 Samuel A. M. Connelly1*
Samuel A. M. Connelly1* Manal AbuOun1
Manal AbuOun1 Nicholas Duggett1,2
Nicholas Duggett1,2 Miranda Kirchner1
Miranda Kirchner1 Indre Navickaite1
Indre Navickaite1 Javier Nunez-Garcia1
Javier Nunez-Garcia1 Susanna Williamson3
Susanna Williamson3 Kelly Vaughan4Christopher Teale5
Kelly Vaughan4Christopher Teale5 Muna F. Anjum1*
Muna F. Anjum1*- 1Animal and Plant Health Agency, Weybridge, United Kingdom
- 2Animal and Plant Health Agency, Thirsk Veterinary Investigation Centre, Thirsk, United Kingdom
- 3Animal and Plant Health Agency, Bury St Edmunds, United Kingdom
- 4APHA Veterinary Investigation Centre, Exeter, United Kingdom
- 5Veterinary Investigation Centre, Animal and Plant Health Agency, Shrewsbury, United Kingdom
Introduction: We characterised the genomes of 208 pathogenic Escherichia coli isolated from pigs diagnosed with enteric colibacillosis (EC), bowel oedema (BO) or colisepticaemia (CS) in England between 2016–2020 to investigate their antimicrobial resistance (AMR) and virulence-associated genes (VAGs).
Methods: AMR and VAGs were identified using the APHA SeqFinder tool, while SNP comparisons were performed with Snippy and RAxML-NG. A subset of isolates were long-read sequencing to examine mobile genetic elements.
Results: The EC group primarily carried fimbrial and toxin genes, including faeG (F4, 33%) and stb (STb, 48%). The BO group primarily encoded fedF (F18, 38%) and stx2e-AB (Stx2e, 38%). The CS group were more homogeneous, with over 70% harbouring genes for curli fibre (csgABGFE), haemolysin, iss, iron transport (sitABCD), and siderophore (iucABCD, iroBCDEN) genes. Overall, 62% of isolates were multidrug resistant (MDR) with the Enterotoxigenic E. coli (ETEC) showing an MDR prevalence of 73%. Phylogenetic analysis identified two high-risk sequence types (STs), ST90 and ST772; both unique to juvenile pigs and previously under-reported in the UK. Plasmid analysis of representative ETECs from these STs revealed large MDR plasmids potentially co-linked with metal and disinfectant resistance genes.
Discussion: This study highlights the predominant AMR and VAGs in pathogenic E. coli from diseased pigs in England, the emergence of high-risk lineages, and the role of mobile genetic elements in resistance dissemination. These findings improve understanding of pathogenic E. coli in pigs and support efforts to improve diagnostics, guide treatment, and control of farm outbreaks.
1 Introduction
Escherichia coli are Gram-negative, rod-shaped bacteria that make up part of the commensal microbial population of the vertebrate gut (Tenaillon et al., 2010). Some types of E.coli are opportunistic pathogens causing various pathologies in their hosts, ranging from diarrhoeal disease to sepsis, depending on the carriage of particular virulence associated genes (VAGs) (Tenaillon et al., 2010).
Pathogenic E. coli can be broadly split into intestinal pathogenic E. coli (InPEC) and extraintestinal pathogenic E. coli (ExPEC), based on location of the host tissue where they induce disease. Both groups can be characterised into the 11 pathotypes described, based on their phenotypic characteristics (Geurtsen et al., 2022).
Within the food chain, pathogenic E. coli causes a particular problem for juvenile pigs. Young pigs (up to 20 weeks) have immature immune systems, and early in life they rely on the passive immunity provided by the sow’s colostrum to protect them from bacterial infections, including pathogenic E. coli (Fairbrother and Nadeau, 2019). But due to naivety of the immune systems of juvenile pigs, there is an increased risk of morbidity and mortality associated with the pre- and post-weaning periods. Resulting outbreaks incur financial costs to farmers, decrease animal welfare, and increase challenges to the UK pork industry (Fairbrother and Nadeau, 2019).
InPEC frequently cause enteric colibacillosis (EC) infections in UK herds (APHA and SRUC, 2024), which is typically caused by the enterotoxigenic E. coli (ETEC) pathotype. This pathotype is characterised by expression of one or more fimbriae or adhesins, which facilitate adherence to epithelial cells of the intestine. In Europe, the predominant fimbriae associated with ETEC are F4 (previously K88) and F18, with much lower abundance of the F5, F6 and F41 fimbriae (Luppi et al., 2016). ETEC also secrete one or more enterotoxins which include the heat-labile (LT) and the heat-stable (STa and STb) toxins (Fairbrother and Nadeau, 2019; Fairbrother et al., 2005). The combination of these VAGs causes the release of ions and water into the gut lumen, resulting in bouts of pre- and post-weaning diarrhoea (PWD) (Duan et al., 2019; Turner et al., 2006), leading to increased morbidity and mortality.
InPEC are also responsible for bowel oedema (BO) due to vascular damage, where oedematous change may be visible in the submucosa of the gastrointestinal tract, subcutis, and brain. This is caused by the Shiga-toxin producing E. coli (STEC), which carry a prophage harbouring genes for the Stx1 and/or Stx2 Shiga-toxins (Rodriguez-Rubio et al., 2021). Typically, porcine STEC carry the Stx2e toxin variant whilst simultaneously expressing the F18 fimbriae on their surface (Fairbrother and Nadeau, 2019). Outbreaks of BO may also occur concurrently with or independently of, outbreaks of EC (Fairbrother and Nadeau, 2019).
There have also been reports of “hybrid” InPECs, incorporating genetic markers from both STEC and ETEC in humans (Lee et al., 2023) and animals (Zhang et al., 2007). The carriage of these genetic markers on mobile genetic elements (MGEs) gives them the potential to move both horizontally and vertically through bacterial populations. These hybrid pathotypes can have variable phenotypes which can make pathotyping more challenging (Robins-Browne et al., 2016).
The ExPEC group are a less well-defined group of disease-causing E. coli and are opportunistically invasive (Robins-Browne et al., 2016). These E. coli are usually indistinguishable from commensal E. coli of the gut, and do not have a well-defined group of virulence factors. Instead, they can harbour many different virulence factors that contribute to disease (Fairbrother and Nadeau, 2019; Geurtsen et al., 2022). In this study, we focus on ExPECs associated with colisepticaemia as it primarily causes disease in neonatal or pre-weaned piglets. CS is often caused by ExPEC that encode virulence factors supporting their survival in the bloodstream, such as the bacteriocin, colicin V and aerobactin (Mokady et al., 2005; Pitout, 2012).
Options for treatment using authorised antimicrobials can be limited or unsuccessful due to antimicrobial resistances that may be present in E. coli from pigs (AbuOun et al., 2020). There is also a requirement to avoid medically or veterinary important antimicrobials in livestock, so future treatments are not compromised (Luppi, 2017; Gale and Velazquez, 2020; World Organisation for Animal Health, 2021; World Health Organisation, 2024). However, antimicrobial sensitivity of the E. coli pathogen may not be known before treatment is initiated, and any antimicrobial use risks selection of AMR bearing organisms.
Previous studies have reported on AMR in commensal E. coli from healthy pigs (AbuOun et al., 2020), but there is limited information on AMR in pathogenic E. coli from diseased pre- and post-weaned pigs, where antimicrobial usage may be higher in response to infection control measures in herds. To address this, we used whole genome sequencing (WGS) to characterise the AMR and VAGs present in 208 E. coli isolated from pigs in England and Wales from between 2016 and 2020, with a confirmed diagnosis for one of the three pig-associated E. coli diseases (EC, BO or CS) described above. We then carried out phylogenetic analysis to compare isolates from this study to understand their genetic relatedness. We then contextualised these isolates with published data from our group for E. coli isolated from healthy pigs and pigs at slaughter. This highlighted some differences between E. coli isolated from difference stages of the food production system. Finally, long-read sequencing was used to elucidate the MGEs from a subset of isolates, allowing us to identify AMR bearing plasmids, and associate these with certain lineages. Together, these data will help improve our understanding of pathogenic E. coli associated with infections in pigs and their treatment.
2 Materials and methods
2.1 Selection, culturing, and DNA extraction from bacterial isolates
Two hundred and eight samples submitted between 2016 and 2020 primarily from England (n = 203/208) and a small number from Wales (n = 5/208), were selected from the APHA pig scanning surveillance archive. Isolates were cultured from samples originating from neonatal (<1 week), pre-weaned (6 days - 8 weeks) and post-weaned (>1 month) pigs that had been diagnosed with either EC, BO, or CS. A full list of isolates can be found in Supplementary Table 1.
For the enteric E. coli, a sterile swab was used to culture bacteria from either the intestines or faeces. For the invasive CS isolates, a tissue sample such as liver, spleen, meninges, and others (excluding intestinal or faecal samples) was seared to sterilise the surface, and a swab was taken from the incision site. Swabs were spread onto MacConkey agar after an overnight incubation at 37 °C, and then a single lactose-fermenting (pink) colony subcultured to MacConkey and grown under the same conditions. Isolates were presumptively identified as E. coli if oxidase negative, catalase positive and indole positive.
Isolates submitted between 2016 and 2017 from animals presenting symptoms of either EC or BO were also subjected to an in-house real-time PCR (qPCR) to validate a real-time PCR that detects nine virulence genes associated with ETEC and STEC (faeG, fedA, fim41a, fanA, fasA, f17A, eltA, sta1 and stx2e). This PCR was implemented in the routine diagnostics of ETEC and STEC at the APHA in 2021 which is why further isolates from this study were not included in the panel. The oligos and reaction conditions for this real-time PCR can be found with document in the Supplementary materials. Comparative analysis of the results from the real-time PCR can be found in Supplementary Table 1. The data from this qPCR was also validated against the WGS data generated in this paper.
Purified isolates were stored on beads at –80 °C until needed. A 1 μL loop of frozen stock was subcultured onto Luria Bertani (LB) agar and grown overnight at 37 °C. A single colony was selected and inoculated into LB broth and grown overnight at 37 °C, whilst shaking at 200 revolutions per minute, for DNA extraction.
DNA was extracted from overnight cultures using the Applied Biosystems™ MagMax™ CORE Nucleic Acid Purification Kit (Thermo Fisher Scientific, United Kingdom) with the Thermo Fisher Scientific KingFisher Flex System as per the manufacturer’s instructions (Thermo Fisher Scientific, United Kingdom). Isolates for long read sequencing were cultured overnight in the same way as above and extracted using the PDQeX nucleic acid extractor (MicroGEM, United Kingdom) as per the manufacturer’s instructions.
2.2 Sequencing and statistical analysis
Short-read sequencing was carried out using the Illumina NextSeq system as previously described (AbuOun et al., 2021). The data for this project is stored in the European Nucleotide Archive (Accession: PRJEB77416). For long-read sequencing, four isolates were sequenced on the Oxford Nanopore Technologies (ONT, United Kingdom) MinION system (Accession: PRJEB97106) using the Rapid barcoding kit (SQK-RBK004; ONT, United Kingdom). The sequencing library was prepared following manufacturer’s instructions with the exception that the AMPure XP bead (Beckman Coulter, United States) incubation was carried out on a Hula mixer™ (ThermoFisher Scientific) for 20 min. Samples were eluted in 10 mM Tris–HCl pH 7.5–8.0 with 50 mM NaCl whilst incubated at room temperature for 5 min. Sequencing was then carried out using R9.4 flow cells and base calling performed using Guppy (v5.0.17) with default parameter settings and the quality threshold set to 9. Long-read sequencing for isolate 149–258 was repeated using the Rapid barcoding kit 24 V14 (SQK-RBK114.24; ONT, United Kingdom) with the R10.4.1 flow cells, and Guppy (v6.5.7) to improve genomic resolution for this isolate.
Genomes were assembled using the Unicycler v0.4.8 pipeline (Wick et al., 2017), and hybrid assemblies created where short and long-read data was available. The quality of the raw and assembled data was checked using the FastQC v0.11.8,1 multiQC v1.10.1 (Ewels et al., 2016), and QUAST v5.1 tools (Gurevich et al., 2013). Kraken2 v1.1.1 (Wood et al., 2019) was used to assign taxonomic groupings to the raw reads and isolates that failed any of the QC checks were re-sequenced.
The APHA SeqFinder software (Duggett et al., 2020) was used to map sequence reads to reference database for AMR (AbuOun et al., 2021) and VAGs (Anjum et al., 2007; Wu et al., 2008). We also used a custom database to identify heavy metal and biocide resistance genes that was adapted from the BacMet database (Pal et al., 2014). ABRicate v0.82 was used to verify the locations of genes within contigs of assembled data.
VAGs included in the main body of text based on their prominence in the literature. When assigning pathotypes, we used a previously published scheme for assigning ETECs and STECs (Luppi, 2017). For the ExPEC, we used the most common genes amongst this group, due to the homogeneity of their VAGs.
AMR genes that confer resistance to antimicrobial classes that were not listed in the World Organisation for Animal Health (WOAH) list of antimicrobial agents of veterinary importance were excluded from this study (World Organisation for Animal Health, 2021), including genes for hygromycin and streptothricin. We did include the catA and cml genes for chloramphenicol resistance in this list due to chloramphenicol’s classification as a highly important antimicrobial (HIA) in humans (World Health Organisation, 2018). Multidrug resistance (MDR) was determined based on the presence of resistance genes to three or more classes of antimicrobials.
Statistical analyses were conducted in Microsoft Excel 365, where Pearson’s chi-squared tests were used to identify significance between observed and expected values. R v4.0.4 was used for data visualisation with all plots generated using the ggplot2 package (Villanueva and Chen, 2019).
2.3 Phylogenetic comparisons between Escherichia coli from this study
The Roary tool was used to estimate the number of genes in the core and accessory genomes of the 208 E. coli from this study (Page et al., 2015). Multi-locus sequence types (MLST) were assigned using SRST2 v0.2.0 (Inouye et al., 2014) as previously described (AbuOun et al., 2021). Any ambiguous sequence types (STs) were confirmed with the pubMLST database. Phylogroups were determined using ClermonTyper (Beghain et al., 2018).
The snippy V3.1 tool (Seemann, 2015) was used to call variants from WGS reads and a core genome single nucleotide polymorphism (SNP) alignment was generated using isolate E. coli MG1655 U00096.3 as a reference. Analysis of SNP differences was carried out in R v4.5.1 (R Core Team, 2025) using the Tidyverse packages (Wickham et al., 2019).
RAxML-NG V1.0.3 (Kozlov et al., 2019) was used to generate a maximum-likelihood (ML) tree file with 100 bootstraps using the GTR + FO + G4m model of nucleotide substitution. We visualised the phylogenetic tree using the ggplot2 v3.5.2 (Villanueva and Chen, 2019) and ggtree v3.16.3 (Yu et al., 2016) packages for R. Phylogenetic and evolutionary analysis between clades was carried out using the ape v5.8-1 package (Paradis and Schliep, 2019).
We used the K-mer based comparison tool, Sourmash v4.8.12 (Pierce et al., 2019), to create a dissimilarity matrix of hash scores. Any isolates that had a dissimilarity score of ≥80% were removed from the analysis due to their likelihood of being an unrelated species. The Pheatmap (Kolde, 2018) and Tidyverse packages were used to hierarchically cluster and visualise the dissimilarity matrix in R. Metadata such as ST, phylogroup, pig age, host diagnosis, MDR genotype, and study were used to identify subclusters within the dataset.
2.4 Selection of publicly available datasets and their relationship to Escherichia coli from this study
To contextualise the pathogenic E. coli within this study with E. coli isolated through various stages of pig production, we chose to utilise two publicly available datasets previously generated through studies carried out by the APHA. E. coli sequences came from two datasets: the ARDIG dataset (Accession: PRJNA750276), containing E. coli sequence data that were initially isolated from five different age groups of healthy pigs (Storey et al., 2022); and the second, MOLSIG (Accession: PRJEB26317) dataset, containing sequence data of E. coli isolated from adult pigs at slaughter (AbuOun et al., 2020). To reduce selection bias, we also ensured that sequences were only used for E. coli genomes that had been grown on non-selective plates, as was the case with the 208 E. coli in this study.
2.5 Analysis of plasmids
Plasmids were annotated using Bakta v1.11.0 (Schwengers et al., 2021), and visualised in BRIG (Alikhan et al., 2011). Linear comparisons of genomic loci were visualised using the Easyfig v2.2.2 software (Sullivan et al., 2011). Images were edited in Inkscape (v1.2).
3 Results
3.1 Host background for pathogenic Escherichia coli
A total of 208 E. coli isolated from diseased pigs submitted to the APHA between 2016 and 2020 through its scanning surveillance programme3 were genomically characterised in this study. All pigs had a confirmed postmortem and laboratory diagnosis of one of the three E. coli related diseases: EC (n = 122, 59%), BO (n = 34, 16%) or CS (n = 52, 25%). A list of isolates and related metadata can be found in Supplementary Table 1.
3.2 Relative proportion of VAGs
Current practice at the APHA is to screen suspected ETEC and STEC isolates associated with EC and BO for some of the known virulence factors associated with enteric disease in juvenile pigs. This high-throughput and cheap alternative to sequencing allows us to make an initial assessment of which isolates should be taken forward for further analysis by WGS. During this study, we validated a qPCR to determine the congruence of genotype and phenotype for virulence (Supplementary Table 1). Approximately 97% (n = 95/98) of the E. coli screened by both the qPCR and WGS showed good congruence, supporting our use of these methods for an initial screen of isolates collected through scanning surveillance.
Further work to broadly characterise the primary VAGs in these E. coli was then carried out. The VAGs associated with ETEC have been well defined in pigs (Luppi, 2017) and a full list of identified genes identified in these isolates is given in Supplementary Table 2. The genes, faeG and fedF, which encode components of the F4 and F18 fimbriae, were the most prevalent fimbrial genes identified in the EC (33%) and BO (38%) isolates, respectively (Supplementary Table 2). The f17A gene, which encodes the F17 fimbriae, was also present in a small population of EC (~2%) and CS isolates (~2%) (Figures 1A,B).
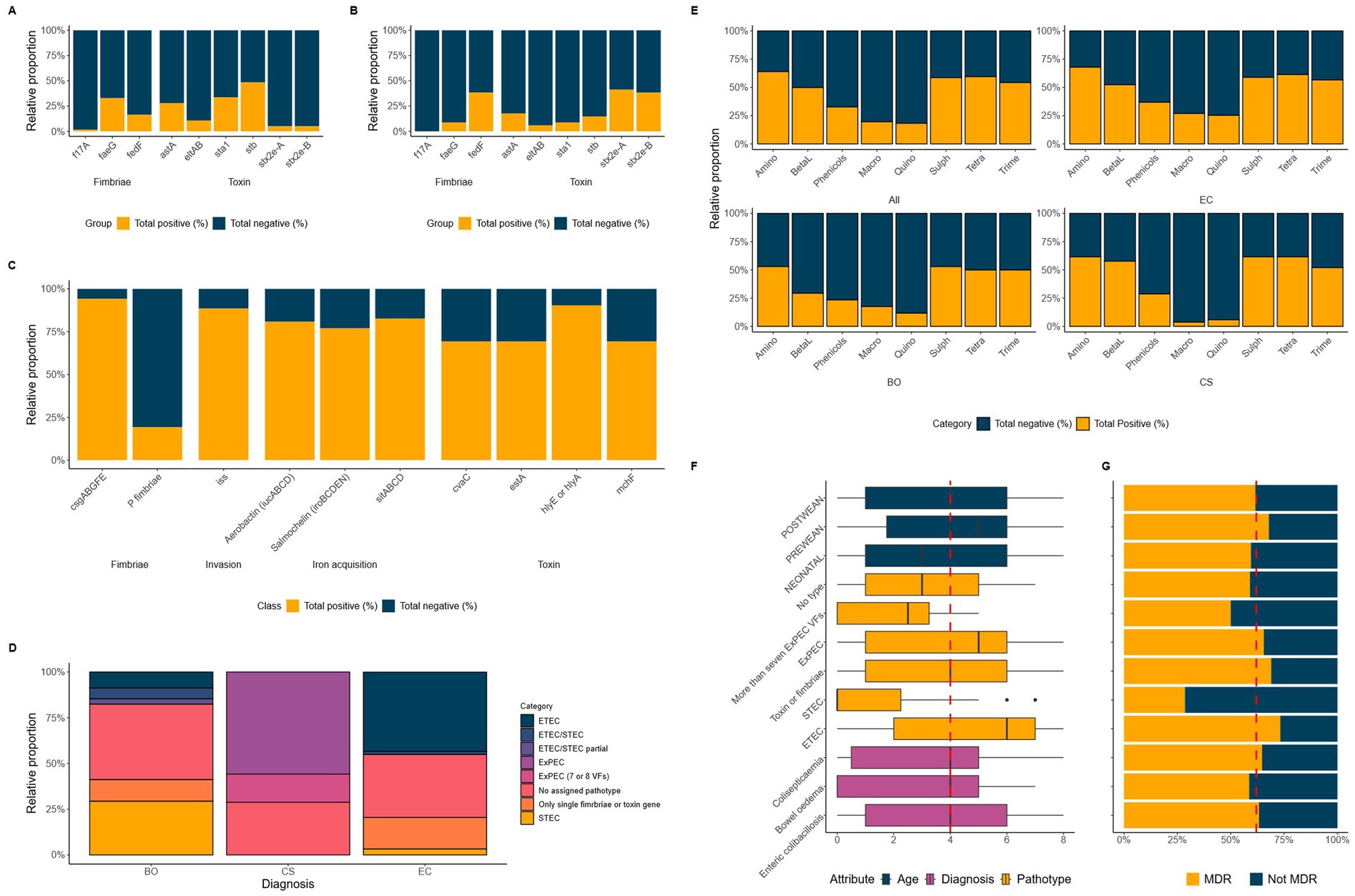
Figure 1. Panels (A–C) show the relative abundance of VAGs associated with ETEC, STEC and ExPEC for EC (A), BO (B) and CS (C) groups of E. coli. Panel (D) shows the relative abundance of each pathotype the E. coli were grouped into based on their respective genotypes. Panel (E) shows the relative abundance of resistance genotypes for CIA and HIA classes for all, EC, BO and CS groups. The final panel (F) shows the median number of HIA and CIA classes for the enteric (EC and BO) and extraintestinal (CS) groups as well as pathotype alongside the prevalence of MDR (G). The red dashed line represents the median or mean value.
The genes encoding the LT (eltAB), STa (sta1), and the Stx2e (stx2e-AB) toxins were only found in the EC and BO associated E. coli. The stb gene (encoding STb) was the most prevalent toxin gene found in EC isolates (48%).
The most common toxin genes in BO isolates were the stx2e-A (41%) and -B (38%) subunit genes, with one isolate lacking the B-subunit gene. Additionally, a small population of the EC isolates also carried the stx2e-AB (5%) genes (Figures 1A,B). We observed that there was a statistically significant relationship between the presence of a complete stx2e-AB genes and disease state of the host (p value = 4.8 × 10−6). The chances of an isolate encoding the Stx2e toxin were higher in E. coli associated with BO than EC (OR = 7.8, CI = 2.9, 20.5). We also note, there was a statistically significant relationship between the presence of the F18 fimbriae genes, fedA and fedF, with the stx2e-AB genes (p value = 7.6 × 10−10).
We included a variety of VAG targets for the CS group of E. coli which included factors for adhesion, invasion, iron acquisition and toxin production. There was less heterogeneity of the VAGs within this group, with >70% of isolates carrying the most common genes, including the csgABGFE (curli fibre) genes present in virtually all CS isolates (94%), haemolysin (90%) and iss (88%) genes. Approximately 80% of all CS isolates encoded the iron transport genes, sitABCD, as well as the genes that encode the salmochelin and aerobactin siderophore systems (Figure 1C).
Using the genomic data, we grouped the E. coli into pathotypes based on their virulotypes (Figure 1D). We were only able to classify 43% of the EC isolates and 29% of BO isolates as ETEC and STEC, respectively. In contrast, over half of the CS isolates could be categorised as ExPEC, due to isolates sharing nine ExPEC VAGs (56%, n = 29/52).
Both EC and BO isolates had a small number of E. coli classified as STEC (3%) or ETECs (9%) within their groups, respectively. This was a particularly surprising outcome for the BO isolates as it is assumed that presence of the stx2e genes is the distinguishing factor for these E. coli. These groups also had a small number of E. coli harbouring both ETEC fimbriae and toxin genes, and the stx2e genes, indicating an ETEC/STEC mixed genotype (Figure 1D).
Finally, we were unable to pathotype just under a third of isolates (32%,) using the chosen VAGs (Figure 1D). We identified three (2.5%) EC isolates (189–250, 36–171, and 46–283) that encoded the intimin gene, eae and the gene for its receptor, tir (Pokharel et al., 2023). This was coupled with presence of genes for the type III secretion system suggesting these isolates were enteropathogenic E. coli (EPEC) (Supplementary Table 2). We also looked at the full complement of VAGs for the other non-typable isolates (n = 63, 30%), identifying a broad variety of genes associated with different pathotypes of E. coli which was outside the scope of this study (Supplementary Table 2).
3.3 Prevalence of AMR and MDR
A total of 61 different AMR genes were identified in 78.8% of isolates (Table 1). Isolates with AMR harboured between 1 and 19 AMR genes, which represented up to eight veterinary critically important and highly important antimicrobial (VCIA and VHIA) classes (World Organisation for Animal Health, 2021). There was no significant difference between any of the three groups encoding at least one AMR gene. AMR was identified in 83% of EC, 77% of CS and 68% of BO isolates. Full AMR genotype information can be found for each isolate included in this study in Table 1. AMR genotype information can be found in Supplementary Table 3.
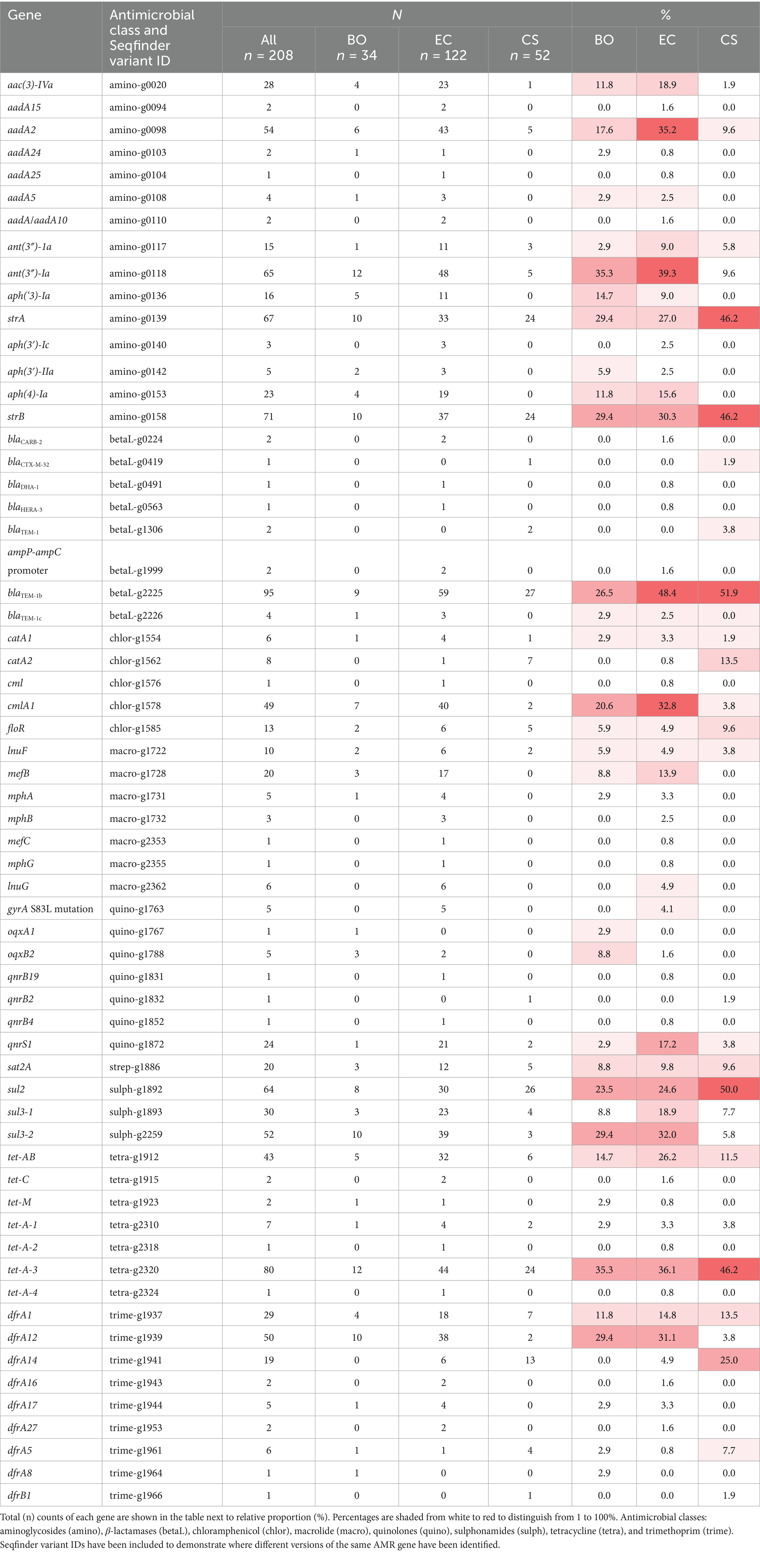
TABLE 1. List of AMR genes detected in this study.
The most common antimicrobial classes across all isolates where a resistance genotype was identified were the aminoglycosides (64%), tetracyclines (60%) and sulphonamides (59%) (Figure 1E). A resistance genotype for trimethoprim was similar between E. coli associated with all three diseased states. Resistance genotypes were also similar between EC and CS associated E. coli for sulphonamides and tetracycline, whilst prevalence was approximately 10% lower in BO isolates to these antimicrobial classes. BO isolates also had much lower prevalence of resistance genotypes to the aminoglycosides and β-lactam antibiotics (Figure 1E).
Out of the 133 isolates that encoded resistance to one of the VCIA aminoglycosides, 13% isolates were positive for the aac(3)-IVa gene (gentamicin/apramycin/tobramycin resistance; Plattner et al., 2020; Figure 1E). The prevalence of this gene was higher in EC and BO groups (17%), compared with the CS group (~2%) (Table 1).
Eighteen percent of EC and BO groups had one of the five genes (mefB, mefC, mphA, mphB or mphG) encoding resistance to the VCIA macrolides azithromycin, erythromycin, spiramycin, and telithromycin (Table 1). These genes were absent in the CS group (Figure 1E). Ten percent of the enteric isolates carried genes associated with resistance for the VHIA, lincomycin (inuF and inuG). Three of these isolates also carried a VCIA macrolide gene as well, which was either mphA (isolate 20–187), or mefB (isolates 80–2 and 99–47).
Genes for fluoroquinolone resistance were predominantly identified in the EC group of E. coli (Figure 1E). The fluoroquinolone resistance gene qnrS1 was predominantly identified in EC isolates (17%), with <4% of the BO and CS groups encoding this gene. A total of 4.1% of EC isolates also had mutations in the quinolone resistance determining regions (QRDRs) of the gyrA gene, giving rise to high level fluoroquinolone resistance. All five isolates (16–170, 20–187, 4–115, 57–120, 65–91) encoded the S83L mutation, with all but 57–120 also encoding a second substitution in D86N. These non-synonymous amino acid changes were not detected in any BO or CS isolates (Table 1).
We identified extended spectrum cephalosporinase (ESC) genes in <2% of isolates (Table 1). A CS associated E. coli harboured the blaCTX-M-32 extended spectrum beta-lactamase (ESBL) gene, and an EC associated E. coli harboured the blaDHA-1 ampC resistance gene. Two additional ETEC isolates harboured mutation in the promoter of ampC gene associated with increased expression of β-lactamase.
The median number of antimicrobial classes where a resistance genotype was identified was four (Figure 1F). Comparing this metric across diagnosis, pathotype, and age exposed differences between some of the groups. This figure was highest in the ETEC (n = 6), and lowest in the STEC pathotype (n = 0). Both BO and neonatal groups had a slightly lower median of 3.
The proportion of all isolates that were MDR was 62% (Figure 1G). There was a statistically significant dependence between MDR and pathotype (p value = 7.29 × 10−3). The highest rate of MDR was seen in the ETEC pathotype (73%) and lowest in the STEC pathotype (29%). The odds of an ETEC isolate being MDR was 6.8 times higher than STEC (CI = 1.9, 25.1), and 2.7 times higher than ExPEC (CI = 0.4, 4.8).
3.4 Exploration of the phylogenetic relationships of Escherichia coli from different stages of pig production using SNP and K-mer based methods
We found that the average assembled genome size was approximately 5.1 Mbp (SD ± 0.29 Mbp), and from this, a core SNP alignment was generated using 102,329 SNPs present in the 208 E. coli. The core genome of these 208 E. coli consisted of approximately 4.1 Mbp (SD ± 0.1 Mbp), equating to 81% of the average full genome which contains approximately 4,000 genes (Blattner et al., 1997). There were an estimated 3,951 genes in the core genome, with 25,247 genes representing the pan-genome. A total of 19 STs were identified in ≥3 isolates (Supplementary Table 4), with the most common being ST10 (n = 27), ST1 (n = 15), ST772 (n = 13), ST90 (n = 13) and ST648 (n = 10).
We constructed a SNP based phylogenetic tree, which had on average, a core genome pairwise SNP distance (referred to as SNP distances from here on out) of 57,093 (SD ± 27,103) across the core genomes the 208 E. coli (Supplementary Table 4). However, SNP distances ranged from 8 to 102,329, highlighting that some isolates were likely clonal, whilst other were incredibly genetically diverse.
We broadly split the tree into two large clades reflecting the branch lengths (Figure 2A). These clades were further subdivided by diagnosis (Figure 2A, ring 1), ST (Figure 2A, ring 3), MDR (Figure 2A, ring 4) and pathotype (Figure 2A, ring 5). We identified that 75% of the tree (n = 155/208) only had an evolutionary divergence of ~2.7% and were grouped into Clade 1, suggesting that much of the diversity in the tree was the result of a smaller number genetically diverse, isolates.
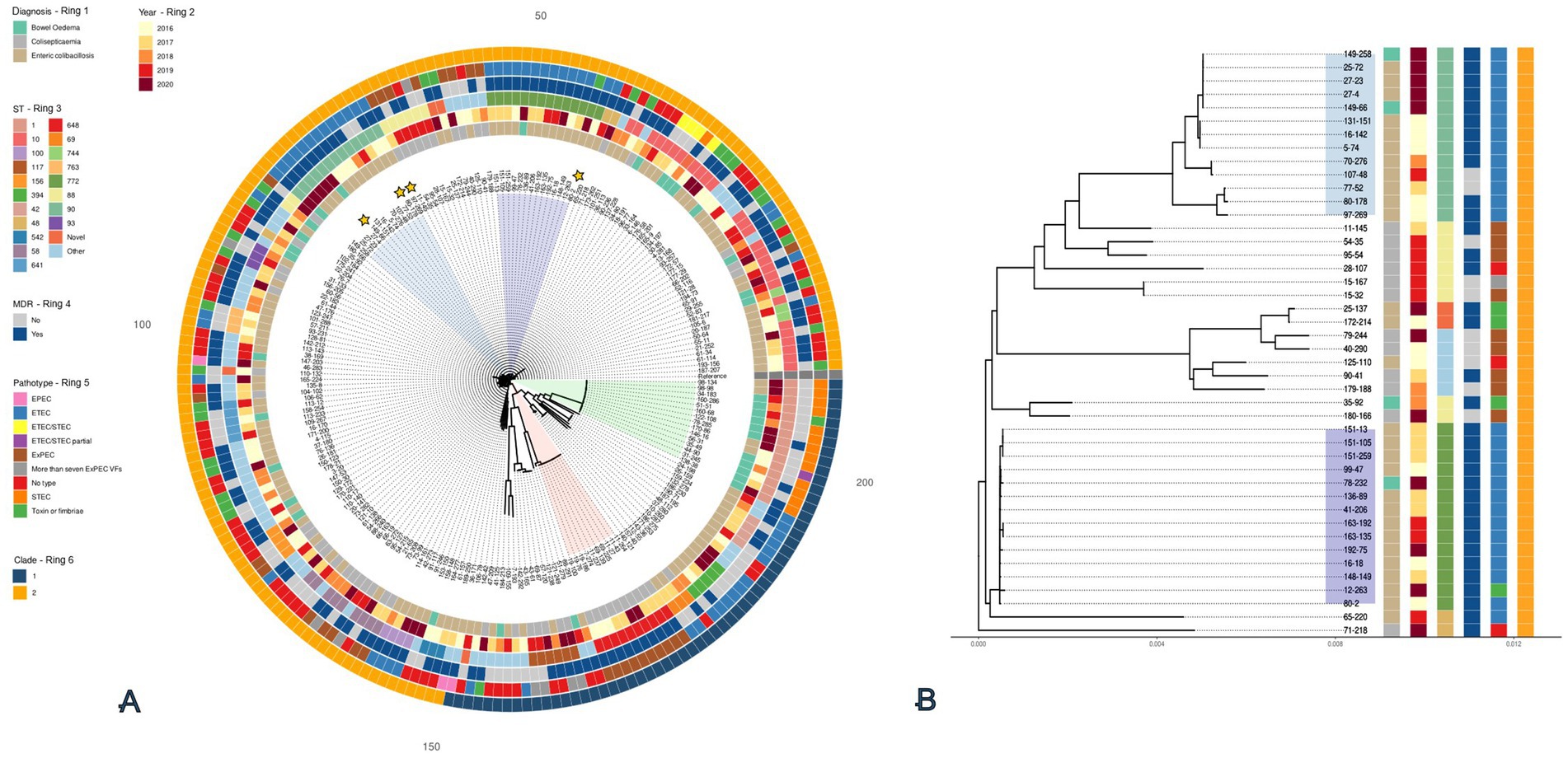
Figure 2. SNP based maximum likelihood tree with 208 E. coli isolated from diseased pigs (A). The tree is based on the core alignment and inferred using RAxML-ng using the GTR + FO + G4m substitution model and rooted on the reference isolate (Reference: E. coli MG1655 U00096.3). Branches are drawn to scale in substitutions per site. A subtree (B) is shown for the 44 isolates that make up the subclades containing ST90 and ST772 as well as the surrounding isolates. Annotation for Diagnosis (Ring 1), Year (Ring 2), ST (Ring 3), MDR (Ring 4), Pathotype (Ring 5), and Clade (Ring 6) are all shown as coloured blocks around both trees. Shaded areas on the tree highlight key clusters for ST90 (blue), ST772 (purple), ST648 (red), and ST1 (green). Yellow stars highlight isolates that have undergone long-read sequencing.
Subclades within Clade 2 included isolates from ST1 and ST648 (Figure 2A, red and green slices). ST1 (n = 15) contained all isolates that encoded the stx2e genes, with most in this group also associated with BO. This subclade had an average SNP distance of 414 SNPs (SD ± 268), showing that these were not clones but likely grouped together because they were more different than other isolates in the comparison. In contrast, ST648 which contained only MDR ExPEC (n = 10), had an average SNP distance of 89 SNPs (SD ± 13) suggesting that these isolates were more closely related, and likely share a more recent common ancestor (MRCA) than isolates in ST1.
Within Clade 1, we identified two of the largest clusters of predominantly EC associated, MDR, ETECs belonging to ST90 (n = 13) and ST772 (n = 14). From the tree we were able to identify that the mean percentage identity between their core genomes was 99.5%. The mean patristic distances within ST772 showed that this cluster was highly homogenous with an average SNP distance of 45 SNPs (SD ± 25, range = 8–143 SNPs). This compared to the average SNP distance of 844 SNPs (SD ± 855, range = 27–1,997 SNPs) within ST90, highlights that ST772 is far more homogeneous with some isolates likely being clonal. To support this, we were able to estimate from the tree that the MRCA between ST90 and ST772 lies at 0.0032 substitutions per site from the root, which corresponds to ~1.2% of the tree’s depth. Alongside this, the average SNP distances between ST90 and ST772 were 45,806 SNPs (SD ± 1,216, range = 41,371 – 47,282), demonstrating that although neighbouring each other, both form distinct subclades (Figure 2B).
To contextualise our dataset with E. coli from pigs during the production process, we generated a K-mer based distance matrix (Supplementary Table 5). We included data from this project with sequences from two other projects in the UK on E. coli from healthy pigs throughout the production process (n = 174; Storey et al., 2022), and pigs at slaughter (n = 248; AbuOun et al., 2020). This increased the total number of sequences compared in this study to N = 630.
Clustering based on STs, diagnosis, and phylogroup (Supplementary Table 5) were observed. A total of 48% (n = 305/630) fell into either phylogroup A or B2 associated with humans, whilst 26% (n = 162/630) fell into phylogroup B1, associated with domestic animals (Tenaillon et al., 2010). In total, there were 179 unique STs, with 26 STs occurring in ≥5 isolates. Interestingly, several of the predominant STs were only found in neonatal, pre- and post-weaned pigs from this study. This included the aforementioned ETEC associated ST90 (n = 13, phylogroup C; Supplementary Figure 1) and ST772 (phylogroup A; Supplementary Figure 2), as well as the ExPEC associated, ST648 (n = 10, phylogroup F, cluster 10).
3.5 Characterisation of the MDR plasmids carried by four isolates from ST90 and ST772
The high rates of MDR genotypes in isolates from ST90 and ST772 warranted further investigation. Long read sequencing of four MDR isolates from these STs was performed: 149–258 (BO, ST90), 80–178 (EC, ST90), 80–2 (EC, ST772), and 97–269 (EC, ST90; Supplementary Table 6). We identified between two and six plasmids encoding either virulence or AMR in these isolates. Plasmids carrying AMR genes had a greater size range (48–285 kb) than those carrying virulence genes (7–136 kb).
One MDR plasmid, p149-258_N2 (Figure 3A), was homologous to p97-269_N4, with both sharing 99% coverage and ~100% identity, although both parental isolates originated from different herds (149–258 and 97–269). Both plasmids encoded resistance to ampicillin (blaTEM-1), gentamicin (aac(3)-IVa), sulphonamides (sul1 and sul3), tetracycline (tet-AB), and trimethoprim (dfrA12) (Figure 3A). Both only differed in a 10 kb region, present in 149–258_N2 but absent from 97–269_N4. It carried resistance genes for kanamycin (aph(3′)-IIa) and fluoroquinolones (oqxB). Both plasmids also carried the silESRCFBAGP, pcoABCDRSE and qacL genes which have been proposed to confer resistance to silver (Elkrewi et al., 2017), copper (Giachino and Waldron, 2020), and quaternary ammonium compounds (QAC) (Boyce, 2023), respectively. Some of these metal resistance genes (MRGs) were also identified in the MDR plasmid, p80-2_N6 (Figure 3B). These included silESRCFBAGP and pcoA. Aligning the MRG regions from these three plasmids (Figure 3C) showed that the gene synteny for the sil operon is the same across all three plasmids, but the pco operon in p80-2_N6 had been truncated. We also note the carriage of the ~4 kb merRTPCADE operon in p80-2_N6, which has been implicated in mercury detoxification (de Mattos D’Avila et al., 2024).
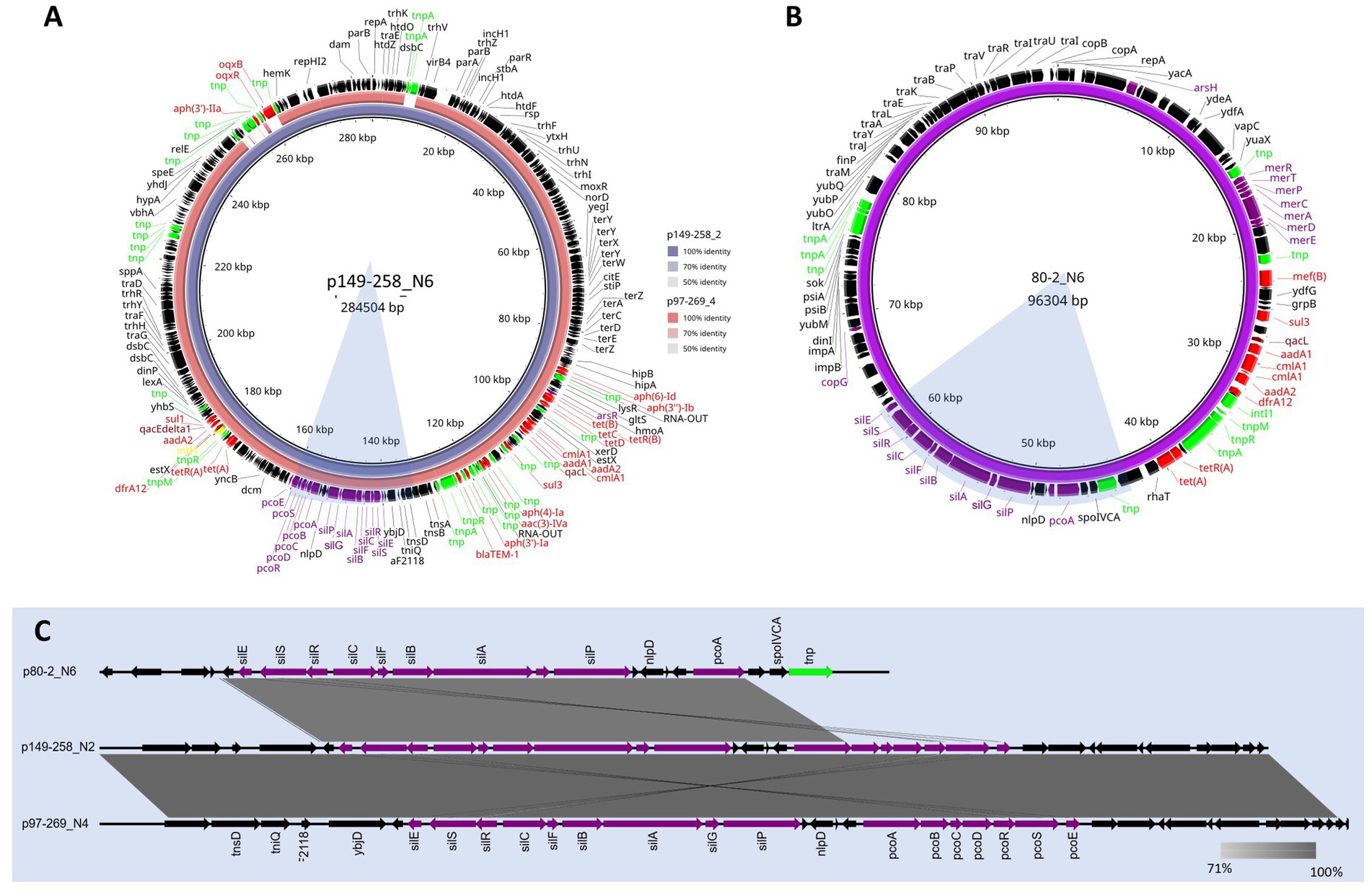
Figure 3. BLAST ring alignments of MDR plasmids containing the sil and pco operons from ST90 and ST772. Alignment (A) shows the comparison between the circular 284 kb plasmid reference, p149-258_N6 (inner ring), and the 273 kb plasmid, p97-269_N4 (outer ring). Identity between the two plasmids is indicated with opacity of the rings. The plasmid map of the circular, 96 kb plasmid, p80-2_N6, is shown in (B). A linear alignment of the sil and pco MRG 30 kb regions of the p149-258_N6 and p97-269_N4, as well as the ~20 kb region in p80-2_N6 is shown in (C). Shaded bars show percent identity (71—100%). The corresponding regions are highlighted in blue in (A,B). Annotations for A–C shown and genes are coloured function: AMR (red), MRGs (purple), disinfectants (maroon), integrons (yellow), transposons (lime), and genes (black).
Both 80–2 and 149–258 carried a second plasmid harbouring AMR which can be viewed in Supplementary Figures 3A,B, respectively. The p80-2_N3 194kb plasmid from 80–2 was found to share 98% coverage and 97% identity with p80-178_N3 (Supplementary Figure 3A). Both p80-2_N3 and p80-178_N3 carried resistance for the VCIAs, apramycin and gentamicin (aac(3)-IVa) in a class I integron. The second AMR plasmid from 149-258 (Figure 3B), was a smaller 77 kb plasmid (p149-258_N6), that encoded three AMR genes for aminoglycoside resistance, aph(4)-la, aac(3)-IVA, and aph(3′)-la. It also encoded resistance for β-lactams (blaTEM-1). All AMR and virulence plasmids are listed in Supplementary Table 6 along with their replicon types.
4 Discussion
Juvenile pigs, including neonatal, pre- or post-weaning pigs, are particularly susceptible to EC, BO or CS diseases, due to factors such as under-developed immune systems, hygiene and the environment. As the risk of disease is a major concern, control measures for pathogenic E. coli in pigs are wide-ranging and include various practices such as: ensuring excellent hygiene; pig management and environment; producing robust healthy pigs at weaning; manipulation of diets; ensuring adequate colostrum intake; weaning at a later age; and use of probiotics (Pan et al., 2017). The use of live oral vaccines for different VAGs such as F4 and the Stx2e toxin, has also been used as a control measure (Johansen et al., 1997; Melkebeek et al., 2013).
The genomes of the 208 E. coli analysed in this study reflect what has been reported previously where the core genome of E. coli is somewhere around 4,000 genes (Geurtsen et al., 2022) and the pan-genome is estimated to exceed 22, 000 (Robins-Browne et al., 2016). Isolates predominantly clustered broadly into phylogroups A, B1, and C, with the latter two commonly associated with livestock in the EU (Kaspersen et al., 2024). Our SNP based analysis showed that there was also a significant amount of genetic variability within these isolates, with some STs having more genetic relatedness.
Despite high core-genome variability, these isolates could be pathotyped using relatively few genetic features that have been reported in the literature. The more homogenous ExPECs (~70% of total) carried VAGs for biofilm production, adherence, iron acquisition, serum survival, the haemolysin toxin and the colicinV bacteriocin. These results are also comparable to reports from other geographic locations (Tan et al., 2011), and likely reflect adaptations facilitating the colonisation by ExPECs of multiple anatomical sites (Russo and Johnson, 2009).
The InPEC were more challenging to categorise using the current pathotyping schemes for ETEC and STEC (Luppi, 2017), due to the absence of well-characterised virulence genes in several isolates in our panel. There were especially few VAGs in the BO group, where only 35% of E. coli encoded both genes required for a functional Stx2e toxin, which is characteristically required for the development of bowel oedema in pigs. For the remaining 65% of E. coli where stx2e was absent but the pigs were diagnosed with BO, it’s possible a mixed infection occurred with both STEC and ETEC, resulting in only the ETEC being cultured. Although there is possibility that other unknown VAGs were present in these cases.
In fact, although the F18 fimbriae has been found in ETEC associated with PWD (Garcia et al., 2020), we identified a statistically significant association between presence of genes encoding the F18 and the Stx2e toxin. This supports previous reports where the Stx2e toxin was almost exclusively found associated with the F18 fimbriae (Luppi et al., 2016).
However, the most common VAGs harboured by ETECs in our study were the F4 (33%) fimbriae and STb (48%) toxin. This mirrors what has been observed in Europe from pigs with diarrhoeal diseases (Luppi et al., 2016). For the isolates that we could not pathotype (32%), uncommon VAGs such as those encoding EaeH and Paa were detected in some isolates and possibly play a role in their pathogenicity. As the virulence mechanisms behind these VAGs are still poorly understood (Ngeleka et al., 2003; Sheikh et al., 2014), further work is required in this area.
We were able to validate our primary screening method using this WGS dataset. One of the main limitations of qPCR is the need for previously characterised genes (Anjum, 2015). We demonstrate that our qPCR method is ~97% accurate, highlighting its accuracy when compared to WGS. However, we do not gain any additional information outside of the nine virulence genes included. Characterising the isolates in this study has highlighted some possible ‘blind-spots’, namely genes such as stb, which was commonly found in ETECs. Additionally, we show that the majority of ETEC and STEC in this study carried genes encoding either F4 or F18, so some of the genes used in this screen may be considered redundant if they are uncommon. Future work will look at using this genomic data to improve our current methods and broaden the scope of our scanning surveillance on pathogenic E. coli to ensure our front-line methods are beneficial to veterinarians requesting these tests.
AMR was present in approximately 80% of E. coli from this study. The InPEC predominantly harboured genes encoding resistance for gentamicin (17%) and azithromycin (10%), compared to CS associated E. coli, where only ~2 and 0% of isolates, respectively, carried the resistance genes. EC associated E. coli predominantly encoded (17%) the fluoroquinolone resistance gene qnrS1, compared to BO and CS isolates (<4%). EC associated E. coli were also the only isolates (4.1%) to harbour mutations in the QRDR that provide resistance to the fluoroquinolones.
MDR genotypes were identified in 62% of the 208 E. coli in this study. Due to differences in the sampling strategy with previous studies, it is hard to make direct comparisons, however, the MDR rate in these E. coli was higher than those detected in healthy adult pigs at slaughter (46.9%) (AbuOun et al., 2020), and significantly higher than those from healthy pigs throughout the production cycle from a farm with history of low antimicrobial usage (3.9%) (Storey et al., 2022). These results suggest that pathogenic E. coli isolated from sick juvenile pigs could be a significant reservoir of AMR.
Nevertheless, the predominant MDR genotypes in our E. coli were to commonly used antimicrobials such as tetracycline, the penicillin β-lactams, sulphonamides, and trimethoprim (Fairbrother and Nadeau, 2019). Genotypic resistance to the HP-VCIAs and HP-CIAs, such as aminoglycosides, macrolides, and the fluoroquinolones were <21%; ESC genes were only detected in ~2% of isolates. These figures likely reflect the very low use of HP-CIAs (0.01%) in pigs in the UK (Broadfoot et al., 2023).
Further investigation demonstrated that MDR, particularly within the ETEC group, was 6.8 times and 2.7 times more likely than in STEC and ExPEC groups, respectively. To investigate this, we carried out a phylogenetic analysis of all 208 E. coli. Within Clade 1, we were able to identify two larger, genetically distinct clusters belonging to ST90 and ST772, that were predominantly characterised as ETECs with MDR genotypes. The over-representation of these STs when compared to other STs provides an explanation for the increased likelihood of MDR in the ETEC. Both STs have previously been reported in Danish and Belgian pig herds (Garcia et al., 2020; De Koster et al., 2023; De Koster et al., 2021), but we were unable to find any reports of these STs within herds in England. With limited survey data on pathogenic E. coli from this age bracket of pigs in England, it is possible that these lineages have been underreported. In support of this, a K-mer based comparison of the genomic data in this study with two other datasets from the APHA on healthy pigs at five different ages (Storey et al., 2022), and adult pigs at slaughter (AbuOun et al., 2020), we were able to show that ST90 and ST772 were only assigned to InPEC from diseased juvenile pigs.
Further investigations into the AMR MGEs carried by some isolates from ST90 and ST772 showed homologous plasmids in isolates from both STs. Additionally, three plasmids carried the sil and pco operons, reported to provide resistance against silver and copper (Munson et al., 2000), respectively. The carriage of metal resistance and AMR genes has been reported in bacteria, especially those associated with agriculture (Pal et al., 2015). However, it is unclear why proposed silver resistance genes would be co-localised on plasmids alongside MDR, as silver is not associated with agricultural pollution (Purcell and Peters, 1998). Previous reports have shown that the sil operon is cryptic in most isolates of E. coli, and it is switched on through spontaneous missense mutations in the silS, sensor kinase gene (Elkrewi et al., 2017). This raises the question that the sil operon could be functionally involved in an unknown process, but whether this may provide a selective advantage to E. coli in this niche is currently unclear.
To conclude, in this retrospective study of pathogenic E. coli isolated from diseased pigs in England, we show common features in E. coli originating from pigs with one of the three recognised E. coli associated disease presentations. We were able to use this data to validate our current front-line methods and highlight our surveillance ‘blind-spots’ for pathogenic E. coli affecting juvenile pigs. Additionally, the genetic diversity in the core genomes of these isolates was found to be quite high, with characteristics for virulence and MDR identified in high-priority lineages such as ST90, ST648, ST772. We found these STs were uniquely isolated from diseased juvenile pigs. Analysis of some of their MGEs identified several MDR plasmids, co-linked with heavy metal resistance genes as well as resistance genes for disinfectants such as QACs. The role these additional factors may play in the spread of AMR is still unclear, but there should be further investigation into these high-priority lineages due to their potential for causing disease and acting as reservoirs for AMR within the ecological niche of the pig industry. With this information, we will be able to develop more robust surveillance methods, provide better support for front-line veterinary workers to improve animal welfare, and reduce the impact of these diseases on swine production.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The APHA undertakes research using animals under the Animal (Scientific Procedures) Act 19 (ASPA) which includes having its own Animal Welfare and Ethics Board (AWERB). The owners of the animals provided written consent to carry out research on isolates sampled from their deceased animals. As this was outside the ASPA, which only concerns live animals, ethical approval was not sought prior to this study.
Author contributions
SC: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. MA: Conceptualization, Funding acquisition, Investigation, Project administration, Writing – review & editing. ND: Methodology, Supervision, Writing – review & editing. MK: Conceptualization, Methodology, Supervision, Writing – review & editing. IN: Software, Supervision, Writing – review & editing. JN-G: Data curation, Investigation, Methodology, Supervision, Writing – review & editing. SW: Conceptualization, Investigation, Methodology, Supervision, Writing – review & editing. KV: Data curation, Formal analysis, Writing – review & editing. CT: Writing – review & editing. MFA: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the Veterinary Medicines Directorate (VMD) to MFA. The APHA laboratory and staff costs of this work were funded by Defra through the “Scanning surveillance for Pig Diseases in England and Wales” (ED1200), and “Understanding the transmission of AMR; its characteristics and methods for control using a molecular toolbox” (VM0533B).
Acknowledgments
We would like to acknowledge the staff at APHA Veterinary Investigation Centres, regional laboratories that collected these samples, and Central Sequencing Unit (CSU), who all have provided their technical skills and expertise to this project. We also thank the farmers that have contributed samples to this study which were sourced through the APHA Scanning Surveillance program. Portions of this research were supported with facilities and consultation from the Scientific Computing Environment at the Animal and Plant Health Agency.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1668327/full#supplementary-material
Footnotes
1. ^https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
2. ^https://github.com/tseemann/abricate
3. ^https://www.gov.uk/guidance/animal-disease-scanning-surveillance-at-apha
References
Abuoun, M., Jones, H., Stubberfield, E., Gilson, D., Shaw, L. P., Hubbard, A. T. M., et al. (2021). A genomic epidemiological study shows that prevalence of antimicrobial resistance in Enterobacterales is associated with the livestock host, as well as antimicrobial usage. Microb. Genom. 7:630. doi: 10.1099/mgen.0.000630
Abuoun, M., O'connor, H. M., Stubberfield, E. J., Nunez-Garcia, J., Sayers, E., Crook, D. W., et al. (2020). Characterizing antimicrobial resistant Escherichia coli and associated risk factors in a cross-sectional study of pig farms in Great Britain. Front. Microbiol. 11:861. doi: 10.3389/fmicb.2020.00861
Alikhan, N. F., Petty, N. K., Zakour, N. L. B., and Beatson, S. A. (2011). Blast ring image generator (Brig) simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Anjum, M. F. (2015). Screening methods for the detection of antimicrobial resistance genes present in bacterial isolates and the microbiota. Future Microbiol. 10, 317–320. doi: 10.2217/fmb.15.2
Anjum, M. F., Mafura, M., Slickers, P., Ballmer, K., Kuhnert, P., Woodward, M. J., et al. (2007). Pathotyping Escherichia coli by using miniaturized DNA microarrays. Appl. Environ. Microbiol. 73, 5692–5697. doi: 10.1128/AEM.00419-07
APHA and SRUC. (2024). Great Britain pig quarterly report: disease surveillance and emerging threats. APHA and SRUC.
Beghain, J., Bridier-Nahmias, A., Le Nagard, H., Denamur, E., and Clermont, O. (2018). Clermontyping: an easy-to-use and accurate in silico method for Escherichia genus strain phylotyping. Microb. Genom. 4:192. doi: 10.1099/mgen.0.000192
Blattner, F. R., Plunkett, G., Bloch, C. A., Perna, N. T., Burland, V., Riley, M., et al. (1997). The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462. doi: 10.1126/science.277.5331.1453
Boyce, J. M. (2023). Quaternary ammonium disinfectants and antiseptics: tolerance, resistance and potential impact on antibiotic resistance. Antimicrob. Resist. Infect. Control 12:32. doi: 10.1186/s13756-023-01241-z
Broadfoot, F., Dewé, T., Glennie, A., Healey, K., Hogan, C., et al. (2023). Uk veterinary antibiotic resistance and sales surveillance report. Veterinary Medicines Directorate and Animal and Plant Health Agency.
De Koster, S., Ringenier, M., Lammens, C., Stegeman, A., Tobias, T., Velkers, F., et al. (2021). Esbl-producing, Carbapenem- and ciprofloxacin-resistant Escherichia coli in Belgian and Dutch broiler and pig farms: a cross-sectional and cross-border study. Antibiotics (Basel) 10:945. doi: 10.3390/antibiotics10080945
De Koster, S., Ringenier, M., Xavier, B. B., Lammens, C., De Coninck, D., and Bruyne, D. (2023). Genetic characterization of Esbl-producing and ciprofloxacin-resistant Escherichia coli from Belgian broilers and pigs. Front. Microbiol. 14:1150470. doi: 10.3389/fmicb.2023.1150470
De Mattos D’avila, D. G., Ferrari, R. G., De Almeida Rodrigues, P., Neves, G. L., Ramos Filho, A. M., Baptista Mano, R. F., et al. (2024). Bacterial resistance to mercury: a mini-review. Appl. Microbiol. 4, 1630–1641. doi: 10.3390/applmicrobiol4040111
Duan, Q., Xia, P., Nandre, R., Zhang, W., and Zhu, G. (2019). Review of newly identified functions associated with the heat-labile toxin of Enterotoxigenic Escherichia coli. Front. Cell. Infect. Microbiol. 9:292. doi: 10.3389/fcimb.2019.00292
Duggett, N., Abuoun, M., Randall, L., Horton, R., Lemma, F., Rogers, J., et al. (2020). The importance of using whole genome sequencing and extended spectrum beta-lactamase selective media when monitoring antimicrobial resistance. Sci. Rep. 10:19880. doi: 10.1038/s41598-020-76877-7
Elkrewi, E., Randall, C. P., Ooi, N., Cottel, J. L., and O'neill, A. J. (2017). Cryptic silver resistance is prevalent and readily activated in certain gram-negative pathogens. J. Antimicrob. Chemother. 72, 3043–3046. doi: 10.1093/jac/dkx258
Ewels, P., Magnusson, M., Lundin, S., and Kaller, M. (2016). Multiqc: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. doi: 10.1093/bioinformatics/btw354
Fairbrother, J. M., and Nadeau, E. (2019). “Colibacillosis” in Diseases of swine. eds. J. J. Zimmerman, E. R. Burrough, L. A. Karriker, K. J. Schwartz, and J. Zhang. 7th ed (New York: John Wiley and Sons).
Fairbrother, J. M., Nadeau, E., and Gyles, C. L. (2005). Escherichia coli in postweaning diarrhea in pigs: an update on bacterial types, pathogenesis, and prevention strategies. Anim. Health Res. Rev. 6, 17–39. doi: 10.1079/AHR2005105
Gale, C., and Velazquez, E. (2020). Oedema disease a review of the disease and control and preventative measures. Livestock 25, 142–147. doi: 10.12968/live.2020.25.3.142
Garcia, V., Gambino, M., Pedersen, K., Haugegaard, S., Olsen, J. E., and Herrero-Fresno, A. (2020). F4- and F18-positive Enterotoxigenic Escherichia coli isolates from diarrhea of Postweaning pigs: genomic characterization. Appl. Environ. Microbiol. 86:20. doi: 10.1128/AEM.01913-20
Geurtsen, J., De Been, M., Weerdenburg, E., Zomer, A., Mcnally, A., and Poolman, J. (2022). Genomics and pathotypes of the many faces of Escherichia coli. FEMS Microbiol. Rev. 46:31. doi: 10.1093/femsre/fuac031
Giachino, A., and Waldron, K. J. (2020). Copper tolerance in bacteria requires the activation of multiple accessory pathways. Mol. Microbiol. 114, 377–390. doi: 10.1111/mmi.14522
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). Quast: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Inouye, M., Dashnow, H., Raven, L.-A., Shultz, M. B., Pope, B. J., Tomita, T., et al. (2014). Srst2: rapid genomic surveillance for public health and hospital microbiology labs-annotated. Genome Med. 6:90. doi: 10.1186/s13073-014-0090-6
Johansen, M., Andresen, L. O., Jorsal, S. E., Thomsen, L. K., Waddell, T. E., and Gyles, C. L. (1997). Prevention of edema disease in pigs by vaccination with verotoxin 2e toxoid. Can. J. Vet. Res. 61, 280–285
Kaspersen, H. P., Brouwer, M. S., Nunez-Garcia, J., Cardenas-Rey, I., Abuoun, M., Duggett, N., et al. (2024). Escherichia coli from six European countries reveals differences in profile and distribution of critical antimicrobial resistance determinants within one health compartments, 2013 to 2020. Euro Surveill. 29:295. doi: 10.2807/1560-7917.ES.2024.29.47.2400295
Kolde, R. (2018). Pheatmap: pretty Heatmaps. R package version 1.0.12. Available online at: https://github.com/raivokolde/pheatmap.
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., and Stamatakis, A. (2019). Raxml-ng: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Lee, W., Kim, M. H., Sung, S., Kim, E., An, E. S., Kim, S. H., et al. (2023). Genome-based characterization of hybrid Shiga toxin-producing and enterotoxigenic Escherichia coli (Stec/Etec) strains isolated in South Korea, 2016-2020. Microorganisms 11:1285. doi: 10.3390/microorganisms11051285
Luppi, A. (2017). Swine enteric colibacillosis: diagnosis, therapy and antimicrobial resistance. Porcine Health Manag. 3:16. doi: 10.1186/s40813-017-0063-4
Luppi, A., Gibellini, M., Gin, T., Vangroenweghe, F., Vandenbroucke, V., Bauerfeind, R., et al. (2016). Prevalence of virulence factors in enterotoxigenic Escherichia coli isolated from pigs with post-weaning diarrhoea in Europe. Porcine Health Manag. 2:20. doi: 10.1186/s40813-016-0039-9
Melkebeek, V., Goddeeris, B. M., and Cox, E. (2013). Etec vaccination in pigs. Vet. Immunol. Immunopathol. 152, 37–42. doi: 10.1016/j.vetimm.2012.09.024
Mokady, D., Gophna, U., and Ron, E. Z. (2005). Virulence factors of septicemic Escherichia coli strains. Int. J. Med. Microbiol. 295, 455–462. doi: 10.1016/j.ijmm.2005.07.007
Munson, G. P., Lam, D. L., Outten, W., and O'halloran, T. V. (2000). Identification of a copper-responsive two-component system on the chromosome of E coli K12. J. Bacteriol. 182, 5864–5871. doi: 10.1128/JB.182.20.5864-5871.2000
Ngeleka, M., Pritchard, J., Appleyard, G., Middleton, D. M., and Fairbrother, J. M. (2003). Isolation and association of Escherichia coli Aida-I Stb, rather than East1 pathotype, with diarrhea in piglets and antibiotic sensitivity of isolates. J. Vet. Diagn. Invest. 15, 242–252. doi: 10.1177/104063870301500305
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pal, C., Bengtsson-Palme, J., Kristiansson, E., and Larsson, D. G. (2015). Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genomics 16:964. doi: 10.1186/s12864-015-2153-5
Pal, C., Bengtsson-Palme, J., Rensing, C., Kristiansson, E., and Larsson, D. G. (2014). BacMet: antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 42, D737–D743. doi: 10.1093/nar/gkt1252
Pan, L., Zhao, P. F., Ma, X. K., Shang, Q. H., Xu, Y. T., Long, S. F., et al. (2017). Probiotic supplementation protects weaned pigs against enterotoxigenic K88 challenge and improves performance similar to antibiotics. J. Anim. Sci. 95, 2627–2639. doi: 10.2527/jas.2016.1243
Paradis, E., and Schliep, K. (2019). Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Pierce, N. T., Irber, L., Reiter, T., Brooks, P., and Brown, C. T. (2019). Large-scale sequence comparisons with sourmash. F1000Res 8:1006. doi: 10.12688/f1000research.19675.1
Pitout, J. D. (2012). Extraintestinal pathogenic Escherichia coli: a combination of virulence with antibiotic resistance. Front. Microbiol. 3:9. doi: 10.3389/fmicb.2012.00009
Plattner, M., Gysin, M., Haldimann, K., Becker, K., and Hobbie, S. N. (2020). Epidemiologic, phenotypic, and structural characterization of aminoglycoside-resistance gene aac(3)-iv. Int. J. Mol. Sci. 21:133. doi: 10.3390/ijms21176133
Pokharel, P., Dhakal, S., and Dozois, C. M. (2023). The diversity of Escherichia coli Pathotypes and vaccination strategies against this versatile bacterial pathogen. Microorganisms 11:344. doi: 10.3390/microorganisms11020344
Purcell, T. W., and Peters, J. J. (1998). Sources of silver in the environment. Environ. Toxicol. Chem. 17, 539–546. doi: 10.1002/etc.5620170404
Robins-Browne, R. M., Holt, K. E., Ingle, D. J., Hocking, D. M., Yang, J., and Tauschek, M. (2016). Are Escherichia coli pathotypes still relevant in the era of whole-genome sequencing? Front. Cell. Infect. Microbiol. 6:141. doi: 10.3389/fcimb.2016.00141
Rodriguez-Rubio, L., Haarmann, N., Schwidder, M., Muniesa, M., and Schmidt, H. (2021). Bacteriophages of Shiga toxin-producing Escherichia coli and their contribution to pathogenicity. Pathogens 10:404. doi: 10.3390/pathogens10040404
Russo, T. A., and Johnson, J. R. (2009). Extraintestinal pathogenic Esherichia coli. Vaccines for the biodefense and emerging and neglected diseases. Amsterdam, Netherlands: Elsiever.
Schwengers, O., Jelonek, L., Dieckmann, M. A., Beyvers, S., Blom, J., and Goesmann, A. (2021). Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 7:685. doi: 10.1099/mgen.0.000685
Sheikh, A., Luo, Q., Roy, K., Shabaan, S., Kumar, P., Qadri, F., et al. (2014). Contribution of the highly conserved EaeH surface protein to enterotoxigenic Escherichia coli pathogenesis. Infect. Immun. 82, 3657–3666. doi: 10.1128/IAI.01890-14
Storey, N., Cawthraw, S., Turner, O., Rambaldi, M., Lemma, F., Horton, R., et al. (2022). Use of genomics to explore AMR persistence in an outdoor pig farm with low antimicrobial usage. Microb. Genom. 8:782. doi: 10.1099/mgen.0.000782
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Tan, C., Tang, X., Zhang, X., Ding, Y., Zhao, Z., Wu, B., et al. (2011). Serotypes and virulence genes of extraintestinal pathogenic Escherichia coli isolates from diseased pigs in China. Vet. J. 192, 483–488. doi: 10.1016/j.tvjl.2011.06.038
Tenaillon, O., Skurnik, D., Picard, B., and Denamur, E. (2010). The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 8, 207–217. doi: 10.1038/nrmicro2298
Turner, S. M., Scott-Tucker, A., Cooper, L. M., and Henderson, I. R. (2006). Weapons of mass destruction: virulence factors of the global killer enterotoxigenic Escherichia coli. FEMS Microbiol. Lett. 263, 10–20. doi: 10.1111/j.1574-6968.2006.00401.x
Villanueva, R. A. M., and Chen, Z. J. (2019). ggplot2: elegant graphics for data analysis measurement. Interdiscip. Res. Perspect. 17:11. doi: 10.1080/15366367.2019.1565254
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wickham, H., Averick, M., Bryan, J., Chang, W., Mcgowan, L., François, R., et al. (2019). Welcome to the tidyverse. J. Open Source Softw. 4:1686. doi: 10.21105/joss.01686
Wood, D. E., Lu, J., and Langmead, B. (2019). Improved metagenomic analysis with kraken 2. Genome Biol. 20:257. doi: 10.1186/s13059-019-1891-0
World Health Organisation (2018). Critically important antimicrobials for human medicine. Geneva: World Health Organisation.
World Health Organisation (2024). Who list of medically important antimicrobials. Geneva: World Health Organisation.
World Organisation for Animal Health (2021). Oie list of antimicrobial agents of veterinary importance. France: World Organisation for Animal Health.
Wu, G., Carter, B., Mafura, M., Liebana, E., Woodward, M. J., and Anjum, M. F. (2008). Genetic diversity among Escherichia coli O157:H7 isolates and identification of genes linked to human infections. Infect. Immun. 76, 845–856. doi: 10.1128/IAI.00956-07
Yu, G., Smith, D. K., Zhu, H., Guan, Y., Lam, T. T. Y., and Mcinerny, G. (2016). Ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. doi: 10.1111/2041-210X.12628
Keywords: Escherichia coli, antimicrobial resistance, virulence, postweaning diarrhoea, pig health, whole genome sequencing
Citation: Connelly SAM, AbuOun M, Duggett N, Kirchner M, Navickaite I, Nunez-Garcia J, Williamson S, Vaughan K, Teale C and Anjum MF (2025) Diversity of antimicrobial resistance and virulence genes of pathogenic Escherichia coli recovered from pigs in England. Front. Microbiol. 16:1668327. doi: 10.3389/fmicb.2025.1668327
Edited by:
Manuel Rodriguez-Iglesias, University of Cádiz, SpainReviewed by:
Charles Martin Dozois, Université du Québec, CanadaXavier Bertrand, Centre Hospitalier Universitaire de Besançon, France
Copyright © 2025 Connelly, AbuOun, Duggett, Kirchner, Navickaite, Nunez-Garcia, Williamson, Vaughan, Teale and Anjum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Samuel A. M. Connelly, U2FtdWVsLmNvbm5lbGx5QGFwaGEuZ292LnVr; Muna F. Anjum, TXVuYS5Bbmp1bUBhcGhhLmdvdi51aw==