 Min Li1†Yun Wang1†
Min Li1†Yun Wang1† Xiaohong Xiang2†Lingling Dong1Qiming Li3
Xiaohong Xiang2†Lingling Dong1Qiming Li3 Zhou Liu4
Zhou Liu4 Wenwen Chu1Ye Naifang1
Wenwen Chu1Ye Naifang1 Zhen Gong1,5*Qiang Zhou1*
Zhen Gong1,5*Qiang Zhou1*- 1Department of Clinical Laboratory, The Second Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China
- 2School of Pharmacy, Chongqing Medical and Pharmaceutical College, Chongqing, China
- 3Department of Clinical Laboratory, The First Affiliated Hospital of Henan University, Henan University, Kaifeng, China
- 4Department of Clinical Laboratory Center, Anhui Chest Hospital, Hefei, China
- 5Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
Introduction: Isoniazid is widely used in the treatment of pulmonary tuberculosis, yet the mechanisms underlying its tolerance remain incompletely understood. The Rv0274 gene of Mycobacterium tuberculosis, presumed to belong to the aldehyde dehydrogenase family, has been hypothesized to contribute to isoniazid tolerance. Mycobacterium smegmatis was used in this study as a surrogate model to investigate this possibility.
Methods: We generated an MSMEG_0608 knockout strain and performed drug susceptibility testing, growth curve analysis, biofilm formation assays, transcriptomic profiling, and RT-qPCR validation. Complementation with Rv0274 and knockdown of MSMEG_0606 were further conducted to substantiate the regulatory relationships observed.
Results: The deletion of MSMEG_0608 significantly impaired isoniazid tolerance. Complementation and gene knockdown experiments supported the involvement of Rv0274/MSMEG_0608 in modulating the expression of genes associated with Rv0273c/MSMEG_0606, ultimately influencing inhA expression. These findings consistently demonstrated that MSMEG_0608 is integral to the isoniazid tolerance phenotype.
Discussion: Our results suggest that Rv0274 (MSMEG_0608) negatively regulates genes linked to Rv0273c (MSMEG_0606), thereby contributing to alterations in inhA expression and influencing isoniazid tolerance. This work provides preliminary mechanistic insight into INH tolerance in mycobacteria and establishes a foundation for further investigations into drug tolerance in Mycobacterium tuberculosis.
1 Introduction
Tuberculosis has become the leading cause of mortality attributable to a single infectious pathogen worldwide, and the global spread of drug-resistant strains poses a formidable public health challenge (WHO, 2024). The widespread administration of anti-tuberculosis drugs has accelerated the emergence and dissemination of multidrug-resistant Mycobacterium tuberculosis (M. tuberculosis), thereby complicating disease management. Elucidating the molecular mechanisms underlying drug tolerance is thus crucial for optimizing therapeutic strategies and curbing the global tuberculosis epidemic.
The Rv0274 gene of Mycobacterium tuberculosis remains functionally uncharacterized. Structurally, it encodes a soluble protein of 193 amino acids with a predicted molecular weight of approximately 21 kDa and features an extracellular loop cleavage near its C-terminus. Rv0274 is highly conserved across various mycobacterial species, and sequence homology analysis suggests that it belongs to the aldehyde dehydrogenase family. Previous studies have shown that mutations within the promoter region of Rv0274 enhance bacterial sensitivity to prothionamide, an industrial precursor for the synthesis of isoniazid (Rawat et al., 2003). Based on these findings, we hypothesize that Rv0274 may play a critical role in the development of isoniazid tolerance.
Recent studies have highlighted that aldehyde metabolism and redox homeostasis are tightly linked to antibiotic stress adaptation and survival in mycobacteria (Limon et al., 2023; Darwin and Stanley, 2022; Chen et al., 2024). Aldehyde-detoxifying enzymes, such as the formaldehyde-degrading MscR in M. smegmatis, and redox-balancing systems in M. tuberculosis have been shown to influence drug tolerance and oxidative stress responses. These findings provide a mechanistic framework supporting the potential involvement of aldehyde dehydrogenase family members, including Rv0274, in INH responsiveness through redox-linked pathways.
Isoniazid (INH) remains a cornerstone in the frontline treatment of tuberculosis (Tiberi et al., 2018). Within host cells, INH is activated by the catalase-peroxidase enzyme KatG to generate an isonicotinyl intermediate, which subsequently conjugates with nicotinamide adenine dinucleotide (NAD) to form an isonicotinyl-NAD adduct. This adduct specifically inhibits InhA, an essential enoyl-acyl carrier protein reductase required for mycolic acid biosynthesis, thereby compromising the integrity of the mycobacterial cell wall and ultimately inducing bacterial death (Van Scoy and Wilkowske, 1992). The predominant mechanism of INH tolerance involves the remodeling of lipid metabolism pathways: under INH-induced selective pressure, M. tuberculosis can downregulate its metabolic activity and lipid synthesis demands, reducing its dependence on lipid precursors (Martinot et al., 2016). Additionally, increasing evidence implicates the regulation of intracellular redox homeostasis in conferring INH tolerance (Jeeves et al., 2015). Nevertheless, beyond these known mechanisms, many molecular determinants and regulatory pathways underlying INH tolerance remain to be fully elucidated.
To elucidate the functional role of Rv0274 in isoniazid tolerance, we employed Mycobacterium smegmatis (M. smegmatis) as a surrogate model. Our findings indicate that Rv0274 may contribute to INH tolerance by negatively regulating Rv0273c, thereby relieving its inhibitory effect on the inhA gene and facilitating its upregulation. M. smegmatis is a well-established surrogate for studying mycobacterial genetics because of its rapid growth, non-pathogenic nature, and high genomic conservation with M. tuberculosis. While this model enables preliminary mechanistic insights, findings require subsequent validation in M. tuberculosis to confirm their translational relevance.
2 Materials and methods
2.1 Bioinformatics analysis
Structural modeling and molecular interaction analyses of Rv0274 were performed using Swiss-model, AutoDockTools, and PyMOL (Choudhary et al., 2023). Data processing and figure preparation were conducted with GraphPad Prism 7, Photoshop, and DNAMAN. Sequence alignment and conservation analyses were performed using MEGA 7, ClustalX, and Espript 3.0 (https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi). Transcriptomic data were analyzed in Rstudio 4.4.1, and statistical significance was determined using Excel's t-test, with thresholds set at *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
2.2 Bacterial strains, plasmids, primers, and culture conditions
Escherichia coli DH5α was used as the host strain for plasmid construction. M. smegmatis was cultured in Middlebrook 7H9 medium supplemented with 0.05% Tween-80, with selective antibiotics (hygromycin, acetamide, and tetracycline) added as required. The primer sequences used in this study are provided in Table 1.

Table 1. Primers used in this study.
2.3 Construction of knockout strains
The MSMEG_0608 knockout strain was constructed using a double-crossover homologous recombination strategy (Zhang et al., 2015). The MSMEG_0608 deletion mutant of M. smegmatis mc2155 was generated using a homologous recombination strategy. Genomic DNA was extracted from freshly cultured bacteria and used as the template to amplify the upstream (834 bp) and downstream (850 bp) homologous arms of MSMEG_0608. The two fragments were fused by overlap-extension PCR and inserted into the pMD19-T vector. After verification by restriction digestion with BamHI and HindIII, the hygromycin resistance cassette (HygR) excised from pAL75 with BglII was ligated into the construct to obtain the recombinant Up-Hyg-Down fragment. The resulting fragment was electroporated into M. smegmatis harboring pJV53, and transformants were selected on 7H9 agar containing kanamycin (25 μg/mL) and hygromycin (50 μg/mL). Positive colonies were verified by PCR using MSMEG_0608 specific primers. To remove the selection marker, the hygromycin-resistant clones were serially passaged in antibiotic-free medium (10–12 passages) and then screened on plates with and without hygromycin. Colonies that grew only on antibiotic-free plates were confirmed by PCR and designated as MSMEG_KO0608.
2.4 Antibiotic susceptibility testing (MIC determination)
Minimum inhibitory concentrations (MICs) were determined using both a 96-well microdilution method (Wiegand et al., 2008) and the Kirby-Bauer disk diffusion assay. Log-phase cultures grown in 7H9 medium supplemented with 0.05% Tween-80 were inoculated into fresh medium and incubated at 37 °C with shaking. For the microdilution method, serial dilutions of the tested antibiotics were prepared in 96-well plates and incubated for 5–7 days, with the lowest concentration showing no visible growth recorded as the MIC (Garbe et al., 1993). In the Kirby-Bauer assay, bacterial suspensions were evenly spread onto blood agar plates, antibiotic-impregnated disks were applied, and inhibition zone diameters were measured. All experiments were performed in triplicate (Brulle et al., 2013).
2.5 Bacterial growth curve
Log-phase cultures were adjusted to an equivalent OD600 (Initial inoculation volume 4.5 × 105 CFU/mL) and inoculated into fresh 7H9 medium. Cultures were incubated at 37 °C with shaking at 170 rpm, and OD600 values were recorded at designated time points. All experiments were performed in triplicate to generate growth kinetic curves (Huang et al., 2024).
2.6 Sliding motility and biofilm assays
For sliding motility assays, log-phase cultures adjusted to an OD600 of 1.5 were spotted onto 7H9 solid medium. Biofilm formation was assessed using 96-well plates: equal volumes of 7H9 medium supplemented with 2% (v/v) glucose were inoculated with 10% of the adjusted bacterial suspension and incubated at 37 °C for 2–3 days. The resulting biofilms were stained with crystal violet and quantified using a microplate reader. All experiments were performed in triplicate (Chakraborty et al., 2021).
2.7 CFU Determination
Bacterial cultures grown in 50 mL of 7H9 medium to an OD600 of 1.5 were serially diluted 10-fold and plated onto 7H9 solid medium containing isoniazid. Colony-forming units (CFUs) were enumerated after incubation for 5 days at 37 °C. All experiments were conducted in triplicate.
2.8 Sample preparation and transcriptomic sequencing
Wild-type (WT) and MSMEG_KO0608 strains were cultured in 7H9 medium. Two experimental conditions were established: an untreated group and a control group supplemented with 8 μg/mL isoniazid. After 2–3 days of incubation, bacterial cells were harvested by low-temperature centrifugation, rapidly frozen in liquid nitrogen, and submitted to Sangon Biotech (Shanghai) Co., Ltd. for high-throughput transcriptomic sequencing and downstream bioinformatics analysis (Deng et al., 2022).
2.9 RNA extraction and RT-qPCR
Total RNA was extracted using the Trizol reagent, and complementary DNA (cDNA) was synthesized by reverse transcription for quantitative real-time PCR (RT-qPCR) analysis, with sigA used as the internal reference gene. Primer sequences are listed in Table 1. PCR amplification was performed under the following conditions: initial denaturation at 95 °C for 5 min; 40 cycles of 95 °C for 15 s and 60 °C for 30 s; followed by melting curve analysis to verify specificity. Relative gene expression levels were calculated using the 2∧−ΔΔCt method, and all experiments were performed in triplicate to ensure reproducibility.
2.10 Complementation and knockdown strain construction
The homologous Rv0274 gene was cloned into the pALACE vector to construct the recombinant plasmid pALACE_Rv0274, which was subsequently transformed into the corresponding M. smegmatis mutant strain. Complemented strains were selected on 7H9 medium supplemented with 0.2% glycerol and 50 μg/mL hygromycin and verified by Western blot analysis. In addition, CRISPR interference (CRISPRi) (Wong and Rock, 2021) was employed to transcriptionally knock down MSMEG_0606 expression, and positive clones were selected for downstream experiments.
2.11 Statistical analysis
All statistical analyses were performed using GraphPad Prism 10.0. Statistical significance was assessed using one-way and two-way ANOVA, with thresholds defined as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Error bars represent the standard deviation (SD).
3 Results
3.1 Bioinformatics analysis and structural features of Rv0274
Bioinformatics analysis revealed that the Rv0274 gene, a putative member of the aldehyde dehydrogenase family, is highly conserved among M. tuberculosis, M. bovis, and M. smegmatis, suggesting an important role in mycobacterial physiology. Comparative genomic analyses demonstrated that, although Rv0274 and its adjacent gene Rv0273c are transcribed in opposite directions, their genomic positions and orientations are highly conserved across diverse mycobacterial species (Figures 1A,B), implying potential functional synergy. Homology analysis revealed that Rv0274 possesses the characteristic three conserved domains typical of the glyoxalase enzyme family (Figure 1C), thereby validating the selection of MSMEG_0608 as a surrogate model for downstream functional studies. Conserved domain prediction revealed that aldehyde dehydrogenase family proteins typically harbor three characteristic domains, although their precise biological roles remain undefined (Figure 1D). To evaluate the evolutionary conservation of Rv0274, multiple sequence alignment was performed among Mycobacterium tuberculosis strains (H37Rv, 18b, MTBC0, M. bovis) and M. smegmatis MSMEG_0608. The analysis revealed a high degree of sequence conservation, with MSMEG_0608 sharing ~74.3% amino acid identity similarity with Rv0274 of M. tuberculosis (Figure 1E). A tertiary structure model of Rv0274 constructed using Swiss-Model (Figure 1F) showed high structural similarity (TM-score > 0.8) to a reference protein (PDB: O53680), supporting its classification as a bona fide aldehyde dehydrogenase. Furthermore, molecular docking analysis suggested a potential interaction between Rv0274 and the frontline anti-tuberculosis drug isoniazid (INH), with a predicted binding energy of −4.72 kcal/mol (Figure 1G). Detailed inspection of the docking interface revealed hydrogen bonds between INH and two key residues, Arg124 (bond length: 2.0 Å) and Gln154 (bond length: 1.8 Å). Although the docking energy indicates only a weak interaction, this result should be interpreted as supportive rather than conclusive. The predicted binding pattern provides preliminary structural insight consistent with the proposed regulatory role of MSMEG_0608/Rv0274, but further biochemical validation is required to establish its physiological relevance. Collectively, these findings imply that Rv0274 may participate in INH metabolism or tolerance mechanisms (Ferreira et al., 2015), warranting further experimental validation.
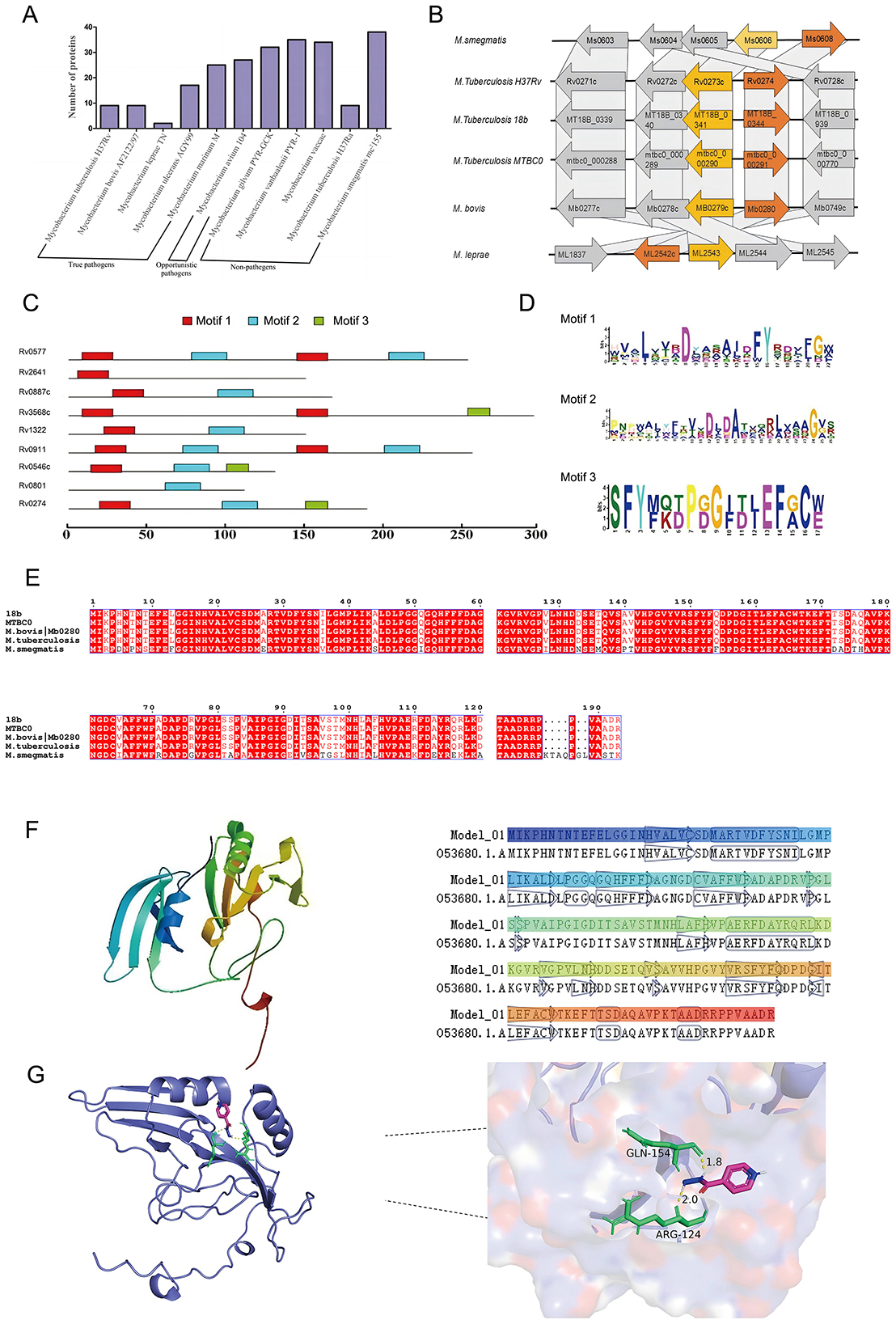
Figure 1. Bioinformatics analysis and structural features of Rv0274. (A, B) Comparative genomic analysis showing conservation of Rv0274 and Rv0273c across mycobacterial species with opposite transcriptional orientations. (C) Homology analysis revealed that Rv0274 possesses the characteristic three conserved domains typical of the glyoxalase enzyme family. (D) Conserved domain prediction identifying three aldehyde dehydrogenase–specific domains. (E) Multiple sequence alignment of Rv0274 and its homologs in different mycobacterial species. (F) Swiss-Model 3D structure of Rv0274 showing high similarity to reference protein O53680 (TM-score > 0.8). (G) Molecular docking showing interaction between Rv0274 and isoniazid (INH) with −4.72 kcal/mol binding energy and hydrogen bonds at Arg124 (2.0 Å) and Gln154 (1.8 Å).
3.2 Construction and phenotypic analysis of the MSMEG_0608 knockout strain
An MSMEG_0608 deletion strain (MSMEG_KO0608) of M. smegmatis was successfully generated using a double-crossover homologous recombination strategy. PCR verification confirmed the deletion, as the wild-type mc2155 strain produced the expected 943 bp fragment encompassing MSMEG_0608, whereas this fragment was absent in the knockout strain (Figures 2A,B). To elucidate the functional role of MSMEG_0608, we systematically evaluated the phenotype of the deletion mutant. Sliding motility assays on 7H9 agar plates demonstrated no significant differences in surface migration between MSMEG_KO0608 and the wild-type strain (Figure 2C). Similarly, biofilm formation assays revealed no marked differences (Figure 2D), and growth curve analysis showed comparable growth rates under nutrient-rich conditions (Figure 2E). Collectively, these results indicate that MSMEG_0608 is not essential for growth, motility, or biofilm formation under standard culture conditions.
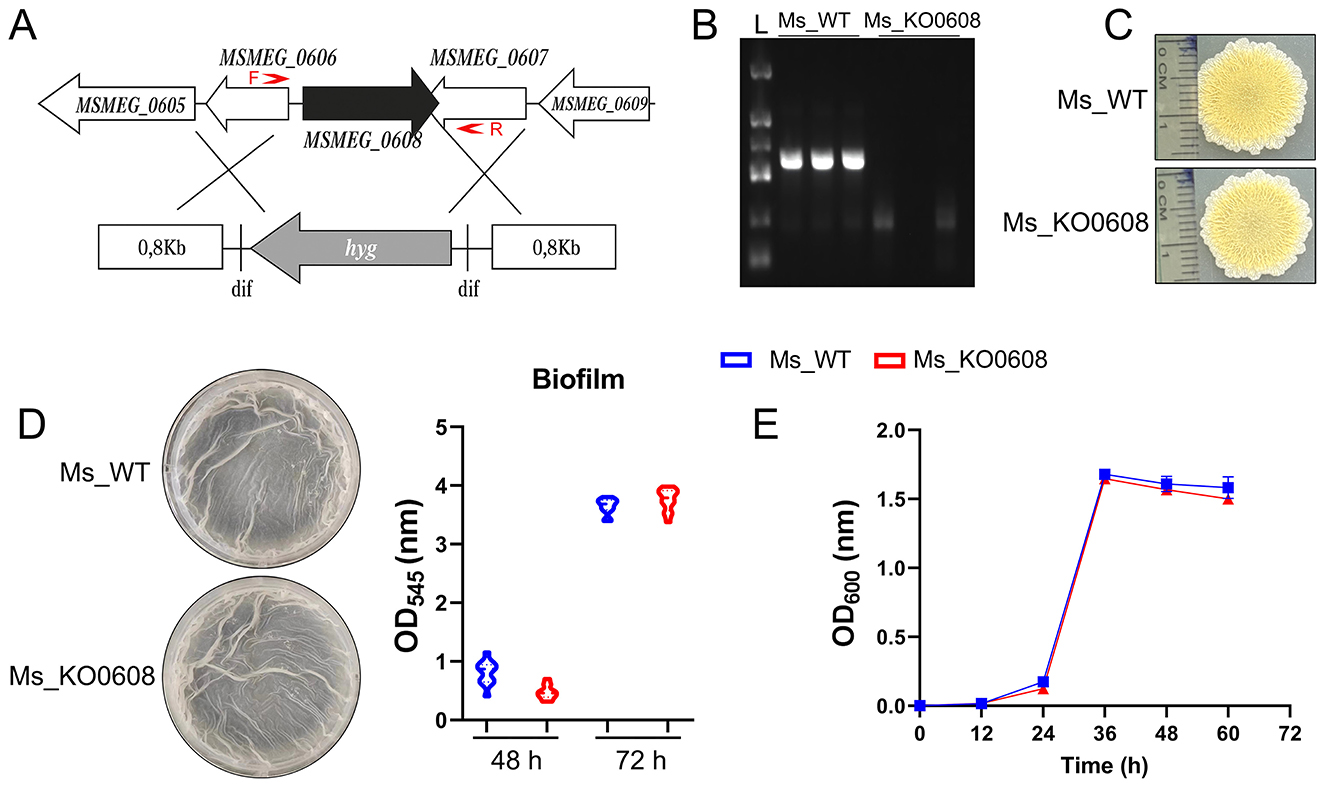
Figure 2. Construction and phenotypic analysis of the MSMEG_0608 knockout strain. (A, B) Schematic representation of the MSMEG_0608 gene deletion strategy. The MSMEG_0608 coding sequence was replaced using a two-step homologous recombination approach with approximately 1 kb upstream and downstream flanking regions. The primer pairs used for PCR verification (F and R) are indicated by red arrows, showing the amplification regions for confirming both the successful recombination and the absence of the MSMEG_0608 gene in the mutant strain. PCR verification showing absence of the 943 bp fragment in MSMEG_KO0608 compared with wild-type (WT). (C) Sliding motility assays showing no differences between WT and MSMEG_KO0608. (D) Biofilm formation assays showing comparable biofilm production in WT and MSMEG_KO0608. (E) Growth curves showing similar growth rates of WT and MSMEG_KO0608 in nutrient-rich medium.
3.3 Impact of MSMEG_0608 deletion on prothionamide tolerance and isoniazid sensitivity
In medium containing 1.75 mM prothionamide, the growth rate of MSMEG_KO0608 was comparable to that of the wild-type mc2155 strain (Figure 3A). However, at a higher concentration of 2.625 mM, the wild-type strain resumed exponential growth at ~30 h, whereas MSMEG_KO0608 exhibited a significant growth delay until around 60 h (Figure 3B), indicating that MSMEG_0608 deletion reduces tolerance to elevated prothionamide levels.
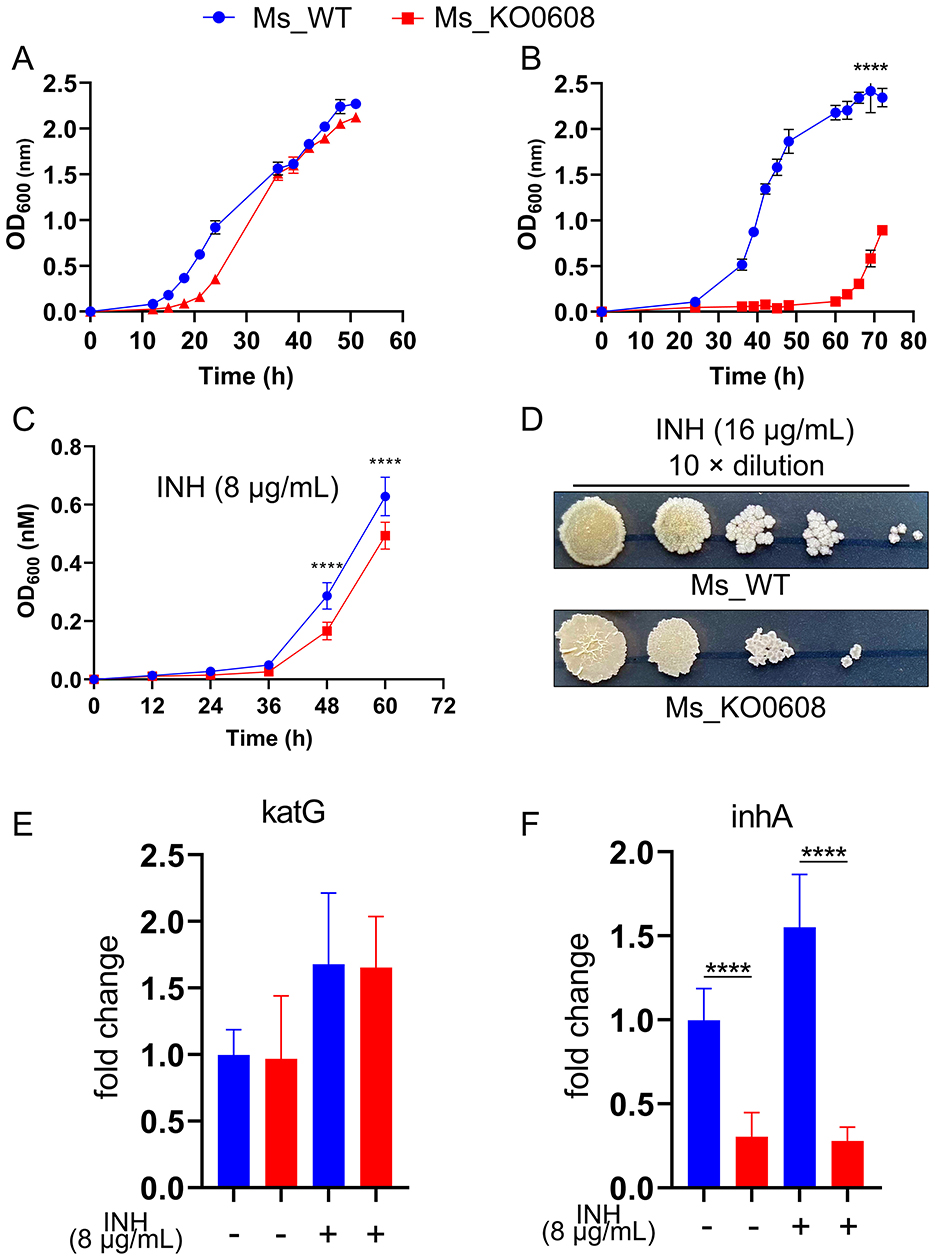
Figure 3. Effect of MSMEG_0608 deletion on prothionamide tolerance and INH susceptibility. (A) Growth curves showing similar proliferation of WT and MSMEG_KO0608 at 1.75 mM prothionamide. (B) Growth curves showing delayed growth of MSMEG_KO0608 compared with WT at 2.625 mM prothionamide. (C) Growth inhibition of MSMEG_KO0608 compared with WT under low INH concentrations. (D) Colony numbers showing reduced survival of MSMEG_KO0608 on plates containing 16 μg/mL INH. (E, F) RT-qPCR showing unchanged katG expression and reduced inhA expression in MSMEG_KO0608 after INH exposure.
Antibiotic susceptibility testing (Table 2) revealed no significant differences in sensitivity to most tested antibiotics (e.g., TE, CIP, TZP) between MSMEG_KO0608 and the wild-type strain. Notably, MIC determination using the microdilution method demonstrated that the MIC of INH for MSMEG_KO0608 was 16 μg/mL—representing a 50% reduction compared with the wild-type value of 32 μg/mL—while the MIC for rifampicin (RIF) remained similar between the two strains (Table 3). Further INH treatment experiments confirmed these findings: under low INH concentrations, logarithmic-phase growth of MSMEG_KO0608 was markedly impaired (Figure 3C), and on 7H9 agar plates containing 16 μg/mL INH, the number of colonies formed by MSMEG_KO0608 was significantly lower than that of the wild-type (Figure 3D). Expression analysis of tolerance-associated genes revealed no significant change in katG expression before and after INH exposure, whereas inhA expression was significantly downregulated following INH treatment (Figures 4E,F). Collectively, these results suggest that MSMEG_0608 may modulate INH sensitivity by regulating inhA expression.
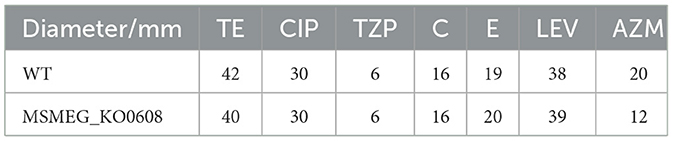
Table 2. Results of Mycobacterium smegmatis drug sensitivity to antibiotics on paper tablets.
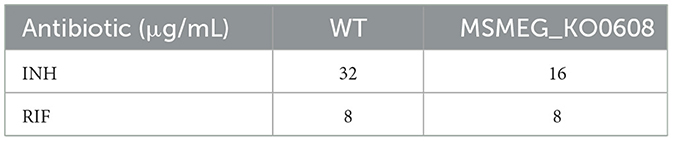
Table 3. MIC testing results for Mycobacterium smegmatis.
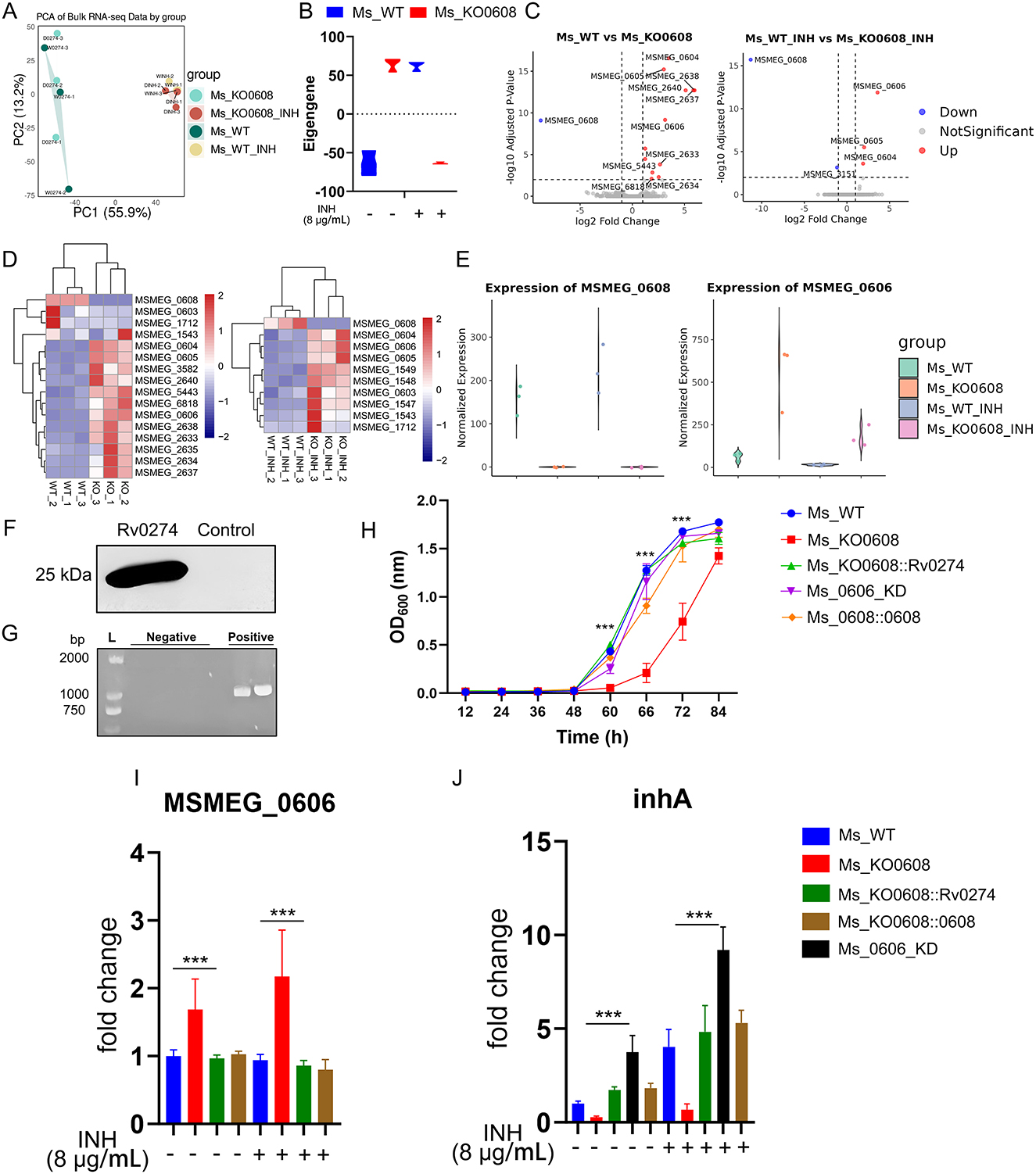
Figure 4. MSMEG_0608 regulates INH tolerance via MSMEG_0606 and inhA. (A) PCA showing distinct clustering under different treatments. (B) Weighted expression analysis showing global upregulation of module genes in MSMEG_KO0608 without INH and overall downregulation under INH. (C) Transcriptomic analysis showing increased MSMEG_0606 expression in MSMEG_KO0608, further elevated by INH. (D) Differential expression analysis identifying multiple genes within the MSMEG_0603–MSMEG_1712 locus. (E) MSMEG_0606 expression upregulated in MSMEG_KO0608 and suppressed in WT. (F) Complementation of Rv0274 in the MSMEG_KO0608 strain successfully restored gene expression. (G) PCR verification of MSMEG_0606 knockdown efficiency. (H) Growth curve analysis showing that, under INH treatment, both complemented and MSMEG_0606 knockdown strains exhibited superior growth during the logarithmic phase compared with MSMEG_KO0608, although the late-stage growth of MSMEG_0606 KD was slightly affected by tetracycline supplementation. (I, J) RT-qPCR showing MSMEG_0608 represses MSMEG_0606 and upregulates inhA.
3.4 MSMEG_0608-mediated regulation of MSMEG_0606 and inhA in INH tolerance
Principal component analysis (PCA) revealed distinct clustering of samples under different treatment conditions, indicating substantial transcriptional variation between groups (Figure 4A). Weighted average analysis showed that, under untreated conditions, global gene expression levels were elevated in the MSMEG_KO0608 strain, whereas overall expression was markedly reduced following INH exposure (Figure 4B).
Transcriptomic profiling demonstrated that deletion of MSMEG_0608 led to significant upregulation of several genes, including MSMEG_0604, MSMEG_0605, and MSMEG_0606; notably, MSMEG_0606 expression was further enhanced under INH treatment (Figure 4C), highlighting a central regulatory role for MSMEG_0608 in INH-associated metabolic pathways. Comparative transcriptomic analysis identified numerous differentially expressed genes between experimental and control groups, including MSMEG_0603, MSMEG_0604, MSMEG_0605, MSMEG_0606, and MSMEG_1712 (Figure 4D). Interestingly, in wild-type strains under INH treatment, MSMEG_0608 was markedly upregulated while MSMEG_0606 expression was repressed; conversely, in the MSMEG_KO0608 strain, MSMEG_0606 expression was strongly upregulated and further enhanced by INH exposure (Figure 4E).
Previous studies reported that the homologous gene of MSMEG_0606, Rv0273c, encodes a putative transcriptional regulator, EtbR, which specifically binds to the upstream regulatory motif of the inhA gene, repressing its expression; under prothionamide induction, enhanced inhA repression potentiates the bactericidal effect of INH on M. tuberculosis (Zhu et al., 2018). To further investigate the role of Rv0274 in INH sensitivity and elucidate the regulatory relationship between MSMEG_0608 and MSMEG_0606, we constructed three strains: complementation strain MSMEG_KO0608::Rv0274, MSMEG_KO0608::0608 and an MSMEG_0606 knockdown strain (MSMEG_0606_KD) (Figures 4F, G).
MIC determination revealed that both MSMEG_KO0608::Rv0274 and MSMEG_0606_KD exhibited an MIC for INH of 64 μg/mL, substantially higher than those of the wild-type (32 μg/mL) and MSMEG_KO0608 (16 μg/mL) strains (Table 4), confirming that both strains displayed markedly enhanced tolerance. Growth curve analysis further demonstrated that, under INH treatment, both strains exhibited superior growth during the logarithmic phase compared to MSMEG_KO0608, although later-stage growth of MSMEG_0606_KD was slightly affected by tetracycline supplementation (Figure 4H).

Table 4. MIC testing results for isoniazid.
Gene expression analysis normalized to sigA revealed that MSMEG_0606 expression was significantly upregulated in MSMEG_KO0608 compared to the wild-type strain, but was strongly suppressed upon complementation with Rv0274 and MSMEG_0608 (Figure 4I), confirming the negative regulatory role of MSMEG_0608 on MSMEG_0606. In contrast, inhA expression displayed the opposite pattern: it was significantly downregulated in MSMEG_KO0608 but markedly elevated in both MSMEG_KO0608::Rv0274, MSMEG_KO0608::0608 and MSMEG_0606_KD strains (Figure 4J).
Taken together, these findings indicate that MSMEG_0608 indirectly enhances bacterial tolerance to INH by negatively regulating MSMEG_0606 and relieving its repression of inhA, thereby positively modulating inhA expression.
4 Discussion
Tuberculosis remains one of the most formidable infectious diseases worldwide, with persistently high incidence and mortality rates posing a substantial public health burden. The widespread use of antibiotics has accelerated the emergence of drug-resistant tuberculosis, further complicating disease management. Chronic infections caused by M. tuberculosis are sustained by a complex genetic regulatory network involving secondary metabolism, cell wall synthesis, stress responses, and signal transduction pathways (Clark-Curtiss and Haydel, 2003). Moreover, transcriptional regulation and biofilm formation are increasingly recognized as key contributors to antibiotic tolerance; however, their precise roles in tuberculosis pathogenesis remain to be fully elucidated.
Bioinformatics analyses identified Rv0274 as a putative member of the aldehyde dehydrogenase family, which is highly conserved across mycobacterial species. Interestingly, aldehyde dehydrogenase family genes in M. tuberculosis share three conserved motifs, suggesting their potential involvement in the metabolism of methylglyoxal or related compounds. In this study, we successfully constructed an MSMEG_0608 knockout strain in M. smegmatis using a homologous recombination strategy. Under nutrient-rich conditions, the growth rate and sliding motility of MSMEG_KO0608 were comparable to those of the wild-type strain, indicating that MSMEG_0608 is non-essential for bacterial survival under such conditions. Biofilm formation, a phenotype closely associated with both mycobacterial growth and tolerance to environmental stress (Richards and Ojha, 2014), was similarly unaffected by the deletion of MSMEG_0608. Although biofilm formation in M. smegmatis is influenced by multiple factors, including genetic changes affecting lipid components such as free mycolic acids (FMA), mycolic acid diesters (MDAG), monothiolated mycolic acid diesters (MMDAG), mycolic acid wax esters (MWE), and glycopeptidolipids (GPL) (Nisbett et al., 2023), single-gene deletion of MSMEG_0608 did not significantly alter biofilm development under standard culture conditions. Although the biofilm and motility assays did not reveal significant differences between the wild-type and MSMEG_KO0608 strains, these negative results provide valuable insight into the functional specificity of MSMEG_0608. The absence of phenotypic changes under non-stress conditions suggests that MSMEG_0608 does not function as a broad stress-response or surface-regulation factor. Instead, its effects appear to be selectively activated under INH-induced stress, consistent with its proposed role in modulating inhA expression and INH tolerance. This specificity further supports the conclusion that MSMEG_0608 participates in targeted redox- and drug-related regulatory pathways rather than in general physiological adaptation.
Our findings further revealed that the Rv0274 gene in M. tuberculosis provides protection against hydrazine-induced stress but exhibits limited responsiveness to other forms of oxidative stress, such as redox-cycling radicals and organic peroxides. Given that isoniazid (INH) is a derivative of hydrazine, we hypothesized that Rv0274 might also contribute to INH tolerance. Integrating bioinformatics predictions with drug susceptibility assays, our results confirmed that Rv0274 functions as an INH tolerance-associated gene.
It is well established that katG and inhA are key genes involved in the regulation of INH activity (Unissa et al., 2016). The inhA gene encodes InhA, a pivotal enzyme in mycolic acid biosynthesis and the direct target of activated INH. In vitro studies have demonstrated that, under low INH concentrations, promoter mutations in inhA (e.g., fabG1–15C>T) represent the predominant tolerance mechanism by increasing InhA protein levels to neutralize active INH. At higher INH concentrations, mutations in katG (e.g., katG S315N, katG N138S, and katG K414N) become the dominant mechanism, leading to reduced or abolished KatG activity and thereby impairing INH activation. Adaptive shifts in tolerance strategies have also been observed: at elevated INH concentrations, the frequency of inhA promoter mutations decreases, whereas katG mutations become more prevalent. Furthermore, compensatory mutations in the ahpC gene may arise to offset the functional loss of KatG (Dokrungkoon et al., 2023).
Our study demonstrates a positive regulatory relationship between MSMEG_0608 and the inhA gene. In the absence of drug treatment, deletion of MSMEG_0608 led to widespread upregulation of numerous genes, suggesting that this transcriptional activation may represent a compensatory response to partial physiological deficits caused by the loss of MSMEG_0608. Under INH treatment, MSMEG_0606 expression was markedly increased, and a negative regulatory interaction between MSMEG_0608 and MSMEG_0606 was observed; elevated MSMEG_0606 levels may suppress the expression or function of MSMEG_0608, thereby attenuating its positive regulatory effect on inhA. Previous studies have reported that Rv0273c, the homolog of MSMEG_0606, encodes the transcriptional regulator EtbR, which specifically binds to an upstream motif of inhA and represses its transcription (Zhu et al., 2018). Consistent with this model, complementation of Rv0274 in the MSMEG_KO0608 strain and knockdown of MSMEG_0606 both confirmed that MSMEG_0608 functions as a key regulatory factor: by indirectly inhibiting MSMEG_0606 expression, it relieves the repression of inhA, thereby upregulating inhA expression and enhancing bacterial tolerance to INH.
It should be noted that the deletion of MSMEG_0608 may exert a certain degree of compensatory transcriptional influence on adjacent genes within the same genomic region, particularly MSMEG_0606, even under basal conditions. However, the difference becomes more pronounced under INH treatment, suggesting that the observed transcriptional changes are primarily stress-induced rather than due to a simple polar effect. This implies that MSMEG_0608 and MSMEG_0606 may participate in a coordinated regulatory network that responds to drug-induced stress, rather than being functionally linked through an operonic structure. To further exclude a classical polar effect caused by gene deletion, we performed transcriptional-strand and operon-prediction analysis (Operon-mapper). The results showed that MSMEG_0608 is located on the opposite strand and is not predicted to form a single operon with MSMEG_0604-0606, supporting a regulatory mechanism rather than operon disruption. These data are provided in Supplementary Table.
Aldehyde dehydrogenases are essential for cellular and bacterial survival, yet their roles in mycobacteria remain poorly understood. As a predicted member of the aldehyde dehydrogenase family, Rv0274 shares conserved structural domains with orthologous genes across mycobacterial species. For example, the putative glyoxalase II enzyme MSMEG_2975 has been reported to influence bacterial growth, biofilm formation, transcriptomic profiles, and antibiotic susceptibility (Haris et al., 2021). Similarly, our findings demonstrate that MSMEG_0608 modulates global transcriptional patterns, regulates INH sensitivity, and affects the expression of tolerance-related genes. These results suggest that aldehyde dehydrogenase family genes may represent promising therapeutic targets for combating mycobacterial pathogens. Despite recent advances linking specific mutations in tolerance-associated genes to INH tolerance, the underlying molecular mechanisms remain incompletely understood. Our experiments, conducted using an M. smegmatis model, require validation of Rv0274′s function in M. tuberculosis. Future studies should elucidate how Rv0274 regulates the expression of Rv0273c and inhA, and assess the impact of altered mycolic acid content on cell wall integrity among the different strains. A key limitation of this study is the use of M. smegmatis as a surrogate model; the results have not yet been validated in M. tuberculosis. Although M. smegmatis exhibits higher intrinsic tolerance to isoniazid than M. tuberculosis, largely due to KatG variants with reduced INH-activating efficiency (Reingewertz et al., 2020), it remains a reliable and mechanistically informative surrogate for dissecting inhA-mediated resistance pathways. The inhA gene and its regulatory network—including Rv0273c/MSMEG_0606-mediated transcriptional repression—are highly conserved across mycobacterial species (Zhu et al., 2018). Therefore, the relative changes in INH susceptibility observed in the MSMEG_KO0608 and complemented strains provide mechanistic insights relevant to the Rv0274–Rv0273c–inhA regulatory axis in M. tuberculosis. Future studies will construct Rv0274 knockout and overexpression strains in M. tuberculosis H37Rv to confirm its role in INH tolerance and its regulatory interactions with Rv0273c and inhA.
Data availability statement
The processed RNA-seq datasets generated and analyzed in this study are publicly available in Zenodo at: https://doi.org/10.5281/zenodo.17636367.
Author contributions
ML: Data curation, Writing – original draft, Formal analysis. YW: Formal analysis, Writing – original draft, Data curation. XX: Data curation, Writing – review & editing. LD: Investigation, Data curation, Formal analysis, Writing – review & editing. QL: Data curation, Writing – review & editing. ZL: Writing – review & editing. WC: Writing – review & editing. YN: Writing – review & editing. ZG: Conceptualization, Investigation, Funding acquisition, Writing – review & editing. QZ: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by National Natural Science Foundation [grant numbers 82302534], Anhui Province Scientific Research Compilation Plan Project [2022AH050730], Major Project of Anhui Provincial Department of Education [Project No. KJ2021ZD0029], Health Research Program of Anhui (AHWJ2024Aa10005), Natural Science Foundation of Chongqing Medical and Pharmaceutical College (YGZZK2025111).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2025.1697416/full#supplementary-material
References
Brulle, J. K., Tschumi, A., and Sander, P. (2013). Lipoproteins of slow-growing Mycobacteria carry three fatty acids and are N-acylated by apolipoprotein N-acyltransferase BCG_2070c. BMC Microbiol. 13:223. doi: 10.1186/1471-2180-13-223
Chakraborty, P., Bajeli, S., Kaushal, D., Radotra, B. D., and Kumar, A. (2021). Biofilm formation in the lung contributes to virulence and drug tolerance of Mycobacterium tuberculosis. Nat. Commun. 12:1606. doi: 10.1038/s41467-021-21748-6
Chen, Y. C., Yang, X., Wang, N., and Sampson, N. S. (2024). Uncovering the roles of Mycobacterium tuberculosis melH in redox and bioenergetic homeostasis: implications for antitubercular therapy. mSphere 9:e0006124. doi: 10.1128/msphere.00061-24
Choudhary, S., Kesavan, A. K., Juneja, V., and Thakur, S. (2023). Molecular modeling, simulation and docking of Rv1250 protein from Mycobacterium tuberculosis. Front. Bioinform. 3:1125479. doi: 10.3389/fbinf.2023.1125479
Clark-Curtiss, J. E., and Haydel, S. E. (2003). Molecular genetics of Mycobacterium tuberculosis pathogenesis. Annu. Rev. Microbiol. 57, 517–549. doi: 10.1146/annurev.micro.57.030502.090903
Darwin, K. H., and Stanley, S. A. (2022). The aldehyde hypothesis: metabolic intermediates as antimicrobial effectors. Open Biol. 12:220010. doi: 10.1098/rsob.220010
Deng, W., Zheng, Z., Chen, Y., Yang, M., Yan, J., Li, W., et al. (2022). Deficiency of GntR family regulator MSMEG_5174 promotes Mycobacterium smegmatis resistance to aminoglycosides via manipulating purine metabolism. Front. Microbiol. 13:919538. doi: 10.3389/fmicb.2022.919538
Dokrungkoon, T., Tulyaprawat, O., Suwannakarn, K., and Ngamskulrungroj, P. (2023). In vitro modeling of isoniazid resistance mechanisms in Mycobacterium tuberculosis H37Rv. Front. Microbiol. 14:1171861. doi: 10.3389/fmicb.2023.1171861
Ferreira, L. G., Dos Santos, R. N., Oliva, G., and Andricopulo, A. D. (2015). Molecular docking and structure-based drug design strategies. Molecules 20, 13384–13421. doi: 10.3390/molecules200713384
Garbe, T., Harris, D., Vordermeier, M., Lathigra, R., Ivanyi, J., and Young, D. (1993). Expression of the Mycobacterium tuberculosis 19-kilodalton antigen in Mycobacterium smegmatis: immunological analysis and evidence of glycosylation. Infect. Immun. 61, 260–267. doi: 10.1128/iai.61.1.260-267.1993
Haris, M., Chen, C., Wu, J., Ramzan, M. N., Taj, A., Sha, S., et al. (2021). Inducible knockdown of Mycobacterium smegmatis MSMEG_2975 (glyoxalase II) affected bacterial growth, antibiotic susceptibility, biofilm, and transcriptome. Arch Microbiol 204:97. doi: 10.1007/s00203-021-02652-5
Huang, Y., Shen, Q., Xu, H., Huang, L., Xiang, S., Li, P., et al. (2024). Mycobacterium smegmatis MfpC is a GEF that regulates mfpA translationally to alter the fluoroquinolone efficacy. Commun. Biol. 7:1035. doi: 10.1038/s42003-024-06737-x
Jeeves, R. E., Marriott, A. A. N., Pullan, S. T., Hatch, K. A., Allnutt, J. C., Freire-Martin, I., et al. (2015). Mycobacterium tuberculosis is resistant to isoniazid at a slow growth rate by single nucleotide polymorphisms in katG codon Ser315. PLoS One 10:e0138253. doi: 10.1371/journal.pone.0138253
Limon, G., Samhadaneh, N. M., Pironti, A., and Darwin, K. H. (2023). Aldehyde accumulation in Mycobacterium tuberculosis with defective proteasomal degradation results in copper sensitivity. mBio 14:e0036323. doi: 10.1128/mbio.00363-23
Martinot, A. J., Farrow, M., Bai, L., Layre, E., Cheng, T.-Y., Tsai, J. H., et al. (2016). Mycobacterial metabolic syndrome: LprG and Rv1410 regulate triacylglyceride levels, growth rate and virulence in Mycobacterium tuberculosis. PLoS Pathog. 12:e1005351. doi: 10.1371/journal.ppat.1005351
Nisbett, L. M., Previti, M. L., and Seeliger, J. C. (2023). A loss of function in LprG-Rv1410c homologues attenuates growth during biofilm formation in Mycobacterium smegmatis. Pathogens 12:1375. doi: 10.3390/pathogens12121375
Rawat, M., Heys, J., and Av-Gay, Y. (2003). Identification and characterization of a diamide sensitive mutant of Mycobacterium smegmatis. FEMS Microbiol Lett 220, 161–169. doi: 10.1016/S0378-1097(03)00127-7
Reingewertz, T. H., Meyer, T., McIntosh, F., Sullivan, J., Meir, M., Chang, Y.-F., et al. (2020). Differential sensitivity of mycobacteria to isoniazid is related to differences in KatG-mediated enzymatic activation of the drug. Antimicrob. Agents Chemother. 64, e01899–e01819. doi: 10.1128/AAC.01899-19
Richards, J. P., and Ojha, A. K. (2014). Mycobacterial biofilms. Microbiol. Spectr. 2. doi: 10.1128/microbiolspec.MGM2-0004-2013
Tiberi, S., du Plessis, N., Walzl, G., Vjecha, M. J., Rao, M., Ntoumi, F., et al. (2018). Tuberculosis: progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 18, e183–e198. doi: 10.1016/S1473-3099(18)30110-5
Unissa, A. N., Subbian, S., Hanna, L. E., and Selvakumar, N. (2016). Overview on mechanisms of isoniazid action and resistance in Mycobacterium tuberculosis. Infect. Genet. Evol. 45, 474–492. doi: 10.1016/j.meegid.2016.09.004
Van Scoy, R. E., and Wilkowske, C. J. (1992). Antituberculous agents. Mayo Clin. Proc. 67, 179–187. doi: 10.1016/S0025-6196(12)61320-2
Wiegand, I., Hilpert, K., and Hancock, R. E. (2008). Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175. doi: 10.1038/nprot.2007.521
Wong, A. I., and Rock, J. M. (2021). CRISPR interference (CRISPRi) for targeted gene silencing in mycobacteria. Methods Mol. Biol. 2314, 343–364. doi: 10.1007/978-1-0716-1460-0_16
Zhang, Z., Wang, R., and Xie, J. (2015). Mycobacterium smegmatis MSMEG_3705 encodes a selective major facilitator superfamily efflux pump with multiple roles. Curr. Microbiol. 70, 801–809. doi: 10.1007/s00284-015-0783-0
Keywords: Mycobacterium tuberculosis, glyoxalase, isoniazid, Rv0274, drug tolerance
Citation: Li M, Wang Y, Xiang X, Dong L, Li Q, Liu Z, Chu W, Naifang Y, Gong Z and Zhou Q (2025) Decoding drug tolerance: insights into the Rv0274 gene's role in isoniazid tolerance. Front. Microbiol. 16:1697416. doi: 10.3389/fmicb.2025.1697416
Received: 02 September 2025; Accepted: 30 October 2025;
Published: 27 November 2025.
Edited by:
Jichan Jang, Gyeongsang National University, Republic of KoreaReviewed by:
Stephane Canaan, Centre National de la Recherche Scientifique (CNRS), FranceSatya Deo Pandey, University of Louisville, United States
Min-Kyoung Shin, Gyeongsang National University, Republic of Korea
Copyright © 2025 Li, Wang, Xiang, Dong, Li, Liu, Chu, Naifang, Gong and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen Gong, emhlbi5nb25nQGtpLnNl; Qiang Zhou, emhvdXFpYW5nMTk3M0AxNjMuY29t
†These authors have contributed equally to this work