 Gulshara Zh Abildinova1
Gulshara Zh Abildinova1 Valeriy V. Benberin1,2
Valeriy V. Benberin1,2 Tamara A. Vochshenkova1,2
Tamara A. Vochshenkova1,2 Nadiar M. Mussin3
Nadiar M. Mussin3 Alireza Afshar1*
Alireza Afshar1* Amin Tamadon4*
Amin Tamadon4*- 1Gerontology Center, Medical Center Hospital of the President’s Affairs Administration of the Republic of Kazakhstan, Astana, Kazakhstan
- 2Corporate Fund “Institute for Innovational and Profilaxy Medicine”, Astana, Kazakhstan
- 3Department of Surgery No. 2, West Kazakhstan Marat Ospanov Medical University, Aktobe, Kazakhstan
- 4Department of Natural Sciences, West Kazakhstan Marat Ospanov Medical University, Aktobe, Kazakhstan
Objectives: Links between gut microbiota and insulin resistance (IR) vary across populations. We profiled the fecal microbiota of Kazakh adults to test whether community composition associates with IR at broad (phylum → genus) and species levels.
Methods: In a cross-sectional case control study (N = 200; IR = 183, controls = 17), TyG indexed IR status. 16S rRNA sequencing (two primer pools; nine hypervariable regions) characterized taxa. After CSS normalization, we compared presence/absence across groups (χ2) and modeled species with univariate and multivariable logistic regressions, using absence of each species as the predictor.
Results: High-level composition did not differ between IR and controls (phylum, class, family, genus; all p > 0.05). In contrast, several species differed. In univariate models, absence of Actinomyces odontolyticus (OR = 25.55, p = 0.010), Bifidobacterium kashiwanohense (OR = 12.69, p = 0.015), Lactobacillus sp. (OR = 5.71, p = 0.020), and Streptococcus lactarius (OR = 6.27, p = 0.044) associated with higher IR odds, suggesting protection when present; whereas absence of Alistipes onderdonkii (OR = 0.30, p = 0.044) and Prevotella copri (OR = 0.19, p = 0.003) associated with lower IR odds, suggesting risk when present. In multivariable models, these signals persisted: absence of P. copri (OR = 0.146, p = 0.003) and Roseburia inulinivorans (OR = 0.143, p = 0.011) predicted lower IR odds (risk alignment), while absence of Lactobacillus sp. (OR = 8.29, p = 0.016) and Coprococcus catus (OR = 7.04, p = 0.004) predicted higher IR odds (protective alignment).
Conclusion: In this Kazakh cohort, no broad compositional signal emerged, but species-specific associations were strong and bidirectional. Findings highlight population-specificity and identify candidate species associated with IR that may serve as hypothesis-generating targets for future validation. Any attempt to modulate these taxa for insulin resistance is unproven and requires function-resolved, diet-measured longitudinal studies and randomized trials before clinical application. The IR:control imbalance (183:17) increases uncertainty for low-prevalence taxa; species-level findings are hypothesis-generating and require validation in a more balanced design. Because 16S rRNA profiling does not measure gene functions or metabolites, these species–IR associations are hypothesis-generating and warrant validation using shotgun metagenomics and metabolomics.
1 Introduction
Insulin resistance (IR) is a metabolic condition characterized by decreased sensitivity to insulin, a hormone crucial for glucose regulation. This condition often precedes more severe metabolic disorders, including type 2 diabetes and cardiovascular diseases, posing a significant burden on public health (Takeuchi et al., 2023). IR is a complex disorder influenced by multiple factors, including genetics, diet, and environmental exposures, which can lead to alterations in metabolic pathways relevant to insulin sensitivity and glucose metabolism (Caricilli and Saad, 2013; Lee et al., 2020). The complications of IR are far-reaching and devastating. According to the World Health Organization (WHO), IR is a major risk factor for developing type 2 diabetes, which affects approximately 422 million people worldwide. Moreover, IR is a significant contributor to cardiovascular disease, which is the leading cause of mortality globally, accounting for approximately 17.9 million deaths annually (Jang and Lee, 2021). The economic burden of IR is substantial, with estimates suggesting that the annual cost of treating diabetes and its complications in the United States alone is over $300 billion (Takeuchi et al., 2023).
The gut microbiota, a complex community of microorganisms residing in the gastrointestinal tract, plays a pivotal role in human health and disease. A healthy gut microbiota is essential for maintaining metabolic homeostasis, modulating the immune system, and influencing the development of various diseases (Caricilli and Saad, 2013; Ghorbani et al., 2021). However, disturbances in the gut microbiota, often referred to as dysbiosis, have been linked to various metabolic disorders, including IR. The gut microbiota’s influence on IR is multifaceted, with alterations in microbial composition and function contributing to the development and progression of this condition (Chen et al., 2021; Gurung et al., 2020; Semo et al., 2024).
The relationship between IR and the gut microbiota is complex and bidirectional. On one hand, alterations in the gut microbiota can contribute to IR by modulating carbohydrate metabolism, influencing the host’s inflammatory response, and altering the expression of genes involved in glucose metabolism (Takeuchi et al., 2023; Semo et al., 2024). On the other hand, IR can also impact the gut microbiota by altering the composition and function of the microbiome, leading to dysbiosis and further exacerbating IR. Understanding the intricate relationship between IR and the gut microbiota is crucial for the development of effective therapeutic strategies aimed at modulating the gut microbiota to manage or prevent IR effectively (Takeuchi et al., 2023; Ebrahimzadeh Leylabadlo et al., 2020).
One of the important factors that has a huge impact on gut microbiota is the region. Studies have shown that the gut microbiota of individuals from distinct ethnic and cultural backgrounds exhibit unique profiles, reflecting the influence of dietary habits, environmental exposures, and genetic factors on the development and maintenance of the intestinal microbial community (Jang and Lee, 2021; Fontana et al., 2019; Senghor et al., 2018). These regional and ethnic variations in gut microbiota composition can have important implications for susceptibility to various diseases (Jang and Lee, 2021; Schnorr et al., 2014). Alterations in the delicate balance of the gut microbiome have been associated with an increased risk of developing metabolic disorders, such as IR and type 2 diabetes (Takeuchi et al., 2023; Caricilli and Saad, 2013). Interestingly, some populations with unique gut microbiota profiles have been observed to have a lower prevalence of IR and related metabolic conditions (Deschasaux et al., 2018; Ley et al., 2006). The relationship between the uniqueness of the gut microbiota and lower rates of IR in specific regions or countries is a complex and multifactorial phenomenon (Fontana et al., 2019; Sonnenburg and Backhed, 2016). Understanding these regional and ethnic differences in gut microbiota composition and their impact on metabolic health could provide valuable insights for the development of personalized interventions aimed at preventing and managing IR and related metabolic disorders (Deschasaux et al., 2018; Sonnenburg and Backhed, 2016).
Despite considerable research linking gut microbiota to various diseases, there is limited information specifically addressing the gut microbiota’s role in IR among the Kazakh population. This population may exhibit unique gut microbiota profiles due to their specific dietary habits and genetic background, which could influence the prevalence and management of metabolic diseases such as IR. Given this context, the study aims are to (i) Characterize the gut microbiota by sequencing the 16S ribosomal RNA of stool samples from insulin-resistant patients and healthy controls in the Kazakh population. This will help identify distinct microbial patterns that may be associated with IR. (ii) Evaluate the differences in microbial composition between insulin-resistant patients and healthy controls to uncover potential microbial indicators of IR. This could further guide personalized medical interventions aimed at modifying the gut microbiota to manage or prevent IR effectively. (iii) In addition to these aims, we will systematically interrogate between-group differences in the gut microbiome across hierarchical taxonomic ranks—from the phylum level down to individual species—to determine whether discriminatory signals emerge only at finer resolution. This taxonomy-spanning analysis is intended to clarify whether broad compositional shifts or species-specific patterns better explain IR in this cohort. This study does not only fill a critical gap in understanding the gut microbiota in the Kazakh population but also potentially inform targeted therapies that could be developed to manage IR more effectively within this group.
Because gut communities vary strongly by geography, diet, ethnicity, and lab methods, we prioritized within-population contrasts and species-level resolution rather than cross-population ‘healthy’ comparisons. Prior work shows that disease classifiers trained in one cohort frequently underperform in external cohorts due to these sources of heterogeneity. Our design therefore benchmarks findings against published healthy-adult profiles for plausibility but avoids direct cross-country performance claims, which can be misleading. This study’s contribution is species-resolved association within a genetically and culturally homogeneous Kazakh cohort (Li et al., 2023).
2 Materials and methods
2.1 Ethical statement
The investigation was conducted in accordance with the ethical principles laid out in the Declaration of Helsinki (1964 and subsequent amendments) and the International Council for Harmonisation Good Clinical Practice (ICH-GCP) guidelines. The protocol received approval from the Local Ethics Committee of the Medical Center Hospital of the President’s Affairs Administration of the Republic of Kazakhstan, Astana (protocol no. 1, dated 05 April 2022). All participants provided written informed consent permitting the use of de-identified data for research and educational purposes; confidentiality safeguards were applied throughout data handling and reporting.
2.2 Population selection
A cross-sectional case–control study was conducted among individuals of Kazakh ethnicity registered at a polyclinic in Astana. The registered population had similar occupational profiles (government employees), lifestyles, and environmental factors, potentially minimizing the influence of external factors on the study results. Participants were randomly selected from individuals attending the polyclinic for preventive purposes between January and March 2023.
Inclusion criteria were: age 30–59, Kazakh ethnicity in the third generation, and voluntary consent to participate. Exclusion criteria included chronic cardiometabolic or autoimmune diseases, cancer, and pregnancy. Participants reporting antibiotic use within eight weeks prior to sampling were excluded to avoid perturbations in gut microbial communities known to persist for up to two months after treatment. Individuals who had consumed probiotic supplements within four weeks preceding enrollment were also excluded, reflecting evidence that transient probiotic strains can influence microbiota composition for several weeks. We documented any antibiotic or probiotic intake during the study period via weekly questionnaires and excluded any samples collected within eight weeks of antibiotic use or four weeks of probiotic use. Ethnicity was self-reported by participants, who identified themselves, their biological parents, and their grandparents as ethnically Kazakh. Dietary intake was not collected in this study; thus, we could not adjust for diet directly and address this as a limitation.
2.3 Participants grouping
Participants were stratified into insulin-resistant (IR+) and control (IR−) categories using the triglyceride–glucose (TyG) index, computed as TyG = Ln([fasting glucose, mg/dL × fasting triglycerides, mg/dL]/2). For primary analyses, individuals with TyG ≥ 4.50 were assigned to the IR + group (n = 183) and those with TyG ≤ 4.49 served as controls (n = 17). For descriptive severity within the IR + group, TyG values of 4.50–4.59 were labeled “moderate IR,” and values ≥ 4.60 were labeled “severe IR”.
2.4 Analyzing serum glucose and triglyceride
Following a 12-h overnight fast, venous blood was drawn from the antecubital vein. Plasma was separated by centrifugation at 1,000 × g for 10 min at 4 °C and stored at −30 °C pending batched analyses; serum aliquots were analyzed the same day. Glucose concentrations were determined using the hexokinase enzymatic method on the Abbott ARCHITECT c8000 platform (Abbott Laboratories, United States). Serum triglycerides were quantified spectrophotometrically on the same analyzer, following manufacturer-recommended procedures.
2.5 Fecal microbiome species composition of population
Fecal microbiome species composition was analyzed using targeted semiconductor sequencing of the 16S rRNA gene, employing next-generation sequencing (NGS) technology with Ion Reporter software. This technique, using a combination of two primer pools, enabled the identification of a wide range of bacterial species in mixed populations.
2.6 Bacterial DNA extraction
Bacterial DNA was extracted from stool samples using the PurLink Genomic DNA Microbiome kit (Invitrogen, United States) according to the manufacturer’s protocol. DNA concentration was measured using the Qubit™ 4 Fluorometer with the Qubit® dsDNA BR Assay Kit. Library preparation involved several stages:
• PCR amplification of the 16S hypervariable region, followed by purification and concentration measurement.
• Library preparation through ligation with barcode adapters and purification of the library adapters.
• Concentration measurement of the resulting DNA libraries using the QuantStudioTM 12 K Flex system with the Ion Library TaqMan® Quantitation Kit (Thermo Fisher Scientific, United States).
2.7 Metagenomic sequencing
Metagenomic sequencing of the nine hypervariable regions was performed using NGS. Gut microbiota structure was determined by sequencing the variable regions V2-4-8 and V3-6, 7–9 V3-V4 of the bacterial 16S rRNA gene. Sequencing reads from each primer pool targeting the V2-4-8 and V3-6,7-9 regions were independently subjected to quality filtering (minimum PHRED score ≥20) and chimera removal using UCHIME to ensure high-fidelity sequence data (Edgar et al., 2011). Amplicon sequence variants (ASVs) were inferred with DADA2, with region-specific ASV tables merged via closed-reference OTU picking at 97% similarity against the SILVA v138 database to standardize taxonomic assignments (Callahan et al., 2016). To integrate data from multiple hypervariable regions into a single phylogenetic framework, we applied SEPP (SATé-Enabled Phylogenetic Placement), allowing consistent biodiversity analyses across V2-4-8 and V3-6,7-9 datasets (Kuhn et al., 2011). Methodological accuracy was validated using the ZymoBIOMICS Microbial Community Standard as a mock community, achieving >95% recovery of expected taxa abundances and confirming sequencing integrity across all variable regions (Kuhn et al., 2011). Sequencing was carried out on the Ion PGM™ system, and bioinformatic analysis was conducted using the Ion Reporter™ software and the Ion 16S™ Metagenomics Kit. The use of two primer pools allowed for the identification of a wide range of bacteria in the mixed population. The advantage of mass parallel sequencing methods lies in their culture-independent approach, enabling the detection of thousands of species. Metagenomic methods include taxonomic profiling, which describes the diversity of the bacterial community, and quantitative profiling. This study used 16S rRNA amplicon profiling only; shotgun metagenomics and metabolomics were not performed, so functional attributions are literature-based and treated as hypothesis-generating.
2.8 Statistical analysis
Statistical analysis was performed using GraphPad Prism software (version 10.0, GraphPad Software, United States). Numerical data were presented as mean values with standard deviation (x̅ ±s). Quantitative comparisons were made using the Independent Samples t test. Spearman’s correlation coefficient was used to determine the relationship between bacterial taxa. Differences were considered statistically significant at p < 0.05. To account for the unbalanced sample sizes between the IR-positive (n = 183) and control (n = 17) groups, we normalized OTU count data using cumulative sum scaling (CSS) implemented in the metagenomeSeq package, thereby mitigating compositional bias (Paulson et al., 2013). In addition, we performed 100 iterations of random subsampling of the IR-positive group to 17 samples and confirmed that alpha- and beta-diversity metrics remained stable, demonstrating robustness to group-size imbalance (Weiss et al., 2017). Differential abundance testing was conducted with ANCOM-BC, which corrects for biases due to unequal group sizes and the compositional nature of microbiome data (Lin and Peddada, 2020). The IR:control ratio (~10.8:1) can inflate variance for control-group estimates and increase susceptibility to small-sample artifacts (e.g., quasi-complete separation). We therefore (i) applied CSS normalization to mitigate compositional bias; (ii) used ANCOM-BC for differential abundance, which corrects bias inherent to compositional data; and (iii) performed random subsampling of the IR group to the control sample size (n = 17) across 100 iterations to verify that alpha- and beta-diversity metrics were not driven by class imbalance. For species screens, we report effect sizes with 95% CIs and explicitly flag taxa exhibiting separation; these signals are interpreted cautiously as hypothesis-generating.
3 Results
3.1 Demographic, anthropometric, and biochemical characteristics in Kazakh population showed differences among IR and control groups
Results of the current study revealed significant differences in demographic, anthropometric, and biochemical characteristics between IR individuals and controls in the Kazakh population (Table 1). Individuals with IR displayed higher fasting blood sugar (FBS) levels (101.67 ± 22.71 mg/dL) compared to controls (89.59 ± 6.06 mg/dL, p = 0.030), with a particularly notable difference among females (p = 0.012), suggesting potential gender-based disparities in glucose metabolism. The TyG index, a marker for IR, was also significantly elevated in the IR group (4.63 ± 0.30) versus controls (4.40 ± 0.17, p < 0.001) for both genders, indicating higher metabolic dysregulation risk across sexes. Anthropometric measures showed that IR individuals had higher body mass index (BMI) and waist circumference, reflecting greater central adiposity. Specifically, BMI in the IR group was significantly higher (27.18 ± 5.04 kg/m2) than in controls (22.71 ± 3.56 kg/m2, p < 0.001), with waist circumference also markedly increased (91.10 ± 13.06 cm in IR vs. 77.52 ± 10.82 cm in controls, p < 0.001), particularly among males. Lipid profile assessments indicated that IR participants had elevated total cholesterol (99.02 ± 18.87 mg/dL vs. 78.19 ± 10.83 mg/dL in controls, p < 0.001) and low-density lipoprotein (LDL) levels (65.63 ± 17.24 mg/dL vs. 44.08 ± 10.55 mg/dL in controls, p < 0.001), alongside lower high-density lipoprotein (HDL) levels (25.38 ± 7.82 mg/dL vs. 29.96 ± 8.20 mg/dL in controls, p = 0.023) and significantly higher triglycerides (128.40 ± 103.50 mg/dL in IR vs. 78.50 ± 27.55 mg/dL in controls, p = 0.049). Collectively, these findings suggest that the IR group exhibits a cluster of metabolic risk factors, including hyperglycemia, central adiposity, and dyslipidemia, which are associated with IR in this population.
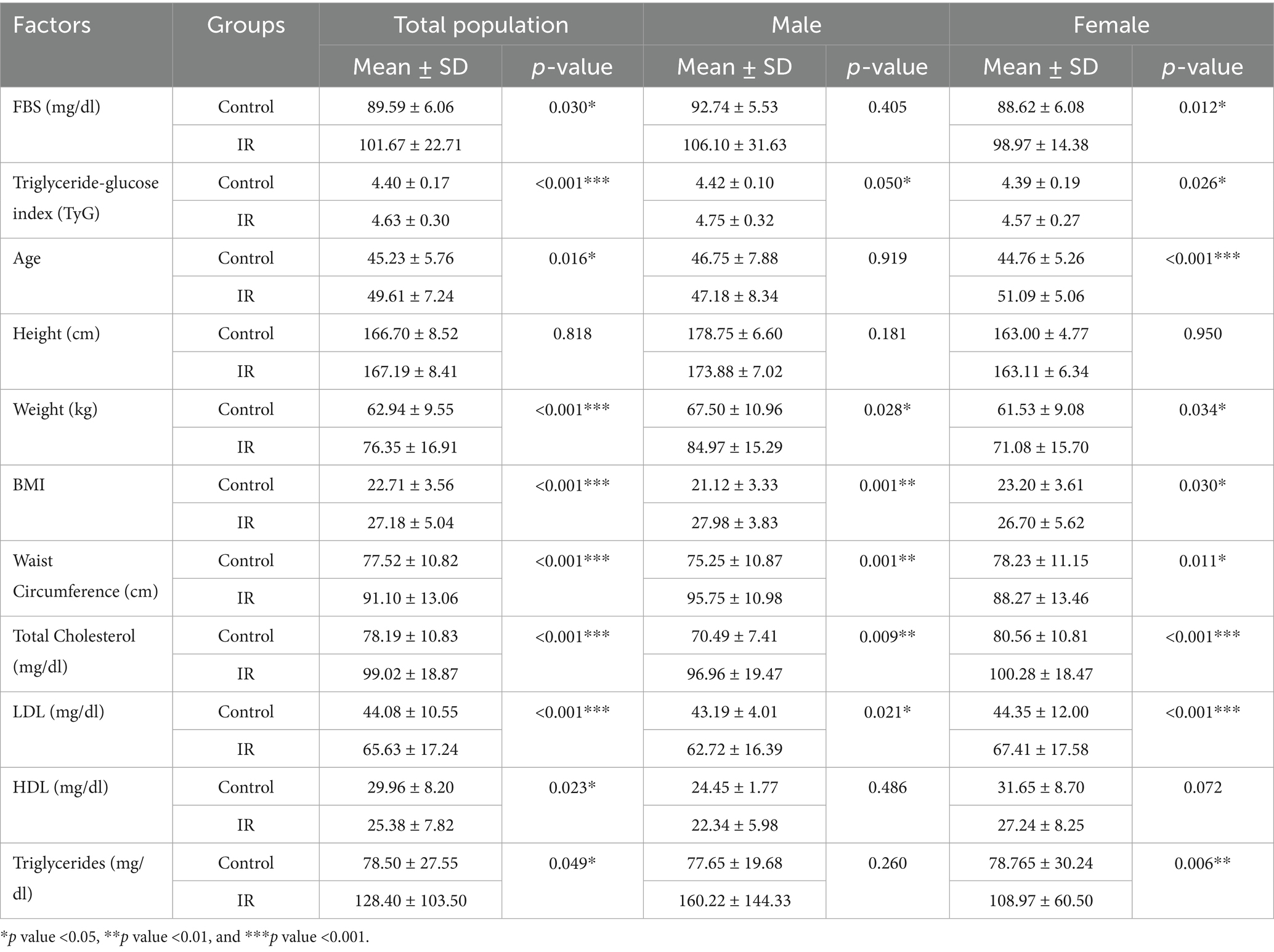
Table 1. Demographic, anthropometric characteristics, and other related factors of the Kazakhstan population analyzed with Independent Samples Test through control and insulin resistance (IR) groups.
3.2 High-level gut microbiota composition (phylum to genus) shows no significant association with IR
The gut microbiota composition of all participants (N = 200) was characterized at multiple taxonomic levels (Figures 1a–d). At the phylum level, Proteobacteria, Firmicutes (Bacillota), and Actinobacteria were dominant, with prevalences of 95.5, 97, and 96.5%, respectively. The most common classes were Betaproteobacteria (73.5%), Deltaproteobacteria (74%), Gammaproteobacteria (76%), Negativicutes (79.5%), and Clostridia (97%). At the family level, Bacteroidaceae (94.5%), Porphyromonadaceae (89%), Eubacteriaceae (80%), Prevotellaceae (61%), and Veillonellaceae (35%) were most prevalent. The leading genera were Bacteroides (98%), Blautia (89.5%), Roseburia (84.5%), Coprococcus (79%), and Alistipes (76%).
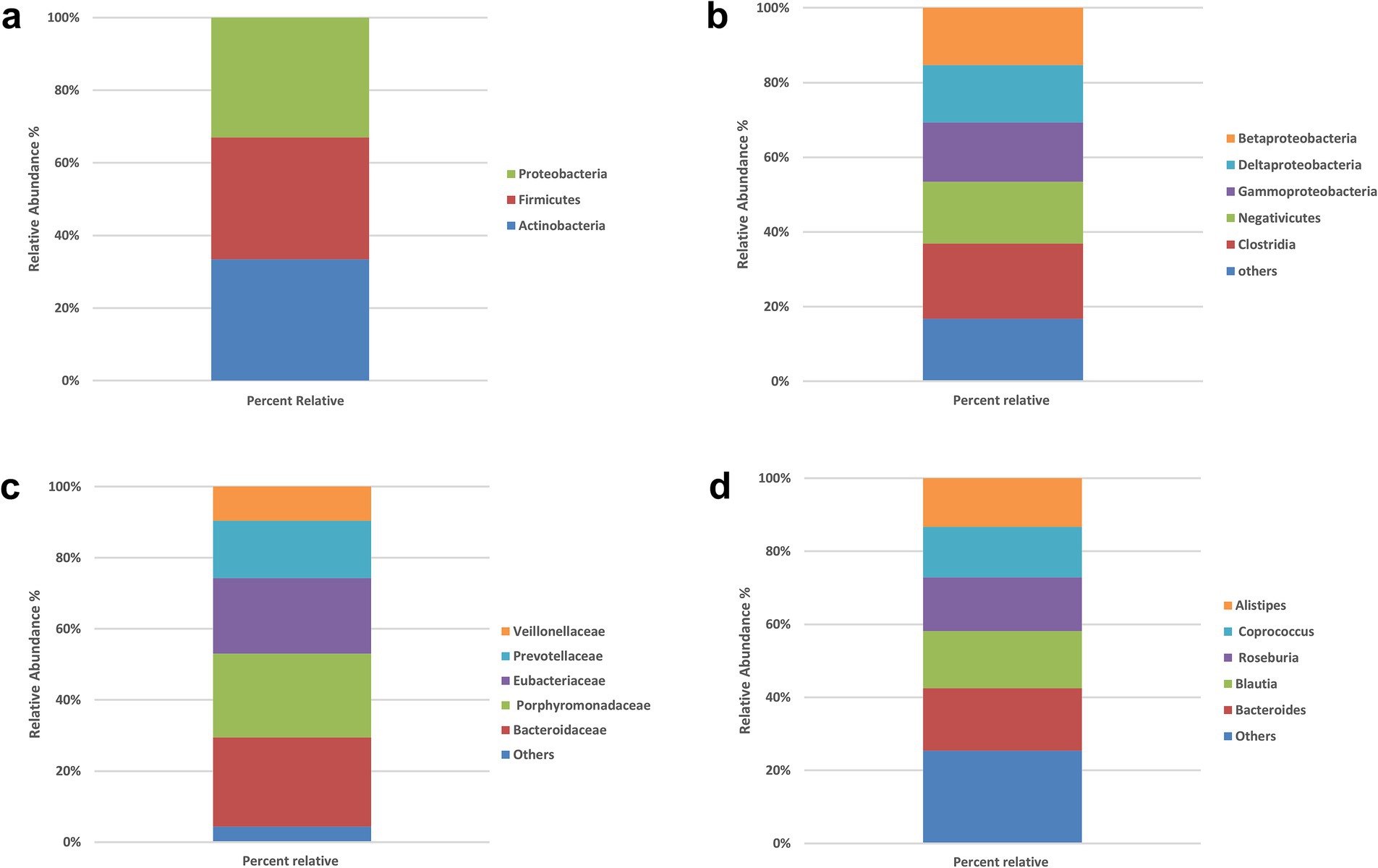
Figure 1. Prevalence of the bacterial types in the stool samples of all participants (N = 200). (a) Phylum level, (b) class level, (c) family level, (d) genus level. No significant between-group differences at phylum or class (χ2, all p > 0.05). Panels show overall prevalence for context; subsequent analyses therefore focus on species-level signals.
To explore whether broad compositional differences in the gut microbiota were associated with IR, we compared the relative abundances of bacterial phyla, classes, families and genera in the IR and control groups using chi-square tests of independence. The detailed frequency distributions are provided in Supplementary Tables S1–S4, and overall taxonomic profiles are visualized in Figures 1, 2a–d.
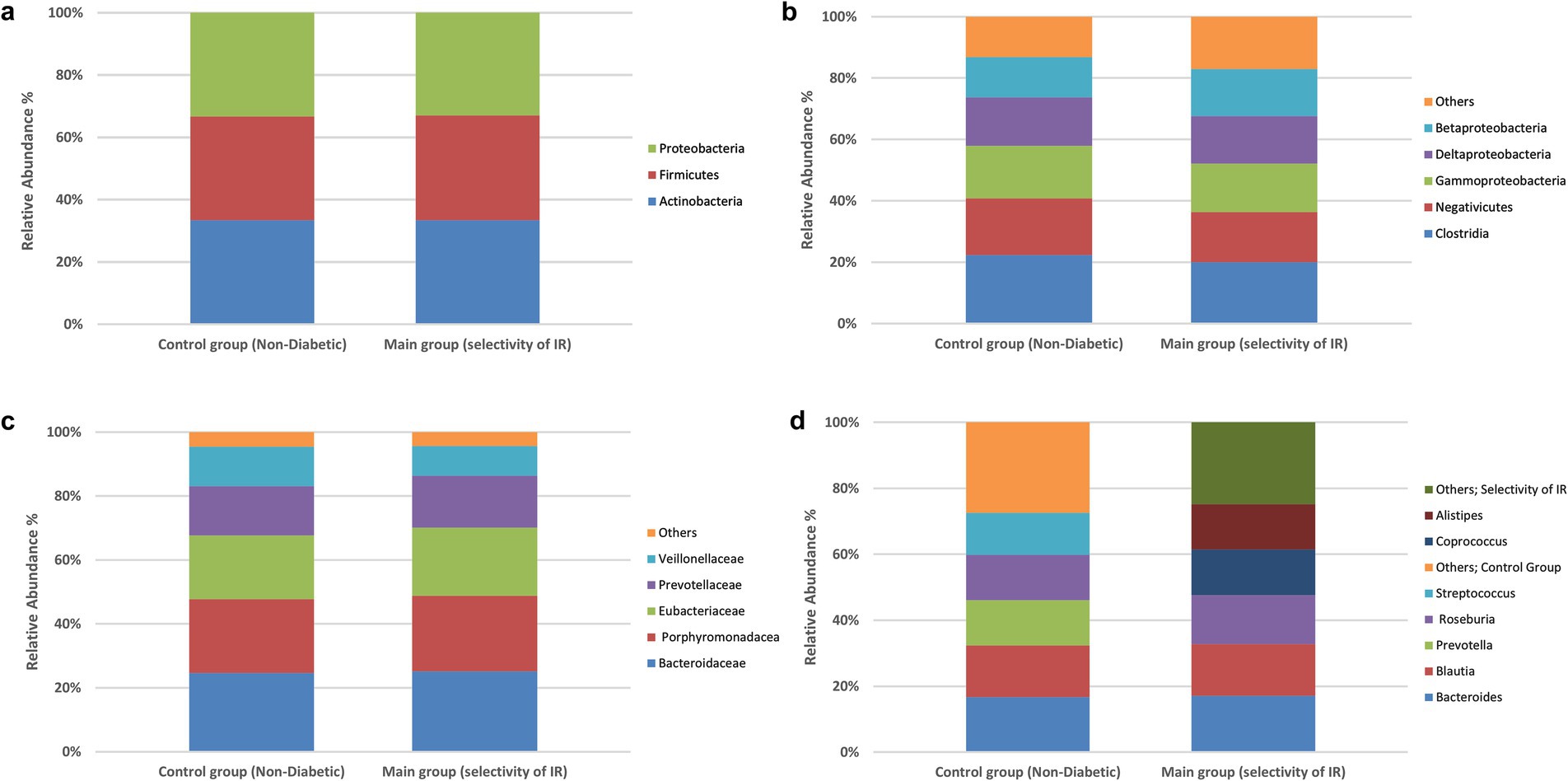
Figure 2. Prevalence of the bacterial types in the stool samples. Differences in species classification between the control and experimental groups. Control group (non-diabetic), main group (selectivity of insulin resistance). (a) Phylum level, (b) class level, (c) family level, (d) genus level. No significant between-group differences at family or genus (χ2, all p > 0.05). Profiles are shown for completeness; inference proceeds at the species level.
At the phylum level (Supplementary Table S1; Figure 1a), virtually all participants were positive for the dominant phyla Firmicutes, Proteobacteria and Actinobacteria. For example, 177 of 183 IR participants and all 17 controls harbored Firmicutes, while Proteobacteria was detected in 174 IR subjects and all controls. Chi-square statistics for Actinobacteria, Proteobacteria and Firmicutes were 0.774, 0.875 and 0.575, respectively, with corresponding p-values of 0.37, 0.34, and 0.44—none approaching the 0.05 significance threshold. These findings indicate no meaningful phylum-level differences between IR and control groups.
At the class level (Supplementary Table S2; Figure 1b), the five most prevalent classes—Betaproteobacteria, Deltaproteobacteria, Gammaproteobacteria, Negativicutes and Clostridia—also showed very similar distributions across groups. In the IR group, 137–177 individuals were positive for these classes, compared with 10–17 in the control group (reflecting the unequal sample sizes). Chi-square values ranged from 0.002 to 2.055 with p-values of 0.15–0.96, again indicating no significant association between class-level composition and IR status.
At the family level (Supplementary Table S3; Figure 1c), we examined Bacteroidaceae, Porphyromonadaceae, Eubacteriaceae, Prevotellaceae and Veillonellaceae. These families were present in 64–173 IR participants and 8–16 controls, but none of the chi-square tests (χ2 = 0.005–0.986) reached significance (all p > 0.32). Thus, family-level abundances did not differ between groups.
At the genus level (Supplementary Table S4; Figure 2d), the seven most common genera—Bacteroides, Blautia, Roseburia, Coprococcus, Alistipes, Streptococcus and Prevotella—were again widely prevalent in both groups (positive in 102–179 IR participants and 10–17 controls). Chi-square statistics ranged from 0.065 to 3.229 with p-values of 0.07–0.79; none were below the 0.05 threshold. Collectively, these analyses show that high-level taxonomic composition (phylum through genus) does not differ significantly between insulin-resistant and control individuals in this Kazakh cohort.
Because no significant associations were found at these broader taxonomic levels, subsequent analyses focused on species-level differences. Across phylum, class, family, and genus, χ2 statistics ranged from 0.002–3.229 with all p > 0.05; thus, no significant between-group differences were observed at these higher taxonomic ranks (Figures 1, 2).
3.3 Species-level differences identified by Chi-square test
The chi-square analysis summarized in Table 2 evaluated differences in prevalence of 336 bacterial species between the insulin-resistant (IR) and control groups. Several taxa showed statistically significant distribution differences (p < 0.05). Species that were more common in controls included Actinomyces odontolyticus, Bifidobacterium kashiwanohense, Bacteroides stercoris, Bulleidia moorei and Megasphaera micronuciformis; these taxa were detected in 13–53% of controls but only 0.6–3% of IR participants. Conversely, Prevotella copri, Alistipes onderdonkii, Alistipes finegoldii, Roseburia inulinivorans, Ruminococcus spp. (including R. gauvreauii), Streptococcus thermophilus and Megamonas funiformis were markedly more prevalent in the IR group (50–86% positive) than in controls (0–40% positive). These disparities suggest that some taxa may confer protection against IR, whereas others may be enriched in individuals with IR. However, these cross-sectional associations do not establish causality and should be explored further using mechanistic or longitudinal studies.
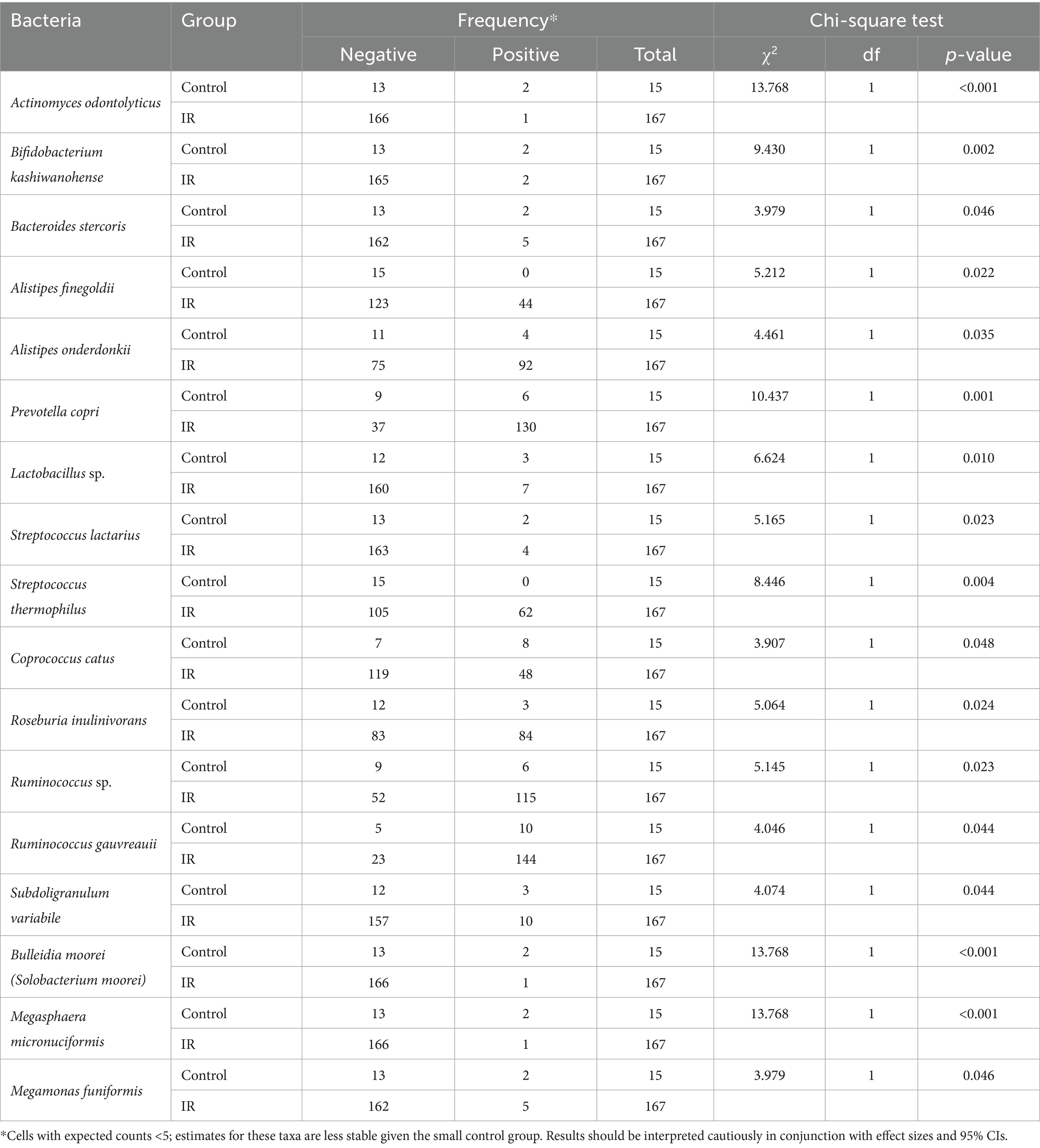
Table 2. Chi-square test for species which become significant among all species.
3.4 Univariate logistic regression identifies protective and risk-associated species
Table 3 presents univariate logistic regression models where the outcome is IR and the predictor is the absence of a given species (i.e., a positive β value indicates that lacking the species increases the odds of IR; see Figure 3A for the forest plot of univariate odds ratios). The absence of A. odontolyticus was associated with a more than 25-fold increase in the odds of IR (OR = 25.55, p = 0.010), implying that harboring this species may be protective. Similar patterns were observed for B. kashiwanohense (OR = 12.69, p = 0.015), Lactobacillus sp. (OR = 5.71, p = 0.020) and S. lactarius (OR = 6.27, p = 0.044), where the absence of these taxa was linked to higher odds of IR. In contrast, the absence of A. onderdonkii and P. copri was associated with lower odds of IR (OR = 0.30, p = 0.044 and OR = 0.19, p = 0.003, respectively), suggesting that these taxa may contribute to IR when present. Estimates for A. finegoldii and S. thermophilus were unstable due to quasi-complete separation and were not statistically significant. These findings highlight that both the presence of potentially protective bacteria and the over-representation of potentially deleterious bacteria can influence metabolic health. Because the control group is small, estimates for low-prevalence species are inherently less precise, and quasi-complete separation can occur; we therefore highlight effect sizes with 95% CIs and interpret such signals cautiously.
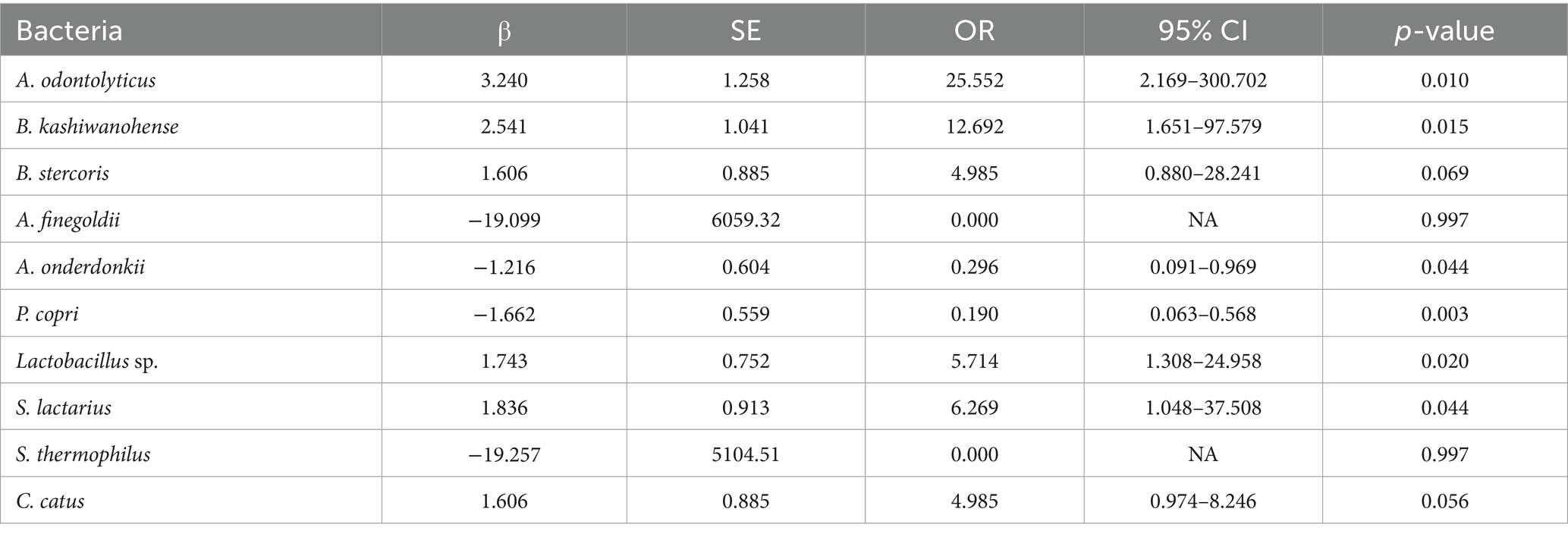
Table 3. Univariate logistic regression of bacterial species according to insulin resistance status in the Kazakh population.
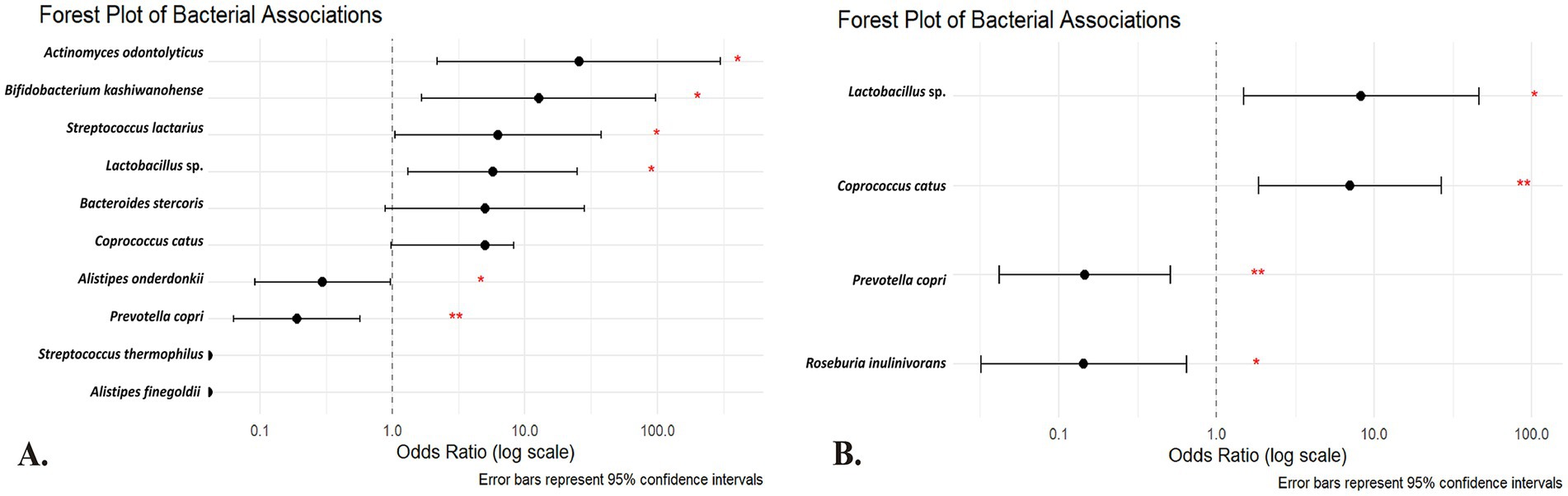
Figure 3. Forest plot of the (A) univariate and (B) multivariate regression analysis of the species of the gut microbiota among Kazakh population. The horizontal axis is on a logarithmic scale; points to the right of the dotted line (OR > 1) indicate that the absence of the species is associated with higher odds of insulin resistance, whereas points to the left (OR < 1) indicate that its absence is associated with lower odds (suggesting a potential deleterious role when present). Red asterisks denote statistical significance (*p < 0.05; **p < 0.01). Predictors are absence-coded (1 = species absent; 0 = present). OR > 1 → higher IR odds when absent (protective when present); OR < 1 → lower IR odds when absent (risk-aligned when present).
3.5 Multivariate logistic regression confirms independent associations
The multivariate logistic regression model (Table 4) simultaneously included taxa that were significant or borderline in the univariate analyses to identify independent predictors (see Figure 3B for the multivariate model results). After adjustment, the absence of P. copri (OR = 0.146; 95% CI 0.042–0.509; p = 0.003) and R. inulinivorans (OR = 0.143; 95% CI 0.032–0.645; p = 0.011) was associated with significantly lower odds of IR, reinforcing the notion that the presence of these species may contribute to IR. Conversely, the absence of Lactobacillus sp. (OR = 8.29; 95% CI 1.48–46.41; p = 0.016) and C. catus (OR = 7.04; 95% CI 1.85–26.78; p = 0.004) was independently associated with higher odds of IR, indicating that these taxa may have protective roles. These multivariate results underscore that specific microbial taxa maintain their associations with IR even when analyzed in combination, and they provide potential targets for future microbiome-based interventions. These associations derive from taxonomic (16S) data and do not by themselves establish functional roles or causality. As a complementary check, ANCOM-BC differential-abundance analysis at the species level generally recapitulated the directionality observed in the absence-coded models for highlighted taxa. Given zero-inflation and compositional constraints, we present presence/absence-based ORs as the primary, clinically interpretable effect estimates.
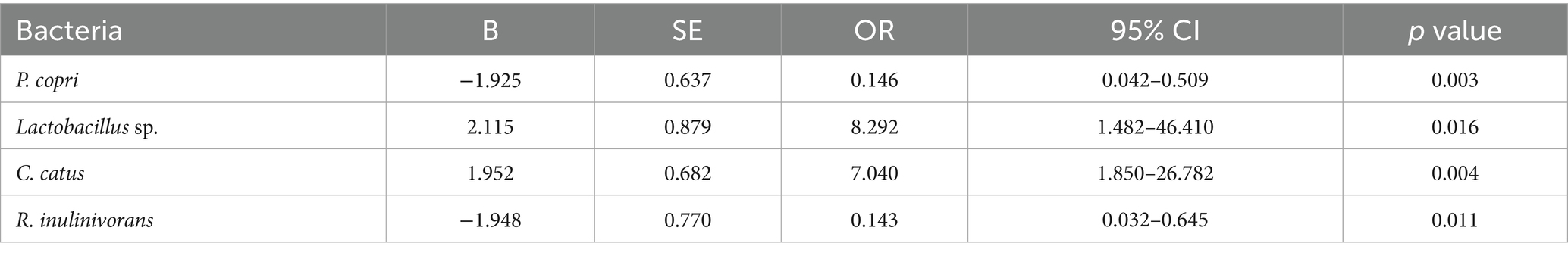
Table 4. Multivariate logistic regression of bacterial species according to insulin resistance status in the Kazakh population.
4 Discussion
4.1 Overview and key findings
IR is a multifactorial condition, and the gut microbiota has been proposed as one of the modifiable drivers of metabolic dysregulation (Abdelsalam et al., 2023; Mohammad and Thiemermann, 2020). In the present study we profiled the fecal microbiota of IR and control Kazakh individuals and observed no significant associations between IR and microbiota composition at the phylum, class, family or genus levels. This absence of high-level associations emphasizes that broad taxonomic summaries may mask important species-specific signals and that microbiome–disease relationships are population-specific (Abdelsalam et al., 2023; Chiang and Ferrell, 2020). By focusing on species-level differences using chi-square and logistic regression analyses, we identified several taxa whose presence was strongly associated with metabolic phenotype. The absence of A. odontolyticus, B. kashiwanohense, Lactobacillus sp., S. lactarius and C. catus was linked to increased odds of IR, whereas the absence of P. copri, R. inulinivorans and A. onderdonkii was associated with lower odds of IR. These findings suggest that a small number of species, rather than broad taxonomic shifts, may influence metabolic status in this cohort (Davis et al., 2021). Interpretation of absence-coded models. Presence/absence modeling targets the occupancy dimension of species–phenotype relationships and is less sensitive to zero inflation and compositional artifacts than raw relative abundances. This choice yields straightforward clinical interpretation (ORs reflect IR odds when a species is absent). Future work will extend these models with quantitative pathways from shotgun metagenomics and metabolomics.
4.2 Species enriched in insulin-resistant individuals
Among the species enriched in IR participants, P. copri stood out as the most consistent risk-associated taxon. Previous studies have shown that strains of P. copri capable of producing branched-chain amino acids (BCAAs) are more common in individuals with type 2 diabetes, and elevated circulating BCAAs correlate with obesity and IR (Abdualkader et al., 2024). In both humans and mouse models, overabundance of P. copri has been linked to higher BCAA concentrations and the development of IR (Abdelsalam et al., 2023; Abdualkader et al., 2024). The outer membrane of P. copri contains lipopolysaccharide, which can translocate into the circulation, causing metabolic endotoxemia and low-grade inflammation that disrupts insulin signaling (Abdelsalam et al., 2023; Mohammad and Thiemermann, 2020; Kim and Sears, 2010; Liang et al., 2022). Our finding that P. copri presence is associated with IR in the Kazakh population aligns with these mechanistic observations and suggests that dietary or environmental factors promoting P. copri expansion may contribute to metabolic dysfunction in this cohort (Abdualkader et al., 2024). A. onderdonkii also showed a positive association with IR. While the genus Alistipes has been implicated in protection against metabolic inflammation in some settings, animal experiments demonstrate that different species can exert divergent effects: oral administration of A. indistinctus reduces intestinal carbohydrate accumulation and ameliorates IR in mice (Zhang, 2024), whereas other Alistipes species have been linked to pro-inflammatory responses. The detrimental association of A. onderdonkii observed here may reflect species-specific metabolite production or interactions with host diet that warrant further investigation.
Interestingly, the presence of R. inulinivorans was positively associated with IR, despite this bacterium being a known butyrate producer. In independent cohorts, R. inulinivorans abundance is significantly lower in individuals with type 2 diabetes than in healthy controls, and the species is inversely correlated with measures of IR (Ge et al., 2022). Butyrate supplementation improves insulin sensitivity and reduces adiposity in mice (Gao et al., 2009), and R. inulinivorans participates in butyrate production alongside other species such as C. catus (Davis et al., 2021). Our finding that R. inulinivorans is enriched in IR subjects therefore contradicts most prior reports and may reflect distinct dietary patterns or functional variants in the Kazakh population. It is possible that strains of R. inulinivorans prevalent in this cohort preferentially metabolize proteins to branched-chain fatty acids rather than fermenting fibre to butyrate, a metabolic shift that has been linked to inflammation and IR (Davis et al., 2021). Further metagenomic and metabolomic analyses are required to clarify the functional capacity of R. inulinivorans in this setting.
4.3 Species enriched in controls and putative protective microbes
Several taxa were less prevalent in IR participants and may exert protective effects. A. odontolyticus is an oral commensal that rarely causes disease (Negrini et al., 2021), and its presence in stool may reflect ingestion and transit of upper-airway microbes. Although we found no prior evidence directly linking A. odontolyticus to insulin sensitivity, our results suggest that individuals harboring this species have substantially lower odds of IR. B. kashiwanohense showed a similar protective association; this is consistent with the broader observation that Bifidobacterium species improve metabolic health. Systematic reviews of probiotic interventions show that supplementation with Bifidobacterium and Lactobacillus species improves IR, lipid profiles and inflammatory markers in animal models, and that mixtures of these genera enhance insulin sensitivity in human trials (Salles et al., 2020; Park et al., 2015). Lactobacillus species ferment dietary carbohydrates to lactic acid and short-chain fatty acids, modulate immune responses and reinforce gut barrier integrity, providing plausible mechanisms for their protective effects (Salles et al., 2020; Carretta et al., 2021; Pham et al., 2024). Similarly, S. lactarius was less common in IR participants; although little is known about this species in human metabolic health, related lactic acid bacteria have been reported to improve glucose tolerance and reduce inflammation in animal models (Salles et al., 2020; Park et al., 2015).
The absence of C. catus was independently associated with increased odds of IR. C. catus is a member of the Lachnospiraceae family and an efficient producer of butyrate (Davis et al., 2021). Butyrate enhances insulin sensitivity by increasing energy expenditure, improving mitochondrial function and suppressing inflammation (Gao et al., 2009; Carretta et al., 2021; Pham et al., 2024). Therefore, the reduced prevalence of C. catus in IR participants is consistent with the notion that diminished butyrate production may contribute to metabolic dysregulation (Tolhurst et al., 2012; Psichas et al., 2015). In contrast to R. inulinivorans, which displayed an unexpected positive association with IR, C. catus exhibited the anticipated protective pattern, suggesting that different butyrate producers may have divergent roles depending on their metabolic outputs and interactions with other community members.
4.4 Implications for regional differences and mechanistic insights
The discrepancy between our findings and previous literature underscores the importance of considering region-specific factors in microbiome studies. The Kazakh diet is rich in fermented dairy products and animal proteins, with relatively low intake of fermentable fibers; such a diet could favor expansion of protein-fermenting bacteria and modulate the production of branched-chain amino acids and short-chain fatty acids (Abdelsalam et al., 2023; Abdualkader et al., 2024). Genetic polymorphisms affecting immune responses to bacterial metabolites may also modify the impact of specific taxa on host metabolism (Abdelsalam et al., 2023; Chiang and Ferrell, 2020). Our observation that A. onderdonkii and R. inulinivorans show risk associations despite some evidence for protective roles in other populations emphasizes that microbial functions, not just taxonomic identities, determine metabolic outcomes. Integrating metagenomic, metabolomic and host transcriptomic analyses will be essential to elucidate these functional differences (Zhang, 2024). The Kazakh diet traditionally includes fermented dairy (e.g., kumis/‘qymyz’; shubat) and relatively higher animal-source foods with comparatively lower fermentable-fiber intake, factors known to shape taxa like Prevotella and butyrate producers. These dietary patterns likely modulate our observed species-phenotype links and should be measured directly in future work (Martuzzi et al., 2024).
4.5 Mechanistic pathways linking gut microbiota to IR
The mechanisms summarized here are biologically plausible but hypothetical given our 16S-only data, which cannot resolve gene content, strain differences, or metabolite profiles. Accordingly, we interpret the species–IR links as functional hypotheses to be tested with multi-omics. Multiple mechanistic pathways have been proposed to explain how gut bacteria influence host insulin sensitivity (Figure 4). Disruption of gut barrier integrity allows lipopolysaccharides (LPS) from Gram-negative bacteria to enter the circulation and activate Toll-like receptor 4 (TLR4) (Mohammad and Thiemermann, 2020; Liang et al., 2022). Elevated plasma LPS levels correlate negatively with muscle insulin sensitivity, and exposure of human myotubes to LPS increases pro-inflammatory cytokine expression while reducing insulin-stimulated IRS-1 and Akt phosphorylation; pharmacological or genetic inhibition of TLR4 abrogates these effects (Kim and Sears, 2010; Liang et al., 2022; Liang et al., 2013). This inflammatory cascade links dysbiosis to chronic low-grade inflammation and impaired insulin signaling (Mohammad and Thiemermann, 2020). Conversely, short-chain fatty acids (SCFAs) produced by fermentation of dietary fiber—particularly acetate, propionate and butyrate—stimulate secretion of glucagon-like peptide 1 (GLP-1) and peptide YY (PYY) from enteroendocrine cells and induce intestinal gluconeogenesis; these actions promote satiety and improve glucose homeostasis (Carretta et al., 2021; Pham et al., 2024; Psichas et al., 2015; Tang and Li, 2021). Reduced fiber intake in the Kazakh diet may limit SCFA production and attenuate these beneficial signaling pathways (Chiang and Ferrell, 2020; Pham et al., 2024).
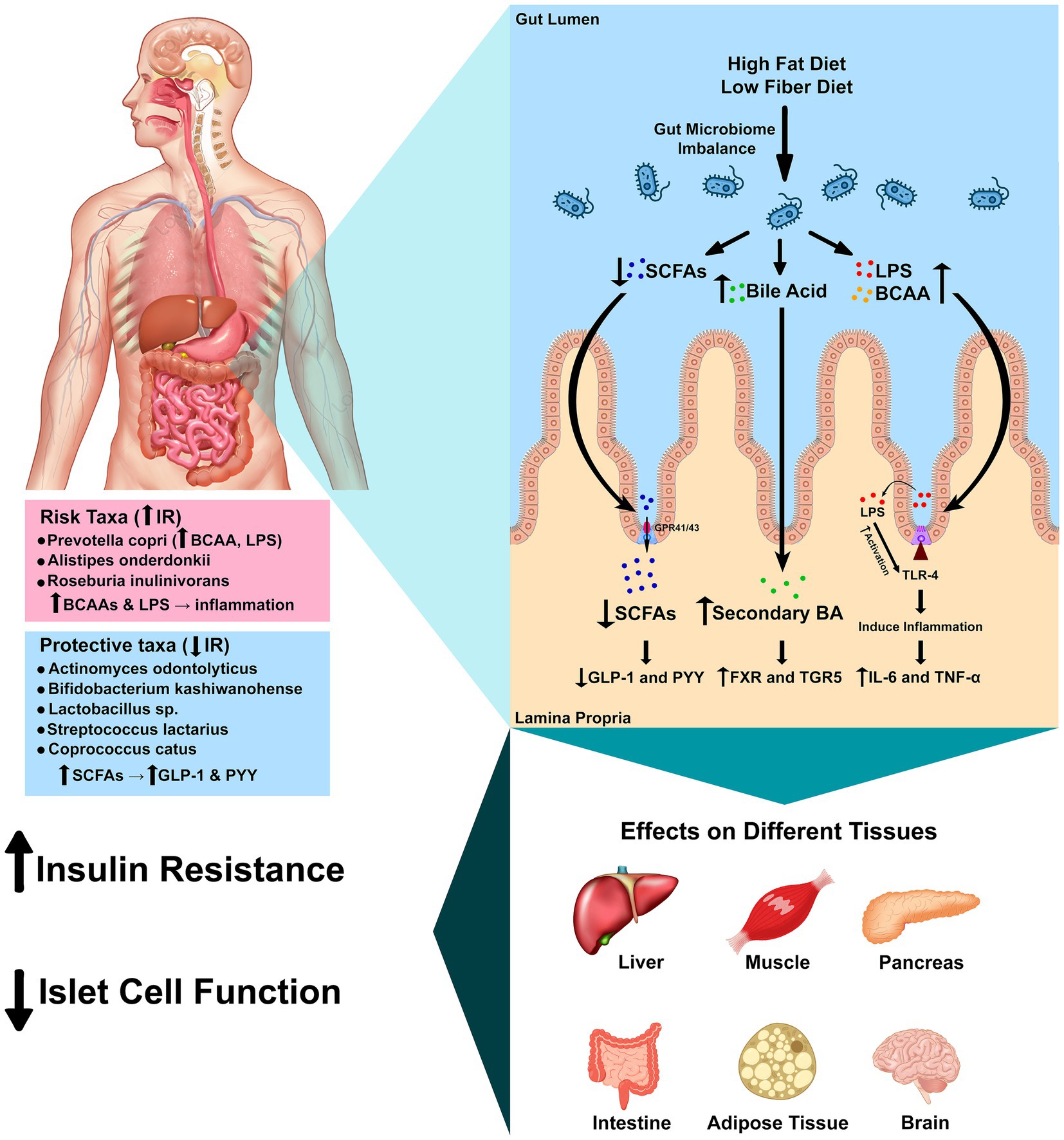
Figure 4. Mechanistic pathways linking gut microbes to insulin resistance. Schematic of four convergent axes: (1) LPS–TLR4 inflammation reduces insulin signaling (IRS-1/Akt); (2) SCFAs (butyrate/propionate) signal via FFAR2/3, increasing GLP-1/PYY and improving glycemic control; (3) Bile acids act through FXR/TGR5 to modulate glucose and inflammation; (4) BCAA production (e.g., Prevotella strains) can activate mTORC1 and promote insulin resistance. Species observed in this study map onto these pathways: P. copri (BCAA/LPS; risk-aligned), C. catus and Lactobacillus spp. (SCFA/barrier; protective-aligned), and R. inulinivorans (butyrate-capable but population-specific association). Arrows indicate direction of effect hypothesized from prior literature. Mechanistic links are hypothesis-generating and require shotgun metagenomic and metabolomic validation in this population.
Bile acids serve not only as emulsifiers but also as metabolic hormones (Chiang and Ferrell, 2020). Primary bile acids are converted to secondary bile acids by intestinal bacteria and activate the nuclear receptor FXR and membrane receptor TGR5 (Chiang and Ferrell, 2020). Activation or inhibition of intestinal FXR improves insulin and glucose sensitivity, and TGR5 signaling has anti-inflammatory effects and stimulates GLP-1 secretion (Chiang and Ferrell, 2020; Chiang et al., 2017). Dietary patterns that alter bile acid pools or the abundance of bile salt hydrolase-producing bacteria could therefore influence metabolic outcomes in this cohort (Chiang and Ferrell, 2020). Other host systems are intertwined with the microbiota. The gut endocannabinoid system, expressed in epithelial and enteroendocrine cells, modulates gut motility, permeability and inflammatory responses; microbiota composition shapes endocannabinoid tone and thereby influences metabolic and behavioral responses (Srivastava et al., 2022; Tagliamonte et al., 2021). Elevated levels of BCAAs, produced by taxa such as P. copri, activate the mammalian target of rapamycin complex 1 (mTORC1) and are associated with obesity and IR (Abdualkader et al., 2024; Yoon, 2016). Defective BCAA catabolism or overabundance of BCAA-producing bacteria may thus contribute to IR via mTORC1 activation and accumulation of toxic intermediates. Figure 4 integrates these pathways and illustrates how microbial metabolites and host receptors converge to modulate inflammation, hormone secretion and energy metabolism.
Multiple, convergent microbe-to-host axes plausibly link our species-level signals to IR. First, metabolic endotoxemia—translocation of Gram-negative LPS—activates TLR4, driving low-grade inflammation that impairs IRS-1/Akt signaling in insulin-responsive tissues (Mohammad and Thiemermann, 2020; Kim and Sears, 2010; Liang et al., 2022; Zhang, 2024; Ge et al., 2022; Gao et al., 2009). Second, SCFAs (particularly butyrate and propionate) engage FFAR2/3, stimulate GLP-1/PYY, and promote intestinal gluconeogenesis, thereby improving glucose homeostasis (Carretta et al., 2021; Pham et al., 2024; Tolhurst et al., 2012; Psichas et al., 2015). Third, bile-acid signaling via FXR/TGR5 modulates glycemic control and inflammation (Chiang and Ferrell, 2020; Chiang et al., 2017). Fourth, microbial BCAA production can elevate circulating BCAAs, activate mTORC1, and worsen insulin sensitivity (Abdualkader et al., 2024; Yoon, 2016). These pathways provide biologically plausible routes through which specific taxa observed here could contribute to metabolic dysfunction.
Species-to-pathway mapping. The risk-aligned association of P. copri in our cohort is consistent with reports of BCAA biosynthesis and potential LPS-mediated inflammation contributing to IR (Abdelsalam et al., 2023; Mohammad and Thiemermann, 2020; Abdualkader et al., 2024; Kim and Sears, 2010; Liang et al., 2022; Zhang, 2024; Yoon, 2016). Conversely, taxa aligned with insulin sensitivity—C. catus and Lactobacillus spp.—are linked to butyrate/SCFA production, barrier support, and immune modulation, mechanisms that improve insulin signaling (Negrini et al., 2021; Park et al., 2015; Carretta et al., 2021; Pham et al., 2024; Tolhurst et al., 2012). The unexpected positive association of R. inulinivorans with IR in this population may reflect strain-level functional variation or diet-dependent metabolic routing, underscoring the need for metagenomic/metabolomic resolution to disambiguate function (Davis et al., 2021; Ge et al., 2022; Carretta et al., 2021; Pham et al., 2024; Tolhurst et al., 2012). Together, these links motivate testing whether diet and microbial metabolites mediate species–IR associations in Kazakh adults.
4.6 External context and generalizability
We contextualized our cohort’s dominant taxa against large healthy-adult references, noting broad agreement at high taxonomic ranks while emphasizing our core finding that only species-level features discriminated IR within this population. Given well-documented cross-study domain shift in microbiome diagnostics, we intentionally refrained from cross-country ‘performance’ claims and instead report effect sizes with CIs for species signals that are most likely to translate. Prospective, diet-measured, multi-site validation is warranted (Li et al., 2023).
4.7 Implications for protecting a metabolically favorable gut ecology
Our species-level signals suggest pragmatic, testable strategies: (1) increase fermentable fiber (whole grains, inulin, resistant starch) to support butyrate producers such as C. catus; (2) consider fermented dairy or probiotics containing Bifidobacterium/Lactobacillus, which have shown improvements in IR in trials; (3) moderate dietary patterns that may favor expansion of BCAA-producing taxa such as P. copri, balancing protein with fiber-rich foods. These proposals are hypothesis-generating and require randomized, diet-measured trials in Kazakh adults (Salles et al., 2020; Pham et al., 2024; Martuzzi et al., 2024).
4.8 Practical implications and hypothesis-generating
Practical implications (hypothesis-generating, population-specific). Our species-resolved findings suggest pragmatic levers to protect a metabolically favorable gut ecology in this population. First, increasing fermentable fiber is expected to support butyrate-producing taxa such as C. catus, with potential downstream benefits via SCFA-mediated enteroendocrine signaling. Second, culturally congruent fermented dairy and/or probiotic products containing Bifidobacterium and Lactobacillus may help sustain taxa aligned with insulin sensitivity in our cohort. Third, maintaining overall diet quality and physical activity—independent determinants of insulin sensitivity—may act synergistically with microbiota-derived metabolites. Conversely, given the risk-aligned signal for P. copri, dietary patterns that balance higher protein intake with adequate fermentable fiber are a reasonable, testable approach. We emphasize that these actions are hypothesis-generating rather than prescriptive and may be population-specific; they require validation in diet-measured randomized trials incorporating stool/serum metabolomics (SCFAs, bile-acid species, BCAAs, LPS-related markers) to test mediation of species–IR associations in Kazakh adults. These ideas are investigational only; causality and efficacy cannot be inferred from 16S associations and require validation via shotgun metagenomics/metabolomics and prospective randomized studies in Kazakh adults.
4.9 Strengths, limitations, and future directions
This study benefits from a well-characterized cohort and comprehensive species-level analysis, yet several limitations must be acknowledged. The cross-sectional design precludes causal inference; longitudinal studies are needed to determine whether changes in specific species precede the onset of IR or result from metabolic alterations. Sample size in the control group was modest, which may limit power to detect associations for less prevalent taxa. Finally, functional inferences are speculative because our 16S rRNA sequencing cannot determine metabolic capacities; metagenomic and metabolomic profiling would clarify whether protective taxa produce beneficial metabolites such as butyrate or lactate, and whether risk taxa increase circulating BCAAs or endotoxin (Abdelsalam et al., 2023; Chiang and Ferrell, 2020; Davis et al., 2021; Abdualkader et al., 2024; Carretta et al., 2021; Pham et al., 2024; Tagliamonte et al., 2021). Despite these limitations, our findings highlight candidate species for microbiome-targeted interventions. Strategies that enrich Bifidobacterium, Lactobacillus and Coprococcus while reducing P. copri may improve insulin sensitivity; this notion is supported by evidence that probiotic supplementation with Bifidobacterium and Lactobacillus improves IR (Salles et al., 2020; Park et al., 2015; Carretta et al., 2021; Pham et al., 2024). The contrasting roles of R. inulinivorans and A. onderdonkii demonstrate that therapeutic approaches must be tailored to the functional characteristics of strains prevalent in a given population. Future studies should incorporate dietary assessments, host genotyping and longitudinal sampling to unravel the complex interplay between diet, microbiota and IR. Diet was not measured; given its strong influence on microbiota and insulin sensitivity, future studies will include validated FFQs and dietary biomarkers (SCFAs, bile acids) to enable diet-adjusted models. Although our mechanistic inferences are biologically grounded, 16S rRNA profiles are taxonomic and not functional. Future work will include validated diet assessment and stool/serum metabolomics (SCFAs, bile-acid species, BCAAs, LPS markers) to test mediation of species–IR associations and to resolve strain-level functional heterogeneity.
A key limitation is the IR:control imbalance (183:17). This asymmetry may increase uncertainty for low-prevalence taxa and contribute to OR instability or separation in logistic models. We mitigated these risks through CSS normalization, ANCOM-BC for bias-corrected differential abundance, and IR-subsampling checks for alpha/beta-diversity. Nonetheless, residual sensitivity to sample size cannot be excluded; accordingly, we emphasize effect sizes with 95% CIs and treat species-level findings as hypothesis-generating. Future studies will prioritize larger and/or matched control sampling, and may employ penalized likelihood or Bayesian shrinkage models to further stabilize estimates.
Another key limitation is that 16S rRNA sequencing lacks functional resolution. To validate and extend these findings, future work will integrate (i) shotgun metagenomics to quantify pathway genes with strain-level resolution, and (ii) targeted metabolomics to measure stool SCFAs (acetate/propionate/butyrate), fecal/serum bile acids, serum BCAAs, and endotoxemia markers. These data will enable mediation analyses to test whether microbial functions/metabolites statistically explain species–IR associations. Because our data are taxonomic (16S), we cannot claim that manipulating specific taxa will improve IR; instead, these species represent prioritized, testable targets for function-resolved and interventional studies.
5 Conclusion
In this Kazakh cohort, broad taxonomic profiles from phylum to genus were not informative for IR, whereas species-level composition was. A restricted set of species differentiated metabolic status: Lactobacillus sp. and C. catus aligned with insulin sensitivity, while P. copri and R. inulinivorans aligned with IR, with additional signals involving A. odontolyticus, B. kashiwanohense, and A. onderdonkii. These findings indicate that coarse community summaries are insufficient for this population and that species-resolved characterization is required to capture metabolically relevant variation. This work reframes microbiome–IR research toward strain- and function-focused analyses tailored to regional diets and host backgrounds and motivates hypothesis-driven trials to test whether modulating community functions or the abundance of candidate taxa affects insulin sensitivity. Causal effects and clinical efficacy cannot be inferred from these 16S data.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: FigShare, accession 30304189.
Ethics statement
This study adheres to the principles of the 1964 Helsinki Declaration and Good Clinical Practice guidelines. Ethical approval was obtained from the Local Ethics Committee of the Medical Center Hospital of the President’s Affairs Administration of the Republic of Kazakhstan, Astana (Local Ethics Committee protocol no. 1 from 05.04.2022). All participants provided informed consent, agreeing to the use of their data for research and educational purposes while maintaining confidentiality. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
GA: Conceptualization, Investigation, Methodology, Writing – review & editing, Resources, Funding acquisition, Project administration, Writing – original draft. VB: Project administration, Data curation, Supervision, Writing – review & editing, Investigation. TV: Investigation, Writing – review & editing, Data curation, Validation, Resources. NM: Project administration, Visualization, Investigation, Supervision, Writing – review & editing. AA: Software, Conceptualization, Investigation, Writing – original draft, Visualization, Writing – review & editing, Validation. AT: Project administration, Methodology, Supervision, Writing – review & editing, Writing – original draft, Conceptualization, Validation, Resources.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research has been funded by the Committee of Science of the Ministry of Science and Higher Education of the Republic of Kazakhstan (grant no. AP14871581).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Gen AI was used in the creation of this manuscript. ChatGPT (GPT-5 Thinking; OpenAI) was used only to refine grammar and wording. It did not generate scientific content, analyze data, create figures/tables, select or interpret references, or influence study design, results, or conclusions. The authors reviewed and take full responsibility for all content.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1683885/full#supplementary-material
References
Abdelsalam, N. A., Hegazy, S. M., and Aziz, R. K. (2023). The curious case of Prevotella copri. Gut Microbes 15:2249152. doi: 10.1080/19490976.2023.2249152
Abdualkader, A. M., Karwi, Q. G., Lopaschuk, G. D., and Al Batran, R. (2024). The role of branched-chain amino acids and their downstream metabolites in mediating insulin resistance. J. Pharm. Pharm. Sci. 27:13040. doi: 10.3389/jpps.2024.13040
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caricilli, A. M., and Saad, M. J. (2013). The role of gut microbiota on insulin resistance. Nutrients 5, 829–851. doi: 10.3390/nu5030829
Carretta, M. D., Quiroga, J., López, R., Hidalgo, M. A., and Burgos, R. A. (2021). Participation of short-chain fatty acids and their receptors in gut inflammation and Colon Cancer. Front. Physiol. 12:662739. doi: 10.3389/fphys.2021.662739
Chen, Z., Radjabzadeh, D., Chen, L., Kurilshikov, A., Kavousi, M., Ahmadizar, F., et al. (2021). Association of Insulin Resistance and Type 2 diabetes with gut microbial diversity: a microbiome-wide analysis from population studies. JAMA Netw. Open 4:e2118811. doi: 10.1001/jamanetworkopen.2021.18811
Chiang, J. Y. L., and Ferrell, J. M. (2020). Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 318, G554–G573. doi: 10.1152/ajpgi.00223.2019
Chiang, J. Y., Pathak, P., Liu, H., Donepudi, A., Ferrell, J., and Boehme, S. (2017). Intestinal farnesoid X receptor and Takeda G protein couple receptor 5 signaling in metabolic regulation. Dig. Dis. 35, 241–245. doi: 10.1159/000450981
Davis, J. A., Collier, F., Mohebbi, M., Pasco, J. A., Shivappa, N., Hebert, J. R., et al. (2021). The associations of butyrate-producing bacteria of the gut microbiome with diet quality and muscle health. Gut Microbiome (Camb). 2:e2. doi: 10.1017/gmb.2021.2
Deschasaux, M., Bouter, K. E., Prodan, A., Levin, E., Groen, A. K., Herrema, H., et al. (2018). Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 24, 1526–1531. doi: 10.1038/s41591-018-0160-1
Ebrahimzadeh Leylabadlo, H., Sanaie, S., Sadeghpour Heravi, F., Ahmadian, Z., and Ghotaslou, R. (2020). From role of gut microbiota to microbial-based therapies in type 2-diabetes. Infect. Genet. Evol. 81:104268. doi: 10.1016/j.meegid.2020.104268
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Fontana, A., Panebianco, C., Picchianti-Diamanti, A., Lagana, B., Cavalieri, D., Potenza, A., et al. (2019). Gut microbiota profiles differ among individuals depending on their region of origin: an Italian pilot study. Int. J. Environ. Res. Public Health 16:4065. doi: 10.3390/ijerph16214065
Gao, Z., Yin, J., Zhang, J., Ward, R. E., Martin, R. J., Lefevre, M., et al. (2009). Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 58, 1509–1517. doi: 10.2337/db08-1637
Ge, X., Zhang, A., Li, L., Sun, Q., He, J., Wu, Y., et al. (2022). Application of machine learning tools: potential and useful approach for the prediction of type 2 diabetes mellitus based on the gut microbiome profile. Exp. Ther. Med. 23:305. doi: 10.3892/etm.2022.11234
Ghorbani, Y., Schwenger, K. J. P., and Allard, J. P. (2021). Manipulation of intestinal microbiome as potential treatment for insulin resistance and type 2 diabetes. Eur. J. Nutr. 60, 2361–2379. doi: 10.1007/s00394-021-02520-4
Gurung, M., Li, Z., You, H., Rodrigues, R., Jump, D. B., Morgun, A., et al. (2020). Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 51:102590. doi: 10.1016/j.ebiom.2019.11.051
Jang, H. R., and Lee, H. Y. (2021). Mechanisms linking gut microbial metabolites to insulin resistance. World J. Diabetes 12, 730–744. doi: 10.4239/wjd.v12.i6.730
Kim, J. J., and Sears, D. D. (2010). TLR4 and insulin resistance. Gastroenterol. Res. Pract. 2010:212563. doi: 10.1155/2010/212563
Kuhn, T. S., Mooers, A. O., and Thomas, G. H. (2011). A simple polytomy resolver for dated phylogenies. Methods Ecol. Evol. 2, 427–436. doi: 10.1111/j.2041-210X.2011.00103.x
Lee, C. J., Sears, C. L., and Maruthur, N. (2020). Gut microbiome and its role in obesity and insulin resistance. Ann. N. Y. Acad. Sci. 1461, 37–52. doi: 10.1111/nyas.14107
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Li, M., Liu, J., Zhu, J., Wang, H., Sun, C., Gao, N. L., et al. (2023). Performance of gut microbiome as an independent diagnostic tool for 20 diseases: cross-cohort validation of machine-learning classifiers. Gut Microbes 15:2205386. doi: 10.1080/19490976.2023.2205386
Liang, H., Hussey, S. E., Sanchez-Avila, A., Tantiwong, P., and Musi, N. (2013). Effect of lipopolysaccharide on inflammation and insulin action in human muscle. PLoS One 8:e63983. doi: 10.1371/journal.pone.0063983
Liang, H., Sathavarodom, N., Colmenares, C., Gelfond, J., Espinoza, S. E., Ganapathy, V., et al. (2022). Effect of acute TLR4 inhibition on insulin resistance in humans. J. Clin. Invest. 132:e162291. doi: 10.1172/JCI162291
Lin, H., and Peddada, S. D. (2020). Analysis of compositions of microbiomes with bias correction. Nat. Commun. 11:3514. doi: 10.1038/s41467-020-17041-7
Martuzzi, F., Franceschi, P., and Formaggioni, P. (2024). Fermented Mare Milk and its microorganisms for human consumption and health. Foods 13:493. doi: 10.3390/foods13030493
Mohammad, S., and Thiemermann, C. (2020). Role of metabolic endotoxemia in systemic inflammation and potential interventions. Front. Immunol. 11:594150. doi: 10.3389/fimmu.2020.594150
Negrini, T. C., Carlos, I. Z., Duque, C., Caiaffa, K. S., and Arthur, R. A. (2021). Interplay among the oral microbiome, oral cavity conditions, the host immune response, diabetes mellitus, and its associated-risk factors-an overview. Front. Oral Health 2:697428. doi: 10.3389/froh.2021.697428
Park, K. Y., Kim, B., and Hyun, C. K. (2015). Lactobacillus rhamnosus GG improves glucose tolerance through alleviating ER stress and suppressing macrophage activation in db/db mice. J. Clin. Biochem. Nutr. 56, 240–246. doi: 10.3164/jcbn.14-116
Paulson, J. N., Stine, O. C., Bravo, H. C., and Pop, M. (2013). Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 10, 1200–1202. doi: 10.1038/nmeth.2658
Pham, N. H. T., Joglekar, M. V., Wong, W. K. M., Nassif, N. T., Simpson, A. M., and Hardikar, A. A. (2024). Short-chain fatty acids and insulin sensitivity: a systematic review and meta-analysis. Nutr. Rev. 82, 193–209. doi: 10.1093/nutrit/nuad042
Psichas, A., Sleeth, M. L., Murphy, K. G., Brooks, L., Bewick, G. A., Hanyaloglu, A. C., et al. (2015). The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int. J. Obes. 39, 424–429. doi: 10.1038/ijo.2014.153
Salles, B. I. M., Cioffi, D., and Ferreira, S. R. G. (2020). Probiotics supplementation and insulin resistance: a systematic review. Diabetol. Metab. Syndr. 12:98. doi: 10.1186/s13098-020-00603-6
Schnorr, S. L., Candela, M., Rampelli, S., Centanni, M., Consolandi, C., Basaglia, G., et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5:3654. doi: 10.1038/ncomms4654
Semo, D., Reinecke, H., and Godfrey, R. (2024). Gut microbiome regulates inflammation and insulin resistance: a novel therapeutic target to improve insulin sensitivity. Signal Transduct. Target. Ther. 9:35. doi: 10.1038/s41392-024-01746-y
Senghor, B., Sokhna, C., and Ruimy, R. (2018). Gut microbiota diversity according to dietary habits and geographical provenance. Hum. Microbiome J. 7, 1–9. doi: 10.1016/j.humic.2018.01.001Lagier J-C
Sonnenburg, J. L., and Backhed, F. (2016). Diet-microbiota interactions as moderators of human metabolism. Nature 535, 56–64. doi: 10.1038/nature18846
Srivastava, R. K., Lutz, B., and Ruiz de Azua, I. (2022). The microbiome and gut endocannabinoid system in the regulation of stress responses and metabolism. Front. Cell. Neurosci. 16:867267. doi: 10.3389/fncel.2022.867267
Tagliamonte, S., Laiola, M., Ferracane, R., Vitale, M., Gallo, M. A., Meslier, V., et al. (2021). Mediterranean diet consumption affects the endocannabinoid system in overweight and obese subjects: possible links with gut microbiome, insulin resistance and inflammation. Eur. J. Nutr. 60, 3703–3716. doi: 10.1007/s00394-021-02538-8
Takeuchi, T., Kubota, T., Nakanishi, Y., Tsugawa, H., Suda, W., Kwon, A. T., et al. (2023). Gut microbial carbohydrate metabolism contributes to insulin resistance. Nature 621, 389–395. doi: 10.1038/s41586-023-06466-x
Tang, R., and Li, L. (2021). Modulation of short-chain fatty acids as potential therapy method for type 2 diabetes mellitus. Can. J. Infect. Dis. Med. Microbiol. 2021, 1–13. doi: 10.1155/2021/6632266
Tolhurst, G., Heffron, H., Lam, Y. S., Parker, H. E., Habib, A. M., Diakogiannaki, E., et al. (2012). Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61, 364–371. doi: 10.2337/db11-1019
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., Gonzalez, A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5:27. doi: 10.1186/s40168-017-0237-y
Yoon, M. S. (2016). The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients 8:405. doi: 10.3390/nu8070405
Keywords: insulin resistance, gut microbiota, 16S ribosomal RNA sequencing, Kazakhstan, bacterium
Citation: Abildinova GZ, Benberin VV, Vochshenkova TA, Mussin NM, Afshar A and Tamadon A (2025) Distinct gut microbial species, but not phylum-to-genus composition, associate with insulin resistance: a unique perspective from the Kazakh population. Front. Microbiol. 16:1683885. doi: 10.3389/fmicb.2025.1683885
Edited by:
Mohamed Ezzat Abdin, Agricultural Research Center, EgyptReviewed by:
Yongsheng Chen, Jinan University, ChinaRicha Dwivedi, Meharry Medical College, United States
Copyright © 2025 Abildinova, Benberin, Vochshenkova, Mussin, Afshar and Tamadon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amin Tamadon, YW1pbnRhbWFkZG9uQHlhaG9vLmNvbQ==; Alireza Afshar, YWxpcmV6YS5hZjIwMTdAZ21haWwuY29t