Abstract
The human gut is one of the most densely populated microbial environments, home to trillions of microorganisms that live in harmony with the body. These microbes help with digestion and play key roles in maintaining a balanced immune system and protecting us from harmful pathogens. However, the crowded nature of this ecosystem makes it easier for harmful bacteria to acquire antimicrobial resistance (AMR) genes, which can lead to multidrug-resistant (MDR) infections. The rise of MDR infections makes treatments harder, leading to more extended hospital stays, relapses, and worse outcomes for patients, ultimately increasing healthcare costs and environmental strain. Since many MDR infections are challenging to treat, nosocomial infection control protocols and infection prevention programmes are frequently the only measures in our hands to stop the spread of these bacteria. New approaches are therefore urgently required to prevent the colonization of MDR infections. This review aims to explore the current understanding of antimicrobial resistance pathways, focusing on how the gut microbiota contributes to AMR. We have also emphasized the potential strategies to prevent the spread and colonization of MDR infections.
1 Introduction
The gut microbiome, a complex ecosystem in the gastrointestinal tract, harbours trillions of commensals, symbiotic organisms, including bacteria, viruses, fungi, archaea, and eukaryotes. These organisms contribute intestinal integrity, immunity, metabolism, digestion, mental health, and pathogen defence to the host (Anto and Blesso, 2022; Lane and Yadav, 2020). The microbial composition of each individual is unique and stable, but the significant phyla remain the same, and an individual will conserve over 60% of the gut microbial phylotypes for 2 years (Manichanh et al., 2010). The microbiome encodes nearly three million genes that produce hundreds of metabolites, outnumbering the roughly 23,000 genes in the host genome (Valdes et al., 2018). However, this ecosystem can serve as a reservoir and epicenter for developing antimicrobial resistance (AMR).
AMR, a global health concern of since the mid-20th century, is the development of resistance by microorganisms to the antimicrobial medications that are used to treat them, reducing clinical efficacy and increasing treatment costs (Penders et al., 2013; Prestinaci et al., 2015). Since antibiotics are not pathogen-specific and are prescribed to treat infections leading to overdose, they impact commensal microbiota present in the same habitat (Bag et al., 2019; Llor and Bjerrum, 2014). The continuous emergence of resistant genes and mechanisms contributes to the global spread of AMR. Several diseases previously treated successfully, with any of the several drug classes have developed resistance, making it difficult to inhibit their growth. The emergence of resistant pathogens such as methicillin-resistant Staphylococcus aureus (MRSA), penicillin-resistant and macrolide-resistant Streptococcus pneumoniae, carbapenem-resistant Enterobacteriaceae, third-generation cephalosporin-resistant Klebsiella pneumoniae, cephalosporin-resistant Escherichia coli, carbapenem-resistant and multidrug-resistant Pseudomonas aeruginosa, have all been classified by the World Health Organization as high or critical priorities for the development of new antibiotics (Kessler et al., 2022). AMR is developed by selecting resistant characteristics, which permits organisms to survive and reproduce, resulting in the persistence of resistant populations (Ferri et al., 2017). Resistance arises because of genetic mutation and horizontal gene transfer. Horizontal gene transfer, a prevalent mechanism, occurs due to the acquisition of resistance genes from environmental and microbial reservoirs. Bacteria employ various mechanisms to achieve antimicrobial resistance namely use of efflux pump, altering the antibiotic target (for example, by altering binding sites in ribosomal RNA), reprogramming metabolic pathways, and production of enzyme to inactivate the antibiotic (Wright, 2005). Antibiotic resistance affects both developed and developing nations equally, therefore, it is essential to examine how antibiotic resistance is spreading over the world. The widespread use of antibiotics in hospitals, the general population, and agriculture has increased the stress on selection, leading to the persistence of resistant microbes in high-income nations, necessitating shifting to more expensive, broad-spectrum antibiotics. The need for antibiotics is rising in low- and middle-income nations due to increased incomes, a more significant hospitalisation rate, and a high prevalence of hospital infections (Laxminarayan et al., 2013). By 2050, according to estimates, antibiotic resistance will cause almost 10 million deaths annually and a loss of $100.2 trillion in GDP (Chokshi et al., 2019). It is essential to investigate the key socioeconomic and political factors that influence how quickly AMR spreads in both developed and developing nations (Chokshi et al., 2019). The direct monetary effects of AMR on health care are high expenses associated with expensive and intensive treatments and an increase in resource consumption (Dadgostar, 2019).
Numerous in silico metagenomics studies have confirmed that the human gastrointestinal tract acts as a reservoir for AMR genes, capable of transferring these genes to transient, pathogenic bacteria (Bag et al., 2019; Cheng et al., 2012; Radovanovic et al., 2023; Ghosh et al., 2013). The spread of antibiotic resistance genes (ARGs) is increased in international human interaction, wherein antibiotic-resistant bacteria from one part of the world are swiftly transferred and spread to far-off nations at great geographic distances (Okeke and Edelman, 2001). Since the development of antibiotic resistance increases the probability of therapeutic failure, relapses, extended hospital stays, and poorer clinical outcomes, treating infections caused by multidrug-resistant organisms provides a significant clinical challenge (Gargiullo et al., 2019). Determining the antimicrobial resistome of the human gastrointestinal microflora will, therefore, be of great importance in evaluating the process of resistance genes being transferred among intestinal microorganisms. The ways through which mutualistic and pathogenic bacteria in the human gut potentially exchange antimicrobial resistance genes have been investigated in the current review.
2 Human gut microbiome
The gut microbiome, plays a vital role in the overall wellbeing of the individual, consists of, principally, of five significant phyla of distinct and complex colony of microorganisms. Firmicutes include Lactobacillus, Bacillus, Clostridium, Enterococcus, and Ruminococcus (Kho and Lal, 2018; Rinninella et al., 2019). Bacteroidetes include Bacteroidia, Flavobacteria, Sphingobacteria, and Cytophagia (Thomas et al., 2011). Actinobacteria include Corynebacterium, Propionibacterium, Rothia, Actinomyces, and Bifidobacterium (Wu, 2013). Proteobacteria include Escherichia coli, Salmonella, and Campylobacter (Moon et al., 2018). Verrucomicrobia is primarily represented by Akkermansia muciniphila (Dubourg et al., 2013).
The human gastrointestinal tract (GI tract), with a surface area of 250–400 m2, forms an interface between the host, environmental factors, and antigens. Over the course of a lifetime, the human GI tract processes about 60 tonnes of food and encounters various pathogens that can be detrimental to gut health (Thursby and Juge, 2017). Initially, the newborn gut is aerobic, but the first colonizers, facultative anaerobes, create a new environment with a low level of oxygen, beneficial for the growth of anaerobes such as Bacteroides, Clostridium, and Bifidobacterium spp. (Breitbart et al., 2008; Fouhy et al., 2012; Rodríguez et al., 2015). The sources of this diversity of gut microbes include nutritional, environmental, and maternal factors, gestational age, delivery technique (vaginal birth vs. assisted delivery), feeding (breast milk vs. formula), sanitation, and antibiotic use (Rodríguez et al., 2015; Townsend et al., 2021). Studies show that vaginal delivery exposes newborns to maternal vaginal microbiota (primarily Lactobacilli), whereas caesarean sections result in significantly different microbial populations (McCann et al., 2018; Firoozeh and Zibaei, 2019). By human anatomy, exposure to the mother’s faecal microbiota after birth is a significant means of transmission. One of the bacterial species with the highest likelihood of direct transmission from mother to newborn through faeces is Enterobacteriaceae (Rodríguez et al., 2015). Mother’s breastmilk acts as a vehicle for the vertical transmission of Bifidobacterium, Streptococcus, and Staphylococcus (Hunt et al., 2011).
The structure, diversity, and functional capabilities of the newborn microbiota increase and resemble those of the adult microbiota by the time the child is 2.5 years old, with temporal patterns that are unique to each newborn (Eckburg et al., 2005; Bäckhed, 2011; Firoozeh and Zibaei, 2019). The Bacteroidetes phylum and Clostridium cluster IV are more prevalent in those over 65 than in younger people (Claesson et al., 2011).
Furthermore, the composition of gut microbiome varies significantly among individuals due to genetics and environmental factors such as routine habits, dietary pattern, personal hygiene, health, medications such as antibiotics, and the use of prebiotics and probiotics (Ahmad et al., 2019; Cunningham et al., 2021). Diet is considered one of the key factors affecting the composition of an individual’s microbiota irrespective of age. Seasonal variation in the gut microbiome, influenced by the consumption of fresh foods, leads to shifts in composition, with Bacteroides common in summer and Actinobacteria in winter, indicating the influence of complex carbohydrate intake on microbiome plasticity (Davenport et al., 2014). Furthermore the composition and heterogeneity can be altered in case of obese and nonobese individuals, where more Firmicutes and fewer Bacteroidetes were observed in obese compared to non-obese adults (Pinart et al., 2021). The composition varies widely among ethnic groups and provides more information about the individual influenced by the same geographical area (Deschasaux et al., 2018; Gaulke and Sharpton, 2018; Schnorr et al., 2014). The gut microbiome greatly influences the health, brain, well-being, stress, and anxiety. Social interactions increase the diversity of the microbiome while anxiety and stress decrease the same (Johnson, 2020).
The gut microbiota coevolved with humans and maintains host health by regulating metabolism, physiology, and immune functions (den Besten et al., 2013; Natividad and Verdu, 2013; Singh et al., 2019). According to estimates, the human microbiota contains roughly 1014 microbial cells, with a microbial cell-to-human cell ratio of 1:1 (Sender et al., 2016; Thursby and Juge, 2017). Colonic bacteria produce carbohydrate-active enzymes that convert complex carbohydrates into short-chain fatty acids (SCFAs) like propionate, butyrate, and acetate (Louis et al., 2014; Thursby and Juge, 2017). These SCFAs are absorbed by epithelial cells, regulating gene expression, inflammation, and cell proliferation (Corrêa-Oliveira et al., 2016). Gut anaerobes create acetate, whereas Bacteroidetes and Firmicutes synthesis propionate, butyrate through glycolytic and acetyl-CoA pathways, as well as succinate or propanediol pathways (Louis and Flint, 2009; Louis and Flint, 2017; Macfarlane and Macfarlane, 2003; Morrison and Preston, 2016). Variations in the composition of the gut microbiome can endanger human health, indicating its critical role in human health (Vandenplas et al., 2020) (see Table 1).
Table 1
| S. No. | Factor | Description | Impact on AMR and host | Examples | References |
|---|---|---|---|---|---|
| 1. | Diet | Nutrient intake | Change in microbial community protect from inflammations and non-infectious colonic diseases | Fiber diet, plant or animal based | David et al. (2014), De Filippo et al. (2010), and Wu et al. (2011) |
| 2. | Age | Microbiome changes across lifespan | Different microbiota depending upon age | Infant vs. young vs. elderly | Ghosh et al. (2022) and Li et al. (2024) |
| 3. | Health status | Presence of diseased condition | Maintaining homeostasis, promotes overall health | Inflammatory bowel disease and metabolic disorders | Afzaal et al. (2022) and Shreiner et al. (2015) |
| 4. | Geographical location | Regional differences | Change in microbial diversity | Urban vs. rural areas, western | Gaulke and Sharpton (2018) |
| 5. | Sanitation and hygiene | Access to clean water and sanitation facilities | Shift in microbial diversity | Hand wash | Monira et al. (2023) |
| 6. | Exposure to antibiotics | Misuse of antibiotics | Selects for resistant strains, reduces diversity | Use of tetracycline, amoxycillin, influencing overall microbial community resilience | Nhu and Young (2023) |
| 7. | Lifestyle factors | Habits and behaviors affecting microbiome | Modify the microbial diversity | Smoking, alcohol consumption, sleep deprivation | Ren et al. (2023) |
| 8. | Genetic factors | Genetic makeup of host influence microbiome composition and function | Genetic predisposition to harbour certain resistant strains | Variations in immune response genes | Blekhman et al. (2015) |
| 9. | Immune system | Immune response of the host | Influence the microbial composition | Inflammatory responses, immune tolerance | Zheng et al. (2020) |
| 10. | Mode of delivery | Vaginal or caesarean | Influences the initial gut colonization | Bifidobacterium, Enterococcus spp. | Brinkac et al. (2017), Wen and Duffy (2017), and Zhang et al. (2021) |
| 11. | Feeding method | Breastfeeding or formula | They are one among the first microbes to enter the infant’s body, and they could play an important role in health | Breast milk contains potential probiotic bacteria and IgA antibody | Davis et al. (2022) and Wen and Duffy (2017) |
| 12. | Gender | Biological differences | Differences in microbial diversity and composition | Hormones influence the microbiota | Niemela et al. (2024) and Yoon and Kim (2021) |
Factors influencing gut microbiome.
3 The gut microbiome and antimicrobial resistance
Antibiotic resistance, a severe threat to public health, signals the end of an era of antibiotics as a “golden therapy” and returns us to a time when effective treatments for microbial infections existed (Huddleston, 2014). Infectious disease remains one of the primary causes of death worldwide, pharmaceutical companies have slowed the drug development process, providing only 0.2% of new drugs (Spellberg et al., 2004). Bacteria develop resistance through mechanisms such as horizontal gene transfer, overexpression of efflux pumps, and protection of the drug target site by designing a specific protein (Munita and Arias, 2016).
The gut microbiome, essential for host wellbeing and a reservoir for ARGs are disrupted by dietary modifications, stress, antibiotic use, causing microbial dysbiosis, having detrimental effect on health and reduces resistance to pathogen colonization (Singh et al., 2019; Gargiullo et al., 2019).
The human gut microbiome, which houses 3.3 million non-reductant genes, is estimated to be 150 times larger than the human host (Qin et al., 2010). The confined environments of the diverse microbiome provide favourable conditions for genetic exchange between transitory and resident bacteria, as well as resident microbes (Brinkac et al., 2017). AMR genes in the gut, collectively termed the resistome, are categorized as intrinsic and mobile (Gargiullo et al., 2019; Singh et al., 2019). Intrinsic AMR genes, relatively stationary, in addition to producing a resistant phenotype, help regulate the physiology and metabolism of bacteria (Cox and Wright, 2013). Mobile AMR genes can rapidly spread by horizontal gene transfer occurring either through transformation, conjugation or transmission (Singh et al., 2019; von Wintersdorff et al., 2016). Mobile genetic elements—plasmids, integrons, transposons, genomic islands, are vehicles for transferring AMR genes in the gut microbiota (Table 2).
Table 2
| S. No. | Mechanism | Description | Examples | References |
|---|---|---|---|---|
| 1. | Enzymatic degradation | Bacteria produce enzymes that degrade the antibiotic | Beta-lactamases, carbapenemases | Bush (2018) and Bush and Jacoby (2010) |
| 2. | Efflux pumps | Remove any potentially dangerous molecules from the anterior of the cell | RND and MATE | Ghotaslou et al. (2018) and Soto (2013) |
| 3. | Target modification | Modification of antibiotic target | Methylation of 16S rRNA or 23S rRNA, MRSA (mecA gene) | Peterson and Kaur (2018) |
| 4. | Reduced permeability | Changes in cell membrane permeability | Porin | Delcour (2009) |
| 5. | Biofilm formation | Bacteria form biofilms that protect them from antibiotics and the immune system | Campylobacter jejuni | Buret and Allain (2023) and Grooters et al. (2024) |
| 6. | Horizontal gene transfer | Transfer of ARGs between bacteria via plasmids, transposons, or phages | Conjugation, transformation, transduction | Groussin et al. (2021) and Huddleston (2014), |
| 7. | Antibiotic modification | Enzymatic alteration of the antibiotic (phosphorylation, acetylation, and adenylation) | N-acetyl transferases, O-phosphotransferases, O-adenyltransferases | Peterson and Kaur (2018) |
Mechanism of antibiotic resistance.
3.1 Mechanism
3.1.1 Horizontal gene transfer
3.1.1.1 Conjugation
Conjugation, known as bacterial sex, is a major horizontal gene transfer mechanism where the donor DNA is transferred to the recipient by direct contact via pilus or pore (Guglielmini et al., 2013; Virolle et al., 2020). Conjugation occurs through a series of events, including cell-to-cell contact, the formation of mating pairs, and the horizontal transfer of genetic material, such as plasmids or transposons, into the recipient cell’s cytoplasm (Peterson et al., 2011). Conjugative transposons integrate into new genome locations, facilitating genetic diversity and responsible for developing AMR and virulence (Salyers et al., 1995; Singh et al., 2019). Genetic flux through conjugation can be observed in inflammatory conditions like inflammatory bowel syndrome or infections caused by E. coli or Salmonella spp. (Stecher et al., 2012). The conjugation efficacy of the β-lactamase plasmid was reduced in research by Machado and Sommer (2014) when clinical isolates of E. coli were co-cultured with human intestinal cells that produce protein-based factors. They concluded that any damage to intestinal cells caused by toxins, drugs, or inflammation reduces the production of peptides, thereby promoting conjugation. A study revealed that a transitory intestinal colonization by an animal-derived E. faecium strain that carries mobile elements with the vanA gene resistance to a human-derived E. faecium isolate poses a risk of infection, particularly in immunocompromised patients (Lester et al., 2006).
Rooney et al. (2019) used a triple stage chemostat model of the human gut to demonstrate the colonization, clonal expansion, and transfer of CRE genes from Klebsiella pneumoniae to the microbiota of CRE-negative human faeces.
A mouse model with human-derived microbiota was created in order to evaluate the conjugative transfer of ARGs by E. coli utilizing fluorescently labeled protein in the gut without the use of antibiotic selection pressure. According to their findings, the ARG-bearing RP4 plasmid from E. coli spread to a variety of bacterial taxa, and the model can be used to comprehend the prerequisites for gene transfer and conjugation (Sher et al., 2025).
Factors such as biofilm formation, the density of donor or recipient bacteria, environmental conditions (availability of nutrients, pH, temperature), exposure to medications and preservatives decides the rate of conjugation (Liu et al., 2023). According to the study, the level of antibiotic-induced dysbiosis affects the colonization of Salmonella species in the gut and the conjugative transfer of the multi-drug resistant IncA/C plasmid to commensal E. coli. They also came to the conclusion that using antibiotics ethically is crucial because the latter may cause the dissemination of ARG (Yilmaz et al., 2024). The antibiotic resistance profile of mucin-degrader Akkermansia muciniphila, ubiquitously present in the adult human gut microbiota is poorly understood. Recent studies revealed resistance to quinolones and horizontal gene transfer of sulphonamide and aminoglycoside resistance genes from Salmonella enterica, indicating the need to access the spread of ARGs (Guo et al., 2017).
3.1.1.2 Transformation
It refers to the ability of the bacterial cell to uptake and integrate extracellular DNA enabled by bacterial competence (Finkel and Kolter, 2001). The primary catalyst for transformation in gut microflora includes conditions like nutrition competition or DNA repair as a result of antibiotic damage (Finkel and Kolter, 2001; Huddleston, 2014). Extracellular DNA maintains the structural integrity of intestinal biofilms, suggesting transformation a crucial mechanism for bacterial persistence and adaptation in the gut environment (Licht et al., 1999).
According to Chowdhury et al. (2024), Enterococcus fecium developed kanamycin resistance by transformation in the presence of antibiotics, demonstrating that bacteria in the gut can absorb eARGs from their surroundings. According to the findings, the degree of gene uptake is correlated with antibiotic levels, suggesting that resistance gene acquisition may be facilitated by higher antibiotic concentrations.
3.1.1.3 Transduction
The transfer of bacterial DNA through bacteriophages and are classified into generalized transduction and specialised transduction (Thierauf et al., 2009). There is little knowledge regarding the transmission of the AMR gene by bacteriophages in the gut. In transduction, phages can transfer genes between bacteria without requiring coexistence and can cross taxonomic boundaries (Muniesa et al., 2013). ARG-carrying phages are prevalent in the human gut and other environment and the number rises following an antibiotic exposure (Fernández-Orth et al., 2019). Studies conducted on mouse models have demonstrated that transduction drives genetic diversity in E. coli strains that colonize the gut and can lead to the development of drug resistance in gut bacteria (Frazão et al., 2019).
Studies have reported that on treatment with β-lactam antibiotics, the expression of phage encoded genes in Staphylococcus aureus, responsible for encoding proteins that regulate cell wall metabolism, stress are upregulated (Maiques et al., 2006). Antibiotic treatment results in the abundance of phage-encoded AMR genes increasing the spread within the gut microflora (Modi et al., 2013). For instance, Streptococcus pyogenes emm12 resistance has emerged in multiples due to the phage element Φ HKU.vir, which carries the superantigen gene ssa as well as the spec and DNase genes spd1 (Davies et al., 2015). In metagenomic research, crAssphage—one of the most prevalent phages in the human gut—has been employed as a marker for faecal contamination. The abundance of resistance genes in the environment must be related to faecal contamination rather than environmental selection, according to Karkman’s et al. (2019) analysis. Therefore, in order to prevent erroneous assumptions regarding environmental selection for antibiotic resistance, the degree of faecal contamination must be taken into account (Dutilh et al., 2014; Karkman et al., 2019) (Figure 1).
Figure 1
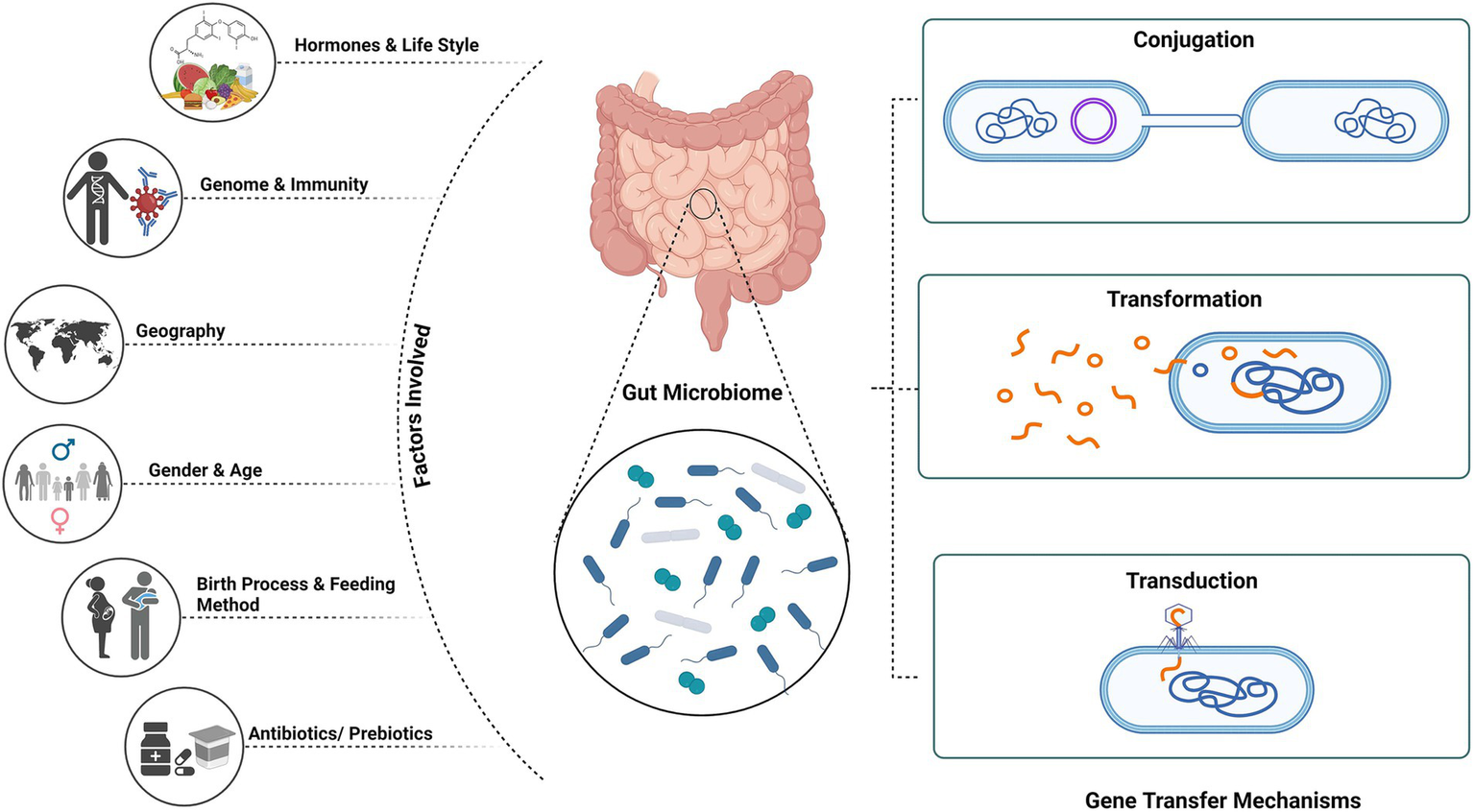
Mechanism of gene transfer and factors affecting gut microbiome. Created using BioRender.com.
3.1.2 Antibiotic and target modification
Exposure protection, a common method of resistance, prevents antibiotic exposure in adjacent sensitive cells by allowing specific bacterial species to degrade antibiotics. The degradation of the antibiotic reduces antibiotic concentrations, which can benefit neighbouring susceptible cells is well recognized and demonstrated using various antimicrobial compounds (Gjonbalaj et al., 2020; Pathak et al., 2023).
The gut microbiota also regulates antibiotic absorption by metabolizing the drug or modifying the intestinal environment, resulting in variations in drug bioavailability, affecting their efficacy and toxicity. Certain bacteria in the gut, for example, can metabolize beta-lactam antibiotics such as penicillin by releasing β-lactamases, rendering them inactive and reducing their potency (Ramirez et al., 2020). The cfxA, cfiA, and cepA genes are associated with resistance to β-lactam antibiotics, while the tetQ gene is associated with resistance to tetracyclines (Lamberte and van Schaik, 2022).
Vancomycin’s interaction with the gut microbiota is one of the most important instances showing how the gut microbiota influences the choice of antibiotic therapy. Vancomycin’s pharmacokinetics and pharmacodynamics can be influenced by the gut microbiota through changes in its distribution, metabolism, and absorption, as well as its capacity to trigger an immunological response. Moreover, vancomycin-induced dysbiosis of the gut microbiota has been linked to heightened vulnerability to Clostridium difficile infection.
Harris et al. (2000) shown that animals express a variety of catecholamine-degrading enzymes throughout the GI tract, particularly in the colon, where the gut microbiome is most abundant.
A study has demonstrated that the intestinal microbiota’s diversity is significantly diminished for at least 28 days following a single dosage of clindamycin, with an ongoing loss of almost 90% of the usual microbial taxa from the cecum. Prior to antibiotic treatment, a fraction of bacterial taxa that contributed only slightly to the microbial consortium experienced rapid sequential expansion and contraction due to the loss of microbial complexity (Buffie et al., 2015).
Adenylyltransferases (ANT) catalyze the adenylation of a hydroxyl group in response to ATP, O-phosphotransferases (APH) catalyze the phosphorylation of a hydroxyl group in response to ATP, and N-acetyltransferases (AAC) catalyze the acetyl-CoA-dependent acetylation of an amino group. These three types of enzymes are known to modify aminoglycosides (Shete et al., 2017). An investigation found that enterococcal isolates had a high frequency of genes modifying aminoglycosides (Shete et al., 2017).
Additionally, bacteria can alter the molecular targets of antibiotics, causing minor structural changes that disrupt the highly precise interaction between the antibiotic and its target molecule. For instance, mutations in 23S rRNA confer resistance to macrolides, lincosamides, and streptogramin B; mutations in DNA topoisomerase II and IV result in resistance to quinolones and fluoroquinolones; and mutations in penicillin-binding proteins decrease the effectiveness of β-lactams. Through the efflux proteins found in their cell membrane, bacteria are able to pump out antimicrobial substances. The majority of these proteins are multidrug transporters, while some may be antibiotic-specific. Reduced permeability of the outer membrane, which lowers antibiotic absorption, is another mechanism of resistance (Ramirez et al., 2020).
3.1.3 Efflux pumps
Efflux pumps actively transport antibiotics out of bacterial cells, lowering their intracellular concentrations and leading to multidrug resistance (Gaurav et al., 2023). The ATP-binding cassette (ABC) superfamily, the major facilitator superfamily (MFS), the multidrug and toxic compound extrusion (MATE) family, the resistance nodulation cell division (RND) family, the small multidrug resistance (SMR) family, and the proteobacterial antimicrobial compound efflux (PACE) family are the six main families of efflux pumps that have been identified in bacteria thus far (Gaurav et al., 2023).
The E. coli genome contains around 20 drug efflux system genes. Previously unknown, E. coli cells survive in the intestine, which has a low oxygen concentration. Anaerobic conditions dramatically increase the expression of the MdtEF drug efflux system in E. coli, and the resulting increase in drug efflux activity results in MDR (Nishino et al., 2021).
Biofilms, as opposed to their planktonic state, are organized group of microorganisms that reside in a matrix of extracellular polymeric substance (EPS) that they produce. They form colonies by adhering to one another on living or non-living surfaces, and they differ in their rates of growth and gene expression (Rather et al., 2021). Additionally, species that are essential for a healthy gut mucosa form biofilm, which can help the host by strengthening defenses, lengthening the time bacteria stay in the body, improving nutrient exchange between the microbiota and the host, increasing plasmid transfer rates, expressing colonization factors, and indicating resistance to colonization by a healthy mucosal biofilm (Miller et al., 2021; Tytgat et al., 2019).
B. thetaiotaomicron accounts for 12% of the gut microbiota and 6% of the faecal microbiome. B. thetaiotaomicron has been found to break down sugar moieties in food particles and in the mucus layer, indicating that biofilm production may play a significant role in their way of life. As a result, biofilms in the human gut can be useful or harmful to the host, depending on whether they are formed by commensal microbiota or enteric pathogens (Béchon and Ghigo, 2022).
Most clinically utilized antibacterial medicines must permeate one or both of the cell envelope membranes in order to reach their required site of action, such as the outer leaflet of the Outer membrane. Loss of porins and other transport systems might alter a drug’s overall capacity to pass through this membrane, which can result in clinical antibacterial resistance, especially in Enterobacteriaceae. Mutations in porin expression reduce expression, limiting nutrients and mediating resistance in bacteria (Masi et al., 2017).
3.2 The progression of colonization and microbial resistance
Overuse of antimicrobial medications, especially in immunocompromised individual, increases the risk of infection from opportunistic pathogens and result in the development of MDR bacteria in the gut microbiome (Dethlefsen and Relman, 2011). Common antimicrobial resistance genes, that are resistant to tetracycline, vancomycin, bacitracin, cephalosporin, and the macrolide-lincosamide-streptogramin (MLS) group have been found in the gut microbiomes globally (Forslund et al., 2014). Gut microbiota plays an important role in host defence by preventing exogenous bacteria and facilitating the growth of indigenous bacteria (Pilmis et al., 2020). This defensive role, referred as colonization resistance, is disturbed by the inappropriate use of broad-spectrum antibiotics (Nasiri et al., 2018; Pilmis et al., 2020). Studies have shown that, oral streptomycin administration altered the gut microbiota in mice increasing the susceptibility to Salmonella infections, with similar findings observed in other animal and human studies (Bartosch et al., 2004; Pecquet et al., 1991; Pilmis et al., 2020).
3.3 Mechanisms responsible for colonization resistance
Colonization resistance, mediated by various mechanisms, is a process where the commensals in a healthy gut from the upper proximal to the intestine guard the host from pathogen invasion (Ducarmon et al., 2019; Ke et al., 2023; Kim et al., 2017). This mechanism was discovered when the depletion of the commensal bacteria due to antibiotic treatment increases the vulnerability to enteric pathogens. The gut microbiota aids in the process by synthesizing and secreting over 500,000 metabolites into the lumen (Chang, 2020). Although the mechanisms underlying colonization resistance are poorly understood, they can be broadly divided into direct and indirect mechanisms (Ducarmon et al., 2019; Khan et al., 2021) (Figure 2).
Figure 2
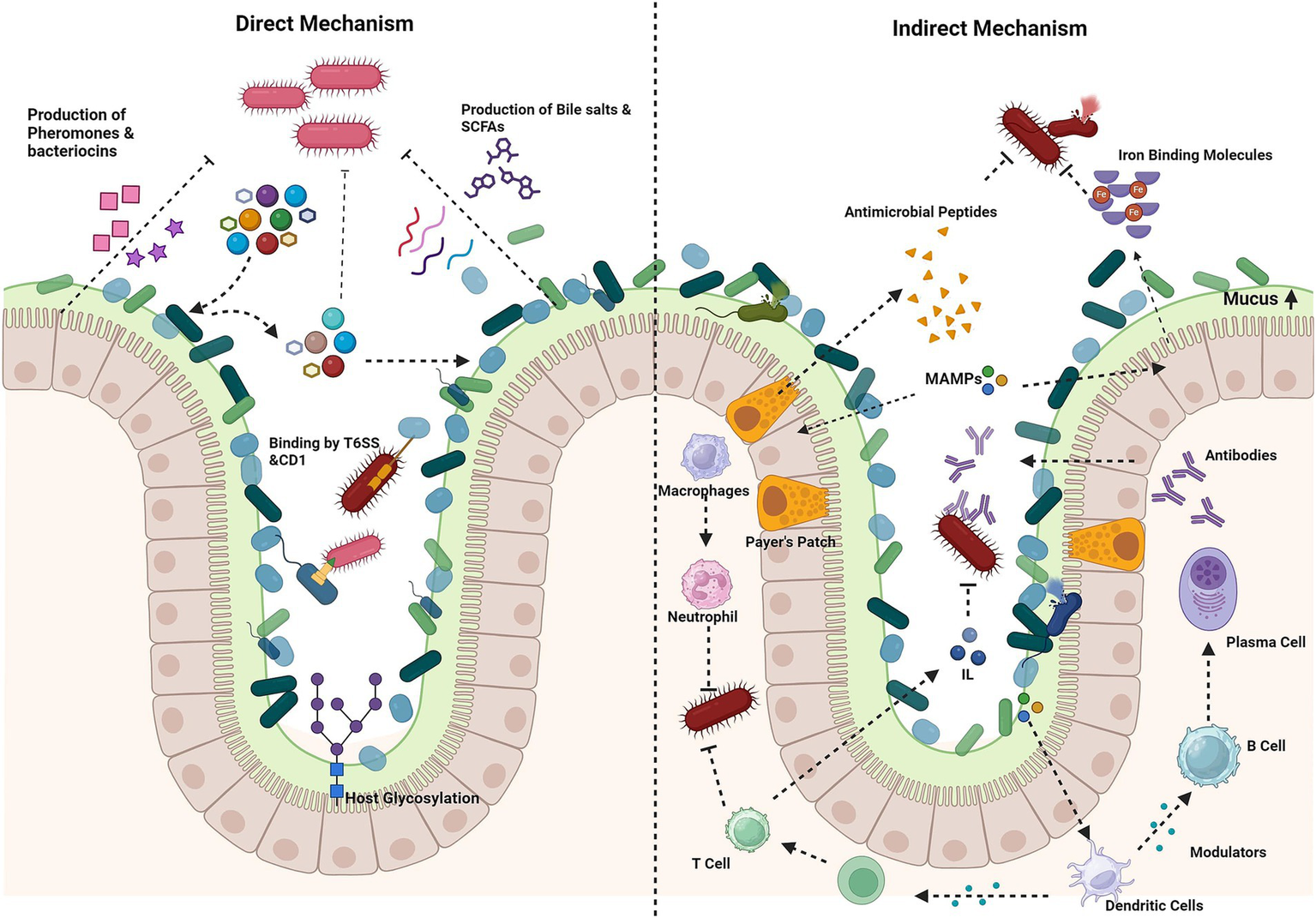
Mechanism of colonization resistance. Direct mechanisms: production of antimicrobial compounds (e.g., bacteriocins, short-chain fatty acids, bile salts), nutrient competition and host glycosylation of epithelial cells by bacteria, for nutrient adhesion, directly kill pathogens via contact-dependent inhibition (CDI), the type VI secretion system (T6SS) or secreted molecules. Indirect mechanisms: stimulation of mucin production by goblet cells forms a protective barrier. Microorganism-associated molecular patterns (MAMPs) trigger the secretion of antimicrobial peptides, which prime macrophages and dendritic cells. Macrophages inhibit pathogens through phagocytosis and the release of reactive oxygen species (ROS). Dendritic cells activate T cells, leading to the activation of immune responses and the stimulation of interleukin production. In Peyer’s patches, dendritic cells stimulate B cells to produce antibodies. Iron-binding proteins limit free iron availability to pathogens. Created using BioRender.com.
3.3.1 Direct mechanism
The microbiota encourages direct colonization resistance, through antagonism and resource competition. Using variety of mechanisms, bacteria compete for both limited physical space and scarce nutrients. Closely related bacterial species that occupy same niches or resources tend to outcompete (Pickard et al., 2017).
3.3.1.1 Nutrient competition
The nutrient niche theory, proposed by Rolf Freter in 1983, states that microorganisms will colonize, multiply and utilize the nutrients as per their requirements. The gut microbiota has a unique nutritional ability enabling it to digest resistant starch, cellulose, inulin, pectin, mucus and bile salts into carbon and nitrogen sources support their growth. Pathogens must compete with gut commensals for nutrition and to colonize (Horrocks et al., 2023; Pickard et al., 2017).
Commensals generally alter the pathogens virulence factor directly by the production of metabolites (Khan et al., 2021). Studies have shown that commensal E. coli with EHEC competes for amino acids, organic acids, and other nutrients (Fabich et al., 2008; Leatham et al., 2009). SCFA such as, butyrate, downregulates the expression of secretion system type 3 proteins (SST3) in Salmonella enteritidis and Salmonella Typhimurium (Gantois et al., 2006). Inhibiting the pathogen growth and colonization requires phylogenetically diverse species to prevent nutrient access and establish colonization resistance (Spragge et al., 2023).
Commensal species have evolved metabolic pathways to utilize mucins and dietary carbohydrates as key intestine nutrition source (Kim and Ho, 2010; Kamada et al., 2012). Citrobacter rodentium and E. coli may compete for monosaccharides while mutualistic Bacteroides species secrete sialic acid and fucose from host glycans, which are essential sugar source for Salmonella Typhimurium and C. difficile invasion. These carbohydrates are only accessible to pathogens when antibiotic therapy decreases the commensal population (Ng et al., 2013; Pickard et al., 2017). Bacterial development requires iron, a crucial trace metal that even the host firmly holds, especially during an inflammatory response. Through siderophores, Salmonella Typhimurium scavenge the host and commensal requirement for iron throughout an infection (Ducarmon et al., 2019; Sassone-Corsi and Raffatellu, 2015). An efficient way to lessen the severity of a Salmonella infection is to use immunization strategies against siderophores (Sorbara and Pamer, 2019). According to studies, two Klebshiella species—K. oxytoca and K. michiganensis—provide colonization resistance against Enterobacteriaceae that are resistant to antibiotics by means of nutritional competition. Colonization resistance was associated with resource utilization, and Klebshiella species reduced the colonization of E. coli and Klebshiella pneumoniae in mice and ex vivo investigations (Horrocks et al., 2023). When the commensal gut microbiota reduces dietary amino acids, it has been shown to increase resistance to Citrobacter rodentium colonization (Caballero-Flores et al., 2020).
3.3.1.2 Bacteriocin
Bacteriocins are short, toxic ribosomal synthesized antimicrobial peptides produced by specific bacterial species that can inhibit the colonization and growth of other species. Their mechanisms of action are multiple including disturbing RNA and DNA metabolism, pore formation in the cell membrane, influence on protein and DNA synthesis (Benítez-Chao et al., 2021; Ducarmon et al., 2019; Pilmis et al., 2020). Peptides are categorized into post-transduction modified (type I) and unmodified peptides (type II), are typically effective against closely related bacteria and exhibit strong specific activity against clinical targets (including MDR strains) (Cotter et al., 2013). Many bacteriocins, from the lactic acid bacteria, human and animal gut microbes, and probiotics like Bifidobacteria, would engage in gastrointestinal competition (Hammami et al., 2013). It has been discovered that the Sactibiotic thuricin CD (bacteriocin type I) is effective against C. difficile. While sactibiotic, subtilosin A, exhibits efficacy against Listeria monocytogenes, Streptococcus pyogenes, and Enterococcus faecalis. In contrast, Pediococcus acidilactici MM33 secretes pediocin PA-1 (bactericin type II), that act against vancomycin-resistant Enterococci (VRE) colonization in the gut (Pilmis et al., 2020). The extent to which bacteriocins contribute to colonization resistance to pertinent intestinal pathogens is still unknown while they support ongoing intraspecies competition in the gut (Pickard et al., 2017). Pediococcus acidilactici produces bacteriocins that hinder the growth of planktonic cells of Salmonella Typhimurium in addition to preventing the formation of biofilms. Probiotic Bacilli, on the other hand, generate bacteriocins such subtilin and subtilosin A, which particularly prevent Salmonella from forming biofilms without harming the planktonic cells (Deng and Wang, 2024).
3.3.1.3 Type VI secretion system
Type VI secretion system (T6SSs) is a mechanism by which bacteria transport proteins into or out of target cells during infection, facilitating interbacterial competition (Russell et al., 2011).
Enteric pathogens use T6SSs to antagonize symbiotic gut E. coli, facilitating colonization and disease progression. T6SS loci are also widely distributed in human gut Bacteroidales including Bacteroides, Parabacteroides, and Prevotella, and exist in three forms: GA1, GA2, and GA3 (Coyne and Comstock, 2019).
The GA1 and GA2 T6SS loci can be transferred between many intestinal species and Bacteroidales families, however the GA3 T6SSs are exclusive to Bacteroides fragilis. The GA3 T6SSs are the only ones that have been demonstrated to target almost every type of Bacteroidales found in the gut (Coyne and Comstock, 2019). Numerous studies have discovered the existence of a T6SS and its related effectors and immune proteins that significantly influences the competitiveness between as it involves a variety of effector and immune protein combinations, and can have a wider target range (Pickard et al., 2017).
3.3.2 Indirect mechanism
Indirect colonization resistance is facilitated by the host-commensal flora interaction, by maintaining the epithelial barrier, regulation of bile acid metabolism, and production of antimicrobial peptides (RegIII and angiotensin-4) (Pilmis et al., 2020).
3.3.2.1 Antimicrobial peptide production
Antimicrobial peptides (AMPs), recognized as a crucial line of defence against infections, are produced by all life forms (Pilmis et al., 2020). AMPs have a multiple mechanism of action, targeting peptidoglycan and bacterial cell membrane (Mookherjee and Hancock, 2007). Bacterial membranes, composed of cardiolipin and phosphatidylglycerol are negatively charged, interact with the positively charged antimicrobial peptides leading to lysis (Pilmis et al., 2020). The host (epithelial and paneth cells) requires taurine or lipopolysaccharide to produce ANG-4 (ribonuclease) and RegIII (type C lectin). Furthermore, the gut bacterium Bacteroides thetaiotaomicron induces ANG-4 expression, which has bactericidal effect against both Gram-negative and Gram-positive bacteria (Pilmis et al., 2020). Lipopolysaccharide-stimulated Toll like receptors (TLRs), notably TLR-4, in the microbiome can trigger RegIII production (Mukherjee et al., 2014). Flagellin also activates the TLR-5 and TLR7 receptors on dendritic cells resulting in the release of IL-23, which prompts innate lymphoid cells to release IL-22, increasing the synthesis of RegIII (Pilmis et al., 2020). Commensal bacteria activate MyD88 signaling in paneth cells and other epithelial cells, which in turn promotes the synthesis of the antimicrobial lectin regenerating islet-derived protein 3γ (REG3γ). By preventing Salmonella Typhimurium from penetrating host tissues, this antimicrobial response promotes gut health and prevents infection (Deng and Wang, 2024).
3.3.2.2 Bile acid metabolism
Bile acids, produced by the liver to breakdown dietary lipids, have antibacterial characteristics. Primary bile acids are linked with glycine or taurine to improve solubility (Ducarmon et al., 2019). Bile acids exhibits dual role in microbial growth where primary bile salts influence germination and vegetative growth of C. difficile spores and Secondary bile acids have been discovered to prevent the growth (Sorg and Sonenshein, 2008). For instance, the symbiotic microbe Clostridium scindens can change the main bile acids (cholic acid and chenodeoxycholic acid) into the secondary bile acids (deoxycholic acid and lithocholic acid). Thus, in both animals and humans, C. scindens increases resistance to C. difficile infections in a secondary bile acid-dependent manner (Buffie et al., 2015).
3.3.2.3 Epithelial barrier maintenance
The inner and outer mucous layers, the epithelial barrier, and its associated immunological barrier make up the physical gut barrier. The inner mucus layer is impermeable and strongly adhered to epithelium thus restricting the movement of bacteria, preventing direct contact between host and commensal bacteria of gut microbiome, thereby avoiding inflammatory reaction (Pilmis et al., 2020). As the thickness of the mucus layer decreases it becomes more vulnerable to pathogen colonization. Therefore, a western-style diet poor in carbohydrates, antibiotic therapy, or other medications that have an impact on the microbiota, alters the thickness of the mucus layer increasing vulnerability to infection (Desai et al., 2016; Pilmis et al., 2020). The NF-κBpathway is activated by the gut bacteria when the mucus layer is altered, encouraging tissue healing by activating innate immunity receptors such as synthesis of anti-apoptotic proteins, increasing cell proliferation, stabilising tight junctions, negatively regulating the production of pro-inflammatory cytokines (Pilmis et al., 2020; Rakoff-Nahoum et al., 2004).
4 Screening for AMR
Antibiotic resistance genes in the gut microbiota can be passed to other bacteria, increasing the risk of evolution of pathogenic strains (Hu et al., 2013; Theophilus and Taft, 2023). AMR, characterised using a variety of techniques, are necessary for the understanding and monitoring of a variety of resistance genes that can contribute to treatment failures and the spread of resistant infections, in complete environmental communities. The screening of AMR, concern for human health and socioeconomic development, helps in the better understanding of the ARGs and identification of novel ARGs (Theophilus and Taft, 2023) (Figure 3 and Table 3).
Figure 3
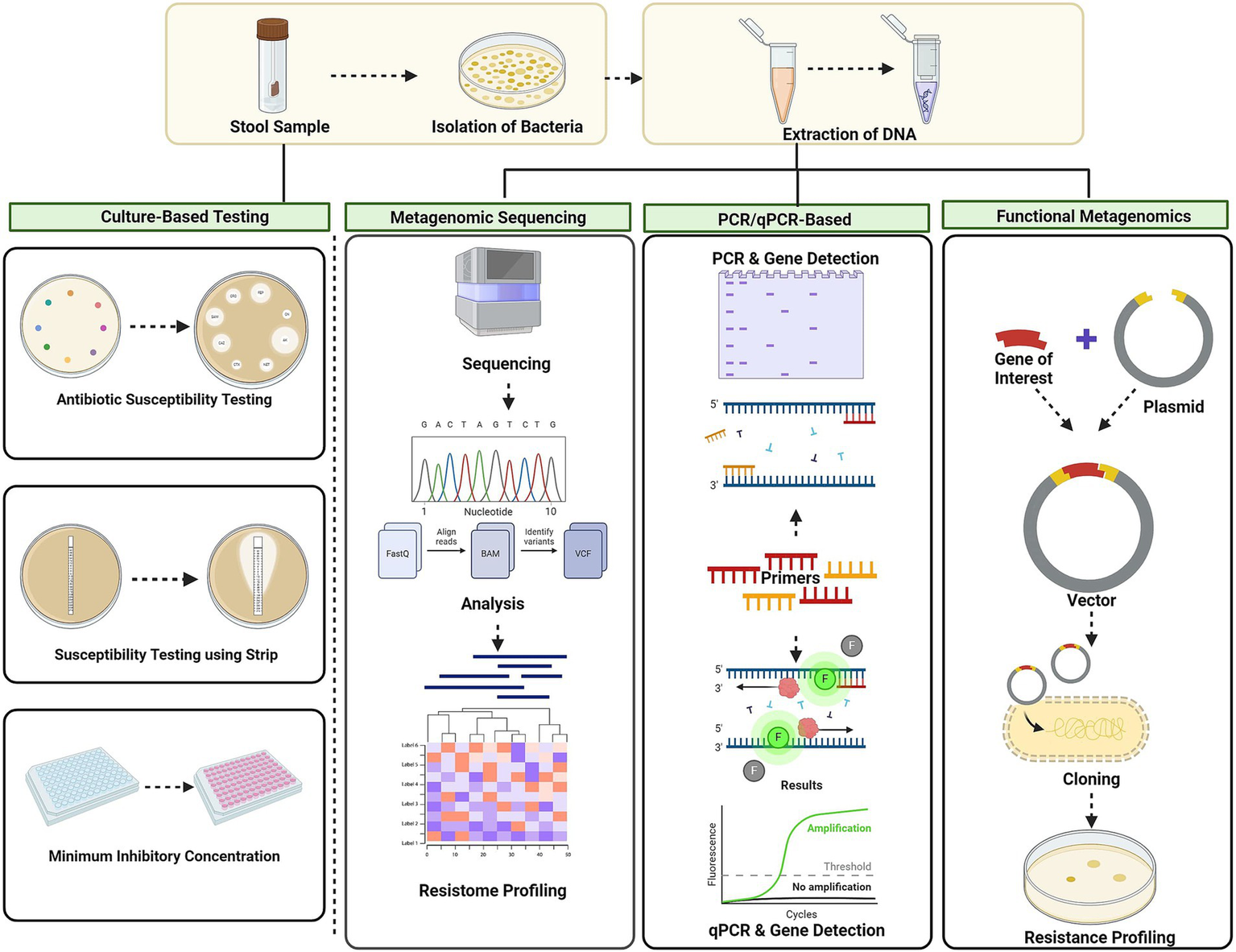
Techniques for AMR screening culture-based methods, metagenomic approaches, PCR-based techniques, and functional metagenomic approach. Culture-based methods involve disc diffusion, MIC. Metagenomic approaches: next-generation sequencing (NGS) and shotgun metagenomic sequencing. PCR-based techniques: conventional PCR and quantitative PCR (qPCR). Functional metagenomics: using vectors involves cloning resistance genes and screening for their functional traits. Created using BioRender.com.
Table 3
| S. No. | Method | Description | Advantages | Limitations | References |
|---|---|---|---|---|---|
| 1. | Metagenomic Sequencing | Sequencing of DNA to find ARGs | Comprehensive | Expensive, requires experts in bioinformatics | Bogri et al. (2024) and Merrick et al. (2023) |
| 2. | qPCR | Quantifies specific genes using primers | Ability to test for multiple antibiotic genes, rapid | Lower specificity compared to culture and staining, expensive, limited to known targets | Liu et al. (2019) |
| 3. | Culture-based methods | Isolates organisms in selective media and antibiotics | Provide reproducible results with minimal error, isolation of specific target organisms, screening at a range of antibiotic concentrations | Turnaround time, contamination | Figdor and Gulabivala (2008) and McLain et al. (2016) |
| 4. | Functional metagenomics | Clones DNA into host to find ARGs | Identifies ARGs based on function, highly sensitive | Limited insert size, require experts | De (2019), Mullany (2014), and Yadav and Kapley (2021) |
| 5. | Resistome profiling | Comprehensive analysis of all resistance genes in a sample | Predicting the possible resistance pattern, determine resistome | Requires advanced sequencing and bioinformatics, limited differentiation | Waskito et al. (2022) |
| 6. | Nanopore sequencing | Real-time sequencing of long DNA fragments | Analysing sequences in real time, size, cost, simple library prep, and portability | High error rate, expensive | Delahaye and Nicolas (2021) and Jain et al. (2016) |
| 7. | CRISPR-based detection | Uses CRISPR-Cas systems to detect specific DNA sequences | Simplicity, sensitivity, and specificity, robust result | Off-target effects, expert required, cost and accessibility | Kaminski et al. (2021), Mayorga-Ramos et al. (2023), and Shin et al. (2024) |
| 8. | Single-cell genomics | Analyzes genetic material from individual cells | High resolution, identifies rare cells | Nosier and more variable data | Chen et al. (2019) |
| 9. | Metaproteomics | Studies the protein composition of microbiomes | Direct functional insights, comprehensive analysis | Limited database, complex data analysis | Kleiner (2019) and Petriz and Franco (2017) |
Current and emerging techniques for AMR screening.
4.1 Culture based techniques
Culture-based analysis, as recommended by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) and the Clinical Laboratory Standards Institute (CLSI), is the gold standard technique for detecting AMR in gut microbiota (Hassall et al., 2024). The diffusion assay and e-test minimum inhibitory concentration approach, part of the standard methods, detects bacterial growth in various antibiotic concentrations after being isolated from selective media (Yamin et al., 2023). Culture-based approaches have several benefits such as targeted isolation, reproducibility, cost effectiveness, quantitative and qualitative measurements (McLain et al., 2016). However, they can be potentially variability in the results, time consuming, limited options for antibiotic testing and ability to detect all potential resistance genes (Hassall et al., 2024). Due to these limitations, the evaluation of the antimicrobial susceptibility (AMS) patterns of the entire microbiome is restricted to indicator bacteria like E. coli, a reservoir of ARGs (Firoozeh et al., 2011; Firoozeh et al., 2013; Nyirabahizi et al., 2020; Penders et al., 2013; Neamati et al., 2015). Studies using enterococci or E. coli as markers have shed light on the occurrence of AMS throughout geographical regions, the effects of hospitalisation and population density, and the link between AMS in humans and food animals (Nyirabahizi et al., 2020). Culture-based analysis of AMS have shown the link between the AMS of faecal E. coli and that of E. coli implicated in diseases including urinary tract infections. A broad range of antibiotics like ampicillin (Amp), amoxicillin (Amx), aztreonam (Azt), cefotaxime (Cefo), ceftriaxone (Ceft), imipenem (Imp), meropenem (Mer), cefepime (Cef), piperacillin (Pip), vancomycin (Van), clindamycin (Cli), colistin (Col), polymyxin B (PolB), daptomycin (Dap), Fosfomycin (Fos), tetracycline (Tet), doxycycline (Dox) is used to test the commensals antimicrobial drug susceptibility. The minimum inhibitory concentration (MIC) of each antibiotic is determined by the observing the bacterial growth with the strip of chosen antibiotic (Bag et al., 2019). Culture-based studies have advanced the understanding of the relationship between antibiotic use and AMS of gut bacteria, revealing an association between antibiotic use and the prevalence of resistant faecal Gram-negative bacteria (Bruinsma et al., 2003; Murray et al., 1982; van der Veen et al., 2009). Individuals who have received medical treatment with antibiotics have been found to harbour these antimicrobial-resistant bacteria (Bartoloni et al., 2004; Grenet et al., 2004).
Other studies have found that people who do not have access to antimicrobial agents, such as those who live in remote areas, can develop faecal antimicrobial-resistant bacteria as a result of environmental exposure to organisms producing antibiotics (mould contaminated food), heavy metal contamination in drinking water (Bartoloni et al., 2004; Calomiris et al., 1984; Timoney et al., 1978).
4.2 Molecular diagnostics used for detection antimicrobial resistance
Molecular diagnostics have recently received attention due to their speed, accuracy, and independence from culture (Anjum et al., 2017). Advanced techniques like polymerase chain reaction assays, sequencing, and various genotyping approaches provide insights into the mechanisms of resistance transfer by identifying resistant-carrying integrons after antibiotic treatment (Geser et al., 2012; Gijón et al., 2012; Overdevest, 2011; van der Veen et al., 2009; Vinue et al., 2008; Vo et al., 2010). Following an outbreak, when phenotypic data is insufficiently precise to prevent potential outbreaks involving resistant bacteria, genetic characterization is sometimes employed as an indirect way to support epidemiological investigations. Also, local, national, or even international surveillance of AMR utilizes molecular characterization of AMR determinants (Anjum et al., 2017).
4.3 Metagenomics to characterize the AMR
Metagenomics involves the study of metagenomes or genetic material directly collected from environmental sources enabling the genomic analysis of every bacterium in a microbial ecosystem without individual identification (Lepage et al., 2013). Targeted PCR-based metagenomics, functional metagenomics, and sequence-based metagenomics are three different metagenomic methods that have been used to explore the resistome (Penders et al., 2013).
4.3.1 PCR-based metagenomics
PCR and qPCR are widely used in vitro techniques that allow the exponential amplification of specific DNA and RNA sequences with high specificity, providing rapid means of identifying bacteria from various environments, including the detection of resistance genes (Galhano et al., 2021). Research have shown that transfer of resistant genes occurs within an ecosystem and across species, emphasizing the real-time applications of PCR-based metagenomics. The relative abundance of the resistant genes can be estimated by analysing the semi-quantitative result of qPCR (Knapp et al., 2011; Koike et al., 2007). PCR was utilized to detect the blaCTX-M gene variations in E. coli isolated from the human and chicken faecal sample, indicating the presence of ARGs in the gut microbiome (Valenzuela et al., 2023). The presence of blaCMY and blaSHV resistance genes in E. coli from migratory birds, indicated the potential for analysing gut microbiota resistance genes (Islam et al., 2022). Vien et al. (2012) using qPCR, showed that higher levels of Plasmid-mediated quinolone resistance genes in gut flora lead to fluoroquinolone resistance. Targeted PCR-based metagenomics remains a valuable technique for identifying the resistome due to the accessibility, and provides high-throughput analysis at reasonable costs. However, the fundamental drawback is that the data are skewed toward known resistance genes and pathways predominantly in case of convergent evolution, where a number of genes perform similar roles. Furthermore, a resistance gene with sequence variation found in numerous species may skew the results in favour of the species (Penders et al., 2013).
4.3.2 Functional metagenomics
Functional metagenomics involves cloning DNA segments into a vector (like a plasmid) and expressing these segments in heterologous hosts, often E. coli. Transformants are cultivated on antibiotic-containing media to assess the expression of resistance genes, with findings relying on each gene’s ability to express in surrogate hosts, allowing for subsequent sequencing (Schmieder and Edwards, 2012). Functional metagenomics DNA screening was used to identify the reservoir for resistance in samples of faeces and saliva from two healthy people. Sequencing and annotation of clones exhibited resistance to 13 different antibiotics resulted in identification of 95 unique inserts encoding functional antibiotic resistance genes. Out of these, 10 previously unknown beta-lactamase gene families where identified indicating an underappreciated barrier separating these unique resistance-producing bacteria from common human pathogens (Schmieder and Edwards, 2012; Sommer et al., 2009). Additionally, a functional screening for seven antibiotics utilizing gut microbiome metagenomic libraries from healthy people revealed novel AMR genes against kanamycin, D-cycloserine, and amoxicillin (Cheng et al., 2012). A recent study using functional metagenomics identified three novel genes, TMSRP1, ABCTPP, and TLSRP1, responsible for the osmotolerance in human gut microbiota (Verma et al., 2018). Despite being commonly used, the functional metagenomics-based approach has several drawbacks. The method depends on individual gene’s ability to express itself in surrogate hosts, leading to false negative results if resistance genes that are not produced by the surrogate host because they require several regulatory elements, or posttranslational modifications. Also, the foreign gene may engage in unique interactions with the surrogate host’s cellular machinery, leading to false positives (Penders et al., 2013; Schmieder and Edwards, 2012).
4.3.3 Sequence based metagenomics
Sequence-based metagenomics eliminates the requirement for culturing by directly sequencing DNA from an environmental sample once it has been extracted, fragmented, and size-separated. Resistance genes are recognized by comparing metagenomic sequences to global sequence databases. The transition from Sanger sequencing to next-generation sequencing technologies, including the Roche 454 sequencer, Illumina’s Genome Analyzer, and Applied Biosystems’ SOLiD system, has significantly reduced the cost of metagenomic sequencing initiatives by producing shorter contiguous reads, higher genome coverage, and fewer consumable costs (Niedringhaus et al., 2011). Sequence-based metagenomics is increasingly used to study the human gut microbiome, but not directly targeting the AMR genes. However, the in-silico identification of resistance components has been made possible by the uploading of these metagenomic libraries to public databases (Penders et al., 2013). The ratio of chromosomal and extra-chromosomal genomes will always be heavily in support of chromosomes, producing a tonne of redundant data when one is only interested in the extra-chromosomal metagenome (Li et al., 2012). Sequence-based metagenomics is often only useful for identifying known genes because it is difficult to discover sequences with little resemblance to known reference sequences (Penders et al., 2013; Schmieder and Edwards, 2012). Additionally, the expression of the discovered genes is not provided by sequence-based metagenomics. Contrarily, sequence-based metagenomics offers a large amount of data not just on AMR genes but also on the whole gene content, making it possible to determine the metabolic profile and community composition. These metagenomic sets of data in particular make it possible to examine which bacteria in the community possess specific functional genes (Penders et al., 2013). A metagenomic study using a sequence data of 2,037 samples concludes that the human gut resistome is influenced by geographical locations and to a lesser extent on the disease conditions (Qiu et al., 2020) According to the metagenome data from mice with UTIs, oral antibiotic therapy led to an enrichment of particular taxa and ARGs and a decrease in the overall diversity of gut microbes. The results of this model demonstrated that after 24 to 72 h of cipro and fosfo treatment, cross-resistance to several types of antibiotics emerged (Xu et al., 2020).
5 Impact of AMR on health
Antimicrobial resistance, a naturally occurring process, has been accelerated due to the inappropriate overuse of antibiotics and poor infection control practices (Salam et al., 2023). Greater patient mobility and movement of carriers have increased the risk of spread of resistant pathogens globally (Findlater and Bogoch, 2018). Delays in appropriate treatments prolong the infection, this in turn puts at risk the immediate contacts of those infected, including health professionals, but it also enhances the dissemination of resistance within communities. Longer duration of disease and treatment due to AMR leads to increased financial costs for families and healthcare systems (O’Neill, 2016; Dadgostar, 2019). Development of new antibiotics has reached an almost complete standstill; no new classes have been discovered after 1987 (Silver, 2011). Drugs for chronic illnesses like those for diabetes and hypertension may provide more profitable economic opportunities for pharmaceutical corporations than newer antibiotics due to their extensive usage in each patient and the lack of problems with resistance. Additionally, smaller pharmaceutical companies struggle to meet the strict requirements for clinical trials involving antibiotics. This puts the development of several potential new agents in danger (Jindal et al., 2015).
This misuse of antibiotics, both in public and private health care facilities, is very common in developing countries like India, where studies indicate that 45–80% of patients suffering from viral respiratory infections and diarrhoea were inappropriately provided with antibiotics without proper diagnosis (Jindal et al., 2015; Kumar et al., 2008). Moreover, the use of antibiotics in agriculture to improve the yield has increased the diversity and abundance of AMR genes in urban, agricultural, and environmental settings (Baquero et al., 2008; Nesme et al., 2014; Wright, 2010; Zhu et al., 2013).
Vaccination plays an important role in blocking the spread of infectious diseases. But as the vaccination rates decline, the unvaccinated population, such as children, and immunocompromised patients are susceptible to the infection, in-turn enhancing the reservoirs of pathogens, some of which may acquire resistance to antibiotics. To effectively address AMR, multiple strategies are necessary, which include enhancing antibiotic stewardship, investing in new drug development, and maintaining a high level of vaccination to prevent the spread of infectious diseases (Muhsen et al., 2012; Zhu et al., 2013).
6 Effects of AMR on environment
Environmental factors have a global impact on development of AMR. Drug-resistant microorganisms and resistance genes could spread into the environment through excreta, water bodies (Konopka et al., 2022; Singer et al., 2016). In agriculture, out of the total antibiotics given to animals, 30–90% are excreted through urine and faeces, leading to environmental pollution and the development of resistance (Berendsen et al., 2015). Animal manure has been identified as one of the significant vectors of both antibiotic-resistant bacteria and residual antibiotics that may persist in the environment (Sarmah et al., 2006; Udikovic-Kolic et al., 2014). Heavy metals also contribute to the dissemination of AMR, often present in WWTPs from urban sources like domestic and commercial effluents, vehicle emissions, and industrial activities. The contamination is further increased by the widespread use of disinfectants, textiles, and common household items containing metal nanoparticles, including those of titanium, copper, and silver. In addition, other metals, including Pb, Cu, Zn, and Cd, were utilized in agriculture and aquaculture as fertilizers and for insecticides, fungicides, and animal growth promotion, thereby producing an optimal ecological environment for the development of AMR.
7 Effect of AMR on economy
The economic burden of AMR includes both direct or indirect costs. These direct medical costs of AMR relate to treatments, including prescription drugs for the disease and hospitalisation costs. Indirect costs are essentially the wider consequences of increased sickness and mortality, leading to decreased productivity and reduced economic output (National Academies of Sciences, Engineering, and Medicine et al., 2018). According to the CDC reports, antibiotic resistance in the United States alone might result in a $1,400 rise in hospital costs for treating patients with any type of bacterial infection (Centers for Disease Control and Prevention, 2013; Thorpe et al., 2018). However, this can sharply rise to more than $2 billion per year. According to a number of estimates, AMR costs would range from $300 billion to over $1 trillion annually globally by 2050. Healthcare is directly impacted financially by AMR, as seen by increased resource use and high costs for complex and expensive treatments (Dadgostar, 2019).
8 Future perspectives
To combat AMR, new solutions are urgently needed. Faecal microbiota transplantation (FMT) is the most advance treatment to tackle AMR and other tactics (such probiotics and bacteriophages) as prospective substitutes for infection prevention solutions (Gargiullo et al., 2019). FMT involves the endoscopic or oral administration of tablet preparations to a patient’s colon for transferring the microbiota from a donor. FMT is being researched for additional uses, however it is now recognized as a clinically extremely successful therapy for persistent Clostridioides difficile infection. FMT has recently been taken into consideration for the elimination of antibiotic-resistant bacteria from their reservoir in the intestine (Pilmis et al., 2020). FMT has been recognized as an effective treatment for additional conditions linked to altered gut microbiome, including intestinal inflammatory diseases like IBD, in addition to its use for MDR infections (Paramsothy et al., 2017; Wang et al., 2016).
Prebiotics, non-digestible food ingredients, favourably influence one or more species of bacteria in the colon by increasing their growth and/or activity. In contrast, probiotics are isolated, live organisms that are given to the host in order to boost their health.
These products have the potential to restore the balance of the gut microbiota by encouraging the recolonization of species, either directly via the action of prebiotics or indirectly through the careful selection of bacterial species in probiotics (Pilmis et al., 2020). When healthy individuals are exposed to antibiotic therapy, human milk oligosaccharides, a typical example of a prebiotic, are known to assist in re-establishing the balance between Firmicutes and Bacteroidetes (Elison et al., 2016). According to Cochrane, use of probiotics have successfully used in prevention diarrhoea (Wei et al., 2018). Using Lactobacillus rhamnosus, patients with vancomycin-resistant enterococci were successfully decolonized in two randomised investigations, while the combination of Lactobacillus bulgaris and Lactobacillus rhamnosus had no effect on the colonization rate in the Gram-negative range (Salomão et al., 2016).
9 Conclusion
Antimicrobial resistance is spreading across the globe and is contributing to an increase in hospital-acquired infections, mortality, and expenditures. Presently, bacterial enteric infections continue to contribute significantly to the global illness burden. Very little is known about the failure of the gut microbiota to give colonization resistance against these enteropathogens, even though the virulence factors involved in infection for many infectious agents are well understood. When established in a human, drug-resistant bacteria and resistance genes could spread into the environment through human waste. The antimicrobial susceptibility among these strains in various hosts, through time, and in various geographic regions has been the focus of extensive prior study on marker gut bacteria. Culture-based investigations are still relevant in the modern era of molecular methods since they are required to determine antibiotic susceptibility. To learn more about the possibility of the human gut microbiome as an AMR reservoir, however, targeted, functional, or sequence-based metagenomics are needed. Strategies based on the microbiota should be considered for MDRO prevention and therapy. Faecal microbial transplantation is a very promising approach, particularly when tested treatment have failed. Faecal microbial transplantation has so far been proven to be reliable and effective. However, in order to use faecal microbial transplantation in MDR clinical therapy, RCTs are required to standardise the methodology and establish regulatory parameters.
Statements
Author contributions
SD: Conceptualization, Investigation, Methodology, Writing – original draft, Data curation. SS: Conceptualization, Investigation, Methodology, Writing – original draft, Data curation. RJ: Validation, Writing – review & editing. SR: Validation, Writing – review & editing. VR: Validation, Writing – review & editing, Supervision, Conceptualization. AS: Validation, Supervision, Writing – review & editing, Conceptualization.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors express their gratitude to Amity University Maharashtra, Mumbai, India, for providing infrastructural support, and to SASTRA University for offering outstanding facilities, access to a wide range of journals, and the opportunity to be part of the Quorum Sensing Lab (QSL).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
AfzaalM.SaeedF.ShahY. A.HussainM.RabailR.SocolC. T.et al. (2022). Human gut microbiota in health and disease: unveiling the relationship. Front. Microbiol.13:999001. doi: 10.3389/fmicb.2022.999001
2
AhmadA. F.DwivediG.O’GaraF.Caparros-MartinJ.WardN. C. (2019). The gut microbiome and cardiovascular disease: current knowledge and clinical potential. Am. J. Phys. Heart Circ. Phys.317, H923–H938. doi: 10.1152/ajpheart.00376.2019
3
AnjumM. F.ZankariE.HasmanH. (2017). Molecular methods for detection of antimicrobial resistance. Microbiol. Spectr.5, 33–50. doi: 10.1128/microbiolspec.ARBA-0011-2017
4
AntoL.BlessoC. N. (2022). Interplay between diet, the gut microbiome, and atherosclerosis: role of dysbiosis and microbial metabolites on inflammation and disordered lipid metabolism. J. Nutr. Biochem.105:108991. doi: 10.1016/j.jnutbio.2022.108991
5
BäckhedF. (2011). Programming of host metabolism by the gut microbiota. Ann. Nutr. Metab.58, 44–52. doi: 10.1159/000328042
6
BagS.GhoshT. S.BanerjeeS.MehtaO.VermaJ.DayalM.et al. (2019). Molecular insights into antimicrobial resistance traits of commensal human gut microbiota. Microb. Ecol.77, 546–557. doi: 10.1007/s00248-018-1228-7
7
BaqueroF.MartínezJ.-L.CantónR. (2008). Antibiotics and antibiotic resistance in water environments. Curr. Opin. Biotechnol.19, 260–265. doi: 10.1016/j.copbio.2008.05.006
8
BartoloniA.BartalesiF.MantellaA.Dell’AmicoE.RoselliM.StrohmeyerM.et al. (2004). High prevalence of acquired antimicrobial resistance unrelated to heavy antimicrobial consumption. J. Infect. Dis.189, 1291–1294. doi: 10.1086/382191
9
BartoschS.FiteA.MacfarlaneG. T.McMurdoM. E. T. (2004). Characterization of bacterial communities in feces from healthy elderly volunteers and hospitalized elderly patients by using real-time PCR and effects of antibiotic treatment on the fecal microbiota. Appl. Environ. Microbiol.70, 3575–3581. doi: 10.1128/AEM.70.6.3575-3581.2004
10
BéchonN.GhigoJ.-M. (2022). Gut biofilms: bacteroides as model symbionts to study biofilm formation by intestinal anaerobes. FEMS Microbiol. Rev.46:fuab054. doi: 10.1093/femsre/fuab054
11
Benítez-ChaoD. F.León-BuitimeaA.Lerma-EscaleraJ. A.Morones-RamírezJ. R. (2021). Bacteriocins: an overview of antimicrobial, toxicity, and biosafety assessment by in vivo models. Front. Microbiol.12:630695. doi: 10.3389/fmicb.2021.630695
12
BerendsenB. J. A.WeghR. S.MemelinkJ.ZuidemaT.StolkerL. A. M. (2015). The analysis of animal faeces as a tool to monitor antibiotic usage. Talanta132, 258–268. doi: 10.1016/j.talanta.2014.09.022
13
BlekhmanR.GoodrichJ. K.HuangK.SunQ.BukowskiR.BellJ. T.et al. (2015). Host genetic variation impacts microbiome composition across human body sites. Genome Biol.16:191. doi: 10.1186/s13059-015-0759-1
14
BogriA.JensenE. E. B.BorchertA. V.BrinchC.OtaniS.AarestrupF. M. (2024). Transmission of antimicrobial resistance in the gut microbiome of gregarious cockroaches: the importance of interaction between antibiotic exposed and non-exposed populations. mSystems9:e0101823. doi: 10.1128/msystems.01018-23
15
BreitbartM.HaynesM.KelleyS.AnglyF.EdwardsR. A.FeltsB.et al. (2008). Viral diversity and dynamics in an infant gut. Res. Microbiol.159, 367–373. doi: 10.1016/j.resmic.2008.04.006
16
BrinkacL.VoorhiesA.GomezA.NelsonK. E. (2017). The threat of antimicrobial resistance on the human microbiome. Microb. Ecol.74, 1001–1008. doi: 10.1007/s00248-017-0985-z
17
BruinsmaN.StobberinghE.de SmetP.van den BogaardA. (2003). Antibiotic use and the prevalence of antibiotic resistance in bacteria from healthy volunteers in the Dutch community. Infection31, 9–14. doi: 10.1007/s15010-002-3035-8
18
BuffieC. G.BucciV.SteinR. R.McKenneyP. T.LingL.GobourneA.et al. (2015). Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature517, 205–208. doi: 10.1038/nature13828
19
BuretA. G.AllainT. (2023). Gut microbiota biofilms: from regulatory mechanisms to therapeutic targets. J. Exp. Med.220:e20221743. doi: 10.1084/jem.20221743
20
BushK. (2018). Past and present perspectives on β-lactamases. Antimicrob. Agents Chemother.62:e01076. doi: 10.1128/AAC.01076-18
21
BushK.JacobyG. A. (2010). Updated functional classification of β-lactamases. Antimicrob. Agents Chemother.54, 969–976. doi: 10.1128/AAC.01009-09
22
Caballero-FloresG.PickardJ. M.FukudaS.InoharaN.NúñezG. (2020). An enteric pathogen subverts colonization resistance by evading competition for amino acids in the gut. Cell Host Microbe28, 526–533.e5. doi: 10.1016/j.chom.2020.06.018
23
CalomirisJ. J.ArmstrongJ. L.SeidlerR. J. (1984). Association of metal tolerance with multiple antibiotic resistance of bacteria isolated from drinking water. Appl. Environ. Microbiol.47, 1238–1242. doi: 10.1128/aem.47.6.1238-1242.1984
24
Centers for Disease Control and Prevention (2013). Antibiotic resistance threats in the United States, 2013. Atlanta, GA: Centers for Disease Control and Prevention.
25
ChangP. V. (2020). Chemical mechanisms of colonization resistance by the gut microbial metabolome. ACS Chem. Biol.15, 1119–1126. doi: 10.1021/acschembio.9b00813
26
ChengG.HuY.YinY.YangX.XiangC.WangB.et al. (2012). Functional screening of antibiotic resistance genes from human gut microbiota reveals a novel gene fusion. FEMS Microbiol. Lett.336, 11–16. doi: 10.1111/j.1574-6968.2012.02647.x
27
ChenG.NingB.ShiT. (2019). Single-cell RNA-seq technologies and related computational data analysis. Front. Genet.10:317. doi: 10.3389/fgene.2019.00317
28
ChokshiA.SifriZ.CennimoD.HorngH. (2019). Global contributors to antibiotic resistance. J. Global Infect. Dis.11, 36–42. doi: 10.4103/jgid.jgid_110_18
29
ChowdhuryN. N.ForryS. P.ServetasS. L.HunterM. E.DootzJ. N.DunkersJ. P.et al. (2024). Measuring microbial community-wide antibiotic resistance propagation via natural transformation in the human gut microbiome. bioRxiv. Available online at: https://doi.org/10.1101/2024.11.26.625464
30
ClaessonM. J.CusackS.O’SullivanO.Greene-DinizR.de WeerdH.FlanneryE.et al. (2011). Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. U.S.A.108, 4586–4591. doi: 10.1073/pnas.1000097107
31
Corrêa-OliveiraR.FachiJ. L.VieiraA.SatoF. T.VinoloM. A. R. (2016). Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol.5:e73. doi: 10.1038/cti.2016.17
32
CotterP. D.RossR. P.HillC. (2013). Bacteriocins—a viable alternative to antibiotics?Nat. Rev. Microbiol.11, 95–105. doi: 10.1038/nrmicro2937
33
CoxG.WrightG. D. (2013). Intrinsic antibiotic resistance: mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol.303, 287–292. doi: 10.1016/j.ijmm.2013.02.009
34
CoyneM. J.ComstockL. E. (2019). Type VI secretion systems and the gut microbiota. Microbiol. Spectr.7, 10–1128. doi: 10.1128/microbiolspec.PSIB-0009-2018
35
CunninghamA. L.StephensJ. W.HarrisD. A. (2021). Intestinal microbiota and their metabolic contribution to type 2 diabetes and obesity. J. Diabetes Metab. Disord.20, 1855–1870. doi: 10.1007/s40200-021-00858-4
36
DadgostarP. (2019). Antimicrobial resistance: implications and costs. Infect. Drug Resist.12, 3903–3910. doi: 10.2147/IDR.S234610
37
DavenportE. R.Mizrahi-ManO.MicheliniK.BarreiroL. B.OberC.GiladY. (2014). Seasonal variation in human gut microbiome composition. PLoS One9:e90731. doi: 10.1371/journal.pone.0090731
38
DavidL. A.MauriceC. F.CarmodyR. N.GootenbergD. B.ButtonJ. E.WolfeB. E.et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature505, 559–563. doi: 10.1038/nature12820
39
DaviesM. R.HoldenM. T.CouplandP.ChenJ. H. K.VenturiniC.BarnettT. C.et al. (2015). Emergence of scarlet fever Streptococcus pyogenes emm12 clones in Hong Kong is associated with toxin acquisition and multidrug resistance. Nat. Genet.47, 84–87. doi: 10.1038/ng.3147
40
DavisE. C.CastagnaV. P.SelaD. A.HillardM. A.LindbergS.MantisN. J.et al. (2022). Gut microbiome and breast-feeding: implications for early immune development. J. Allergy Clin. Immunol.150, 523–534. doi: 10.1016/j.jaci.2022.07.014
41
de FilippoC.CavalieriD.di PaolaM.RamazzottiM.PoulletJ. B.MassartS.et al. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A.107, 14691–14696. doi: 10.1073/pnas.1005963107
42
DelahayeC.NicolasJ. (2021). Sequencing DNA with nanopores: troubles and biases. PLoS One16:e0257521. doi: 10.1371/journal.pone.0257521
43
DelcourA. H. (2009). Outer membrane permeability and antibiotic resistance. Biochim. Biophys. Acta1794, 808–816. doi: 10.1016/j.bbapap.2008.11.005
44
den BestenG.van EunenK.GroenA. K.VenemaK.ReijngoudD.-J.BakkerB. M. (2013). The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res.54, 2325–2340. doi: 10.1194/jlr.R036012
45
DengL.WangS. (2024). Colonization resistance: the role of gut microbiota in preventing Salmonella invasion and infection. Gut Microbes16:2424914. doi: 10.1080/19490976.2024.2424914
46
DeR. (2019). Metagenomics: aid to combat antimicrobial resistance in diarrhea. Gut Pathogens11:47. doi: 10.1186/s13099-019-0331-8
47
DesaiM. S.SeekatzA. M.KoropatkinN. M.KamadaN.HickeyC. A.WolterM.et al. (2016). A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell167, 1339–1353.e21. doi: 10.1016/j.cell.2016.10.043
48
DeschasauxM.BouterK. E.ProdanA.LevinE.GroenA. K.HerremaH.et al. (2018). Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med.24, 1526–1531. doi: 10.1038/s41591-018-0160-1
49
DethlefsenL.RelmanD. A. (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. U.S.A.108, 4554–4561. doi: 10.1073/pnas.1000087107
50
DubourgG.LagierJ.-C.ArmougomF.RobertC.AudolyG.PapazianL.et al. (2013). High-level colonisation of the human gut by Verrucomicrobia following broad-spectrum antibiotic treatment. Int. J. Antimicrob. Agents41, 149–155. doi: 10.1016/j.ijantimicag.2012.10.012
51
DucarmonQ. R.ZwittinkR. D.HornungB. V. H.van SchaikW.YoungV. B.KuijperE. J. (2019). Gut microbiota and colonization resistance against bacterial enteric infection. Microbiol. Mol. Biol. Rev.83:e00007. doi: 10.1128/MMBR.00007-19
52
DutilhB. E.CassmanN.McNairK.SanchezS. E.SilvaG. G. Z.BolingL.et al. (2014). A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun.5:4498. doi: 10.1038/ncomms5498
53
EckburgP. B.BikE. M.BernsteinC. N.PurdomE.DethlefsenL.SargentM.et al. (2005). Diversity of the human intestinal microbial flora. Science308, 1635–1638. doi: 10.1126/science.1110591
54
ElisonE.VigsnaesL. K.Rindom KrogsgaardL.RasmussenJ.SørensenN.McConnellB.et al. (2016). Oral supplementation of healthy adults with 2′-O-fucosyllactose and lacto-N-neotetraose is well tolerated and shifts the intestinal microbiota. Br. J. Nutr.116, 1356–1368. doi: 10.1017/S0007114516003354
55
FabichA. J.JonesS. A.ChowdhuryF. Z.CernosekA.AndersonA.SmalleyD.et al. (2008). Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect. Immun.76, 1143–1152. doi: 10.1128/IAI.01386-07
56
Fernández-OrthD.MiróE.Brown-JaqueM.Rodríguez-RubioL.EspinalP.Rodriguez-NavarroJ.et al. (2019). Faecal phageome of healthy individuals: presence of antibiotic resistance genes and variations caused by ciprofloxacin treatment. J. Antimicrob. Chemother.74, 854–864. doi: 10.1093/jac/dky540
57
FerriM.RanucciE.RomagnoliP.GiacconeV. (2017). Antimicrobial resistance: a global emerging threat to public health systems. Crit. Rev. Food Sci. Nutr.57, 2857–2876. doi: 10.1080/10408398.2015.1077192
58
FigdorD.GulabivalaK. (2008). Survival against the odds: microbiology of root canals associated with post-treatment disease. Endod. Top.18, 62–77. doi: 10.1111/j.1601-1546.2011.00259.x
59
FindlaterA.BogochI. I. (2018). Human mobility and the global spread of infectious diseases: a focus on air travel. Trends Parasitol.34, 772–783. doi: 10.1016/j.pt.2018.07.004
60
FinkelS. E.KolterR. (2001). DNA as a nutrient: novel role for bacterial competence gene homologs. J. Bacteriol.183, 6288–6293. doi: 10.1128/JB.183.21.6288-6293.2001
61
FiroozehF.ShahcheraghiF.Zahraei-SalehiT.AslaniM. M.BanisaeedR. (2013). First CTX-M type ß-lactamase-producing and ciprofloxacin resistant Salmonella infection acquired by a child in Iran. Int. J. Enteric Pathog.1, 76–78. doi: 10.17795/ijep13774
62
FiroozehF.ShahcheraghiF.Zahraei SalehiT.KarimiV.AslaniM. M. (2011). Antimicrobial resistance profile and presence of class I integrongs among Salmonella enterica serovars isolated from human clinical specimens in Tehran, Iran, Iran. J. Microbiol., 3, 112–117. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/22347592
63
FiroozehF.ZibaeiM. (2019). The role of gut microbiota in antimicrobial resistance: a mini-review. Anti-Infect. Agents.18, 201–206. doi: 10.2174/2211352517666190716154013
64
ForslundK.SunagawaS.CoelhoL. P.BorkP. (2014). Metagenomic insights into the human gut resistome and the forces that shape it. BioEssays36, 316–329. doi: 10.1002/bies.201300143
65
FouhyF.RossR. P.FitzgeraldG. F.StantonC.CotterP. D. (2012). Composition of the early intestinal microbiota. Gut Microbes3, 203–220. doi: 10.4161/gmic.20169
66
FrazãoN.SousaA.LässigM.GordoI. (2019). Horizontal gene transfer overrides mutation in Escherichia coli colonizing the mammalian gut. Proc. Natl. Acad. Sci. U.S.A.116, 17906–17915. doi: 10.1073/pnas.1906958116
67
GalhanoB. S. P.FerrariR. G.PanzenhagenP.de JesusA. C. S.Conte-JuniorC. A. (2021). Antimicrobial resistance gene detection methods for bacteria in animal-based foods: a brief review of highlights and advantages. Microorganisms9:923. doi: 10.3390/microorganisms9050923
68
GantoisI.DucatelleR.PasmansF.HaesebrouckF.HautefortI.ThompsonA.et al. (2006). Butyrate specifically down-regulates Salmonella pathogenicity island 1 gene expression. Appl. Environ. Microbiol.72, 946–949. doi: 10.1128/AEM.72.1.946-949.2006
69
GargiulloL.Del ChiericoF.D’ArgenioP.PutignaniL. (2019). Gut microbiota modulation for multidrug-resistant organism decolonization: present and future perspectives. Front. Microbiol.10:1704. doi: 10.3389/fmicb.2019.01704
70
GaulkeC. A.SharptonT. J. (2018). The influence of ethnicity and geography on human gut microbiome composition. Nat. Med.24, 1495–1496. doi: 10.1038/s41591-018-0210-8
71
GauravA.BakhtP.SainiM.PandeyS.PathaniaR. (2023). Role of bacterial efflux pumps in antibiotic resistance, virulence, and strategies to discover novel efflux pump inhibitors. Microbiology169:001333. doi: 10.1099/mic.0.001333
72
GeserN.StephanR.KorczakB. M.BeutinL.HächlerH. (2012). Molecular identification of extended-spectrum-β-lactamase genes from Enterobacteriaceae isolated from healthy human carriers in Switzerland. Antimicrob. Agents Chemother.56, 1609–1612. doi: 10.1128/AAC.05539-11
73
GhoshT. S.GuptaS. S.NairG. B.MandeS. S. (2013). In silico analysis of antibiotic resistance genes in the gut microflora of individuals from diverse geographies and age-groups. PLoS One8:e83823. doi: 10.1371/journal.pone.0083823
74
GhoshT. S.ShanahanF.O’TooleP. W. (2022). The gut microbiome as a modulator of healthy ageing. Nat. Rev. Gastroenterol. Hepatol.19, 565–584. doi: 10.1038/s41575-022-00605-x
75
GhotaslouR.YekaniM.MemarM. Y. (2018). The role of efflux pumps in Bacteroides fragilis resistance to antibiotics. Microbiol. Res.210, 1–5. doi: 10.1016/j.micres.2018.02.007
76
GijónD.CuriaoT.BaqueroF.CoqueT. M.CantónR. (2012). Fecal carriage of carbapenemase-producing Enterobacteriaceae: a hidden reservoir in hospitalized and nonhospitalized patients. J. Clin. Microbiol.50, 1558–1563. doi: 10.1128/JCM.00020-12
77
GjonbalajM.KeithJ. W.DoM. H.HohlT. M.PamerE. G.BecattiniS. (2020). Antibiotic degradation by commensal microbes shields pathogens. Infect. Immun.88:e00012. doi: 10.1128/IAI.00012-20
78
GrenetK.GuillemotD.JarlierV.MoreauB.DubourdieuS.RuimyR.et al. (2004). Antibacterial resistance, Wayampis Amerindians, French Guyana. Emerg. Infect. Dis.10, 1150–1153. doi: 10.3201/eid1006.031015
79
GrootersK. E.KuJ. C.RichterD. M.KrinockM. J.MinorA.LiP.et al. (2024). Strategies for combating antibiotic resistance in bacterial biofilms. Front. Cell. Infect. Microbiol.14:1352273. doi: 10.3389/fcimb.2024.1352273
80
GroussinM.PoyetM.SistiagaA.KearneyS. M.MonizK.NoelM.et al. (2021). Elevated rates of horizontal gene transfer in the industrialized human microbiome. Cell184, 2053–2067.e18. doi: 10.1016/j.cell.2021.02.052
81
GuglielminiJ.de la CruzF.RochaE. P. C. (2013). Evolution of conjugation and type IV secretion systems. Mol. Biol. Evol.30, 315–331. doi: 10.1093/molbev/mss221
82
GuoX.LiS.ZhangJ.WuF.LiX.WuD.et al. (2017). Genome sequencing of 39 Akkermansia muciniphila isolates reveals its population structure, genomic and functional diversity, and global distribution in mammalian gut microbiotas. BMC Genomics18:800. doi: 10.1186/s12864-017-4195-3
83
HammamiR.FernandezB.LacroixC.FlissI. (2013). Anti-infective properties of bacteriocins: an update. Cell. Mol. Life Sci.70, 2947–2967. doi: 10.1007/s00018-012-1202-3
84
HarrisR. M.PictonR.SinghS.WaringR. H. (2000). Activity of phenolsulfotransferases in the human gastrointestinal tract. Life Sci.67, 2051–2057. doi: 10.1016/S0024-3205(00)00791-8
85
HassallJ.CoxonC.PatelV. C.GoldenbergS. D.SergakiC. (2024). Limitations of current techniques in clinical antimicrobial resistance diagnosis: examples and future prospects. npj Antimicrob. Resist.2:16. doi: 10.1038/s44259-024-00033-8
86
HorrocksV.KingO. G.YipA. Y. G.MarquesI. M.McDonaldJ. A. K. (2023). Role of the gut microbiota in nutrient competition and protection against intestinal pathogen colonization. Microbiology169:001377. doi: 10.1099/mic.0.001377
87
HuddlestonJ. R. (2014). Horizontal gene transfer in the human gastrointestinal tract: potential spread of antibiotic resistance genes. Infect. Drug Resist.7, 167–176. doi: 10.2147/IDR.S48820
88
HuntK. M.FosterJ. A.ForneyL. J.SchütteU. M. E.BeckD. L.AbdoZ.et al. (2011). Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS One6:e21313. doi: 10.1371/journal.pone.0021313
89
HuY.YangX.QinJ.LuN.ChengG.WuN.et al. (2013). Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat. Commun.4:2151. doi: 10.1038/ncomms3151
90
IslamM. S.SoburM. A.RahmanS.BallahF. M.IevyS.SiddiqueM. P.et al. (2022). Detection of blaTEM, blaCTX-M, blaCMY, and blaSHV genes among extended-spectrum beta-lactamase-producing Escherichia coli isolated from migratory birds travelling to Bangladesh. Microb. Ecol.83, 942–950. doi: 10.1007/s00248-021-01803-x
91
JainM.OlsenH. E.PatenB.AkesonM. (2016). The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol.17:239. doi: 10.1186/s13059-016-1103-0
92
JindalA. K.PandyaK.KhanI. D. (2015). Antimicrobial resistance: a public health challenge. Med. J. Armed Forces India71, 178–181. doi: 10.1016/j.mjafi.2014.04.011
93
JohnsonK. V.-A. (2020). Gut microbiome composition and diversity are related to human personality traits. Hum Microb. J.15:100069. doi: 10.1016/j.humic.2019.100069
94
KamadaN.KimY.-G.ShamH. P.VallanceB. A.PuenteJ. L.MartensE. C.et al. (2012). Regulated virulence controls the ability of a pathogen to compete with the gut microbiota. Science336, 1325–1329. doi: 10.1126/science.1222195
95
KaminskiM. M.AbudayyehO. O.GootenbergJ. S.ZhangF.CollinsJ. J. (2021). CRISPR-based diagnostics. Nat. Biomed. Eng.5, 643–656. doi: 10.1038/s41551-021-00760-7
96
KarkmanA.PärnänenK.LarssonD. G. J. (2019). Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun.10:80. doi: 10.1038/s41467-018-07992-3
97
KesslerC.HouJ.NeoO.BucknerM. M. C. (2022). In situ, in vivo and in vitro approaches for studying AMR plasmid conjugation in the gut microbiome. FEMS Microbiol. Rev.47:fuac044. doi: 10.1093/femsre/fuac044
98
KeS.XiaoY.WeissS. T.ChenX.KellyC. P.LiuY.-Y. (2023). A computational method to dissect colonization resistance of the gut microbiota against pathogens. Cell Rep. Methods3:100576. doi: 10.1016/j.crmeth.2023.100576
99
KhanI.BaiY.ZhaL.UllahN.UllahH.ShahS. R. H.et al. (2021). Mechanism of the gut microbiota colonization resistance and enteric pathogen infection. Front. Cell. Infect. Microbiol.11:716299. doi: 10.3389/fcimb.2021.716299
100
KhoZ. Y.LalS. K. (2018). The human gut microbiome—a potential controller of wellness and disease. Front. Microbiol.9:1835. doi: 10.3389/fmicb.2018.01835
101
KimS.CovingtonA.PamerE. G. (2017). The intestinal microbiota: antibiotics, colonization resistance, and enteric pathogens. Immunol. Rev.279, 90–105. doi: 10.1111/imr.12563
102
KimY. S.HoS. B. (2010). Intestinal goblet cells and mucins in health and disease: recent insights and Progress. Curr. Gastroenterol. Rep.12, 319–330. doi: 10.1007/s11894-010-0131-2
103
KleinerM. (2019). Metaproteomics: much more than measuring gene expression in microbial communities. mSystems4:e00115. doi: 10.1128/mSystems.00115-19
104
KnappC. W.McCluskeyS. M.SinghB. K.CampbellC. D.HudsonG.GrahamD. W. (2011). Antibiotic resistance gene abundances correlate with metal and geochemical conditions in archived Scottish soils. PLoS One6:e27300. doi: 10.1371/journal.pone.0027300
105
KoikeS.KrapacI. G.OliverH. D.YannarellA. C.Chee-SanfordJ. C.AminovR. I.et al. (2007). Monitoring and source tracking of tetracycline resistance genes in lagoons and groundwater adjacent to swine production facilities over a 3-year period. Appl. Environ. Microbiol.73, 4813–4823. doi: 10.1128/AEM.00665-07
106
KonopkaJ. K.ChatterjeeP.LaMontagneC.BrownJ. (2022). Environmental impacts of mass drug administration programs: exposures, risks, and mitigation of antimicrobial resistance. Infect. Dis. Poverty11:78. doi: 10.1186/s40249-022-01000-z
107
KumarR.IndiraK.RizviA.RizviT.JeyaseelanL. (2008). Antibiotic prescribing practices in primary and secondary health care facilities in Uttar Pradesh, India. J. Clin. Pharm. Ther.33, 625–634. doi: 10.1111/j.1365-2710.2008.00960.x
108
LamberteL. E.van SchaikW. (2022). Antibiotic resistance in the commensal human gut microbiota. Curr. Opin. Microbiol.68:102150. doi: 10.1016/j.mib.2022.102150
109
LaneM.YadavV. (2020). “Multiple sclerosis” in Textbook of natural medicine (St. Louis, MO: Elsevier), 1587–1599.e3.
110
LaxminarayanR.DuseA.WattalC.ZaidiA. K. M.WertheimH. F. L.SumpraditN.et al. (2013). Antibiotic resistance—the need for global solutions. Lancet Infect. Dis.13, 1057–1098. doi: 10.1016/S1473-3099(13)70318-9
111
LeathamM. P.BanerjeeS.AutieriS. M.Mercado-LuboR.ConwayT.CohenP. S. (2009). Precolonized human commensal Escherichia coli strains serve as a barrier to E. coli O157:H7 growth in the streptomycin-treated mouse intestine. Infect. Immun.77, 2876–2886. doi: 10.1128/IAI.00059-09
112
LepageP.LeclercM. C.JoossensM.MondotS.BlottièreH. M.RaesJ.et al. (2013). A metagenomic insight into our gut’s microbiome. Gut62, 146–158. doi: 10.1136/gutjnl-2011-301805
113
LesterC. H.Frimodt-MøllerN.SørensenT. L.MonnetD. L.HammerumA. M. (2006). In vivo transfer of the vanA resistance gene from an Enterococcus faecium isolate of animal origin to an E. faecium isolate of human origin in the intestines of human volunteers. Antimicrob. Agents Chemother.50, 596–599. doi: 10.1128/AAC.50.2.596-599.2006
114
LichtT. R.ChristensenB. B.KrogfeltK. A.MolinS. (1999). Plasmid transfer in the animal intestine and other dynamic bacterial populations: the role of community structure and environment. Microbiology145, 2615–2622. doi: 10.1099/00221287-145-9-2615
115
LiL. L.NormanA.HansenL. H.SorensenS. J. (2012). Metamobilomics—expanding our knowledge on the pool of plasmid encoded traits in natural environments using high-throughput sequencing. Clin. Microbiol. Infect.18, 8–11. doi: 10.1111/j.1469-0691.2012.03862.x
116
LiuH. Y.HoppingG. C.VaidyanathanU.RonquilloY. C.HoopesP. C.MoshirfarM. (2019). Polymerase chain reaction and its application in the diagnosis of infectious keratitis. Med. Hypothesis Discov. Innov. Ophthalmol.8, 152–155
117
LiuW.HuangY.ZhangH.LiuZ.HuanQ.XiaoX.et al. (2023). Factors and mechanisms influencing conjugation in vivo in the gastrointestinal tract environment: a review. Int. J. Mol. Sci.24:5919. doi: 10.3390/ijms24065919
118
LiX.BrejnrodA.TrivediU.RusselJ.ThorsenJ.ShahS. A.et al. (2024). Co-localization of antibiotic resistance genes is widespread in the infant gut microbiome and associates with an immature gut microbial composition. Microbiome12:87. doi: 10.1186/s40168-024-01800-5
119
LlorC.BjerrumL. (2014). Antimicrobial resistance: risk associated with antibiotic overuse and initiatives to reduce the problem. Ther. Adv. Drug Saf.5, 229–241. doi: 10.1177/2042098614554919
120
LouisP.FlintH. J. (2009). Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett.294, 1–8. doi: 10.1111/j.1574-6968.2009.01514.x
121
LouisP.FlintH. J. (2017). Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol.19, 29–41. doi: 10.1111/1462-2920.13589
122
LouisP.HoldG. L.FlintH. J. (2014). The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol.12, 661–672. doi: 10.1038/nrmicro3344
123
MacfarlaneS.MacfarlaneG. T. (2003). Regulation of short-chain fatty acid production. Proc. Nutr. Soc.62, 67–72. doi: 10.1079/PNS2002207
124
MachadoA. M. D.SommerM. O. A. (2014). Human intestinal cells modulate conjugational transfer of multidrug resistance plasmids between clinical Escherichia coli isolates. PLoS One9:e100739. doi: 10.1371/journal.pone.0100739
125
MaiquesE.ÚbedaC.CampoyS.SalvadorN.LasaI.NovickR. P.et al. (2006). β-lactam antibiotics induce the SOS response and horizontal transfer of virulence factors in Staphylococcus aureus. J. Bacteriol.188, 2726–2729. doi: 10.1128/JB.188.7.2726-2729.2006
126
ManichanhC.ReederJ.GibertP.VarelaE.LlopisM.AntolinM.et al. (2010). Reshaping the gut microbiome with bacterial transplantation and antibiotic intake. Genome Res.20, 1411–1419. doi: 10.1101/gr.107987.110
127
MasiM.RéfregiersM.PosK. M.PagèsJ.-M. (2017). Mechanisms of envelope permeability and antibiotic influx and efflux in gram-negative bacteria. Nat. Microbiol.2:17001. doi: 10.1038/nmicrobiol.2017.1
128
Mayorga-RamosA.Zúñiga-MirandaJ.Carrera-PachecoS. E.Barba-OstriaC.GuamánL. P. (2023). CRISPR-Cas-based antimicrobials: design, challenges, and bacterial mechanisms of resistance. ACS Infect Dis.9, 1283–1302. doi: 10.1021/acsinfecdis.2c00649
129
McCannA.RyanF. J.StockdaleS. R.DalmassoM.BlakeT.RyanC. A.et al. (2018). Viromes of one year old infants reveal the impact of birth mode on microbiome diversity. PeerJ6:e4694. doi: 10.7717/peerj.4694
130
McLainJ. E.CytrynE.DursoL. M.YoungS. (2016). Culture-based methods for detection of antibiotic resistance in agroecosystems: advantages, challenges, and gaps in knowledge. J. Environ. Qual.45, 432–440. doi: 10.2134/jeq2015.06.0317
131
MerrickB.SergakiC.EdwardsL.MoyesD. L.KertanegaraM.ProssomaritiD.et al. (2023). Modulation of the gut microbiota to control antimicrobial resistance (AMR)—a narrative review with a focus on faecal microbiota transplantation (FMT). Infect. Dis. Rep.15, 238–254. doi: 10.3390/idr15030025
132
MillerA. L.BesshoS.GrandoK.TükelÇ. (2021). Microbiome or infections: amyloid-containing biofilms as a trigger for complex human diseases. Front. Immunol.12:638867. doi: 10.3389/fimmu.2021.638867
133
ModiS. R.LeeH. H.SpinaC. S.CollinsJ. J. (2013). Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature499, 219–222. doi: 10.1038/nature12212
134
MoniraS.BarmanI.JubydaF. T.AliS. I.IslamA.KMZR.et al. (2023). Gut microbiota shifts favorably with delivery of handwashing with soap and water treatment intervention in a prospective cohort (CHoBI7 trial). J. Health Popul. Nutr.42:146. doi: 10.1186/s41043-023-00477-0
135
MookherjeeN.HancockR. E. W. (2007). Cationic host defence peptides: innate immune regulatory peptides as a novel approach for treating infections. Cell. Mol. Life Sci.64, 922–933. doi: 10.1007/s00018-007-6475-6
136
MoonC. D.YoungW.MacleanP. H.CooksonA. L.BerminghamE. N. (2018). Metagenomic insights into the roles of Proteobacteria in the gastrointestinal microbiomes of healthy dogs and cats. Microbiologyopen7:e00677. doi: 10.1002/mbo3.677
137
MorrisonD. J.PrestonT. (2016). Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes7, 189–200. doi: 10.1080/19490976.2015.1134082
138
MuhsenK.Abed El-HaiR.Amit-AharonA.NehamaH.GondiaM.DavidovitchN.et al. (2012). Risk factors of underutilization of childhood immunizations in ultraorthodox Jewish communities in Israel despite high access to health care services. Vaccine30, 2109–2115. doi: 10.1016/j.vaccine.2012.01.044
139
MukherjeeS.ZhengH.DerebeM. G.CallenbergK. M.PartchC. L.RollinsD.et al. (2014). Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature505, 103–107. doi: 10.1038/nature12729
140
MullanyP. (2014). Functional metagenomics for the investigation of antibiotic resistance. Virulence5, 443–447. doi: 10.4161/viru.28196
141
MuniesaM.Colomer-LluchM.JofreJ. (2013). Could bacteriophages transfer antibiotic resistance genes from environmental bacteria to human-body associated bacterial populations?Mob. Genet. Elem.3:e25847. doi: 10.4161/mge.25847
142
MunitaJ. M.AriasC. A. (2016). Mechanisms of antibiotic resistance. Microbiol. Spectr.4, 481–511. doi: 10.1128/microbiolspec.VMBF-0016-2015
143
MurrayB. E.RensimerE. R.DuPontH. L. (1982). Emergence of high-level trimethoprim resistance in fecal Escherichia coli during oral administration of trimethoprim or trimethoprim-sulfamethoxazole. N. Engl. J. Med.306, 130–135. doi: 10.1056/NEJM198201213060302
144
NasiriM. J.GoudarziM.HajikhaniB.GhaziM.GoudarziH.PouriranR. (2018). Clostridioides (Clostridium) difficile infection in hospitalized patients with antibiotic-associated diarrhea: a systematic review and meta-analysis. Anaerobe50, 32–37. doi: 10.1016/j.anaerobe.2018.01.011
145
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Global Health; Forum on Microbial Threats (2018). Understanding the economics of microbial threats: proceedings of a workshop. Washington, DC: National Academies Press.
146
NatividadJ. M. M.VerduE. F. (2013). Modulation of intestinal barrier by intestinal microbiota: pathological and therapeutic implications. Pharmacol. Res.69, 42–51. doi: 10.1016/j.phrs.2012.10.007
147
NeamatiF.FiroozehF.SaffariM.ZibaeiM. (2015). Virulence genes and antimicrobial resistance pattern in uropathogenic Escherichia coli isolated from hospitalized patients in Kashan, Iran. Jundishapur J. Microbiol.8:e17514. doi: 10.5812/jjm.17514
148
NesmeJ.CécillonS.DelmontT. O.MonierJ.-M.VogelT. M.SimonetP. (2014). Large-scale metagenomic-based study of antibiotic resistance in the environment. Curr. Biol.24, 1096–1100. doi: 10.1016/j.cub.2014.03.036
149
NgK. M.FerreyraJ. A.HigginbottomS. K.LynchJ. B.KashyapP. C.GopinathS.et al. (2013). Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature502, 96–99. doi: 10.1038/nature12503
150
NhuN. T. Q.YoungV. B. (2023). The relationship between the microbiome and antimicrobial resistance. Clin. Infect. Dis.77, S479–S486. doi: 10.1093/cid/ciad641
151
NiedringhausT. P.MilanovaD.KerbyM. B.SnyderM. P.BarronA. E. (2011). Landscape of next-generation sequencing technologies. Anal. Chem.83, 4327–4341. doi: 10.1021/ac2010857
152
NiemelaL.LamouryG.CarrollS.MorgiaM.YeungA.OhB. (2024). Exploring gender differences in the relationship between gut microbiome and depression—a scoping review. Front. Psychiatry15:1361145. doi: 10.3389/fpsyt.2024.1361145
153
NishinoK.YamasakiS.NakashimaR.ZwamaM.Hayashi-NishinoM. (2021). Function and inhibitory mechanisms of multidrug efflux pumps. Front. Microbiol.12:737288. doi: 10.3389/fmicb.2021.737288
154
NyirabahiziE.TysonG. H.DessaiU.ZhaoS.KaberaC.CrareyE.et al. (2020). Evaluation of Escherichia coli as an indicator for antimicrobial resistance in Salmonella recovered from the same food or animal ceca samples. Food Control115:107280:107280. doi: 10.1016/j.foodcont.2020.107280
155
O’NeillJ. (2016). Review on antimicrobial resistance: tackling drug-resistant infections globally: final report and recommendations. London: Wellcome Trust, 1–84.
156
OkekeI. N.EdelmanR. (2001). Dissemination of antibiotic-resistant bacteria across geographic borders. Clin. Infect. Dis.33, 364–369. doi: 10.1086/321877
157
OverdevestI. (2011). Extended-spectrum B-lactamase genes of Escherichia coli in chicken meat and humans, the Netherlands. Emerg. Infect. Dis.17, 1216–1222. doi: 10.3201/eid1707.110209
158
ParamsothyS.ParamsothyR.RubinD. T.KammM. A.KaakoushN. O.MitchellH. M.et al. (2017). Faecal microbiota transplantation for inflammatory bowel disease: a systematic review and meta-analysis. J. Crohn’s Colitis11, 1180–1199. doi: 10.1093/ecco-jcc/jjx063
159
PathakA.AngstD. C.León-SampedroR.HallA. R. (2023). Antibiotic-degrading resistance changes bacterial community structure via species-specific responses. ISME J.17, 1495–1503. doi: 10.1038/s41396-023-01465-2
160
PecquetS.ChachatyE.TancrèdeC.AndremontA. (1991). Effects of roxithromycin on fecal bacteria in human volunteers and resistance to colonization in gnotobiotic mice. Antimicrob. Agents Chemother.35, 548–552. doi: 10.1128/AAC.35.3.548
161
PendersJ.StobberinghE. E.SavelkoulP. H. M.WolffsP. F. G. (2013). The human microbiome as a reservoir of antimicrobial resistance. Front. Microbiol.4:87. doi: 10.3389/fmicb.2013.00087
162
PetersonE.KaurP. (2018). Antibiotic resistance mechanisms in bacteria: relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front. Microbiol.9:2928. doi: 10.3389/fmicb.2018.02928
163
PetersonG.KumarA.GartE.NarayananS. (2011). Catecholamines increase conjugative gene transfer between enteric bacteria. Microb. Pathog.51, 1–8. doi: 10.1016/j.micpath.2011.03.002
164
PetrizB. A.FrancoO. L. (2017). Metaproteomics as a complementary approach to gut microbiota in health and disease. Front. Chem.5:4. doi: 10.3389/fchem.2017.00004
165
PickardJ. M.ZengM. Y.CarusoR.NúñezG. (2017). Gut microbiota: role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev.279, 70–89. doi: 10.1111/imr.12567
166
PilmisB.Le MonnierA.ZaharJ.-R. (2020). Gut microbiota, antibiotic therapy and antimicrobial resistance: a narrative review. Microorganisms8:269. doi: 10.3390/microorganisms8020269
167
PinartM.DötschA.SchlichtK.LaudesM.BouwmanJ.ForslundS. K.et al. (2021). Gut microbiome composition in obese and non-obese persons: a systematic review and meta-analysis. Nutrients14:12. doi: 10.3390/nu14010012
168
PrestinaciF.PezzottiP.PantostiA. (2015). Antimicrobial resistance: a global multifaceted phenomenon. Pathog. Glob. Health109, 309–318. doi: 10.1179/2047773215Y.0000000030
169
QinJ.LiR.RaesJ.ArumugamM.BurgdorfK. S.ManichanhC.et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature464, 59–65. doi: 10.1038/nature08821
170
QiuQ.WangJ.YanY.RoyB.ChenY.ShangX.et al. (2020). Metagenomic analysis reveals the distribution of antibiotic resistance genes in a large-scale population of healthy individuals and patients with varied diseases. Front. Mol. Biosci.7:590018. doi: 10.3389/fmolb.2020.590018
171
RadovanovicM.KekicD.GajicI.KabicJ.JovicevicM.KekicN.et al. (2023). Potential influence of antimicrobial resistance gene content in probiotic bacteria on the gut resistome ecosystems. Front. Nutr.10:1054555. doi: 10.3389/fnut.2023.1054555
172
Rakoff-NahoumS.PaglinoJ.Eslami-VarzanehF.EdbergS.MedzhitovR. (2004). Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell118, 229–241. doi: 10.1016/j.cell.2004.07.002
173
RamirezJ.GuarnerF.Bustos FernandezL.MaruyA.SdepanianV. L.CohenH. (2020). Antibiotics as major disruptors of gut microbiota. Front. Cell. Infect. Microbiol.10:572912. doi: 10.3389/fcimb.2020.572912
174
RatherM. A.GuptaK.MandalM. (2021). Microbial biofilm: formation, architecture, antibiotic resistance, and control strategies. Braz. J. Microbiol.52, 1701–1718. doi: 10.1007/s42770-021-00624-x
175
RenY.WuJ.WangY.ZhangL.RenJ.ZhangZ.et al. (2023). Lifestyle patterns influence the composition of the gut microbiome in a healthy Chinese population. Sci. Rep.13:14425. doi: 10.1038/s41598-023-41532-4
176
RinninellaE.RaoulP.CintoniM.FranceschiF.MiggianoG.GasbarriniA.et al. (2019). What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms7:14. doi: 10.3390/microorganisms7010014
177
RodríguezJ. M.MurphyK.StantonC.RossR. P.KoberO. I.JugeN.et al. (2015). The composition of the gut microbiota throughout life, with an emphasis on early life. Microb. Ecol. Health Dis.26:26050. doi: 10.3402/mehd.v26.26050
178
RooneyC. M.SheppardA. E.ClarkE.DaviesK.HubbardA. T. M.SebraR.et al. (2019). Dissemination of multiple carbapenem resistance genes in an in vitro gut model simulating the human colon. J. Antimicrob. Chemother.74, 1876–1883. doi: 10.1093/jac/dkz106
179
RussellA. B.HoodR. D.BuiN. K.LeRouxM.VollmerW.MougousJ. D. (2011). Type VI secretion delivers bacteriolytic effectors to target cells. Nature475, 343–347. doi: 10.1038/nature10244
180
SalamM. A.Al-AminM. Y.SalamM. T.PawarJ. S.AkhterN.RabaanA. A.et al. (2023). “Antimicrobial resistance: a growing serious threat for global public health,” in Healthcare.Multidisciplinary Digital Publishing Institute, 11:1946.
181
SalomãoM. C. C.Heluany-FilhoM. A.MeneguetiM. G.De KrakerM. E. A.MartinezR.Bellissimo-RodriguesF. (2016). A randomized clinical trial on the effectiveness of a symbiotic product to decolonize patients harboring multidrug-resistant gram-negative bacilli. Rev. Soc. Bras. Med. Trop.49, 559–566. doi: 10.1590/0037-8682-0233-2016
182
SalyersA. A.ShoemakerN. B.StevensA. M.LiL. Y. (1995). Conjugative transposons: an unusual and diverse set of integrated gene transfer elements. Microbiol. Rev.59, 579–590. doi: 10.1128/mr.59.4.579-590.1995
183
SarmahA. K.MeyerM. T.BoxallA. B. A. (2006). A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere65, 725–759. doi: 10.1016/j.chemosphere.2006.03.026
184
Sassone-CorsiM.RaffatelluM. (2015). No vacancy: how beneficial microbes cooperate with immunity to provide colonization resistance to pathogens. J. Immunol.194, 4081–4087. doi: 10.4049/jimmunol.1403169
185
SchmiederR.EdwardsR. (2012). Insights into antibiotic resistance through metagenomic approaches. Future Microbiol.7, 73–89. doi: 10.2217/fmb.11.135
186
SchnorrS. L.CandelaM.RampelliS.CentanniM.ConsolandiC.BasagliaG.et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat. Commun.5:3654. doi: 10.1038/ncomms4654
187
SenderR.FuchsS.MiloR. (2016). Revised estimates for the number of human and bacteria cells in the body. PLoS Biol.14:e1002533. doi: 10.1371/journal.pbio.1002533
188
SherA. A.Whitehead-TilleryC. E.PeerA. M.BellJ. A.VocelleD. B.DippelJ. T.et al. (2025). Dynamic spread of antibiotic resistance determinants by conjugation to a human-derived gut microbiota in a transplanted mouse model. Antibiotics14:152. doi: 10.3390/antibiotics14020152
189
SheteV.GroverN.KumarM. (2017, 2017). Analysis of aminoglycoside modifying enzyme genes responsible for high-level aminoglycoside resistance among enterococcal isolates. J. Pathog.2017:3256952. doi: 10.1155/2017/3256952
190
ShinJ.KimS. R.XieZ.JinY.-S.WangY.-C. (2024). A CRISPR/Cas12a-based system for sensitive detection of antimicrobial-resistant genes in carbapenem-resistant Enterobacterales. Biosensors14:194. doi: 10.3390/bios14040194
191
ShreinerA. B.KaoJ. Y.YoungV. B. (2015). The gut microbiome in health and in disease. Curr. Opin. Gastroenterol.31, 69–75. doi: 10.1097/MOG.0000000000000139
192
SilverL. L. (2011). Challenges of antibacterial discovery. Clin. Microbiol. Rev.24, 71–109. doi: 10.1128/cmr.00030-10
193
SingerA. C.ShawH.RhodesV.HartA. (2016). Review of antimicrobial resistance in the environment and its relevance to environmental regulators. Front. Microbiol.7:1728. doi: 10.3389/fmicb.2016.01728
194
SinghS.VermaN.TanejaN. (2019). The human gut resistome: current concepts & future prospects. Indian J. Med. Res.150, 345–358. doi: 10.4103/ijmr.IJMR_1979_17
195
SommerM. O. A.DantasG.ChurchG. M. (2009). Functional characterization of the antibiotic resistance reservoir in the human microflora. Science325, 1128–1131. doi: 10.1126/science.1176950
196
SorbaraM. T.PamerE. G. (2019). Interbacterial mechanisms of colonization resistance and the strategies pathogens use to overcome them. Mucosal Immunol.12:840. doi: 10.1038/s41385-018-0053-0
197
SorgJ. A.SonensheinA. L. (2008). Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol.190, 2505–2512. doi: 10.1128/JB.01765-07
198
SotoS. M. (2013). Role of efflux pumps in the antibiotic resistance of bacteria embedded in a biofilm. Virulence4, 223–229. doi: 10.4161/viru.23724
199
SpellbergB.PowersJ. H.BrassE. P.MillerL. G.EdwardsJ. E. (2004). Trends in antimicrobial drug development: implications for the future. Clin. Infect. Dis.38, 1279–1286. doi: 10.1086/420937
200
SpraggeF.BakkerenE.JahnM. T.AraujoB. N. E.PearsonC. F.WangX.et al. (2023). Microbiome diversity protects against pathogens by nutrient blocking. Science382:eadj3502. doi: 10.1126/science.adj3502
201
StecherB.DenzlerR.MaierL.BernetF.SandersM. J.PickardD. J.et al. (2012). Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc. Natl. Acad. Sci. U.S.A.109, 1269–1274. doi: 10.1073/pnas.1113246109
202
TheophilusR. J.TaftD. H. (2023). Antimicrobial resistance genes (ARGs), the gut microbiome, and infant nutrition. Nutrients15:3177. doi: 10.3390/nu15143177
203
ThieraufA.PerezG.MaloyS. (2009). Generalized transduction. Methods Mol. Biol.501, 267–286. doi: 10.1007/978-1-60327-164-6_23
204
ThomasF.HehemannJ.-H.RebuffetE.CzjzekM.MichelG. (2011). Environmental and gut bacteroidetes: the food connection. Front. Microbiol.2:93. doi: 10.3389/fmicb.2011.00093
205
ThorpeK. E.JoskiP.JohnstonK. J. (2018). Antibiotic-resistant infection treatment costs have doubled since 2002, now exceeding $2 billion annually. Health Aff.37, 662–669. doi: 10.1377/hlthaff.2017.1153
206
ThursbyE.JugeN. (2017). Introduction to the human gut microbiota. Biochem. J.474, 1823–1836. doi: 10.1042/BCJ20160510
207
TimoneyJ. F.PortJ.GilesJ.SpanierJ. (1978). Heavy-metal and antibiotic resistance in the bacterial flora of sediments of New York bight. Appl. Environ. Microbiol.36, 465–472. doi: 10.1128/aem.36.3.465-472.1978
208
TownsendE. M.KellyL.MuscattG.BoxJ. D.HargravesN.LilleyD.et al. (2021). The human gut phageome: origins and roles in the human gut microbiome. Front. Cell. Infect. Microbiol.11:643214. doi: 10.3389/fcimb.2021.643214
209
TytgatH. L. P.NobregaF. L.van der OostJ.de VosW. M. (2019). Bowel biofilms: tipping points between a healthy and compromised gut?Trends Microbiol.27, 17–25. doi: 10.1016/j.tim.2018.08.009
210
Udikovic-KolicN.WichmannF.BroderickN. A.HandelsmanJ. (2014). Bloom of resident antibiotic-resistant bacteria in soil following manure fertilization. Proc. Natl. Acad. Sci. U.S.A.111, 15202–15207. doi: 10.1073/pnas.1409836111
211
ValdesA. M.WalterJ.SegalE.SpectorT. D. (2018). Role of the gut microbiota in nutrition and health. BMJ361:k2179. doi: 10.1136/bmj.k2179
212
ValenzuelaX.HedmanH.VillagomezA.CardenasP.EisenbergJ. N. S.LevyK.et al. (2023). Distribution of blaCTX-M-gene variants in E. coli from different origins in Ecuador. Med. Microecol.18:100092. doi: 10.1016/j.medmic.2023.100092
213
VandenplasY.CarnielliV. P.KsiazykJ.LunaM. S.MigachevaN.MosselmansJ. M.et al. (2020). Factors affecting early-life intestinal microbiota development. Nutrition78:110812. doi: 10.1016/j.nut.2020.110812
214
van der VeenE. L.SchilderA. G. M.TimmersT. K.RoversM. M.FluitA. C.BontenM. J.et al. (2009). Effect of long-term trimethoprim/sulfamethoxazole treatment on resistance and integron prevalence in the intestinal flora: a randomized, double-blind, placebo-controlled trial in children. J. Antimicrob. Chemother.63, 1011–1016. doi: 10.1093/jac/dkp050
215
VermaM. K.AhmedV.GuptaS.KumarJ.PandeyR.MandhanV.et al. (2018). Functional metagenomics identifies novel genes ABCTPP, TMSRP1 and TLSRP1 among human gut enterotypes. Sci. Rep.8:1397. doi: 10.1038/s41598-018-19862-5
216
VienL. T. M.MinhN. N. Q.ThuongT. C.KhuongH. D.NgaT. V. T.ThompsonC.et al. (2012). The co-selection of fluoroquinolone resistance genes in the gut flora of Vietnamese children. PLoS One7:e42919. doi: 10.1371/journal.pone.0042919
217
VinueL.SaenzY.SomaloS.EscuderoE.MorenoM. A.Ruiz-LarreaF.et al. (2008). Prevalence and diversity of integrons and associated resistance genes in faecal Escherichia coli isolates of healthy humans in Spain. J. Antimicrob. Chemother.62, 934–937. doi: 10.1093/jac/dkn331
218
VirolleC.GoldlustK.DjermounS.BigotS.LesterlinC. (2020). Plasmid transfer by conjugation in Gram-negative bacteria: from the cellular to the community level. Genes11:1239. doi: 10.3390/genes11111239
219
VoA. T. T.van DuijkerenE.GaastraW.FluitA. C. (2010). Antimicrobial resistance, class 1 integrons, and genomic island 1 in Salmonella isolates from Vietnam. PLoS One5:e9440. doi: 10.1371/journal.pone.0009440
220
von WintersdorffC. J. H.PendersJ.van NiekerkJ. M.MillsN. D.MajumderS.van AlphenL. B.et al. (2016). Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front. Microbiol.7:173. doi: 10.3389/fmicb.2016.00173
221
WangA. Y.PopovJ.PaiN. (2016). Fecal microbial transplant for the treatment of pediatric inflammatory bowel disease. World J. Gastroenterol.22, 10304–10315. doi: 10.3748/wjg.v22.i47.10304
222
WaskitoL. A.RezkithaY. A. A.VilaichoneR.WibawaI. D. N.MustikaS.SugihartonoT.et al. (2022). Antimicrobial resistance profile by metagenomic and metatranscriptomic approach in clinical practice: opportunity and challenge. Antibiotics11:654. doi: 10.3390/antibiotics11050654
223
WeiD.HeusP.van de WeteringF. T.van TienhovenG.VerleyeL.ScholtenR. J. (2018). Probiotics for the prevention or treatment of chemotherapy- or radiotherapy-related diarrhoea in people with cancer. Cochrane Database Syst. Rev.2018:CD008831. doi: 10.1002/14651858.CD008831.pub3
224
WenL.DuffyA. (2017). Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J. Nutr.147, 1468S–1475S. doi: 10.3945/jn.116.240754
225
WrightG. (2005). Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev.57, 1451–1470. doi: 10.1016/j.addr.2005.04.002
226
WrightG. D. (2010). Antibiotic resistance in the environment: a link to the clinic?Curr. Opin. Microbiol.13, 589–594. doi: 10.1016/j.mib.2010.08.005
227
WuC. (2013). “Human microbiome, Actinobacteria in” in Encyclopedia of metagenomics (New York: Springer), 1–7.
228
WuG. D.ChenJ.HoffmannC.BittingerK.ChenY.-Y.KeilbaughS. A.et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science334, 105–108. doi: 10.1126/science.1208344
229
XuL.SurathuA.RapleeI.ChockalingamA.StewartS.WalkerL.et al. (2020). The effect of antibiotics on the gut microbiome: a metagenomics analysis of microbial shift and gut antibiotic resistance in antibiotic treated mice. BMC Genomics21:263. doi: 10.1186/s12864-020-6665-2
230
YadavS.KapleyA. (2021). Antibiotic resistance: global health crisis and metagenomics. Biotechnol. Rep.29:e00604. doi: 10.1016/j.btre.2021.e00604
231
YaminD.UskokovićV.WakilA.GoniM.ShamsuddinS.MustafaF.et al. (2023). Current and future technologies for the detection of antibiotic-resistant bacteria. Diagnostics13:3246. doi: 10.3390/diagnostics13203246
232
YilmazG.ChanM.LauC. H.-F.CapitaniS.KangM.CharronP.et al. (2024). How gut microbiome perturbation caused by antibiotic pre-treatments affected the conjugative transfer of antimicrobial resistance genes. Microorganisms12:2148. doi: 10.3390/microorganisms12112148
233
YoonK.KimN. (2021). Roles of sex hormones and gender in the gut microbiota. J. Neurogastroenterol. Motil.27, 314–325. doi: 10.5056/jnm20208
234
ZhangC.LiL.JinB.XuX.ZuoX.LiY.et al. (2021). The effects of delivery mode on the gut microbiota and health: state of art. Front. Microbiol.12:724449. doi: 10.3389/fmicb.2021.724449
235
ZhengD.LiwinskiT.ElinavE. (2020). Interaction between microbiota and immunity in health and disease. Cell Res.30, 492–506. doi: 10.1038/s41422-020-0332-7
236
ZhuY.-G.JohnsonT. A.SuJ.-Q.QiaoM.GuoG.-X.StedtfeldR. D.et al. (2013). Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. U.S.A.110, 3435–3440. doi: 10.1073/pnas.1222743110
Summary
Keywords
gut microbiome, antimicrobial resistance, multidrug resistance, screening of AMR, microbiome
Citation
Deshpande SP, Sujith S, Jobby R, Rajasekharan SK, Ravichandran V and Solomon AP (2025) The gut microbiome: an emerging epicenter of antimicrobial resistance?. Front. Microbiol. 16:1593065. doi: 10.3389/fmicb.2025.1593065
Received
14 March 2025
Accepted
29 April 2025
Published
20 May 2025
Volume
16 - 2025
Edited by
Avinash Karpe, Commonwealth Scientific and Industrial Research Organisation (CSIRO), Australia
Reviewed by
Selvaraj Anthonymuthu, University of California, Irvine, United States
Noble K. Kurian, B. S. Abdur Rahman Crescent Institute of Science and Technology, India
Updates
Copyright
© 2025 Deshpande, Sujith, Jobby, Rajasekharan, Ravichandran and Solomon.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinothkannan Ravichandran, vrvinothan@gmail.com; vravichandran@mum.amity.edu; Adline Princy Solomon, adlineprinzy@sastra.ac.in
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.