 Roberta Loddo1†
Roberta Loddo1† Matteo Incerti2*†
Matteo Incerti2*† Elda Favari2
Elda Favari2 Valeria Manca1
Valeria Manca1 Marta Cogoni1
Marta Cogoni1 Rebecca Piras1Luca Virdis1Vanessa Palmas1
Rebecca Piras1Luca Virdis1Vanessa Palmas1 Elena Tamburini1Paolo La Colla1
Elena Tamburini1Paolo La Colla1 Giuseppina Sanna1*
Giuseppina Sanna1*- 1Dipartimento di Scienze Biomediche, Sezione di Microbiologia e Virologia, Università degli Studi di Cagliari, Monserrato, Italy
- 2Dipartimento di Scienze degli Alimenti e del Farmaco, Università degli Studi di Parma, Parma, Italy
A new class of 1,2-benzisothiazol-3(2H)-one benzenesulfonamides has been synthesized. In cell-based assays, the lead compound 6 inhibits the replication of HIV-1, HIV-2, and HIV-1 variants carrying clinically relevant mutations against non-nucleoside, nucleoside, and protease inhibitors. In enzyme assays, compound 6 does not inhibit HIV-1 reverse transcriptase and integrase. Genome sequencing of HIV-1 mutants selected for resistance to compound 6 reveals no mutations in the pol or env genes. Instead, two mutations are mapped in the gag region, which encodes nucleocapsid (NC) proteins involved in early and late key processes of retrovirus replication, suggesting that NC proteins are the target of the title compounds. Compound 6 shows concentration-dependent virucidal activity against cell-free HIV-1 and HIV-2. Benzisothiazol-3(2H)-one benzenesulfonamides are a new class of antiretroviral agents with an intriguing spectrum and mode of action.
1 Introduction
HIV infection leads to acquired immunodeficiency syndrome (AIDS) through progressive destruction of the immune system and degeneration of the central and peripheral nervous systems. One of the greatest achievements of modern medicine has been the development of antiretroviral therapy (ART) to prevent and treat HIV infection. However, the HIV pandemic is still increasing. Approximately 1.3 million people become infected with HIV each year, and about 630,000 die from HIV-related complications (World Health Organization, 2025). There is continued interest in the development of therapeutics against old and new targets in the HIV replication cycle, despite significant progress in the treatment of HIV/AIDS (Landovitz et al., 2023; Sever et al., 2024).
A large number of viral targets susceptible to selective inhibition or host-dependent factors have been identified (Puhl et al., 2019; Kinch and Patridge, 2014). These include the envelope CD4 binding protein gp120 and the fusion protein gp41; the key enzymes in HIV replication, reverse transcriptase (RT), integrase (IN), and protease (PR); regulatory proteins, such as TAT and C; and core proteins, such as the nucleocapsid NCp7 (De Clercq, 2002; Musah, 2004; Druillennec and Roques, 2000); however, the intrinsic high genetic variability of HIV combined with the error-prone nature of RT, which continuously generates mutants capable of overcoming inhibition by all known drugs, requires a combination of antiretroviral drugs with different modes of action.
The ability to target RT with two different classes of inhibitors, nucleoside (NRTIs) and non-nucleoside (NNRTIs), has made it possible to prove the efficacy of highly active antiretroviral therapy (HAART), initially based on triple combinations of drugs belonging to NRTIs, NNRTIs, and PR inhibitors (PRIs) (Flexner, 2007). Currently, NNRTIs, protease inhibitors, integrase inhibitors, and entry inhibitors are the five main classes of antiretroviral agents used in combination with ART (Melo et al., 2020; Shin et al., 2021).
For reasons such as virological failure (development of drug resistance and drug–drug interaction), adverse effects, convenience, or cost, regimens may need to be constantly updated (Gandhi et al., 2025; Battini and Bollini, 2019; Gill et al., 2019). Therefore, novel anti-HIV-1 agents are required to overcome these drawbacks.
This study is an attempt to explore the potential of a new class of small molecule HIV inhibitors, 1,2-benzisothiazol-3(2H)-one benzenesulfonamides, which emerged from our research into biologically active benzisothiazole derivatives (Vicini et al., 2006; Vicini et al., 2007; Vicini et al., 2008; Vicini et al., 2009; Zani et al., 2009). The synthetic modulation of the basic scaffold, aimed at elucidating SAR analysis, led to structures 6–24 (Figure 1), which are endowed with antiretroviral activity in vitro at the micromolar level. Their potency is in the range reported by other researchers for benzisothiazolones and nucleocapsid (NC) inhibitors belonging to other classes (Goel et al., 2002; Sancineto et al., 2018). However, in addition to being capable of preventing the replication of HIV type-1 (HIV-1), type-2 (HIV-2), and HIV-1 strains resistant to the major drug classes currently used in clinics, these compounds show concentration-dependent inactivation of cell-free HIV-1 and HIV-2 infectivity in cell-based assays.
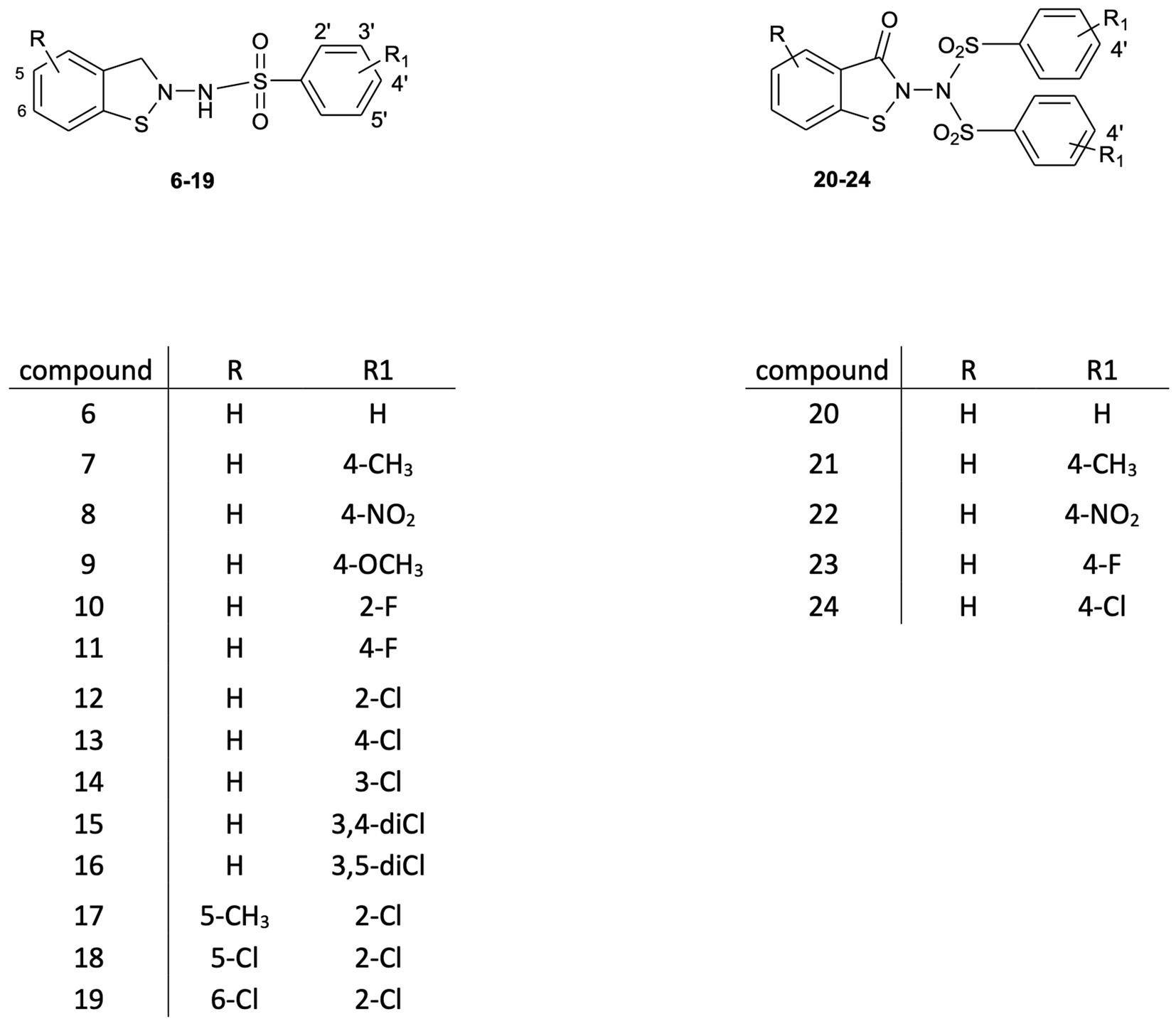
Figure 1. Chemical structure of 1,2-benzisothiazol-3(2H)-one benzensulfonamides 6–24.
2 Materials and methods
2.1 Chemistry
Unless otherwise noted, reagents and solvents were purchased from commercial suppliers and used without purification. The progress of the reaction was monitored by thin-layer chromatography using F254 silica gel-precoated sheets, and spots were detected under a UV lamp at 254 nm. Flash chromatography was performed using Merck Silica Gel 60 (Si 60, 40–63 μm, 230–400 mesh ASTM; Merck KGaA, Darmstadt, Germany). Melting points were determined using a Gallenkamp melting point apparatus (Gallenkamp Labs, Cambridge, UK) and were not corrected. Infrared (IR) spectra were recorded as KBr pellets on a Jasco FT-IR 300E spectrophotometer (Jasco Ltd., Tokyo, Japan). Absorbance values are reported as ν (cm−1). 1H NMR spectra were recorded on a Bruker Avance 400 spectrometer (400 MHz; Bruker BioSpin GmbH, Rheinstetten, Germany). Chemical shifts (δ scale) are reported in parts per million (ppm). 1H NMR spectra were reported in the following order: multiplicity, approximate coupling constant (J value) in hertz (Hz), and number of protons. Signals were characterized as follows: s (singlet), d (doublet), t (triplet), and m (multiplet). Mass spectra were recorded on an API-150 EX system spectrometer (Applied Biosystems, MA, USA) with an electrospray ionization (ESI) interface. The final compounds were analysed for C, H, and N, and the percentages found were within ±0.4% of the theoretical values.
2.2 General experimental procedures
Bis(2-carboxyphenyl)disulphide (1a) was purchased from Sigma-Aldrich, while Bis(4-methyl-2-carboxyphenyl)disulphide (1b), Bis(4-chloro-2-carboxyphenyl)disulphide (1c), and Bis(5-chloro-2-carboxyphenyl)disulphide (1d) were prepared according to a modified procedure (Saha et al., 2017; Wu et al., 2023) outlined in Scheme 1. The suitably substituted 2-aminobenzoic acid was diazotized and then converted into an ester intermediate by a Sandmeyer reaction using potassium ethyloxantogenate. Next, upon alkaline hydrolysis and subsequent acidification, 2-mercaptoarylcarboxylic acid was obtained. Oxidation of the thiol group with iodine afforded key intermediates 1b-1d.
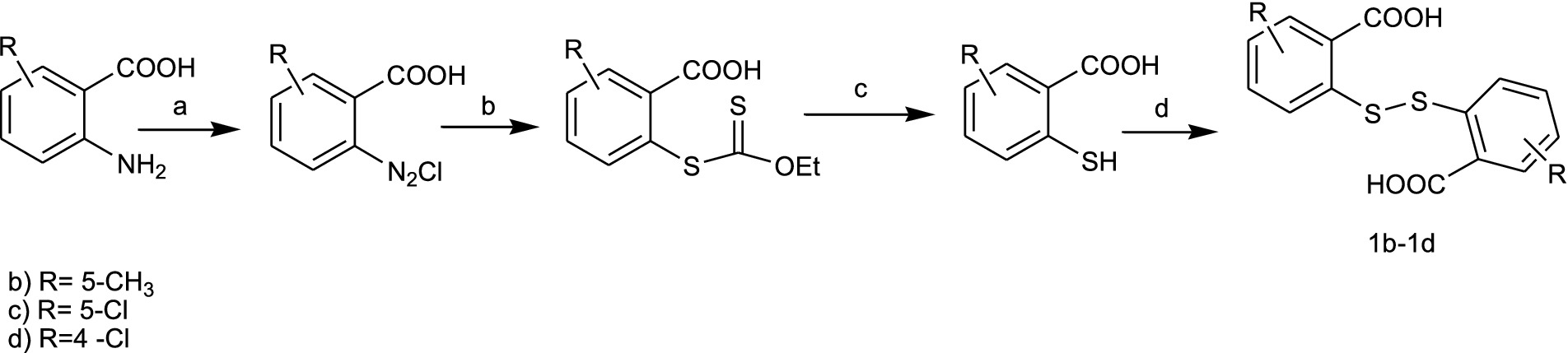
SCHEME 1. Synthesis of the key intermediates 1b–1d. Reagents and conditions: (a) NaNO2, HCl, 0°C; (b) C3H5KOS2; (c) NaOHaq 10%, reflux, 4h; HClconc; (d) I2. ethanol, 50 °C.
From a modified procedure previously described (Yevich et al., 1986) and outlined in Scheme 2, the target compound 6 was synthesized, starting from Bis(2-carboxyphenyl)disulphide 1a and treated with thionyl chloride to afford intermediate 2, which was then converted into chlorocarbonylphenylsulfenylchloride 3 by treatment with dry chlorine. Tert-butoxycarbonylhydrazine reacted with intermediate 3, affording tert-Butyl-(3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4a, which, after hydrolysis with trichloroacetic acid, yielded 2-amino-1,2-benzisothiazol-3(2H)-one 5a, according to the method previously described by Vicini et al. (1997). Reaction with the appropriate benzenesulfonyl chloride, in pyridine, in a cooling bath, afforded a mixture of N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamides 6 and of the respective N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)-(phenylsulfonyl)benzenesulfonamide 20 (Figure 1).
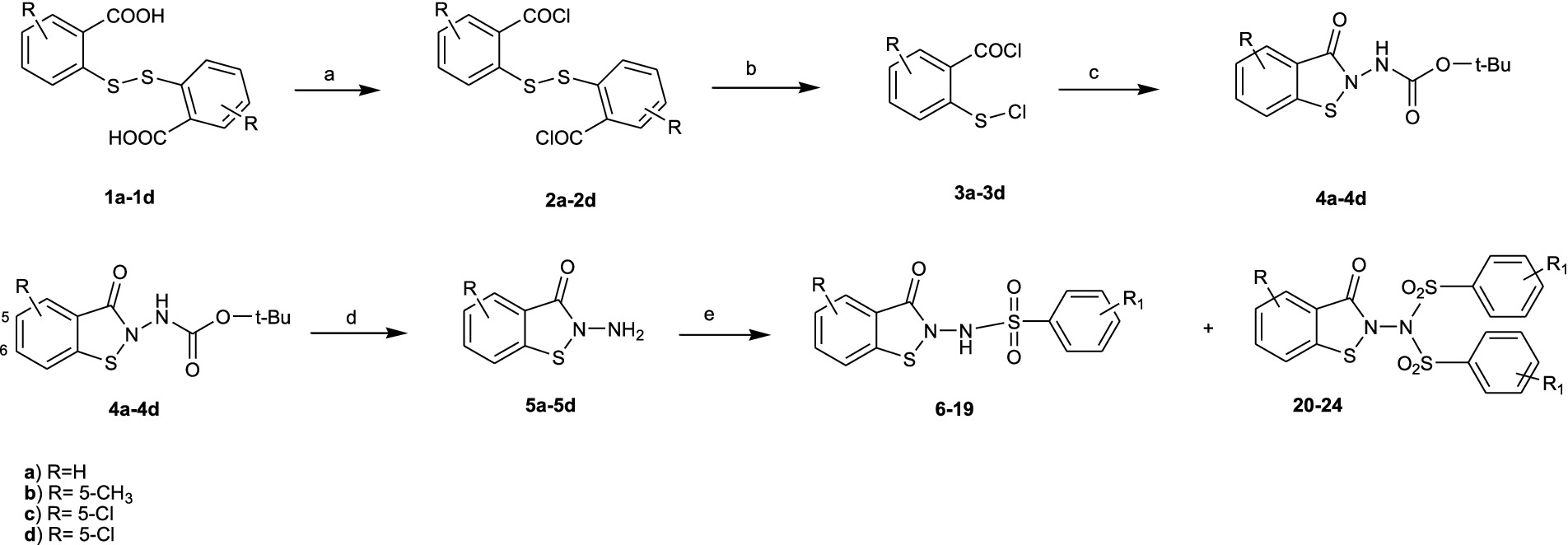
SCHEME 2. Synthesis of the target compounds 6-24. Reagents and conditions: (a) SOCl2, reflux 3h; (b) Cl2, 3h, RT; (c) NH2-NH-Boc, CH2Cl2/pyridine,60 min. 10 °C; (d) CCl3COOH-H2O,150 min, rt; (e) R1- Phenyl-SO2Cl, pyridine –5 °C, 60 min.
Compounds 7–19 and 21–24 were synthesized following the previously described procedure for compounds 6 and 20, starting from the suitably substituted Bis(2-carboxyphenyl)disulphide 1b–1d.
2.2.1 Bis(4-methyl-2-carboxyphenyl)disulphide (1b)
Compound 1b was synthesized following a previously described procedure (Saha et al., 2017; Wu et al., 2023), starting from 5-methyl-2-aminobenzoic acid.
2.2.2 Bis(4-chloro-2-carboxyphenyl)disulphide (1c)
Compound 1c was synthesized following a previously described procedure (Saha et al., 2017; Wu et al., 2023), starting from 5-chloro-2-aminobenzoic acid.
2.2.3 Bis(5-chloro-2-carboxyphenyl)disulphide (1d)
Compound 1d was synthesized following a previously described procedure (Saha et al., 2017; Wu et al., 2023), starting from 4-chloro-2-aminobenzoic acid.
2.2.4 tert-Butyl N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4a
Compound 4a was synthesized following a previously described procedure (Vicini et al., 1997), starting from Bis(2-carboxyphenyl)disulphide (1a).
2.2.5 tert-Butyl N- (5-methyl-3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4b
Compound 4b was synthesized following the procedure described for compound 4a, starting from Bis(4-methyl-2-carboxyphenyl)disulphide (1b). Yield: 77%: White Crystals, mp 182–184 °C (ethyl acetate). 1H NMR (DMSO-d6): δ 10.02 (d, 1H, NH), 7.99 (d, 1H, J = 8.1, H-7), 7.71 (s, 1H, H-4), 7.54 (d, 1H, J = 8.1, H-6), 2.40 (s, 3H, CH3), 1.44 (s, 9H, CH3). IR (KBr): 3178, 2,979, 1745, 1,643. MS (ESI) calcd. For C13H17N2O3S [(M + 1)+]: 281.0960; found: 281.1. Anal. calcd. For C13H16N2O3S C,55.70; H,5.75; N,9.99; found C,55.85; H,5.46; N,9.67.
2.2.6 tert-Butyl N-(5-chloro-3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4c
Compound 4c was synthesized following the procedure described for compound 4a, starting from Bis(4-chloro-2-carboxyphenyl)disulphide (1c). Yield: 87%: White, pale yellowish solid, mp 164–165 °C (ethanol-water). 1H NMR (DMSO-d6): δ 10.11 (s, 1H, NH), 8.01 (d, 1H, J = 8.7, H-7), 7.89 (s, 1H, J = 1.8, H-4), 7.80 (dd, 1H, J = 1.8, J = 8.7, H-6), 1.44 (s, 9H, CH3). IR (KBr): 3186, 2,980, 1751, 1,643. MS (ESI) calcd. For C12H14ClN2O3S [(M + 1)+]: 301.0414; found: 301.7. Anal. calcd. For C12H13ClN2O3S C,47.92; H,4.36; N,9.31; found C,47.89; H,4.43; N,9.14.
2.2.7 tert-Butyl N-(6-chloro-3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4d
Compound 4d was synthesized following the procedure described for compound 4a, starting from Bis(5-chloro-2-carboxyphenyl)disulphide (1d). Yield: 60%, White solid, mp 156–157 °C (ethanol-water). 1H NMR (DMSO-d6): δ 10.08 (s, 1H, NH), 8.08 (s,1H, J = 2.1, H-7), 7.88 (d,1H, J = 8.4, H-4), 7.50 (d, 1H, J = 2.1, J = 8.7, H-5), 1.44 (s, 9H, CH3). IR (KBr): 3209, 2,978, 1755, 1,650. MS (ESI) calcd. For C12H14ClN2O3S [(M + 1)+]: 301.0414; found: 301.7. Anal. calcd. For C12H13ClN2O3S C,47.92; H,4.36; N,9.31; found C,48.02; H,4.47; N,9.08.
2.2.8 2-Amino-1,2-benzisothiazol-3(2H)-one 5a
Compound 5a was synthesized following a previously described procedure (Vicini et al., 1997), starting from tert-Butyl N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4a.
2.2.9 2-Amino-5-methyl-1,2-benzisothiazol-3(2H)-one 5b
Compound 5b was synthesized following a previously described procedure (Vicini et al., 1997), starting from tert-Butyl N-(5-methyl-3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4b.
2.2.10 2-Amino-5-chloro-1,2-benzisothiazol-3(2H)-one 5c
Compound 5c was synthesized following a previously described procedure (Vicini et al., 1997), starting from tert-Butyl N-(5-chloro-3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4c. Yield: 47%, white solid, mp 140–141 °C (ethanol). 1H NMR (DMSO-d6): δ 7.93 (d, 1H, J = 8.4, H-7), 7.82 (s, 1H, J = 2.1, H-4), 7.72 (d, 1H, J = 2.1, J = 8.7, H-6), 5.65 (s, 2H, NH2). IR (KBr): 3305, 3,188, 1,676. MS (ESI) calcd. For C7H6ClN2OS [(M + 1)+]: 200.9889; found: 201.6. Anal. calcd. For C7H5ClN2OS C,41.90; H,2.51; N,13.96; found C,42.02; H,4.2.56; N,13.83.
2.2.11 2-Amino-6-chloro-1,2-benzisothiazol-3(2H)-one 5d
Compound 5d was synthesized following a previously described procedure (Vicini et al., 1997), starting from tert-Butyl N-(6-chloro-3-oxo-1,2-benzisothiazol-2(3H)-yl)carbamate 4d. Yield: 22%, pale yellow-pinkish crystals, mp 170–172 °C (ethanol). 1H NMR (DMSO-d6): δ 8.04 (s, 1H, J = 1.8, H-7), 7.82 (d, 1H, J = 1.8, H-4), 7.43 (dd, 1H, J = 1.8, J = 8.4, H-5), 5.07 (s, 2H, NH2). IR(KBr): 3296, 3,176, 1,653. MS (ESI) calcd. For C7H6ClN2OS [(M + 1)+]: 200.9889; found: 201.3. Anal. calcd. For C7H5ClN2OS C,41.90; H,2.51; N,13.96; found C,42.18; H,0.2.81; N,13.56.
2.2.12 N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 6 and N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)-N-(phenylsulfonyl)benzenesulfonamide 20
2-amino-1,2-benzisothiazol-3(2H)-one (5a) (10 mmol) was dissolved in anhydrous pyridine (8 mL) and cooled to 0 °C. Benzenesulfonyl chloride (11 mmol) was slowly added to the stirred solution under a nitrogen atmosphere. After the addition was complete, the reaction mixture was warmed to room temperature and stirred for 2 h. Crushed ice was added, and the mixture was acidified with concentrated hydrochloric acid until a precipitate was formed. The resulting suspension was filtered under vacuum, and the residue was washed with water. The obtained solid was purified by flash chromatography[SiO2, CH2Cl2: EtOH 95:5] to obtain, in order of elution, the first fraction, which was identified as compound 20, and the second fraction, which was identified as compound 6.
N-(3-Oxo-1,2-benzisothiazol-2(3H)-yl)-N-(phenylsulfonyl) benzenesulfonamide 20. Yield: 40%. Pale yellow solid, mp 195–197 °C (ethanol). 1H NMR (DMSO-d6): δ 8.00–7.86 (m, 8H, H-4, H-7, H-2′, H-4′, H-6′, H-2″, H-4″, H-6″), 7.80 (td, 1H, J = 1.2, J = 8.1, H-5), 7.72 (t, 4H, J = 1.6, H-3′, H-5′, H-3″, H-5″), 7.47 (t, 1H, J = 1.6, H-6). 13C NMR (151 MHz, DMSO-d6) δ 164.10, 141.63, 137.12, 135.59, 134.15, 129.67, 128.76, 126.84, 126.13, 122.65, 119.62. IR (KBr): 3065, 1,688, 1,381, 1,173. MS (ESI) calcd. For C19H15N2O5S3 [(M + 1)+]: 447.0143; found: 447.1. Anal. calcd. For: C19H14N2O5S3 C,51.11; H,3.16; N,6.27; found C,51.27; H,3.22; N,5.88.
N-(3-Oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 6. Yield: 30%. White solid, mp 162–164 °C (ethanol). 1H NMR (DMSO-d6): δ 11.34 (s, 1H, NH), 7.90–7.80 (m, 4H, H-4, H-7, H-2′, H-6′), 7.76–7.60 (m, 4H, H- H-5, H-6, H-3′, H-5′), 7.41 (t, 1H, J = 7.5, H-4′). 13C NMR (151 MHz, DMSO-d6) δ 163.17, 140.32, 138.85, 133.78, 133.00, 129.35, 127.65, 126.14, 126.72, 122.13, 121.24. IR (KBr): 3187, 3,062, 1,686, 1,348, 1,173. MS (ESI) calcd. For C13H9N2O3S2 [(M-1)−]: 305.0055; found: 305.5. Anal. calcd. For C13H10N2O3S2 C,50.97; H,3.29; N,9.14; found C,51.16; H,3.37; N,9.14.
2.2.13 4-Methyl-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 7 and 4-methyl-N-[(4-methylphenyl)sulfonyl]-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 21
Compounds 7 and 21 were synthesized following the same procedure previously described for compounds 6 and 20.
4-Methyl-N-[(4-methylphenyl)sulfonyl]-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 21. Yield: 40%. White solid, mp 203–204 °C (ethanol). 1H NMR (DMSO-d6): δ 7.98 (d, 1H, J = 8.1, H-4), 7.92 (d, 1H, J = 7.8, H-7), 7.85–7.76 (m, 5H, H-5, H-2′, H-6′, H-2″, H-6″), 7.53–7.41 (m, 5H, H-6, H-3′, H-5′, H-3″, H-5″), 2.46 (s, 6H, CH3). 13C NMR (151 MHz, DMSO-d6) δ 164.12, 146.50, 141.61, 134.33, 134.07, 130.06, 128.82, 126.83, 126.07, 122.61, 119.70, 21.26. IR (KBr), 3,056, 2,920, 2,854, 1705, 1,361, 1,170. MS (ESI) calcd. For C21H19N2O5S3 [(M + 1)+]: 475.0456; found: 475.0. Anal. calcd. For C21H18N2O5S3 C,53.15; H,3.82; N,5.90; found C,53.53; H,3.90; N,5.90.
4-Methyl-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 7. Yield: 28%. Pale yellow crystals, mp 168–170 °C (ethanol). 1H NMR (DMSO-d6): δ 11.21 (s, 1H, NH), 7.87 (d, 1H, J = 8.1, H-4), 7.81 (d, 1H, J = 8.1, H-7), 7.77–7.67 (m, 3H, H-5, H-2′, H-6′), 7.44–7.38 (m, 3H, H-6, H-3′, H-5′), 2.41 (s, 3H, CH3). 13C NMR (151 MHz, DMSO-d6) δ 163.20, 144.22, 140.31, 135.99, 132.96, 129.81, 127.72, 126.14, 125.69, 122.13, 121.29, 21.12. IR (KBr): 3057, 2,850, 1,656, 1,340, 1,158. MS (ESI) calcd. For C14H11N2O3S2 [(M-1)−]: 319.0211; found: 319.2. Anal. calcd. For C14H12N2O3S2 C,52.49; H,3.78; N,8.74; found C,52.43; H,3.72; N,8.48.
2.2.14 4-Nitro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 8 and 4-Nitro-N-[(4-nitrophenyl)sulfonyl]-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 22
Compounds 8 and 22 were synthesized following the same procedure described for compounds 6 and 20.
4-Nitro-N-[(4-nitrophenyl)sulfonyl]-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 22. Yield: 30%. Yellow solid, mp 194–195 °C (acetic acid). 1H NMR (DMSO-d6): δ 8.53 (d, 4H, J = 8.7, H-3′, H-5′, H-3″, H-5″), 8.24 (d, 4H, J = 9.3, H-2′, H-6′, H-2″, H-6″), 8.02 (d, 1H, J = 8.1, H-4), 7.92 (d, 1H, J = 7.5, H-7), 7.84 (t, 1H, J = 8.1, H-5), 7.50 (t, 1H, J = 7.5, H-6). 13C NMR (151 MHz, DMSO-d6) δ 164.29, 154.33, 151.45, 147.26, 141.89, 141.77, 134.52, 130.72, 127.07, 126.91, 126.58, 125.07, 123.32, 122.87, 119.38. IR (KBr): 3112, 1704, 1,535, 1,376, 1,348, 1,182. MS (ESI) calcd. For C19H13N4O9S3 [(M + 1)+]: 536.9845; found: 536.1. Anal. calcd. For C19H12N4O9S C,42.53; H,2.25; N,10.44; found C,42.44; H,2.26; 10.45.
2.2.15 4-Nitro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 8
Yield: 47%. Yellow solid, mp 176–178 °C (ethanol). 1H NMR (DMSO-d6): δ 11.34 (s, 1H, NH), 8.43 (d, 2H, J = 9.0, H-3′, H-5′), 8.12 (d, 2H, J = 8.4, H-2′, H-6′), 7.91 (d, 1H, J = 8.1, H-4), 7.79 (d, 1H, J = 8.1, H-7), 7.72 (t, 1H, J = 8.1, H-5), 7.42 (t, 1H, J = 7.0, H-6). 13C NMR (151 MHz, DMSO-d6) δ 163.20, 150.30, 144.66, 140.47, 133.18, 129.42, 126.22, 125.84, 124.57, 122.21, 121.06. IR (KBr): 3033, 2,846, 1,655, 1,354, 1,169. MS (ESI) calcd. For C13Hc.8N3O5S2 [(M-1)−]: 349.9905; found: 350.8. Anal. calcd. For C13H9N3O5S2 C,44.44; H,2.58; N,11.96; found C,44.57; H,2.74; 11.61.
2.2.16 4-Methoxy-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 9
Compound 9 was synthesized following the same procedure described for compound 6.
Yield: 27%. Pale yellow solid, mp 167 °C (ethanol-water). 1H NMR (DMSO-d6): δ 11.13 (s, 1H, NH), 7.88 (d, 1H, J = 8.1, H-4), 7.84–7.76 (m, 3H, H-7, H-2′, H-6′), 7.70 (t, 1H, J = 7.8, H-5), 7.41 (t, 1H, J = 7.8, H-6), 7.15 (d, 2H, J = 9.0, H-3′, H-5′), 3.85 (s, 3H, CH3). 13C NMR (151 MHz, DMSO-d6) δ 163.23, 163.16, 140.31, 132.93, 130.06, 126.13, 125.67, 122.11, 121.31, 114.61, 114.55, 55.74. IR (KBr): 3060, 2,841, 1,670, 1,346, 1,155. MS (ESI) calcd. For C14H11N2O4S2 [(M-1)−]: 335.0160; found: 335.1. Anal. calcd. For C14H12N2O4S C,49.99; H,3.60; N,8.33; found C,50.34; H,3.70; N 8.31.
Anal: C14H12N2O4S2 (336.39).
2.2.17 2-Fluoro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 10
Compound 10 was synthesized following the same procedure described for Compound 6. Yield: 22%. Ivory solid, mp 159–161 °C (ethanol). 1H NMR (DMSO-d6): δ 11.67 (s, 1H, NH), 7.88 (d, 1H, J = 7.5, H-4), 7.82–7.68 (m, 4H, H-7, H-3′, H-4′, H-6′), 7.50 (t, 1H, H-5), 7.43–7.33 (m, 2H, H-6, H-5′). IR (KBr): 3060, 2,829, 1,664, 1,356, 1,176. MS (ESI) calcd. For C13H8FN2O3S2 [(M-1)−]: 322.9960; found: 323.8. Anal. calcd. For C13H9FN2O3S2C,48.14; H,2.80; N,8.64; found C,48.01; H,3.08; N 8.36.
2.2.18 4-Fluoro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 11 and 4-Fluoro-N-[(4-fluorolphenyl)sulfonyl]-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 23
Compounds 11 and 23 were synthesized following the same procedure described for compounds 6 and 20.
2.2.19 4-Fluoro-N-[(4-fluorolphenyl)sulfonyl]-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 23
Yield: 33%. Ivory solid, mp 195–196 °C (ethanol). 1H NMR (DMSO-d6): δ 8.10–8.01 (m, 4H, H-2′, H-6′, H-2″, H-6″), 7.98 (d, 1H, J = 8.1, H-4), 7.91 (d, 1H, J = 8.4, H-7), 7.81 (td, 1H, J = 8.1, J = 7.2, H-5), 7.64–7.56 (m, 4H, H-3′, H-5′, H-3″, H-5″), 7.48 (td, 1H, J = 0.9, J = 7.8, H-6). 13C NMR (151 MHz, DMSO-d6) δ 166.84, 165.15, 164.20, 141.66, 134.23, 133.27, 132.31, 127.89, 126.19, 122.71, 119.58, 117.15. IR (KBr): 3187, 3,062, 1,686, 1,348, 1,173. MS (ESI) calcd. For C19H13F2N2O5S3 [(M + 1)+]: 482.9955; found: 483.6. Anal. calcd. For C19H12F2N2O5S3 C,47.30; H,2.51; N, 5.81; found C,47.65; H,3.2.52; N 5.65.
2.2.20 4-Fluoro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 11
Yield: 50%. Ivory solid, mp 150–151 °C (methanol–water). 1H NMR (DMSO-d6): δ 11.39 (s, 1H, NH), 7.96–7.88 (m, 3H, H-4, H-2′, H-6′), 7.80 (d, 1H, J = 8.4, H-7), 7,71 (td, 1H, J = 1.5, J = 7.2, H-5), 7.51–7.38 (m, 3H, H-6, H-3′, H-5′). 13C NMR (151 MHz, DMSO-d6) δ 165.49, 163.15, 140.37, 135.16, 135.14, 133.04, 130.95, 125.95, 122.16, 121.21, 116.59. IR (KBr): 3048, 2,848, 1,656, 1,355, 1,161. MS (ESI) calcd. For C13H8FN2O3S2 [(M-1)−]: 322.9960; found: 323.2. Anal. calcd. For C13H9FN2O3S2C,48.14; H,2.80; N,8.64; found C,48.56; H,2.81; N 8.22.
2.2.21 2-Chloro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 12
Compound 12 was synthesized following the same procedure described for compound 6. Yield: 33%. Ivory solid, mp 170–172 °C (ethanol). 1H NMR (DMSO-d6): δ 11.57 (s, 1H, NH), 7.98 (d, 1H, J = 8.7, H-4), 7.87 (d, 1H, J = 7.8, H-6′), 7.82 (d, 1H, J = 8.4, H-7), 7.77–7.67 (m, 3H, H-5, H-3′, H-5′), 7.51 (t, 1H, H-4′), 7.40 (t, 1H, J = 7.5, H-6). 13C NMR (151 MHz, DMSO-d6) δ 163.32, 140.41, 136.56, 135.16, 133.05, 132.01, 131.83, 131.40, 127.70, 126.17, 125.74, 122.14, 121.15. IR (KBr): 3018, 2,850, 1,672, 1,344, 1,170. MS (ESI) calcd. For C13H8ClN2O3S2 [(M-1)−]: 338.9665; found: 339.2. Anal. calcd. For C13H9FN2O3S2C,45.81; H,2.66; N,8.22; found C,45.41; H,2.82; N 8.10.
2.2.22 4-Chloro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 13 and 4-chloro-N-[(4-chlorophenyl)sulfonyl]-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 24
Compounds 13 and 24 were synthesized following the same procedure described for compounds 6 and 20.
2.2.23 4-Chloro-N-[(4-chlorophenyl)sulfonyl]-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 24
Yield: 80%. Pale yellow solid, mp 222–224 °C dec.(ethanol). 1H NMR (DMSO-d6): δ 8.04–7.98 (m, 5H, H-4, H-2′, H-6′, H-2″, H-6″), 7.92 (d, 1H, J = 8.1, H-7), 7.86–7.78 (m, 5H, H-5 H-3′, H-5′, H-3″, H-5″), 7.49 (t, 1H, J = 7.8, H-6). IR (KBr): 3057, 2,850, 1,656, 1,340, 1,158. MS (ESI) calcd. For C19H13Cl2N2O5S3 [(M + 1)+]: 514.9364; found: 516.5. Anal. calcd. For C19H12Cl2N2O5S3C,44.36; H,2.35; N,5.45; found C,44.47; H,2.38; N 5.44.
2.2.24 4-Chloro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 13
Yield: 28%. Pale yellow solid, mp 158–159 °C (ethanol). 1H NMR (DMSO-d6): δ 11.58 (s, 1H, NH), 7.91–7.81 (m, 4H, H-4, H-7, H-2′, H-6′), 7.74–7.68 (m, 3H, H-5, H-3′, H-5′), 7.42 (t, 1H, J = 7.1, 6). 13C NMR (151 MHz, DMSO-d6) δ 163.17, 140.38, 138.73, 137.81, 133.06, 129.66, 129.52, 126.18, 125.76, 122.18, 121.19. IR (KBr): 3058, 2,852, 1,652, 1,342, 1,161. MS (ESI) calcd. For C13H8ClN2O3S2 [(M-1)−]: 338.9665; found: 339.6. Anal. calcd. For C13H9FN2O3S2C,45.81; H,2.66; N,8.22; found C,45.63; H,2.98; N 8.04.
2.2.25 3-Chloro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 14
Compound 14 was synthesized following the same procedure described for compound 6. Yield: 23%. Tan solid, mp 149–150 °C (Ethanol). 1H NMR (DMSO-d6): δ 11.48 (s,1H, NH), 7.91–7.81 (m, 4H, H-4, H-7, H-2′, H-6′), 7.74–7.73 (m, 3H, H-5, H-3’ H-5′), 7.42 (t, 1H, J = 7.6, H-6). 13C NMR (151 MHz, DMSO-d6) δ 163.18, 140.84, 140.39, 133.86, 133.75, 133.09, 131.38, 127.11, 126.43, 126.18, 125.79, 122.20, 121.16. IR (KBr): 3074, 2,870, 1,664, 1,365, 1,176. MS (ESI) calcd. For C13H8ClN2O3S2 [(M-1)−]: 338.9665; found: 339.6. Anal. calcd. For C13H9FN2O3S2C,45.81; H,2.66; N,8.22; found C,45.88; H,2.61; N 8.07.
2.2.26 3,4-Dichloro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 15
Compound 15 was synthesized following the same procedure described for compound 6. Yield: 35%. Ivory solid, mp 183–184 °C (ethanol) 1H NMR (DMSO-d6): δ 11.69 (s,1H, NH), 8.05 (s, 1H, J = 7.6, H-2′), 7.92–7.89 (m, 2H, H-4, H-6′), 7.84–7.79 (m, 2H, H-7, H-5′), 7.72 (td, 1H, J = 1.2, J = 8.2, H-5), 7.42 (t, 1H, J = 7.3, H-6). HS17 (15). 13C NMR (151 MHz, DMSO-d6) δ 163.20, 140.45, 139.36, 136.90, 133.14, 132.19, 131.72, 129.32, 127.84, 126.21, 125.82, 122.23, 121.12. IR (KBr): 3031, 2,852, 1,657, 1,327, 1,169. MS (ESI) calcd. For C13H7Cl2N2O3S2 [(M-1)−]: 372.9275; found: 373.4. Anal. calcd. For C13H8Cl2N2O3S2 C,41.61; H,2.15; N,7.47; found C,41.96; H,2.20; N 7.42.
2.2.27 3,5-Dichloro-N-(3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 16
Compound 16 was synthesized following the same procedure above for compound 6.
Yield: 28%. Pale yellow solid, mp 178–180 °C (ethanol-water). 1H NMR (DMSO-d6): δ 11.50 (s, 1H, NH), 8.06 (t, 1H, J = 1.9, H-4′), 7.92 (d, 1H, J = 8.4, H-4), 7.84–7.82 (m, 3H, H-7, H-2′, H-6′), 7.73 (td, 1H, J = 1.5, J = 8.7, H-5), 7.42 (t, 1H, J = 7.2, H-6). IR (KBr): 3216, 3,085, 1,672, 1,362, 1,178. MS (ESI) calcd. For C13H7Cl2N2O3S2 [(M-1)−]: 372.9275; found: 373.2. Anal. calcd. For C13H8Cl2N2O3S2 C,41.61; H,2.15; N,7.47; found C,41.23; H,2.53; N 7.04.
2.2.28 2-Chloro-N-(5-methyl-3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 17
Compound 17 was synthesized following the same procedure described for compound 6. Yield: 35%. Ivory solid, mp 174–176 °C (ethanol). 1H NMR (DMSO-d6): δ 11.51 (s, 1H, NH), 7.94 (d, 1H, J = 7.5, H-6′), 7.76–7.68 (m, 3H, H-6, H-7, H-3′), 7.62 (s,1H, 4), 7.55–7.47 (m, 2H, H-3′, H-4′), 2.37 (s, 3H, CH3). 13C NMR (151 MHz, DMSO-d6) δ 163.29, 137.45, 136.58, 135.46, 135.12, 134.38, 131.99, 131.84, 131.38, 127.67, 125.84, 121.89, 121.17. IR (KBr): 3022, 2,781, 1,668, 1,342, 1,169. MS (ESI) calcd. For C14H10ClN2O3S2 [(M-1)−]: 352.9821; found: 352.1. Anal. calcd. For C14H11ClN2O3S2 C,47.39; H,3.12; N,7.89; found C,47.46; H,3.25; N,7.80.
2.2.29 2-Chloro-N-(5-chloro-3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 18
Compound 18 was synthesized following the same procedure described for compound 6. Yield: 23%. Tan solid, mp 174–175 °C (Ethanol). 1H NMR (DMSO-d6): δ 11.57 (s, 1H, NH), 7.99 (d, 1H, J = 8.1, H-6′), 7.93 (d, 1H, J = 8.7, H-7), 7.80 (d, 1H, J = 2.2, H-4), 7.76–7.69 (m, 3H, H-6, H-5′, H-3′), 7.50 (td, 1H, J = 1.6, J = 8.1, H-4′). IR (KBr): 3035, 2,852, 1,662, 1,350, 1,167. MS (ESI) calcd. For C13H7Cl2N2O3S2 [(M-1)−]: 372.9275; found: 373.7. Anal. calcd. For C13H8Cl2N2O3S2 C,41.61; H,2.15; N,7.47; found C,41.90; H,2.47; N 7.11.
2.2.30 2-Chloro-N-(6-chloro-3-oxo-1,2-benzisothiazol-2(3H)-yl)benzenesulfonamide 19
Compound 19 was synthesized following the same procedure described for compound 6. Yield: 23%. Pale yellow solid, mp 153–155 °C (Ethanol). 1H NMR (DMSO-d6): δ 11.61 (s, 1H, NH), 8.00–7.96 (m, 2H, H-6′, H-7), 7.79 (d, 1H, J = 8.1, H-4), 7.75–7.67 (m, 2H, H-5, H-5′), 7.53–7.42 (m, 2H, H-3′, H-4′). 13C NMR (151 MHz, DMSO-d6) δ 162.49, 141.93, 137.96, 136.57, 135.16, 132.02, 131.80, 131.41, 127.72, 126.26, 121.79, 120.23. IR (KBr): 3091, 2,850, 1,664, 1,358, 1,176. MS (ESI) calcd. For C13H7Cl2N2O3S2 [(M-1)−]: 372.9275; found: 373.1. Anal. calcd. For C13H8Cl2N2O3S2 C,41.61; H,2.15; N,7.47; found C,41.90; H,2.50; N 7.05.
2.3 Biology
2.3.1 Compounds
Compounds were solubilized in DMSO at 100 mM and then diluted in a culture medium. The final concentration of DMSO in samples and controls employed in biological assays was less than 0.02%.
2.3.2 Cells and viruses
MT-4, C8166, H9/IIIB, and CEM cells, as well as laboratory-adapted HIV-1 strains, were obtained from the NIH AIDS Research &Reference Reagent Program, USA. Cell cultures were grown in RPMI 1640 medium, supplemented with 10% foetal calf serum (FCS), 100 IU/mL penicillin G, and 100 μg/mL streptomycin, and incubated at 37 °C in a 5% CO2 atmosphere. Cell cultures were checked periodically for the absence of mycoplasma contamination using the MycoTect Kit (Gibco, Thermo Fisher Scientific, MA, USA).
HIV-1IIIB and HIV-2 CBL-20 strains were obtained from the supernatants of persistently infected H9/IIIB and CEM cells, respectively. HIV-1 and HIV-2 stock solutions had titres of 4.5 × 107 and 1.4 × 106 50% cell culture infectious doses (CCID50)/ml, respectively. The Y181C mutant (NIH N119) was derived from an AZT-sensitive clinical isolate passaged initially in CEM and then in MT-4 cells in the presence of Nevirapine (up to 10 μM). The K103N + Y181C mutant (NIH A17) was derived from an IIIB strain passaged in H9 cells in the presence of BI-RG 587 (up to 1 μM). The K103R + V179D + P225H mutant (EFVR) was derived from an IIIB strain passaged in MT-4 cells in the presence of Efavirenz (up to 2 μM). N119, A17 and EFVR stock solutions had titres of 1.2 × 108, 2.1 × 107, and 4.0 × 107 CCID50/ml, respectively. Mutants carrying NRTI mutations, such as the AZTR strain (67N, 70R, 215F, 219Q) and the MDR strain (74V, 41L, 106A, 215Y) or PRI mutations, such as SAQR (L10F, G48V, L90M), were also tested.
2.3.3 Biological evaluation of antiviral activities
2.3.3.1 Anti-HIV assays
The activity of the test compounds against the replication of HIV-1 wt and mutant strains (N119, A17, EFVR, AZTR, MDR, SAQR) and HIV-2 wt in acutely infected cells was based on the inhibition of virus-induced cytopathogenicity in MT-4 and C8166 cells, respectively. Briefly, 50 μL of culture medium containing 1 × 104 cells was added to each well of flat-bottom microtitre trays containing 50 μL of culture medium with or without different serial concentrations of test compounds. Then, 20 μL of an HIV suspension (containing the appropriate amount of CCID50 required to cause complete cytopathogenicity on day 4, final multiplicity of infection [m.o.i.] 0.01) was added. After incubation at 37 °C, cell viability was determined using the 3-(4,5-dimethylthiazol-1-yl)-2,5-diphenyltetrazolium bromide (MTT) method (Pauwels et al., 1988). The cytotoxicity of the test compounds was evaluated in parallel with their antiviral activity through the viability of mock-infected, treated cells, as monitored by the MTT method and described elsewhere (Morellet et al., 1994).
2.3.4 Mode of action studies
2.3.4.1 Reverse transcriptase assay
Assays using recombinant HIV-1 reverse transcriptase were performed as previously described (Costi et al., 2004; Tramontano and Cheng, 1992).
2.3.4.2 Integrase assays
Expression and purification of HIV-1 recombinant integrase (rIN) and rIN assays were performed as previously reported (Costi et al., 2004).
2.3.5 p24 determination
Quantitation of the p24 Gag protein present in supernatants of HIV-1-infected MT-4 cells was assessed using the Alliance HIV-1 P24 ANTIGEN ELISA Kit (Perkin Elmer), according to the manufacturer’s protocol, on culture supernatant using antigen-capture enzyme-linked immunosorbent assay test (ELISA), Perkin Elmer.
2.3.5.1 H9/C8166 cocultures
Chronically infected H9 cells were washed and cocultured with uninfected C8166 cells (ratio 1:500) in the absence or presence of test inhibitors. Following 36 h of incubation at 37 °C, cocultures were monitored by optical microscopy, syncytia were counted, and those found in drug-treated cocultures were reported as a percentage of those counted in untreated cocultures (Wang et al., 2004).
2.3.5.2 Time of addition assay
2 × 104 MT-4 cells were infected for 1 h at 20 °C with HIV-1 at a high multiplicity of infection (m.o.i. = 5), corresponding to 5 cell culture infectious dose 50 per cell (CCID50/cell). Test compounds were added in duplicate at the beginning of the 1 h infection period (and then removed with the inoculum by extensive washing) or immediately after the end of infection, 1, 2, 4, 6, 8, 12, or 24 h later. The amounts of p24 and infectious virus present in the supernatants were measured at 42 h post-infection (p.i.).
2.3.5.3 Selection of drug-resistant mutants
Drug-resistant variants were selected by serial passages of HIV-1IIIB in the presence of progressively doubling drug concentrations, starting from a cell culture infected with an m.o.i. of 0.01 and treated with a drug concentration equal to the EC50. Usually, the amount of virus obtained after each passage was sufficient to determine the infection of the next cell culture, which, after infection and washing, was incubated with double the amount of the selected drug. In the case of compound 6, the drug-resistant virus population was selected up to a drug concentration 8-fold greater than the EC50. Resistant virus preparations were subjected to RNA extraction, RT-PCR, and genome sequencing to identify the mutational patterns responsible for resistance.
2.3.5.4 Molecular analysis of resistant viruses
Viral RNAs from wt and drug-resistant mutants were obtained using the QIAamp viral RNA minikit (Qiagen), starting from 140 μL of cell-free viral suspensions containing about 5 × 104 PFU/ml, to determine the nucleotide sequence of the pol, gag, and env regions of the HIV-1 genome. Reverse transcriptions were carried out using the Superscript II enzyme (Invitrogen), and cDNAs were amplified by PCR using Platinum Pfx polymerase (Invitrogen) following the manufacturer’s protocol. Details of the RT-PCR conditions are provided in the Supplementary Data. PCR fragments were purified using the QIAquick PCR Purification kit (Qiagen) and analyzed using the cycle-sequencing method (C. I. B. I. A. C. I. service of the University of Florence). Both DNA strands were sequenced using specific primers. The comparative analysis of received chromatograms allowed us to obtain the nucleotide sequences of the gag, pol, and env regions for wild-type HIVIIIB and HIV-16 resistant.
2.3.5.5 Virucidal activity assays
Cell-free, high-titre HIV-1 and HIV-2 stock solutions were exposed to the test compounds for 2 h at 0 °C or 37 °C. At the end of incubation, residual infectivity was determined using the Reed–Muench end-point titration method, as described below.
2.3.5.6 HIV titration
Titration was performed in C8166 cells using the standard limiting dilution method (dilution 1:2, four replica wells/dilution) in 96-well plates. The infectious virus titre was determined by light microscope scoring of syncytia (multinucleated giant cells) after 4 days of incubation (Tondelli et al., 1996). Virus titres were expressed as 50% cell culture infectious dose per ml using the Reed and Muench (1938) method.
3 Results
3.1 Antiviral activities of benzenesulfonamide derivatives against HIV-1, HIV-2, and mutant strains
The cytotoxicity (CC50), potency (EC50), and spectrum of antiretroviral activity of 1,2- benzisothiazol-3(2H)-one benzenesulfonamides (derivatives 6–24) are reported in Table 1. The HIV-1 strains tested included wild-type IIIB and mutants resistant to NNRTIs, NRTIs, and PRIs, as detailed in the Materials and Methods. Activity against HIV-2 (CBL-20 strain) in C8166 cells was also reported. In this study, benzenesulfonamides were evaluated in comparison with the selected reference drugs: efavirenz (EFV), azidothymidine (AZT), and saquinavir (SAQ). Most of the title derivatives were effective in the micromolar concentration range against HIV-1 wild-type and variants carrying clinically relevant mutations that confer resistance to the three representative classes of antiretrovirals. Furthermore, in contrast to EFV, compounds 6, 7, 17, and 19 inhibited HIV-2 as well as HIV-1 (Table 1), demonstrating their broad-spectrum antiretroviral activity and Selectivity Index (SI) of around 10 against all tested strains.
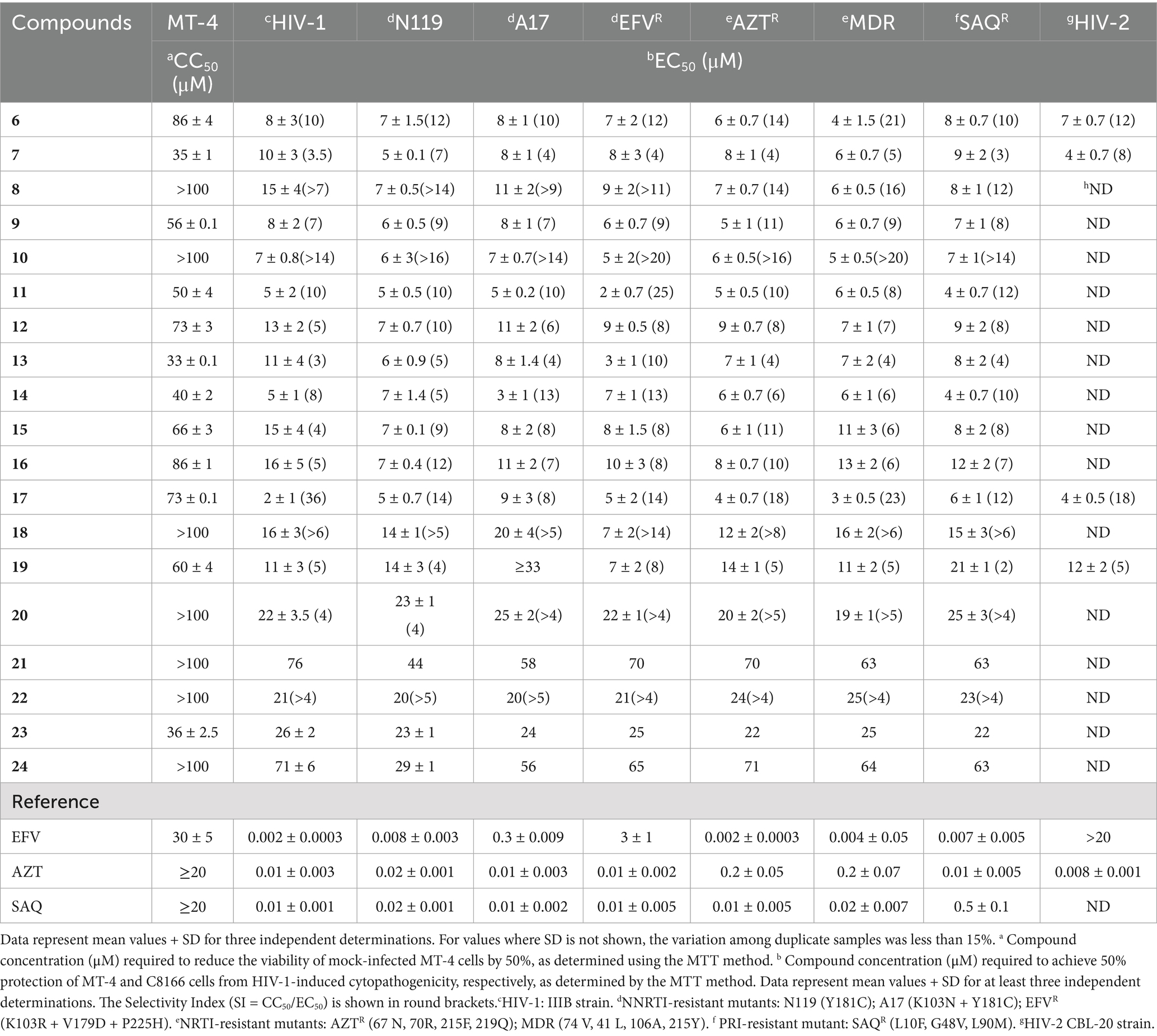
Table 1. Cytotoxicity and antiretroviral activity of compounds 6–24 and reference drugs.
The comparative activities of derivative 6, EFV, and Amphotericin B (AMPH-B) against HIV-2 are summarized in Table 2. Compound 6, similar to AMPH-B (Waheed et al., 2008; Otake et al., 1991; Pleskoff et al., 1995; Hansen et al., 1990), was equally potent against HIV-2 and HIV-1 wt, but also against variants containing mutations conferring resistance to NNRTIs (Table 1).
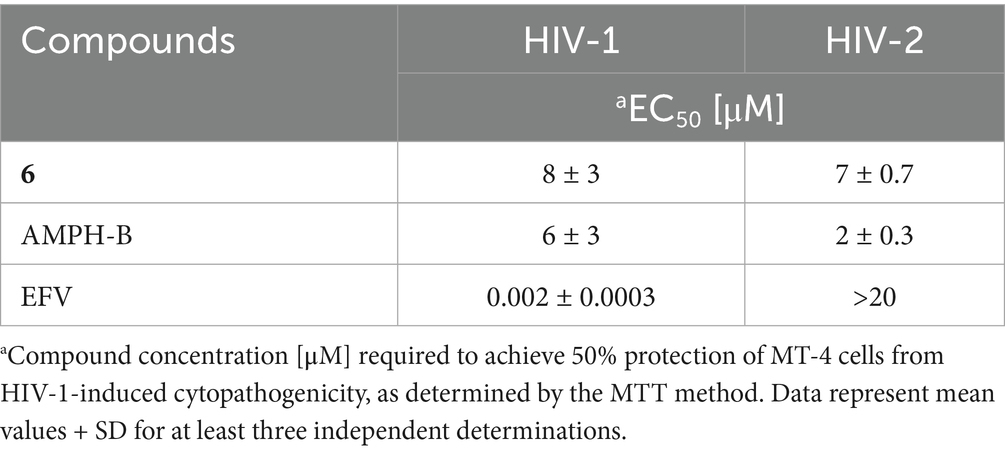
Table 2. Comparative activity of compound 6 and reference compounds against HIV-1 and HIV-2 in cell-based assays.
3.2 Study of the mechanism of action of benzenesuphonamide derivatives
3.2.1 Enzymatic assays
Compounds 6, 7, and 19 were selected and subjected to further evaluation, as described in our previous studies (Costi et al., 2004; Tramontano and Cheng, 1992), in enzyme assays aimed at investigating their capability to inhibit recombinant HIV-1 RT and IN. Interestingly, unlike reference drugs [EFV (IC50 on rRT = 0.06 μM) and the diketoacid inhibitor of rIN, L-731,988 (internal control) (IC50s on 3′ processing and strand transfer = 2.5 and 0.35 μM, respectively)], none of the benzenesulfonamides were active at concentrations of 30 μM (Supplementary Tables S1, S2), thus making it unlikely that RT or IN might be their target enzymes.
3.2.2 Entry assay
The potential of compound 6 to interfere with HIV entry mechanisms is being investigated using coculture experiments between chronically HIV-1-infected H9 cells and uninfected C8166 cells (Wang et al., 2004). Compound 6 was selected as the lead candidate based on its favorable antiviral profile and cytotoxicity data, as summarized in the EC50 curves in Figure 2.
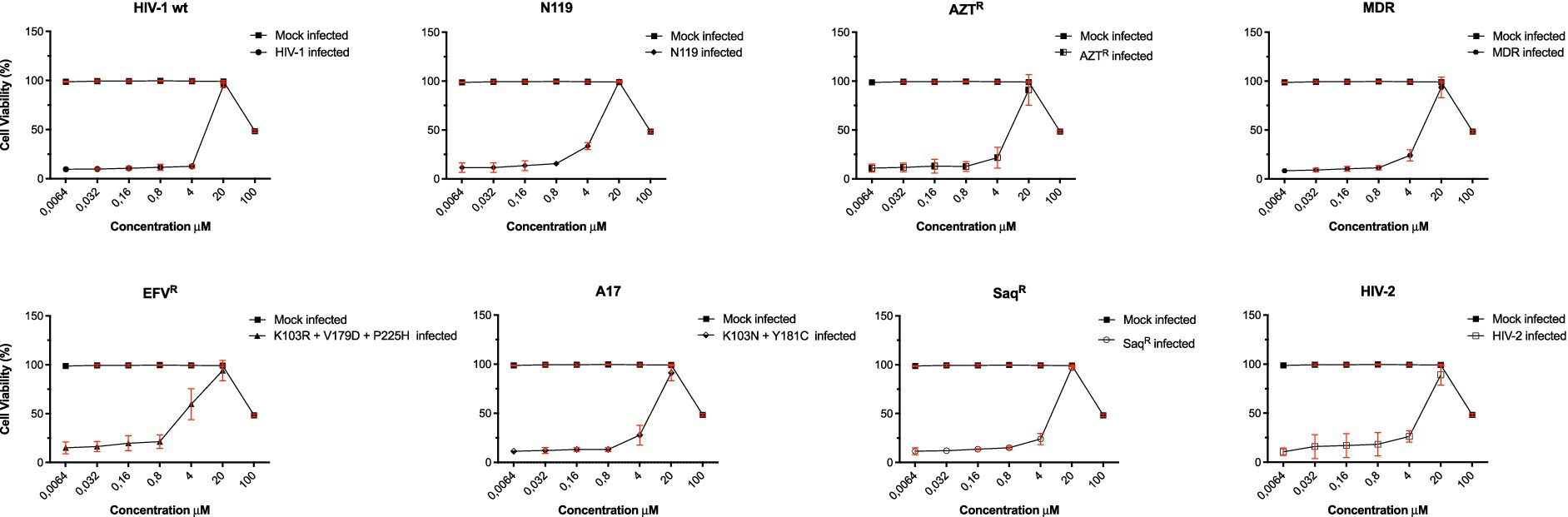
Figure 2. Cytotoxicity and anti-HIV-1 activity of derivative 6. To evaluate antiviral activities of compound 6 in HIV-1 wt and drug-resistant strains, several concentrations of compound 6 (100–0.0064 μM) were used to inhibit wt IIIB and clinically relevant RT-inhibitor-resistant strains (EFVR, N119, A17, AZTR, MDR, SaqR, and HIV-2). The viability of all HIV-1-infected MT-4 cells or C8166 for HIV-2 was estimated by MTT assay 4 days post-infection. The number of live cells was expressed as a percentage of mock-infected, untreated control cells. Data are expressed as the mean ± S.D. of at least three independent measurements.
In this assay, syncytia were formed as a result of the interaction of env-encoded glycoproteins (present on the outer membrane of chronically infected H9 cells) with the CD4 of cocultured C8166 cells. In contrast to dextran sulphate, 0.5 μM, which prevented syncytia formation by 98%, 6, tested at 30 μM, did not interfere with the very early events (adsorption/attachment/fusion) in the HIV replication cycle triggered by the gp120-CD4 interaction (Figure 3) (Baba et al., 1988; Mitsuya et al., 1988).
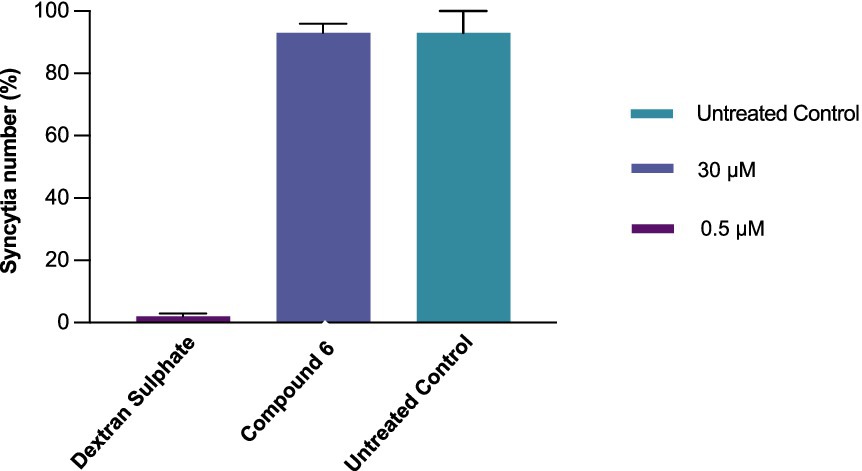
Figure 3. Comparative activity of compound 6 (30 μM) and dextran sulphate (0.5 μM) in preventing syncytium formation in chronically infected H9/C8166 cocultures. The H9/C8166 ratio was 1:550. The cells were incubated for 36 h at 37 °C. Data represent mean values of at least three independent determinations of syncytial numbers using an optical microscope. Bars are mean values, and whiskers are standard deviations (SD) of at least three independent experiments.
To identify the step(s) of the HIV-1 infection cycle targeted by compound 6, a time-of-addition study was performed in MT-4 cells under a single round of viral replication, in comparison with EFV, a drug representative of the NNRTI antiretroviral class (Figure 4).
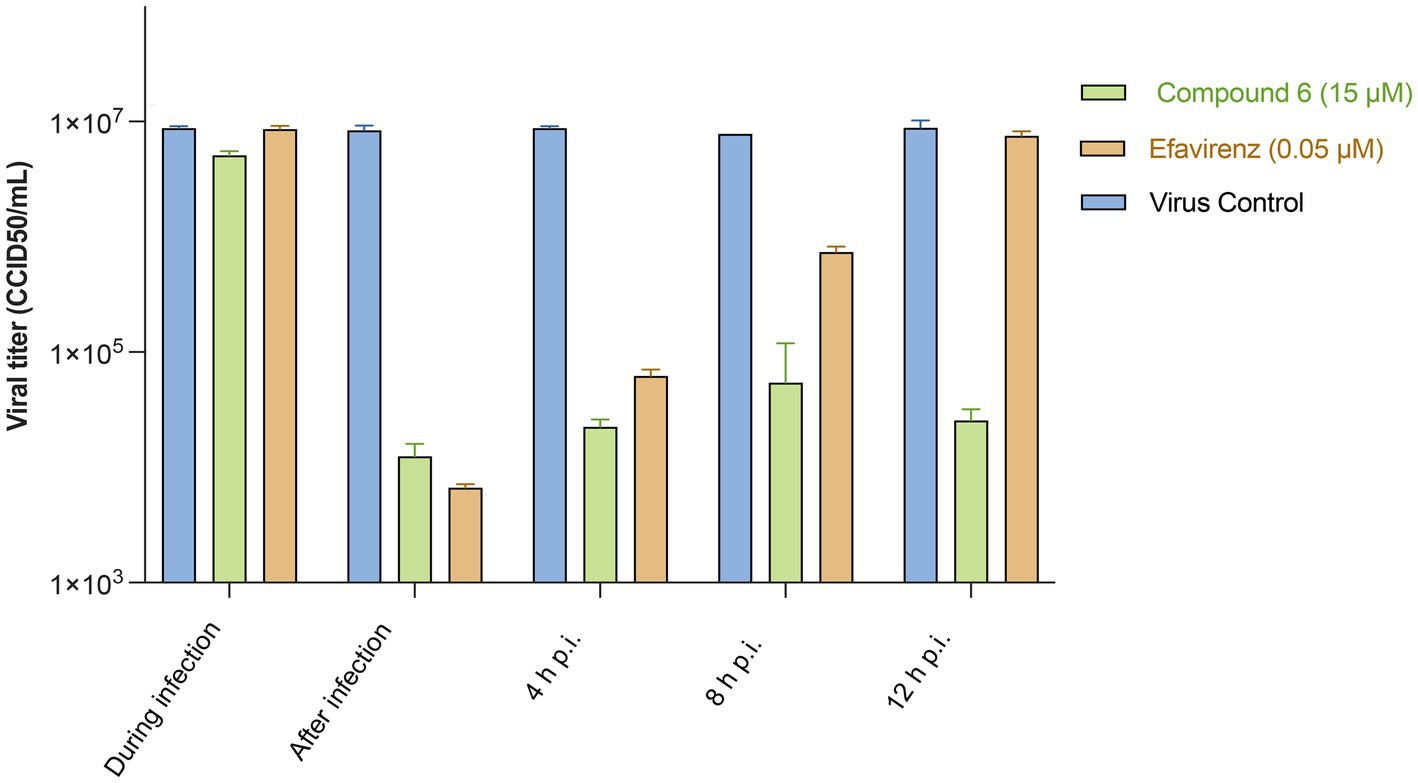
Figure 4. HIV-1 titres in supernatants of cultures treated with compound 6 and EFV during infection or at different times p.i. In a time-of-addition assay, MT-4 cells were inoculated with HIV-1 wt (MOI = 5), and then compound 6 (15 μM) or EFV (0.05 μM) was added at the indicated time points. Virus yields were determined using the Reed–Muench method. Bars are mean values, and whiskers are standard deviations (SD) of three independent experiments.
None of the tested compounds inhibited HIV-1 multiplication when present only during the 1-h infection period, as revealed by the viral yield. In particular, both EFV and compound 6 produced an infectious virus yield similar to that of the untreated infected controls (Figure 3), suggesting that, in addition to not affecting HIV-1 entry, compound 6 (15 μM) did not accumulate and persist in the cells. EFV, which is capable of targeting RT without prior metabolism, was still inhibitory even if added within 4 h p.i. (Figure 4), suggesting that under the experimental conditions used, reverse transcription was essentially completed by compound 6 h p.i. Interestingly, compound 6 resulted in effective prevention of virus yield (regardless of whether it was added to the cultures immediately at the end of the infection period or any time) up to 12 h later. These data suggest that compound 6 target events can occur both early and late in the HIV infection cycle.
3.2.3 Benzenesuphonamide derivatives’ direct infectivity inactivation of virions
Thus, to investigate the potential virucidal activity of our candidate compound, compound 6 was incubated for 2 h at 37 °C with a high-titre HIV-1 stock solution, whose residual infectivity was then determined using the Reed–Muench endpoint titration method.
Amphotericin B, a cholesterol-depleting compound capable of inhibiting HIV-1 infectivity, was used as a reference drug because of its direct virucidal properties (Waheed et al., 2008). When used at concentrations approximately 10- and 15-fold higher than the respective EC50s, both compound 6 (80 μM) and AMPH-B (100 μM) were capable of inactivating HIV-1 infectivity by more than 3 log10 (Figure 5). Interestingly, when the incubation was carried out at 0 °C, compound 6 failed to affect HIV-1 infectivity, while AMPH-B was still able to reduce it by a high percentage. When the same experiment was carried out with HIV-2, both compound 6 and AMPH-B inactivated virion infectivity by approximately 2 log10 (Figure 5). Again, if the incubation was carried out at 0 °C, compound 6 failed to significantly affect HIV-2 infectivity, whereas AMPH-B reduced it by more than 1 log10 (Figure 5). Infectivity inactivation of both HIV-1 and HIV-2 was determined by exposing cell-free stock solutions to serial dilutions of 6. As shown in Figure 6, concentrations of 100 and 50 μM strongly inactivated the infectivity of both HIV types in a concentration-dependent manner; conversely, lower concentrations (25 and 12.5 μM) had only marginal or no effects on HIV infectivity.
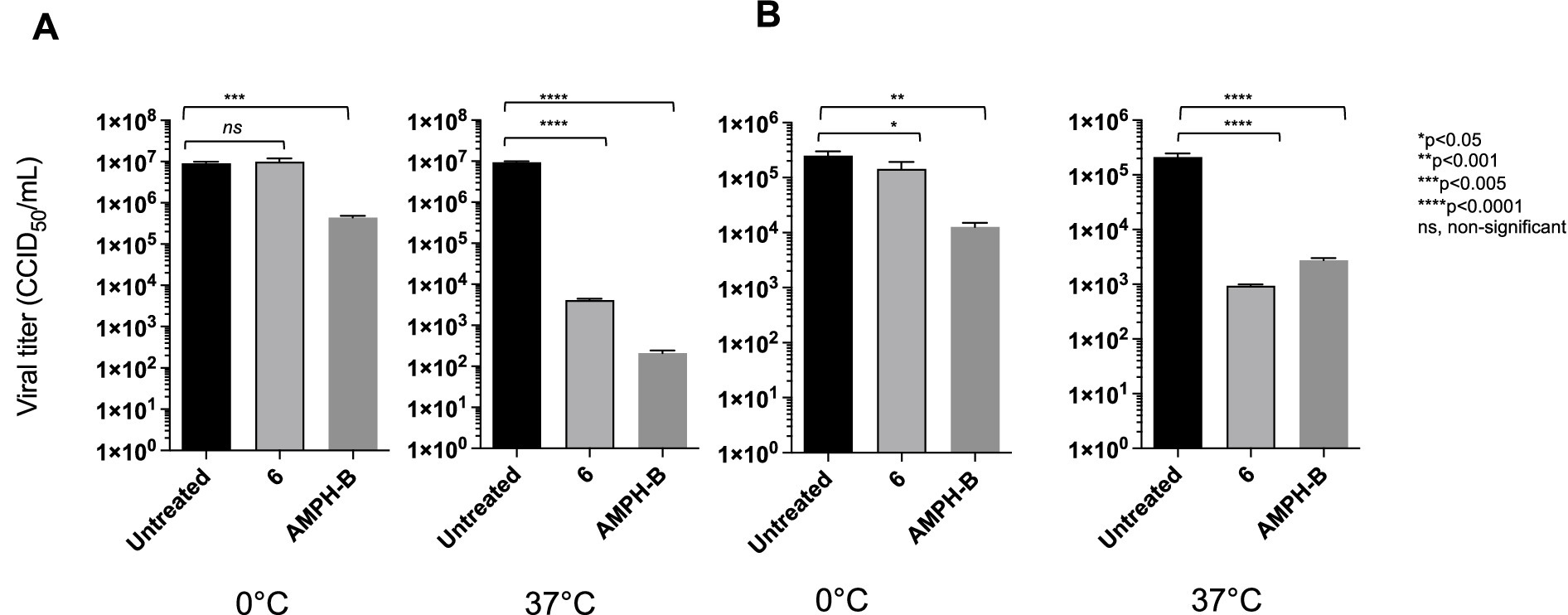
Figure 5. Comparative virucidal effects of the derivative against HIV-1 (A) and HIV-2 (B). For virucidal activity evaluation, virions were treated with compound 6 at either 0 °C or 37 °C for 1 h. Black columns for viral titre for untreated viral solution and light grey columns for viral solution treated with derivative 6. Dark-grey columns viral solution treated with AMPH-B. Viral yields were determined using the Reed–Muench method (CCID50/ml). ANOVA post-tests were performed for all groups. ****p < 0.0001, ***p < 0.001, **p < 0.005, ns, non-significant.
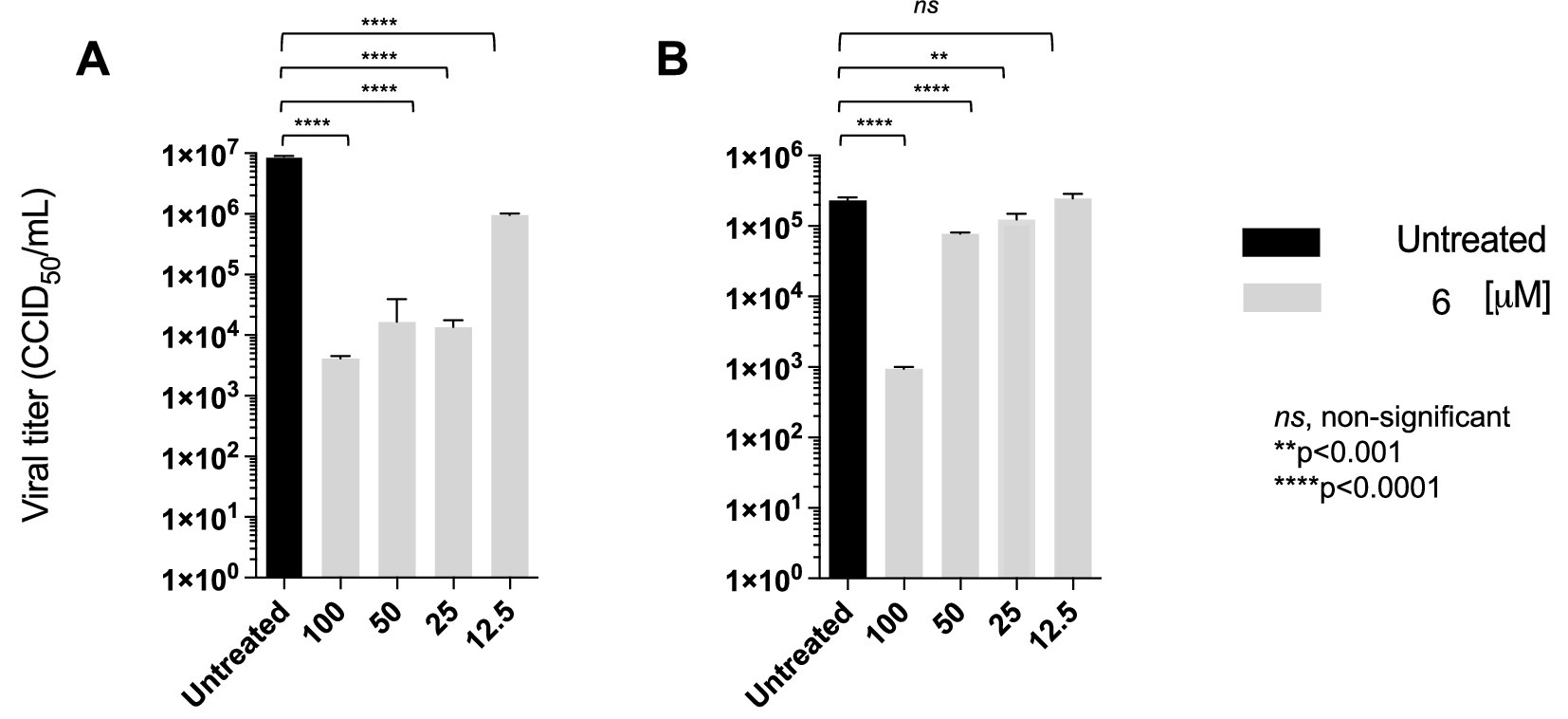
Figure 6. Compound 6 concentration-dependent inactivation of cell-free infectious HIV-1 (A) and HIV-2 (B). Viral particles were directly treated with different concentrations (100–12.5 μM) of compound 6 at 37 °C. Quantification of viral titres was performed, and results for each treatment group are presented CCID50/ml. ANOVA post-tests were performed on all groups. **** p < 0.0001 (higher concentrations of 6), compared to the HIV-1 and HIV-2 control group (untreated).
3.2.4 In vitro selection of a resistant mutant to compound 6
Mutant viruses resistant to compound 6 were selected by serial passages of HIV-1IIIB in the presence of stepwise doubling of drug concentrations, starting from a cell culture infected with an m.o.i. of 0.01, and treated with a drug concentration equal to the EC50 of the drug. The amount of virus obtained after each passage was usually sufficient to continue infection of the next cell culture, which, after infection and washing of the unadsorbed inoculum, was incubated with double the amount of the selected drug. However, in the case of 6, we had to keep the drug concentrations unchanged at passage 3 in order to obtain enough infectious virus to propagate through the next passages. With this single restriction, compound 6-resistant mutants (HIV-16) were grown to drug concentrations 16 times higher (16x) than the EC50. The selected virus population was then subjected to RNA extraction, RT-PCR, and sequencing of the gag, pol, and env genes to identify the mutation pattern responsible for drug resistance. Comparative genomic analysis of wild-type HIV and HIV-16 mutants showed that neither had mutations in the RT and PR genes. The same was true for the env gene. On the contrary, two mutations, F6C and R32K, were selected in the NCp7 sequence of the gag gene (Figure 7). Since the presence of two well-conserved zinc-finger domains of the Cys-X2-Cys-X4-His-X4-C (CCHC) type is crucial for the activity of NCp7, it is reasonable to foresee that compound 6 binds to this domain.
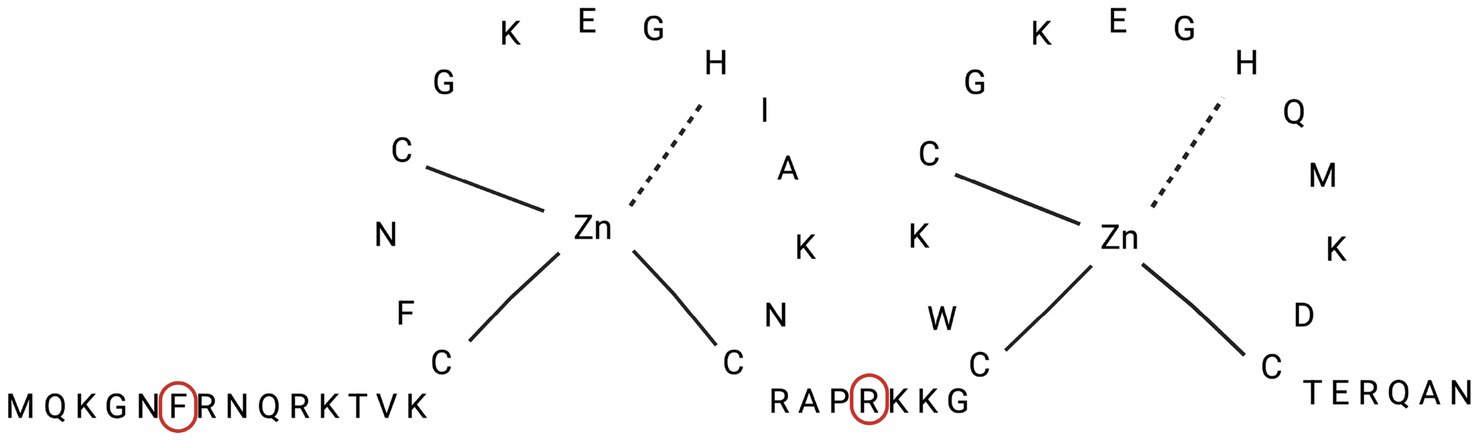
Figure 7. Amino acid sequence of NCp7 protein, with localization of amino acids (Phe-6 and Arg-32) involved in mutations of HIV-1 6 strain.
4 Discussion
Novel synthetic 1,2-benzisothiazol-3(2H)-one benzenesulfonamides with a wide spectrum of antiretroviral activity against HIV-1, HIV-2, and HIV-1 variants carrying clinically relevant mutations conferring resistance to NNRTIs, NRTIs, and PRIs are described. They neither interfere with the gp120-CD4 interaction leading to HIV entry and syncytium formation nor inhibit RT or IN in enzyme assays. Nevertheless, they affect events occurring in both early and late stages of the HIV multiplication cycle and inactivate the infectivity of cell-free HIV virions in a concentration-dependent manner.
Two mutations, F6C and R32K, were detected in the gag region coding for NC proteins in mutants selected for resistance to compound 6. Therefore, since mutations evolve to counteract the binding of the drug to its target, it is reasonable to suggest that in wt viruses, NC proteins are the target of 6. The fact that NC proteins are involved in both early reverse transcription (Carteau et al., 1997; Zybarth and Carter, 1995), integration (Aldovini and Young, 1990), late protease processing (Thomas and Gorelick, 2008), and packaging of viral genomic RNA (Arner et al., 1996) stages of the HIV-1 replication cycle, in addition to being structural components of mature virions, is consistent with both inhibition of HIV multiplication in infected cells and direct inactivation of virion infectivity.
NC retrovirus proteins are synthesized as functional domains of the Gag precursor. In HIV-1, mature NC is generated late in the assembly before virus budding takes place by a cascade of PR-mediated cleavages of Pr55Gag. End-products are matrix (p17MA), capsid (p24CA), and nucleocapsid (NCp7) proteins, p6, and smaller peptides (Clerici et al., 2007).
NC proteins, whether as domains of Pr55Gag, intermediate cleavage products (p15NC and p9NC), or mature NCp7, are characterized by two zinc finger sequences, C-X2-C-X4-H-X4-C (CCHC), which are highly conserved among retroviruses. Zinc finger sequences are linked by a central spacer, RAPRKKG, and flanked by the NH2-terminal (MQRGNFRNQRKIVK) and COOH-terminal (TERQAN) regions, which are highly conserved within a particular retrovirus species. As a matter of fact, HIV-1 NC proteins function as nucleic acid chaperones; in fact, they destabilize nucleic acid helices (through zinc fingers) and elicit nucleic acid aggregation (through basic residues). NC proteins are absolutely required for viral replication, and genetic analyses have proven that many, even minor, alterations can affect virus assembly by disrupting genomic RNA (gRNA) recognition and packaging or formation of infectious particles (Clerici et al., 2007), consistent with the results described in this paper. Evidence for the binding of isothiazolones to cysteine (Cys) has been claimed (Arner et al., 1996), as well as for the liability of the N-S bond that involves a nucleophilic attack by the sulfur atom of Cys at the sulfur of the isothiazolone moiety, followed by the cleavage of the N-S bond (Clerici et al., 2007). When binding to cysteine occurs in the HIV-1 NCp7 Zn-finger domain, zinc extrusion (with subsequent denaturation of the viral protein) occurs for several thio-compounds bearing proper electrophilic groups (Musah, 2004; Schito et al., 2006; Basrur et al., 2000; Ranganathan et al., 2002; Topol et al., 2001; Tummino et al., 1997; Loo et al., 1996).
While our study provides valuable initial insights, we are aware that several significant limitations prevent definitive conclusions about the resistance mechanism and target identification. Genetic analysis was constrained to only three genomic regions (gag, pol, and env), which represent narrow targets that may miss critical resistance-conferring mutations in other viral genes. This limited sequencing approach leaves substantial gaps in our understanding of the complete mutational landscape associated with the development of resistance.
This approach makes it unclear whether the identified F6C and R32K substitutions occur within the same viral genome or whether they are independent mutations in different viral particles within the population.
Without experimental validation, the functional significance of the F6C and R32K mutations remains speculative. Site-directed mutagenesis studies are essential to definitively establish the causal relationship between these specific substitutions and the development of resistance. Without this, we cannot rule out the possibility that other undetected mutations are primarily responsible for resistance.
This study also presents additional constraints: the observed resistance level of 16 × EC₅₀, while significant, represents a relatively modest fold-change, and the lack of cross-resistance profiling against related nucleocapsid inhibitors further restricts our ability to evaluate broader therapeutic implications.
The novel finding of this study is the establishment of the ability of compound 6 to induce F6C and R32K mutations that could be responsible for drug resistance by inducing a conformational change in the NCp7 structure, preventing its interaction with compound 6. This is consistent with the activity shown by compound 6 against HIV-1 laboratory strains carrying clinically relevant NNRTI mutations and HIV-2, which occurs at concentrations (7–8 μM) similar to those active against the wt strain.
These observations lead us to suggest an inhibitory mechanism involving the NCp7 protein as a target. Further analysis of site-directed mutagenesis will be performed to (i) confirm the correlation between the observed mutations and the degree of resistance to compound 6 and (ii) investigate the contribution of each mutation to resistance.
5 Conclusion
A new class of molecules with an unusual spectrum of antiretroviral activity has been identified. The experiments clearly show that the selected compound 6 does not inhibit HIV-1 reverse transcriptase, integrase, or viral attachment and fusion to host cells. Notably, compound 6 was equally potent and selective against HIV-2 and HIV-1 variants, with mutations conferring resistance to NNRTIs. In a comparative assay for virucidal activity, AMPH-B and compound 6 turned out to be active in inactivating the HIV-1 infectivity of an HIV-1 stock solution, showing virucidal activity, the former at both 0 °C and 37 °C, and the latter only after incubation at 37 °C. Consistent with this, genome analysis of 6-resistant mutants (selected up to 16-fold EC50) shows that no significant mutation was selected in the RT and protease genes, as well as in the env gene, while two mutations (F6C and R32K) were selected in the NCp7 gene of the gag polyprotein, which is involved in genomic RNA packaging and virus particle morphogenesis, suggesting that NCp7 may be the target of the inhibitory action of compound 6.
Although we recognize that compound 6 is not very potent, its moderate selectivity index suggests that the therapeutic window may be narrow; however, this does not rule out further development. Although we acknowledge this as a limitation, it also presents an opportunity for targeted optimization in future studies. This compound has an unusual spectrum of activity and an interesting mode of action. It is an antiviral that targets a fundamental part of the viral structure rather than an enzyme; therefore, it merits further investigation.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used. Ethical approval was not required for the studies on animals in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
RL: Data curation, Funding acquisition, Investigation, Writing – review & editing, Supervision. MI: Data curation, Formal analysis, Investigation, Writing – review & editing. EF: Investigation, Writing – review & editing. VM: Data curation, Investigation, Writing – review & editing. MC: Data curation, Investigation, Writing – review & editing. RP: Data curation, Investigation, Writing – review & editing. LV: Investigation, Writing – review & editing. VP: Investigation, Writing – review & editing. ET: Data curation, Writing – review & editing. PL: Data curation, Writing – review & editing, Conceptualization, Formal analysis. GS: Conceptualization, Data curation, Formal analysis, Investigation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the Istituto Superiore di Sanità, “Programma Nazionale di Ricerca sull’AIDS” (Grant number RF-CAD-2009-1306044). We are also grateful to the Centro Interdipartimentale Misure (CIM) of the University of Parma for the NMR instrumental facilities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1664231/full#supplementary-material
References
Aldovini, A. , and Young, R. A. (1990). Mutations of RNA and protein sequences involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J. Virol. 64, 1920–1926. doi: 10.1128/jvi.64.5.1920-1926.1990
Arner, E. C. , Pratta, M. A. , Freimark, B. , Lischwe, M. , Trzaskos, J. M. , Magolda, R. L., et al. (1996). Isothiazolones interfere with normal matrix metalloproteinase activation and inhibit cartilage proteoglycan degradation. Biochem. J. 318, 417–424. doi: 10.1042/bj3180417
Baba, M. , Nakajima, M. , Schols, D. , Pauwels, R. , Balzarini, J. , and De Clercq, E. (1988). Pentosan polysulfate, a sulfated oligosaccharide, is a potent and selective anti-HIV agent in vitro. Antivir. Res. 9, 335–343. doi: 10.1016/0166-3542(88)90035-6
Basrur, V. , Song, Y. , Mazur, S. J. , Higashimoto, Y. , Turpin, J. A. , Rice, W. G., et al. (2000). Inactivation of HIV-1 nucleocapsid protein P7 by pyridinioalkanoyl thioesters. Characterization of reaction products and proposed mechanism of action. J. Biol. Chem. 275, 14890–14897. doi: 10.1074/jbc.275.20.14890
Battini, L. , and Bollini, M. (2019). Challenges and approaches in the discovery of human immunodeficiency virus type-1 non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev. 39, 1235–1273. doi: 10.1002/med.21544
Carteau, S. , Batson, S. C. , Poljak, L. , Mouscadet, J. F. , de Rocquigny, H. , Darlix, J. L., et al. (1997). Human immunodeficiency virus type 1 Nucleocapsid protein specifically stimulates Mg2+−dependent DNA integration in vitro. J. Virol. 71, 6225–6229. doi: 10.1128/jvi.71.8.6225-6229.1997
Clerici, F. , Gelmi, M. L. , Pellegrino, S. , and Pocar, D. (2007). “Chemistry of biologically active Isothiazoles” in Bioactive heterocycles III. Topics in heterocyclic chemistry. New York: Springer.
Costi, R. , Di Santo, R. , Artico, M. , Massa, S. , Ragno, R. , Loddo, R., et al. (2004). 2,6-Bis(3,4,5-trihydroxybenzylydene) derivatives of cyclohexanone: novel potent HIV-1 integrase inhibitors that prevent HIV-1 multiplication in cell-based assays. Bioorg. Med. Chem. 12, 199–215. doi: 10.1016/j.bmc.2003.10.005
De Clercq, E. (2002). New anti-HIV agents and targets. Med. Res. Rev. 22, 531–565. doi: 10.1002/med.10021
Druillennec, S. , and Roques, B. P. (2000). HIV-1 NCp7 as a target for the design of novel antiviral agents. Drug News Perspect. 13, 337–49.
Flexner, C. (2007). HIV drug development: the next 25 years. Nat. Rev. Drug Discov. 6, 959–966. doi: 10.1038/nrd2336
Gandhi, R. T. , Landovitz, R. J. , Sax, P. E. , Smith, D. M. , Springer, S. A. , Günthard, H. F., et al. (2025). Antiretroviral drugs for treatment and prevention of HIV in adults: 2024 recommendations of the international antiviral society-USA panel. JAMA 333, 609–628. doi: 10.1001/jama.2024.24543
Gill, M. S. A. , Hassan, S. S. , and Ahemad, N. (2019). Evolution of HIV-1 reverse transcriptase and integrase dual inhibitors: recent advances and developments. Eur. J. Med. Chem. 179, 423–448. doi: 10.1016/j.ejmech.2019.06.058
Goel, A. , Mazur, S. J. , Fattah, R. J. , Hartman, T. L. , Turpin, J. A. , Huang, M., et al. (2002). Benzamide-based Thiolcarbamates: a New class of HIV-1 NCp7 inhibitors. Bioorg. Med. Chem. Lett. 12, 767–770. doi: 10.1016/S0960-894X(02)00007-0
Hansen, J. E. , Witzke, N. M. , Nielsen, C. , Mathiesen, L. R. , Teglbjaerg, L. S. , Nielsen, C. M., et al. (1990). Derivatives of amphotericin inhibit infection with human immunodeficiency virus in vitro by different modes of action. Antivir. Res. 14, 149–159. doi: 10.1016/0166-3542(90)90031-2
Kinch, M. S. , and Patridge, E. (2014). An analysis of FDA-approved drugs for infectious disease: HIV/AIDS drugs. Drug Discov. Today 19, 1510–1513. doi: 10.1016/j.drudis.2014.05.012
Landovitz, R. J. , Scott, H. , and Deeks, S. G. (2023). Prevention, treatment and cure of HIV infection. Nat. Rev. Microbiol. 21, 657–670. doi: 10.1038/s41579-023-00914-1
Loo, J. A. , Holler, T. P. , Sanchez, J. , Gogliotti, R. , Maloney, L. , and Reily, M. D. (1996). Biophysical characterization of zinc ejection from HIV nucleocapsid protein by anti-HIV 2,2′-dithiobis[benzamides] and benzisothiazolones. J. Med. Chem. 39, 4313–4320. doi: 10.1021/jm960253w
Melo, R. , Lemos, A. , Preto, A. J. , Bueschbell, B. , Matos-Filipe, P. , Barreto, C., et al. (2020). An overview of antiretroviral agents for treating HIV infection in Paediatric population. Curr. Med. Chem. 27, 760–794. doi: 10.2174/0929867325666180904123549
Mitsuya, H. , Looney, D. J. , Kuno, S. , Ueno, R. , Wong-Staal, F. , and Broder, S. (1988). Dextran sulfate suppression of viruses in the HIV family: inhibition of virion binding to CD4+ cells. Science 240, 646–649. doi: 10.1126/science.2452480
Morellet, N. , De Rocquigny, H. , Mély, Y. , Jullian, N. , Déméné, N. , Ottmann, M., et al. (1994). Conformational behaviour of the active and inactive forms of the nucleocapsid NCp7 of HIV-1 studied by H NMR. J. Mol. Biol. 235, 287–301. doi: 10.1016/s0022-2836(05)80033-6
Musah, R. A. (2004). The HIV-1 nucleocapsid zinc finger protein as a target of antiretroviral therapy. Curr. Top. Med. Chem. 4, 1605–1622. doi: 10.2174/1568026043387331
Otake, T. , Miyano, K. , Mori, H. , Morimoto, M. , Ueba, N. , Kunita, N., et al. (1991). Anti-HIV-1 activity of sulfated amphotericin B in vitro. Antivir. Res. 16, 243–255. doi: 10.1016/0166-3542(91)90004-b
Pauwels, R. , Balzarini, J. , Baba, M. , Snoeck, R. , Schols, D. , Herdewijn, P., et al. (1988). Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 20, 309–321. doi: 10.1016/0166-0934(88)90134-6
Pleskoff, O. , Seman, M. , and Alizon, M. (1995). Amphotericin B derivative blocks human immunodeficiency virus type 1 entry after CD4 binding: effect on virus-cell fusion but not on cell-cell fusion. J. Virol. 69, 570–574. doi: 10.1128/JVI.69.1.570-574.1995
Puhl, A. C. , Demo, A. G. , Makarov, V. A. , and Ekins, S. (2019). New targets for HIV drug discovery. Drug Discov. Today 24, 1139–1147. doi: 10.1016/j.drudis.2019.03.013
Ranganathan, S. , Muraleedharan, K. M. , Bharadwaj, P. , Chatterji, D. , and Karle, I. (2002). The design and synthesis of redox core-alpha amino acid composites based on thiol-disulfide exchange mechanism and a comparative study of their zinc abstraction potential from [CCXX] boxes in proteins. Tetrahedron 58, 2861–2874. doi: 10.1016/S0040-4020(02)00159-X
Reed, L.J. , and Muench, H. (1938). A simple method of estimating fifty per cent endpoints. Baltimore, Maryland: Johns Hopkins Press.
Saha, M. , Scerba, M. T. , Shank, N. I. , Hartman, T. L. , Buchholz, C. A. , Buckheit, R. W. Jr., et al. (2017). Probing mercaptobenzamides as HIV inactivators via nucleocapsid protein 7. ChemMedChem 12, 714–721. doi: 10.1002/cmdc.201700141
Sancineto, L. , Iraci, N. , Tabarrini, O. , and Santi, C. (2018). NCp7: targeting a multitasking protein for next-generation anti-HIV drug development part 1: covalent inhibitors. Drug Discov. Today 23, 260–271. doi: 10.1016/j.drudis.2017.10.017
Schito, M. L. , Soloff, A. C. , Slovitz, D. , Trichel, A. , Inman, J. K. , Appella, E., et al. (2006). Preclinical evaluation of a zinc finger inhibitor targeting lentivirus nucleocapsid protein in SIV-infected monkeys. Curr. HIV Res. 4, 379–386. doi: 10.2174/157016206777709492
Sever, B. , Otsuka, M. , Fujita, M. , and Ciftci, H. (2024). A review of FDA-approved anti-HIV-1 drugs, anti-gag compounds, and potential strategies for HIV-1 eradication. Int. J. Mol. Sci. 25:3659. doi: 10.3390/ijms25073659
Shin, Y. H. , Park, C. M. , and Yoon, C. H. (2021). An overview of human immunodeficiency virus-1antiretroviral drugs: general principles and current status. Infect. Chemother. 53:29. doi: 10.3947/ic.2020.0100
Thomas, J. A. , and Gorelick, R. J. (2008). Nucleocapsid protein function in early infection processes. Virus Res. 134, 39–63. doi: 10.1016/j.virusres.2007.12.006
Tondelli, L. , Colonna, F. P. , Garbesi, A. , Zanella, S. , Marongiu, M. E. , Corrias, S., et al. (1996). Native oligodeoxynucleotides specifically active against human immunodeficiency virus type 1 in vitro: a G-quartet-driven effect? Antimicrob. Agents Chemother. 40, 2034–2038. doi: 10.1128/aac.40.9.2034
Topol, I. A. , Nemukhin, A. V. , Dobrogorskaya, Y. I. , and Burt, S. K. (2001). Interactions of Azodicarbonamide (ADA) species with the model zinc finger site: theoretical support of the zinc finger domain destruction in the HIV-1 Nucleocapsid protein (NCp7) by ADA. J. Phys. Chem. B 105, 11341–11350. doi: 10.1021/jp011734g
Tramontano, E. , and Cheng, Y. (1992). HIV-1 reverse transcriptase inhibition by a dipyridodiazepinone derivative: BI-RG-587. Biochem. Pharmacol. 43, 1371–1376. doi: 10.1016/0006-2952(92)90515-K
Tummino, P. J. , Harvey, P. J. , McQuade, T. , Domagala, J. , Gogliotti, R. , Sanchez, J., et al. (1997). The human immunodeficiency virus type 1 (HIV-1) nucleocapsid protein zinc ejection activity of disulfide benzamides and benzisothiazolones: correlation with anti-HIV and virucidal activities. Antimicrob. Agents Chemother. 41, 394–400. doi: 10.1128/AAC.41.2.394
Vicini, P. , Geronikaki, A. , Incerti, M. , Zani, F. , Dearden, J. , and Hewitt, M. (2008). 2-Heteroarylimino-5-benzylidene-4-thiazolidinones analogues of 2-thiazolylimino-5-benzylidene-4-thiazolidinones with antimicrobial activity: synthesis and structure-activity relationship. Bioorg. Med. Chem. 16, 3714–3724. doi: 10.1016/j.bmc.2008.02.001
Vicini, P. , Incerti, M. , Cardile, V. , Garufi, F. , Ronsisvalle, S. , and Panico, A. M. (2007). Benzo[d]isothiazol-3-yl-benzamidines: a class of protective agents on culture of human cartilage and chondrocytes stimulated by IL-1β. ChemMedChem 2, 113–119. doi: 10.1002/cmdc.200600157
Vicini, P. , Incerti, M. , Doytchinova, I. A. , La Colla, P. , Busonera, B. , and Loddo, R. (2006). Synthesis and antiproliferative activity of benzo[d]isothiazole hydrazones. Eur. J. Med. Chem. 41, 624–632. doi: 10.1016/j.ejmech.2006.01.010
Vicini, P. , Incerti, M. , La Colla, P. , and Loddo, R. (2009). Anti-HIV evaluation of benzo[d]isothiazole hydrazones. Eur. J. Med. Chem. 44, 1801–1807. doi: 10.1016/j.ejmech.2008.05.030
Vicini, P. , Manotti, C. , Caretta, A. , and Amoretti, L. (1997). Synthesis and antiplatelet effects of 2-amino-1,2-benzisothiazolin-3-one. Arzneimittelforschung. 47, 1218–21.
Waheed, A. A. , Ablan, S. D. , Soheilian, F. , Nagashima, K. , Ono, A. , Schaffner, C. P., et al. (2008). Inhibition of human immunodeficiency virus type 1 assembly and release by the cholesterol-binding compound amphotericin B methyl ester: evidence for Vpu dependence. J. Virol. 82, 9776–9781. doi: 10.1128/JVI.00917-08
Wang, Q. , Wang, Y. T. , Pu, S. P. , and Zheng, Y. T. (2004). Zinc coupling potentiates anti HIV-1 activity of Baicalin. Biochem. Biophys. Res. Commun. 324, 605–610. doi: 10.1016/j.bbrc.2004.09.093
World Health Organization . (2025). HIV and AIDS. Available online at: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (Accessed January 08, 2025)
Wu, Q. , Wu, S. , Zou, J. , Wang, Q. , Mou, C. , Zheng, P., et al. (2023). Carbene-catalyzed access to Thiochromene derivatives: control of reaction pathways via slow release of thiols from disulfides. Org. Lett. 25, 3967–3971. doi: 10.1021/acs.orglett.3c01414
Yevich, J. P. , New, J. S. , Smith, D. W. , Lobeck, W. G. , Catt, J. D. , Minielli, J. L., et al. (1986). Synthesis and biological evaluation of 1-(1,2-benzisothiazol-3-yl)- and (1,2-benzisoxazol-3-yl)piperazine derivatives as potential antipsychotic agents. J. Med. Chem. 29, 359–369. doi: 10.1021/jm00153a010
Zani, F. , Incerti, M. , Ferretti, R. , and Vicini, P. (2009). Hybrid molecules between benzenesulfonamides and active antimicrobial benzo[d]isothiazol-3-ones. Eur. J. Med. Chem. 44, 2741–2747. doi: 10.1016/j.ejmech.2008.07.022
Keywords: 1,2-benzisothiazol-3(2H)-one benzenesulfonamides, HIV, nucleocapsid proteins, NCp7, broad spectrum antiretroviral activity, mutant strains
Citation: Loddo R, Incerti M, Favari E, Manca V, Cogoni M, Piras R, Virdis L, Palmas V, Tamburini E, La Colla P and Sanna G (2025) HIV nucleocapsid proteins as targets for novel 1,2-benzisothiazol-3(2H)-one benzenesulfonamides: synthesis and antiretroviral activity. Front. Microbiol. 16:1664231. doi: 10.3389/fmicb.2025.1664231
Edited by:
Yoshinao Kubo, Nagasaki University, JapanReviewed by:
Djamal Brahim Belhaouari, Texas A&M University, United StatesTsutomu Fukuda, Nagasaki University, Japan
Copyright © 2025 Loddo, Incerti, Favari, Manca, Cogoni, Piras, Virdis, Palmas, Tamburini, La Colla and Sanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matteo Incerti, bWF0dGVvLmluY2VydGlAdW5pcHIuaXQ=; Giuseppina Sanna, Zy5zYW5uYUB1bmljYS5pdA==
†These authors have contributed equally to this work