 Lingzhi Dong1
Lingzhi Dong1 Yu Du2
Yu Du2 Feifei Qiu1Meng Zhang3
Feifei Qiu1Meng Zhang3 Xiaoxia Wang1Xiaming Zhu4Yuntao Yao2Jinyi Li1
Xiaoxia Wang1Xiaming Zhu4Yuntao Yao2Jinyi Li1 Xiang Ji5*Xiong Zhu1*
Xiang Ji5*Xiong Zhu1*- 1Clinical and Central Laboratory of Sanya People’s Hospital, Sanya, China
- 2Hainan Key Laboratory for Herpetological Research, College of Fisheries and Life Sciences, Hainan Tropical Ocean University, Sanya, China
- 3Department of Radiology, Sanya People’s Hospital, Sanya, China
- 4Herpetological Research Center, College of Life and Environmental Sciences, Hangzhou Normal University, Hangzhou, China
- 5Zhejiang Provincial Key Laboratory for Water Environment and Marine Biological Resources Protection, College of Life and Environmental Sciences, Wenzhou University, Wenzhou, China
Introduction and methods: Sea turtles have been proposed as health indicators of marine ecosystems for their characteristic of longevity and migratory, but they are facing serious threats due to various factors. The microbial communities within animals play an important role in health and disease. Our study aims to explore a thorough evaluation of the sea turtle microbiome by examining the oral, nasal, and cloacal microbial communities of three species: green turtles, hawksbills, and loggerheads, through metagenomic sequencing.
Results: Utilizing approximately 705.81 GB of metagenomic sequencing data from 63 samples collected from different turtle species and tissue regions, we created a nonredundant sea turtle microbial gene catalog (STMGC) containing 10,733,232 unique genes through the de-redundancy of open reading frames (ORFs). Our findings revealed that the sea turtle microbiomes were primarily composed of Pseudomonadota (formerly Proteobacteria) and Bacteroidota (formerly Bacteroidetes). The tissue region was a key factor affecting the variability in the sea turtle microbiome, with green turtles showing notable differences among the three turtle species. Pseudomonadota was significantly more abundant in oral samples, while Bacteroidota was more prevalent in nasal samples. Campylobacterota was identified as significantly more abundant in cloacal samples. Importantly, we discovered 389 genera and 1,445 species of potential pathogens within the sea turtle microbiome, indicating potential pathogenic risks that warrant further investigation alongside culturomics. Additionally, our study highlighted significant functional differences among the three turtles and tissue regions. It is worth noting that among the three sea turtles, antibiotic resistance genes are more prevalent in hawksbills, while virulence genes are more abundant in loggerheads. Moreover, within the three tissue regions, antibiotic resistance genes are higher in oral samples, while virulence genes are more extensive in cloacal samples.
Conclusion: The findings in our study demonstrate that the microbial composition and function in these sea turtles exhibit both species-specific and region-specific variations. The implications of these associations and the underlying mechanisms not only provide valuable insights for future studies on the microbial communities of turtles, but also lay the foundation for further research on the health interrelationships among sea turtles, marine and terrestrial animals, humans and the environment, and for defining “One Health” factors.
Introduction
Sea turtles, a type of marine reptile, have complex life cycles as a long-lived species. As they grow and migrate across oceans, they experience significant changes in their habitats and diets, which could reflect the long-term effects of environmental changes and pollution (Drane et al., 2021). The characteristic makes them ideal biological indicators for monitoring environmental pollutants, and various biological samples such as blood, fat tissue, and eggs have been used for environmental pollutant monitoring (Fraga et al., 2018; Figgener et al., 2019). In addition, the position of sea turtles in the food chain and their extensive distribution in the marine environment enable them to accumulate and reflect heavy metals, organic pollutants, pathogenic infection, and antibiotic resistance (Kraemer et al., 2019; Al-Bahry et al., 2011; Al-Bahry et al., 2009; Conrad et al., 2023). Their migratory habits mean that they may act as a medium for the transmission of pathogens and resistance between different geographical regions, thereby affecting the extensive marine ecosystem. The detection of opportunistic pathogens and antibiotic resistance in sea turtles provides valuable information for the protection of sea turtles and the safety of marine environments (Ebani, 2023; Conrad et al., 2023; Ahasan et al., 2018; Al-Bahry et al., 2011).
Research increasingly shows that sea turtles are threatened by a variety of factors, such as consuming marine debris, the loss and pollution of nesting and feeding habitats due to urban development, predation on nests and hatchlings by both wildlife and domestic animals, collisions with vessels, traditional hunting and egg harvesting, the effects of climate change on marine and land environments, and entanglement in fishing gear (Stanford et al., 2020; Gong et al., 2017). These issues have led to a notable decline in sea turtle populations (Mazaris et al., 2017; Stanford et al., 2020; Mazaris et al., 2023; Rivas et al., 2023). According to the IUCN Red List classification and criteria guidelines,1 six of the seven sea turtle species are currently listed as vulnerable, endangered, or critically endangered, and five of which could found in China, including the green turtles (Chelonia mydas), hawksbills (Eretmochelys imbricata), loggerheads (Caretta caretta), olive ridleys (Lepidochelys olivacea), and leatherbacks (Dermochelys coriacea) (Chan et al., 2009). According to the research, approximately 90% of the sea turtles in China inhabit the South Sea, and the estimated proportion of each species is approximately as follows: 87% for green turtles, 10% for hawksbills, and the remaining 3% is the combined proportion of leatherback turtles, loggerhead turtles, and olive ridley turtles (Chan et al., 2009; Mou et al., 2013). To alleviate some of the current pressures on sea turtles, global conservation efforts for sea turtles have started to take shape (Mazaris et al., 2017). The China Sea Turtle Conservation Action Plan (2019–2033) outlines a strategy to boost wild populations through captive breeding, to speed up the recovery of sea turtle numbers. However, releasing captive-bred turtles requires careful planning, particularly should avoid the release of turtles carrying pathogens, which could potentially pose a threat to the health, behavior and genetic diversity of the wild population (Kock et al., 2010). Thus, analyzing the microbial composition of captive sea turtles and assessing the presence of pathogens is crucial for effective turtle conservation and management strategies.
Advanced genomic technologies and bioinformatics approaches have exponentially increased the diversity of known microbes in recent years (Wensel et al., 2022; Forde and O’Toole, 2013; Ursell et al., 2012). The microbial community associated with the animal host plays a critical role in host development, physiology, immune response, metabolism, and reproduction, and may have an impact on the evolutionary success of the host (McFall-Ngai et al., 2013; Henry et al., 2021). Previous sea turtle microbiota studies have focused on the gastrointestinal tract (Abdelrhman et al., 2016; Chen et al., 2022; Kittle et al., 2018; Campos et al., 2018; Bloodgood et al., 2020; Arizza et al., 2019), with a small portion of the microbiota research found in the cloacal, oral, and nasal (Santoro et al., 2006; Zavala-Norzagaray et al., 2015; McNally et al., 2021; Price et al., 2017; Ahasan et al., 2017) regions. Additionally, most current research on the sea turtle microbiome relies on culture methods and 16S rRNA high-throughput sequencing, which limits our understanding of the functional aspects of the turtle microbiome. However, sea turtles have been proposed as health indicators of marine habitats and carriers of antibiotic resistant, for their longevity and migratory lifestyle (Blasi et al., 2020; Alduina et al., 2020). The spread of antibiotic resistance genes (ARGs) not only poses a threat to the original microbial communities in the marine environment, but also directly poses a threat to marine organisms, as there are multiple antibiotic-resistant bacteria (MDR bacteria) in the marine environment, which can cause harm to marine organisms. Moreover, these multiple antibiotic-resistant bacteria may serve as a platform for transmitting antibiotic resistance genes from the environment to humans (Drane et al., 2021). Furthermore, microbial virulence factors encompass a wide range of molecules produced by pathogenic microorganisms, enhancing their ability to evade their host defenses and cause disease. A deep understanding of the biological characteristics of microbial pathogens and their pathogenic factors is crucial for developing new therapeutic molecules and strategies to combat microbial infections (Leitão, 2020).
Therefore, we performed a detailed analysis of the oral, nasal, and cloacal microbiota composition and functional characteristics in three species of sea turtles, including green turtles (Chelonia mydas), loggerheads (Caretta caretta), and hawksbills (Eretmochelys imbricata), using shotgun metagenomic sequencing. Our objectives are (1) to investigate the microbial composition and identify the potential pathogens; (2) to discover the functional genes including ARGs and virulence factors, aiming to provide basic knowledge for the work related to assessing the potential pathogenic risks of sea turtles, minimize interference with wild sea turtle populations, and protect the marine ecosystem, thereby further strengthening the efforts in sea turtle conservation.
Materials and methods
Sample source
All samples in our study were collected from the Hainan Province Sea Turtle Rescue and Conservation Center (18.5°N, 108.5°E), which is a provincial-level aquatic wildlife rescue institution established by the Hainan Provincial Department of Agriculture and Rural Affairs with the support of Hainan Tropical Marine University. It mainly receives live sea turtles confiscated by government-designated law enforcement departments. Since its establishment, it has received nearly 600 live sea turtles involved in cases from over a dozen provinces and cities across the country. At present, there are still 61 sea turtles remaining, which are mainly used for artificial breeding and scientific research. These turtles have been kept in captivity for 4 to 5 years. Most of them are in health status (i.e., without any disease symptoms or external injuries), and have not been treated with antibiotics. These turtles are separately placed in different ponds, with approximately two to three turtles in each pond. All the turtles share the circulating seawater. The main food of the turtles is fish, including squid and various other types of fish.
Sample collection and DNA extraction
The samples were collected from August 20th to 24th, 2024. A total of 61 sea turtles were collected, including 48 green turtles, ten hawksbills, and three loggerheads. We documented essential information, including biomarkers, sex, and injury status, straight carapace length/width, weight, and the time of arrival at the rescue center of the sea turtles (Supplementary Table S1). For each turtle, oral, cloacal, and nasal swabs were obtained, with three swabs collected from each region. Before sample collection, sterile water was used to continuously flush around the sampling site until no obvious contaminants were present. Oral swab samples were collected by gently rotating a sterile dry cotton or synthetic swab over the mucous membranes of the tongue and palate. Cloacal and nasal samples were collected by inserting a swab approximately 10 cm into the respective cavity, applying gentle pressure in a circular motion, and rubbing the inner circumference two to three times (Ahasan et al., 2018; McNally et al., 2021; Trotta et al., 2021). The swab head was then broken off and placed in a sterile 5 mL. After collection, samples were immediately frozen using liquid nitrogen, transported to the laboratory, and stored at − 80 °C until DNA extraction.
Genomic DNA was extracted using the Qiagen AllPrep DNA/RNA Mini Kit (Qiagen, Germany) following the manufacturer’s guidelines. The concentration and quality of all DNA samples were evaluated using the NanoDrop-1000 spectrophotometer and agarose gel electrophoresis. To improve both the concentration and quality of the DNA, samples extracted from the same anatomical regions of two to four turtles were pooled, resulting in a total of 63 DNA samples prepared for subsequent sequencing. This mixing strategy was based on the premise that DNA samples are pooled from the same species of sea turtles, from the same anatomical region, of the same gender or with the same injury condition.
Shotgun metagenomic sequencing and assembly
Genomic DNA was subjected to random fragmentation into segments of approximately 350 base pairs using a Covaris ultrasonic disruptor for library construction. Subsequently, DNA libraries were constructed, purified, analyzed, and quantified, followed by sequencing on an Illumina PE150 platform at Novogene in Beijing, China. The raw data generated from the Illumina sequencing platform were processed using Fastp2 to eliminate paired reads that exhibited adapter contamination, contained more than 10% ambiguous nucleotides, or had over a specified percentage of low-quality nucleotides (with base quality scores below 5). Bowtie2 software3 was employed with default parameters set to --end-to-end, --sensitive, -I 200, and -X 400 to filter out reads potentially originating from the host. For assembly analysis of the cleaned data, MEGAHIT software was utilized with assembly parameters set to --presets meta-large (--end-to-end, --sensitive, -I 200, -X 400) (Li et al., 2015). Scaffolds devoid of N were generated by segmenting the resulting scaffolds at the N junction.
Gene prediction and taxonomy annotation
MetaGeneMark4 was employed to predict open reading frames (ORFs) for shafts of at least 500 base pairs from each sample, followed by filtering to exclude any predicted sequences shorter than 100 nucleotides. The CD-HIT software (Li and Godzik, 2006; Fu et al., 2012) was utilized to remove redundancy and generate a non-redundant initial gene catalog, defined as the nucleic acid sequences encoded by successive non-redundant genes. The following parameter settings were applied: -c 0.95, -G 0, -a 0.9, -g 1, -d 0. Clean data from each sample was then aligned to the initial gene catalog using Bowtie2 to quantify the number of reads corresponding to each gene in the sample alignment, with parameter settings of --end-to-end, --sensitive, -I 200, -x 400. Genes with read counts of two or fewer in each sample were excluded, resulting in a refined gene catalog (Unigenes) for further analysis.
DIAMOND software5 (Buchfink et al., 2015) was utilized to align unigene sequences with the Micro_NR database, which comprises sequences from bacteria, fungi, archaea, and viruses extracted from NCBI NR database.6 The alignment was conducted using the Blastp algorithm with a parameter setting of 1e-5. From the alignment results for each sequence, the one with a value <= min value *10 was selected. Since each sequence may yield multiple alignment results, the Lowest Common Ancestor (LCA) algorithm, implemented in the MEGAN software (Huson et al., 2007), was employed to determine the species annotation information for each sequence. Based on the results of the LCA annotation and the gene abundance table, the abundance of each sample at each taxonomic level, along with the corresponding gene abundance tables, was obtained.
In addition, it should be noted in advance that in 2021, the International Committee on Prokaryotic Taxonomy changed the classification names of 42 Prokaryotic phyla to be consistent with the International Nomenclature for Prokaryotes (Oren and Garrity, 2021). The new names are used for the phyla within this paper, such as Pseudomonadota (formerly Proteobacteria), Bacteroidota (formerly Bacteroidetes), Bacillota (formerly Firmicutes), Actinomycetota (formerly Actinobacteria), Fusobacteriota (formerly Fusobacteria), and Spirochaetota (formerly Spirochaetes).
Functional annotation
DIAMOND software was utilized to align unigenes with entries in the functional database, using the following parameter settings: blast, -e 1e-5. The functional databases included the Kyoto Encyclopedia of Genes and Genomes (KEGG7) (Kanehisa et al., 2017), Nonsupervised Orthologous Groups (eggNOG8) (Huerta-Cepas et al., 2016), Carbohydrate-Active enZYmes (CAZy9) (Cantarel et al., 2009), and the Virulence Factor Databases (VFDB10). Based on the alignment results, the relative abundance at various functional levels was calculated (the relative abundance at each functional level is defined as the sum of the relative abundance of genes annotated at that functional level). Furthermore, unigenes were aligned with the Comprehensive Antibiotic Resistance Database (CARD11) (Jia et al., 2017) using the Resistance Gene Identifier (RGI) software (RGI built-in blast, default value < 1e-30) (McArthur et al., 2013). Utilizing the RGI alignment result and unigenes abundance information, the relative abundance of each Antimicrobial Resistance Ontology (ARO) was determined.
Statistical analysis
Based on the computations outlined above, we obtained several datasets, including an overview gene catalog and a table detailing microbiome composition and functional abundance at each taxonomic level. Utilizing these data, we analyzed the diversity and differences in microbiome composition and function across various samples, turtle species (green turtles, hawksbills, and loggerheads), and tissue region (oral, nasal, and cloacal). We used two alpha diversity indexes (Chao 1 and Shannon diversity index) to assess the community richness and diversity. The Wilcoxon rank sum test was used to examine the pairwise differences among three turtle species and tissue regions, respectively. We used principal coordinate analysis (PCoA) based on Bray–Curtis distance to visualize sample clustering patterns. Furthermore, we used the adonis function in vegan package to perform the permutational multivariate analysis of variance (PERMANOVA, permutation = 9,999) to examine the effects of host species and tissue regions on microbiota. Additionally, we performed the ANOSIM analysis to examine the differences between and within groups. The Kruskal-Wallis test was used to detect taxa and function with differences in abundance among different groups. The linear discriminant analysis (LDA) effect size (LEfSe) (Segata et al., 2011) was performed to obtain the biomarker bacterial with LDA > 3, p < 0.05 in each host species and tissue regions. And we used statistical analysis of metagenomic profiles (STAMP) (Parks et al., 2014) to visualize the top 20 differential functions among different groups. Furthermore, we analyzed the pathogens associated with these marine reptiles to evaluate the potential pathogenic risks and enhance the survival rates of sea turtles. We utilized the human pathogen lists (Bartlett et al., 2022), which enumerates 1,513 bacterial pathogens known to affect humans before 2021, comprising 1,100 established and 403 putative pathogens. These pathogens were classified into 327 genera. Additionally, we incorporated data from the BacDive database,12 encompassing 2,292 bacterial strain pathogens affecting animals, 1,611 affecting humans, and 384 affecting plants, linked to 809 species and 210 genera. By integrating these two data sources, we identified a total of 412 pathogen genera and 2,134 pathogen species.
Results
Sea turtle microbiome gene catalog constructed using metagenomic analysis
The shotgun metagenomic sequencing produced a total of 705.81 gigabytes (GB) of raw Illumina data from 63 genomic DNA. After quality control, 697.98 Gb of clean, high-quality data remained, achieving an effective quality control rate of about 98.89% (Supplementary Table S2). The metagenomic assembly and open reading frame (ORF) prediction yielded 24.50 million contigs and 43.37 million ORFs (Supplementary Table S2). Ultimately, a nonredundant sea turtle microbial gene catalog (STMGC) was created, containing 10,733,232 unique genes after de-redundancy of the ORFs. To assess the validity and completeness of the identified genes, a rarefaction analysis was conducted with 50 random samples, and the core-pan gene rarefaction curve approached a plateau (Supplementary Figures S1A,B). A heatmap was generated to analyze the correlations between samples based on the gene table (Supplementary Figure S1C). A box plot illustrating the differences in gene numbers among groups revealed that the oral region had the highest gene counts, followed by the nasal and the cloacal across three turtle species (Supplementary Figure S1D).
Overview of the sea turtle microbiome taxonomic composition
A total of 9,067,768 (84.48%) nonredundant genes were annotated in the NCBI NR database (Supplementary Table S3). As shown in Figure 1A, from kingdom to species level, there are, respectively, annotated in these NR genes at 77.36% (7,015,215), 71.14% (6,450,983), 61.41% (5,568,232), 54.16% (4,910,861), 48.40% (4,388,677), 42.03% (3,811,134), and 46.11% (4,181,434). At the kingdom level, 6,883,406 (98.12%) genes were identified as bacteria, while eukaryotes accounted for only 80,269 genes (1.14%), viruses for 29,726 genes (0.42%), and archaea for 21,814 genes (0.31%) (Figure 1B). Our research primarily concentrated on the bacterial component, which constituted 98.12% of the microbial community. For the bacterial, we identified 165 phyla (including 45 non-Candidatus and 120 Candidatus phyla, total 6,352,686 genes), 145 classes (5,486,773 genes), 315 orders (4,858,058 genes), 782 families (4,337,212 genes), 3,922 genera (3,758,642 genes), and 36,709 species (4,107,141 genes) of microorganisms (Figure 1A). At the phylum level, the microbiome of sea turtles was predominantly composed of Pseudomonadota (formerly Proteobacteria, 44.05%) and Bacteroidota (formerly Bacteroidetes, 26.40%), which together made up 70.45% of the total. Other prevalent phyla included Myxococcota (5.51%), Campylobacterota (3.28%), Actinomycetota (formerly Actinobacteria, 2.95%), Bacillota (formerly Firmicutes, 2.59%), Thermodesulfobacteriota (2.27%), Planctomycetota (2.25%), Chloroflexota (1.65%), and Acidobacteriota (1.63%) (Figure 1C, phylum). At the genus level, the most ten abundant bacteria were Tenacibaculum (Flavobacteriaceae of Bacteroidota), Marinicella (Alcanivoracaceae of Campylobacterota), Aliiroseovarius (Paracoccaceae of Pseudomonadota), Aquimarina (Flavobacteriaceae of Bacteroidota), Ruegeria (Roseobacteraceae of Pseudomonadota), Vibrio (Vibrionaceae of Pseudomonadota), Aureispira (Saprospiraceae of Bacteroidota), Psychrobacter (Moraxellaceae of Pseudomonadota), Arcobacter (Arcobacteraceae of Campylobacterota), and Paracoccus (Paracoccaceae of Pseudomonadota), collectively representing 16.29% of the total counts (Figure 1C, genus). Additionally, we examined the microbial composition at various taxonomic levels across all samples, finding that Pseudomonadota and Bacteroidota were the most prevalent phyla, while the distribution of other phyla varied in different samples (Supplementary Figure S2A). These differences were also apparent at other taxonomic levels, suggesting that microbiomes may vary among different turtle species and tissue regions (Supplementary Figure S2).
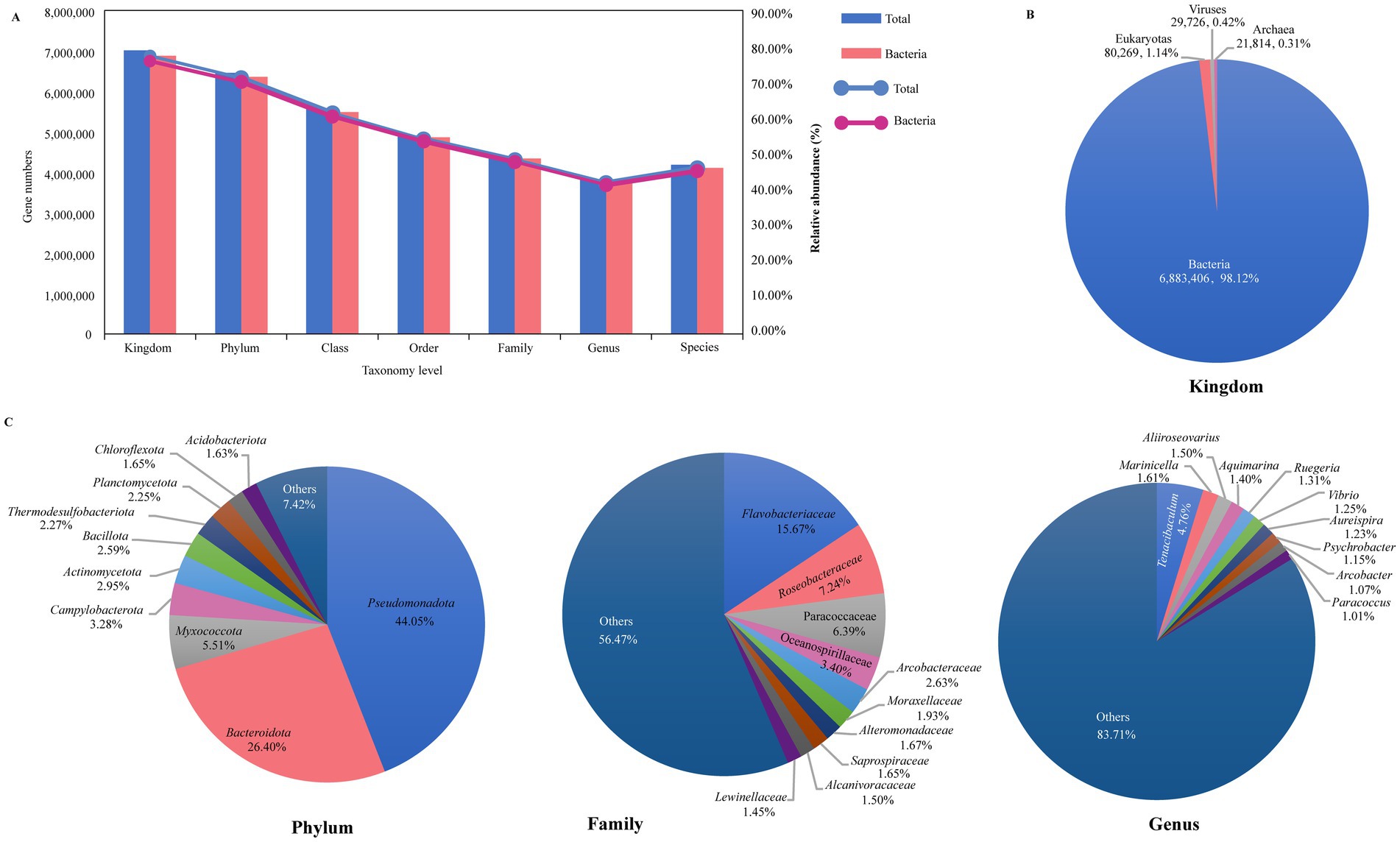
Figure 1. Overview of the sea turtle microbiome taxonomic composition. (A) The gene counts and relative abundance of different taxonomic levels (from kingdom to species level). Blue represents total taxonomic taxa, and pink represents bacterial taxa. (B) The relative abundance of bacteria, eukaryotes, viruses, and archaea. (C) The relative abundance of the top 10 bacteria at phylum, family, and genus level.
Diversity analysis of the sea turtle microbiome
The alpha diversity analysis showed that there were no significant differences in loggerheads and green turtles using Chao 1 and Shannon index, but a significant difference was noted between loggerheads and hawksbills at the phylum and order levels when using the Shannon index (Figures 2A,B; Supplementary Figures S3A,B). Furthermore, significant differences were observed between green turtles and hawksbills at the phylum level when using the Chao 1 index (Figure 2A). In terms of tissue region, the Wilcoxon test applied to the Chao 1 index showed significant differences across all taxonomic levels at the three tissue regions (Figure 2C; Supplementary Figure S3C). Additionally, significant differences in the Shannon index between oral and cloacal samples were found at all taxonomic levels (Figure 2D; Supplementary Figure S3D).
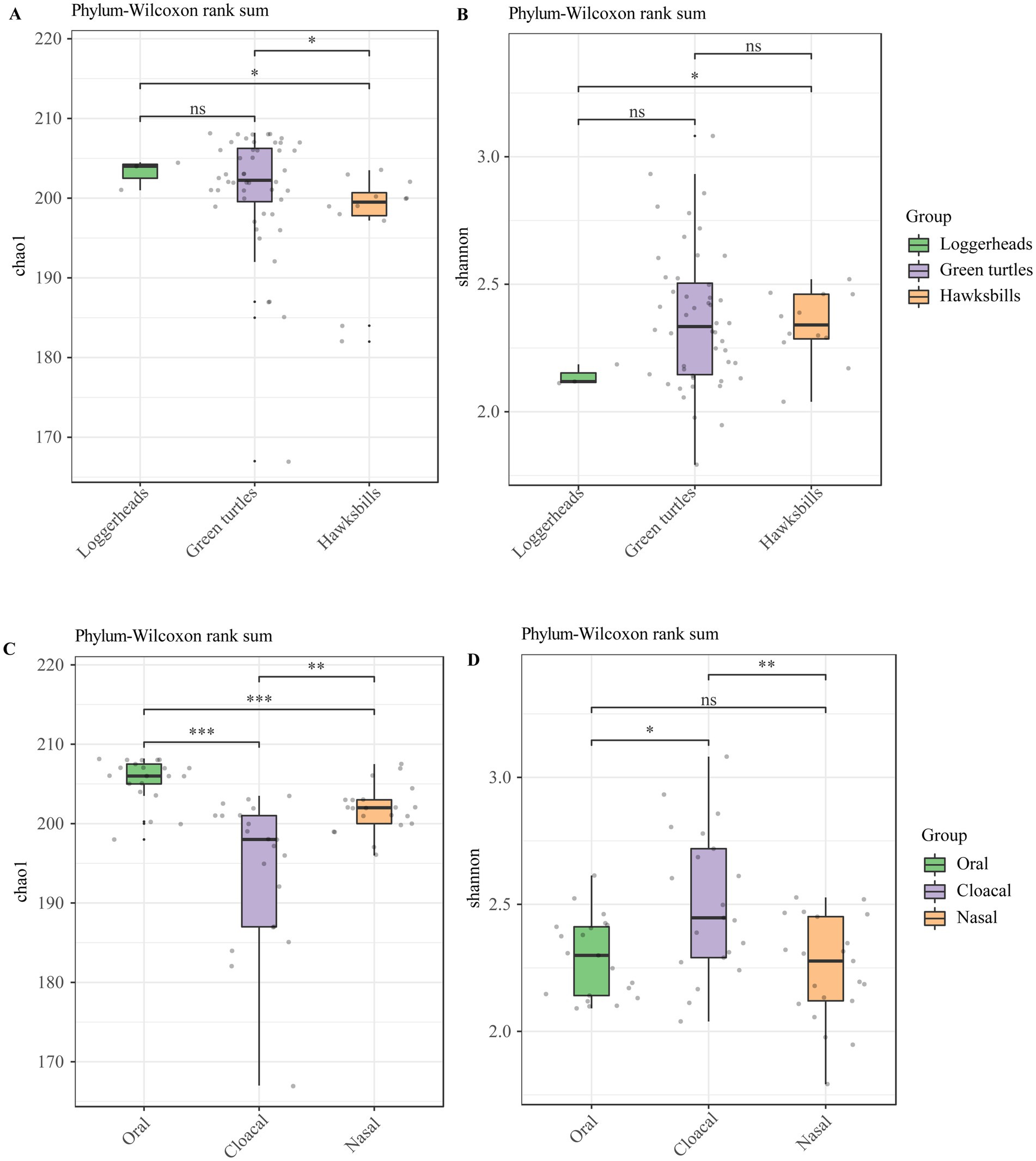
Figure 2. The alpha diversity analysis of the sea turtle bacterial community at the phylum level based on host species and tissue region. (A,B) The Chao 1 index and Shannon diversity index across three host species. (C,D) The Chao 1 index and Shannon diversity index across three tissue regions. p-values indicate the confidence level of statistical analyses, with p < 0.05 indicating statistically significant differences. ns represents no significant differences, * represents p < 0.05, **represents p < 0.01, and ***represents p < 0.001.
Principal coordinates analysis (PCoA) clearly distinguished tissue regions, while the differences among host species were less marked (Figures 3A,C; Supplementary Figures S4A,B). PERMANOVA analysis revealed significant statistical differences in microbial profiles across the three species and tissue regions at the phylum level, accounting for 6.8% (R2 = 0.068, p = 0.047) and 44.6% (R2 = 0.446, p = 0.001) of the variation, respectively (Figures 3A,C). This pattern was also evident at other taxonomic levels (Supplementary Figures S4A,B). Pairwise comparisons of the three sea turtles indicated that the differences between loggerheads and hawksbills were not statistically significant at any taxonomic level, while significant differences were found between loggerheads and green turtles at the order and species levels (Supplementary Table S4). Additionally, significant differences were noted between green turtles and hawksbills from the order to species levels. In the analysis of tissue region, all comparisons from phylum to species levels showed significant differences (Supplementary Table S4). ANOSIM analysis further confirmed significant differences among the three tissue regions at all taxonomic levels (Figures 3B,D; Supplementary Figures S4C,D). When comparing same tissue region from different species, PCoA results revealed that the same tissue regions of different turtle species always clustered together, and the cloacal was clearly separated from the oral and nasal regions (Supplementary Figure S5). Pairwise comparisons showed that there were significant differences in the same tissue regions between green turtles and hawksbill from class to species levels (Supplementary Table S4).
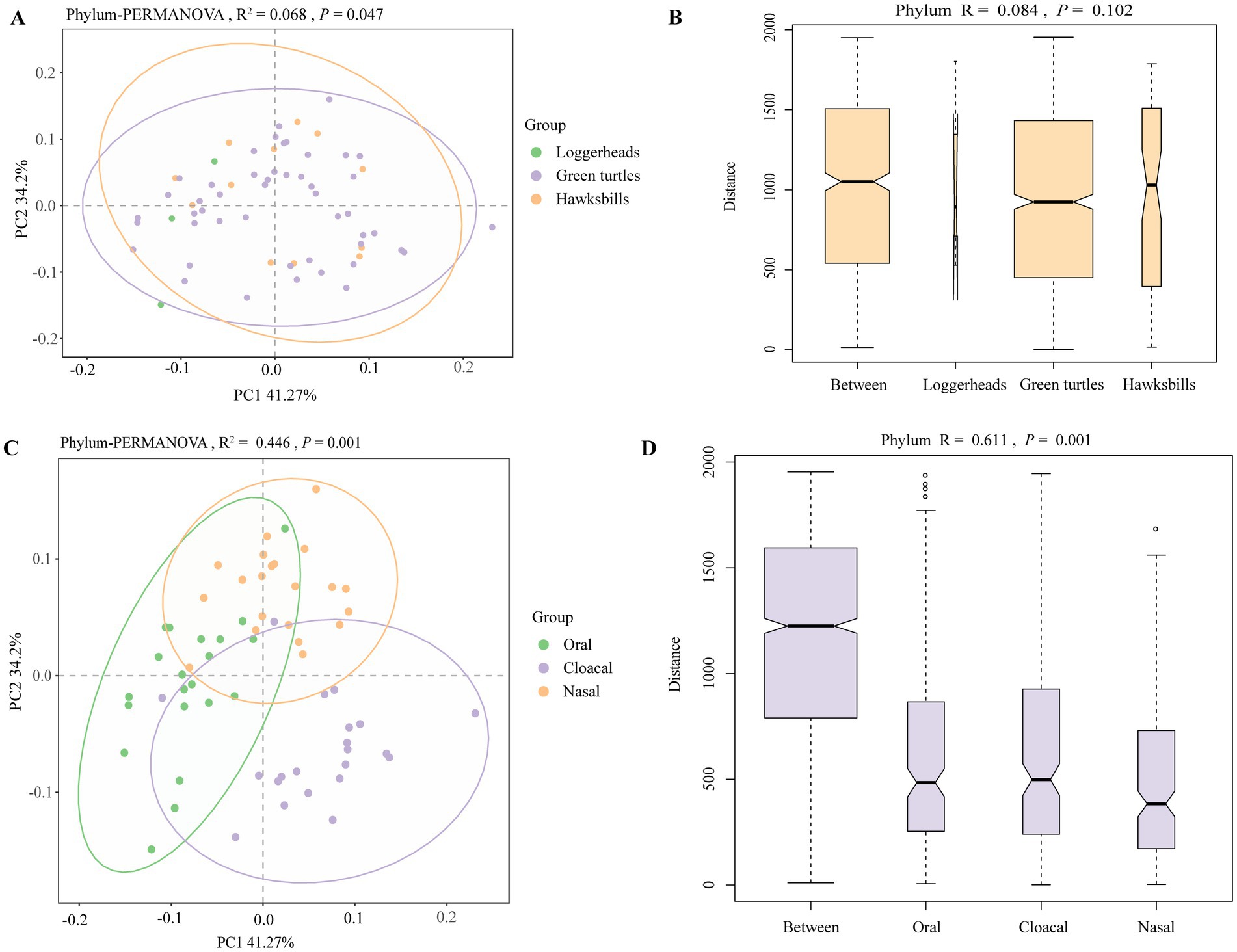
Figure 3. The beta diversity analysis of the sea turtle bacterial community at the phylum level based on host species and tissue region. (A,C) The PCoA results of bacteria at the phylum level based on three turtle species and tissue regions. (B,D) ANOSIM results of bacteria at the phylum level based on three turtle species and tissue regions.
Major differences in the sea turtle microbiome based on host species and tissue region
To investigate the major differences in the sea turtle microbiome, we conducted correlation analyses referring to the host species and tissue region, respectively. The comparative analysis of bacterial composition across various taxonomic levels highlighted both similarities and differences among host species and tissue regions, respectively (Supplementary Figures S6A,B). Within the three host species, green turtles had more unique bacterial species. In three tissue regions, oral samples contained a greater number of distinct species (Supplementary Figures S6C,D).
We identified a total of 54 bacterial taxa that were differentially abundant across the three host species by linear discriminant analysis (Figures 4A–E). Notably, Deinococcota and Actinomycetota were more prevalent in hawksbills, while Thermodesulfobacteriota and Chloroflexota were more abundant in green turtles and loggerheads, respectively, at the phylum level (LDA > 3, p < 0.05; Figure 4A). At the class level, the most differentially abundant taxa in hawksbills were identified as Deinococci and Actinomycetes, whereas Bacteroidia and Thermodesulfobiia were predominant in green turtles, and Flavobacteriia, Acidimicrobiia, and Desulfobacteria were more abundant in loggerheads (Figure 4B). At the order level, hawksbills exhibited six significantly abundant bacterial orders, whereas green turtles and loggerheads displayed three each (Figure 4C). The number of significantly abundant families varied, with five in hawksbills, four in green turtles, and six in loggerheads (Figure 4D). The significantly abundant genera in hawksbills included Deinococcus within the Deinococcota and Kangiella within the Pseudomonadota. The significantly abundant genera comprised Balneicella, Psychrobacter, Campylobacter, Terasakiispira, and Paracoccus in green turtles, while loggerhead exhibited Lutibacter, Arcobacter, and five more (Figure 4E).
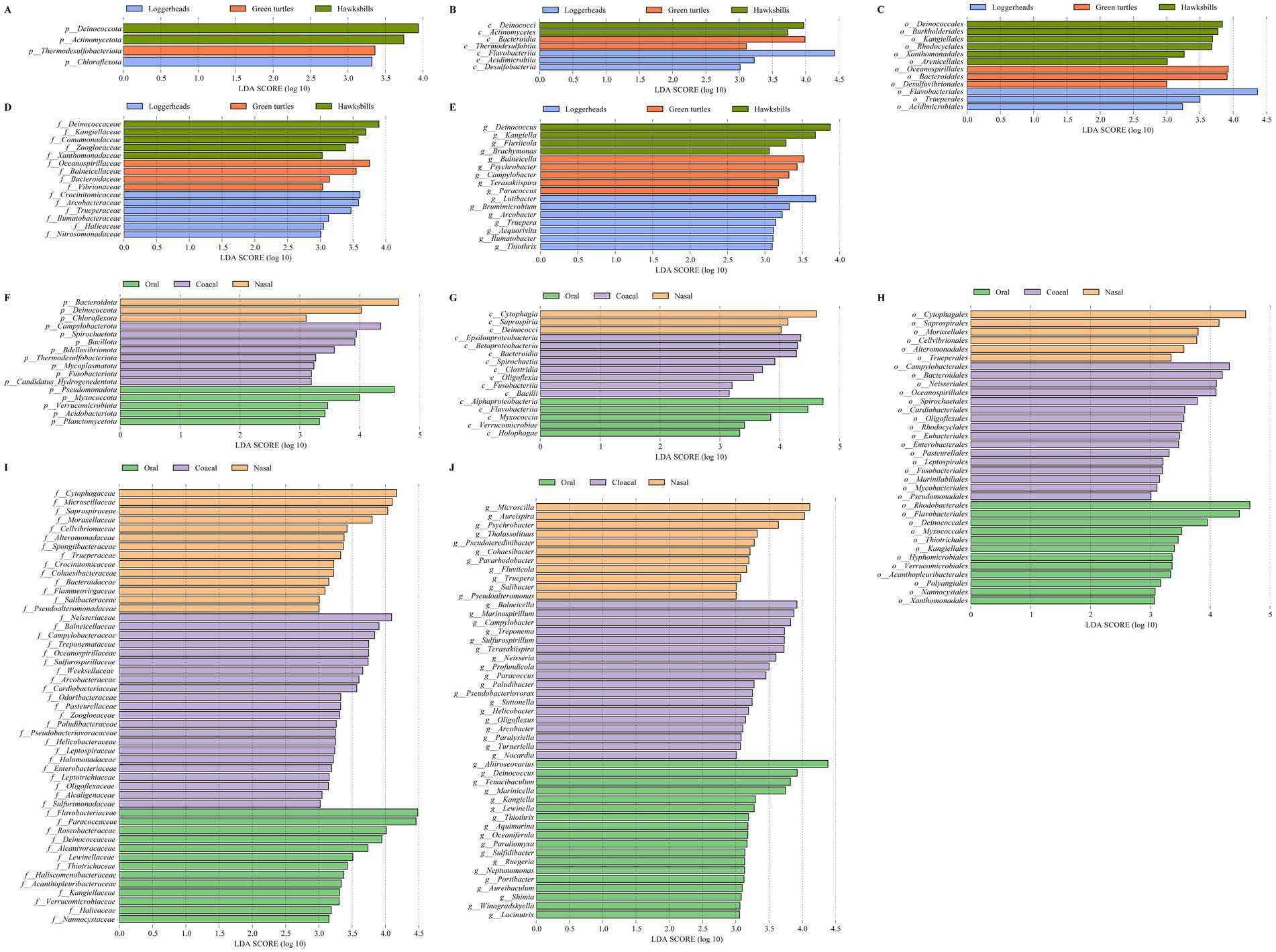
Figure 4. The LEfSe analysis of the sea turtle bacterial community based on turtle species and tissue region. (A–E) The differential bacteria from phylum to genus level among the three turtle species. (F–J) The differential bacteria from phylum to genus level among the three tissue regions.
In the analysis of tissue region, we identified 16, 16, 34, 49, and 47 differentially abundant bacterial taxa from phylum to genus level (LDA > 3). Cloacal samples had the highest number of differentially abundant bacteria (72), followed by oral samples (53) and nasal samples (37) (Figures 4F–J). Pseudomonadota was significantly more abundant in oral samples (LDA = 4.58), and Bacteroidota were present a greater abundance in nasal samples (LDA = 4.65) (Figure 4F). Additionally, Myxococcota, Verrucomicrobiota, Acidobacteriota, and Planctomycetota were found to be more abundant in oral samples, while Deinococcota and Chloroflexota were significantly abundant in nasal samples. In cloacal samples, Campylobacterota was identified as the most significantly abundant phylum (LDA = 4.35), followed by Spirochaetota, Bacillota, Bdellovibrionota, Thermodesulfobacteriota, Mycoplasmatota, Fusobacteriota, and Candidatus Hydrogenedentota (Figure 4F). The differential patterns observed at the phylum level were consistent across other taxonomic levels. For instance, differentially abundant bacteria in oral samples included Alphaproteobacteria, five orders (Rhodobacterales, LDA > 4), six families (Paracoccaceae, Roseobacteraceae, LDA > 4), and seven genera (Aliiroseovarius, LDA > 4) within Pseudomonadota, as well as Myxococcia, three orders, Nannocystaceae, and Paraliomyxa in Myxococcota, along with various taxa in Verrucomicrobiota and Acidobacteriota. The nasal samples contained two classes, two orders, seven families, and four genera within Bacteroidota. The cloacal samples included Epsilonproteobacteria, Campylobacterales, five families, and four genera (Campylobacter, Arcobacter) within Campylobacterota, along with seven bacteria in Spirochaetota, 13 bacteria in Bacillota, and six bacteria in Bdellovibrionota (Figures 4G–J).
Potential pathogenic bacteria detected in the sea turtle microbiome
Our investigation ultimately identified 389 pathogen genera and 1,445 pathogen species, with total gene counts of 25.24% and 3.85%, respectively (Supplementary Table S5). Notably, 11 genera exhibited gene counts exceeding 0.5%, including Tenacibaculum, Aliiroseovarius, Aquimarina, Vibrio, Psychrobacter, Arcobacter, Paracoccus, Flavobacterium, Pseudoalteromonas, Pseudomonas, and Campylobacter (Figure 5A). Additionally, 17 species demonstrated gene counts greater than 0.05%, such as Helicobacter pylori, Escherichia coli, Nocardia nova, Tenacibaculum maritimum, Klebsiella pneumoniae, Ornithobacterium rhinotracheale, Alcaligenes faecalis, Morganella morganii, Pasteurella skyensis, Edwardsiella tarda, Vibrio parahaemolyticus, Aliiroseovarius crassostreae, Pseudomonas aeruginosa, Sneathia sanguinegens, Salmonella enterica, Shewanella algae, and Listeria monocytogenes (Figure 5B).
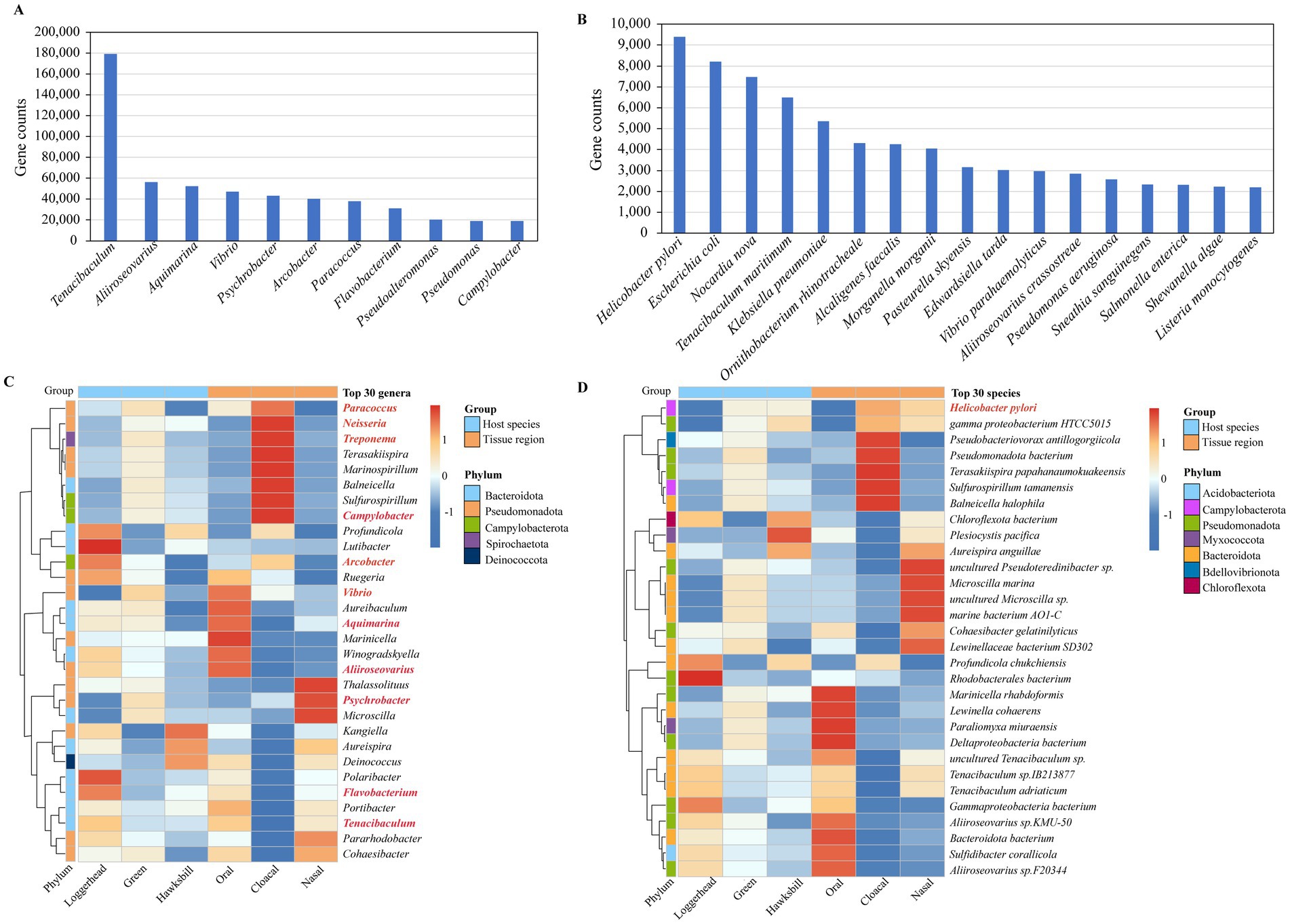
Figure 5. The potential pathogens detected in sea turtle microbiomes. (A) The potential pathogenic genera with gene counts exceeding 0.5%. (B) The potential pathogenic species with gene counts greater than 0.05%. (C) The pathogenic genera detected in the top 30 genera. The red mark is a pathogenic genus. (D) The pathogenic species detected in the top 30 species. The red mark is a pathogenic species.
Among the top 30 genera identified across all samples, 11 were classified as pathogenic bacterial genera, as indicated in red in Figure 5C. Of these 11 pathogen genera, with three turtles, Paracoccus, Treponema, Campylobacter, Vibrio, and Psychrobacter were found to be more abundant in green turtles, while Arcobacter, Aliiroseovarius, Flavobacterium, and Tenacibaculum were more prevalent in loggerheads. With three tissue regions, Paracoccus, Neisseria, Treponema, Campylobacter, and Arcobacter were predominantly detected in cloacal samples, while Vibrio, Aquimarina, and Aliiroseovarius were more abundant in oral samples, and Psychrobacter was more abundant in nasal samples (Figure 5C). Among the top 30 species identified in all samples, only H. pylori was identified as a pathogenic bacterium. Among the three turtles, it is mainly found in green turtles and hawksbills, while in the three tissue regions, it is mainly present in the cloacal samples, followed by nasal samples (Figure 5D).
Functional characteristics of the sea turtle microbiome
The functional gene annotation results showed that the KEGG, eggNOG and CAZY databases, respectively, annotated 29.35% (3,150,254), 76.11% (8,169,033) and 3.73% (400,661) of the non-redundant genes. Through the CARD and VFDB databases, 1,904 and 344,839 genes were annotated, and 157 antibiotic resistance genes (ARGs) and 697 virulence genes were discovered (Supplementary Table S3; Supplementary Figure S7A).
An analysis of KEGG metabolic pathways revealed that microbial functions were predominantly linked to metabolic processes, with a particular emphasis on “Amino acid metabolism” and “Carbohydrate metabolism” (Supplementary Figure S7B). In the Clusters of Orthologous Groups (COG) database, “Amino acid transport and metabolism” emerged as the most enriched functional category, aside from the “Function unknown” category (Supplementary Figure S7C). In the CAZY database, the “Glycosyl Transferases” category was found to be significantly enriched (Supplementary Figure S7D). Furthermore, the CARD and VFDB databases identified “Antibiotic Efflux” and “Immune Modulation” as enriched functions, respectively (Supplementary Figures S7E,F). To illustrate the overall distribution of functions across different host species and tissue region, the top 20 most abundant results were selected and represented in bar plots (Supplementary Figures S8A–H).
Additionally, we employed STAMP analysis to visualize the top 20 differential functions of KO pathways, antibiotic resistance genes (ARGs), and virulence factors across three host species and tissue regions (Figure 6). Across three host species, a total of 17 KO pathways exhibited significantly differential abundance, with seven of these pathways ranking among the top 20 most abundant. Notably, functions related to metabolism, such as “metabolism of terpenoids and polyketides,” “Metabolism of cofactors and vitamins,” “Metabolism of other amino acids,” “Amino acid metabolism,” and “Lipid metabolism” were found to be more abundant in loggerheads (Figure 6A). In comparison to tissue region, eight of the top 20 abundant KOs functions demonstrated significantly differential abundance, with “Cell motility,” “Membrane transport,” “Glycan biosynthesis and metabolism,” “Infection disease: bacterial,” “Translation,” and “Drug resistance: antimicrobial” being significantly more prevalent in cloacal samples (Figure 6B).

Figure 6. The differential functions of the microbiome across three turtle species and tissue regions by STAMP analysis. (A,B) The top 20 differential functions of KO pathways across three turtle species and tissue regions, respectively. (C,D) The top 20 differential ARGs across turtle species and tissue regions. (E,F) The top 20 differential virulence factors across turtle species and tissue regions.
Furthermore, we analyzed the differences in ARGs and virulence factors across three host species and tissue regions. Notably, 55 ARGs and 535 virulence factors were identified as common among the three host species, while 81 ARGs and 597 virulence factors were shared across the three tissue regions (Supplementary Figures S8I–L). The green turtles and cloacal samples contained more unique ARGs and virulence genes. The top 20 abundant ARGs and virulence factors were consistently found across all samples, and the top 20 ARGs exhibited higher abundance in hawksbills and oral samples (Supplementary Figures S8E,F), whereas the top 20 virulence factors were more abundant in loggerheads and cloacal samples (Supplementary Figures S8G,H). Of the top 20 differential abundance ARGs identified in three host species, 12 ARGs were common across all host species but most of them had a lower abundance in green turtles, while eight ARGs were not detected in loggerheads, including the vanX gene in the vanA cluster, TMB-1, acrD, MexR, sdiA, mdsA, E. coli AcrAB-TolC with MarR mutations conferring resistance to ciprofloxacin and tetracycline, and E. coli soxR with mutation conferring antibiotic resistance (Figure 6C). When comparing the three tissue regions, 14 ARGs were common in three tissue regions and most of them had lower abundance in cloacal samples, while six ARGs were not detected in nasal samples, including TriA, MexB, MexJ, MexD, MexD, and basS (Figure 6D). In an analysis of virulence factors, 14 of the top 20 differential abundance virulence factors were shared among three host species, and six were not detected in green turtles, such as LenF, Per, IcsA (VirG), Gelatinase, Ebp pili, and ESX-2 (T7SS) (Figure 6E). In comparing across tissue region, 11 virulence factors were significantly more abundant in cloacal samples, such as Pseudomonine, Petrobactin, Fur, O-linked flagellar glycosylation, Glutamine synthesis, CheA / CheY, Siderophore-dependent iron uptake system, Ton system, ComE1, IlpA, and VctPDGC system. Conversely, eight virulence factors were found to be more abundant in oral samples, including Xps, BvrR-BvrS, PDH-B, Mrx fimbriae, cytolysin, PlaB, Beta-haemolysin/cytolysin, and GPL locus (Figure 6F).
Discussion
Sea turtles are regarded as “flagship” and “umbrella” species in marine ecosystems, appreciated for their ecological importance and vital role in preserving biodiversity and assessing ecosystem health (Senko et al., 2022; Lovich et al., 2018; Bouchard and Bjorndal, 2000), but they are seriously endangered due to factors mainly related to human activities and climate change such as pollution, temperature increase, and predation (Gong et al., 2017; Stanford et al., 2020). Infectious and parasitic diseases may contribute to reducing the number of sea turtles. Bacteria are widespread in marine environments, and depending on the species, may act as primary or opportunistic pathogens. Most of them can infect other animal species, including humans, and may cause mild or severe diseases. Therefore, any direct or indirect contact between humans and sea turtles, their products, and environment where they live represent a One Health threat (Mashkour et al., 2020). Our study is the first to combine the oral, nasal, and cloacal of green turtles, loggerheads, and hawksbills for comparative analysis. The objective is to investigate the oral, nasal, and cloacal microbiota composition and functional characteristics of three threatened sea turtles, intending to gain knowledge and understanding to improve husbandry in rehabilitation settings, improve treatment and prevention of disease processes, and increase the survival rate of sea turtles. In addition, we hope to identify the microorganisms that could serve as potential human pathogens, such as multidrug-resistant bacteria, to provide theoretical knowledge, support for potential turtle and human health, disease prevention and treatment, and promote future conservation efforts.
Our results suggested that the Pseudomonadota was the most abundant phylum across all samples. There was no significant difference in its abundance among the three species, but in the three tissue regions, it was significantly higher in oral samples. In 2020, Scheelings et al. have reported microbiota data obtained from wild nesting females of all seven extant sea turtle species via distal colon swab, and their results showed that the Proteobacteria was the predominant bacterial phylum across all sea turtle species, which is consistent with our results (Scheelings et al., 2020). Some previous studies have also reported that Proteobacteria was the dominant phylum in stranded sea turtles, such as loggerheads of Tuscany and Liguria regions and green turtles of Great Barrier Reef regions (Abdelrhman et al., 2016; Ahasan et al., 2017; Ahasan et al., 2018). Additionally, Campos et al. and Samuelson et al. noted that a significant increase in the abundance of Proteobacteria was associated with prolonged captivity and treatment with antibiotics (Campos et al., 2018; Samuelson et al., 2020). Campos et al. and Bloodgood et al. also have found that sea turtles which consumed more animal protein had a higher relative abundance of the Proteobacteria (Bloodgood et al., 2020; Campos et al., 2018). These studies suggest that in our research, the Proteobacteria was dominant phylum among all sea turtles, possibly because sea turtles have been in captivity and consuming seafood for a long period. Furthermore, previous studies have shown that the high prevalence of Proteobacteria is one of the recognized signatures of dysbiosis as well as an indication of disease in animals, including humans (Shin et al., 2015). Our study demonstrated that among the 389 potential pathogenic genera detected, 167 belong to the Proteobacteria, particularly Aliiroseovarius, Vibrio, Psychrobacter, Paracoccus, and Neisseria, which are affiliated with the top 30 abundant genera. Aliiroseovarius crassostreae, formerly known as Roseovarius crassostreae, is the causative agent of Roseovarius oyster disease (ROD), which results in high mortality rates in hatchery-raised juvenile eastern oysters in the northeast United States (Kessner et al., 2016). The abundance was significantly higher in oral samples, which may be because the diet origin of turtles was fish, and they might have been exposed to shellfish in the breeding facility and thus contracted the infection. Additionally, sea turtles have been frequently identified as carriers of Vibrio spp. (Zavala-Norzagaray et al., 2015; Fichi et al., 2016; Andrews et al., 2003), becoming potential sources of infections for humans. Vibrio spp. has been isolated from lesions of sea turtles of different species, including olive ridley sea turtles, green turtles, and loggerheads (Acuña et al., 1999; Fichi et al., 2016; Trotta et al., 2021). Although the Vibrio spp. naturally present in marine ecosystems and estuarine waters, indicating these bacteria may be some that coexist with the environment and sea turtles (Baker-Austin et al., 2018). However, among Vibrio spp., 12 species can cause infections in humans. These bacteria pose potential risks to the health of sea turtles and human.
In contrast, some studies have reported that Firmicutes was the dominant phylum, whether wild or stranded sea turtles, especially in green turtles (Abdelrhman et al., 2016; Price et al., 2017; Ahasan et al., 2017; Arizza et al., 2019; Chen et al., 2022). This may be due to the key role of Firmicutes in the digestion of complex polysaccharides (Hong et al., 2011). However, in our study, the abundance of the Firmicutes was relatively lower, which was similar to the research of Scheelings et al. (2020). They found that the abundance of the Firmicutes in green turtles and loggerheads was the lowest among all sea turtle species, suggesting that this phylum may not be as important for cellulose digestion in herbivorous reptiles as previously reported. However, considering that all the female sea turtles in their investigation were likely fasting for a long period, which led them to believe that this might have changed the bacterial phylum that is very important for digestive function, thereby affecting the turtle microbiota. Also, there are numerous examples in vertebrates, both terrestrial and aquatic, which indicate that the abundance of the Firmicutes is lower than that of other phyla in herbivorous species (Hong et al., 2011; Ishaq and Wright, 2012; Pope et al., 2012; Sullam et al., 2012). Therefore, considering that the microbiota of sea turtles may be affected by factors such as species, health conditions, captivity, and feeding situations, we should view these results with caution.
The Bacteroidota (formerly Bacteroidetes) was another dominant phylum across all samples. LEfSe analysis revealed no significant difference in its abundance among the three host species, but considering the three tissue regions, its abundance was significantly higher in nasal samples. Previous studies on the microbiota of sea turtles have shown that Bacteroidota is always a core bacterial phylum in sea turtles (Campos et al., 2018; Ahasan et al., 2018; Ahasan et al., 2017; Abdelrhman et al., 2016; Al-Bahry et al., 2009). Part of the reason might be that the members of the Bacteroides possess a variety of genes encoding carbohydrate activity, enabling them to easily switch between different energy sources through complex regulatory mechanisms based on the availability of different energy sources in the gastrointestinal tract (Xu et al., 2007; Thomas et al., 2011). Another reason of the decrease in abundance of Firmicutes and increase in Bacteroidetes is likely because of abundant seafood. Previous studies have shown that Bacteroidetes consists of many bile-tolerant organisms that aid in protein digestion, and, in humans that switched from a fiber-rich diet to an animal protein-based diet, there was a decrease in Firmicutes and an increase in Bacteroidetes in as little as 4 days (Wu et al., 2011; David et al., 2014). However, its specific functions in the nasal and oral of sea turtles remain to be further studied. Notably, the members of Campylobacterota were significantly higher in cloacal samples in our study, particularly Arcobacteriaceae, Campylobacteraceae, and Helicobacteraceae, which have recently been found to form unique, cold-adapted communities in ectothermic reptiles (Gilbert et al., 2019). Previous studies have also reported that loggerhead cloacal communities are a promising source of novel Campylobacterota (Filek et al., 2024). In our study, Campylobacter was significantly more abundant in green turtles. This might be because the number of green sea turtles in our sample was the highest, while the number of loggerheads was the lowest. Campylobacterota is also recognized as an important human pathogen, and half of the human population is colonized with the ulcer-causing stomach bacterium H. pylori (Eusebi et al., 2014). The H. pylori is the most abundant pathogenic species detected in our study. It reminds us to pay more attention to the intestinal problems of sea turtles.
In terms of the functions of the sea turtle microbiome, three turtle species and tissue regions displayed partly similar patterns and partly different features, which are consistent with the microbiome composition patterns. For example, the most abundant KEGG pathways and CAZymes across all samples were linked to metabolic processes and glycosyl transferases, which are similar with previous study on gut communities between hawksbills and green turtles, suggesting that sea turtles may hydrolyze and ferment complex carbohydrates or polysaccharides in the food by utilizing the enzymes expressed by gut microbes (Chen et al., 2022). Additionally, it is worth our attention that previous studies have discovered resistant bacteria in the gut microbes of wild loggerheads and green turtles (Alduina et al., 2020; Blasi et al., 2020; Conrad et al., 2023). And Yu et al. have discovered that the ARGs are significantly higher in artificially bred green turtles than in wild turtles (Niu et al., 2024). In our study, the glycopeptide and tetracycline resistance genes were dominant ARGs, and a higher abundance of ARGs was detected in hawksbills and oral samples, which is consistent with the study of Chen et al. (2022). Glycopeptide antibiotics are frequently used to treat life-threatening infections caused by multidrug-resistant Gram-positive pathogens, such as Staphylococcus aureus, Enterococcus spp., and Clostridium difficile (Marcone et al., 2018). Tetracycline antibiotics have been widely detected in terrestrial and aquatic environments (Chen et al., 2011; Li et al., 2011). Considering that the spread of drug-resistant microorganisms can occur through direct contact or the food chain, the large number of antibiotic resistance genes (ARGs) present in the sea turtles and their oral samples may mainly result from the fact that the sea turtles in this study mainly feed on farmed fish and have lived in recirculating seawater for a long time. This might introduce drug-resistant microorganisms into the microbial communities of the sea turtles. Furthermore, virulence genes with higher abundance were detected in cloacal samples, such as Fur, O-linked flagellar glycosylation, CheA / CheY, Siderophore-dependent iron uptake system, Ton system, and VctPDGC system. All these virulence genes encoding proteins were related to flagellar, iron uptake, and transport. Flagella are typically regarded as significant virulence factors that can facilitate motility and chemotaxis, allowing bacteria to travel to more favorable environments (Duan et al., 2013; Nakamura and Minamino, 2019). Iron is an important element for survival and colonization by bacteria since it plays a crucial role in the electron transport chain to produce energy (Andrews et al., 2003). Iron acquisition systems are used by bacteria to scavenge iron from the environment under iron-restricted conditions (Andrews et al., 2003; Krewulak and Vogel, 2008). Therefore, successful competition for iron is crucial for pathogenicity. These virulence factors may be important potential factors causing diseases in sea turtles.
This study also has its shortcomings. Although we collected three sea turtles for comparative analysis, due to their different distributions in the study area, the numbers of each type of sea turtle were uneven, which might be the reason for the differences in the bacterial communities of sea turtles. In addition, we have detected pathogenic bacteria in sea turtles, but have not yet successfully isolated potential pathogenic bacteria. This can serve as one of the directions for further research in the future.
Conclusion
Taken together, we first performed a detailed analysis of the oral, nasal, and cloacal microbiota composition and functional characteristics in three species of sea turtles, including green turtles, loggerheads, and hawksbills, using shotgun metagenomic sequencing. Our study suggested that the microbiota compositions and functions are significantly different among the three host species and tissue regions. It is worth noting that we have identified some potential pathogenic bacteria, detected several ARGs and virulence genes associated with pathogenicity. These may pose potential risks to the health of sea turtles and could also be transmitted to the marine environment and even humans through sea turtles as vectors, which deserves our attention. The findings in our study demonstrate that the microbial composition and function in these sea turtles exhibit both species-specific and region-specific variations. The implications and underlying mechanisms of these associations offer valuable insights for future research on the sea turtle microbiome.
Data availability statement
The raw metagenomic datasets in this study are publicly available in the NCBI BioProject (https://www.ncbi.nlm.nih.gov/bioproject) with the accession PRJNA1232438.
Ethics statement
The animal study was approved by Animal Research Ethics Committee of the College of Fisheries and Life Sciences, Hainan Tropical Ocean University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
LD: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. YD: Conceptualization, Data curation, Resources, Writing – review & editing. FQ: Investigation, Writing – review & editing. MZ: Conceptualization, Investigation, Writing – review & editing. XW: Investigation, Writing – review & editing. XiaZ: Investigation, Writing – review & editing. YY: Investigation, Writing – review & editing. JL: Investigation, Writing – review & editing. XJ: Conceptualization, Data curation, Methodology, Supervision, Writing – review & editing. XioZ: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the specific research fund of the Innovation Platform for Academicians of Hainan Province (YSPTZX202515), the Nanhai Innovation Talent Project of Hainan Province, and the fund of Sanya Talent Development Special Fund: Cultivation of Innovative Talents for Academicians in Etiology.
Acknowledgments
We would like to appreciate Professor Yu Du and his team for providing help on sea turtle sampling.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fpubh.2025.1675896.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1652229/full#supplementary-material
FIGURE S1 | Overview of commonalities and differences among all samples. (A,B) Core-pan gene rarefaction curve. The curve nearly plateaued when sufficient sequence data were input. (C) Heatmap showing the correlation between the samples and the gene table. (D) The gene numbers among different groups. We divided all samples into six groups, HG. G represents cloacal samples of green turtles, HG. B represents nasal samples of green turtles, HG. K represents oral samples of green turtles, DM. G, DM. B, DM. K represent cloacal, nasal, and oral samples of hawksbills, XG. G, XG. B, XG. K represent cloacal, nasal, oral samples of loggerheads.
FIGURE S2 | The top 30 bacteria from phylum to species level across all samples. From (A–F) are from the phylum to the species level.
FIGURE S3 | The alpha diversity analysis of the sea turtle bacterial community from the class to species levels based on turtle species and tissue regions. (A,B) The Chao 1 index and Shannon diversity index across three turtle species. (C,D) The Chao 1 index and Shannon diversity index across three tissue regions. p-values indicate the confidence level of statistical analyses, with p < 0.05 indicating statistically significant differences. ns represents no significant differences, * represents p < 0.05, ** represents p < 0.01, and *** represents p < 0.001.
FIGURE S4 | The beta diversity analysis of the sea turtle bacterial community from the class to species levels based on turtle species and tissue regions. (A,B) PCoA results based on three species and tissue regions, respectively. (C,D) ANOSIM results based on three species and tissue regions, respectively.
FIGURE S5 | The beta diversity analysis of the sea turtle bacterial community from phylum to species levels (A–F) in the same tissue area based on different sea turtles. HG. G represents cloacal samples of green turtles, HG. B represents nasal samples of green turtles, HG. K represents oral samples of green turtles, DM. G, DM. B, DM. K represent cloacal, nasal, and oral samples of hawksbills, XG. G, XG. B, XG. K represent cloacal, nasal, oral samples of loggerheads.
FIGURE S6 | (A,B) The relative abundance of the top 10 bacterial phyla and top 30 classes, orders, families, genera, and species based on three turtle species and tissue regions. (C,D) The shared and unique bacterial taxa from phylum to species based on three turtle species and tissue regions.
FIGURE S7 | The overview functional characteristics of the sea turtle microbiome. (A) The gene counts annotated in KEGG, eggNOG, CAZY, CARD, and VFDB databases. (B) The KEGG pathway annotation results. (C) The COG function annotation results. (D) The CAZY family annotation results. (E) The ARO resistance mechanism prediction results. (F) The virulence factors prediction results
FIGURE S8 | (A,B) The abundance of the top 20 KEGG pathways based on three turtle species and sampling locations. (C,D) The abundance of the CAZY families based on three turtle species and tissue regions. (E,F) The abundance of the top 20 ARGs based on three turtle species and tissue regions. (G,H) The abundance of the top 20 virulence factors based on three turtle species and tissue regions. (I,J) The shared and unique ARGs based on three turtle species and tissue regions. (K,L) The shared and unique virulence factors based on three turtle species and tissue regions.
Footnotes
1. ^https://www.iucnredlist.org
2. ^https://github.com/OpenGene/fastp
3. ^http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
4. ^http://topaz.gatech.edu/GeneMark/
5. ^https://github.com/bbuchfink/diamond/
6. ^https://www.ncbi.nlm.nih.gov/
8. ^http://eggnogdb.embl.de/#/app/home
10. ^http://www.mgc.ac.cn/VFs/main.htm
References
Abdelrhman, K. F., Bacci, G., Mancusi, C., Mengoni, A., Serena, F., and Ugolini, A. (2016). A first insight into the gut microbiota of the sea turtle Caretta caretta. Front. Microbiol. 7:1060. doi: 10.3389/fmicb.2016.01060
Acuña, M. T., Díaz, G., Bolaños, H., Barquero, C., Sánchez, O., Sánchez, L. M., et al. (1999). Sources of Vibrio mimicus contamination of turtle eggs. Appl. Environ. Microbiol. 65, 336–338. doi: 10.1128/AEM.65.1.336-338.1999
Ahasan, M. S., Waltzek, T. B., Huerlimann, R., and Ariel, E. (2017). Fecal bacterial communities of wild-captured and stranded green turtles (Chelonia mydas) on the great barrier reef. FEMS Microbiol. Ecol. 93:fix139. doi: 10.1093/femsec/fix139
Ahasan, M. S., Waltzek, T. B., Huerlimann, R., and Ariel, E. (2018). Comparative analysis of gut bacterial communities of green turtles (Chelonia mydas) pre-hospitalization and post-rehabilitation by high-throughput sequencing of bacterial 16S rrna gene. Microbiol. Res. 207, 91–99. doi: 10.1016/j.micres.2017.11.010
Al-Bahry, S. N., Mahmoud, I. Y., Al-Zadjali, M., Elshafie, A., Al-Harthy, A., and Al-Alawi, W. (2011). Antibiotic resistant bacteria as bio-indicator of polluted effluent in the green turtles, Chelonia mydas in Oman. Mar. Environ. Res. 71, 139–144. doi: 10.1016/j.marenvres.2010.12.005
Al-Bahry, S., Mahmoud, I., Elshafie, A., Al-Harthy, A., Al-Ghafri, S., Al-Amri, I., et al. (2009). Bacterial flora and antibiotic resistance from eggs of green turtles Chelonia mydas: an indication of polluted effluents. Mar. Pollut. Bull. 58, 720–725. doi: 10.1016/j.marpolbul.2008.12.018
Alduina, R., Gambino, D., Presentato, A., Gentile, A., Sucato, A., Savoca, D., et al. (2020). Is Caretta caretta a carrier of antibiotic resistance in the Mediterranean Sea? Antibiotics (Basel) 9:116. doi: 10.3390/antibiotics9030116
Andrews, S. C., Robinson, A. K., and Rodríguez-Quiñones, F. (2003). Bacterial iron homeostasis. FEMS Microbiol. Rev. 27, 215–237. doi: 10.1016/S0168-6445(03)00055-X
Arizza, V., Vecchioni, L., Caracappa, S., Sciurba, G., Berlinghieri, F., Gentile, A., et al. (2019). New insights into the gut microbiome in loggerhead sea turtles Caretta caretta stranded on the Mediterranean coast. PLoS One 14:e0220329. doi: 10.1371/journal.pone.0220329
Baker-Austin, C., Oliver, J. D., Alam, M., Ali, A., Waldor, M. K., Qadri, F., et al. (2018). Vibrio spp. infections. Nat. Rev. Dis. Primers 4:8. doi: 10.1038/s41572-018-0005-8
Bartlett, A., Padfield, D., Lear, L., Bendall, R., and Vos, M. (2022). A comprehensive list of bacterial pathogens infecting humans. Microbiology (Reading) 168:001269. doi: 10.1099/mic.0.001269
Blasi, M. F., Migliore, L., Mattei, D., Rotini, A., Thaller, M. C., and Alduina, R. (2020). Antibiotic resistance of gram-negative bacteria from wild captured Loggerhead Sea turtles. Antibiotics (Basel) 9:162. doi: 10.3390/antibiotics9040162
Bloodgood, J. C. G., Hernandez, S. M., Isaiah, A., Suchodolski, J. S., Hoopes, L. A., Thompson, P. M., et al. (2020). The effect of diet on the gastrointestinal microbiome of juvenile rehabilitating green turtles (Chelonia mydas). PLoS One 15:e0227060. doi: 10.1371/journal.pone.0227060
Bouchard, S. S., and Bjorndal, K. A. (2000). Sea turtles as biological transporters of nutrients and energy from marine to terrestrial ecosystems. Ecology 81, 2305–2313. doi: 10.2307/177116
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using Diamond. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Campos, P., Guivernau, M., Prenafeta-Boldú, F. X., and Cardona, L. (2018). Fast acquisition of a polysaccharide fermenting gut microbiome by juvenile green turtles Chelonia mydas after settlement in coastal habitats. Microbiome 6:69. doi: 10.1186/s40168-018-0454-z
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2009). The carbohydrate-active EnZymes database (Cazy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–D238. doi: 10.1093/nar/gkn663
Chan, S., Cheng, I. J., Zhou, T., Wang, H.-J., Gu, H.-X., and Song, X.-J. (2009). A comprehensive overview of the population and conservation status of sea turtles in China. Chelonian Conserv. Biol. 6, 185–198. doi: 10.2744/1071-8443(2007)6[185:ACOOTP]2.0.CO;2
Chen, Y., Xia, Z., and Li, H. (2022). Metagenomic comparison of gut communities between hawksbills (Eretmochelys imbricata) and green sea turtles (Chelonia mydas). Arch. Microbiol. 204:450. doi: 10.1007/s00203-022-03073-8
Chen, G., Zhao, L., and Dong, Y. H. (2011). Oxidative degradation kinetics and products of chlortetracycline by manganese dioxide. J. Hazard. Mater. 193, 128–138. doi: 10.1016/j.jhazmat.2011.07.039
Conrad, C., Inglis, J., Wende, A., Sanborn, M., Mukundan, N., Price, A., et al. (2023). Anthropogenic uranium signatures in turtles, tortoises, and sea turtles from nuclear sites. Pnas Nexus 2:pgad241. doi: 10.1093/pnasnexus/pgad241
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. doi: 10.1038/nature12820
Drane, K., Huerlimann, R., Power, M., Whelan, A., Ariel, E., Sheehan, M., et al. (2021). Testudines as sentinels for monitoring the dissemination of antibiotic resistance in marine environments: an integrative review. Antibiotics (Basel) 10:775. doi: 10.3390/antibiotics10070775
Duan, Q., Zhou, M., Zhu, L., and Zhu, G. (2013). Flagella and bacterial pathogenicity. J. Basic Microbiol. 53, 1–8. doi: 10.1002/jobm.201100335
Ebani, V. V. (2023). Bacterial infections in sea turtles. Vet. Sci. 10:333. doi: 10.3390/vetsci10050333
Eusebi, L. H., Zagari, R. M., and Bazzoli, F. (2014). Epidemiology of Helicobacter pylori infection. Helicobacter 19, 1–5. doi: 10.1111/hel.12165
Fichi, G., Cardeti, G., Cersini, A., Mancusi, C., Guarducci, M., Di Guardo, G., et al. (2016). Bacterial and viral pathogens detected in sea turtles stranded along the coast of Tuscany, Italy. Vet. Microbiol. 185, 56–61. doi: 10.1016/j.vetmic.2016.02.003
Figgener, C., Bernardo, J., and Plotkin, P. T. (2019). Beyond trophic morphology: stable isotopes reveal ubiquitous versatility in marine turtle trophic ecology. Biol. Rev. Camb. Philos. Soc. 94, 1947–1973. doi: 10.1111/brv.12543
Filek, K., Vuković, B. B., ŽIŽek, M., Kanjer, L., Trotta, A., Di Bello, A., et al. (2024). Loggerhead Sea turtles as hosts of diverse bacterial and fungal communities. Microb. Ecol. 87:79. doi: 10.1007/s00248-024-02388-x
Forde, B. M., and O’Toole, P. W. (2013). Next-generation sequencing technologies and their impact on microbial genomics. Brief. Funct. Genomics 12, 440–453. doi: 10.1093/bfgp/els062
Fraga, N. S., Martins, A. S., Faust, D. R., Sakai, H., Bianchini, A., Da Silva, C. C., et al. (2018). Cadmium in tissues of green turtles (Chelonia mydas): a global perspective for marine biota. Sci. Total Environ. 637-638, 389–397. doi: 10.1016/j.scitotenv.2018.04.317
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). Cd-hit: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gilbert, M. J., Duim, B., Zomer, A. L., and Wagenaar, J. A. (2019). Living in cold blood: Arcobacter, Campylobacter, and Helicobacter in reptiles. Front. Microbiol. 10:1086. doi: 10.3389/fmicb.2019.01086
Gong, S.-P., Shi, H.-T., Jiang, A.-W., Fong, J. J., Gaillard, D., and Wang, J.-C. (2017). Disappearance of endangered turtles within China’s nature reserves. Curr. Biol. 27, R170–R171. doi: 10.1016/j.cub.2017.01.039
Henry, L. P., Bruijning, M., Forsberg, S. K. G., and Ayroles, J. F. (2021). The microbiome extends host evolutionary potential. Nat. Commun. 12:5141. doi: 10.1038/s41467-021-25315-x
Hong, P. Y., Wheeler, E., Cann, I. K., and Mackie, R. I. (2011). Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galápagos Islands using 16S rrna-based pyrosequencing. ISME J. 5, 1461–1470. doi: 10.1038/ismej.2011.33
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). Eggnog 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). Megan analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Ishaq, S. L., and Wright, A. D. (2012). Insight into the bacterial gut microbiome of the north American moose (Alces alces). BMC Microbiol. 12:212. doi: 10.1186/1471-2180-12-212
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). Card 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–d573. doi: 10.1093/nar/gkw1004
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). Kegg: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–d361. doi: 10.1093/nar/gkw1092
Kessner, L., Spinard, E., Gomez-Chiarri, M., Rowley, D. C., and Nelson, D. R. (2016). Draft genome sequence of Aliiroseovarius crassostreae Cv919-312, the causative agent of Roseovarius oyster disease (formerly juvenile oyster disease). Genome Announc. 4:e00148-16. doi: 10.1128/genomeA.00148-16
Kittle, R. P., Mcdermid, K. J., Muehlstein, L., and Balazs, G. H. (2018). Effects of glyphosate herbicide on the gastrointestinal microflora of Hawaiian green turtles (Chelonia mydas) Linnaeus. Mar. Pollut. Bull. 127, 170–174. doi: 10.1016/j.marpolbul.2017.11.030
Kock, R. A., Woodford, M. H., and Rossiter, P. B. (2010). Disease risks associated with the translocation of wildlife. Rev. Sci. Tech. 29, 329–350. doi: 10.20506/rst.29.2.1980
Kraemer, S. A., Ramachandran, A., and Perron, G. G. (2019). Antibiotic pollution in the environment: from microbial ecology to public policy. Microorganisms 7:180. doi: 10.3390/microorganisms7060180
Krewulak, K. D., and Vogel, H. J. (2008). Structural biology of bacterial iron uptake. Biochim. Biophys. Acta 1778, 1781–1804. doi: 10.1016/j.bbamem.2007.07.026
Leitão, J. H. (2020). Microbial virulence factors. Int. J. Mol. Sci. 21:5320. doi: 10.3390/ijms21155320
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). Megahit: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, R., Zhang, Y., Lee, C. C., Liu, L., and Huang, Y. (2011). Hydrophilic interaction chromatography separation mechanisms of tetracyclines on amino-bonded silica column. J. Sep. Sci. 34, 1508–1516. doi: 10.1002/jssc.201100130
Lovich, J. E., Ennen, J. R., Agha, M., and Gibbons, J. W. (2018). Where have all the turtles gone, and why does it matter? Bioscience 68, 771–781. doi: 10.1093/biosci/biy095
Marcone, G. L., Binda, E., Berini, F., and Marinelli, F. (2018). Old and new glycopeptide antibiotics: from product to gene and back in the post-genomic era. Biotechnol. Adv. 36, 534–554. doi: 10.1016/j.biotechadv.2018.02.009
Mashkour, N., Jones, K., Kophamel, S., Hipolito, T., Ahasan, S., Walker, G., et al. (2020). Disease risk analysis in sea turtles: a baseline study to inform conservation efforts. PLoS One 15:e0230760. doi: 10.1371/journal.pone.0230760
Mazaris, A. D., Dimitriadis, C., Papazekou, M., Schofield, G., Doxa, A., Chatzimentor, A., et al. (2023). Priorities for Mediterranean marine turtle conservation and management in the face of climate change. J. Environ. Manag. 339:117805. doi: 10.1016/j.jenvman.2023.117805
Mazaris, A. D., Schofield, G., Gkazinou, C., Almpanidou, V., and Hays, G. C. (2017). Global Sea turtle conservation successes. Sci. Adv. 3:e1600730. doi: 10.1126/sciadv.1600730
Mcarthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Mcfall-Ngai, M., Hadfield, M. G., Bosch, T. C., Carey, H. V., Domazet-Lošo, T., Douglas, A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 110, 3229–3236. doi: 10.1073/pnas.1218525110
Mcnally, K. L., Innis, C. J., Kennedy, A., and Bowen, J. L. (2021). Characterization of oral and cloacal microbial communities in cold-stunned Kemp's ridley sea turtles (Lepidochelys kempii) during the time course of rehabilitation. PLoS One 16:e0252086. doi: 10.1371/journal.pone.0252086
Mou, J., Tao, X. H., Wu, R. E., Lozupone, F. X., Miao, X., Wang, X. Y., et al. (2013). Investigations on the distribution of sea turtle species in the coastal water of China (in Chinese). J. Oceanogr. 32, 238–242. doi: 10.3969/J.ISSN.2095-4972.2013.02.013
Nakamura, S., and Minamino, T. (2019). Flagella-driven motility of bacteria. Biomolecules 9:279. doi: 10.3390/biom9070279
Niu, X., Lin, L., Zhang, T., An, X., Li, Y., Yu, Y., et al. (2024). Research on antibiotic resistance genes in wild and artificially bred green turtles (Chelonia mydas). Sci. Total Environ. 954:176716. doi: 10.1016/j.scitotenv.2024.176716
Oren, A., and Garrity, G. M. (2021). Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 71:005056. doi: 10.1099/ijsem.0.005056
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). Stamp: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Pope, P. B., Mackenzie, A. K., Gregor, I., Smith, W., Sundset, M. A., Mchardy, A. C., et al. (2012). Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS One 7:e38571. doi: 10.1371/journal.pone.0038571
Price, J. T., Paladino, F. V., Lamont, M. M., Witherington, B. E., Bates, S. T., and Soule, T. (2017). Characterization of the juvenile green turtle (Chelonia mydas) microbiome throughout an ontogenetic shift from pelagic to neritic habitats. PLoS One 12:e0177642. doi: 10.1371/journal.pone.0177642
Rivas, M. L., Rodríguez-Caballero, E., Esteban, N., Carpio, A. J., Barrera-Vilarmau, B., Fuentes, M., et al. (2023). Uncertain future for global sea turtle populations in face of sea level rise. Sci. Rep. 13:5277. doi: 10.1038/s41598-023-31467-1
Samuelson, M., Pulis, E., Ray, C., Arias, C. R., Samuelson, D., Mattson, E. E., et al. (2020). Analysis of the fecal microbiome in Kemp’s ridley Lepidochelys kempii undergoing rehabilitation. Endangered Species Res. 43, 121–131. doi: 10.3354/esr01043
Santoro, M., Hernández, G., and Caballero, M. (2006). Aerobic bacterial flora of nesting green turtles (Chelonia mydas) from Tortuguero National Park, Costa Rica. J. Zoo Wildl. Med. 37, 549–552. doi: 10.1638/05-118.1
Scheelings, T. F., Moore, R. J., Van, T. T. H., Klaassen, M., and Reina, R. D. (2020). Microbial symbiosis and coevolution of an entire clade of ancient vertebrates: the gut microbiota of sea turtles and its relationship to their phylogenetic history. Anim. Microbiome 2:17. doi: 10.1186/s42523-020-00034-8
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Senko, J. F., Burgher, K. M., Del Mar Mancha-Cisneros, M., Godley, B. J., Kinan-Kelly, I., Fox, T., et al. (2022). Global patterns of illegal marine turtle exploitation. Glob. Chang. Biol. 28, 6509–6523. doi: 10.1111/gcb.16378
Shin, N. R., Whon, T. W., and Bae, J. W. (2015). Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503. doi: 10.1016/j.tibtech.2015.06.011
Stanford, C. B., Iverson, J. B., Rhodin, A. G. J., Paul Van Dijk, P., Mittermeier, R. A., Kuchling, G., et al. (2020). Turtles and tortoises are in trouble. Curr. Biol. 30, R721–R735. doi: 10.1016/j.cub.2020.04.088
Sullam, K. E., Essinger, S. D., Lozupone, C. A., O'connor, M. P., Rosen, G. L., Knight, R., et al. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol. Ecol. 21, 3363–3378. doi: 10.1111/j.1365-294X.2012.05552.x
Thomas, F., Hehemann, J. H., Rebuffet, E., Czjzek, M., and Michel, G. (2011). Environmental and gut bacteroidetes: the food connection. Front. Microbiol. 2:93. doi: 10.3389/fmicb.2011.00093
Trotta, A., Marinaro, M., Sposato, A., Galgano, M., Ciccarelli, S., Paci, S., et al. (2021). Antimicrobial resistance in Loggerhead Sea turtles (Caretta caretta): a comparison between clinical and commensal bacterial isolates. Animals (Basel) 11:2435. doi: 10.3390/ani11082435
Ursell, L. K., Metcalf, J. L., Parfrey, L. W., and Knight, R. (2012). Defining the human microbiome. Nutr. Rev. 70, S38–S44. doi: 10.1111/j.1753-4887.2012.00493.x
Wensel, C. R., Pluznick, J. L., Salzberg, S. L., and Sears, C. L. (2022). Next-generation sequencing: insights to advance clinical investigations of the microbiome. J. Clin. Invest. 132:e154944. doi: 10.1172/JCI154944
Wu, G. D., Chen, J., Hoffmann, C., Bittinger, K., Chen, Y. Y., Keilbaugh, S. A., et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. doi: 10.1126/science.1208344
Xu, J., Mahowald, M. A., Ley, R. E., Lozupone, C. A., Hamady, M., Martens, E. C., et al. (2007). Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 5:e156. doi: 10.1371/journal.pbio.0050156
Zavala-Norzagaray, A. A., Aguirre, A. A., Velazquez-Roman, J., Flores-Villaseñor, H., León-Sicairos, N., Ley-Quiñonez, C. P., et al. (2015). Isolation, characterization, and antibiotic resistance of Vibrio spp. in sea turtles from northwestern Mexico. Front. Microbiol. 6:635. doi: 10.3389/fmicb.2015.00635
Keywords: sea turtles, Chelonia mydas , Caretta caretta , Eretmochelys imbricata , metagenomic microbiome
Citation: Dong L, Du Y, Qiu F, Zhang M, Wang X, Zhu X, Yao Y, Li J, Ji X and Zhu X (2025) Metagenomic insights reveal the differences in the community composition and functional characteristics of the sea turtle microbiomes based on host species and tissue region. Front. Microbiol. 16:1652229. doi: 10.3389/fmicb.2025.1652229
Edited by:
Valerio Mazzella, Anton Dohrn Zoological Station Naples, ItalyReviewed by:
Enrico Nanetti, University of Bologna, ItalyVanessa Maria Bachmann, University of Pavia, Italy
Copyright © 2025 Dong, Du, Qiu, Zhang, Wang, Zhu, Yao, Li, Ji and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Ji, eGppQHd6dS5lZHUuY24=; Xiong Zhu, emh1eGlvbmc2QDE2My5jb20=