 Sascha D. Braun1,2*
Sascha D. Braun1,2* Martin Reinicke1,2
Martin Reinicke1,2 Celia Diezel1,2
Celia Diezel1,2 Elke Müller1,2Katrin Frankenfeld3Thomas Schumacher4
Elke Müller1,2Katrin Frankenfeld3Thomas Schumacher4 Hugo Arends4
Hugo Arends4 Stefan Monecke1,2
Stefan Monecke1,2 Ralf Ehricht1,2,5
Ralf Ehricht1,2,5- 1Leibniz Institute of Photonic Technology, Member of the Research Alliance “Leibniz Health Technologies” and the Leibniz Centre for Photonics in Infection Research (LPI), Jena, Germany
- 2InfectoGnostics Research Campus Jena, Center for Applied Research, Jena, Germany
- 3INTER-ARRAY Part of fzmb GmbH, Bad Langensalza, Germany
- 4Institut Virion\Serion GmbH, Würzburg, Germany
- 5Institute of Physical Chemistry, Friedrich Schiller University Jena, Jena, Germany
Introduction: Carbapenemase-producing bacteria undermine the efficacy of carbapenems, a class of last-resort antibiotics used primarily to treat infections caused by multidrug-resistant Gram-negative pathogens. Carbapenemases are among the most alarming antimicrobial resistance mechanisms because they inactivate all β-lactam antibiotics leaving clinicians with few or no therapeutic options. The genes encoding these enzymes are typically located on mobile genetic elements (MGE), which facilitate rapid horizontal gene transfer among different bacterial species. These MGE’s often additionally carry toxin-antitoxin systems that promote long-term persistence in bacterial populations. Carbapenem-resistant Enterobacteriaceae (CRE) often colonize the gastrointestinal tract without symptoms, serving as silent reservoirs for further dissemination. Infections caused by CRE are associated with high morbidity and mortality and are frequently resistant to multiple drug classes. Given the urgent clinical need for rapid diagnostics, immunochromatographic assays represent a promising and urgently needed approach for economic and available point-of-care detection. However, the development of such assays is often hindered by the time-consuming process of identifying high-affinity antibody pairs.
Methods: To accelerate this process, we evaluated a protein microarray platform as a high-throughput screening tool to identify optimal monoclonal antibody (mAb) pairs targeting the most clinically relevant carbapenemases. Monoclonal antibodies derived from hybridoma libraries and commercial sources were spotted in triplicates and tested in a single experiment against lysates from reference strains expressing the carbapenemase enzymes KPC, NDM, IMP, VIM, OXA-23/48/58, and MCR-1, an enzyme conferring resistance to colistin. Signal intensities were quantified, and diagnostic performance was assessed across four thresholds.
Results: A cut-off > 0.2 yielded the best balance, with approximately 61% balanced accuracy and ≥99% specificity. Around 22% of tested antibodies showed strong, reproducible reactivity. For several targets–such as KPC, IMP, VIM, OXA-58, and MCR-1–100% sensitivity was achieved. The array allowed simultaneous mapping of cross-reactivity, a key advantage over conventional ELISA workflows.
Discussion: Our findings confirm that protein-based microarrays offer a robust, efficient platform for antibody pair selection, reducing reagent use while accelerating assay development. The validated antibody pairs are directly applicable to ELISA or lateral flow test formats and provide a strong foundation for next-generation diagnostics capable of detecting an evolving panel of carbapenemases in clinical settings.
1 Introduction
Carbapenem-resistant Gram-negative bacteria are recognized as a critical threat to global health because carbapenemases inactivate almost all β-lactams and frequently act together with outer-membrane porin loss, producing pan-resistant phenotypes (van Duin and Doi, 2017; Tacconelli et al., 2018; Alvisi et al., 2025). Clinical infections caused by these organisms lead to limited therapeutic options, prolonged hospitalization and excess mortality (Perez and Bonomo, 2019), prompting their classification in the World Health Organization’s highest “critical” AMR priority tier (Govindaraj Vaithinathan and Vanitha, 2018).
The enzymatic landscape of carbapenemases is highly heterogeneous. Class A KPC, class B metallo-β-lactamases (NDM, VIM, IMP) and class D oxacillinases (OXA-48-like, OXA-23, OXA-58) account for most documented cases of carbapenem resistance, yet >1,000 allelic variants have been deposited in public databases (Alcock et al., 2020; Feldgarden et al., 2022; Gschwind et al., 2023). Many genes reside on broad-host-range plasmids equipped with toxin–antitoxin modules, ensuring stable maintenance even in absence antimicrobial pressure and facilitating rapid dissemination across Enterobacterales, Acinetobacter baumannii and Pseudomonas aeruginosa populations (Partridge et al., 2018; Rana et al., 2024).
Timely identification of carbapenemase producers is essential for targeted therapy and infection-control interventions, but existing diagnostics have important shortcomings. Phenotypic assays (e.g., RAPIDEC® CARBA NP, ETEST®, or double disk synergy tests) are inexpensive but time-consuming to perform, typically requiring overnight incubation, and may miss OXA variants; nucleic-acid amplification panels provide high sensitivity but require costly instrumentation and continuous redesign to keep pace with emerging alleles; single-plex lateral-flow immunoassays detect at most five enzyme families and depend on a limited set of antibody pairs (Boutal et al., 2017). These constraints hamper large-scale surveillance and leave laboratories poorly equipped to accommodate the accelerating diversity of resistance determinants.
An ideal platform for test development should therefore (i) simultaneously screen for multiple carbapenemases, (ii) support high-throughput optimization of capture and detector monoclonal antibodies (mAbs) and (iii) generate quantitative metrics that guide transfer to user-friendly formats. Protein-based microarrays satisfy these criteria and offer several advantages over the ELISA gold standard. First, they miniaturize sandwich immunoassays into micrometer-scale spots, allowing thousands of antibody combinations to be interrogated on a single slide while using only a fraction of the reagents and sample volume required by ELISA. Second, the parallel layout exposes every candidate capture antibody to every potential detector in one experiment, enabling rapid identification of both high-affinity pairs and undesirable cross-reactivity patterns that would otherwise emerge only after weeks of sequential ELISA testing. Third, because all targets are printed side-by-side, the array provides an internal reference framework: cross-family reactivity is revealed instantly, streamlining specificity optimization. Collectively, these attributes reduce development time, lower costs and provide more detailed and quantitative insights into antibody–antigen interactions than traditional well-based methods (Kingsmore, 2006). However, protein microarrays also have limitations. They require specialized equipment for fabrication and scanning, may be sensitive to variations in antibody stability and spotting quality, and often require well-characterized monoclonal antibodies with high specificity and sensitivity. Additionally, the transition from array data to user-friendly diagnostic formats still necessitates further assay development and validation (Sauer, 2017).
Here, we evaluate a protein-based microarray comprising 49 mAbs directed against KPC, NDM, IMP, VIM, OXA-23/48/58 and the mobilized colistin-resistance factor MCR-1 (Liu et al., 2016). The specific aims were two-fold: (i) to identify high-affinity capture–detector pairs for each carbapenemase family and (ii) to quantify the diagnostic performance of the microarray across a range of signal thresholds. By establishing a streamlined discovery workflow that rapidly pinpoints optimal antibody pairs–and simultaneously unmasks cross-reactivity–this study lays the groundwork for next-generation multiplex lateral-flow or ELISA diagnostics capable of keeping pace with the evolving carbapenemase repertoire.
2 Materials and methods
2.1 Strains
Bacterial strains used in this study are listed in Table 1. The isolates were characterized by whole-genome sequencing using Oxford Nanopore Technologies (ONT) for genetic analysis and MALDI-TOF (Bruker, Bremen, Germany) for species identification. Prior to species identification, DNA extraction and long-read sequencing, all bacterial isolates were grown on Columbia blood agar plates and incubated overnight at 37 °C under aerobic conditions.
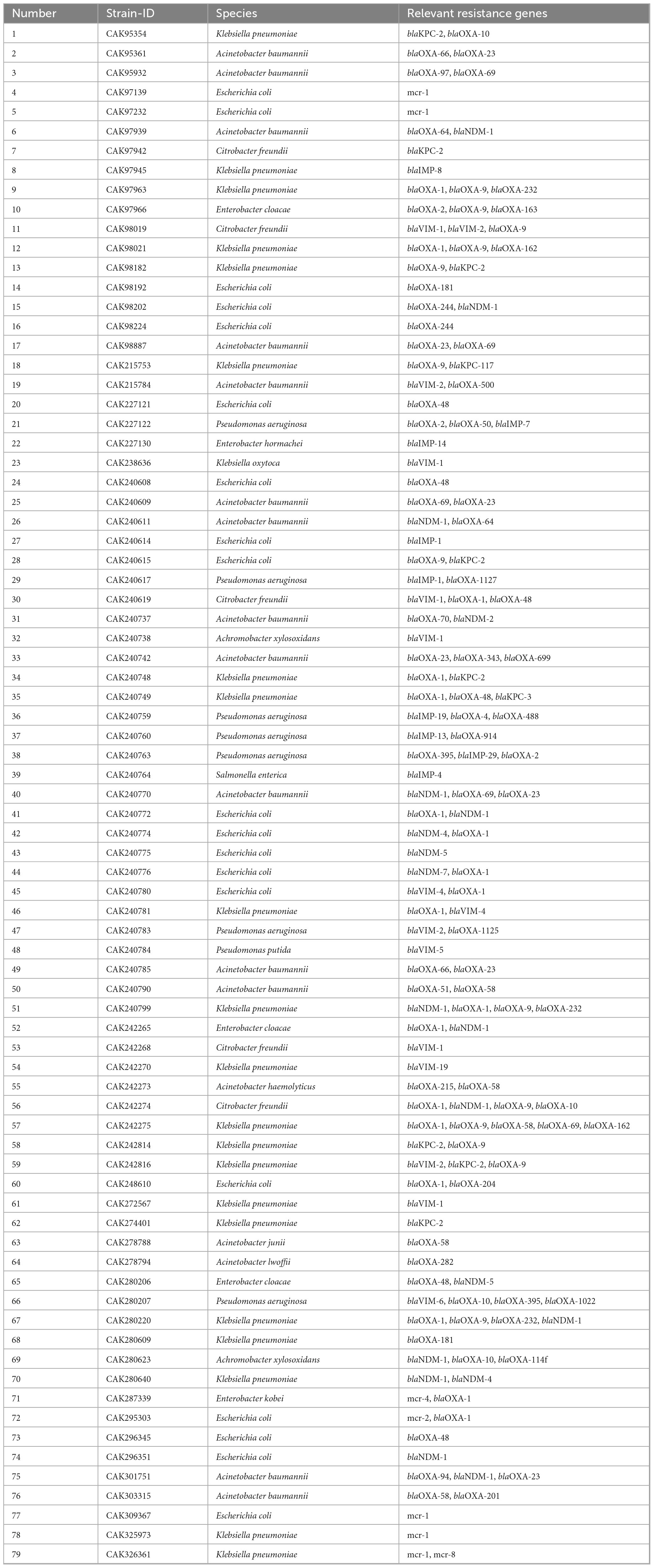
Table 1. Fully sequenced reference strains used for protein-based microarray experiments.
2.2 Sequencing and resistance genotyping
Whole-genome sequencing of all strains listed in Table 1 was performed using the Oxford Nanopore Technologies (ONT) MinION platform to confirm species identity and characterize resistance gene profiles. Genomic DNA was extracted using the NucleoSpin Microbial DNA Kit (Macherey-Nagel, Düren, Germany) with minor protocol modifications. Bacterial isolates were cultured overnight on Columbia blood agar (Becton Dickinson, Heidelberg, Germany), and biomass was collected using a full inoculation loop. Cells were suspended in 500 μL PBS (pH 7.4), pelleted by centrifugation, and resuspended in 100 μL buffer BE. Mechanical lysis was achieved using a BeatBeater (Biozym, Hessisch Oldendorf, Germany) for 5 min at maximum speed. Proteinase K digestion was followed by heat inactivation at 70 °C for 5 min. RNase A (100 mg/mL; Sigma-Aldrich, Steinheim, Germany) was then added and incubated at 37 °C for 5 min. DNA was purified and eluted in 70 μL of nuclease-free water (Carl Roth, Karlsruhe, Germany).
Library preparation was conducted using the SQK-NBD114.24 Native Barcoding Kit (Oxford Nanopore Technologies), and sequencing was performed exclusively on R10.4.1 flow cells (FLO-MIN114). DNA was size-selected using AMPure XP beads (Beckman Colter, Krefeld, Germany) at a 1:1 ratio to enrich for high-molecular-weight fragments. Sequencing runs were executed for 72 h using MinKNOW (v22.12.7), and raw signal data were recorded in POD5 format.
Basecalling was performed using Dorado (v0.9.1, Oxford Nanopore Technologies) with the high-accuracy model res_dna_r10.4.1_e8.2_400bps_sup@2023-09-22_bacterial-methylation. De novo assembly was conducted with Flye (v2.9.1-b1780), followed by four rounds of polishing with Racon (v1.5.0) using optimized parameters (match = 8, mismatch = 6, gap = 8, window = 500). Final polishing was completed with Medaka (v1.7.3) using the model r1041_e82_400bps_bacterial_methylation. The resulting high-quality assemblies were used to confirm resistance gene content and guide recombinant antigen selection. Resistance genotyping was performed using abricate (v1.0.0) with curated resistance gene databases (ResFinder, CARD, NCBI) to identify acquired antimicrobial resistance determinants (Supplementary File 4 Data Sheet 2.zip).
2.3 Antigen and antibody production
All recombinant antigens used in this study were produced and developed by our partner VIRION\SERION GmbH (Würzburg, Germany). Reference genes and protein sequences used for expression are listed in Table 2. These reference sequences were selected based on their prevalence and clinical relevance in antimicrobial resistance surveillance. All genes were cloned into Novagen’s pET-16b expression vector (Merck, Darmstadt, Germany) for recombinant protein expression. The gene sequences were codon-optimized for Escherichia coli when necessary, while genes of E. coli origin were used in their native form. Cloning was performed using NcoI and BamHI restriction sites, ensuring precise integration into the vector. To facilitate protein purification, the expressed antigens were designed with an N-terminal 10× His-tag. The Factor Xa cleavage site present in the pET16b vector was removed to prevent unwanted proteolytic processing. The resulting construct included only the mature protein sequence, excluding signal peptides. Expression was carried out in E. coli BL21 (DE3) using auto-induction media to optimize yield. Protein purification was performed under native conditions via Ni-NTA affinity chromatography, followed by SDS-PAGE and Western blot analysis to confirm integrity and purity. Purified antigens were stored in PBS at a minimum concentration of 2 mg/ml. These antigens were then utilized for subsequent applications, including antibody production, microarray development, and lateral flow assay optimization.
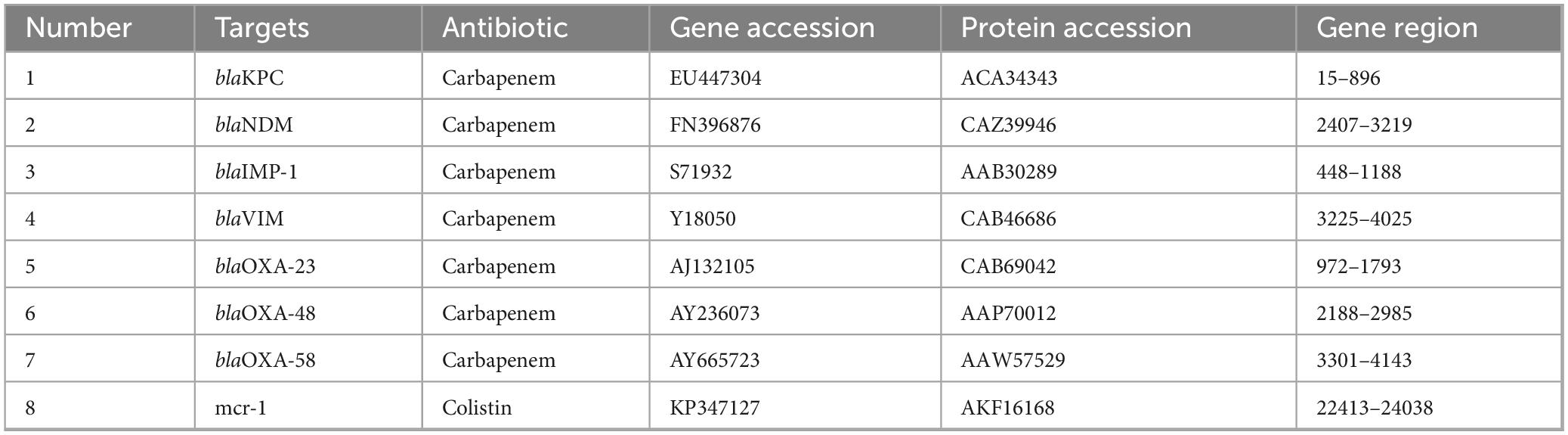
Table 2. Resistance targets, antibiotic class, GenBank accessions, and nucleotide coordinates of the mature coding regions cloned for recombinant-antigen production.
Monoclonal antibodies were produced by our partner fzmb GmbH (Bad Langensalza, Germany) using hybridoma technology according to Köhler and Milstein (1975). Briefly, purified antigens were used to immunize mice, with multiple injections administered over several weeks to elicit a strong immune response. To enhance immunogenicity, adjuvants were included in the immunization protocol; however, the specific formulation remains undisclosed due to confidentiality agreements with the industrial partner. Finally, spleen cells were harvested and fused with immortal myeloma cells using polyethylene glycol, generating hybridoma cells capable of continuous antibody production in vitro. These hybridomas were then cultured in hypoxanthine-aminopterin-thymidine (HAT) medium to select for successfully fused cells, while unfused myeloma and B cells were eliminated. The resulting hybridomas were screened for specific antibody production using enzyme-linked immunosorbent assays (ELISA), and positive clones cloned and re-cloned. For each antigen, at least ten distinct monoclonal antibodies were generated, with each hybridoma cell clone being expanded and cultured for large-scale production. Antibodies were purified from the culture supernatant using protein A/G affinity chromatography, ensuring high purity. Finally, the purified antibodies underwent extensive characterization, including ELISA, Western blotting, and affinity determination, to confirm specificity and suitability for downstream applications such as microarray development and lateral flow assays.
Additionally, commercially available antibodies from various manufacturers were incorporated into the study to complement the panel of monoclonal antibodies generated via hybridoma technology. These antibodies were commercially obtained from different suppliers to ensure broad coverage and validated specificity for the targeted resistance determinants. These antibodies are listed in Table 3, detailing their origin, target-specificity, and order number. The inclusion of commercially available antibodies allowed for comparative validation and facilitated the development of a robust detection platform suitable for microarray and lateral flow assay applications.
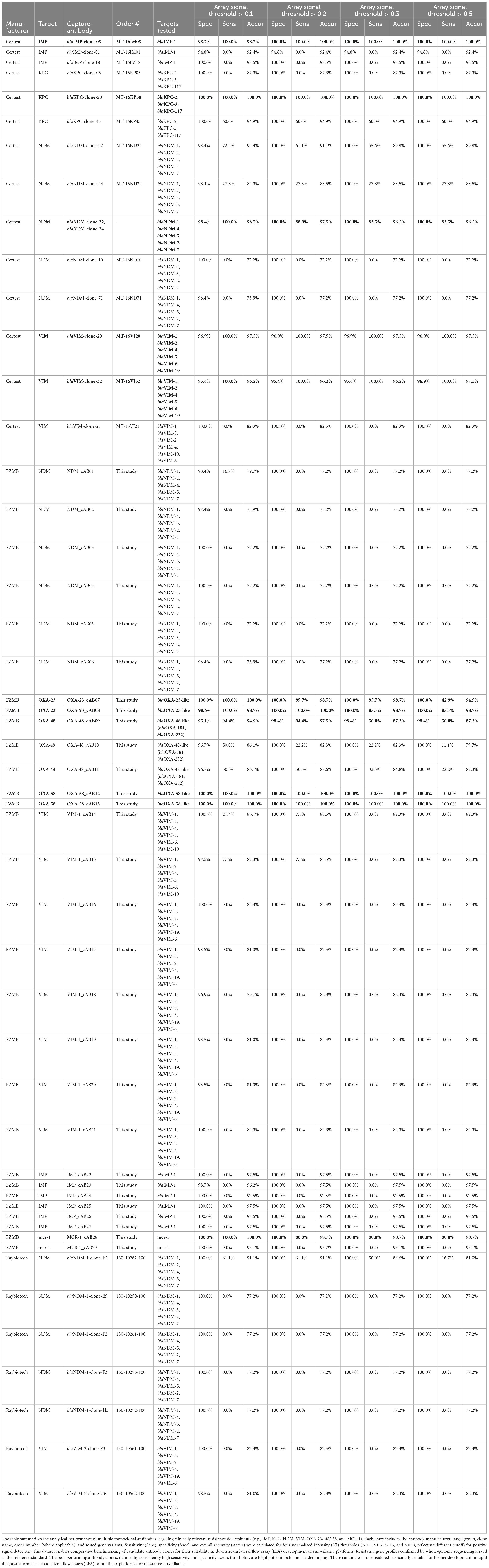
Table 3. Diagnostic performance of capture antibodies using the detection antibody mix against various carbapenemase and resistance targets at increasing array signal thresholds.
2.4 Microarray production and procedure
The microarrays used in this study were manufactured by INTER-ARRAY (Part of fzmb GmbH, Research Center for Medical Technology and Biotechnology, Bad Langensalza, Germany). All antibodies were covalently immobilized directly onto functionalized plastic microarray strips (Scienion, Germany) using a fully automated M2 spotter (M2 Automation, Berlin, Germany). The spotted area measured 3.5 mm by 3.5 mm, accommodating 196 spots with a diameter of 80–120 μm (for the microarray layout, see Supplementary File 1 Table1.xlsx). The spotting process ensured uniform distribution and optimal binding conditions for each antibody, which were applied at a final concentration of 0.25 μg/μL. All antibodies were provided at 1 mg/mL by FZMB or commercial suppliers and diluted accordingly prior to spotting. Following manufacturing, each 8-well strip was sealed under an argon atmosphere to maintain stability and stored at room temperature until use. This approach preserved antibody functionality and ensured reproducibility in downstream applications.
The detection of resistance-associated proteins using antibody-based microarrays was carried out according to an optimized protocol (Figure 1). For all microarray experiments described in this study, bacterial lysates were used as the antigen source. Recombinant antigens were not applied to the arrays during screening. The strains were incubated on Columbia Blood agar (BD, Germany) at 37 °C for 18–24 h. One loop of cells was inoculated directly from the agar into 300 μl buffer (1xPBS; 0.05% Tween20; 0.25% TritonX-100; 1% fetal calf serum) and vortexed. The arrays were washed twice with 150 μl buffer for 3 min at 37 °C and 400 rpm using an Eppendorf Thermomixer (Eppendorf, Germany), followed by 100 μl blocking solution (10% fetal calf serum diluted in 1xPBS; 0.05% Tween20; 0.25% TritonX-100) for 5 min at 37 °C and 300 rpm. Then 100 μl of the cell suspensions were added to the microarray strip and incubated at 37 °C and 300 rpm for 30 min. The arrays were then washed with 150 μl buffer for 5 min at 37 °C and 400 rpm. The specifically bound proteins were detected by the addition of 100 μl of an antibody-detection-mix (Table 4), including eight biotin-labeled antibodies, one for each target, and incubate at 37 °C and 300 rpm for 30 min. After a washing step 100 μl of streptavidin-horseradish peroxidase (HRP) was added and incubated for 15 min at 37 °C and 300 rpm. After two final washing steps (37 °C, 400 rpm, 3 min each), the microarrays were incubated with SeramunBlue substrate (Seramun Diagnostica GmbH, Heidesee, Germany) for exactly 10 min at 25 °C without shaking to visualize antibody-antigen interactions.
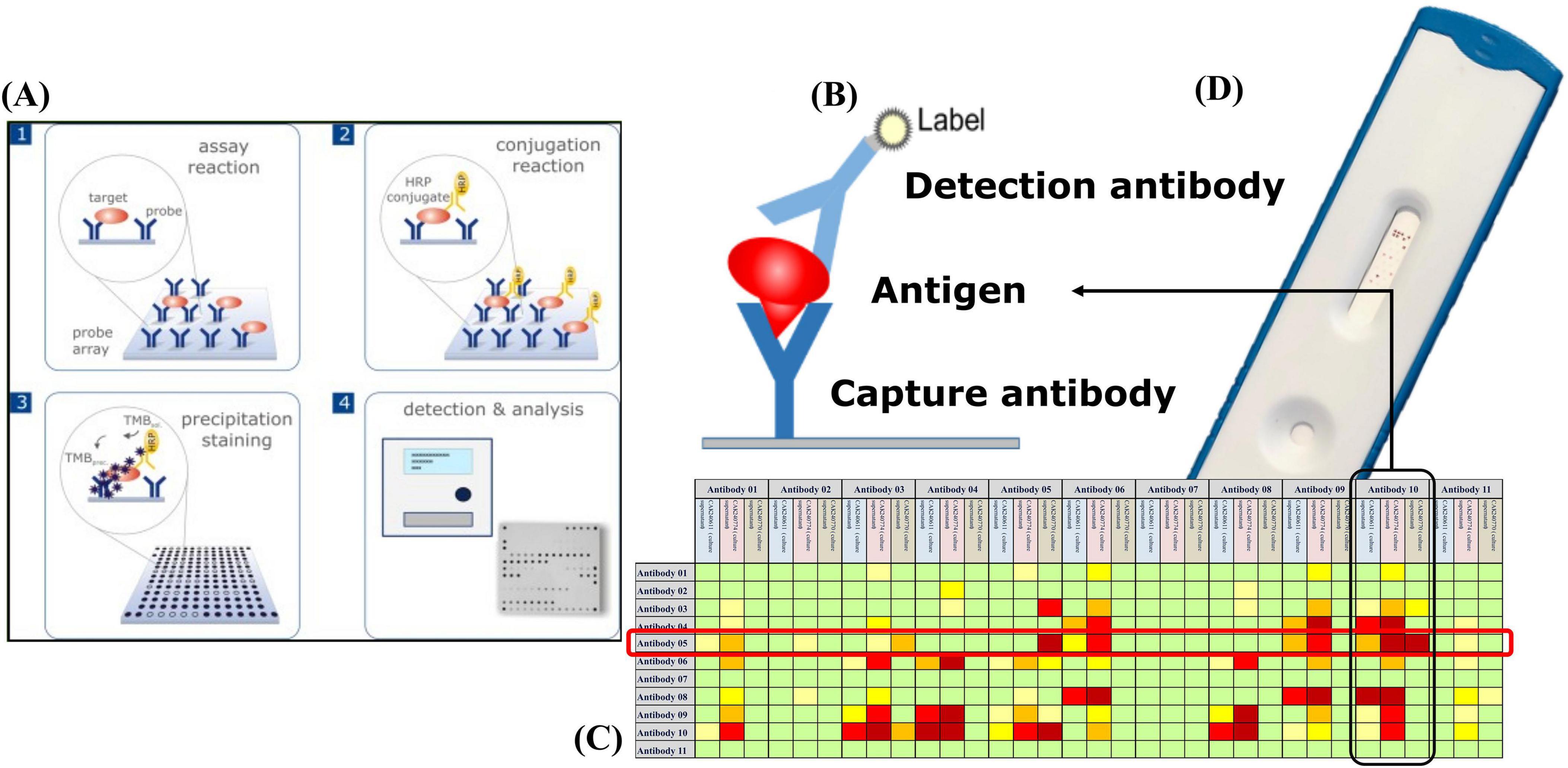
Figure 1. High-throughput microarray screening for perfectly matched antibody pairs: (A) Microarray workflow: an array of immobilized capture antibodies first binds the native antigen, after which streptavidin-horseradish peroxidase (HRP)–tagged detection antibodies are added; enzymatic conversion of the insoluble Seramun-Blue substrate (TMB) then produces a dark spot that is quantified by an automated reader. (B) Sandwich geometry in which the surface-bound capture antibody holds the antigen in place for recognition by the HRP-labeled detector. (C) Heat map ranking every possible capture-detector combination (rows versus columns) by signal intensity (green, none; yellow, moderate; red, strong), highlighting the intersection of capture Antibody 05 and detector Antibody 10 as the optimal choice for device translation. (D) Prototype lateral-flow strip built with the top-ranked pair; the distinct test line confirms a robust signal in the point-of-care format.
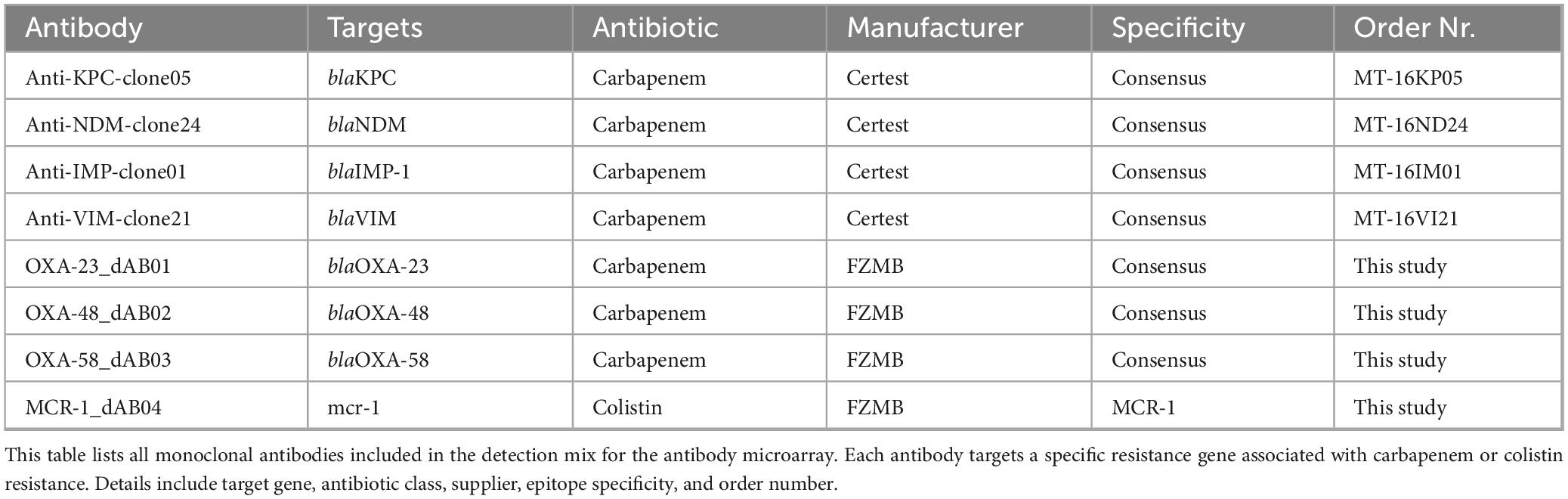
Table 4. Composition of the detection antibody mix used in the microarray assay.
2.5 Microarray image analysis
The antibody-based microarrays were scanned and automatically analyzed using the INTER-VISION device following the manufacturer’s guidelines (INTER-ARRAY part of fzmb GmbH, Bad Langensalza, Germany). Signal intensities were quantified based on predefined spot coordinates, ensuring precise evaluation of antibody-antigen interactions.
During analysis, the relative signal intensities were calculated by determining the normalized intensity (NI) for each spot. The NI values were computed using the formula NI = 1−(M/BG), where M represents the average intensity of the spot, and BG corresponds to the intensity of the local background. This normalization method ensured that NI values ranged between 0 (no detectable signal) and 1 (maximum intensity), allowing for accurate and reproducible data interpretation.
2.6 Statistics
For each antibody spot, diagnostic performance was assessed across four predefined array signal thresholds: >0.1, >0.2, >0.3, and >0.5. The classification outcomes were determined by comparing the microarray-based detection signals to known resistance gene profiles. These profiles, established through whole-genome sequencing using the Oxford Nanopore MinION platform (as detailed in Section “2.3 Antigen and antibody production”), served as the reference standard for evaluating antibody signal classifications. Sensitivity was defined as TP/(TP + FN); specificity as TN/(TN + FP); accuracy as (TP + TN)/(TP + TN + FP + FN); and balanced accuracy as (sensitivity + specificity)/2, where TP = true positives, TN = true negatives, FP = false positives, and FN = false negatives. All values were expressed as percentages.
To assess the effect of signal threshold variation on diagnostic performance, the Friedman test was applied, which is appropriate for comparing multiple related samples. This test was used to evaluate differences in sensitivity, specificity, and accuracy across the four signal thresholds. Post hoc comparisons were performed using the Wilcoxon signed-rank test to identify statistically significant differences between threshold pairs. A p-value < 0.05 was considered indicative of statistical significance. All statistical analyses were conducted using Python (v3.11), employing functions from the scipy and pandas packages. The corresponding analysis script is provided as Supplementary File 2 Data Sheet 1.csv.
3 Results
3.1 Overview of antibody screening performance
Microarray-based screening revealed several high-sensitivity antibody pairs, with distinct capture-detection combinations displaying strong interactions across all signal thresholds tested. Out of the 49 antibodies analyzed, approximately 22% demonstrated consistent and reliable signal intensities across all microarray layouts, indicating their suitability for carbapenemase detection applications. Notably, multiple antibodies from Certest and FZMB exhibited robust detection sensitivity. These included Certest clones blaIMP-clone-05, blaKPC-clone-58, blaNDM-clone-22, blaNDM-clone-24, blaVIM-clone-20 and blaVIM-clone-32, as well as FZMB clones OXA-23_cAB07, OXA-23_cAB08, OXA-48_cAB09, OXA-58_cAB12, OXA-58_cAB13 and MCR-1_cAB28 all of which showed very high sensitivity and specificity across all experimental thresholds. Their stable and reproducible performance highlights their potential as reliable components for downstream diagnostic development. All raw data are available at Supplementary File 3 Table2.xlsx.
3.2 Performance of the antibodies on the microarray
This study assessed the sensitivity of various antibodies targeting carbapenemase genes across signal thresholds of >0.1, >0.2, >0.3, and >0.5 to evaluate their effectiveness in detecting carbapenemase-producing bacteria (Table 3). Among the antibodies tested, the following 11 pairings demonstrated the highest sensitivity across all signal thresholds, making them reliable choices for diagnostic applications. BlaIMP-clone-05, which targets blaIMP-1, achieved 100% sensitivity across all thresholds, showing excellent detection consistency and robustness at any signal level. BlaKPC-clone-58, specific to blaKPC-2, blaKPC-3, and blaKPC-117, also reached 100% sensitivity across all thresholds, underscoring its strong and consistent performance in detecting KPC-related resistance, a critical target due to the clinical significance of KPC enzymes. OXA-58_cAB12 and OXA-58_cAB13, both targeting blaOXA-58-like genes, also maintained 100% sensitivity across all thresholds, making them highly effective for identifying blaOXA-58, which is an important variant in resistance profiling.
Another good performing antibody, blaKPC-clone-43, targeting blaKPC-2, blaKPC-3, and blaKPC-117, achieved 100% sensitivity at lower thresholds (>0.1 and >0.2) but showed a slight reduction in sensitivity to 60% at higher thresholds (>0.3 and >0.5), indicating strong efficacy in early-stage detection scenarios. BlaVIM-clone-20, which targets a broad spectrum of blaVIM variants (blaVIM-1, blaVIM-2, blaVIM-4, blaVIM-5, blaVIM-6, and blaVIM-19), demonstrated 100% sensitivity across all thresholds, offering comprehensive detection capability for blaVIM genes, which are associated with significant antibiotic resistance challenges.
OXA-23_cAB08, targeting blaOXA-23-like genes, exhibited 100% sensitivity at >0.2 and >0.3 thresholds, with a high sensitivity of 85.7% at >0.5, making it highly effective in detection at moderate and high signal thresholds. BlaNDM-clone-22, targeting multiple blaNDM gene variants, achieved 72.2% sensitivity at the >0.1 threshold and demonstrated gradually decreasing sensitivity at higher thresholds, reflecting its particular utility for lower-threshold detection. BlaNDM-1-clone-E2, targeting blaNDM-1, blaNDM-2, blaNDM-4, blaNDM-5, and blaNDM-7, showed sensitivity of 61.1% at the >0.1 and >0.2 thresholds, though its sensitivity decreased at higher thresholds, suggesting its optimal application in early detection settings. Finally, blaNDM-clone-24, targeting various blaNDM variants, displayed consistent sensitivity of 27.8% across thresholds >0.2, >0.3, and >0.5, providing stable detection performance at moderate and higher signal levels.
Conversely, some antibodies exhibited significantly lower sensitivity, limiting their diagnostic utility. For instance, VIM-1_cAB14 (FZMB), specific to blaVIM variants, showed a maximum sensitivity of just 21.4% at >0.1, decreasing further at higher thresholds, suggesting it may not be effective for consistent blaVIM gene detection. BlaNDM-1-clone-F3 and blaNDM-clone-71, targeting blaNDM-1, blaNDM-4, blaNDM-5, blaNDM-2, and blaNDM-7, exhibited no sensitivity at any threshold beyond >0.1, indicating limited applicability in most diagnostic settings that require higher detection levels, thus offering limited utility in broader diagnostic contexts.
3.3 Threshold optimization
Performance metrics were calculated for four array-signal cut-offs (>0.1, >0.2, >0.3 and >0.5) (Table 5). Mean specificity was ≥99% for all thresholds, whereas mean sensitivity dropped from 25.8% at >0.1% to 18.9% at >0.5, reducing balanced accuracy from 62.4% to 59.4%. A Friedman test confirmed that threshold choice significantly affected specificity (χ2 = 71.2, p < 10–14), sensitivity (χ2 = 38.4, p < 10–7) and accuracy (χ2 = 8.4, p = 0.039). Post hoc Wilcoxon comparisons showed that specificity at >0.1 was significantly lower than at any higher threshold (p ≤ 1.6 × 10–5), whereas results based on thresholds >0.2, >0.3 and >0.5 did not differ from one another (p ≈ 0.32).
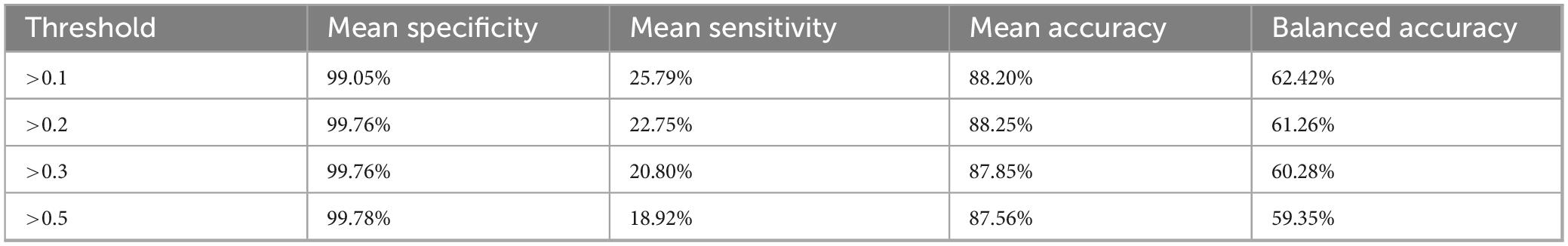
Table 5. Effect of array-signal threshold on diagnostic performance.
Raising the cut-off above 0.2 provided no additional gain in specificity but incurred a steady loss of sensitivity. Thus, the >0.2 threshold yielded the highest overall accuracy (88.3%) and it was the best compromise between false-positive control and true-positive yield. We therefore recommend >0.2 as the default diagnostic threshold for this microarray platform and our specific test setup; a lower cut-off (>0.1) may be chosen only when maximal analytical sensitivity is essential and a slight increase in false positive rates is acceptable.
4 Discussion
The rise of carbapenemase-producing organisms represents a critical global health threat, compounding the broader crisis of antimicrobial resistance (AMR). Recent global estimates suggest that antimicrobial resistance (AMR) was directly responsible for approximately 1.27 million deaths in 2019, with nearly 5 million deaths associated with drug-resistant infections (Caliskan-Aydogan and Alocilja, 2023; Murray et al., 2024). The latter figure includes cases in which AMR contributed to poor clinical outcomes or treatment failure, even if it was not the primary cause of death. Carbapenem-resistant Enterobacterales (CRE) and other carbapenemase-producing pathogens represent some of the most critical threats to public health, frequently causing severe infections that are difficult to treat due to limited therapeutic options (Hong et al., 2021). Infections by these organisms are associated with significantly higher mortality rates and prolonged hospital stays, especially when appropriate therapy is delayed (Caliskan-Aydogan and Alocilja, 2023). Rapid identification of carbapenemase producers is therefore paramount to guide timely effective therapy and implement infection control measures (Richter and Marchaim, 2017). However, diagnosing carbapenemase production remains challenging. Traditional culture-based phenotypic assays (e.g., modified Hodge test (Girlich et al., 2012), RAPIDEC® CARBA NP, etc.) can be laborious and slow, while molecular tests require expensive equipment and skilled personnel (Rabaan et al., 2022). Moreover, the diversity of carbapenemase genes (KPC, NDM, VIM, OXA-variants, IMP, etc.) complicates single-test detection – no single conventional assay easily covers all variants. Indeed, recent reviews emphasize the lack of cost-effective methods to broadly screen for all carbapenemase-producing species and the ongoing need for new diagnostic tools with high sensitivity across diverse enzymes (Rabaan et al., 2022). The World Health Organization has classified CRE as critical priority pathogens, underscoring the urgent need for innovative diagnostics as well as therapeutics (World-Health-Organisation, 2017).
In response to these challenges, immunochromatographic lateral flow assays (LFAs) have emerged in the past few years as rapid, practical diagnostics for the most prevalent carbapenemases. Notably, the NG-Test CARBA 5 (NG Biotech, Guipry, France), the RESIST-5 O.K.N.V.I. (Coris BioConcept, Gembloux, Belgium) and the KarbaDia (GaDIA SA, Mothey, Switzerland) kits can each detect five common carbapenemase families (KPC, OXA-48-like, NDM, VIM, IMP) within minutes. Clinical evaluations of these assays have shown excellent performance: for example, CARBA 5 achieved ∼100% sensitivity and 98%–100% specificity compared to PCR gold-standard in multicenter studies (Boutal et al., 2018; Hong et al., 2021; Kon et al., 2021). Similarly, a four-target immunochromatographic strip (K-SeT for KPC, NDM, VIM, OXA-48) reported 99.2% sensitivity and 100% specificity when testing bacterial isolates (Greissl et al., 2019). These commercially available assays have demonstrated that antibody-based detection of carbapenemase proteins can rival molecular methods in accuracy while being faster and simpler. However, immunoassays also have limitations. First, they typically target only the most common enzyme types – emerging or rare carbapenemases (e.g., GES, OXA-23 variants) may escape detection. Second, since they rely on antigen–antibody binding, sufficient protein expression is required; in practice this often means testing from a cultured isolate or an 18–24 h enrichment step to accumulate detectable enzyme levels (Wareham et al., 2016). As a result, sensitivity can drop in complex clinical specimens. Furthermore, mutations in carbapenemase genes might alter antibody binding sites and thus compromise the binding efficacy of the antibodies used for an assay (Decousser et al., 2017), necessitating continual development of new antibodies as resistance evolves. These constraints highlight a crucial bottleneck in immunoassay development – the fast, economic and complete identification of high-affinity, specific monoclonal antibody pairs for defined sample types for each relevant carbapenemase target.
Our work addresses this bottleneck by leveraging a high-throughput protein microarray to streamline the screening of monoclonal antibody pairs. Conventional ELISA-based pairing studies require testing each capture–detection antibody combination in separate wells, a low-throughput approach that becomes impractical and costly for large antibody libraries (e.g., hundreds of candidates), and that consumes considerable amounts of antibody preparations. In contrast, antibody microarrays can miniaturize and parallelize these immunoassays, allowing thousands of interactions to be evaluated simultaneously on a single slide under the same set of reaction conditions (Garcia et al., 2007). The microarray format offers clear advantages over single-plex ELISA: it is faster, conserves precious reagents and sample, and enables massive scaling of pairwise binding experiments in one experiment (Haab et al., 2001). In our prototype, 49 antibodies (from multiple sources) were spotted in an array and tested in parallel, effectively condensing what would amount to hundreds of individual ELISA tests into a single high-throughput screening assay. This approach dramatically increases the efficiency of identifying promising antibody pairs. Indeed, antibody microarrays have shown success in other fields for multiplex protein detection and antibody profiling, demonstrating high sensitivity (pg/mL levels) and robust performance when properly optimized (Garcia et al., 2007). By printing replicates on array and using a standardized readout, we achieved a platform that is both scalable and reproducible for antibody screening. Notably, the small spot size and arrayed format mean that only microgram quantities of each antibody are needed per test – an important practical benefit when screening early-stage hybridoma supernatants or precious monoclonal antibodies.
Using this microarray prototype, we surveyed a broad panel of monoclonal antibodies (mAbs) against various carbapenemase targets and rapidly pinpointed a subset with superior performance. Approximately 22% of the 49 tested antibodies emerged as top performers, displaying consistently strong signals across all array layouts and conditions. These high-sensitivity antibodies included multiple clones against the major enzyme families. For example, antibodies against blaIMP (blaIMP-clone-05) and bla_KPC (e.g., blaKPC-clone-58) achieved 100% detection sensitivity on the array at all signal thresholds evaluated, indicating robust binding to their target epitopes. Similarly, clones targeting OXA-58 and VIM carbapenemases showed uniformly high responses–often achieving 100% sensitivity even at the strictest criteria. Notably, the combination of blaNDM-clone-22 and blaNDM-clone-24 exhibited a particularly favorable diagnostic profile, yielding both high sensitivity and specificity across thresholds, and thus represent a strong candidate pair for downstream assay development targeting blaNDM variants. The fact that these known high-prevalence targets (KPC, NDM, VIM, OXA variants) yielded strong antibody hits is an encouraging outcome, as it aligns with clinical priorities–suggesting that the microarray effectively identified candidate detector pairs for the enzymes most needed in diagnostics. In contrast, several antibodies failed to generate appreciable signals or responded only under the most lenient threshold conditions. The weak or absent signal observed for some antibodies may reflect low binding affinity, suboptimal antigen presentation in lysates, or batch-dependent loss of antibody activity. Additionally, structural differences between recombinant immunogens and native proteins may have contributed to reduced epitope recognition. Notably, some clones that were designed to target blaNDM variants exhibited minimal reactivity, with sensitivity declining from 100% at the lowest cut-off to nearly 0% at more stringent thresholds. These would likely be poor choices for any diagnostic application without further optimization. The variability in performance underscores the importance of high-throughput screening: it is difficult to predict a priori which monoclonal antibody pairs will work well in a sandwich assay, so empirical testing of many candidates is essential. Our results provide a filtered shortlist of the most promising capture–detector pairs for each carbapenemase target, winnowing down the pool from hundreds of antibodies to a manageable handful per target enzyme.
To translate microarray data into practical diagnostic use, we analyzed the platform’s performance in terms of sensitivity and specificity at different signal thresholds. The array outputs were expressed as normalized intensity (NI) values (0–1 scale), and we evaluated four potential cut-off criteria (>0.1, >0.2, >0.3, >0.5) for calling a spot “positive.” This analysis simulates how one might set an analytical threshold in a clinical assay to balance false negatives vs. false positives. We observed that specificity was exceptionally high across all thresholds – on average ≥ 99% even at the lowest cut-off. This indicates a low false-positive rate inherent to the microarray, likely owing to the stringent washing conditions and the requirement for both capture and detection antibody binding for a signal. Increasing the stringency of the cut-off did further reduce false positives (specificity improved from ∼99.0% at NI > 0.1% to 99.8% at NI > 0.5), and this drop between >0.1 and >0.2 was statistically significant (p ≪ 0.001). However, raising the threshold came at the cost of sensitivity. Mean sensitivity fell from 25.8% at >0.1% to 18.9% at >0.5, reflecting the fact that weak true signals were increasingly filtered out at higher cut-offs. As a result, the balanced accuracy of the assay actually declined slightly as the threshold grew stricter (from ∼62% at >0.1 down to ∼59% at >0.5). We determined that a moderate cut-off of NI > 0.2 provides the best compromise for this microarray platform. At >0.2, overall accuracy was highest (∼88.3%) and balanced accuracy (∼61%) was close to maximal, with specificity near 99.8% while retaining ∼23% sensitivity. In practical terms, using >0.2 as the signal threshold would minimize the number of false positives without substantially sacrificing the detection of true positives. Little additional benefit was gained by harsher criteria (no significant specificity gains beyond >0.2, per post-hoc tests), whereas sensitivity and true-positive yield would continue to erode. We thus recommend NI > 0.2 as an optimal cut-off for interpreting this microarray’s results, though a more lenient > 0.1 threshold could be chosen in settings where maximizing sensitivity is critical and a slight increase in false positives is tolerable. This kind of threshold tuning, enabled by high throughput data, is valuable in guiding downstream diagnostic design – it establishes the signal level that differentiates specific binding from background noise under various conditions.
While the average sensitivity of the microarray (only ∼20%–25% at best) may appear low in a diagnostic sense, it is crucial to recognize that this metric was calculated across all 49 antibodies, the majority of which were non-performers under the given set of conditions. In practice, one would not use the entire array as a clinical test; instead, the few top-performing antibody pairs would be selected and developed into a focused assay. Those pairs, as demonstrated, can individually achieve 95%–100% sensitivity for their intended targets (e.g., the anti-IMP, -KPC, -VIM clones identified showed 100% sensitivity on the array). We expect that a multiplex lateral-flow device or diagnostic ELISA built from these optimal pairs would have performance on par with existing commercial kits. For example, using the selected antibodies, a combined test strip could potentially detect KPC, NDM, VIM, IMP, and OXA-48-like enzymes with near 100% sensitivity each, analogous to the RESIST-5 and CARBA-5 products. The role of the microarray described in this study is not to serve as a final diagnostic format, but rather to enable rapid identification of antibody combinations suitable for reliable diagnostic applications. This efficient screening is especially valuable as new carbapenemase variants emerge. Rather than laboriously developing one antibody pair at a time, a library of new candidate monoclonal antibodies can be arrayed and tested against the target antigen (or panel of antigens) in parallel. The power of this approach is evidenced not only by our study but also by similar high throughput pairing efforts addressing other targets. For instance, Chabi et al. (2023) employed a LFA system to screen 84 phage-display monoclonal antibody pairs for SARS-CoV-2 nucleocapsid protein and identified an optimal pair achieving an impressive 25 pg/mL limit of detection. Such examples illustrate how massively parallel antibody screening can drastically shorten the development cycle for immunoassays, yielding ultra-sensitive pairs that might be missed by smaller-scale testing. Our protein microarray provides a comparable screening pipeline for carbapenemase diagnostics – it can quickly pinpoint antibodies with the affinity and specificity needed for sensitive detection, which can then be integrated into user-friendly formats like lateral flow strips, ELISA plates, or biosensor chips.
A limitation of our study is that carbapenemase expression levels were not directly measured. While resistance genotyping via whole-genome sequencing served as the gold standard, protein expression can vary significantly under clinical conditions. It is well established that bacterial isolates may carry carbapenemase genes but express them poorly in patient samples, leading to false negatives in protein-based assays (Banerjee and Humphries, 2017). This discrepancy can be influenced by environmental factors, such as iron availability, which has been shown to downregulate NDM expression and reduce detection sensitivity (Hamprecht et al., 2018). These findings underscore the need to validate immunoassays under physiologically relevant conditions.
It is important to emphasize that our microarray is a prototype research tool for antibody pair discovery and test development, rather than a deployable diagnostic device in itself. The current platform requires laboratory instrumentation (array reader and analysis software) and skilled operators, which would not be practical in routine clinical settings. Instead, the microarray’s value lies in its ability to efficiently screen a large pool of antibody candidates and pinpoint those with the highest diagnostic potential for further development. Once top pairs are selected, they can be produced in bulk and incorporated into conventional test formats. For example, the best capture–detector combinations from this study could next be evaluated in a sandwich ELISA or a dipstick lateral flow context, where factors like antibody orientation, membrane properties, and sample matrix effects will be optimized. The knowledge gained from the microarray (e.g., relative binding strengths, cross-reactivity profiles, optimal signal thresholds) provides a strong starting point for those optimizations. In the longer term, we envision maintaining a dynamic pipeline: as new carbapenemase variants (or entirely new resistance enzymes) arise, one could rapidly screen new monoclonal antibodies on the microarray to find those that recognize the novel epitopes. This agility is essential given the evolutionary plasticity of carbapenemase genes – a point mutation could potentially hinder an antibody’s binding, but alternative antibodies might accommodate it. Our approach allows quick modifications including the addition of new capture/detector pairs to cover such mutations. The same high-density array can also serve as a post-market-surveillance tool: newly encountered clinical isolates that trigger false-positive or false-negative signals in the deployed lateral-flow test can be re-screened against the full antibody panel in a single run, rapidly revealing whether epitope drift or cross-reactivity is responsible and guiding the introduction of updated capture/detector pairs. Additionally, while we focused on carbapenemase proteins, the general microarray strategy could be extended to cover other resistance determinants (e.g., ESBLs, AmpC β-lactamases or other mcr allelic variants) or even to non-enzymatic biomarkers of resistance, making it a versatile platform in the fight against AMR. Finally, as microarray fabrication and detection technologies continue to advance (with increasing automation and lower costs), it is conceivable that array-based tests could eventually find a role in clinical laboratories for multiplexed pathogen or resistance detection. For now, however, the most immediate impact of our high-throughput protein microarray is as an enabling tool in the development pipeline – accelerating the creation of next-generation carbapenemase diagnostics. In an era where rapid detection of quickly evolving superbugs is critical, such tools that bridge the gap between antibody discovery and practical point-of-care tests will be invaluable in safeguarding global health.
5 Conclusion
This study demonstrates that a miniaturized protein-microarray provides an efficient discovery platform for sandwich antibody pairs directed against the major carbapenemase families. Screening 49 monoclonal antibodies in a single experiment rapidly narrowed the field to fewer than ten capture–detector combinations with outstanding analytical performance. Antibodies against KPC, IMP, VIM and OXA-58 enzymes displayed 100% sensitivity at all evaluated signal thresholds, while the combined use of blaNDM-clone-22 and blaNDM-clone-24 delivered the best overall balance of sensitivity and specificity for NDM variants. Statistical optimization showed that a normalized-intensity cut-off of >0.2 maximizes accuracy without compromising the very high specificity inherent to the array. These findings confirm that high-throughput microarrays can efficiently replace serial ELISA pairing studies, saving time and reagents and enabling the systematic inclusion of emerging resistance determinants. Although the present platform is a research prototype, the identified antibody pairs are immediately transferable to lateral-flow or ELISA formats, paving the way for rapid, multiplex point-of-care tests that can detect the predominant carbapenemase enzymes in minutes. The approach is readily scalable: as new carbapenemase alleles appear, additional monoclonal antibodies can be integrated into the array and evaluated in parallel, ensuring diagnostic coverage keeps pace with evolutionary changes. In summary, the study delivers a rigorously validated shortlist of high-performance antibodies and establishes a versatile methodology for continuous diagnostic innovation against carbapenemase-mediated resistance. By accelerating assay development and fostering universal access to rapid resistance testing, this strategy ultimately supports evidence-based antimicrobial stewardship and helps safeguard the dwindling effectiveness of last-line β-lactams worldwide today.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
SB: Software, Investigation, Funding acquisition, Conceptualization, Writing – original draft, Data curation, Project administration, Visualization, Methodology. MR: Formal analysis, Validation, Writing – review & editing, Methodology. CD: Methodology, Writing – review & editing, Data curation. EM: Methodology, Writing – review & editing, Data curation, Visualization. KF: Methodology, Software, Writing – review & editing, Funding acquisition. TS: Resources, Methodology, Writing – review & editing. HA: Writing – review & editing, Resources, Methodology. SM: Validation, Data curation, Writing – review & editing, Supervision. RE: Resources, Supervision, Funding acquisition, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Federal Ministry of Research, Technology and Space (BMFTR), the Funding Project RESISTOVAC (13GW0458D), and the Funding Program Photonics Research Germany (LPI-BT5 Leibniz IPHT 13N15717). It is integrated into the Leibniz Center for Photonics in Infection Research (LPI), which was initiated by Leibniz-IPHT, Leibniz-HKI, UKJ, and FSU Jena, and is part of the BMFTR National Roadmap for Research Infrastructures.
Acknowledgments
We gratefully acknowledge the dedicated support of the staff at fzmb GmbH and all members of the research group led by Ralf Ehricht for their valuable contributions to this work. Special thanks go to my wife, Christina Braun, for her unwavering support throughout the course of this project.
Conflict of interest
KF was employed by INTER-ARRAY Part of fzmb GmbH. TS, HA were employed by Institut Virion\Serion GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1650094/full#supplementary-material
Supplementary File 1 | Microarray spotting layout of monoclonal antibodies. This table provides the layout and positional arrangement of monoclonal antibodies (mAbs) spotted on the microarray surface used in the high-throughput screening assay. Each row represents a well position on the spotting platform, indicating the unique well ID, numerical spot identifier, antibody designation (if applicable), and the spotting concentration in μg/μL. The layout corresponds to the Resistovac array prototype (Version 5) and includes both experimental and control positions. This layout was used to generate the antibody microarrays evaluated in the study. Additionally, a representative microarray image acquired with the INTER-ARRAY reader is included. The image shows results from strain CAK240772 (E. coli, blaNDM-1), with marked spots highlighting both a weak signal (blaNDM-clone-71) and a strong signal (blaNDM-clone-24) targeting NDM.
Supplementary File 2 | Python script for statistical analysis of signal thresholds. This file contains the full Python script used to perform the statistical evaluation of antibody microarray performance across different signal intensity thresholds (>0.1, >0.2, >0.3, and >0.5). The script reads threshold-wise specificity, sensitivity, and accuracy values from a Tables–delimited input file and applies the Friedman test to detect global differences across thresholds. Pairwise comparisons between thresholds are conducted using the Wilcoxon signed-rank test to identify statistically significant differences. Output includes test statistics and p-values, which were used to support conclusions regarding optimal diagnostic thresholds in the main study.
Supplementary File 3 | Raw microarray signal data for all reference strains. This dataset contains the raw gray values obtained from the protein microarray experiments for all tested reference strains listed in Table 1. Each row corresponds to an individual microarray spot, detailing the capture antibody, spot ID, and signal intensity (gray value) measured across all biological replicates. The dataset includes both positive and negative control samples and serves as the primary source for calculating normalized intensities (NI) and diagnostic performance metrics (sensitivity, specificity, accuracy). These raw values underpin all subsequent statistical analyses and threshold evaluations described in the main text.
Supplementary File 4 | Resistance gene profiles of reference strains. This file contains the results of resistance gene identification using the ResFinder, Card and NCBI databases as implemented in the latest version of abricate. Whole-genome assemblies of all reference strains used in this study were screened to detect acquired antimicrobial resistance genes. For each isolate, detected resistance genes, sequence identity, coverage, and associated antibiotic classes are listed. These data provide a comprehensive overview of the genotypic resistance background of the strain panel used for assay development and validation.
References
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, 517–525. doi: 10.1093/nar/gkz935
Alvisi, G., Curtoni, A., Fonnesu, R., Piazza, A., Signoretto, C., Piccinini, G., et al. (2025). Epidemiology and genetic traits of carbapenemase-producing enterobacterales: A global threat to human health. Antibiotics 14:141. doi: 10.3390/antibiotics14020141
Banerjee, R., and Humphries, R. (2017). Clinical and laboratory considerations for the rapid detection of carbapenem-resistant Enterobacteriaceae. Virulence 8, 427–439. doi: 10.1080/21505594.2016.1185577
Boutal, H., Naas, T., Devilliers, K., Oueslati, S., Dortet, L., Bernabeu, S., et al. (2017). Development and validation of a lateral flow immunoassay for rapid detection of NDM-producing Enterobacteriaceae. J. Clin. Microbiol. 55, 2018–2029. doi: 10.1128/JCM.00248-17
Boutal, H., Vogel, A., Bernabeu, S., Devilliers, K., Creton, E., Cotellon, G., et al. (2018). A multiplex lateral flow immunoassay for the rapid identification of NDM-, KPC-, IMP- and VIM-type and OXA-48-like carbapenemase-producing Enterobacteriaceae. J. Antimicrob. Chemother. 73, 909–915. doi: 10.1093/jac/dkx521
Caliskan-Aydogan, O., and Alocilja, E. C. (2023). A review of carbapenem resistance in enterobacterales and its detection techniques. Microorganisms 11:1491. doi: 10.3390/microorganisms11061491
Chabi, M., Vu, B., Brosamer, K., Smith, M., Chavan, D., Conrad, J. C., et al. (2023). Smartphone-read phage lateral flow assay for point-of-care detection of infection. Analyst 148, 839–848. doi: 10.1039/d2an01499h
Decousser, J. W., Poirel, L., and Nordmann, P. (2017). Recent advances in biochemical and molecular diagnostics for the rapid detection of antibiotic-resistant Enterobacteriaceae: A focus on ss-lactam resistance. Expert Rev. Mol. Diagn. 17, 327–350. doi: 10.1080/14737159.2017.1289087
Feldgarden, M., Brover, V., Fedorov, B., Haft, D. H., Prasad, A. B., and Klimke, W. (2022). Curation of the AMRFinderPlus databases: Applications, functionality and impact. Microb. Genom 8:mgen000832. doi: 10.1099/mgen.0.000832
Garcia, B. H., Hargrave, A., Morgan, A., Kilmer, G., Hommema, E., Nahrahari, J., et al. (2007). Antibody microarray analysis of inflammatory mediator release by human leukemia T-cells and human non small cell lung cancer cells. J. Biomol. Tech. 18, 245–251.
Girlich, D., Poirel, L., and Nordmann, P. (2012). Value of the modified Hodge test for detection of emerging carbapenemases in Enterobacteriaceae. J. Clin. Microbiol. 50, 477–479. doi: 10.1128/JCM.05247-11
Govindaraj Vaithinathan, A., and Vanitha, A. (2018). WHO global priority pathogens list on antibiotic resistance: An urgent need for action to integrate One Health data. Perspect. Public Health 138, 87–88. doi: 10.1177/1757913917743881
Greissl, C., Saleh, A., and Hamprecht, A. (2019). Rapid detection of OXA-48-like, KPC, NDM, and VIM carbapenemases in Enterobacterales by a new multiplex immunochromatographic test. Eur. J. Clin. Microbiol. Infect. Dis. 38, 331–335. doi: 10.1007/s10096-018-3432-2
Gschwind, R., Ugarcina Perovic, S., Weiss, M., Petitjean, M., Lao, J., Coelho, L. P., et al. (2023). ResFinderFG v2.0: A database of antibiotic resistance genes obtained by functional metagenomics. Nucleic Acids Res. 51, W493–W500. doi: 10.1093/nar/gkad384
Haab, B. B., Dunham, M. J., and Brown, P. O. (2001). Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2:research0004. doi: 10.1186/gb-2001-2-2-research0004
Hamprecht, A., Vehreschild, J. J., Seifert, H., and Saleh, A. (2018). Rapid detection of NDM, KPC and OXA-48 carbapenemases directly from positive blood cultures using a new multiplex immunochromatographic assay. PLoS One 13:e0204157. doi: 10.1371/journal.pone.0204157
Hong, J., Kang, D., and Kim, D. (2021). Performance evaluation of the newly developed in vitro rapid diagnostic test for detecting OXA-48-Like, KPC-, NDM-, VIM- and IMP-type carbapenemases: The RESIST-5 O.K.N.V.I. multiplex lateral flow assay. Antibiotics 10:460. doi: 10.3390/antibiotics10040460
Kingsmore, S. F. (2006). Multiplexed protein measurement: Technologies and applications of protein and antibody arrays. Nat. Rev. Drug Discov. 5, 310–320. doi: 10.1038/nrd2006
Köhler, G., and Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497. doi: 10.1038/256495a0
Kon, H., Abramov, S., Frenk, S., Schwartz, D., Shalom, O., Adler, A., et al. (2021). Multiplex lateral flow immunochromatographic assay is an effective method to detect carbapenemases without risk of OXA-48-like cross reactivity. Ann. Clin. Microbiol. Antimicrobials 20:61. doi: 10.1186/s12941-021-00469-0
Liu, Y. Y., Wang, Y., Walsh, T. R., Yi, L. X., Zhang, R., Spencer, J., et al. (2016). Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168. doi: 10.1016/S1473-3099(15)00424-7
Murray, C. J. L., Ikuta, K. S., and Sharara, F. (2024). Global burden of bacterial antimicrobial resistance 1990-2021: A systematic analysis with forecasts to 2050. Lancet 404, 1199–1226. doi: 10.1016/S0140-6736(24)01867-1
Partridge, S. R., Kwong, S. M., Firth, N., and Jensen, S. O. (2018). Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31, 88–17. doi: 10.1128/CMR.00088-17
Perez, F., and Bonomo, R. A. (2019). Carbapenem-resistant Enterobacteriaceae: Global action required. Lancet Infect. Dis. 19, 561–562. doi: 10.1016/S1473-3099(19)30210-5
Rabaan, A. A., Eljaaly, K., Alhumaid, S., Albayat, H., Al-Adsani, W., Sabour, A. A., et al. (2022). An overview on phenotypic and genotypic characterisation of carbapenem-resistant enterobacterales. Medicina 58:1675. doi: 10.3390/medicina58111675
Rana, C., Vikas, V., Awasthi, S., Gautam, D., Vats, A., Rajput, S., et al. (2024). Antimicrobial resistance genes and associated mobile genetic elements in Escherichia coli from human, animal and environment. Chemosphere 369:143808. doi: 10.1016/j.chemosphere.2024.143808
Richter, S. S., and Marchaim, D. (2017). Screening for carbapenem-resistant Enterobacteriaceae: Who, When, and How? Virulence 8, 417–426. doi: 10.1080/21505594.2016.1255381
Sauer, U. (2017). Analytical protein microarrays: Advancements towards clinical applications. Sensors 17:256. doi: 10.3390/s17020256
Tacconelli, E., Carrara, E., Savoldi, A., Harbarth, S., Mendelson, M., Monnet, D. L., et al. (2018). Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. doi: 10.1016/S1473-3099(17)30753-3
van Duin, D., and Doi, Y. (2017). The global epidemiology of carbapenemase-producing Enterobacteriaceae. Virulence 8, 460–469. doi: 10.1080/21505594.2016.1222343
Wareham, D. W., Shah, R., Betts, J. W., Phee, L. M., and Momin, M. H. (2016). Evaluation of an immunochromatographic lateral flow assay (OXA-48 K-SeT) for rapid detection of OXA-48-like carbapenemases in Enterobacteriaceae. J. Clin. Microbiol. 54, 471–473. doi: 10.1128/JCM.02900-15
World-Health-Organisation. (2017). “Guidelines for the prevention and control of carbapenem-resistant Enterobacteriaceae, Acinetobacter baumannii and Pseudomonas aeruginosa in health care facilities,” in WHO guidelines approved by the guidelines review committee, (WHO: Geneva). Available online at: https://www.yunbaogao.cn/index/partFile/5/who/2022-04/5_26182.pdf
Keywords: carbapenemase, protein microarray, monoclonal antibody, antimicrobial resistance, lateral flow assay, high-throughput antibody screening, point-of-care diagnostics
Citation: Braun SD, Reinicke M, Diezel C, Müller E, Frankenfeld K, Schumacher T, Arends H, Monecke S and Ehricht R (2025) High-throughput screening of monoclonal antibodies against carbapenemases using a multiplex protein microarray platform. Front. Microbiol. 16:1650094. doi: 10.3389/fmicb.2025.1650094
Received: 19 June 2025; Accepted: 06 August 2025;
Published: 26 August 2025;
Corrected: 27 August 2025.
Edited by:
Artur J. Sabat, University Medical Center Groningen, NetherlandsReviewed by:
Jason L. Cantera, Global Health Labs, United StatesAndre Valencio Siqueira, Universidade Federal de Sao Paulo, Brazil
Kali Kishore Reddy Tetala, VIT University, India
Copyright © 2025 Braun, Reinicke, Diezel, Müller, Frankenfeld, Schumacher, Arends, Monecke and Ehricht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sascha D. Braun, c2FzY2hhLmJyYXVuQGxlaWJuaXotaXBodC5kZQ==