 Misheck Shawa1,2*
Misheck Shawa1,2* Herman Chambaro3Harvey K. Kamboyi4Clement Sulwe5
Herman Chambaro3Harvey K. Kamboyi4Clement Sulwe5 Joseph Y. Chizimu6Situmbeko J. Nasilele7Shohei Ogata1,2Mulemba Samutela8Tuvshinzaya Zorigt9
Joseph Y. Chizimu6Situmbeko J. Nasilele7Shohei Ogata1,2Mulemba Samutela8Tuvshinzaya Zorigt9 Steward Mudenda6Manyando Simbotwe10Mwamba Nsofwa10Jedidiah Chanda6
Steward Mudenda6Manyando Simbotwe10Mwamba Nsofwa10Jedidiah Chanda6 Freeman Chabala11
Freeman Chabala11 Mike Nundwe1,7Joseph Ndebe2
Mike Nundwe1,7Joseph Ndebe2 Msangwa Sinjani10
Msangwa Sinjani10 Kyoko Hayashida1,12Naganori Nao1,2,13Roma Chilengi6Hirofumi Sawa1,2,13,14,15,16,17
Kyoko Hayashida1,12Naganori Nao1,2,13Roma Chilengi6Hirofumi Sawa1,2,13,14,15,16,17 Yasuhiko Suzuki14,18
Yasuhiko Suzuki14,18 Bernard Hang'ombe16,19
Bernard Hang'ombe16,19 Masahiro Kajihara1,2,13
Masahiro Kajihara1,2,13 Hideaki Higashi1,9
Hideaki Higashi1,9- 1Hokudai Center for Zoonosis Control in Zambia, Hokkaido University, Lusaka, Zambia
- 2Division of International Research Promotion, International Institute for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 3Department of Pathobiology, College of Veterinary Medicine, University of Illinois Urbana-Champaign, Champaign, IL, United States
- 4Department of Clinical Medicine, School of Medicine, Eden University, Lusaka, Zambia
- 5Institute of Biochemistry, Molecular Biology and Biotechnology, University of Colombo, Colombo, Sri Lanka
- 6Zambia National Public Health Institute, Ministry of Health, Lusaka, Zambia
- 7Department of Biomedical Sciences, School of Veterinary Medicine, University of Zambia, Lusaka, Zambia
- 8Department of Biomedical Sciences, School of Health Sciences, University of Zambia, Lusaka, Zambia
- 9Division of Infection and Immunity, International Institute for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 10Department of Disease Control, School of Veterinary Medicine, University of Zambia, Lusaka, Zambia
- 11Institute of Basic and Biomedical Sciences, Levy Mwanawasa Medical University, Lusaka, Zambia
- 12Division of Collaboration and Education, International Institute for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 13One Health Research Center, Hokkaido University, Sapporo, Japan
- 14Hokkaido University, Institute for Vaccine Research and Development, Sapporo, Japan
- 15Division of Molecular Pathobiology, International Institute for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 16Africa Centre of Excellence for Infectious Diseases of Humans and Animals, School of Veterinary Medicine, University of Zambia, Lusaka, Zambia
- 17Global Virus Network, Baltimore, MD, United States
- 18Division of Bioresources, International Institute for Zoonosis Control, Hokkaido University, Sapporo, Japan
- 19Department of Research and Innovation, Copperbelt University, Kitwe, Zambia
Antimicrobial resistance (AMR) mediated by extended-spectrum β-lactamases (ESBLs) is a growing global concern, particularly among Enterobacteriaceae. The CTX-M-type ESBLs, encoded by the blaCTX-M gene, are of significant public health importance due to their high prevalence and broad geographic distribution. Typically located on plasmids and often co-occurring with other AMR genes, blaCTX-M contributes to multidrug resistance (MDR). However, increasing evidence suggests secondary chromosomal integration of blaCTX-M, sometimes alongside other resistance determinants. The extent and implications of this mechanism remain poorly characterized, especially in Africa, where genomic surveillance is limited. In this study, we retrieved 295 chromosomal sequences of Enterobacteriaceae of African origin from the GenBank and performed in silico predictions of blaCTX-M and other AMR genes. blaCTX-M-carrying sequences were further characterized by in silico multilocus sequence typing and genome annotation. Chromosomal insertions were identified through alignment with reference genomes. Overall, 47 of 295 sequences (15.9%) harbored the blaCTX-M gene, with the highest prevalence in Klebsiella pneumoniae (29/157, 18.5%), followed by Escherichia coli (13/72, 18.1%), Enterobacter spp. (4/38, 10.5%), and Shigella spp. (1/12, 8.3%). The most common allele was blaCTX-M-15 (31/47, 66.0%), followed by blaCTX-M-14 (12/47, 25.5%), blaCTX-M-55 (3/47, 6.4%), and blaCTX-M-27 (1/27, 3.7%). Co-occurrence of blaCTX-M with additional AMR genes was frequently observed, with integration events often associated with mobile genetic elements such as ISEcp1 and IS26. Notably, strains from the same hospital setting were phylogenetically related and shared sequence types and AMR gene profiles, suggesting local clonal dissemination. These findings reveal a notable presence of chromosomally integrated blaCTX-M among African Enterobacteriaceae, frequently in association with other resistance genes, thereby facilitating stable MDR propagation independent of plasmid maintenance. This evolutionary adaptation may have significant implications for the persistence and spread of MDR in clinical settings.
Introduction
Despite significant strides in developing novel antimicrobials, the treatment of bacterial infections has become increasingly challenging due to the widespread emergence of multidrug resistance (MDR), particularly among Enterobacteriaceae. MDR pathogens represent a clear and present danger that affects every populated continent of the globe (Bassetti et al., 2015). For instance, MDR bacteria are estimated to contribute to 45% of deaths in Africa (WHO, 2014), and in 2019 alone, MDR Escherichia coli and Klebsiella pneumoniae were associated with over 100,000 deaths globally (Murray et al., 2022). In Enterobacteriaceae, MDR is often mediated by extended-spectrum β-lactamases (ESBLs) (Zhang et al., 2015; Beyene et al., 2024). While TEM and SHV variants were historically predominant, the CTX-M-type ESBLs, which hydrolyze third-generation cephalosporins like cefotaxime, have now become the most prevalent globally (Castanheira et al., 2021). CTX-M type ESBLs are classified into five groups, CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9, and CTX-M-25, with the CTX-M-1 group being the most predominant. Recent meta-analyses in sub-Saharan Africa confirm CTX-M-15 (CTX-M-1 group) as the most reported ESBL type across human and animal health sectors (Olaitan et al., 2025). The pandemic clone E. coli ST131 producing CTX-M-15 type ESBL remains a key driver in Africa, mirroring global trends (Nicolas-Chanoine et al., 2014). The blaCTX-M gene, which encodes CTX-M types ESBLs, is typically located on mobile genetic vectors called plasmids that often harbor multiple antimicrobial resistance (AMR) genes (Mikhayel et al., 2024). This facilitates the rapid dissemination of MDR phenotypes.
Although plasmids contribute significantly to AMR gene spread, they may impose a fitness cost on the bacteria due to the energetic burden of their replication and maintenance, thus increasing the chances of instability or loss (Carroll and Wong, 2018). Additionally, plasmids are susceptible to loss during cell division through segregational instability. To overcome these limitations and ensure stable inheritance of critical traits, plasmid-encoded genes, including blaCTX-M, may integrate into the bacterial chromosome, provided such insertions do not truncate essential genes or alter vital regulatory pathways (San Millan et al., 2015). Chromosomal integration of blaCTX-M is frequently mediated by mobile elements such as ISEcp1 and IS26 (Gomi et al., 2022; Komori et al., 2024). Most studies have reported chromosomal blaCTX-M gene insertions as small segments (<5,000 bp) containing few or no additional resistance genes (Mshana et al., 2015). However, emerging evidence suggests that larger chromosomal islands harboring blaCTX-M alongside multiple AMR genes [e.g., aac(3)-IIa, qnrB1, aac(6′)-Ib-cr5, blaOXA-1, dfrA14, catB3, tet(A)] also exist. These blaCTX-M-carrying islands are usually mobilized by transposable elements, which facilitate their insertion at various chromosomal sites. Notably, Yoon et al. (2020) described blaCTX-M-carrying chromosomal segments in K. pneumoniae, while Goswami et al. (2020) reported similar findings in E. coli.
Previously, our research group reported large MDR chromosomal islands carrying blaCTX-M in E. coli and Enterobacter cloacae from Zambia (Shawa et al., 2021; Shawa et al., 2025). However, the overall prevalence and genetic landscape of chromosomal blaCTX-M across the African continent remain poorly characterized. With the growing availability of whole genome sequencing (WGS) data from African countries submitted to the National Center for Biotechnology Information (NCBI), this study used in silico analysis to estimate the prevalence of chromosomally integrated blaCTX-M among Enterobacteriaceae of African origin and characterize the genetic context of these chromosomal insertions.
Methodology
In September 2024, whole genome sequences of Enterobacteriaceae chromosomes (E. coli, K. pneumoniae, Enterobacter spp., Salmonella spp., and Shigella spp.) from African clinical sources were downloaded from the NCBI nucleotide database. To this end, we explored the NCBI using search terms including the bacterial species, host species, and country (e.g., “Escherichia coli, Homo sapiens, Kenya”) and filtered the results using a customized sequence length range of 1,000,000 to 6,000,000 bp. Fasta files of the output sequences were collected as a single folder for each country through a bulk download. The number of unique sequences per dataset was determined using Seqkit (rmdup function) (Shen et al., 2016).
In silico prediction of AMR genes was performed by ResFinder (Florensa et al., 2022), and blaCTX-M-harboring genomes were annotated using dfast version 1.3.2 (Tanizawa et al., 2018). The chromosome sequences were also subjected to in silico multilocus sequence typing (MLST) using the mlst database,1 which partly uses the PubMLST database2 (Jolley and Maiden, 2010). Chromosomal insertions harboring the blaCTX-M gene were detected by aligning the annotated files to appropriate reference genomes using Mauve (Darling et al., 2004), and the results were visualized in genoPlotR version 0.8.11 (Guy et al., 2010). To further characterize the insertions, nucleotide sequences were subjected to BLASTn against the NCBI database, and the resulting hit tables were filtered in R using the dplyr package version 1.1.4 (Wickham et al., 2023), with filtering criteria set to “% identity > 99.” Additionally, plasmid replicons were detected using the PlasmidFinder database (Carattoli et al., 2014), while genomic islands of horizontal origin were predicted using IslandViewer 4 (Bertelli et al., 2017), which includes IslandPath-DIMOB (Hsiao et al., 2003), SIGI-HMM (Waack et al., 2006), and IslandPick (Langille et al., 2008). Previously published Zambian sequences were excluded from this analysis, as they have been characterized in prior studies (Shawa et al., 2021).
To distinguish between E. coli and Shigella spp., one sequence was subjected to a local BLAST using lacY and ipaH genes. To this end, lacY (from E. coli str. K-12 substr. MG1655, Accession Number: NC_000913.3) and ipaH (from S. flexneri 2a str. 301, Accession Number: NC_004337.2) were downloaded from the NCBI and concatenated into one fasta file. A local database was then created from the merged fasta file using the “makeblastdb” command while specifying the “-dbtype nucl” and “-parse_seqids” arguments. Finally, the query sequence was subjected to a BLAST search against the local database using the “blastn” command. S. flexneri 2a str. 2,457 T (Accession Number: AE014073.1) was used as a positive control for ipaH.
Core-SNP-based phylogenetic trees for the blaCTX-M-carrying genome sequences of each species were created using parsnp version 2.1.4 (Treangen et al., 2014) using E. fergusonii (Accession Number NZ_CP083638.1) and K. quasipneumoniae (Accession Number NZ_LR588411.1) as outgroups for E. coli and K. pneumoniae, respectively. Finally, the output parsnp tree files were visualized and edited in iTOL version 7 (Letunic and Bork, 2024).
Results
Chromosomal blaCTX-M detected in ~ 16% of Enterobacteriaceae genomes
A total of 295 Enterobacteriaceae chromosomal sequences, each exceeding 1,000,000 bp, were retrieved from the NCBI nucleotide database (Table 1). The sequences originated from clinical isolates in 18 African countries and represented five genera: K. pneumoniae (n = 157), E. coli (n = 72), Enterobacter (n = 38), Salmonella (n = 16), and Shigella (n = 12) (Table 1). The genome sizes ranged from 1,000,114 to 5,772,140 bp. Out of 295 genomes analyzed, 47 (47/295, 15.9%) harbored the blaCTX-M gene. The highest prevalence was observed in K. pneumoniae (29/157, 18.5%), followed by E. coli (13/72, 18.1%), Enterobacter (4/38, 10.5%), and Shigella (1/12, 8.3%) (Table 1). Geographically, chromosomal blaCTX-M was detected in nine out of 18 countries (50%). The country-specific prevalence was; Sudan 94.4% (17/18), Ethiopia 20.9% (9/43), South Africa 6.0% (5/83), Uganda 40% (4/10), Ghana 42.9% (3/7), Nigeria 6.5% (3/46), Tanzania 14.3% (2/14), Malawi 25.0% (2/8), Niger 50% (1/2), and Egypt 5.6% (1/18) (Table 1). Among the blaCTX-M-carrying sequences, the most frequently detected allele was blaCTX-M-15 (31/47, 66.0%), followed by blaCTX-M-14 (12/47, 25.5%), blaCTX-M-55 (3/47, 6.4%), and blaCTX-M-27 (1/27, 3.7%). Six out of 29 K. pneumoniae sequences possessed two copies of blaCTX-M, while other species had a single copy of the gene.
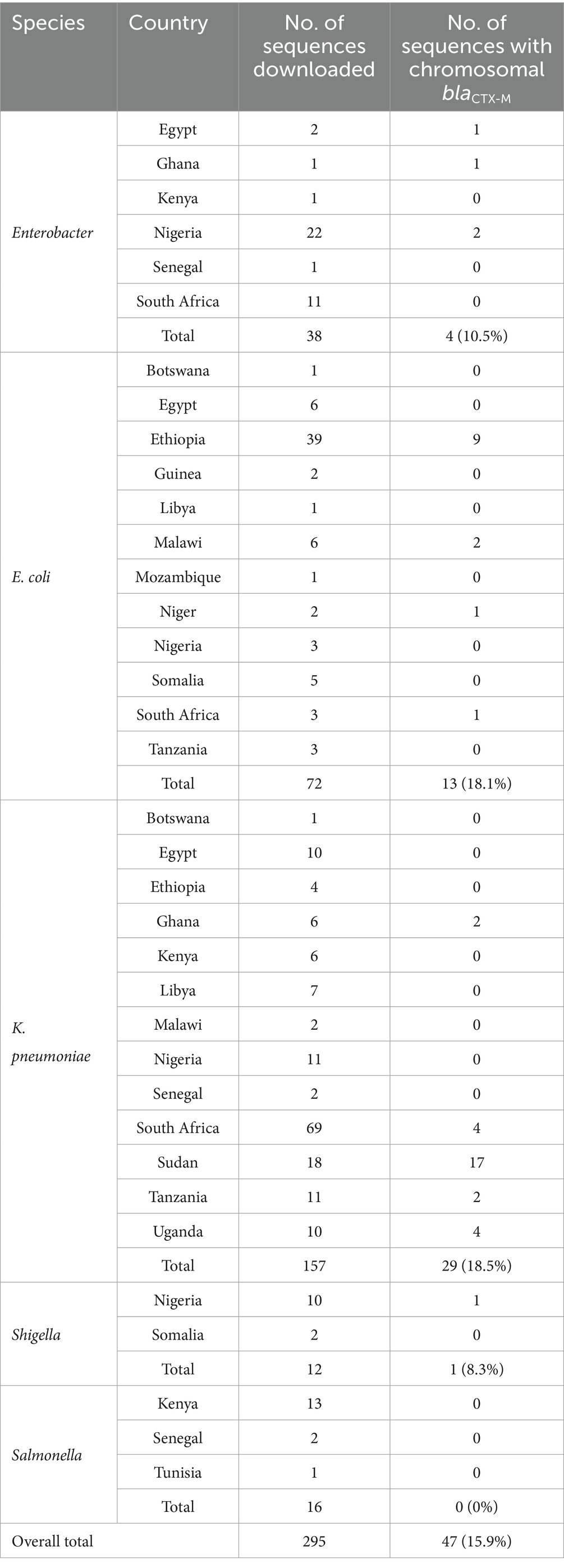
Table 1. Distribution of chromosomal Enterobacteriaceae sequences downloaded from the GenBank.
Of the 47 blaCTX-M-harboring sequences, 45 were larger than 4.6 Mbp, while two Enterobacter sequences were 1,274,920 bp and 1,023,858 bp (Supplementary Table S1). Interestingly, plasmid replicons were detected in some chromosomes of blaCTX-M-carrying K. pneumoniae (19/29, 65.5%) and E. coli (2/13, 15.4%), all of which were larger than 4.9 Mbp (Table 2). The replicons included p0111, IncFIB(AP001918), IncFII, Col440I, Col(BS512), ColpVC, IncR, IncFIB(pKPHS1), IncA/C2, IncFII_1_pKP91, and IncFIB(K)_1_Kpn3 (Table 2).
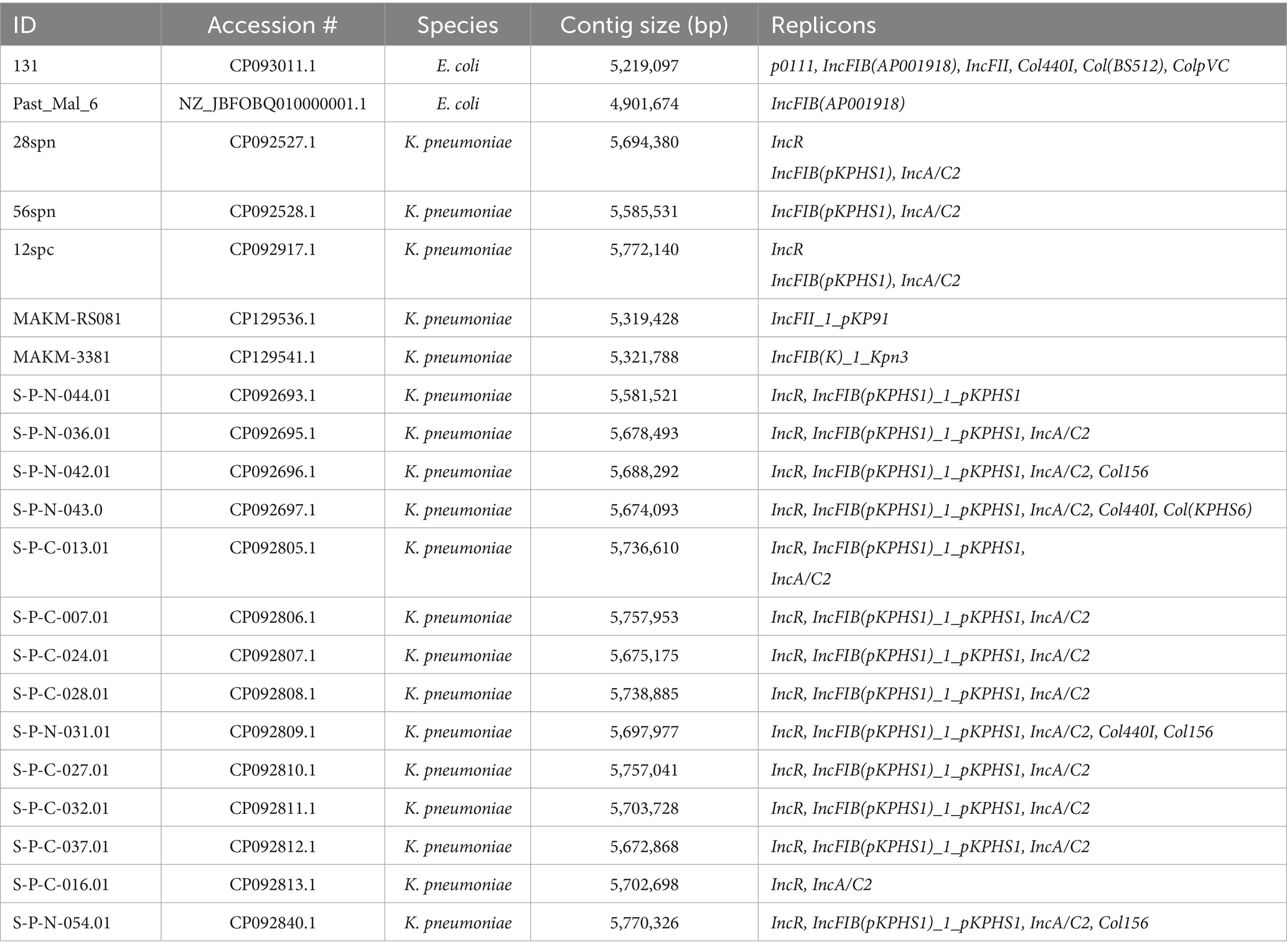
Table 2. Plasmid replicons detected among chromosomal sequences.
Multidrug-resistant chromosomal insertion (~12.8 kbp) identified in a Malawian E. coli sequence
Among the 13 E. coli genomes analyzed, nine distinct sequence types (STs) were identified, with ST38 and ST450 observed twice. The remaining STs were unique to individual strains (Supplementary Table S2). Two isolates could not be assigned to an ST due to uncertainties in the adk allele. Notably, strain NW-MR1609 (Accession Number NZ_JASATV010000001.1), previously identified as S. sonei, was reassigned to E. coli ST484. This strain possessed lacY, an E. coli hallmark gene (Horakova et al., 2008), but lacked ipaH, which is present in all Shigella (Ashida and Sasakawa, 2015). Analysis of genomic regions flanking the blaCTX-M gene revealed the presence of ISEcp1 insertion sequence 255 bp upstream in all but three strains. Furthermore, a 273 bp gene encoding the WbuC family cupin fold metalloprotein was located 46 bp downstream of the blaCTX-M gene in all but two strains (Figure 1). Four strains exhibited additional AMR genes within 10,000 bp of blaCTX-M. Strains NW-MR1609 (ST484, Nigeria), Past_Dab_2 (ST8130, Ethiopia), and Past_Mal_15 (ST38, Ethiopia) each carried the qnrS1 gene 4,640 bp downstream of blaCTX-M. The qnrS1 gene was part of a 5,250 bp genomic island that was immediately adjacent to blaCTX-M and harbored the Tn3-like element Tn3 family transposase gene (Supplementary Figure S1). The CAC124 strain from Malawi (ST5640, Accession Number NZ_JAWZSZ010000002.1) harbored blaOXA-1, two aminoglycoside-encoding genes [aac(6′)-Ib-cr and aac(3)-IIa], and catB for chloramphenicol resistance, all located within a 12,837 bp chromosomal insertion that lacked ISEcp1 (Figure 1). Compared to the reference strain (EC958, Accession Number HG941718.1), the CAC124 insertion was a composite transposon flanked by directly oriented IS26 elements, but the insertion point lacked target-site duplications (TSDs). Additional interspersed IS26 copies were observed adjacent to AMR genes or other transposable elements (Figure 1). A BLAST search revealed that this insertion was present in multiple sequences, including chromosomes from clinical E. coli ST131 strains isolated in over 30 countries worldwide (Supplementary Table S3; Figure 2). Furthermore, this sequence was part of a 13,577 bp genomic island bounded by hypothetical protein-encoding genes immediately external to the insertion’s flanking IS26 elements (Supplementary Figure S2).

Figure 1. Genetic environment of blaCTX-M in Shigella and E. coli chromosomes from Africa. From a total of 14 genomes, the ISEcp1 was observed upstream of blaCTX-M in 11 strains, while wbuC existed downstream of blaCTX-M in strains. CAC124 from Malawi exhibited multiple AMR genes on a composite transposon bracketed by directly oriented IS26 elements. Yellow; mobile genetic elements. Red; β-lactamase gene. Cyan; wbuC. Green; aminoglycoside resistance gene. Brown; chloramphenicol resistance gene.
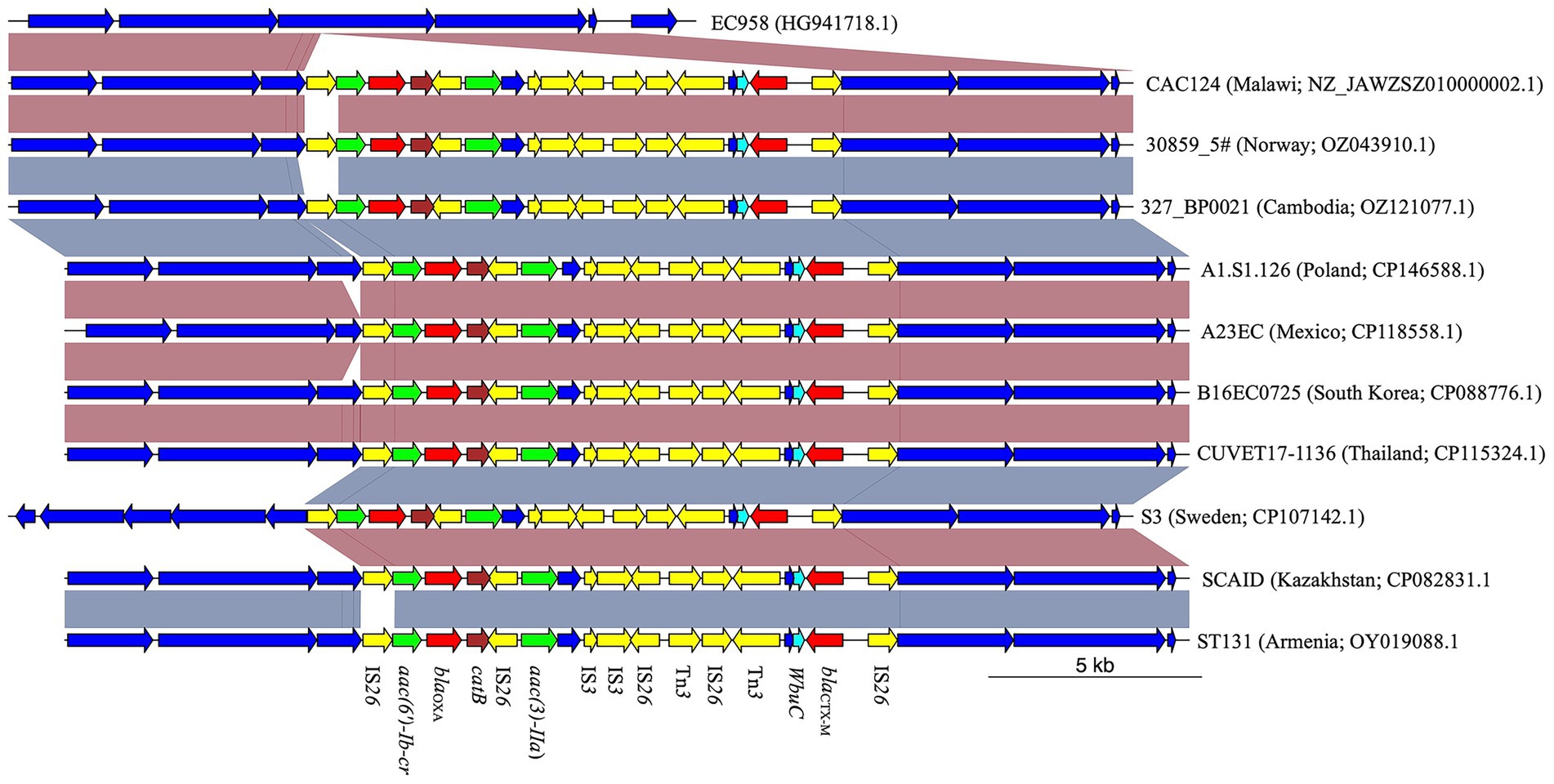
Figure 2. Comparison analysis of CAC124 from Malawi with E. coli ST131 strains from various countries across the world. The IS26-flanked composite transposon in CAC124 was found in several E. coli ST131. Yellow; mobile genetic elements. Red; β-lactamase gene. Cyan; wbuC. Green; aminoglycoside resistance gene. Brown; chloramphenicol resistance gene.
MDR chromosomal insertions identified in two E. cloacae strains from West Africa
Of the four E. cloacae strains, two STs (ST456 and ST544) were identified, while the remaining two could not be typed due to the absence of the seven housekeeping genes (Supplementary Table S4). All four strains carried ISEcp1 upstream of blaCTX-M and wbuC downstream. However, two strains (50%) carried additional AMR genes near blaCTX-M. For instance, strain EFN743 (ST456, Ghana) possessed genes conferring resistance to trimethoprim (dfrA14), quinolones [qnrB1, aac(6′)-Ib-cr], aminoglycosides [aac(3)-IIa, aac(6′)-Ib-cr, aph(6)-Id, aph(3″)-Ib, ant(3″)-Ia], chloramphenicol (catB), β-lactams (blaOXA-1, blaTEM-1B), sulfonamides (sul2), and tetracycline (tetA). When compared to E. cloacae ST456 (ST456ECL1 KP1759_1, Accession Number NZ_JBBFWY010000001.1), these genes were located within a 75,439 bp chromosomal insertion flanked by directly oriented IS26 (Figure 3A). The two peripheral IS26 copies were flanked by putative 8 bp TSDs (GACCACAC) at positions 66,296–66,301 and 141,741–141,748. Notably, rearranged versions were observed in GeneBank sequences, including plasmid p23_A-OXA140 (Accession Number CP048350.1) (Figure 3B). Furthermore, the abovementioned insertion harbored multiple genomic islands rich in virulence factors, mobile elements, and AMR genes (Supplementary Figure S3).
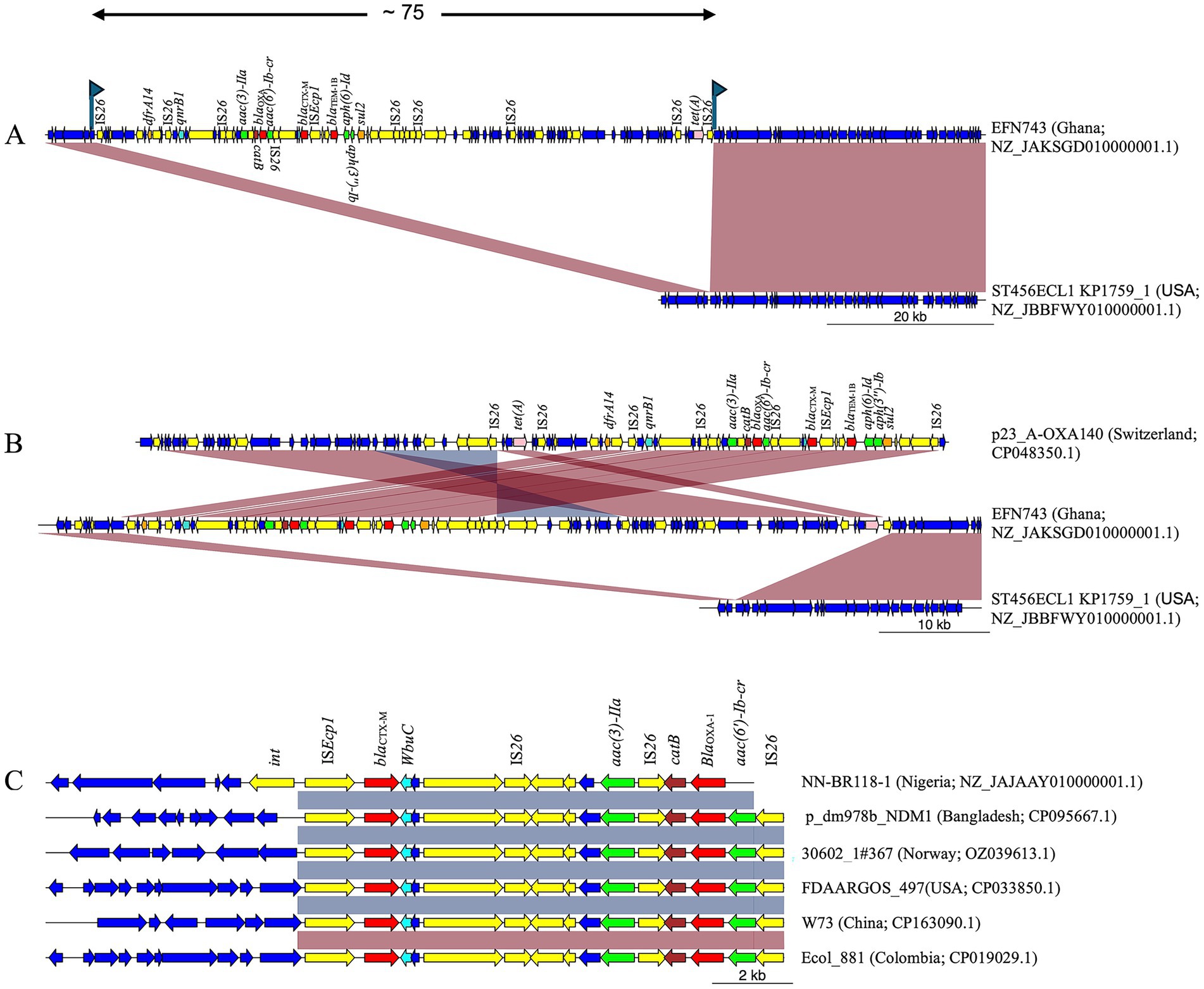
Figure 3. MDR chromosomal insertions in E. cloacae strains from West Africa. (A) Strain EFN743 from Ghana harbored blaCTX-M on a ~ 75 kbp IS26-flanked chromosomal insertion containing multiple AMR genes. The insertion was bracketed by putative 8 bp TSDs (represented by flags). (B) The insertion in strain EFN743 was similar to plasmid p23_A-OXA140 from Switzerland, though the gene arrangement was different. (C) Nigerian strain NN-BR118-1 carried blaCTX-M on a ~ 13 kbp MDR insertion bounded by the int. gene on one end. This insertion was found in several sequences, though they lacked the int gene upstream, but harbored IS26 on the opposite end. Yellow; mobile genetic elements. Red; β-lactamase gene. Cyan; wbuC. Green; aminoglycoside resistance gene. Brown; chloramphenicol resistance gene. Pink; tetracycline resistance gene. Orange; folate pathway antagonist.
Meanwhile, a comparison of strain NN-BR118-1 (ST544, Nigeria) to E. cloacae ATCC13047 (Accession Number CP001918.1) showed that NN-BR118-1 had the blaCTX-M gene on an insertion >13 kbp at position 4,685,679–4,698,752 bp. This insertion contained a site-specific integrase gene positioned immediately upstream of ISEcp1, and an 8,284 bp genomic island downstream of the blaCTX-M gene. This genomic island included blaOXA, as well as genes for aminoglycoside [aac(3)-IIa and aac(6′)-Ib-cr], quinolone [aac(6′)-Ib-cr], and chloramphenicol (catB) resistance (Supplementary Figure S4). A BLAST analysis of the 13 kbp insertion revealed >100 high-identity matches (>99.9% identity, >11.5 kbp sequence alignment), although none retained the site-specific integrase gene, suggesting rearrangement via ISEcp1 or IS26 located on the opposite end (Figure 3C).
Clonally related K. pneumoniae strains share STs and AMR genes
Chromosomally integrated blaCTX-M was identified in K. pneumoniae strains from Ghana, South Africa, Uganda, and Tanzania (Table 1). Ghana strains EFN299 (Accession Number NZ_CP092589.1) and MIN-106 (Accession Number NZ_JAJBHB010000001.1) belonged to ST11 and ST152, respectively (Supplementary Table S5). In addition to the ISEcp1/blaCTX-M-15/wbuC unit, the chromosome of strain EFN299 possessed oqxA, oqxB, blaSHV-182, and fosA6. However, none of these genes were in the immediate vicinity (i.e., within 10,000 bp) of blaCTX-M. In contrast, MIN-106 possessed dfrA14 8,616 bp downstream of blaCTX-M-15, along with mobile elements and additional AMR genes [blaOXA-1, aac(6′)-Ib-cr, and tet(A)]. When compared to another K. pneumoniae ST152 (strain HZKP1, Accession Number CP139932.1), MIN-106 showed a 139,735 bp chromosomal insertion bounded by ISEcp1/blaCTX-M-15/wbuC on one end. BLAST analysis of this insertion, filtered for matches with “percent identity > 99%” and “alignment length > 60,000,” yielded 24 hits. However, these alignments covered approximately 60,300 bp of the insertion. For instance, p2247421-T20-ESBL_2, an IncF plasmid from a clinical ESBL-producing K. pneumoniae isolate in Switzerland, showed 60,380 bp of aligned sequence. Notably, these aligned regions did not include the AMR genes observed in MIN-106 (Figure 4A). However, the entire insertion in MIN-106 included intermittently distributed genomic islands associated with virulence, mobility, and metal resistance (Supplementary Figure S5).
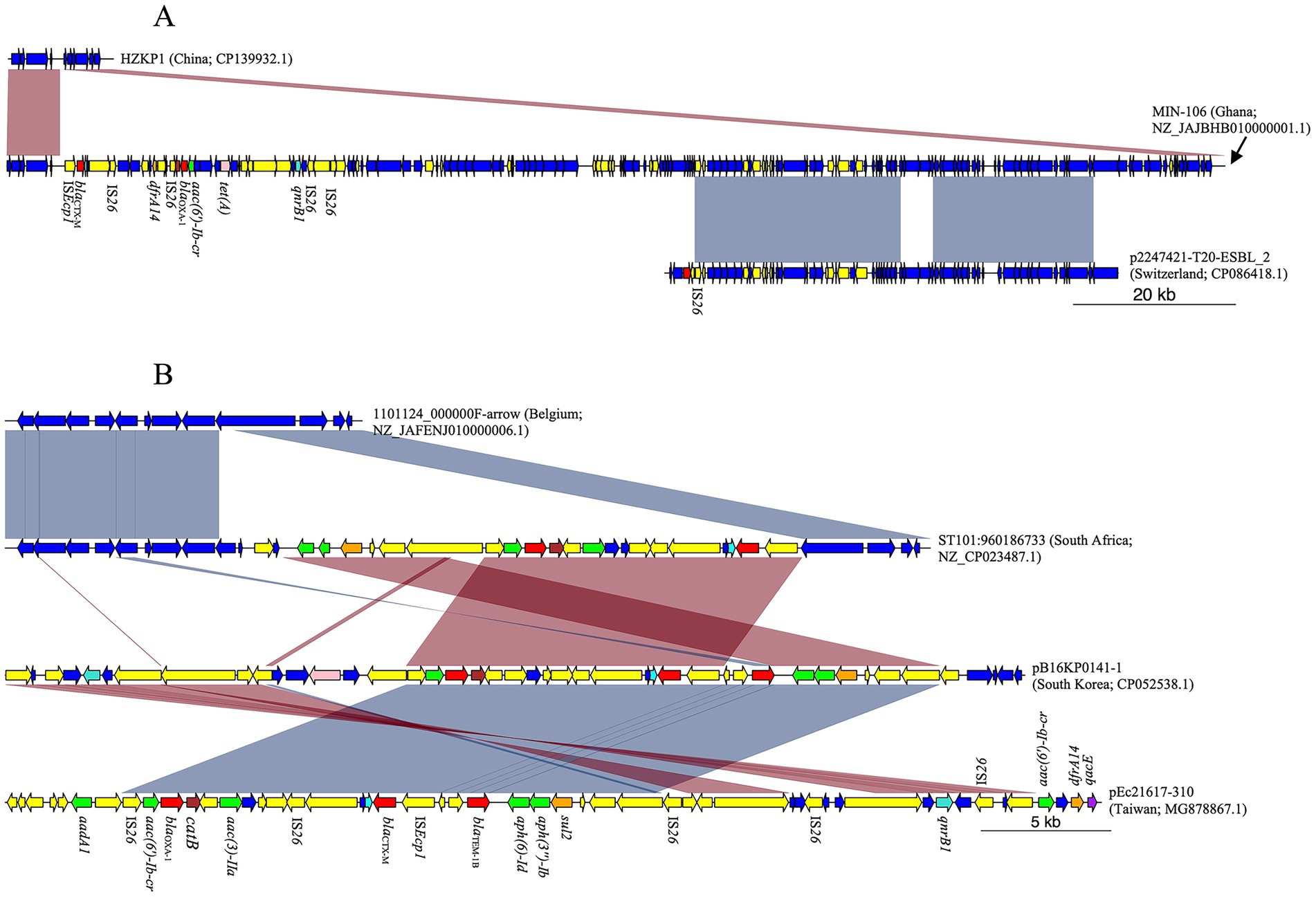
Figure 4. MDR chromosomal insertions in K. pneumoniae strains from West Africa. (A) Ghanaian strain MIN-106 possessed a 139,735 bp chromosomal insertion bounded by ISEcp1/blaCTX-M-15/wbuC on one end. About 60,300 bp of this insertion was similar to plasmid p2247421-T20-ESBL_2, but the aligned regions did not include any AMR genes. (B) South African strain ST101:960186733 harbored multiple AMR genes on a 22,270 bp insertion bounded by ISEcp1/blaCTX-M-15/wbuC on one end. The insertion was similar to the AMR gene cluster, which was observed in plasmids pB16KP0141–1 and pEc21617-310.
From South Africa, K. pneumoniae strains were assigned to ST101 (n = 1) and ST2497 (n = 3), all of which carried blaCTX-M-15 flanked by upstream ISEcp1 and downstream wbuC. The three ST2497 strains, KLEB-CRE-TBH-0080 (Accession Number NZ_JAWQUU010000001.1), KLEB-CRE-M09-0012 (Accession Number NZ_JAWQWY010000001.1), and KLEB-CRE-M09-0005 (Accession Number NZ_JAWQXC010000001.1) harbored an identical AMR gene repertoire (oqxA, oqxB, blaSHV-182, fosA), but none of these genes was in the immediate vicinity of blaCTX-M-15. In contrast, ST101:960186733 strain (ST101, Accession Number NZ_CP023487.1) possessed the aminoglycoside resistance gene aac(3)-IIa positioned 5,153 bp downstream of the blaCTX-M-15 gene, followed by other AMR genes including catB, blaOXA-1, aac(6′)-Ib-cr, sul2, aph(3″)-Ib, and aph(6)-Id. Comparative analysis with another K. pneumoniae ST101 genome (1101124_000000F-arrow, Accession Number NZ_JAFENJ010000006.1) revealed that ST101:960186733 harbored the blaCTX-M gene on a 22,270 bp insertion bounded by ISEcp1 on one end (Figure 4B) and a 20,394 bp genomic island immediately downstream of ISEcp1 (Supplementary Figure S6). BLAST analysis of this insertion revealed several plasmids with >85% coverage and 100% sequence identity. Notably, the AMR gene cluster was also identified in plasmids pB16KP0141–1 (IncF, Accession Number CP052538.1) and pEc21617-310 (IncH, Accession Number MG878867.1) (Figure 4B).
Four K. pneumoniae strains from Uganda were assigned to ST39 (n = 2), ST231 (n = 1), and ST1119 (n = 1) (Supplementary Table S5). All carried the ISEcp1/blaCTX-M-15/wbuC unit. Two strains, both annotated as MAKM-3381 but with distinct accession numbers and genome lengths, were designated as MAKM-3381A (ST39, Accession Number CP129122.1) and MAKM-3381B (ST1119, Accession Number CP129541.1) to distinguish them. Along with MAKM-5490 (ST39, Accession Number CP130492.1), these strains shared an identical resistance architecture featuring blaTEM-1B located 2,821 bp upstream of blaCTX-M-15 in the opposite orientation. Immediately downstream of blaTEM-1B was the aminoglycoside-encoding gene aac(3)-IIa. These AMR genes existed on a 45,632 bp genomic island containing virulence genes encoding the type VI secretion system (hcp, tssL, and tssK), mercury resistance (merR, merT, merP, merC, merA), and various IS elements (Supplementary Figure S7). Notably, this genomic island also exhibited the repA gene that encodes the IncFII family plasmid replication initiator RepA.
On the other hand, MAKM-RS081 (ST231, Accession Number CP129536.1) lacked blaTEM-1B and did not possess any additional AMR genes in the vicinity of blaCTX-M-15. Both K. pneumoniae strains from Tanzania belonged to ST437 and carried blaCTX-M-15 flanked by upstream ISEcp1 and downstream wbuC. No additional AMR genes were identified in the vicinity of the ESBL gene.
Among the 17 K. pneumoniae strains from Sudan, two (S-P-N-044.01, Accession Number CP092693.1 and S-P-C-024.01, Accession Number CP092807.1) were assigned to ST11, while the remaining 15 could not be definitively typed due to partial or low-identity alleles in the MSLT housekeeping genes. Yet, all strains carried ISEcp1 upstream of blaCTX-M but lacked the downstream wbuC element. Instead, all the strains featured the IS903B insertion sequence immediately downstream of blaCTX-M-14. In addition, all the strains harbored several resistance genes, including blaSHV-182, blaTEM-1B, and the carbapenemase-encoding blaKPC-2, although none of these were co-localized with blaCTX-M.
blaCTX-M-harboring MDR insertions existed in clonally unrelated strains
Analysis of Core-SNP-based phylogenetic trees showed a tendency for regional clustering, while the presence of blaCTX-M-harboring MDR insertions was not related to strain clonality (Figure 5). For instance, E. coli strains from Ethiopia formed two distinct clades, and Malawi strains were closely related (Figure 5A). Yet, E. coli Past_Mal_15 (ST38, Ethiopia) harbored the qnrS1 gene on a large blaCTX-M-carrying insertion, while the insertion in the clonally related E. coli Past_Mal_10 (ST38, Ethiopia) (Figure 5A) lacked AMR genes other than blaCTX-M (Figure 1). Similarly, Malawian E. coli strain CAC124 (ST5640) had a 12,837 bp blaCTX-M-carrying MDR insertion (Figure 1), but its closest relative from South Africa (strain 131) lacked AMR genes around blaCTX-M. Also, most Sudanese K. pneumoniae strains clustered together, while three out of four South African strains formed a clade (Figure 5B). South African K. pneumoniae strain ST101:960186733, with a 22,270 bp blaCTX-M-harboring MDR insertion, was closely related to Ugandan strain MAKM-RS081 (ST231) (Figure 5B), which had no AMR genes near blaCTX-M. These discrepancies were also observed for closely related K. pneumoniae strains MAKM-3381B (with a large blaCTX-M-carrying insertion) and S-P-N-031.01 (lacking a large blaCTX-M-carrying insertion), as well as MIN-106 (with a large blaCTX-M-carrying insertion) and S-P-N-042.01 (lacking a large blaCTX-M-carrying insertion).
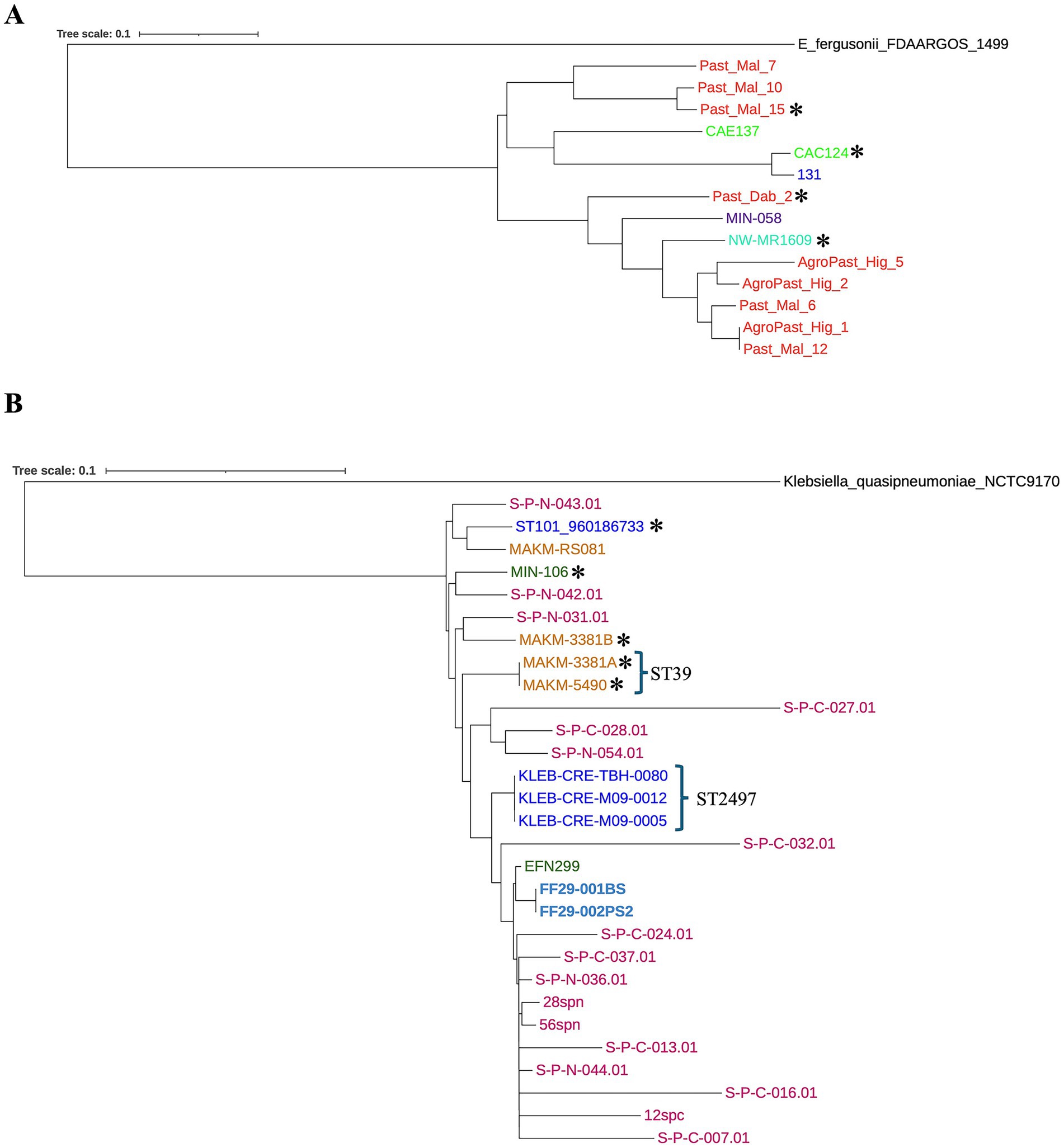
Figure 5. Core-SNP-based phylogenetic trees for E. coli (A) and K. pneumoniae (B). Clustering was seen among strains from the same geographic location. There was no obvious clustering based on the presence large blaCTX-M-carrying MDR chromosomal insertions. Strains from the same country are indicated by the same color. Asterisks (*) represent the presence of large blaCTX-M-carrying MDR chromosomal insertions.
Discussion
In this study, we utilized publicly available WGS data to investigate the burden and genetic context of chromosomally integrated blaCTX-M genes among Enterobacteriaceae species from Africa. Chromosomal blaCTX-M was detected in ~16% of genomes analyzed, spanning nine out of 18 countries. The most frequently identified allele was blaCTX-M-15, found in two-thirds of the sequences. The blaCTX-M gene was located within the chromosomal insertions flanked by ISEcp1 or IS26 elements, highlighting the central role of these mobile elements in facilitating stable chromosomal integration of this clinically significant ESBL determinant. Notably, some of these insertions also harbored additional AMR genes, underscoring their potential to encode chromosomal MDR.
The high prevalence of chromosomal blaCTX-M may reflect strong selection pressure arising from widespread antimicrobial use in clinical settings. Chromosomal integration offers a potential evolutionary advantage by reducing fitness costs and structural instability typically associated with plasmid carriage, as well as mitigating the risk of plasmid loss through segregational events during cell division (Gu et al., 2015). The strains examined in this study appear to have acquired secondary chromosomal integration of plasmid-derived blaCTX-M via two main mechanisms. In the majority of sequences, the ISEcp1 gene was located upstream of blaCTX-M, consistent with ISEcp1-mediated transposition (Poirel et al., 2005). The presence of MDR islands bounded by ISEcp1 further suggests that this element can transpose large DNA segments. These MDR islands often included additional IS elements, indicating that chromosomal integration may occur through a multistep process involving successive transposition events. Nevertheless, the identification of nearly identical insertions in both plasmid and chromosomal sequences supports the possibility of a single-step integration.
Notably, in strains lacking ISEcp1, insertions were flanked by IS26 elements. Previous studies have shown that IS26-mediated transposition can occur via a cointegration mechanism, leading to duplication of IS26 and the formation of 8 bp TSDs (Harmer et al., 2014). Alternatively, a single IS26 element with adjacent “passenger” genes can form a translocatable unit, which may insert next to another IS26 copy through conservative transposition that does not generate TSDs (Harmer et al., 2014). We identified 8 bp TSDs flanking two directly oriented IS26 elements on either end of a 75 kbp chromosomal insertion encoding MDR. This observation is consistent with IS26-mediated replicative transposition. However, comparison with matching GeneBank sequences revealed notable differences in the arrangement of genes within these insertions. Given the presence of multiple IS copies across the segment, it is likely that the genes were integrated through a multistep process involving several mobile gene elements. We speculate that the initial event may have been a copy-in transposition involving a single IS26 element and associated genes, leading to the formation of a second IS26 and TSDs. This could have been followed by a series of conservative (non-replicative) transposition events, whereby translocatable units targeted pre-existing IS26 sites within the growing composite island (Harmer and Hall, 2024).
Meanwhile, in one E. cloacae strain, the MDR insertion was flanked by int immediately upstream of ISEcp1, suggesting that chromosomal integration may have occurred via site-specific recombination. However, the involvement of ISEcp1 cannot be excluded, given its role in mobilizing resistance elements. Notably, this MDR segment was detected in numerous plasmid and chromosome sequences in the GeneBank, all of which lacked the flanking int gene but possessed ISEcp1 and IS26 on opposite ends. This pattern suggests that structural rearrangement, potentially mediated by ISEcp1 and/or IS26, may have occurred following initial integration.
The observation of similar genetic architectures shared by clonally unrelated strains highlights the frequency with which chromosomal integration occurs by horizontal gene transfer. For instance, the qnrS1 gene was found precisely 4,640 bp downstream of blaCTX-M in two genetically distinct E. coli strains (ST38 and ST8130), as well as a strain previously identified as S. sonei but reassigned to E. coli ST484. This suggests that an identical DNA segment was horizontally acquired by diverse genetic backgrounds. In parallel, we also observed evidence of the spread of chromosomal blaCTX-M by clonal expansion. Three of the four K. pneumoniae strains from South Africa belonged to the same ST (2497) and carried the same AMR profiles. These three strains were recovered from patients admitted to the same hospital in Cape Town (Marais et al., 2024), supporting a likely scenario of patient-to-patient transmission. Similarly, the two K. pneumoniae ST39 from Uganda and the two ST437 strains from Tanzania shared indistinguishable resistance gene arrangements, consistent with local clonal dissemination. In Sudan, while only two of the 17 K. pneumoniae strains were typed (both as ST11), the remaining 15 untyped strains also harbored the same set of AMR genes, suggesting either the expansion of a single dominant clone or recurrent horizontal acquisition of a common resistance island by unrelated strains. Generally, most strains from different countries did not share STs, and when the same ST was observed across borders, the associated AMR profiles differed. For example, K. pneumoniae ST11 from Ghana and Sudan had blaCTX-M-15 and blaCTX-M-14, respectively, and the two strains exhibited distinct resistance architectures, indicating independent acquisition events and no evidence of inter-country clonal spread. Phylogenetic analysis revealed regional clustering of E. coli and K. pneumoniae strains, but the presence of blaCTX-M-harboring MDR insertions did not correlate with strain clonality, highlighting the role of horizontal gene transfer in MDR dissemination.
Interestingly, the MDR region identified in the Malawian strain CAC124 (ST5640) was detected in the chromosomes of over 30 E. coli ST131 strains (Supplementary Table S3). While CAC124 most likely acquired this segment through horizontal gene transfer, its consistent presence within the ST131 lineage suggests subsequent clonal dissemination. The broad geographic distribution of E. coli ST131 carrying a chromosomally integrated MDR-encoding genetic island could suggest an emerging clonal wave of extraintestinal pathogenic E. coli, particularly given that ST131 is already recognized as a pandemic clone.
The presence of multiple genomic islands in the analyzed genomes highlights the high frequency of horizontal gene transfer events and the potential role of factors beyond AMR genes for virulence and fitness enhancement. Furthermore, detecting plasmid replicons on chromosomes confirms the horizontal origins of these factors. While exceptionally large plasmids (>1.7 Mbp) have been reported in rare cases (Kothari et al., 2019), all the replicon-harboring contigs in this study were larger than 4.9 Mbp (Table 2), ruling out their possibility of being plasmids.
This study is not without limitations. First, some strains could not be assigned STs due to missing loci or nucleotide ambiguities within the MLST regions. Second, we assumed that all genomic sequences exceeding 1,000,000 bp represented chromosomal sequences, which may not always be true, as described above. Third, our approach may have excluded chromosomal segments <1,000,000 bp, as many genome assemblies are incomplete. Lastly, a strain previously reported as S. sonei by Microflex LT MALDI-TOF MS (Bruker Daltonik, GmbH, United Kingdom) (Portal et al., 2024) was reassigned to E. coli ST484 based on our WGS analysis. This discrepancy is not unexpected, as Shigella species are phylogenetically nested within E. coli, prompting ongoing discussions to reclassify Shigella as a sublineage of E. coli (Lan and Reeves, 2002).
Conclusion
We used publicly available WGS data to characterize the genetic landscape of chromosomally integrated blaCTX-M among Enterobacteriaceae strains from Africa. Approximately 16% of the analyzed genomes carried chromosomally encoded blaCTX-M, many of which could be linked to plasmid-derived origins. In several cases, the blaCTX-M-containing chromosomal insertions also harbored additional AMR genes, resulting in genotypically MDR chromosomal segments. While widespread clonal dissemination was observed in the globally dominant E. coli ST131 lineage, clonal spread among African strains appeared localized, often limited to individual hospital settings. Our findings underscore an evolutionary strategy by which Enterobacteriaceae stabilize and preserve MDR traits through chromosomal integration, potentially ensuring long-term persistence even in the absence of plasmid-mediated transmission.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
MSh: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Validation, Visualization, Writing – original draft. HC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. HK: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. CS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. JC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. SN: Conceptualization, Data curation, Formal analysis, Methodology, Software, Validation, Visualization, Writing – original draft. SO: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. MSa: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Validation, Visualization, Writing – original draft. TZ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. SM: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft. MSim: Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft. MNs: Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft. JYC: Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Visualization, Writing – original draft. FC: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft. MNu: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. JN: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. MSin: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. KH: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Visualization, Writing – review & editing. NN: Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing – review & editing. RC: Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – review & editing. HS: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing. YS: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing. BH: Conceptualization, Formal analysis, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing. MK: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Writing – review & editing. HH: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research project was supported in part by the Japan Program for Infectious Diseases Research and Infrastructure (JP20wm0125008) under the Japan Agency for Medical Research and Development (AMED). This work was also partly supported by the Japan Agency for Medical Research and Development (AMED) (grant no. JP223fa627005) to HS and YS. The funders had no role in study design, data collection and analysis, publication decisions, or manuscript preparation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1655907/full#supplementary-material
Footnotes
References
Ashida, H., and Sasakawa, C. (2015). Shigella Ipa H family effectors as a versatile model for studying pathogenic bacteria. Front. Cell. Infect. Microbiol. 5:100. doi: 10.3389/fcimb.2015.00100
Bassetti, M., Pecori, D., Sibani, M., Corcione, S., and De Rosa, F. G. (2015). Epidemiology and treatment of MDR Enterobacteriaceae. Curr. Treat. Options Infect. Dis. 7, 291–316. doi: 10.1007/s40506-015-0065-1
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing GroupLau, B. Y., Hoad, G., et al. (2017). Islandviewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Beyene, A. M., Gizachew, M., Yousef, A. E., Haileyesus, H., Abdelhamid, A. G., Berju, A., et al. (2024). Multidrug-resistance and extended-spectrum beta-lactamase-producing lactose-fermenting Enterobacteriaceae in the human-dairy interface in Northwest Ethiopia. PLoS One 19:e0303872. doi: 10.1371/journal.pone.0303872
Carattoli, A., Zankari, E., García-Fernández, A., Voldby Larsen, M., Lund, O., Villa, L., et al. (2014). In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Carroll, A. C., and Wong, A. (2018). Plasmid persistence: costs, benefits, and the plasmid paradox. Can. J. Microbiol. 64, 293–304. doi: 10.1139/cjm-2017-0609
Castanheira, M., Simner, P. J., and Bradford, P. A. (2021). Extended-spectrum β-lactamases: an update on their characteristics, epidemiology and detection. JAC Antimicrob Resist. 3:dlab092. doi: 10.1093/jacamr/dlab092
Darling, A. C., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D., and Aarestrup, F. M. (2022). Res finder - an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. 8:000748. doi: 10.1099/mgen.0.000748
Gomi, R., Yamamoto, M., Tanaka, M., and Matsumura, Y. (2022). Chromosomal integration of blaCTX-M genes in diverse Escherichia coli isolates recovered from river water in Japan. Curr. Res. Microb. Sci. 3:100144. doi: 10.1016/j.crmicr.2022.100144
Goswami, C., Fox, S., Holden, M. T. G., Connor, M., Leanord, A., and Evans, T. J. (2020). Origin, maintenance and spread of antibiotic resistance genes within plasmids and chromosomes of bloodstream isolates of Escherichia coli. Microb Genom. 6:e000353. doi: 10.1099/mgen.0.000353
Gu, P., Yang, F., Su, T., Wang, Q., Liang, Q., and Qi, Q. (2015). A rapid and reliable strategy for chromosomal integration of gene (s) with multiple copies. Sci. Rep. 5:9684. doi: 10.1038/srep09684
Guy, L., Kultima, J. R., and Andersson, S. G. (2010). GenoPlotR: comparative gene and genome visualization in R. Bioinformatics 26, 2334–2335. doi: 10.1093/bioinformatics/btq413
Harmer, C. J., and Hall, R. M. (2024). IS26 and the IS26 family: versatile resistance gene movers and genome reorganizers. Microbiol. Mol. Biol. Rev. 88:e0011922. doi: 10.1128/mmbr.00119-22
Harmer, C. J., Moran, R. A., and Hall, R. M. (2014). Movement of IS26-associated antibiotic resistance genes occurs via a translocatable unit that includes a single IS26 and preferentially inserts adjacent to another IS26. MBio 5, e01801–e01814. doi: 10.1128/mBio.01801-14
Horakova, K., Mlejnkova, H., and Mlejnek, P. (2008). Specific detection of Escherichia coli isolated from water samples using polymerase chain reaction targeting four genes: cytochrome bd complex, lactose permease, beta-D-glucuronidase, and beta-D-galactosidase. J. Appl. Microbiol. 105, 970–976. doi: 10.1111/j.1365-2672.2008.03838.x
Hsiao, W., Wan, I., Jones, S. J., and Brinkman, F. S. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420. doi: 10.1093/bioinformatics/btg004
Jolley, K. A., and Maiden, M. C. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Komori, K., Aoki, K., Harada, S., Ishii, Y., and Tateda, K. (2024). Plasmid-mediated acquisition and chromosomal integration of Bla (CTX-M-14) in a subclade of Escherichia coli ST131-H30 clade C1. Antimicrob. Agents Chemother. 68:e0081724. doi: 10.1128/aac.00817-24
Kothari, A., Wu, Y. W., Chandonia, J. M., Charrier, M., Rajeev, L., Rocha, A. M., et al. (2019). Large circular plasmids from groundwater Plasmidomes span multiple incompatibility groups and are enriched in multimetal resistance genes. MBio 10:e02899-18. doi: 10.1128/mBio.02899-18
Lan, R., and Reeves, P. R. (2002). Escherichia coli in disguise: molecular origins of Shigella. Microbes Infect. 4, 1125–1132. doi: 10.1016/S1286-4579(02)01637-4
Langille, M. G., Hsiao, W. W., and Brinkman, F. S. (2008). Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9:329. doi: 10.1186/1471-2105-9-329
Letunic, I., and Bork, P. (2024). Interactive tree of life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–w82. doi: 10.1093/nar/gkae268
Marais, G., Moodley, C., Claassen-Weitz, S., Patel, F., Prentice, E., Tootla, H., et al. (2024). Carbapenem-resistant Klebsiella pneumoniae among hospitalized patients in Cape Town, South Africa: molecular epidemiology and characterization. JAC Antimicrob. Resist. 6:dlae050. doi: 10.1093/jacamr/dlae050
Mikhayel, M., Praud, K., Leclercq, S., Karam Sarkis, D., and Doublet, B. (2024). Genomic insights into epidemic plasmids carrying Bla (CTX-M) and mcr-1 genes in Escherichia coli from Lebanese broiler production. JAC Antimicrob. Resist. 6:dlae149. doi: 10.1093/jacamr/dlae149
Mshana, S. E., Fritzenwanker, M., Falgenhauer, L., Domann, E., Hain, T., Chakraborty, T., et al. (2015). Molecular epidemiology and characterization of an outbreak causing Klebsiella pneumoniae clone carrying chromosomally located blaCTX-M-15 at a German university-hospital. BMC Microbiol. 15:122. doi: 10.1186/s12866-015-0460-2
Murray, C. J. L., Ikuta, K. S., Sharara, F., Swetschinski, L., Robles Aguilar, G., Gray, A., et al. (2022). Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655. doi: 10.1016/S0140-6736(21)02724-0
Nicolas-Chanoine, M. H., Bertrand, X., and Madec, J. Y. (2014). Escherichia coli ST131, an intriguing clonal group. Clin. Microbiol. Rev. 27, 543–574. doi: 10.1128/CMR.00125-13
Olaitan, M. O., Orababa, O. Q., Shittu, R. B., Obunukwu, G. M., Kade, A. E., Arowolo, M. T., et al. (2025). Prevalence of ESBL-producing Escherichia coli in sub-Saharan Africa: a meta-analysis using a one health approach. One Health 20:101090. doi: 10.1016/j.onehlt.2025.101090
Poirel, L., Lartigue, M. F., Decousser, J. W., and Nordmann, P. (2005). ISEcp1B-mediated transposition of blaCTX-M in Escherichia coli. Antimicrob. Agents Chemother. 49, 447–450. doi: 10.1128/AAC.49.1.447-450.2005
Portal, E. A. R., Sands, K., Farley, C., Boostrom, I., Jones, E., Barrell, M., et al. (2024). Characterisation of colistin resistance in gram-negative microbiota of pregnant women and neonates in Nigeria. Nat. Commun. 15:2302. doi: 10.1038/s41467-024-45673-6
San Millan, A., Toll-Riera, M., Qi, Q., and Mac Lean, R. C. (2015). Interactions between horizontally acquired genes create a fitness cost in Pseudomonas aeruginosa. Nat. Commun. 6:6845. doi: 10.1038/ncomms7845
Shawa, M., Furuta, Y., Mulenga, G., Mubanga, M., Mulenga, E., Zorigt, T., et al. (2021). Novel chromosomal insertions of IS Ecp1-Bla CTX-M-15 and diverse antimicrobial resistance genes in Zambian clinical isolates of Enterobacter cloacae and Escherichia coli. Antimicrob. Resist. Infect. Control 10, 1–16. doi: 10.1186/s13756-021-00941-8
Shawa, M., Hayashida, K., Nao, N., Paudel, A., Kamboyi, H., Chambaro, H., et al. (2025). Development of a PCR-dipstick DNA chromatography-based tool for the detection of CTX-M- and TEM-producing Escherichia coli and Klebsiella pneumoniae isolated from patients in Kafue and Katete districts of Zambia. BMC Infect. Dis. 25:541. doi: 10.1186/s12879-025-10628-9
Shen, W., Le, S., Li, Y., and Hu, F. (2016). SeqKit: a cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS One 11:e0163962. doi: 10.1371/journal.pone.0163962
Tanizawa, Y., Fujisawa, T., and Nakamura, Y. (2018). DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34, 1037–1039. doi: 10.1093/bioinformatics/btx713
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142. doi: 10.1186/1471-2105-7-142
WHO. Antimicrobial resistance: global report on surveillance (2014). Available online at: https://www.who.int/publications/i/item/9789241564748.
Wickham, H., François, R., Henry, L., Müller, K., and Vaughan, D. (2023). Dplyr: A grammar of data manipulation. R package version, 1.1.4. Available online at: https://CRAN.R-project.org/package=dplyr.
Yoon, E.-J., Gwon, B., Liu, C., Kim, D., Won, D., Park, S. G., et al. (2020). Beneficial chromosomal integration of the genes for CTX-M extended-spectrum β-lactamase in Klebsiella pneumoniae for stable propagation. mSystems 5, e00459–e00420. doi: 10.1128/mSystems.00459-20
Keywords: blaCTX-M , Enterobacteriaceae , Africa, chromosomal, ISEcp1, IS26
Citation: Shawa M, Chambaro H, Kamboyi HK, Sulwe C, Chizimu JY, Nasilele SJ, Ogata S, Samutela M, Zorigt T, Mudenda S, Simbotwe M, Nsofwa M, Chanda J, Chabala F, Nundwe M, Ndebe J, Sinjani M, Hayashida K, Nao N, Chilengi R, Sawa H, Suzuki Y, Hang'ombe B, Kajihara M and Higashi H (2025) In silico characterization of chromosomally integrated blaCTX-M genes among clinical Enterobacteriaceae in Africa: insights from whole-genome analysis. Front. Microbiol. 16:1655907. doi: 10.3389/fmicb.2025.1655907
Edited by:
Hazem Ramadan, Mansoura University, EgyptReviewed by:
Ahmed Mahrous Soliman, Kafrelsheikh University, EgyptAngeliki Mavroidi, General University Hospital of Patras, Greece
Copyright © 2025 Shawa, Chambaro, Kamboyi, Sulwe, Chizimu, Nasilele, Ogata, Samutela, Zorigt, Mudenda, Simbotwe, Nsofwa, Chanda, Chabala, Nundwe, Ndebe, Sinjani, Hayashida, Nao, Chilengi, Sawa, Suzuki, Hang’ombe, Kajihara and Higashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Misheck Shawa, bWlzaGVja3NoYXdhQGN6Yy5ob2t1ZGFpLmFjLmpw