Abstract
Introduction:
Reproductive health in mares is pivotal for the sustainability of the equine industry, yet vaginal microbiota dysbiosis remains an underrecognized contributor to infections such as endometritis and bacterial vaginosis. While Lactobacillus spp. dominate healthy vaginal ecosystems in humans and livestock, their role in equine reproductive health, particularly in resilient breeds like Mongolian mares, is poorly understood. This study aimed to isolate and characterize a novel Lactobacillus strain from the vaginal microbiota of healthy Mongolian mares and evaluate its probiotic potential for mitigating equine reproductive disorders.
Methods:
A polyphasic approach integrating phenotypic, biochemical, and 16S rRNA gene sequencing was employed to isolate and identify a novel Lactobacillus salivarius strain (Y20) from the vaginal microbiota of healthy Mongolian mares. The probiotic potential of Y20 was assessed through in vitro assays, including tolerance to low pH (pH 2.5) and bile salts (0.3%), antagonistic activity against equine pathogens (Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa), and antioxidant capacity (DPPH radical scavenging assay). Whole-genome sequencing (1.74 Mb, 33.01% GC content) was performed to analyze genes related to carbohydrate metabolism, adhesion factors, bacteriocin biosynthesis, and stress response pathways. Comparative genomics was used to explore phylogenetic relationships and genomic adaptations for vaginal colonization.
Results:
Lactobacillus salivarius Y20 demonstrated robust tolerance to low pH and bile salts, potent antagonistic activity against key equine pathogens, and significant antioxidant capacity (82.4% DPPH radical scavenging). Genomic analysis revealed genes encoding carbohydrate metabolism, adhesion factors, bacteriocin biosynthesis (including a novel putative bacteriocin cluster), and stress response pathways. Comparative genomics confirmed Y20’s close phylogenetic relationship with horse-derived Lactobacillus salivarius strains and identified unique genomic adaptations for vaginal colonization.
Discussion:
These findings identify Lactobacillus salivarius Y20 as a promising candidate probiotic for mitigating equine reproductive disorders. Its multifaceted probiotic properties, including pathogen inhibition, antioxidant activity, and genomic adaptations for vaginal colonization, suggest potential applications in sustainable equine health management. This study advances microbiome-based strategies as viable alternatives to antibiotics, offering new avenues for improving reproductive health in mares.
1 Introduction
The equine industry, a vital component of global agriculture, recreation, and cultural heritage, hinges on the reproductive health of mares to sustain productivity, genetic diversity, and economic viability. However, reproductive tract infections (RTIs) in mares remain a persistent and costly challenge, with far-reaching consequences for equine welfare and breeding programs. RTIs are frequently linked to dysbiosis of the vaginal microbiota-a complex ecosystem that, when disrupted, can precipitate adverse outcomes such as infertility, recurrent abortion, chronic endometritis, and premature ovarian failure (Edwards, 2008; Sharifzadeh and Shokri, 2021). These conditions reduce foaling rates, increase veterinary costs and culling risks, and necessitate reliance on assisted reproductive technologies, threatening the long-term sustainability of equine operations.
The vaginal microbiota of healthy mares acts as a dynamic, species-specific barrier against pathogens. However, its composition and functional role are less understood compared to those in humans and livestock (Gil-Miranda et al., 2024). Studies in humans and rodents have underscored the critical importance of Lactobacilli-dominated microbiota in maintaining vaginal pH homeostasis, producing antimicrobial compounds (e.g., lactic acid, hydrogen peroxide, bacteriocins), and competing with pathogens for adhesion sites (Kovachev, 2018; Malaluang et al., 2024). In contrast, equine vaginal microbiota research has historically prioritized pathogenic microorganisms-such as Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, and Streptococcus zooepidemicus-which are frequently implicated in endometritis, pyometra, and neonatal sepsis (Barba et al., 2020; da Silva-Álvarez et al., 2025). While this pathogen-centric approach has advanced diagnostic and therapeutic strategies, it has inadvertently overshadowed investigations into the role of commensal bacteria in conferring resilience against RTIs.
Among Lactobacilli, Lactobacillus salivarius stands out as a model probiotic with documented benefits in humans, poultry, and ruminants. In humans, Lactobacillus salivarius strains have been shown to mitigate urogenital infections by modulating immune responses, producing broad-spectrum bacteriocins (e.g., salivaricin A/B), and enhancing epithelial barrier integrity (Messaoudi et al., 2013; Wang et al., 2022). Similarly, in livestock, Lactobacillus salivarius has demonstrated efficacy in preventing gastrointestinal disorders, reducing pathogen colonization, and improving weight gain (Yang et al., 2023; Yang et al., 2024). Despite these parallels, the role of Lactobacillus salivarius in the equine vaginal ecosystem remains largely unexplored, particularly in indigenous breeds adapted to extreme environments.
Mongolian horses, renowned for their endurance, cold tolerance, and foraging adaptability, represent a unique genetic resource for studying host-microbiome interactions under harsh conditions (Wen et al., 2022). Their reproductive physiology, shaped by millennia of natural selection in the steppe ecosystem, may favor microbiota profiles distinct from those of domesticated breeds raised in controlled environments. However, modernization of equine husbandry-including intensified breeding practices, antibiotic overuse, and reduced genetic diversity-has introduced novel stressors that could destabilize the vaginal microbiota. For instance, artificial insemination, while increasing breeding efficiency, may disrupt microbial transmission and expose the reproductive tract to exogenous pathogens (Cazales et al., 2020). Similarly, antibiotic therapies, while critical for treating RTIs, can inadvertently erode protective microbiota, fostering antibiotic resistance and recurrent infections (de la Fuente-Nunez et al., 2023).
Probiotic interventions offer a sustainable alternative to antibiotics by restoring microbial balance and enhancing host defenses. In equine medicine, probiotics have shown promise in preventing diarrhea, modulating gut-brain axis signaling, and reducing post-surgical complications (Lai et al., 2023; Boucher et al., 2024). However, their application to the vaginal microbiota is nascent, with limited studies investigating strain-specific traits (e.g., acid/bile tolerance, adhesion, antimicrobial activity) essential for colonization and efficacy. For instance, specific lactic acid bacteria combinations have been shown to regulate the vaginal environment in postpartum cows, although the exact strains and mechanisms remain unclear (Genís et al., 2017).
This study addresses these gaps by isolating and characterizing vaginal Lactobacillus strains from healthy Mongolian mares, with a focus on Lactobacillus salivarius—a species hypothesized to thrive in acidic, nutrient-poor niches due to its metabolic versatility. Using a polyphasic approach integrating phenotypic, biochemical, and 16S rRNA gene sequencing, we identified a novel strain, Lactobacillus salivarius Y20, and evaluated its probiotic potential through in vitro assays of acid/bile tolerance, antimicrobial activity against equine pathogens, and antioxidant capacity. To elucidate the genetic basis of these traits, we performed whole-genome sequencing and comparative genomic analysis, comparing Y20 with publicly available Lactobacillus salivarius genomes from diverse hosts and ecological niches. Our findings not only advance understanding of equine vaginal microbiota resilience but also introduce Y20 as a candidate probiotic for mitigating RTIs in mares. By bridging microbiology, genomics, and veterinary medicine, this work underscores the translational potential of equine-derived probiotics in sustainable livestock management and One Health approaches.
2 Materials and methods
2.1 Headings ethics statement
This study was approved by the Ethics Committee of the Inner Mongolia Agricultural University (NND2022046), dated 2 March 2022. All experimental procedures adhered to the guidelines for the care and use of experimental animals.
2.2 Isolation and screening of vaginal Lactobacilli
2.2.1 Sample collection
In this study, the sampling subjects were female Mongolian horses (aged 2–3 years old) from the Salaqi Horse Farm in Baotou City, Inner Mongolia. All horses were confirmed to be healthy and disease-free through veterinary examination. After cleaning the vulva of each mare, vaginal secretions were collected using vaginal swabs. The secretions were then placed into sterile anaerobic bags, transported in an icebox, and processed in a sterile laminar flow hood at the laboratory of Inner Mongolia Agricultural University.
2.2.2 Isolation and purification
2.2.2.1 Dilution and plating
Swab samples were suspended in 50 mL phosphate-buffered saline (PBS), vortexed, and serially diluted (10−1 to 10−6). Aliquots (200 μL) of 10−4, 10−5, and 10−6 dilutions were spread onto MRS agar plates containing bromocresol purple (0.04 g/L). The MRS Broth (Qingdao Hopebio, HB0384-1) and Bromocresol Purple Agar (Qingdao Hopebio, HB8616-2) used in this step were sourced from commercial suppliers. The plates were incubated anaerobically at 37°C for 24–36 h.
2.2.2.2 Preservation
Yellow-pigmented colonies (indicative of lactic acid production) were streaked onto fresh MRS agar and subcultured in MRS broth (37°C, 18–24 h) before storage in 20% glycerol at −80°C.
2.2.3 Phenotypic characterization
2.2.3.1 Litmus milk test
Twenty microliters of activated Lactobacilli (OD600 = 1.0) was inoculated into litmus milk medium and incubated anaerobically at 37°C for 24–36 h. Pink coagulation indicated acid production and casein precipitation.
2.2.3.2 Acid tolerance
Lactobacilli were grown in MRS broth adjusted to pH 2.0, 3.0, or 4.0. OD600 was measured at 0, 2, 4, 6, 8, 12, 16, 24, and 32 h using the Infinite 200 PRO Microplate Reader (Tecan).
2.2.3.3 Growth curve
Lactobacilli were incubated in MRS broth, and OD600 was recorded at the same time intervals.
2.2.3.4 Probiotic traits
(1) Antimicrobial activity: Assessed using the Oxford cup method against quality control strains. The quality control strains used in this study and their sources are as follows: Salmonella enterica (CMCC (B) 50071), Escherichia coli (ATCC 25922), Staphylococcus aureus (CMCC (B) 26003), Gardnerella vaginalis (ATCC 49145), Streptococcus equi subsp. zooepidemicus (ATCC 39920), Streptococcus pyogenes (CMCC (B) 32210), inhibition zones were measured after 24–36 h of incubation. (2) Hydrogen peroxide production: Lactobacilli were streaked onto TMB-HRP-MRS agar and incubated anaerobically (36 h) followed by aerobic exposure (1 h). Blue-green coloration indicated H2O2 production. (3) Antioxidant capacity: Total superoxide dismutase (T-SOD), total antioxidant capacity (T-AOC), and DPPH radical scavenging were quantified using commercial kits (Nanjing Jiancheng Bioengineering Institute).
2.2.4 Morphological and biochemical identification
2.2.4.1 Gram staining
Lactobacilli were Gram-stained and examined under a light microscope (Nikon NI-U).
2.2.4.2 Biolog GEN III MicroPlate
Pure cultures were suspended in IF-A inoculating fluid (Biolog) to 95% transmittance and incubated at 37°C for 24 h. The Biolog GEN III MicroPlate (Biolog Inc.) was used for this analysis. Metabolic profiles were analyzed using Biolog MicroLog software (Cao et al., 2025).
2.3 Genomic DNA extraction and preparation
The genomic investigation of Lactobacillus salivarius Y20 began with the isolation and cultivation of the strain. Single colonies were obtained by streak-inoculating Lactobacillus salivarius Y20 onto LB agar plates, followed by incubation at 37°C for 12–18 h. These colonies were then transferred to LB liquid medium and cultured overnight at 37°C with shaking at 110 rpm. Genomic DNA extraction was performed using the Biomed Gene Genomic DNA Extraction Kit (Model DL111-01), adhering to the manufacturer’s protocol to ensure high-quality DNA for downstream sequencing applications (Liang, 2021).
2.4 Whole-genome sequencing and data quality control
A hybrid sequencing approach was employed to comprehensively characterize the Lactobacillus salivarius Y20 genome, utilizing both second-generation (Illumina MiSeq) and third-generation (PacBio) sequencing technologies. The Whole Genome Shotgun (WGS) strategy was used for library construction, ensuring broad genomic coverage.
The Illumina sequencing data underwent rigorous quality control using Fast QC software. Parameters evaluated included GC content distribution, base quality distribution, sequence base quality, and base content distribution to ensure data integrity and suitability for downstream assembly and analysis (Liang, 2021).
2.5 Genome assembly and refinement
Given the extended read lengths provided by third-generation sequencing, the Falcon assembler was selected for de novo assembly of the PacBio reads. This approach simplified the assembly process and enhanced genome continuity.
2.5.1 Assembly assessment and refinement
The resulting assembled genome was statistically analyzed, and second-generation sequencing data were used to refine the assembly, correcting potential errors. Assembly quality was assessed by aligning the original reads to the assembled genome, with coverage depth and GC distribution serving as key metrics.
2.5.2 Genome visualization
A circular genome map was generated to provide a holistic and intuitive overview of the genomic architecture, facilitating the identification of key genomic features and structural elements (Liang, 2021).
2.6 Functional annotation and bacteriocin gene analysis
Functional annotation of the coding proteins within the Lactobacillus salivarius Y20 genome was conducted using a curated set of databases, including Nr, SwissProt, KEGG, GO, COG, Pfam Scan, and CAZy.
2.6.1 Multi-database functional annotation
This approach enabled a comprehensive understanding of gene functions and metabolic pathways encoded within the genome, providing insights into the strain’s physiological and biochemical capabilities.
2.6.2 Bacteriocin gene prediction and validation
Bacteriocin gene clusters were predicted using AntiSMASH, and the gene sequences encoding bacteriocins were further validated using BAGEL4, offering insights into the strain’s antimicrobial potential and ecological role (Blin et al., 2019).
2.7 Comparative genomics and evolutionary relationships
To contextualize the genomic features of Lactobacillus salivarius Y20 within its broader taxonomic framework, comparative genomics analyses were undertaken.
2.7.1 Reference genome selection
Existing genome assembly and annotation reports were sourced from the NCBI database1 to serve as reference genomes for comparative analysis.
2.7.2 SNP and InDel detection
Single nucleotide polymorphisms (SNPs) were detected using the MUMmer alignment software, with statistical analysis conducted to elucidate their positional relationships with genes and mutation outcomes. Small-scale insertions and deletions (InDels, <50 bp) were identified using MUMme software, providing a detailed account of genomic variability.
2.7.3 Synteny and structural variation analysis
Synteny analysis was performed to compare the evolutionary distances between Lactobacillus salivarius Y20 and reference genomes, with MUMmer used to identify large-scale syntenic relationships and SyRI for fine-scale alignment to detect chromosomal rearrangements (translocations, inversions, and combined translocations with inversions). Structural variations (SVs), encompassing insertions, deletions, inversions, and translocations of genomic segments >50 bp, were detected using SyRI software, offering insights into large-scale genomic rearrangements (Kurtz et al., 2004).
2.8 Population evolutionary analysis
To further elucidate the evolutionary relationships of Lactobacillus salivarius Y20, population evolutionary analyses were conducted.
2.8.1 Gene family analysis and coreness
Gene family analysis involved aligning protein (or nucleotide) sequences from all analyzed species using diamond, followed by similarity clustering with OrthoMCL to generate lists of homologous genes (clusters). The species distribution within each cluster was statistically analyzed to infer evolutionary patterns. Gene coreness analysis focused on identifying species-specific genes in Lactobacillus salivarius Y20 and closely related strains, based on gene family clustering results, highlighting unique genomic features.
2.8.2 Phylogenetic tree construction
A maximum-likelihood phylogenetic tree was constructed for the sequenced and reference strains using single-copy gene families identified in their whole genomes. The tree, built using iqtree, provided a robust framework for inferring evolutionary relationships among the strains.
2.8.3 Average nucleotide identity analysis
The average nucleotide identity (ANI) between the sequenced and reference strains was calculated using pyani, offering a quantitative measure of genomic relatedness and further supporting the phylogenetic inferences.
2.9 Statistical analysis
The statistical analysis of the data was conducted using MEGA version 6.06 and SPSS version 23.0. For comparing means across multiple groups, a one-way analysis of variance (ANOVA) was employed, followed by Tukey’s post hoc test to identify specific pairwise differences. Visual representations of the data were generated using GraphPad Prism version 8 (Zhang et al., 2025).
3 Results
3.1 Isolation and screening of vaginal Lactobacilli from Mongolian mares
Through a systematic approach, this part of the results describes the isolation, screening, and identification of an optimal vaginal Lactobacillus strain (Y20) from Mongolian mares. This was achieved by evaluating acid production, resistance, growth kinetics, and morphological traits (Figures 1–3).
Figure 1
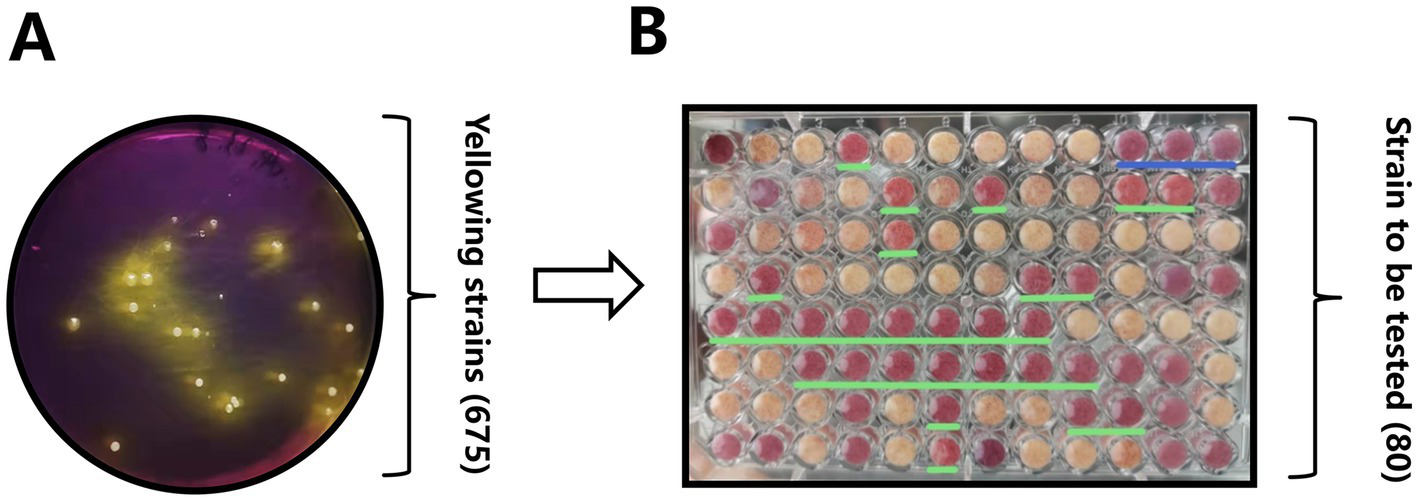
Initial screening pipeline for vaginal Lactobacillus strains from Mongolian mares. (A) An example of a bromocresol violet screening plate, where colonies producing lactic acid turn the medium yellow. (B) Results of the litmus milk color test, highlighting colonies with proteolytic activity and acidification potential.
Figure 2
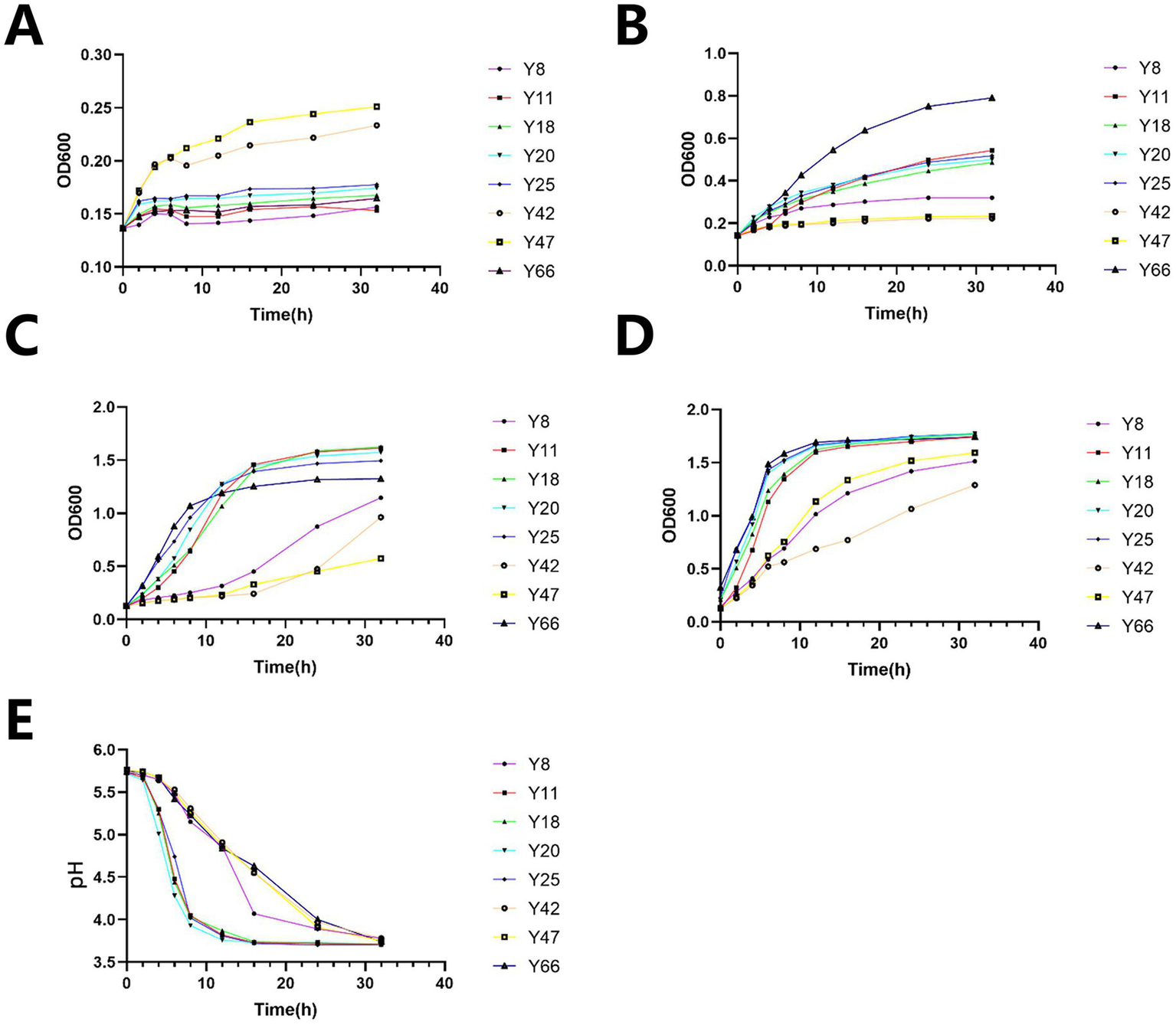
Results of acid resistance tests, growth assays, and acid production experiments. (A) Growth manifestation of bacterial strains under pH 2 condition: temporal variation of OD600 values. (B) Growth manifestation of bacterial strains under pH 2 condition: temporal variation of OD600 values. (C) Growth manifestation of bacterial strains under pH 4 condition: temporal variation of OD600 values. (D) Growth manifestation of bacterial strains: comprehensive temporal OD600 profiles. (E) Acid production manifestation of bacterial strains: temporal variation of pH values.
Figure 3
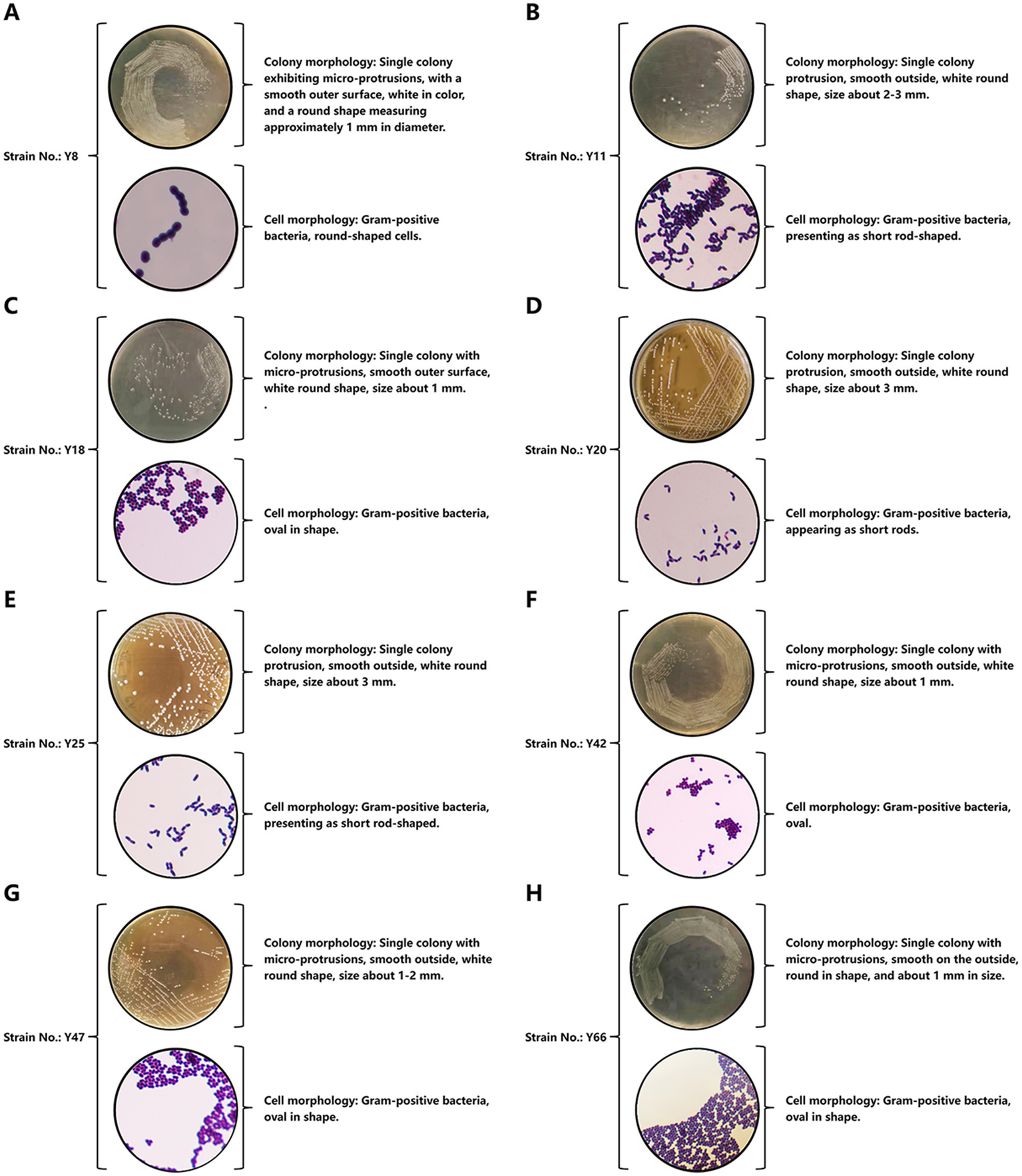
Morphological identification results. (A) Colony morphology and cell morphology of bacterial strain Y8. (B) Colony morphology and cell morphology of bacterial strain Y11. (C) Colony morphology and cell morphology of bacterial strain Y18. (D) Colony morphology and cell morphology of bacterial strain Y20. (E) Colony morphology and cell morphology of bacterial strain Y25. (F) Colony morphology and cell morphology of bacterial strain Y42. (G) Colony morphology and cell morphology of bacterial strain Y47. (H) Colony morphology and cell morphology of bacterial strain Y66.
Initially, 675 acid-producing colonies were isolated through dilution-plating on a bromocresol purple-containing medium. Color changes from purple to yellow (Figure 1A) served as an indicator of successful lactic acid production. Among these colonies, 80 strains (Y1-Y80) were selected using a litmus milk color test. In this test, a pink curd-like morphology (highlighted in the green-lined regions of Figure 1B) was found to correlate with proteolytic activity and acidification potential.
Subsequent assays were conducted to assess the strains’ growth under acidic stress (pH 2–4). The results showed that Y42 and Y47 exhibited superior growth at pH 2, with OD600 values of 0.25–0.23 (Figure 2A). Meanwhile, Y66 demonstrated excellent growth at pH 3, reaching an OD600 of 0.8 (Figure 2B). At pH 4, Y11, Y18, and Y20 achieved comparable growth, with OD600 values around 1.7 (Figure 2C), while Y25 had a slightly lower OD600 of 1.5. In the blank medium, Y20, Y25, and Y66 entered the logarithmic phase by 6 h, with an OD600 of 1.8 at 32 h (Figure 2D), outperforming the other strains. Notably, Y20 acidified the medium most rapidly, reaching a pH of 3.7 within 12 h, with a ΔpH of 2.0 (Figure 2E). It also maintained lower pH values (≤4.1 at 16 h) compared to all its competitors.
Morphological analysis (Figure 3) classified Y8, Y18, Y42, Y47, and Y66 as round or oval in shape (Figures 3A,C,F–H), while Y11, Y20, and Y25 were short-rod-shaped (Figures 3B,D,E). This morphological classification was consistent with their growth and acidification profiles. Given Y20’s exceptional acid production capabilities, its resilience to pH values ranging from 2 to 4, and its rapid growth, it was prioritized for advanced investigation into equine vaginal microbiome dynamics and probiotic applications. This highlights its critical role in maintaining vaginal health and contributing to the overall well-being of the host.
3.2 Identification results of vaginal Lactobacilli in Mongolian mares
The identification results of the vaginal Lactobacillus strain Y20 from Mongolian mares are as follows (Figure 4 and Tables 1–3).
Figure 4
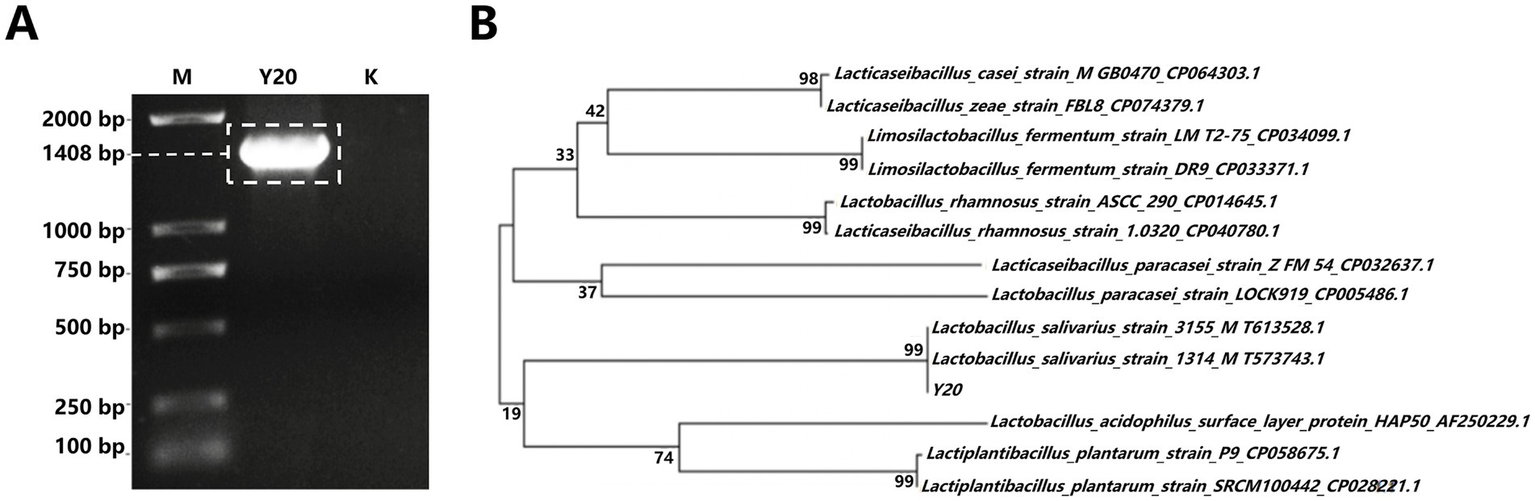
Electrophoresis diagram and phylogenetic tree. (A) Gel electrophoresis diagram of 16S rRNA amplification products, where “M” denotes the DNA Marker (DL2000) and “K” indicates the negative control. (B) Phylogenetic tree constructed based on 16S rRNA sequence homology, showing the evolutionary relationship of the isolated strain with other Lactobacillus species.
In the physiological and biochemical tests, strain Y20 was capable of fermenting salicin, sorbitol, sucrose, raffinose, inulin, and lactose, and it might also ferment cellobiose and mannitol. It did not produce indole, gelatinase, or H2S, nor could it hydrolyze starch (Table 1). Based on these results and in conjunction with the data from Bergey’s Manual of Determinative Bacteriology (9th edition), strain Y20 was preliminarily identified as belonging to the genus Lactobacillus.
In the Biolog microwell plate bacterial identification, after the Biolog microwell plate reaction, there were 38 positive reactions, 47 negative reactions, and 11 intermediate-value reactions (Supplementary Figure 1). Positive reactions were observed for substrates such as raffinose, maltose, trehalose, and sucrose, while negative reactions were observed for substrates like arabitol and inositol. Intermediate-value reactions were noted for substrates such as cellobiose and turanose. After integrating all the reactions and using the Biolog bacterial identification system, the highest similarity index (SIM) was 0.682 (>0.5), indicating reliable data. The strain with the highest SIM value was identified as Lactobacillus salivarius. Specifically, the top four strains with the highest SIM values were Lactobacillus salivarius, Lactobacillus salivarius ss araffinosus, Leuconostoc mesenteroides ss dextranicum, and Lactobacillus brevis (Table 2).
Table 1
| Physiological and biochemical indicator | Y20 |
|---|---|
| Esculin | − |
| Cellobiose | −/+ |
| Maltose | + |
| Mannitol | −/+ |
| Salicin | + |
| Sorbitol | + |
| Sucrose | + |
| Raffinose | + |
| Inulin | + |
| Lactose | + |
| 1% Sodium hippurate | − |
| Indole | − |
| Starch hydrolysis | − |
| Catalase | − |
| Gelatin | − |
| H₂S | − |
| Motility | − |
Biochemical test result table of Y20.
Table 2
| Comparison number | PROB value | SIM value | DIST value | Biological type | Species |
|---|---|---|---|---|---|
| 1 | 0.967 | 0.682 | 4.523 | GP-Rod | Lactobacillus salivarius |
| 2 | 0.018 | 0.139 | 5.566 | GP-Rod | Lactobacillus salivarius ss araffinosus |
| 3 | 0.008 | 0.094 | 6.207 | GP-Coccus | Leuconostoc mesenteroides ss dextranicum |
| 4 | 0.007 | 0.085 | 6.292 | GP-Rod | Lactobacillus brevis |
Biolog microwell plate identification results.
For the 16S rRNA gene identification, during the 16S rRNA gene PCR electrophoresis detection, analysis of the amplified products via 1.2% agarose gel electrophoresis revealed a distinct and singular target band of approximately 1,408 bp for strain Y20, aligning with the anticipated size (Figure 4A); subsequently, in the 16S rRNA gene PCR product sequencing and homology comparison, the PCR products were dispatched to BGI Genomics for sequencing, and BLAST software was employed to assess the homology of the 16S rRNA gene sequence of strain Y20 (Table 3), with the construction of a phylogenetic tree using MEGA6.06 indicating that strain Y20 exhibited the closest phylogenetic relationship to Lactobacillus salivarius, sharing a homology exceeding 99% (Figure 4B).
Table 3
| Serial number | English name | Coverage | Similarity | Gene amplification fragment size |
|---|---|---|---|---|
| Y20 | Lactobacillus salivarius | 100% | 100% | 1,408 bp |
16S rRNA sequence homology alignment.
3.3 Analysis of biological characteristics of Lactobacillus salivarius Y20 isolated from the vagina of Mongolian mares
A meticulous and multifaceted evaluation of the biological attributes of Lactobacillus salivarius Y20, isolated from the vaginal microenvironment of Mongolian mares, was undertaken utilizing a series of rigorously designed and executed assays (Figures 5–9 and Tables 4, 5).
Figure 5
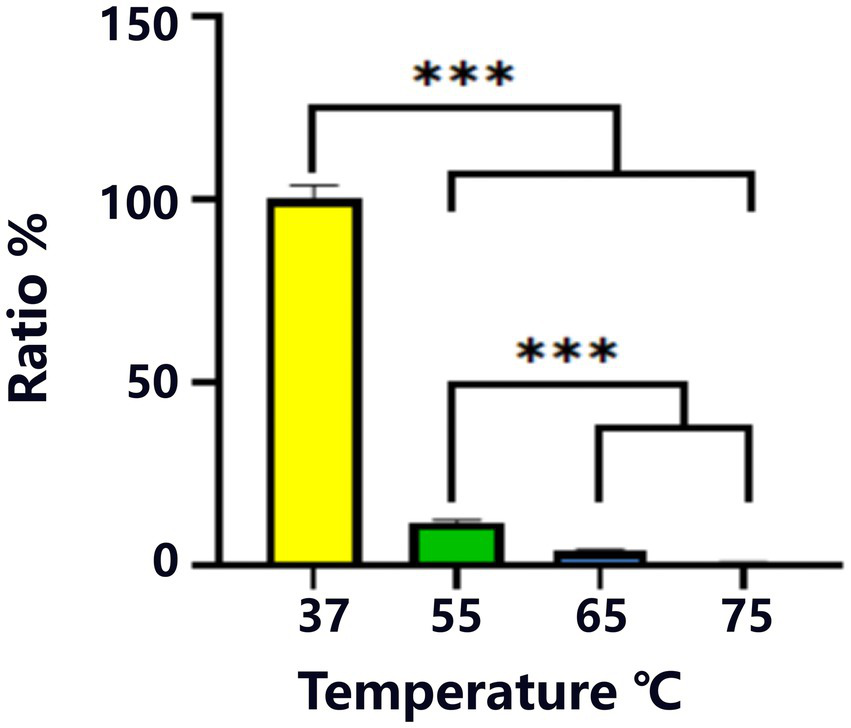
Heat resistance test for strain Y20. ***Indicates statistical significance at p < 0.001.
Figure 6
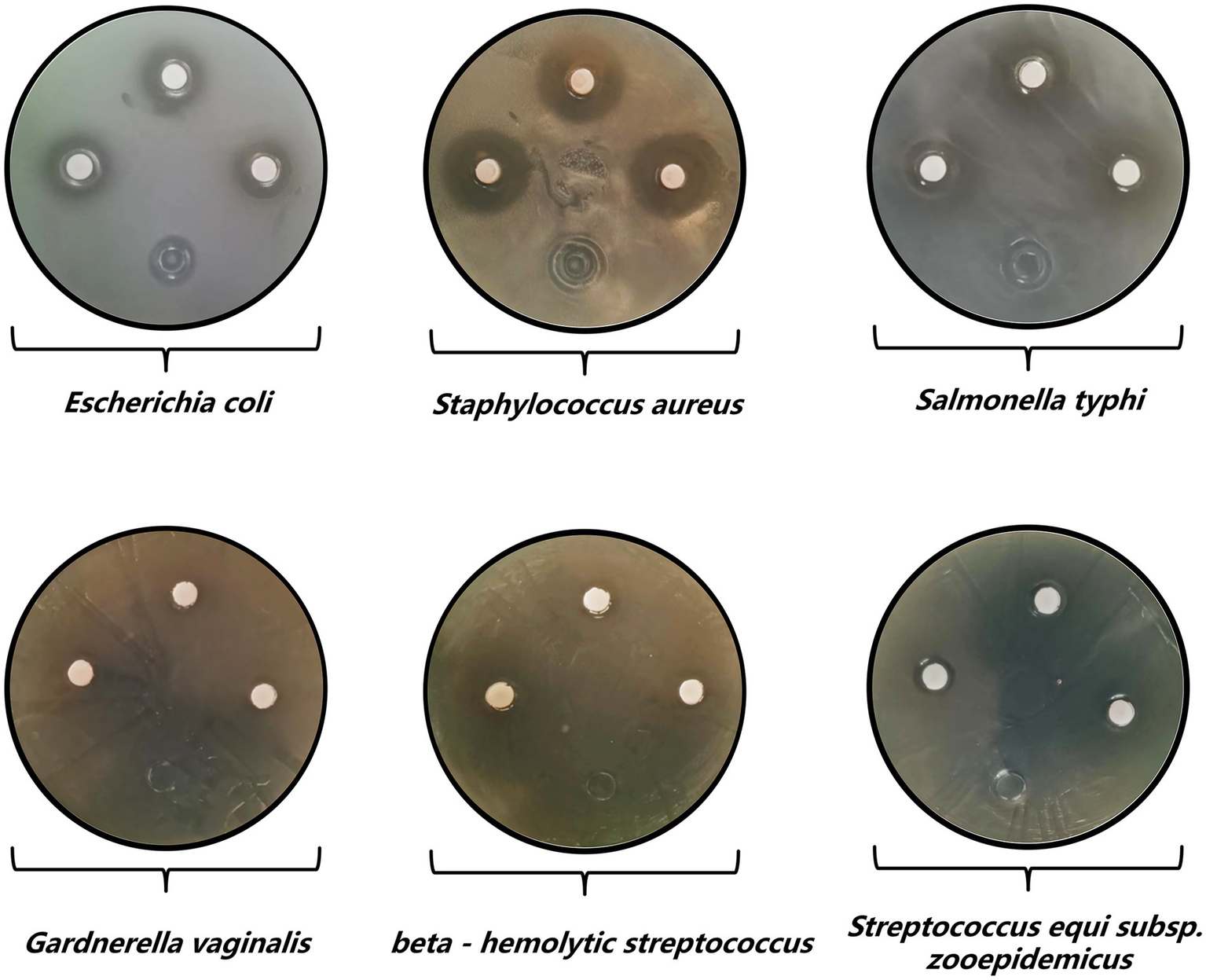
Antibacterial activity results of strain Y20.
Figure 7
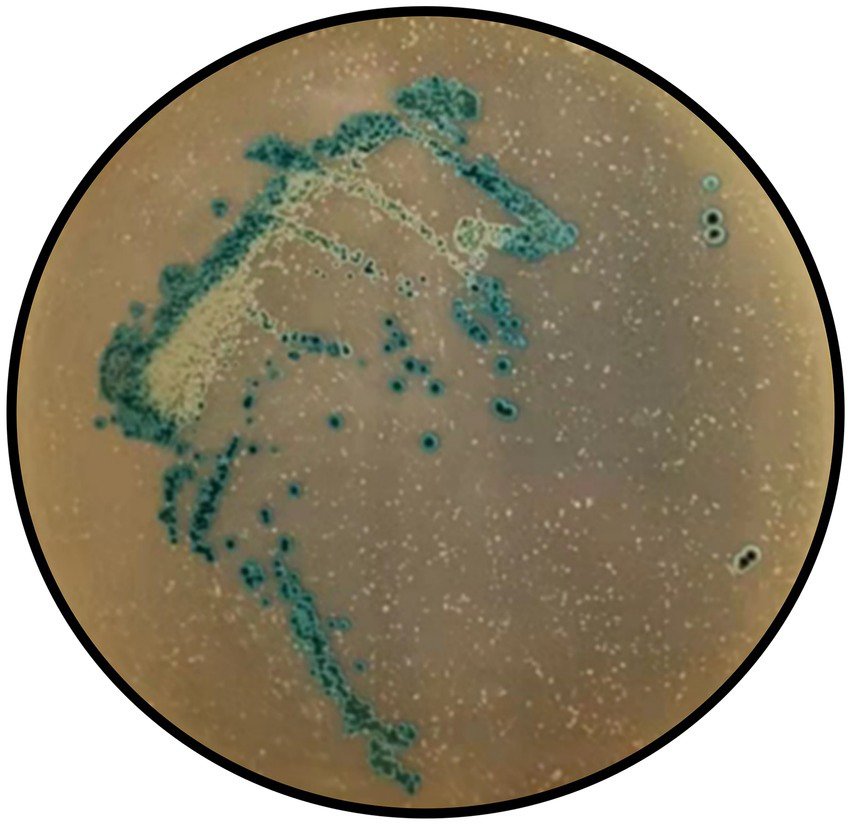
Hydrogen peroxide production test results of Y20.
Figure 8
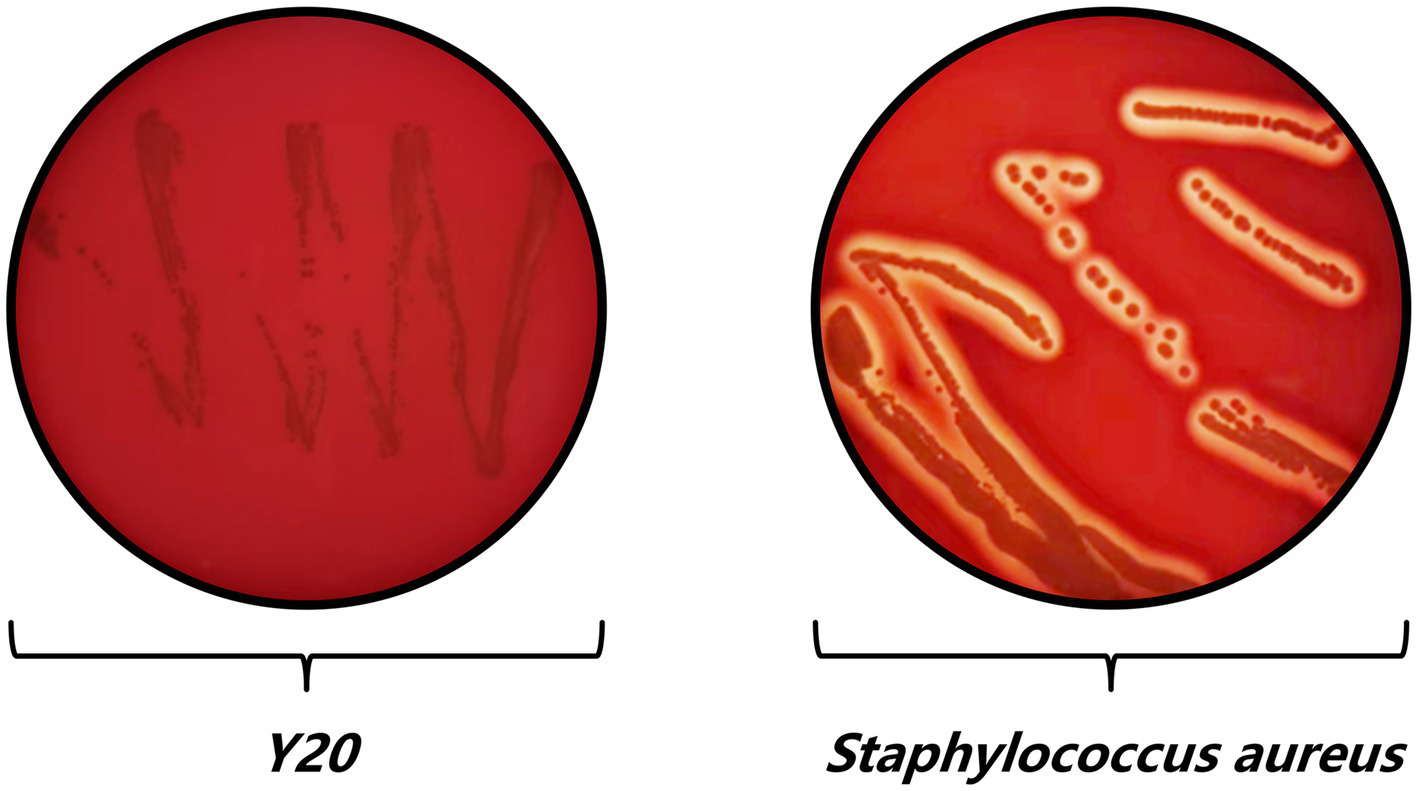
Hemolysis test results of Y20.
Figure 9
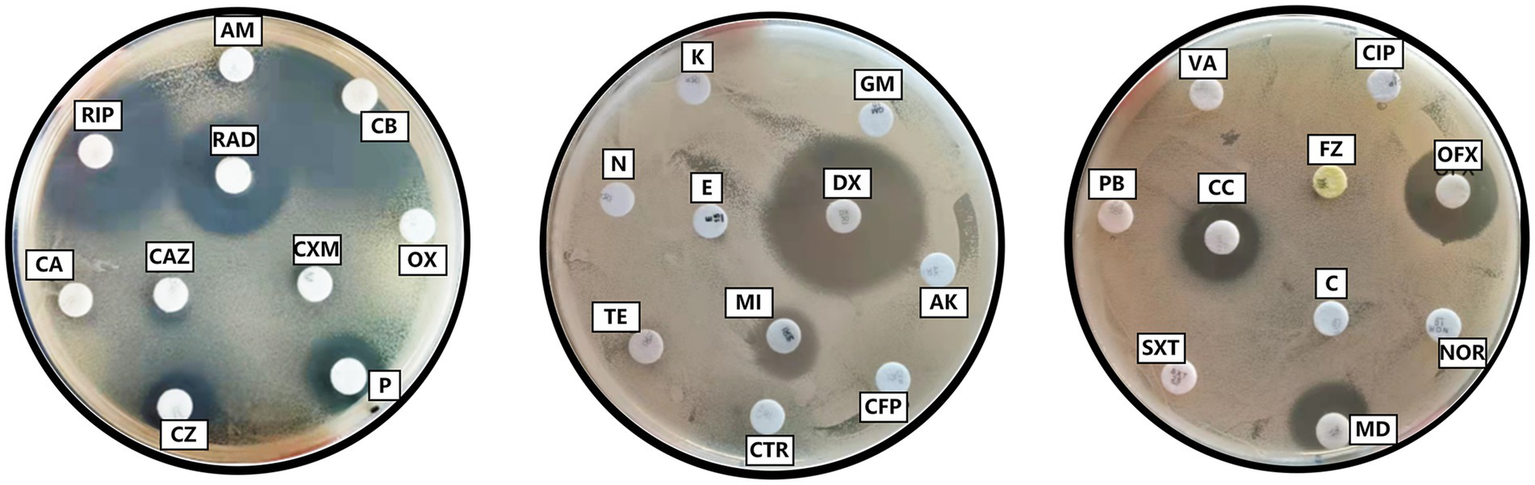
Antibiotic susceptibility test results of Y20. Penicillin (P), oxacillin (OX), carbenicillin (CB), ampicillin (AM), piperacillin (PIP), cefalexin (CA), cefazolin (CZ), cefradine (RAD), cefuroxime (CXM), ceftazidime (CAZ), ceftriaxone (CTR), cefoperazone (CFP), amikacin (AK), gentamicin (GM), kanamycin (K), neomycin (N), tetracycline (TE), doxycycline (DX), minocycline (MI), erythromycin (E), midecamycin (MD), norfloxacin (NOR), ofloxacin (OFX), ciprofloxacin (CIP), vancomycin (VA), polymyxin B (PB), sulfamethoxazole-trimethoprim (SXT), furazolidone (FZ), chloramphenicol (C), clindamycin (CC).
During the heat-resistance assessment, the heat-survival ratio of strain Y20 was precisely determined, with 37°C serving as the reference survival rate (Figure 5). A striking and statistically robust decline (p < 0.001) in the survival rate was observed as the temperature was elevated to 55°C, 65°C, and 75°C. At the extreme temperature of 75°C, the survival rate of Y20 experienced a precipitous drop to less than 1%, clearly indicating its relatively limited thermotolerance.
Shifting focus to the antibacterial testing, strain Y20 demonstrated a broad-spectrum antimicrobial activity against six well-established and thoroughly characterized quality-control strains (Table 4 and Figure 6). Among these, the most pronounced inhibitory effect was exerted against Staphylococcus aureus, as evidenced by an exceptionally large inhibition zone diameter of 20.42 mm. A hierarchical order of inhibitory efficacy was established, with Escherichia coli exhibiting the second-highest level of inhibition, succeeded by Salmonella typhi, Streptococcus equi subsp. zooepidemicus, Gardnerella vaginalis, and finally Streptococcus hemolyticus group B, which displayed the least inhibitory response among the tested strains.
Table 4
| Bacterial strain | Inhibition zone diameter (mm) ± standard deviation |
|---|---|
| Escherichia coli | 15.1600 ± 0.8795 |
| Staphylococcus aureus | 20.4233 ± 0.1297 |
| Salmonella enterica serovar Typhi | 14.0333 ± 0.4288 |
| Gardnerella vaginalis | 11.0066 ± 0.0903 |
| Streptococcus pyogenes (Group A β-hemolytic) | 10.4100 ± 0.0294 |
| Streptococcus equi subsp. zooepidemicus | 12.3333 ± 0.1271 |
Antibacterial activity test results.
In the hydrogen peroxide production assay, all colonies of Y20 exhibited a characteristic blue reaction (Figure 7), unequivocally indicating a positive result and underscoring its capacity to synthesize hydrogen peroxide, a crucial antimicrobial metabolite with significant implications for microbial competition and host defense. The antioxidant capacity analysis revealed that strain Y20 possesses remarkable antioxidant properties. It achieved a DPPH radical-scavenging rate of 73.77%, a total antioxidant capacity (T-AOC) of 67.49 U/mL, and a total superoxide dismutase activity (T-SOD) of 59.66 U/mL. These findings suggest its potential to effectively counteract oxidative stress within the vaginal environment, which is a critical factor in maintaining vaginal health and preventing oxidative-damage-related pathologies.
The hemolysis test provided compelling evidence of the safety profile of Lactobacillus salivarius Y20. In contrast to Staphylococcus aureus, which produced a distinct and well-defined hemolysis ring, strain Y20 did not generate any such ring (Figure 8). This observation unequivocally indicates its non-hemolytic nature, thereby offering a high level of assurance regarding its safety for potential therapeutic or probiotic applications.
In the drug-sensitivity analysis, a panel of 10 antibiotics, including penicillin, carbenicillin, piperacillin, cefazolin, cefradine, doxycycline, minocycline, midecamycin, ofloxacin, and clindamycin, exhibited inhibitory effects on strain Y20. The strain demonstrated a high degree of susceptibility to carbenicillin, piperacillin, cefradine, and doxycycline, while displaying moderate susceptibility to midecamycin, ofloxacin, and clindamycin. Conversely, it exhibited resistance to penicillin, cefazolin, and minocycline, and showed no susceptibility to the remaining 20 antibiotics included in the test panel (Table 5 and Figure 9).
Table 5
| Antibiotic disk | Inhibition zone diameter (cm)/interpretation |
|---|---|
| Penicillin (P) | 1.33/R |
| Oxacillin (OX) | -/R |
| Carbenicillin (CB) | 3.24/S |
| Ampicillin (AM) | -/R |
| Piperacillin (PIP) | 2.30/S |
| Cefalexin (CA) | -/R |
| Cefazolin (CZ) | 1.26/R |
| Cefradine (RAD) | 2.07/S |
| Cefuroxime (CXM) | -/R |
| Ceftazidime (CAZ) | -/R |
| Ceftriaxone (CTR) | -/R |
| Cefoperazone (CFP) | -/R |
| Amikacin (AK) | -/R |
| Gentamicin (GM) | -/R |
| Kanamycin (K) | -/R |
| Neomycin (N) | -/R |
| Tetracycline (TE) | -/R |
| Doxycycline (DX) | 2.91/S |
| Minocycline (MI) | 1.13/R |
| Erythromycin (E) | -/R |
| Midecamycin (MD) | 1.45/I |
| Norfloxacin (NOR) | -/R |
| Ofloxacin (OFX) | 1.69/I |
| Ciprofloxacin (CIP) | -/R |
| Vancomycin (VA) | -/R |
| Polymyxin B (PB) | -/R |
| Sulfamethoxazole-trimethoprim (SXT) | -/R |
| Furazolidone (FZ) | -/R |
| Chloramphenicol (C) | -/R |
| Clindamycin (CC) | 1.42/I |
Drug sensitivity test results of Y20.
S (Sensitive): Bacterial growth is fully inhibited within the susceptible breakpoint range. I (Intermediate): Bacterial growth is partially inhibited, requiring higher doses or alternative therapies. R (Resistant): Bacterial growth is not inhibited within the susceptible breakpoint range. “-”: No measurable inhibition zone observed (indicating resistance or methodological limitation).
With regard to the auto-aggregation and biofilm-formation capacity assessments, strain Y20 displayed an outstanding auto-aggregation rate (A%) of 86.8127%. In the biofilm-formation evaluation, the OD595 value of strain Y20 was measured at 1.2834, with an ODC of 0.4244. Given the criterion of 2 ODc < OD ≤ 4 ODc, this indicates a moderate (++) biofilm-formation ability. These attributes are of paramount significance for Lactobacilli, as they facilitate robust adhesion to vaginal epithelial cells, enhance their adaptability and survival within the complex vaginal ecosystem, and enable them to effectively compete with pathogenic bacteria, thereby playing a pivotal role in maintaining vaginal homeostasis and preventing infections.
3.4 Whole-genome analysis of Lactobacillus salivarius strain Y20
3.4.1 Genome-wide visualization map
The genome of strain Y20 was assembled using Falcon, a long-read sequencing assembler, yielding a final genome size of 1,735,042 base pairs (bp) with a GC content of 33.01%. Detailed assembly metrics are provided in Supplementary Table 1. To assess GC bias, a GC-depth distribution plot was generated (Supplementary Figure 2), revealing a homogeneously clustered scatter pattern within a narrow range, indicating the absence of species contamination or GC skew in the Y20 genome. The 16S rRNA gene sequence of Lactobacillus salivarius Y20 is presented in Supplementary Table 2.
A circular genome map of Y20 was constructed, demonstrating that the assembly achieved a complete genome level (Figure 10A), with plasmids P1 and P2 visualized in Figures 10B,C, respectively. This integrated visualization—combining genomic architecture, plasmid content, and GC-depth profiling—provides a comprehensive and intuitive understanding of the structural and compositional features of the Y20 genome.
Figure 10
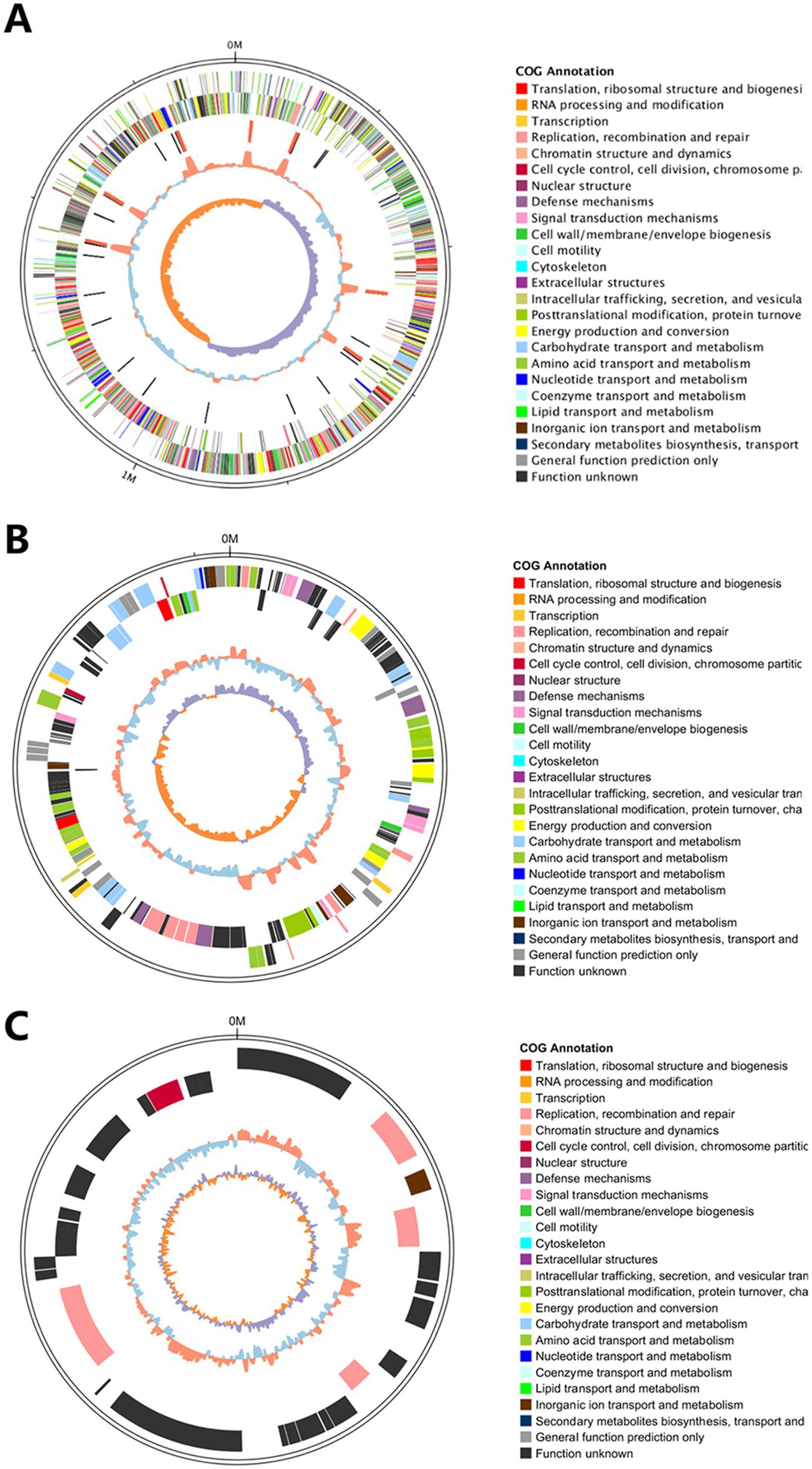
Circular genome and plasmid maps of Y20. (A) Genome circle map of Y20. From the outermost to the innermost layer: the outermost solid circle represents the genomic sequence position coordinates. Moving inward, the first circle illustrates the distribution of genes annotated by COG on the positive strand. The second circle inward depicts the distribution of genes annotated by COG on the negative strand. The third circle inward displays the distribution of ncRNAs, including tRNAs marked in black and rRNAs marked in red. The fourth circle inward represents the GC content, where orange indicates regions with GC content above the mean, and blue denotes regions with GC content below the mean. The fifth and innermost circle corresponds to the GC skew values, with specific color coding: orange for regions where the GC skew value is greater than 0 (GC skew >0), and purple for regions where the GC skew value is less than 0 (GC skew <0). (B) The outermost circle represents the positional coordinates of the genomic sequence. From the outermost to the innermost circles, the annotations are as follows: genes on the positive strand, genes on the negative strand, ncRNAs (where tRNAs are denoted in black and rRNAs in red), GC content (with red indicating values above the mean and blue indicating values below the mean), and GC skew (which reflects the relative abundance of G and C nucleotides and is used to identify the origin and terminus in circular chromosomes; it is calculated as GC skew = (G − C)/(G + C); purple denotes values greater than 0, while orange denotes values less than 0). (C) The outermost track displays the positional coordinates along the genomic sequence. Progressing inward from this outermost track, the subsequent circles depict, in order: genes located on the positive (forward) strand, genes on the negative (reverse) strand, non-coding RNAs (ncRNAs; with tRNAs represented in black and rRNAs in red), GC content (where red indicates regions with a GC proportion exceeding the mean, and blue denotes regions below the mean), and GC skew (a measure of the relative abundance of guanine (G) versus cytosine (C) nucleotides, which is instrumental in identifying the origin and terminus of replication in circular chromosomes. GC skew is calculated as (G − C)/(G + C); purple regions signify a positive GC skew value (>0), while orange regions indicate a negative skew value (<0)).
3.4.2 Genomic composition analysis
Predictive analyses of the Y20 genome were conducted using NCBI and Prokka, yielding the following results: in the coding gene prediction, a total of 1,602 genes were identified, with a cumulative gene length of 1,505,322 base pairs (bp). The longest gene spans 5,856 bp, while the shortest is 99 bp, with gene lengths predominantly clustering within the 200–1,400 bp range (Figure 11A). The overall genomic GC content was determined to be 33.27%. Regarding non-coding RNA prediction (Table 6), a total of 79 tRNAs and 22 rRNAs were identified. Predictive analysis of interspersed and tandem repeats (Figure 11B) revealed the presence of seven short interspersed nuclear elements (SINEs), each 471 bp in length, nine long interspersed nuclear elements (LINEs), each 818 bp in length, and two DNA transposons (DNA elements), each 87 bp in length. Transposon and CRISPR sequence predictions (Table 7) further indicated the discovery of six transposons and two CRISPR arrays. Predictions of genomic islands (GIs) and prophages (Table 8) demonstrated the existence of three GIs in the Y20 genome, with a cumulative length of 74,287 bp and an average length of 24,762.33 bp. Additionally, one prophage with a total length of 64,184 bp was detected.
Figure 11
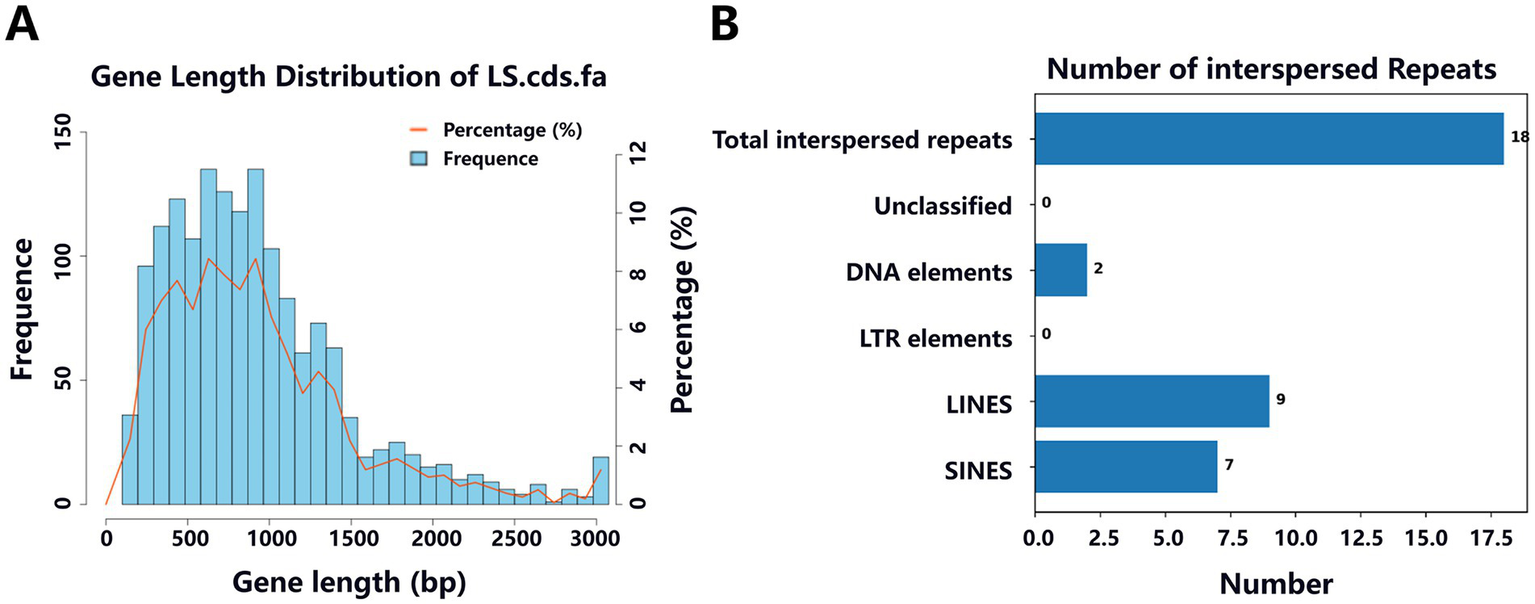
Length distribution of coding sequences and histogram analysis of interspersed repeats in the Y20 genome. (A) Length distribution of coding gene sequence. (B) Interspersed repeat histogram.
Table 6
| RNA type | Gene count |
|---|---|
| tRNA (transfer RNA) | 79 |
| rRNA (ribosomal RNA) | 22 |
| 16S rRNA (16S ribosomal RNA) | 7 |
| 5S rRNA (5S ribosomal RNA) | 8 |
| 23S rRNA (23S ribosomal RNA) | 7 |
Statistics of non-coding RNA prediction results.
Table 7
| Genomic element type | Count |
|---|---|
| Transposons | 6 |
| CRISPR arrays | 2 |
Transposon prediction and CRISPR sequence prediction results.
Table 8
| Genomic element | Count | Total length (bp) | Average length (bp) |
|---|---|---|---|
| Genomic islands | 3 | 74,287 | 24,762.33 |
| Prophages | 1 | 64,184 | 64,184.00 |
Gene island and prophage prediction results.
3.4.3 Gene functional annotation
The statistical data from the annotation functional databases reveal (Figure 12A) that among the genes of Lactobacillus salivarius Y20, 1,602 genes could be annotated, while the total number of unannotated genes was 2. In terms of annotation results from various databases, 1,600 genes were annotated in the Nr database, 1,122 genes in the Swissprot database, 1,241 genes in the KOG database, and 1,009 genes in the KEGG database. Notably, 896 genes were jointly annotated by all four databases. The Nr annotation results (Figure 12B) indicate that the highest number of genes (1,555) in Lactobacillus salivarius Y20 were homologous to those in Lactobacillus salivarius.
Figure 12
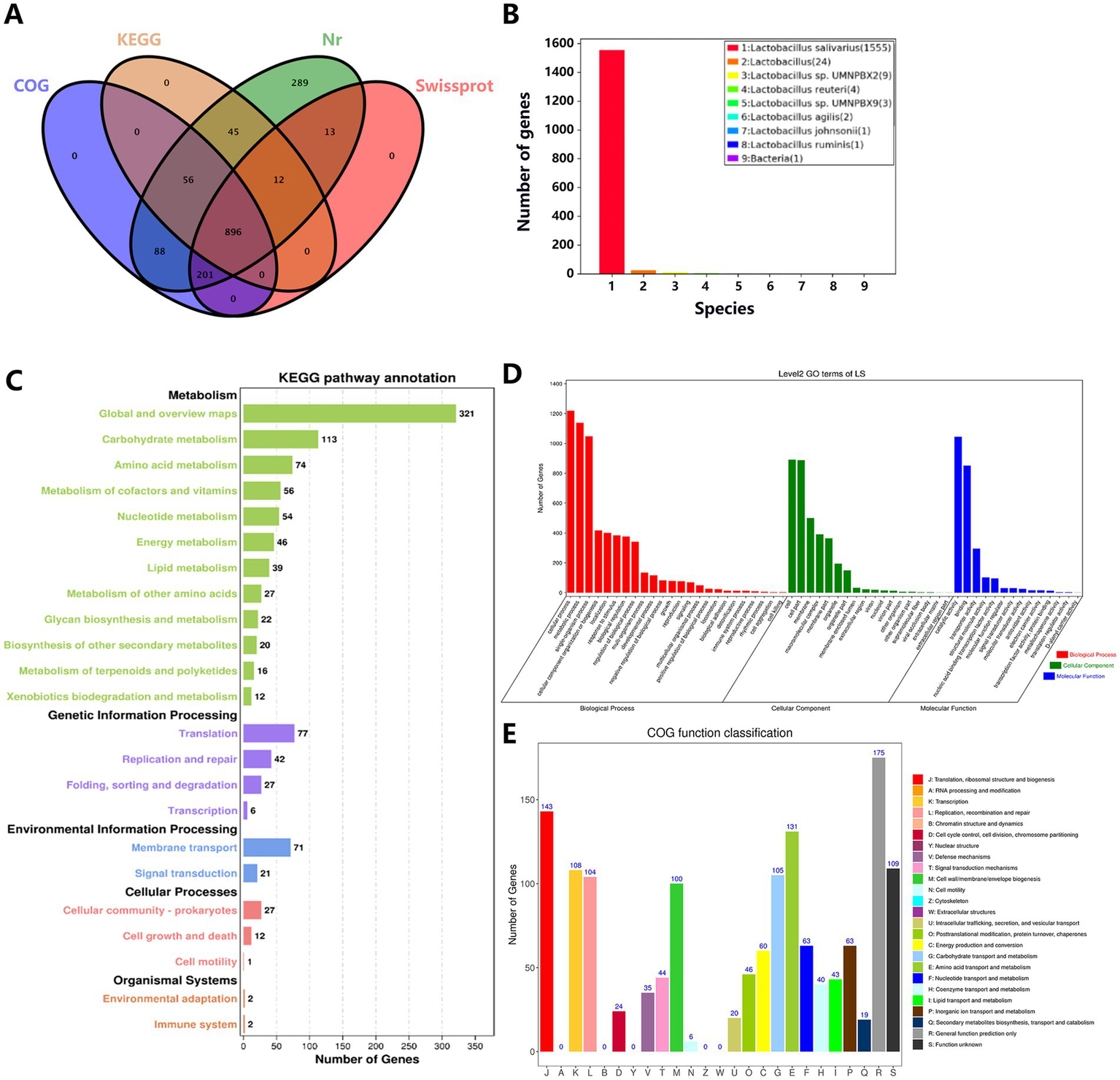
A comprehensive overview of functional annotation for Y20. (A) Four database annotation Venn diagrams. (B) Nr database annotated species statistics chart (Top 9 Species). (C) Statistics of KEGG pathway annotation results. (D) GO database function classification diagram. (E) COG annotation map.
Based on the KEGG annotation information (Figure 12C), the functional classification of Lactobacillus salivarius Y20 comprises five hierarchical levels (KEGG A class). There are 800 genes associated with metabolism, 152 genes involved in genetic information processing, 92 genes related to environmental information processing, 40 genes linked to cellular processes, and four genes associated with organismal systems. Among them, immune-related pathways (such as the NOD-like receptor signaling pathway) are closely associated with the occurrence and development of multiple immune diseases. These pathways can also regulate the structure and function of the intestinal microbiota, thereby maintaining intestinal immune homeostasis. Additionally, the trxA gene, which encodes thioredoxin reductase, can modulate the redox state and protect DNA from oxidative damage.
Through GO database analysis (Figure 12D), a total of 12,076 gene annotation sequences were obtained, categorized into three major categories and 56 sub-entries. Specifically, 6,040 sequences were classified under biological process, 3,503 under cellular component, and 2,533 under molecular function. There are 17 genes associated with antioxidant activity, including thioredoxin-disulfide reductase and dihydrolipoamide dehydrogenase. Thioredoxin-disulfide reductase primarily participates in regulating the intracellular redox balance, while dihydrolipoamide dehydrogenase is a key enzyme that can convert organic matter into energy and provide power for vital activities.
The COG annotation results of Lactobacillus salivarius Y20 are classified into 26 functional categories (Figure 12E). Among them, there are 143 genes related to translation, ribosomal structure, and biogenesis; 104 genes involved in replication, recombination, and repair; 105 genes associated with carbohydrate transport and metabolism; and 131 genes related to amino acid transport and metabolism.
3.4.4 Analysis of special functional elements
The CAZy annotation results of Lactobacillus salivarius Y20 revealed the presence of 241 enzymes belonging to five carbohydrate-enzyme-related categories, including 120 glycosyl transferases (GTs), 71 glycoside hydrolases (GHs), 32 carbohydrate-binding modules (CBMs), 17 carbohydrate esterases (CEs), and 1 auxiliary activity (AA) enzyme (Figure 13A). For the prediction of secretory proteins in Y20, 62 proteins with signal peptide structures, 35 proteins with transmembrane domains, and 27 proteins predicted to be secretory proteins were identified (Table 9). In the prediction of bacteriocin gene clusters in Lactobacillus salivarius Y20, one bacteriocin gene cluster was found on the P1 plasmid. Sequence alignment showed that this bacteriocin gene cluster exhibited a high degree of similarity (83%) to the bacteriocin-encoding gene cluster in Lactobacillus salivarius UCC118 (Figure 13B). Through AntiSMASH analysis, three bacteriocins were identified within a single bacteriocin gene cluster on the plasmid genome. BAGEL analysis identified one bacteriocin hotspot region (Figure 13C). After database comparison, it was found that two of the bacteriocins showed high similarity to previously discovered bacteriocins, while one exhibited low similarity, suggesting that this bacteriocin might be a novel one (Table 10). Based on these findings, it can be inferred that Lactobacillus salivarius Y20 may produce a diverse range of bacteriocins.
Figure 13
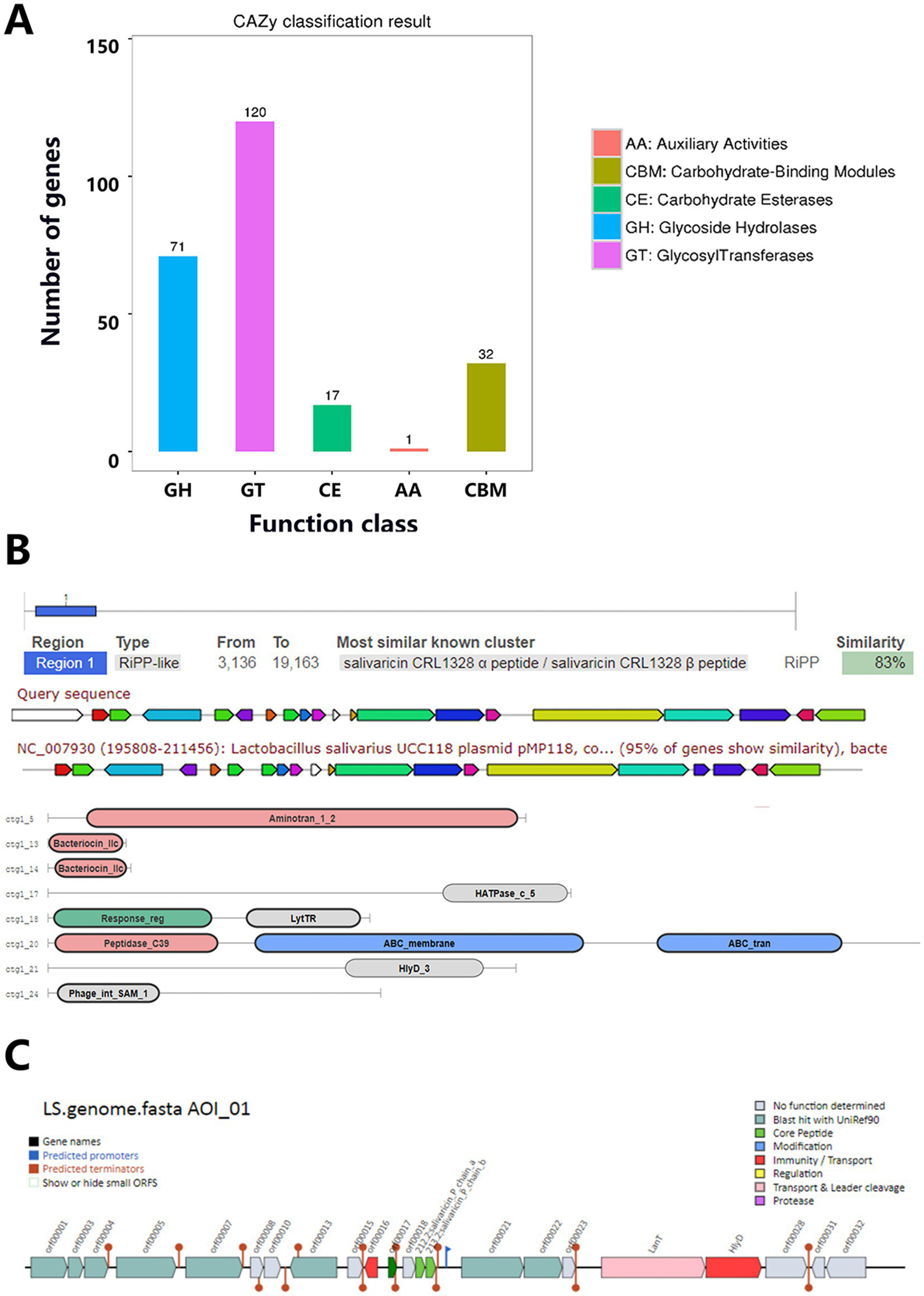
Special functional elements analysis for Y20. (A) CAZy classification chart. (B) Prediction of Y20 gene cluster. (C) Area of interest from Y20.
Table 9
| Sample name | Total protein | Signal peptide | Protein with transmembrane structure | Secreted protein |
|---|---|---|---|---|
| Y20 | 1,602 | 62 | 35 | 27 |
Secreted protein prediction statistics.
Table 10
| Name | Size | Nucleotide sequence | Amino acid sequence |
|---|---|---|---|
| Bacteriocin_IIc1 | 195 bp | ATGATGAAGGAATTTACAGTATTGACAGAATGTGAATTAGCAAAGGTTGATGGTGGGAAACGTGGACCTAACTGTGTAGGTAACTTCTTAGGTGGTCTATTTGCTGGAGCAGCTGCAGGAGTTCCACTTGGACCAGCTGGTATCGTAGGTGGGGCAAATCTAGGAATGGTAGGCGGAGCGCTTACTTGTTTATAA | MMKEFTVLTECELAKVDGGKRGPNCVGNFLGGLFAGAAAGVPLGPAGIVGGANLGMVGGALTCL |
| Bacteriocin_IIc2 | 207 bp | ATGAAAAATCTAGATAAAAGATTCACAATTATGACTGAAGATAACTTAGCATCAGTAAATGGTGGTAAAAATGGTTATGGTGGTAGCGGAAATCGCTGGGTTCACTGTGGAGCTGGCATCGTAGGTGGAGCTTTAATTGGAGCTATCGGTGGACCCTGGTCAGCCGTAGCGGGTGGAATTTCTGGTGGATTTGCAAGTTGCCATTAA | MKNLDKRFTIMTEDNLASVNGGKNGYGGSGNRWVHCGAGIVGGALIGAIGGPWSAVAGGISGGFASCH |
| Bacteriocin_IIc3 | 174 bp | ATGAATAATAATTTTGTACAAGTTGATAAGAAAGAATTGGCACATATAATTGGTGGAAGAAATTCTTATGATTATATAGATAGCGGACAGTTTGGTTACGATATAGGATGTACAATTGCTAATACTAAATTTTTCAAAAGATTAAGACACTCAAATCAAAATATTTGTAGTTAA | MNNNFVQVDKKELAHIIGGRNSYDYIDSGQFGYDIGCTIANTKFFKRLRHSNQNICS |
Y20 bacteriocin gene situation table.
3.4.5 Comparative genomics analysis
After gaining insights into the gene composition and functional profile of Lactobacillus salivarius Y20, we aimed to further elucidate its taxonomic status and its relationships with closely-related species. To this end, complete genomes of Lactobacillus salivarius from diverse sources were retrieved from the NCBI database, and comprehensive analyses including whole-genome synteny, gene family, pangenome, and phylogenetic relationships were conducted. The reference strains selected are listed in Table 11.
Table 11
| Sequenced genome | Reference genome | Size (Mb) | Source |
|---|---|---|---|
| Y20 | Lsa_IBB3154 | 2.17 | Hen feces |
| Lsa_JCM1046 | 2.32 | Pig intestine | |
| Lsa_LPM01 | 2.03 | Human milk | |
| Lsa_2D | 1.98 | Horse feces | |
| Lsa_S01 | 1.94 | Fish intestine |
Genome summary of Lactobacillus salivarius from different sources.
Parallel synteny plots were employed to display the syntenic relationships between the reference genomes and the target species (Y20) genome. By comparing the evolutionary distance disparities between pairwise species, we could analyze the phylogenetic relationships between the sample species (Y20) and the reference species. The synteny alignment results between Lactobacillus salivarius Y20 and the reference genomes indicated that IBB3154, LPM01, S01, and JCM1046 had fewer reverse-matching regions with Y20. In contrast, 2D exhibited more reverse-matching regions with Y20, with sizes ranging from 0.5–1.5 Mbp, suggesting that Y20 is most closely related to the equine-derived Lactobacillus salivarius 2D (Table 12 and Figure 14A).
Table 12
| Index | Strain ID | ||||
|---|---|---|---|---|---|
| Lsa_IBB3154 | Lsa_JCM1046 | Lsa_LPM01 | Lsa_2D | Lsa_S01 | |
| Total length of aligned target sequence region (bp) | 1,526,770 | 1,526,969 | 1,578,263 | 1,595,818 | 1,503,801 |
| Total length of target sequence (bp) | 1,735,042 | 1,735,042 | 1,735,042 | 1,735,042 | 1,735,042 |
| Percentage of aligned target sequence region in whole genome (%) | 88 | 88.01 | 90.96 | 91.98 | 86.67 |
| Total length of aligned query sequence region (bp) | 1,528,844 | 1,537,891 | 1,615,186 | 1,599,332 | 1,516,105 |
| Total length of query sequence (bp) | 1,921,419 | 1,836,297 | 1,788,723 | 1,700,858 | 1,737,623 |
| Percentage of aligned query sequence region in whole genome (%) | 79.57 | 83.75 | 90.3 | 94.03 | 87.25 |
| Number of alignment block | 12 | 18 | 17 | 4 | 8 |
Statistics of coverage of collinearity comparison between reference strain and Y20.
Figure 14
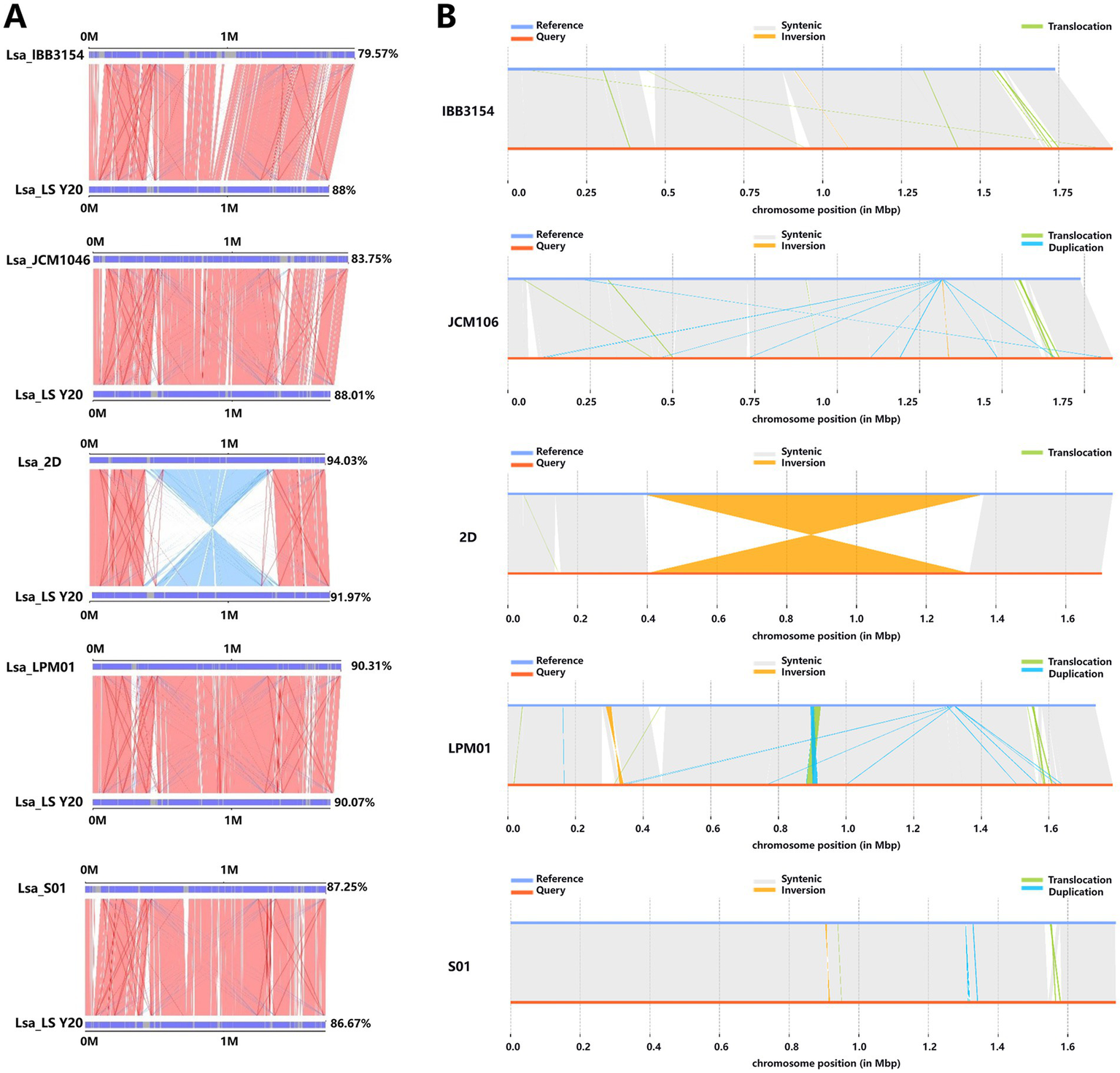
Comprehensive analysis of parallel collinearity and variation types. (A) Results of parallel collinearity. The upper axis represents the target species genome, and the lower axis represents the reference genome. Red lines: forward matches of corresponding regions. Blue lines: reverse matches of corresponding regions. (B) Display of variation types. Synthetic: collinear region. Translocation: translocation region. Inversion: inversion area. Insertion: insertion area. Deletion: deletion of region. Duplication: repeated area.
Statistical analysis of single-nucleotide variations (SNVs) and InDels in Lactobacillus salivarius Y20 revealed that the number of insertion and deletion sites between Y20 and 2D was lower compared to those between Y20 and Lactobacillus salivarius strains from chicken, pig, human, and fish sources (Table 13).
Table 13
| Strain name | SNP count statistics | Insertion | Deletion |
|---|---|---|---|
| Lsa_IBB3154 | 29,845 | 334 | 418 |
| Lsa_JCM1046 | 25,888 | 338 | 334 |
| Lsa_LPM01 | 25,547 | 310 | 351 |
| Lsa_2D | 22,524 | 287 | 276 |
| Lsa_S01 | 25,323 | 467 | 493 |
Statistics of SNP/InDel.
When it came to the number of long-fragment sequences (≥50 bp) on the Lactobacillus salivarius Y20 genome, it was higher compared to IBB3154, JCM106, and LPM106, similar to S01, and lower compared to 2D. Specifically, IBB3154 had very few insertion, deletion, inversion, and translocation sites with Y20, mostly translocation sites. JCM106 had more insertion, deletion, inversion, and translocation sites with Y20 compared to IBB3154, mostly translocation and duplication sites. 2D had few insertion, deletion, and translocation sites with Y20, mostly inversion sites, with regions ranging from 0.4–1.4 Mbp. LPM01 had more insertion, deletion, inversion, and translocation sites with Y20 compared to IBB3154 and JCM106, mostly translocation, inversion, and duplication regions, with significant overlap between duplication and translocation regions in the 0.8–1.0 Mbp range. S01 only had duplication, translocation, and inversion sites with Y20 (Figure 14B).
Gene family analysis, which helps understand the correlation between unique genes and strain-specific traits, revealed 1,303 core genes. Y20 had 65 unique genes, IBB3154 had 220, JCM1046 had 158, LPM01 had 61, 2D had 53, and S01 had 77 (Figure 15A).
Figure 15

Outcomes from population evolutionary analysis. (A) Wayne diagram of homologous gene family. Each ellipse represents a genome. The number displayed above each region indicates the count of gene families within the species corresponding to this region. The number within parentheses below each region denotes the total number of genes across all gene families in the species of this region. (B) Species evolution tree. The phylogenetic tree illustrates the evolutionary relationships and topological structure among different species. At the terminal ends of the tree diagram lie the individual species, while the nodes (branching points) represent their common ancestors. The branch lengths depict the relative genetic distances between species. Numbers displayed on the branches indicate the bootstrap support values (branch confidence scores), with values closer to 100 signifying higher confidence. (C) Thermal diagram of ANI analysis.
By analyzing the gene commonality between Lactobacillus salivarius Y20 and five other Lactobacillus salivarius strains, we also examined immune-related unique and core genes (Table 14). Among them, there was one immune-related unique gene, LRS64_05055 (RAMP protein csm5), which is involved in antiviral immune defense and may also participate in certain cellular autophagy mechanisms, protein synthesis regulation, and post-transcriptional modifications. Four immune-related genes were common to all six strains: LRS64_01800 (thioredoxin trxA), LRS64_03595 (thioredoxin trxA), LRS64_05635 (GTPase), and (bacteriocin immunity protein mesI). Thioredoxin trxA plays a crucial reductive role in cells, primarily participating in intracellular redox reactions. mesI is considered a pleiotropic immunity protein that can resist external bacterial invasions while also regulating internal bacterial gene expression to adapt to different environmental stresses.
Table 14
| Immune gene | Strain | |||||
|---|---|---|---|---|---|---|
| Y20 | IBB3154 | JCM1046 | LPM01 | 2D | S01 | |
| trxA* | + | + | + | + | + | + |
| mesI* | + | + | + | + | + | + |
| trxA* | + | + | + | + | + | + |
| Cas2 | + | + | + | + | + | |
| Cas1 | + | + | + | + | ||
| RAMP protein Csm5# | + | |||||
| Csm5 | + | + | ||||
| Csm4 | + | + | ||||
| Csm3 | + | + | ||||
| Csm2 | + | + | ||||
| Csm1 | + | |||||
| Cas6 | + | |||||
| GTPase* | + | + | + | + | + | + |
| Cas2 | + | + | ||||
| casC | + | |||||
| amtB | + | + | + | + | + | + |
Immunological related gene proportion map.
Genes with a * suffix after their names are common immune genes shared by the six strains. Genes with a # suffix are unique immune genes of strain Y20. A “+” indicates that the strain possesses the corresponding immune gene.
Additionally, Y20 and 2D shared two immune-related genes: LRS64_06545 (CRISPR-associated protein Cas2) and LRS64_06565 (CasC protein). The SNVs of Cas2 and CasC are listed in Tables 15, 16. Cas2 protein is important in DNA repair and genome stability. The CRISPR-Cas system is a natural immune system that resists the invasion of foreign genetic sequences.
Table 15
| Reference base | Mutant base | Mutation type | Location type |
|---|---|---|---|
| G | A | SNP | Exonic |
| G | A | SNP | Exonic |
| G | A | SNP | Exonic |
| T | C | SNP | Exonic |
| C | T | SNP | Exonic |
| G | A | SNP | Exonic |
| A | G | SNP | Exonic |
| C | T | SNP | Exonic |
Immune shared gene SNP (Cas2) of Y20 and 2D.
Table 16
| Reference base | Mutant base | Mutation type | Location type |
|---|---|---|---|
| A | G | SNP | Exonic |
| A | G | SNP | Exonic |
| A | G | SNP | Exonic |
| T | G | SNP | Exonic |
| A | G | SNP | Exonic |
| G | A | SNP | Exonic |
| A | G | SNP | Exonic |
| T | C | SNP | Exonic |
Immune co existing gene SNP (CasC) of Y20 and 2D.
Sequence alignment and phylogenetic tree construction showed that Lactobacillus salivarius Y20 and 2D were on the same branch with a high branch confidence value of 100. Y20, 2D, and LPM01 were on the same branch, and Y20, 2D, LPM01, and JCM1046 were on the same branch. IBB3145 and S01 had a close phylogenetic relationship but were distant from Y20, 2D, LPM01, and JCM1046 (Figure 15B).
The average nucleotide identity (ANI) values between the alignment regions of Lactobacillus salivarius Y20 and the reference genomes showed a high degree of discrimination among closely related species. Typically, an ANI value of 95% is used as a taxonomic threshold to distinguish different species. The results indicated that Y20 was most closely related to 2D and LPM01, with S01 being the most distant, consistent with the phylogenetic tree in 5.5.3 (Figure 15C).
4 Discussion
4.1 Isolation, screening and biological characterization analysis of vaginal lactic acid bacteria from Mongolian horses
The equine industry, while experiencing significant growth, concurrently confronts numerous reproductive health challenges, with bacterial vaginosis (BV) in mares emerging as a critical disease that severely compromises their reproductive health. This condition markedly reduces conception and fertility rates, imposing substantial economic losses on production and breeding efficiency within the equine sector and thus posing a significant bottleneck to its further industrial transformation, upgrading, and sustainable development (Coudray and Madhivanan, 2020; Marnach et al., 2022).
In traditional livestock production practices, antibiotic therapy remains the predominant approach for the treatment of bacterial vaginosis in mares. However, the prolonged and widespread use of antibiotics has triggered a series of severe problems. Antimicrobial resistance (AMR) has escalated dramatically, with many pathogens, such as Staphylococcus aureus, progressively developing resistance to multiple antibiotic agents and evolving into concerning “superbugs” (Liu et al., 2021; Mlynarczyk-Bonikowska et al., 2022). S. aureus colonizes not only the host’s gastrointestinal tract, skin, and perineal regions but also secretes diverse toxins, including alpha-hemolysin (Hla), enterotoxins, and leukocidins, which collectively endanger host health. The emergence of resistance undermines the efficacy of conventional antibiotic treatments, thereby increasing the complexity and cost of disease management (references omitted for brevity). Beyond vaginal infections, Escherichia coli is capable of cause severe systemic illnesses, such as diarrhea, sepsis, hemolytic uremic syndrome, and urinary tract infections, posing threats to both animal and human health (Ahmad-Mansour et al., 2021). Salmonella spp., as zoonotic pathogens, present substantial risks in livestock production and public health, with outbreaks potentially leading to large-scale infections in animals and humans, severely impacting the economic viability of breeding operations and public health safety (Takaya et al., 2020). Streptococcus pyogenes can induce severe invasive diseases, damaging local tissues and causing conditions such as scarlet fever and erysipelas, with significant implications for animal welfare (Yu et al., 2023). Although primarily a commensal bacterium in horses, Streptococcus equi subsp. zooepidemicus (SEZ) is a common pathogen in bacterial endometritis in mares and can also infect dogs, pigs, and other animals, particularly in high-value breeding sectors like horse racing, resulting in substantial economic losses (Ikhuoso et al., 2020; Boyle, 2023). Gardnerella vaginalis is widely implicated in bacterial vaginosis and associated with adverse reproductive health outcomes, adversely affecting mare reproductive function (Joseph et al., 2024). The AMR, drug residues, and complex pathogenicity mechanisms induced by these pathogens have emerged as critical global health threats, necessitating urgent exploration of safe, efficient, and sustainable alternative therapeutic strategies (McDonald and Linde, 2002).
Probiotics, as potential alternatives to antibiotics, have garnered considerable research attention due to their safety, controllability, eco-friendliness, and sustainability (Mnisi et al., 2025). Probiotics exhibit immense potential in modulating microecological balance, enhancing host immunity, and inhibiting pathogenic growth, offering novel solutions to combat antibiotic misuse (Suez et al., 2019; Ahire et al., 2023). Through a rigorous screening strategy, this study successfully isolated Lactobacillus salivarius Y20, a strain characterized by robust growth performance, strong acid tolerance, and high acid production efficiency. In preliminary screening, Y20 demonstrated rapid growth kinetics and achieved high cell density. Subsequent acid tolerance and acid production assays confirmed its ability to thrive under acidic conditions while efficiently producing organic acids. Critically, Y20 exhibited potent inhibitory effects against multiple pathogens, including S. aureus, E. coli, Salmonella enterica serovar Typhi, G. vaginalis, SEZ, and S. pyogenes. Theoretically, Y20’s superior growth traits enable rapid colonization and proliferation within the mare’s vaginal microenvironment, establishing dominant microbiota. Its acid tolerance ensures survival and sustained functionality in the acidic vaginal milieu, while its high acid production capacity further lowers vaginal pH, reinforcing pathogen suppression. Collectively, these attributes underscore the potential of Y20 as a probiotic for preventing and treating bacterial vaginosis in mares, offering a viable alternative to antibiotic therapy and mitigating the risks associated with AMR and drug residues. Future studies should focus on elucidating Y20’s colonization dynamics in the mare’s vagina, its long-term impact on vaginal microecology, practical therapeutic efficacy, and safety profiles. Such investigations will provide a robust theoretical foundation and empirical support for its clinical application in the equine industry, advancing the sector toward healthier, more sustainable practices.
4.2 Genomic characterization and functional dissection of Lactobacillus salivarius Y20
Whole-genome high-throughput sequencing technology has been widely employed in microbiology-related research. For the genomic study of Lactobacillus salivarius, the first published genome of Lactobacillus salivarius was that of Lactobacillus salivarius UCC118 (Raftis et al., 2014). The genome of Lactobacillus salivarius UCC118 comprises a 1.83 Mb chromosome, a large plasmid of 242 kb (pMP118), and two smaller plasmids of 20 kb (pSF118-20) and 44 kb (pSF118-44) (Fang et al., 2008). With the advancement of sequencing technology, related research has continued to increase. For instance, Ayala et al. (2017) isolated Lactobacillus salivarius L28 from ground beef. The genome size of L28 is approximately 2,028,405 bp, with an average GC content of 32.7%. A total of 1982 coding sequences, 64 tRNAs, and 31 rRNAs were predicted. Kergourlay et al. (2012) isolated Lactobacillus salivarius SMXD51 from chickens. Using the RAST server, they achieved functional annotation of predicted genes. SMXD51 contains 1795 coding sequences, 103 RNAs, and 78 tRNA genes. Raftis et al. (2014) isolated Lactobacillus salivarius JCM1046 from pigs, and its genome comprises a 1.83 Mb chromosome and four plasmids. Mackenzie et al. (2014) isolated Lactobacillus salivarius cp400 from pigs and performed automated gene prediction using GeneMark and Glimmer3. Cho et al. (2011) isolated Lactobacillus salivarius GJ-24 from human feces. By utilizing the RAST server and comparing with the KEGG and COG databases, they achieved functional annotation of predicted genes. Sun et al. (2015) isolated Lactobacillus salivarius LsR from the feces of a centenarian. Its genome contains a 1,751,565 bp circular chromosome and two plasmids. This study also identified several important genes that enable LsR to survive in the gastrointestinal tract and adhere to the intestine. Additionally, it was found that LsR can inhibit the occurrence of oral cancer and colorectal cancer caused by some factors. Jiménez et al. (2010) determined the complete genome sequence of Lactobacillus salivarius CECT 5713 isolated from a mother-infant pair (breast milk and infant feces) using whole-genome shotgun sequencing. Its genome consists of a 1,828,169 bp circular chromosome and two plasmids. They observed that CECT 5713 has anti-inflammatory, immunomodulatory, and anti-infective properties. In this study, the circular genome size of Lactobacillus salivarius Y20 is approximately 1.74 Mb, which is smaller than that of Lactobacillus salivarius IBB3154 (from hens), JCM1046 (from pigs), LPM01 (from humans), 2D (from horses), and S01 (from fish). Lee et al. (2017) found that the genome size of human-derived Lactobacillus salivarius strains is smaller than that of pig-derived strains. This could be attributed to the internal environment of humans is more attractive to strains than that of pigs and chickens, and humans are exposed to harsh environments (such as diseases, toxins, etc.) less frequently than livestock. Symbiotic bacteria accompany the evolution of the host, and in non-harsh environments, symbiotic bacteria lose more genes than in harsh environments. The genome size of Lactobacillus salivarius Y20 derived from the horse vagina is much smaller than that of many other sources of Lactobacillus salivarius. It is hypothesized that the vaginal environment of Mongolian horses is more attractive to bacteria, and the strains are exposed to fewer harsh conditions in the Mongolian horse vaginal environment.
In the analysis of the GO annotation of the Y20 genome, several antioxidant-related genes were found, such as thioredoxin, which, together with nicotinamide adenine dinucleotide phosphate and thioredoxin reductase, plays a positive role in stabilizing the redox state of organisms (Ball et al., 2022; Bradford et al., 2024). In the KEGG annotation, immune-related pathways (NOD-like receptor signaling pathway) and the TrxA gene for regulating redox were identified. Comparative genomics analysis revealed the unique genes of Lactobacillus salivarius Y20. One immune-related unique gene, LRS64_05055 (RAMP protein Csm5), possesses an immune defense function. Three immune-related genes are shared among six strains, namely LRS64_01800 (thioredoxin TrxA), LRS64_03595 (thioredoxin TrxA), and LRS64_05635 (GTPase). The aforementioned results collectively indicate that Y20 possesses a robust foundation for immunity and antioxidant properties.
Based on the findings of this study, several avenues for future research emerge. First, in vivo studies are warranted to evaluate the efficacy of Y20 in preventing or treating equine reproductive disorders. These studies should investigate the optimal dosage, duration of treatment, and potential interactions with other vaginal microorganisms. Second, further exploration of the molecular mechanisms underlying the probiotic effects of Y20, such as the production of antimicrobial substances and modulation of the host immune response, would provide deeper insights into its therapeutic potential. Additionally, comparative studies with other Lactobacillus salivarius strains from different hosts could identify strain-specific traits that contribute to their ecological success in specific niches. Finally, long-term safety studies are essential to ensure the absence of any adverse effects associated with the prolonged use of Y20 as a probiotic.
5 Conclusion
This study successfully isolated and identified a novel strain of Lactobacillus salivarius (Y20) from the vaginal microbiota of healthy Mongolian mares. The probiotic potential of Y20 was evaluated through a series of in vitro assays, demonstrating robust tolerance to low pH and bile salts, potent antagonistic activity against equine pathogens, and significant antioxidant capacity. Whole-genome sequencing and comparative genomics revealed that Y20 shares a close phylogenetic relationship with horse-derived Lactobacillus salivarius strains and possesses unique genomic adaptations for vaginal colonization. These findings identify Lactobacillus salivarius Y20 as a candidate probiotic for mitigating equine reproductive disorders, offering a sustainable alternative to antibiotics and advancing microbiome-based strategies for equine health management.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Ethics Committee of the Inner Mongolia Agricultural University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
YZ: Writing – review & editing, Conceptualization, Funding acquisition. YuL: Writing – original draft, Writing – review & editing. JT: Methodology, Writing – original draft. JC: Investigation, Writing – original draft, Data curation. YaL: Writing – original draft, Formal analysis. QH: Resources, Writing – original draft. XF: Formal analysis, Writing – original draft. SY: Writing – original draft, Investigation. MiD: Investigation, Writing – original draft. SS: Resources, Writing – original draft. TB: Resources, Writing – original draft. DB: Writing – original draft, Project administration. XZ: Writing – original draft, Funding acquisition. MaD: Funding acquisition, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Key Research and Development Project of the Inner Mongolia Autonomous Region (2023YFDZ0002); the Special Project for Capacity Building of Scientific Research Innovation Platforms under the Basic Scientific Research Fund for Directly Affiliated Universities of the Inner Mongolia Autonomous Region (BR221018 and BR251007).
Acknowledgments
The authors thank the Key Laboratory of Genetics, Breeding, and Reproduction of Horse Genus in the Inner Mongolia Autonomous Region for their help and support in this experiment.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1635639/full#supplementary-material
Footnotes
References
1
AhireJ. J.RohillaA.KumarV.TiwariA. (2023). Quality management of probiotics: ensuring safety and maximizing health benefits. Curr. Microbiol.81:1. doi: 10.1007/s00284-023-03526-3
2
Ahmad-MansourN.LoubetP.PougetC.Dunyach-RemyC.SottoA.LavigneJ. P.et al. (2021). Staphylococcus aureus toxins: an update on their pathogenic properties and potential treatments. Toxins13:677. doi: 10.3390/toxins13100677
3
AyalaD. I.CookP. W.CamposD. L.BrashearsM. M.den BakkerH.NightingaleK. K. (2017). Draft genome sequence of Lactobacillus salivarius L28 isolated from ground beef. Genome Announc.5:e00955-17. doi: 10.1128/genomeA.00955-17
4
BallD. P.TsamouriL. P.WangA. E.HuangH. C.WarrenC. D.WangQ.et al. (2022). Oxidized thioredoxin-1 restrains the NLRP1 inflammasome. Sci. Immunol.7:eabm7200. doi: 10.1126/sciimmunol.abm7200
5
BarbaM.Martínez-BovíR.QueredaJ. J.MocéM. L.Plaza-DávilaM.Jiménez-TrigosE.et al. (2020). Vaginal microbiota is stable throughout the estrous cycle in Arabian Maress. Animals10:2020. doi: 10.3390/ani10112020
6
BlinK.ShawS.SteinkeK.VillebroR.ZiemertN.LeeS. Y.et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res.47, W81–W87. doi: 10.1093/nar/gkz310
7
BoucherL.LeducL.LeclèreM.CostaM. C. (2024). Current understanding of equine gut dysbiosis and microbiota manipulation techniques: comparison with current knowledge in other species. Animals14:758. doi: 10.3390/ani14050758
8
BoyleA. G. (2023). Streptococcus equi subspecies equi. Vet. Clin. North Am. Equine Pract.39, 115–131. doi: 10.1016/j.cveq.2022.11.006
9
BradfordH. F.McDonnellT. C. R.StewartA.SkeltonA.NgJ.BaigZ.et al. (2024). Thioredoxin is a metabolic rheostat controlling regulatory B cells. Nat. Immunol.25, 873–885. doi: 10.1038/s41590-024-01798-w
10
CaoJ.ZhangJ.WuH.LinY.FangX.YunS.et al. (2025). Probiotic potential of Pediococcus pentosaceus M6 isolated from equines and its alleviating effect on DSS-induced colitis in mice. Microorganisms13:957. doi: 10.3390/microorganisms13050957
11
CazalesN.EstradéM. J.PereyraF.Fiala-RechsteinerS. M.MattosR. C. (2020). Sperm transport and endometrial inflammatory response in mares after artificial insemination with cryopreserved spermatozoa. Theriogenology158, 180–187. doi: 10.1016/j.theriogenology.2020.09.021
12
ChoY. J.ChoiJ. K.KimJ. H.LimY. S.HamJ. S.KangD. K.et al. (2011). Genome sequence of Lactobacillus salivarius GJ-24, a probiotic strain isolated from healthy adult intestine. J. Bacteriol.193, 5021–5022. doi: 10.1128/JB.05616-11
13
CoudrayM. S.MadhivananP. (2020). Bacterial vaginosis-a brief synopsis of the literature. Eur. J. Obstet. Gynecol. Reprod. Biol.245, 143–148. doi: 10.1016/j.ejogrb.2019.12.035
14
da Silva-ÁlvarezE.Gómez-ArronesV.Correa-FizF.Martín-CanoF. E.Gaitskell-PhillipsG.CarrascoJ. J.et al. (2025). Metagenomic and proteomic analyses reveal similar reproductive microbial profiles and shared functional pathways in uterine immune regulation in mares and jennies. PLoS One20:e0321389. doi: 10.1371/journal.pone.0321389
15
de la Fuente-NunezC.CesaroA.HancockR. E. W. (2023). Antibiotic failure: beyond antimicrobial resistance. Drug Resist. Updat.71:101012. doi: 10.1016/j.drup.2023.101012
16
EdwardsJ. F. (2008). Pathologic conditions of the stallion reproductive tract. Anim. Reprod. Sci.107, 197–207. doi: 10.1016/j.anireprosci.2008.05.002
17
FangF.FlynnS.LiY.ClaessonM. J.van PijkerenJ. P.CollinsJ. K.et al. (2008). Characterization of endogenous plasmids from Lactobacillus salivarius UCC118. Appl. Environ. Microbiol.74, 3216–3228. doi: 10.1128/AEM.02631-07
18
GenísS.BachÀ.ArísA. (2017). Effects of intravaginal lactic acid bacteria on bovine endometrium: implications in uterine health. Vet. Microbiol.204, 174–179. doi: 10.1016/j.vetmic.2017.04.025
19
Gil-MirandaA.MacnicolJ.Orellana-GuerreroD.SamperJ. C.GomezD. E. (2024). Reproductive tract microbiota of mares. Vet. Sci.11:324. doi: 10.3390/vetsci11070324
20
IkhuosoO. A.MonroyJ. C.Rivas-CaceresR. R.Cipriano-SalazarM.Barbabosa PliegoA. (2020). Streptococcus equi in equine: diagnostic and healthy performance impacts. J. Equine Vet. Sci.85:102870. doi: 10.1016/j.jevs.2019.102870
21
JiménezE.MartínR.MaldonadoA.MartínV.Gómez de SeguraA.FernándezL.et al. (2010). Complete genome sequence of Lactobacillus salivarius CECT 5713, a probiotic strain isolated from human milk and infant feces. J. Bacteriol.192, 5266–5267. doi: 10.1128/JB.00703-10
22
JosephA.AntonL.GuanY.FergusonB.MirroI.MengN.et al. (2024). Extracellular vesicles from vaginal Gardnerella vaginalis and Mobiluncus mulieris contain distinct proteomic cargo and induce inflammatory pathways. npj Biofilms Microbiomes.10:28. doi: 10.1038/s41522-024-00502-y
23
KergourlayG.MessaoudiS.DoussetX.PrévostH. (2012). Genome sequence of Lactobacillus salivarius SMXD51, a potential probiotic strain isolated from chicken cecum, showing anti-campylobacter activity. J. Bacteriol.194, 3008–3009. doi: 10.1128/JB.00344-12
24
KovachevS. (2018). Defence factors of vaginal Lactobacilli. Crit. Rev. Microbiol.44, 31–39. doi: 10.1080/1040841X.2017.1306688
25
KurtzS.PhillippyA.DelcherA. L.SmootM.ShumwayM.AntonescuC.et al. (2004). Versatile and open software for comparing large genomes. Genome Biol.5:R12. doi: 10.1186/gb-2004-5-2-r12
26
LaiH.LiY.HeY.ChenF.MiB.LiJ.et al. (2023). Effects of dietary fibers or probiotics on functional constipation symptoms and roles of gut microbiota: a double-blinded randomized placebo trial. Gut Microbes15:2197837. doi: 10.1080/19490976.2023.2197837
27
LeeJ. Y.HanG. G.KimE. B.ChoiY. J. (2017). Comparative genomics of Lactobacillus salivarius strains focusing on their host adaptation. Microbiol. Res.205, 48–58. doi: 10.1016/j.micres.2017.08.008
28
LiangH. X. (2021). “Whole-genome sequencing of a multi-drug resistant Klebsiella Pneumoniae ST37 of swine origin and comparative genomic analysis” in Master’s thesis (Guangzhou: South China University of Technology).
29
LiuW. T.ChenE. Z.YangL.PengC.WangQ.XuZ.et al. (2021). Emerging resistance mechanisms for 4 types of common anti-MRSA antibiotics in Staphylococcus aureus: a comprehensive review. Microb. Pathog.156:104915. doi: 10.1016/j.micpath.2021.104915
30
MackenzieD. A.McLayK.RoosS.WalterJ.SwarbreckD.DrouN.et al. (2014). Draft genome sequence of a novel Lactobacillus salivarius strain isolated from piglet. Genome Announc.2:e01231-13. doi: 10.1128/genomeA.01231-13
31
MalaluangP.ÅkerholmT.NymanG.LindahlJ.HanssonI.MorrellJ. M. (2024). Bacteria in the healthy equine vagina during the estrous cycle. Theriogenology213, 11–18. doi: 10.1016/j.theriogenology.2023.09.011
32
MarnachM. L.WygantJ. N.CaseyP. M. (2022). Evaluation and management of vaginitis. Mayo Clin. Proc.97, 347–358. doi: 10.1016/j.mayocp.2021.09.022
33
McDonaldB. A.LindeC. (2002). Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol.40, 349–379. doi: 10.1146/annurev.phyto.40.120501.101443
34
MessaoudiS.ManaiM.KergourlayG.PrévostH.ConnilN.ChobertJ. M.et al. (2013). Lactobacillus salivarius: bacteriocin and probiotic activity. Food Microbiol.36, 296–304. doi: 10.1016/j.fm.2013.05.010
35
Mlynarczyk-BonikowskaB.KowalewskiC.Krolak-UlinskaA.MaruszaW. (2022). Molecular mechanisms of drug resistance in Staphylococcus aureus. Int. J. Mol. Sci.23:8088. doi: 10.3390/ijms23158088
36
MnisiC. M.NjeriF. M.MainaA. N.WaliaulaP. K.ChengV.KumaloI.et al. (2025). A review on the potential use of eubiotics in non-chicken poultry species. Trop. Anim. Health Prod.57:213. doi: 10.1007/s11250-025-04466-9
37
RaftisE. J.FordeB. M.ClaessonM. J.O’TooleP. W. (2014). Unusual genome complexity in Lactobacillus salivarius JCM1046. BMC Genomics15:771. doi: 10.1186/1471-2164-15-771
38
SharifzadehA.ShokriH. (2021). In vitro synergy of eugenol on the antifungal effects of voriconazole against Candida tropicalis and Candida krusei strains isolated from the genital tract of mares. Equine Vet. J.53, 94–101. doi: 10.1111/evj.13268
39
SuezJ.ZmoraN.SegalE.ElinavE. (2019). The pros, cons, and many unknowns of probiotics. Nat. Med.25, 716–729. doi: 10.1038/s41591-019-0439-x
40
SunE.RenF.LiuS.GeS.ZhangM.GuoH.et al. (2015). Complete genome sequence of Lactobacillus salivarius Ren, a probiotic strain with anti-tumor activity. J. Biotechnol.210, 57–58. doi: 10.1016/j.jbiotec.2015.06.399
41
TakayaA.YamamotoT.TokoyodaK. (2020). Humoral immunity vs. Salmonella. Front. Immunol.10:3155. doi: 10.3389/fimmu.2019.03155
42
WangC. H.YenH. R.LuW. L.HoH. H.LinW. Y.KuoY. W.et al. (2022). Adjuvant probiotics of Lactobacillus salivarius subsp. salicinius AP-32, L. johnsonii MH-68, and Bifidobacterium animalis subsp. lactis CP-9 attenuate glycemic levels and inflammatory cytokines in patients with type 1 diabetes mellitus. Front. Endocrinol13:754401. doi: 10.3389/fendo.2022.754401
43
WenX.LuoS.LvD.JiaC.ZhouX.ZhaiQ.et al. (2022). Variations in the fecal microbiota and their functions of thoroughbred, Mongolian, and hybrid horses. Front. Vet. Sci.9:920080. doi: 10.3389/fvets.2022.920080
44
YangJ.ShangP.ZhangB.WangJ.DuZ.WangS.et al. (2023). Genomic and metabonomic methods reveal the probiotic functions of swine-derived Ligilactobacillus salivarius. BMC Microbiol.23:242. doi: 10.1186/s12866-023-02993-9
45
YangY.SongX.WangG.XiaY.XiongZ.AiL. (2024). Understanding Ligilactobacillus salivarius from probiotic properties to omics technology: a review. Foods13:895. doi: 10.3390/foods13060895
46
YuD.GuoD.ZhengY.YangY. (2023). A review of penicillin binding protein and group A Streptococcus with reduced-β-lactam susceptibility. Front. Cell. Infect. Microbiol.13:1117160. doi: 10.3389/fcimb.2023.1117160
47
ZhangX.LiuY.LiL.MaW.BaiD.DugarjaviinM. (2025). Physiological and metabolic responses of Mongolian horses to a 20 km endurance exercise and screening for new oxidative-imbalance biomarkers. Animals15:1350. doi: 10.3390/ani15091350
Summary
Keywords
Lactobillus salivarius, probiotics, vaginal microbiota, comparative genomics, Mongolian horse
Citation
Zhao Y, Liu Y, Tao J, Cao J, Lin Y, He Q, Fang X, Yun S, Du M, Su S, Bao T, Bai D, Zhang X and Dugarjaviin M (2025) Isolation, identification and comparative genomic analysis of Lactobacillus salivarius from Mongolian horse vagina. Front. Microbiol. 16:1635639. doi: 10.3389/fmicb.2025.1635639
Received
26 May 2025
Accepted
09 July 2025
Published
31 July 2025
Volume
16 - 2025
Edited by
Renpeng Du, Heilongjiang University, China
Reviewed by
Hao Lizhuang, Qinghai University, China
Liu Guiqin, Liaocheng University, China
Updates
Copyright
© 2025 Zhao, Liu, Tao, Cao, Lin, He, Fang, Yun, Du, Su, Bao, Bai, Zhang and Dugarjaviin.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manglai Dugarjaviin, dmanglai@163.com
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.