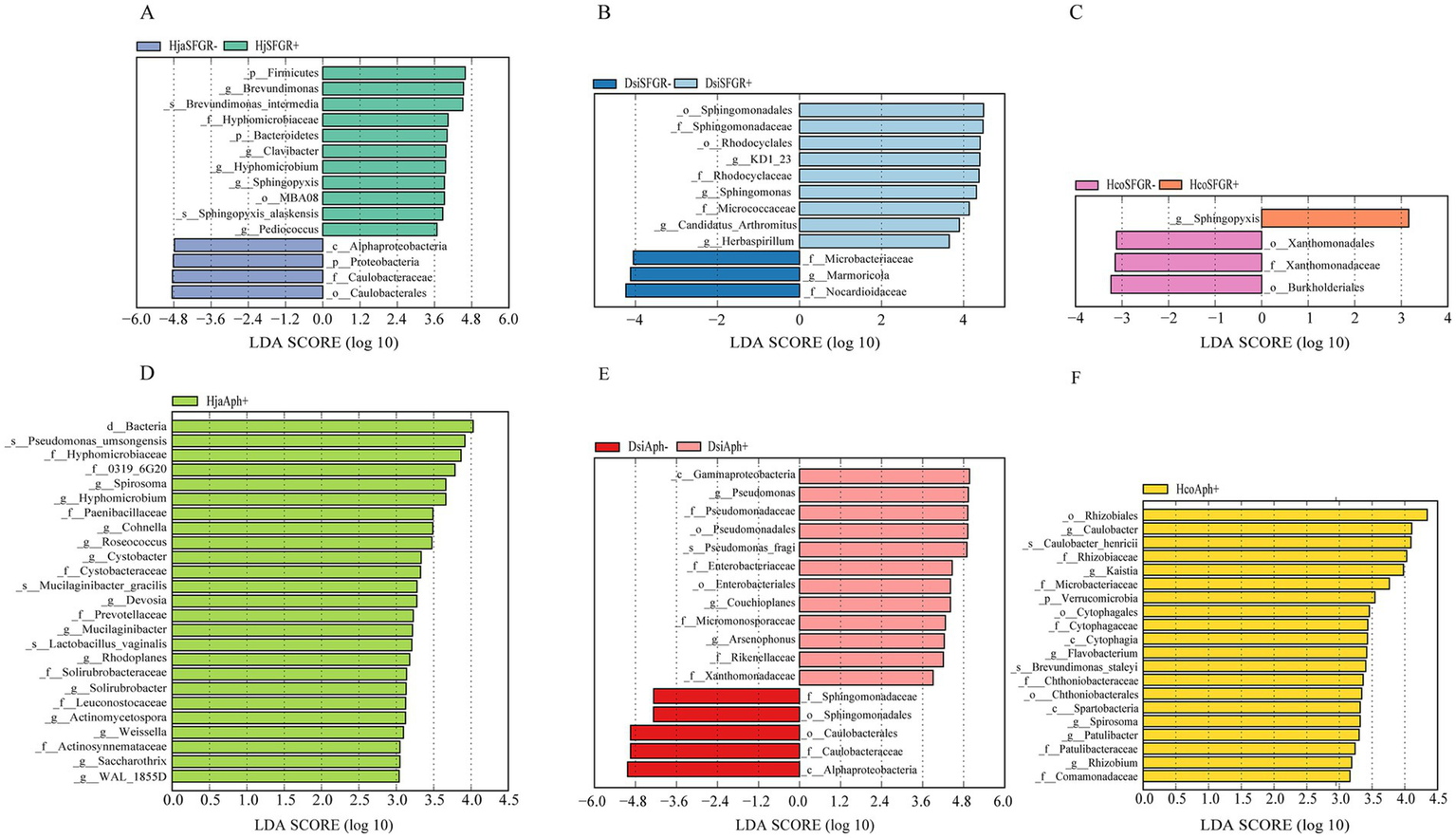
Genus-level biomarkers identified by LEfSE analysis in three tick species infected with distinct pathogens. (A–F) Differential genera corresponding to infection of Haemaphysalis japonica, Dermacentor silvarum, and Haemaphysalis concinna with spotted fever group Rickettsia or Anaplasma. Dermacentor silvarum; Hco, Haemaphysalis concinna; Hja, Haemaphysalis japonica.
4 Discussion
In recent years, there has been growing interest in the microbial communities associated with disease vectors. This interest stems from the understanding that bacterial interactions can influence the survival and transmission of pathogens (Wu et al., 2019; Laukaitis and Macaluso, 2021). Despite this, many arthropod-associated human diseases remain undiagnosed; furthermore, our knowledge about the prevalence, diversity, and pathogenicity of novel arthropod-borne pathogens is limited. This highlights the necessity for ongoing microbiological surveillance of vectors. Differences in microbiota composition have been extensively documented in other vector arthropods, including those between vector species, sexes, organs, and different developmental stages (Muturi et al., 2017; Strand, 2018; Choubdar et al., 2021). This study focuses on the differences in microbiota between ticks with regard to two factors: species and pathogens.
Our integrated approach—PCR identification, BLAST alignment, and phylogenetic analysis—revealed two distinct pathogen species: Anaplasma and SFG Rickettsia. This finding aligns with integrated tick surveillance data from the China-Russia-North Korea border regions, which reported a 35.05% prevalence of Anaplasma in five tick species (Min et al., 2025), with phylogenetic clusters showing 99.2–99.7% identity to A. capra strains from domestic ruminants in central China (GenBank MT799937, MG869594). Three SFG Rickettsia genotypes were concurrently identified, including strains demonstrating 98.4–99.1% sequence homology to Siberian tick isolates (MK304548) and Turkish human clinical variants (MG920563). While these cross-jurisdictional genotypic patterns suggest potential pathogen dispersal across the Tumen River delta, conclusive validation remains constrained by the exclusive sampling of Chinese border territories in existing datasets. A. capra exhibits a global distribution pattern (Altay et al., 2022; Numan et al., 2023), with China being both its initial discovery site and major endemic area (Liu et al., 2012). This pathogen infects diverse hosts, including goats, sheep, cattle, and wildlife (Yang et al., 2016; Sahin et al., 2022). Since the first report of human infection in 2015 (Li et al., 2015), its pathogenicity has been confirmed by clinical manifestations such as fever, headache, fatigue, and occasional neurological involvement. A. capra strains from China exhibit significant genetic diversity, while retaining >99% 16S rRNA homology with East Asian isolates (Zhang et al., 2020)—a pattern consistent with our phylogenetic analyses, suggesting trans-species transmission networks (Shi et al., 2019; Altay et al., 2024). Intriguingly, South Korean surveillance detected a 17.7% prevalence in Hydropotes inermis argyropus (water deer) populations, along with two novel variants (Cheongju and Chungbuk isolates) (Amer et al., 2019). Of particular epidemiological significance, co-infection with A. capra and R. raoultii was identified in H. longicornis parasitizing wildlife hosts, highlighting the complexity of potential mixed pathogen transmission. R. raoultii exhibits a pan-Eurasian distribution that is strongly associated with specific tick vectors. Chinese border surveillance confirms its distribution in the analyzed region to be as follows: 32.25% prevalence in D. silvarum along the Sino-Russian border (Wen et al., 2014), 6.25% in H. erinacei at the Sino-Kazakh border (Guo et al., 2015), and 4% in D. silvarum from Mudanjiang, China (Wang et al., 2021). Notably, Northeast China reported two human cases presenting tender eschars without lymphadenopathy (Jia et al., 2014). In South Korea, H. longicornis exhibited a remarkably high R. raoultii infection rate of 40.9%, with dogs identified as a potential mammalian reservoir. These findings underscore the critical role of this tick species as a zoonotic transmission vector (Seo et al., 2020; Tariq et al., 2021). In Europe (including France, Spain, and Germany), Dermacentor marginatus and Dermacentor reticulatus serve as the principal vectors, with infection rates ranging from 2 to 80% across studies. Clinical cases typically present with hallmark TIBOLA/DEBONEL manifestations (Parola et al., 2009). The high genetic conservation observed across geographical isolates, as demonstrated by ≥99.4% sequence similarity in the ompA gene, underscores their evolutionary stability (Mediannikov et al., 2008). Both A. capra and R. raoultii demonstrate transboundary endemicity influenced by vector distribution, wildlife reservoirs, and geographic factors, necessitating enhanced multinational surveillance and clinical vigilance.
Our results revealed significant differences in the microbial composition of hematophagous ticks infected by various pathogens. This finding aligns with previous studies that have demonstrated how different pathogens can shape the microbial communities within their vectors, potentially influencing vector competence and disease transmission dynamics (Lacroux et al., 2023; Qiu et al., 2024). The intergroup analysis further elucidated the distinct microbial profiles associated with specific pathogens, suggesting that the presence of certain pathogens may drive the selection of particular microbial taxa. For instance, the dominance of specific bacterial genera in arthropods infected with pathogens such as Plasmodium or the dengue virus indicates a potential role for these microbes in modulating the host immune response or enhancing pathogen survival (Wu et al., 2019).
Our integrated analysis revealed that the genus Spirosoma—recently reclassified under Mycoplasmatota—showed significant enrichment in Anaplasma-infected Haemaphysalis ticks (LDA >2, p < 0.05), but was undetectable in Anaplasma-positive Dermacentor cohorts. This host-specific association suggests that the biomarker potential of Spirosoma for Anaplasma surveillance may be restricted to Haemaphysalis ticks. Similarly, Sphingopyxis enrichment was only seen in the Haemaphysalis species and showed no correlation with Rickettsia infection status in the analyzed tick populations (Oren and Garrity, 2021). This bacterium is found in various arthropods and has been extensively documented in ticks (Binetruy et al., 2019a). Spiroplasmataceae is transmitted between arthropods by maternal inheritance and horizontal transfer (Bell-Sakyi et al., 2015).
5 Conclusion
This study investigated the prevalence of SFGR and Anaplasma in ticks collected from the Yanbian region and examined the changes in the microbiome that occur following infection with these pathogens. The results showed that SFGR and Anaplasma had high positivity rates in Yanbian, whereas D. silvarum was the tick species with the highest prevalence of infection and the dominant tick species in the region. Notably, certain bacterial taxa were significantly enriched in infected ticks, suggesting their potential role as biomarkers of pathogen presence. The study highlights the complex interactions between tick-borne pathogens and the tick microbiome, providing insights into the ecological dynamics of pathogen transmission. The findings underscore the importance of monitoring tick microbiomes as part of integrated vector management strategies. Future research should focus on elucidating the functional roles of the identified microbial taxa in pathogen transmission and exploring their potential as targets for tick-borne disease control.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1225783.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
J-qW: Data curation, Writing – original draft, Writing – review & editing. TY: Writing – original draft, Validation. H-yQ: Data curation, Writing – review & editing, Writing – original draft. S-wJ: Writing – review & editing, Methodology. Z-qX: Writing – review & editing, Methodology. Q-cC: Writing – review & editing, Formal analysis. H-fL: Writing – review & editing, Formal analysis. W-fL: Writing – review & editing, Formal analysis. SF: Writing – review & editing, Resources. C-tF: Writing – review & editing, Resources. XG: Writing – review & editing. Z-zH: Writing – review & editing, Data curation. W-nT: Writing – review & editing, Validation. J-xL: Writing – review & editing, Resources. S-jX: Funding acquisition, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by National Natural Science Foundation of China (32360886) and Natural Science Foundation of Jilin Province (YDZJ202201ZYTS616).
Acknowledgments
We would like to express our gratitude to the staff and workers of Yanbian CDC who assisted in the collection of ticks.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1589263/full#supplementary-material
References
1
AbrahamN. M.LiuL.JutrasB. L.YadavA. K.NarasimhanS.GopalakrishnanV.et al. (2017). Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc. Natl. Acad. Sci. U.S.A.114, E781–E790. doi: 10.1073/pnas.1613422114
2
AdrionE. R.AucottJ.LemkeK. W.WeinerJ. P. (2015). Health care costs, utilization and patterns of care following Lyme disease. PLoS One10:e0116767. doi: 10.1371/journal.pone.0116767
3
AltayK.ErolU.SahinO. F. (2022). The first molecular detection of Anaplasma capra in domestic ruminants in the central part of Turkey, with genetic diversity and genotyping of Anaplasma capra. Trop. Anim. Health Prod.54:129. doi: 10.1007/s11250-022-03125-7
4
AltayK.ErolU.SahinO. F. (2024). Anaplasma capra: a new emerging tick-borne zoonotic pathogen. Vet. Res. Commun.48, 1329–1340. doi: 10.1007/s11259-024-10337-9
5
AmerS.KimS.YunY.NaK. J. (2019). Novel variants of the newly emerged Anaplasma capra from Korean water deer (Hydropotes inermis argyropus) in South Korea. Parasit. Vectors12:365. doi: 10.1186/s13071-019-3622-5
6
AndersonM. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol.26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
7
AndreottiR.Perez de LeonA. A.DowdS. E.GuerreroF. D.BendeleK. G.ScolesG. A. (2011). Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol.11:6. doi: 10.1186/1471-2180-11-6
8
AsnicarF.WeingartG.TickleT. L.HuttenhowerC.SegataN. (2015). Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ3:e1029. doi: 10.7717/peerj.1029
9
Bell-SakyiL.PalomarA. M.KazimirovaM. (2015). Isolation and propagation of a Spiroplasma sp. from Slovakian Ixodes ricinus ticks in Ixodes spp. cell lines. Ticks Tick Borne Dis.6, 601–606. doi: 10.1016/j.ttbdis.2015.05.002
10
BenjaminiY.HochbergY. (2018). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
11
BinetruyF.BaillyX.ChevillonC.MartinO. Y.BernasconiM. V.DuronO. (2019a). Phylogenetics of the spiroplasma ixodetis endosymbiont reveals past transfers between ticks and other arthropods. Ticks Tick Borne Dis.10, 575–584. doi: 10.1016/j.ttbdis.2019.02.001
12
BinetruyF.DuprazM.BuysseM.DuronO. (2019b). Surface sterilization methods impact measures of internal microbial diversity in ticks. Parasit. Vectors12:268. doi: 10.1186/s13071-019-3517-5
13
BlackW. C. T.PiesmanJ. (1994). Phylogeny of hard-and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc. Natl. Acad. Sci. U.S.A.91, 10034–10038. doi: 10.1073/pnas.91.21.10034
14
BokulichN. A.KaehlerB. D.RideoutJ. R.DillonM.BolyenE.KnightR.et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome6:90. doi: 10.1186/s40168-018-0470-z
15
BokulichN. A.SubramanianS.FaithJ. J.GeversD.GordonJ. I.KnightR.et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods10, 57–59. doi: 10.1038/nmeth.2276
16
BonnetS. I.BinetruyF.Hernández-JarguínA. M.DuronO. (2017). The tick microbiome: why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front. Cell. Infect. Microbiol.7:236. doi: 10.3389/fcimb.2017.00236
17
BoulangerN.InsonereJ. L.Van BlerkS.BarthelC.SerresC.RaisO.et al. (2023). Cross-alteration of murine skin and tick microbiome concomitant with pathogen transmission after Ixodes ricinus bite. Microbiome11:250. doi: 10.1186/s40168-023-01696-7
18
BudachetriK.BrowningR. E.AdamsonS. W.DowdS. E.ChaoC.-C.ChingW.-M.et al. (2014). An insight into the microbiome of the Amblyomma maculatum (acari: Ixodidae). J. Med. Entomol.51, 119–129. doi: 10.1603/ME12223
19
BudachetriK.KumarD.CrispellG.BeckC.DaschG.KarimS. (2018). The tick endosymbiont Candidatus midichloria mitochondrii and selenoproteins are essential for the growth of Rickettsia parkeri in the Gulf Coast tick vector. Microbiome6:141. doi: 10.1186/s40168-018-0524-2
20
CallahanB. J.McMurdieP. J.RosenM. J.HanA. W.JohnsonA. J.HolmesS. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods13, 581–583. doi: 10.1038/nmeth.3869
21
CaporasoJ. G.KuczynskiJ.StombaughJ.BittingerK.BushmanF. D.CostelloE. K.et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods7, 335–336. doi: 10.1038/nmeth.f.303
22
ChoubdarN.KarimianF.KooshaM.OshaghiM. A. (2021). An integrated overview of the bacterial flora composition of Hyalomma anatolicum, the main vector of CCHF. PLoS Negl. Trop. Dis.15:e0009480. doi: 10.1371/journal.pntd.0009480
23
ClaessonM. J.O’SullivanO.WangQ.NikkiläJ.MarchesiJ. R.SmidtH.et al. (2009). Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One4:e6669. doi: 10.1371/journal.pone.0006669
24
Dantas-TorresF.ChomelB. B.OtrantoD. (2012). Ticks and tick-borne diseases: a one health perspective. Trends Parasitol.28, 437–446. doi: 10.1016/j.pt.2012.07.003
25
DeSantisT. Z.HugenholtzP.LarsenN.RojasM.BrodieE. L.KellerK.et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with arb. Appl. Environ. Microbiol.72, 5069–5072. doi: 10.1128/aem.03006-05
26
DuL. F.ZhangM. Z.YuanT. T.NiX. B.WeiW.CuiX. M.et al. (2023). New insights into the impact of microbiome on horizontal and vertical transmission of a tick-borne pathogen. Microbiome11:50. doi: 10.1186/s40168-023-01485-2
27
DuronO.GottliebY. (2020). Convergence of nutritional symbioses in obligate blood feeders. Trends Parasitol.36, 816–825. doi: 10.1016/j.pt.2020.07.007
28
DuronO.MorelO.NoelV.BuysseM.BinetruyF.LancelotR.et al. (2018). Tick-bacteria mutualism depends on b vitamin synthesis pathways. Curr. Biol.28:e1895, 1896–1902.e5. doi: 10.1016/j.cub.2018.04.038
29
FinneyC. A.KamhawiS.WasmuthJ. D. (2015). Does the arthropod microbiota impact the establishment of vector-borne diseases in mammalian hosts?PLoS Pathog.11:e1004646. doi: 10.1371/journal.ppat.1004646
30
GallC. A.ReifK. E.ScolesG. A.MasonK. L.MouselM.NohS. M.et al. (2016). The bacterial microbiome of Dermacentor andersoni ticks influences pathogen susceptibility. ISME J.10, 1846–1855. doi: 10.1038/ismej.2015.266
31
Gonzalez-CeronL.SantillanF.RodriguezM. H.MendezD.Hernandez-AvilaJ. E. (2003). Bacteria in midguts of field-collected Anopheles albimanus block plasmodium vivax sporogonic development. J. Med. Entomol.40, 371–374. doi: 10.1603/0022-2585-40.3.371
32
GrandiG.ChiappaG.UllmanK.LindgrenP. E.OlivieriE.SasseraD.et al. (2023). Characterization of the bacterial microbiome of Swedish ticks through 16S rRNA amplicon sequencing of whole ticks and of individual tick organs. Parasit. Vectors16:39. doi: 10.1186/s13071-022-05638-4
33
GuoL. P.MuL. M.XuJ.JiangS. H.WangA. D.ChenC. F.et al. (2015). Rickettsia raoultii in Haemaphysalis erinacei from marbled polecats, China-Kazakhstan border. Parasit. Vectors8:461. doi: 10.1186/s13071-015-1065-1
34
HuangW.WangS.Jacobs-LorenaM. (2020). Use of microbiota to fight mosquito-borne disease. Front. Genet.11:196. doi: 10.3389/fgene.2020.00196
35
JiaN.WangJ.ShiW.DuL.SunY.ZhanW.et al. (2020). Large-scale comparative analyses of tick genomes elucidate their genetic diversity and vector capacities. Cell182, 1328–1340.e13. doi: 10.1016/j.cell.2020.07.023
36
JiaN.ZhengY. C.MaL.HuoQ. B.NiX. B.JiangB. G.et al. (2014). Human infections with Rickettsia raoultii, China. Emerg. Infect. Dis.20, 866–868. doi: 10.3201/eid2005.130995
37
KlindworthA.PruesseE.SchweerT.PepliesJ.QuastC.HornM.et al. (2013). Evaluation of general 16s ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res.41:e1. doi: 10.1093/nar/gks808
38
LacrouxC.BonnetS.PouydebatE.BuysseM.RaholaN.RakotobeS.et al. (2023). Survey of ticks and tick-borne pathogens in wild chimpanzee habitat in western Uganda. Parasit. Vectors16:22. doi: 10.1186/s13071-022-05632-w
39
LaukaitisH. J.MacalusoK. R. (2021). Unpacking the intricacies of Rickettsia-vector interactions. Trends Parasitol.37, 734–746. doi: 10.1016/j.pt.2021.05.008
40
LiH.ZhengY. C.MaL.JiaN.JiangB. G.JiangR. R.et al. (2015). Human infection with a novel tick-borne Anaplasma species in China: a surveillance study. Lancet Infect. Dis.15, 663–670. doi: 10.1016/s1473-3099(15)70051-4
41
LiuZ.MaM.WangZ.WangJ.PengY.LiY.et al. (2012). Molecular survey and genetic identification of Anaplasma species in goats from central and southern China. Appl. Environ. Microbiol.78, 464–470. doi: 10.1128/AEM.06848-11
42
Madison-AntenucciS.KramerL. D.GebhardtL. L.KauffmanE. (2020). Emerging tick-borne diseases. Clin. Microbiol. Rev.33:e00083. doi: 10.1128/cmr.00083-18
43
Maldonado-RuizL. P.Montenegro-CadenaL.BlattnerB.MenghwarS.ZurekL.Londono-RenteriaB. (2019). Differential tick salivary protein profiles and human immune responses to lone star ticks (Amblyomma americanum) from the wild vs. a laboratory colony. Front. Immunol.10:1996. doi: 10.3389/fimmu.2019.01996
44
MediannikovO.MatsumotoK.SamoylenkoI.DrancourtM.RouxV.RydkinaE.et al. (2008). Rickettsia raoultii sp. nov., a spotted fever group Rickettsia associated with Dermacentor ticks in Europe and Russia. Int. J. Syst. Evol. Microbiol.58, 1635–1639. doi: 10.1099/ijs.0.64952-0
45
MinP.SongJ.ZhaoS.MaZ.MengY.TangZ.et al. (2025). Tick species, tick-borne pathogen distribution and risk factor analysis in border areas of China, Russia and North Korea. Front. Vet. Sci.12:1529253. doi: 10.3389/fvets.2025.1529253
46
MoreiraL. A.Iturbe-OrmaetxeI.JefferyJ. A.LuG.PykeA. T.HedgesL. M.et al. (2009). A wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and plasmodium. Cell139, 1268–1278. doi: 10.1016/j.cell.2009.11.042
47
MuturiE. J.RamirezJ. L.RooneyA. P.KimC. H. (2017). Comparative analysis of gut microbiota of mosquito communities in central Illinois. PLoS Negl. Trop. Dis.11:e0005377. doi: 10.1371/journal.pntd.0005377
48
NovakovaE.WoodhamsD. C.Rodriguez-RuanoS. M.BruckerR. M.LeffJ. W.MaharajA.et al. (2017). Mosquito microbiome dynamics, a background for prevalence and seasonality of west nile virus. Front. Microbiol.8:526. doi: 10.3389/fmicb.2017.00526
49
NumanM.AlouffiA.AlmutairiM. M.TanakaT.AhmedH.AkbarH.et al. (2023). First detection of Theileria sinensis-like and Anaplasma capra in Ixodes kashmiricus: with notes on cox1-based phylogenetic position and new locality records. Animals13:3232. doi: 10.3390/ani13203232
50
OrenA.GarrityG. M. (2021). Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol.71:e005056. doi: 10.1099/ijsem.0.005056
51
ParolaP.RoveryC.RolainJ. M.BrouquiP.DavoustB.RaoultD. (2009). Rickettsia slovaca and R. raoultii in tick-borne rickettsioses. Emerg. Infect. Dis.15, 1105–1108. doi: 10.3201/eid1507.081449
52
PavaneloD. B.SchroderN. C. H.Pin VisoN. D.MartinsL. A.MalossiC. D.GallettiM.et al. (2020). Comparative analysis of the midgut microbiota of two natural tick vectors of Rickettsia rickettsii. Dev. Comp. Immunol.106:103606. doi: 10.1016/j.dci.2019.103606
53
QiuH. Y.LvQ. B.WangC. R.JuH.LuoC. F.LiuS. S.et al. (2024). Microbiota profile in organs of the horseflies (Diptera: Tabanidae) in northeastern China. Front. Microbiol.15:1467875. doi: 10.3389/fmicb.2024.1467875
54
RaiS. N.QianC.PanJ.RaiJ. P.SongM.BagaitkarJ.et al. (2021). Microbiome data analysis with applications to pre-clinical studies using QIIME2: statistical considerations. Genes Dis.8, 215–223. doi: 10.1016/j.gendis.2019.12.005
55
SahinO. F.ErolU.AltayK. (2022). Buffaloes as new hosts for Anaplasma capra: molecular prevalence and phylogeny based on gtlA, groEL, and 16S rRNA genes. Res. Vet. Sci.152, 458–464. doi: 10.1016/j.rvsc.2022.09.008
56
SegataN.IzardJ.WaldronL.GeversD.MiropolskyL.GarrettW. S.et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol.12:R60. doi: 10.1186/gb-2011-12-6-r60
57
SeoM. G.KwonO. D.KwakD. (2020). High prevalence of Rickettsia raoultii and associated pathogens in canine ticks, South Korea. Emerg. Infect. Dis.26, 2530–2532. doi: 10.3201/eid2610.191649
58
ShiK.LiJ.YanY.ChenQ.WangK.ZhouY.et al. (2019). Dogs as new hosts for the emerging zoonotic pathogen Anaplasma capra in China. Front. Cell. Infect. Microbiol.9:394. doi: 10.3389/fcimb.2019.00394
59
SimoL.KazimirovaM.RichardsonJ.BonnetS. I. (2017). The essential role of tick salivary glands and saliva in tick feeding and pathogen transmission. Front. Cell. Infect. Microbiol.7:281. doi: 10.3389/fcimb.2017.00281
60
SperlingJ. L.Silva-BrandaoK. L.BrandaoM. M.LloydV. K.DangS.DavisC. S.et al. (2017). Comparison of bacterial 16S rRNA variable regions for microbiome surveys of ticks. Ticks Tick Borne Dis.8, 453–461. doi: 10.1016/j.ttbdis.2017.02.002
61
StrandM. R. (2018). Composition and functional roles of the gut microbiota in mosquitoes. Curr. Opin. Insect Sci.28, 59–65. doi: 10.1016/j.cois.2018.05.008
62
TariqM.SeoJ. W.KimD. Y.PanchaliM. J. L.YunN. R.LeeY. M.et al. (2021). First report of the molecular detection of human pathogen Rickettsia raoultii in ticks from the Republic of Korea. Parasit. Vectors14:191. doi: 10.1186/s13071-021-04695-5
63
TokarzR.LipkinW. I. (2021). Discovery and surveillance of tick-borne pathogens. J. Med. Entomol.58, 1525–1535. doi: 10.1093/jme/tjaa269
64
Van TreurenW.PonnusamyL.BrinkerhoffR. J.GonzalezA.ParobekC. M.JulianoJ. J.et al. (2015). Variation in the microbiota of Ixodes ticks with regard to geography, species, and sex. Appl. Environ. Microbiol.81, 6200–6209. doi: 10.1128/AEM.01562-15
65
WangQ.PanY. S.JiangB. G.YeR. Z.ChangQ. C.ShaoH. Z.et al. (2021). Prevalence of multiple tick-borne pathogens in various tick vectors in northeastern China. Vector Borne Zoonotic Dis.21, 162–171. doi: 10.1089/vbz.2020.2712
66
WenJ.JiaoD.WangJ. H.YaoD. H.LiuZ. X.ZhaoG.et al. (2014). Rickettsia raoultii, the predominant Rickettsia found in Dermacentor silvarum ticks in China-Russia border areas. Exp. Appl. Acarol.63, 579–585. doi: 10.1007/s10493-014-9792-0
67
WikelS. (2013). Ticks and tick-borne pathogens at the cutaneous interface: host defenses, tick countermeasures, and a suitable environment for pathogen establishment. Front. Microbiol.4:337. doi: 10.3389/fmicb.2013.00337
68
WuP.SunP.NieK.ZhuY.ShiM.XiaoC.et al. (2019). A gut commensal bacterium promotes mosquito permissiveness to arboviruses. Cell Host Microbe25:e105, 101–112.e5. doi: 10.1016/j.chom.2018.11.004
69
YangJ.LiuZ.NiuQ.LiuJ.HanR.LiuG.et al. (2016). Molecular survey and characterization of a novel Anaplasma species closely related to Anaplasma capra in ticks, northwestern China. Parasit. Vectors9:603. doi: 10.1186/s13071-016-1886-6
70
ZhangY.CuiY.SunY.JingH.NingC. (2020). Novel Anaplasma variants in small ruminants from central China. Front. Vet. Sci.7:580007. doi: 10.3389/fvets.2020.580007
71
ZhangL.HanJ.ZhouQ.HeZ.SunS. W.LiR.et al. (2023). Differential microbial composition in parasitic vs. questing ticks based on 16s next-generation sequencing. Front. Microbiol.14:1264939. doi: 10.3389/fmicb.2023.1264939
72
ZhangL.WangD.ShiP.LiJ.NiuJ.ChenJ.et al. (2024). A naturally isolated symbiotic bacterium suppresses flavivirus transmission by Aedes mosquitoes. Science384:eadn9524. doi: 10.1126/science.adn9524
73
ZhongZ.WangK.WangJ. (2024). Tick symbiosis. Curr. Opin. Insect Sci.62:101163. doi: 10.1016/j.cois.2024.101163
Summary
Keywords
tick, Rickettsia, anaplasmosis, microbiota, 16S rRNA gene, microbiome analysis
Citation
Wang J-q, Yu T, Qiu H-y, Ji S-w, Xu Z-q, Cui Q-c, Li H-f, Liang W-f, Feng S, Fu C-t, Gao X, Han Z-z, Tian W-n, Li J-x and Xue S-j (2025) Differential impact of spotted fever group rickettsia and anaplasmosis on tick microbial ecology: evidence from multi-species comparative microbiome analysis. Front. Microbiol. 16:1589263. doi: 10.3389/fmicb.2025.1589263
Received
07 March 2025
Accepted
22 April 2025
Published
13 May 2025
Volume
16 - 2025
Edited by
Lei Deng, Chinese Academy of Agricultural Sciences, China
Reviewed by
Benjamin Cull, University of Minnesota Twin Cities, United States
Roland Eric Yessinou, University of Abomey-Calavi, Benin
Ratree Takhampunya, Armed Forces Research Institute of Medical Science, Thailand
Artem Rogovsky, Michigan State University, United States
Updates
Copyright
© 2025 Wang, Yu, Qiu, Ji, Xu, Cui, Li, Liang, Feng, Fu, Gao, Han, Tian, Li and Xue.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-jiang Xue, sjxue@ybu.edu.cn
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.