 Kristin F. Phillips
Kristin F. Phillips Laxmikant S. Deshpande
Laxmikant S. Deshpande Robert J. DeLorenzo1,2*
Robert J. DeLorenzo1,2*- 1Department of Neurology, Virginia Commonwealth University, Richmond, VA, United States
- 2Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, VA, United States
Status Epilepticus (SE) is a major neurological emergency and is considered a leading cause of Acquired Epilepsy (AE). We have shown that SE produces neuronal injury and prolonged alterations in hippocampal calcium levels ([Ca2+]i) that may underlie the development of AE. Interventions preventing the SE-induced Ca2+ plateau could therefore prove to be beneficial in lowering the development of AE after SE. Hypothermia is used clinically to prevent neurological complications associated with Traumatic Brain Injury, cardiac arrest, and stroke. Here, we investigated whether hypothermia prevented the development of Ca2+ plateau following SE. SE was induced in hippocampal neuronal cultures (HNC) by exposing them to no added MgCl2 solution for 3 h. To terminate SE, low Mg2+ solution was washed off with 31°C (hypothermic) or 37°C (normothermic) physiological recording solution. [Ca2+]i was estimated with ratiometric Fura-2 imaging. HNCs washed with hypothermic solution exhibited [Ca2+]i ratios, which were significantly lower than ratios obtained from HNCs washed with normothermic solution. For in vivo SE, the rat pilocarpine (PILO) model was used. Moderate hypothermia (30–33°C) in rats was induced at 30-min post-SE using chilled ethanol spray in a cold room. Hypothermia following PILO-SE significantly reduced mortality. Hippocampal neurons isolated from hypothermia-treated PILO SE rats exhibited [Ca2+]i ratios which were significantly lower than ratios obtained from PILO SE rats. Hypothermia also provided significant neuroprotection against SE-induced delayed hippocampal injury as characterized by decreased FluoroJade C labeling in hypothermia-treated PILO SE rats. We previously demonstrated that hypothermia reduced Ca2+ entry via N-methyl-D-aspartate and ryanodine receptors in HNC. Together, our studies indicate that by targeting these two receptor systems hypothermia could interfere with epileptogenesis and prove to be an effective therapeutic intervention for reducing SE-induced AE.
Introduction
Status Epilepticus (SE) is a major clinical emergency associated with significant mortality and severe neurological morbidities amongst the survivors (1, 2). One of the major morbidities associated with survival from SE is the development of acquired epilepsy (AE) (3, 4). Epilepsy is a common neurological condition characterized by recurring spontaneous seizures. It affects approximately 1–2% of the population worldwide (1, 4). Acquired epilepsy (AE) results from a previous neurological insult such as a SE or a stroke, or a traumatic brain injury (TBI) and accounts for at least 40% of all epilepsy cases (1–4). In AE, a known cause or injury damages the brain and produces a plasticity change that leads to the development of epilepsy (5, 6). The transformation of normal brain tissue into a hyperexcitable neuronal population manifesting spontaneous recurrent epileptic discharges, or seizures, is called epileptogenesis (5–7).
It has been demonstrated in both in vivo and in vitro models of SE, stroke, and TBI that neuronal calcium (Ca2+) dynamics are severely altered following injury (8–10). One such alteration relevant to this study is a persistent elevation in intracellular calcium concentrations ([Ca2+]i) following SE, termed the “Ca2+ plateau” (8, 11, 12). The formation of the Ca2+ plateau has been implicated in playing a major role in the development of AE (13). Ca2+ is responsible for an array of cellular effects (14, 15), and alterations in a neuron's ability to regulate Ca2+ could substantially contribute to neuronal plasticity changes (16) that lead to epileptogenesis and subsequently AE (5, 13, 15). Therefore, preventing the rise in [Ca2+]i immediately after SE may prevent the Ca2+-mediated signaling effects that lead to the development of AE. Targeting the molecular alterations observed in epileptogenesis such as elevated [Ca2+]i may therefore offer new avenues to developing anti-epileptogenic approaches (5, 13, 15).
One such possible intervention is hypothermia. Hypothermia is used clinically to reduce neurological injury following a variety of insults including cardiac arrest (17–19), TBI (20, 21) and stroke (22, 23). Hypothermia exerts its neuroprotective effects through a variety of mechanisms including a reduction in cerebral metabolism, apoptosis, and inflammation (24, 25). One particular mechanism of interest is its ability to modulate neuronal Ca2+entry (26). Evidence has shown that the N-methyl-D-aspartate (NMDA) receptor mediates the majority of Ca2+ influx during SE (8, 11). Inhibition of the NMDA receptors during SE blocks the formation of the Ca2+ plateau and prevents the development of AE (5, 8). The excessive entry of Ca2+ into the cell during SE stimulates Ca2+-induced Ca2+ release via activation of ryanodine receptors. Evidence has suggested ryanodine receptors may be responsible for maintaining the Ca2+ plateau following SE (27, 28). Our lab has demonstrated in hippocampal neuronal cultures that hypothermia reduces neuronal Ca2+ entry through NMDA and ryanodine receptors following a high-potassium or glutamate stimulation (26). Therefore, hypothermia may prevent the formation of the Ca2+ plateau following SE and thus may serve as a valuable therapeutic option in preventing epileptogenesis.
This study was initiated to investigate whether hypothermia prevents the formation of the Ca2+ plateau following SE and decreases neuronal injury and mortality. We utilized two widely used in vivo (29) and in vitro (27) models of SE in this study. The results of this study demonstrate that hypothermia prevents the development of the Ca2+ plateau when administered following SE. Based on previous studies showing that blocking the Ca2+ plateau prevents the development of AE, the results of this study offer promising evidence that hypothermia induced following SE prevents the long lasting rise in [Ca2+]i, neuronal loss and decreases mortality and may prevent the development of AE.
Materials and Methods
All reagents were purchase from Sigma Chemical Co. (St. Louis, MO) unless otherwise specified. Cell culture media was purchased from Invitrogen (Carlsbad, CA). All animal use procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by Virginia Commonwealth University's Institutional Animal Care and Use Committee.
Hippocampal Neuronal Culture Preparation
Studies were conducted on primary mixed hippocampal neuronal cultures (HNC) prepared as described previously (27, 30, 31). In brief, hippocampal cells were obtained from 2-day post-natal Sprague-Dawley rats (Harlan, Frederick, MD) and plated at a density of 2.0 × 104 cells/cm2 onto a glial support layer previously plated onto poly-L-lysine coated (0.05 mg/ml) Lab-Tek® two-well cover glass chambers (Nunc, Naperville, IL). Cultures were maintained at 37°C in a 5% CO2/95% air atmosphere and fed twice weekly with MEM enriched with N3 supplement containing 25 mM HEPES buffer (pH 7.4), 2 mM L-glutamine, 3 mM glucose, 100 μg/ml transferrin, 5 μg/ml insulin, 100 μM putrescine, 3 nM sodium selenite, 200 nM progesterone, 1 mM sodium pyruvate, 0.1% ovalbumin, 0.2 ng/ml triiodothyroxine, 0.4 ng/ml corticosterone and supplemented with a glial bed-condition media (20%). These mixed cultures were used for experiments between 15 and 21 days in vitro following neuronal plating.
In Vitro SE in Hippocampal Neuronal Cultures and Hypothermia Induction
In vitro SE was generated using a low Mg2+-containing solution (27, 30, 32). Hippocampal neuronal culture media was replaced with physiological basal recording solution (pBRS) containing (in mm): 145 NaCl, 2.5 KCl, 10 HEPES, 2 CaCl2, 10 glucose, 1 MgCl2 and 0.002 glycine, pH 7.3, and osmolarity adjusted to 325 ± 5 mOsm with sucrose or pBRS without any added MgCl2 (referred to hereafter as low Mg2+). The cells were then incubated at 37°C under 5% CO2 / 95% O2 atmosphere for 3 h. During this time, neurons in low Mg2+ demonstrate high-frequency spiking, characterized as in vitro SE. Neurons treated with pBRS for 3 h served as sham-controls.
SE was terminated by replacing the low Mg2+ solution with either 31°C (moderate hypothermia) or 37°C (physiological temperature) pBRS. [Ca2+]i was measured at the end of the 3 h treatment every 30 s for 20 min to get a measurement of [Ca2+]i.
Pilocarpine Model of SE and Induction of Moderate Hypothermia in Vivo
Sprague-Dawley male rats (Envigo, formerly Harlan) weighing 200–250 g were administered methyl scopolamine nitrate (1 mg/kg, i.p.) followed by pilocarpine nitrate (PILO) (375 mg/kg, i.p.) 30 min later. Sixty minutes after the onset of SE, rats were administered diazepam (5 mg/kg, i.p.) followed by additional diazepam injections at 3 and 5 h after the onset of SE to control seizure activity (8, 29, 33).
Moderate hypothermia was rapidly induced at 30 min post-SE onset by gently spraying rats with chilled ethanol (17°C) to speed the process of cooling. Rats were then placed in a cold room (5–8°C) for 8–10 min. Surface cooling methods were used because they are non-invasive and cost-effective. Core body temperature was determined every 2–3 min using a rectal probe (2100 Tele-thermometer; YSI, Inc., Yellow Springs, OH USA). Once core temperatures reached a 32-33°C, the rats were returned to their home cages in a room temperature environment. Core temperatures were continuously monitored and maintained between 30 and 33°C (moderate hypothermia range) with the intermittent use of ice packs and heating pads. Moderate hypothermia was maintained for 4 h at which point all the active cooling procedures were stopped and the animal was allowed to naturally rewarm to physiological temperature at room temperatures (see Figures 1, 3A).
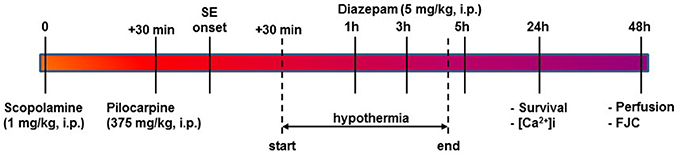
Figure 1. Timeline for PILO SE, induction of HYPO, and experimental endpoints. SE was induced using PILO injection and terminated with diazepam injections as per established procedures. At 30-mins into SE, moderate HYPO was induced and then maintained for 4 h using procedures described in the Materials and Methods section. At 24 h post-SE, survival and ratiometric Ca2+ imaging studies were conducted. At 48 h post-SE, rats were perfused and brain sections from these rats were labeled with FJC for assessing neuronal injury as described.
At 24 h post-SE, gross behavior was noted to assess recovery from SE. Rats were assigned a score of 0 or 1 on three parameters: mobility, posture, and alertness. For mobility, a score of 1 indicated rat being ambulatory, while a score of 0 indicated rat sitting in the cage, lethargic, and not moving about. For posture, a score of 1 indicated normal posture while 0 indicated a hunched posture. Alertness as indicated by being responsive to handling and approach was scored as 1 while 0 score indicated being non-responsive to handle and approach. Sum of all the scores gave the gross behavioral score and was indicative of physical recovery following SE.
Acute Isolation of Hippocampal Neurons
Hippocampal CA1 neurons were acutely isolated by a modification of the methods described previously (8, 10–12). The brain was rapidly dissected and placed in a 4°C chilled oxygenated (95% O2-5% CO2) artificial cerebrospinal fluid solution (aCSF) composed of (in mM) 201 sucrose, 3 KCl, 1.25 NaHPO4, 6 MgCl2, 0.2 CaCl2, 26 NaHCO3, MK-801 (1 μM) and 10 glucose (solution A). Hippocampal slices of 450 μm were cut with a vibratome sectioning system (Series 3000, Technical Products International, St. Louis, MO) and incubated for 10 min in an oxygenated medium at 34°C containing (in mM) 120 NaCl, 5 KCl, 6 MgCl2, 0.2 CaCl2, 25 glucose, and 20 PIPES, pH adjusted to 7.2 with NaOH (solution B). Slices were then treated with 8 mg/ml of Protease XXIII (Sigma Chemical Co.) in solution B for 6–8 min and then thoroughly rinsed with solution B. The CA1 region was visualized in a dark background with the help of a dissecting microscope and tissue chunks were excised. These tissue preparations were then triturated in solution B with a series of Pasteur pipettes of decreasing diameter at 4°C in the presence of acetoxymethyl (AM) form of high affinity Ca2+ indicator Fura-2AM (1 μM) in order to load the cells prior to [Ca2+]i measurements. The resulting cell suspension was then placed in the center of poly-L-lysine coated Lab-Tek® two-well cover glass chambers (Nalge-Nunc International, Naperville, IL) and immediately placed in a humidified oxygenated dark chamber at 37°C for 45 min. Fura-2 was washed off with solution B and the loaded cells were allowed to equilibrate for 15 min allowing the cellular esterase to cleave the dyes from their AM forms.
Calcium Microfluorimetry
Fura-2AM was loaded in the neurons as described above and then transferred to a heated stage (37°C) of an Olympus IX-70 inverted microscope coupled to an ultra-high-speed fluorescence imaging system (Olympus/ Perkin-Elmer). Ratio images were acquired by using alternating excitation wavelengths (340/380 nm) with a filter wheel (Sutter Instruments, Novato, CA) and Fura filter cube at 510/540 nm emissions with a dichroic mirror at 400 nm. Image pairs were captured and digitized every 15 s, and the images at each wavelength were averaged over four frames and corrected for background fluorescence by imaging a non-indicator loaded field.
In neuronal cultures, ratio measurements for individual neurons were taken at 30 s intervals for 20 min. For acutely isolated neurons, ratio measurements for individual neurons were taken at 5 s intervals for 30 s. The resulting 340/380 ratios correspond directly to the total concentration of Ca2+ inside the cell (8, 10–12).
Fluoro-Jade Staining
Rats were sacrificed 48 h following SE. Briefly, deep anesthesia was induced in rats with ketamine/xylazine (75 mg/kg/7.5 mg/kg i.p.) mixture. Anesthetized animals were flushed transcardially with saline and perfused with 4% paraformaldehyde in a 100 mM sodium phosphate buffer (pH 7.4). Fixed brains were removed and post-fixed in 4% paraformaldehyde/phosphate buffer overnight, cryoprotected in 30% sucrose/phosphate buffer (pH 7.4) (48 h), flash frozen in isopentane and stored at −80°C until used for sectioning. Coronal sections (40 μm) were cut on a cryostat (Leica Microsystems, Wetzlar, Germany) and mounted onto microscope slides (Trubond 380; Tru Scientific LLC, Bellingham, WA). Slides were dried in a desiccant chamber at 55°C for 30 min prior to staining. Slides were first incubated in a solution of 1% NaOH in 80% ethanol for 5 min followed by hydration in a 70% ethanol and then ddH2O for 2 min each. Slides were then incubated in a 0.06% KMnO4 solution for 10 min followed by washing in ddH2O for 2 min. Slides were then stained in a 0.0004% Fluoro-Jade C (FJC) solution in 0.1% acetic acid for 20 min (12, 28, 34). Stained slides underwent 3x washes in ddH2O for 2 min each and then dried in a desiccant chamber at 55°C for 30 min. Stained slides were then cleared with xylene for 5 min and cover slipped with DPX mounting agent. Stained sections were evaluated with a fluorescent inverted microscope with a 20X (UApo 340, 0.7 n.a., water) objective and excitation/emission filters for visualization of FITC. Grayscale digital images (1,324 × 1,024, 16-bit, 1X1 binning) of FJC staining for select brain regions were acquired.
Data Analyses
For experiments performed in cell cultures, a sample size (n) of at least 6 plates per treatment group was used. Experiments in cultures were performed over several weeks so that results were representative of multiple cultures. For experiments performed in whole animal, a sample size of n = 15 rats per treatment group were used. For each rat, 20–30 hippocampal neurons were isolated and imaged. Individual neurons from multiple experiments were pooled to calculate average and standard error of the mean (SEM). Data is presented as mean ± SEM. To determine statistical significance between treatment groups, student's t-test or one-way analysis of variance (ANOVA) were used followed by Tukey post-hoc analysis when appropriate. A p-value of less than 0.05 (p < 0.05) was considered statistically significant. Analysis of digital images to count FJC positive cell staining was carried out with Image-J (NIH, Bethesda, MD) by thresholding for specific stain and obtaining positive cell counts using the particle analysis component (size range in pixel: 25–1,000). Digital acquisition and staining analysis parameters remained constant throughout. Statistical analysis and graphs were drawn using SigmaPlot 13 (Systat Software, San Jose, CA).
Results
Hypothermia Blocked the Development of the Ca2+ Plateau in the HNC Model of SE
Figure 2A depicts ratiometric pseudocolor images of hippocampal neurons from the cultures under different temperature conditions. Fura-2 ratios were measured at 20-min following 3 h of low Mg2+ treatment. As illustrated in Figure 2B, hippocampal neurons washed with physiological 37°C buffer solution exhibited 340/380 ratios of 0.49 ± 0.03 compared to control ratios of 0.26 ± 0.02, indicating that [Ca2+]i was elevated after in vitro SE under physiological temperature. In contrast, hypothermia treated hippocampal neurons (31°C pBRS) quickly recovered the low Mg2+ SE induced elevated [Ca2+]i to baseline levels and exhibited 340/380 ratios of 0.25 ± 0.01 which were significantly lower than SE-only ratios but not significantly different from 340/380 ratios in control neurons (one-way ANOVA, p < 0.05).
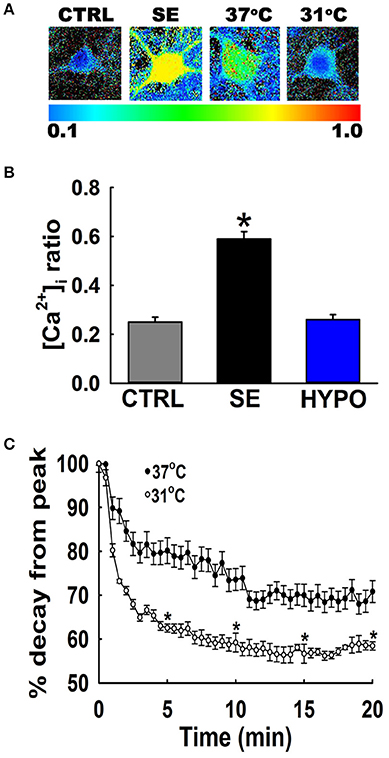
Figure 2. Hypothermia blocked the Ca2+ plateau after in vitro SE. (A) Pseudocolor ratiometric images of representative hippocampal neurons in culture from control, low Mg2+ SE, 37°C pBRS, and 31°C pBRS. All the ratiometric images are at the 20-min time point following respective treatments. (B) Fura-2 340/380 ratiometric values at the end of 20-min of treatment with hypothermic solution (31°C pBRS). (C) Ca2+ decay curves in the presence and absence of hypothermic intervention. Following 3 h of in vitro SE (time = 0), cells were washed with either 31°C pBRS or 37°C pBRS. 340/380 ratios were recorded every 30 s for 20 min and normalized to percent of the peak ratio observed at time = 0. n = 6 plates per treatment group with 40–60 neurons imaged per group. *p < 0.05, Student's t-test for all time points after 5 min.
To evaluate the dynamics of this [Ca2+]i decay, at the end of 3 h of low Mg2+ treatment, [Ca2+]i of neurons washed with either 31 or 37°C pBRS was measured over the course of 20 min. The ratio values for 31°C (hypothermia) and 37°C (normothermia) wash groups were normalized to the peak ratio (Figure 2C). After 5 min post-treatment, neurons washed with 37°C pBRS showed a slight decay in 340/380 ratios. However, cells exposed to hypothermic treatment exhibited a larger decrease in 340/380 ratios that were 62% of the peak observed at the end of in vitro SE. At 10 min post-treatment, the 340/380 ratio values reduced by 41 and 26% of the post-SE peak in cells washed with 31 and 37°C pBRS, respectively. At 15 min post-treatment, the ratio values fell by 44% of the post-SE peak for cells washed with 31°C pBRS and 30% of the peak for cells washed with 37°C solution. After 20 min, in the cells treated with hypothermia ratio values fell by 42% of the peak. These reductions were significantly higher than the cells washed with 37°C solution whose 340/380 ratio values fell by only 29% of the peak. Within 20 min of hypothermia treatment, [Ca2+]i had returned to baseline ratio values of 0.25 ± 0.01 which were not significantly different from values observed in control neurons (0.26 ± 0.02). In comparison, [Ca2+]i remained significantly elevated in cells washed with 37°C pBRS with values of 0.36 ± 0.02 (Figure 2B). There was a significant difference in the 340/380 ratio values between the two groups at each time point after 5 min of treatment (Student's t-test, p < 0.05).
Hypothermia Reduces Mortality and Improves Recovery From SE
PILO administration induced SE in 14 out of 15 rats, with an average seizure onset latency of 17 ± 3 min. Seizure severity as indicated by Racine score was 4.0 ± 0.2. There were no deaths during the first hour of SE, at which point diazepam regime was initiated to terminate the seizures. At 24 h post-SE, 3 out of 14 rats died giving a mortality rate of 21%. The surviving rats displayed hunched posture, were less ambulatory, and were not responsive to handling or approach. Their average gross physical recovery score was 0.72 ± 0.2 (see section Materials and Methods).
In separate group of 15-rats PILO was used to induce SE using established procedures. Moderate hypothermia (31–33°C) was induced in 14-rats 30 min after PILO SE and maintained for 4 h (Figure 3A. Also see materials and methods). No differences were observed in the severity of seizures during the remainder of 30 min of SE. However, mortality rate assessed at 24 h post-SE was only 7% in hypothermia treated group. Surviving rats in the hypothermia group (n = 13) were alert, mobile, displayed normal posture, and were responsive to handing and approach. Fifty-percent of the hypothermia-SE survivors exhibited a complete behavioral recovery score of 3.0. Their mean gross recovery score was 2.53 ± 0.3, which was significantly higher than PILO-SE group (n = 13, t-test, p < 0.05).
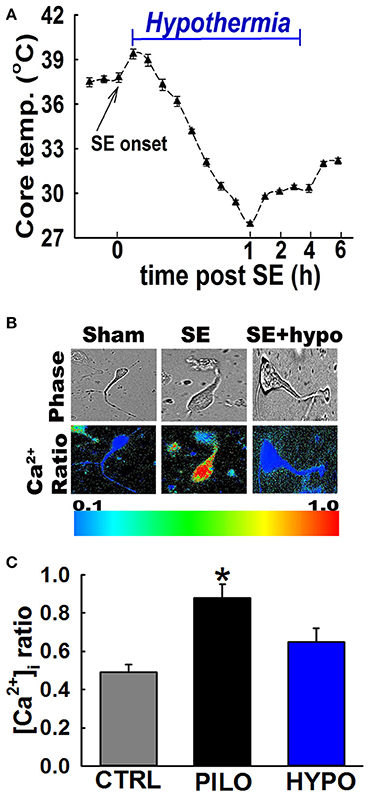
Figure 3. Hypothermia blocked the Ca2+ plateau after in vivo SE. (A) Hypothermia in rats was induced using surface cooling methods. Core body temperatures at various time points depicts rapid induction and steady maintenance of moderate hypothermia in the therapeutic range. (B) Pseudocolor ratiometric images of representative acutely isolated hippocampal neurons from control, PILO and hypothermia (HYPO) treated SE rats. Control and HYPO neurons had bluish color that corresponds to lower Fura-2 ratio while SE neurons had orange-red color that corresponds to higher Fura-2 ratio. (C) Elevated [Ca2+]i in hippocampal neurons acutely isolated from animals at 24 h following 1 h of PILO induced SE compared to neurons from control and hypothermia (HYPO) treated rats. (*p < 0.001, t-test, n = 6 and 5 animals respectively). Data are represented as mean ± SEM.
Hypothermia Blocked the Development of the Ca2+ Plateau Following SE
In order to investigate whether hypothermia was able to lower hippocampal neuronal [Ca2+]i, in vivo, moderate hypothermia (31–33°C) was induced in rats 30 min after PILO SE and maintained for 4 h (Figure 3A. Also see materials and methods). Hippocampal neurons were acutely isolated 24 h after SE to evaluate [Ca2+]i using established procedures.
Figure 3B depicts ratiometric pseudocolor images of CA1 hippocampal neurons acutely isolated from rats treated with and without therapeutic hypothermia. As illustrated in Figure 3C, hippocampal neurons isolated 24 h after PILO-induced SE exhibited significant elevations in [Ca2+]i compared to naïve controls. The average Fura-2 ratio values increased from 0.49 ± 0.04 in controls neurons to 0.88 ± 0.07 in neurons isolated from PILO-SE rats (n = 8 rats, t-test, p < 0.05). Thus, these results confirm our previous findings that PILO-SE results causes significant elevations in [Ca2+]i 24 h after SE. In contrast, neurons isolated from rats treated with 4 h moderate hypothermia exhibit an average ratio value of 0.65 ± 0.07, which was significantly lower than ratios obtained from PILO-SE only rats (n = 12 rats, one-way ANOVA, p < 0.05).
Hypothermia Reduces Neuronal Injury Following SE
To assess neuronal injury in rats surviving SE, brain sections from SE animals treated with and without hypothermia were labeled with Fluoro-Jade C (FJC), which is an early marker of neurodegeneration (12, 28, 34). We focused on dentate-gyrus and CA1 region of the hippocampus since neurons in these regions are sensitive to SE-induced neuronal injury. Across brain regions examined, there was negligible FJC labeling in sections obtained from control rats. In contrast, after PILO-SE, within the hippocampus, FJC-positive staining was observed in the polymorphic layer and along the hilus/granule cell border of the dentate gyrus and CA1 region. An average of 8.4 ± 2.4 and 10.6 ± 3.2 FJC positive cells/ 100 μM2 were quantified in dentate gyrus and CA1 region respectively. Brain sections from hypothermia-treated SE rats exhibited minimal FJC staining within these regions. The quantification of the neuronal injury expressed as FJC positive cells revealed approximately a 60% reduction in FJC labeling in the dentate gyrus and CA1 region of hypothermia-SE slices with an average of 3.0 ± 1.2 and 4.4 ± 0.8 FJC positive cells/ 100 μM2 respectively (Figures 4A,B).
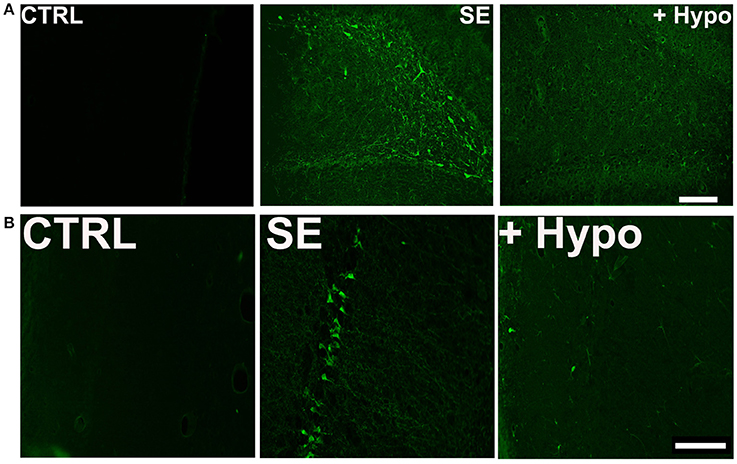
Figure 4. Neuroprotective effect of hypothermia. Representative photomicrographs of Fluoro-Jade C (FJC) staining in the (A) dentate gyrus hilus and (B) CA1 region of a control (CTRL), pilocarpine-SE (SE), and hypothermia (+HYPO) treated rats. Scale bars, 100 μm.
Discussion
The results of this study demonstrate that hypothermia administered following SE not only reduces mortality but improves the recovery from SE. Hypothermia was effective at blocking the development of the Ca2+ plateau, and also reduced the hippocampal neuronal injury following SE.
In both the in vivo and in vitro models of SE, [Ca2+]i remains elevated following injury, and this persistent elevation in [Ca2+]i is believed to contribute to the pathological consequences associated with epileptogenesis. Blocking the development of the plateau has been shown to prevent the development of epilepsy in both in vivo and in vitro models of SE-induced AE (5, 8, 13, 27). The novel finding that hypothermia not only reduces mortality but also reduces elevated [Ca2+]i and prevent the formation of the Ca2+ plateau offers additional information about the neuroprotective mechanisms of hypothermia as well as evidence that it could serve as an alternative intervention to pharmacological agents in preventing epileptogenesis after SE. This study has enormous translational potential for clinical application since hypothermia is already used in hospitals and ambulances settings to provide neuroprotection following cardiac arrest and hypoxia (18, 19, 23, 35, 36).
It has been demonstrated in the PILO model of SE-induced AE that SE causes a significant rise in [Ca2+]i and these elevations persist in animals that eventually develop epilepsy (8, 37), which suggests that the Ca2+ plateau is involved the progression of epileptogenesis to the epileptic phenotype (5, 13). Ca2+ is a ubiquitous second messenger involved in a variety of cellular processes including neurotransmitter release and protein transcription, as well as long-term potentiation (14, 16, 38). Excessive elevations in [Ca2+]i activate several downstream effectors leading to plasticity changes and eventually activation neuronal death pathways (38–41). Therefore, modulating the rise in [Ca2+]i may be effective in blocking the Ca2+-mediated cascade that leads to plasticity changes associated with epileptogenesis, thus preventing epilepsy.
Blocking the formation of the Ca2+ plateau with the use of NMDA receptor antagonists prior to and during SE prevents the development of AE (8, 11, 27), further suggesting that elevations in [Ca2+]i are involved in the pathophysiology associated with epileptogenesis. However, NMDA antagonists are ineffective at preventing AE if administered after the injury (8, 11, 27). Currently, there are no anti-epileptogenic drugs that can be administered following a neurological insult (42). Thus, it is important to develop a therapy that can block epileptogenesis when administered post-SE injury (6).
There are several postulated mechanisms for neuroprotective actions of hypothermic interventions (24, 25). Hypothermia is known to reduce cerebral metabolism and improve post-ischemic glucose utilization (43). Studies have reported beneficial effects of hypothermia on cellular redox regulation. For example, under ischemia/reperfusion conditions, therapeutic hypothermia suppresses enhanced oxidative stress and enhances anti-oxidative potency (44). Hypothermia is also reported to inhibit several aspects of apoptosis including increased production of anti-apoptotic protein Bcl2 (45) and inhibition of cytochrome C (46). In addition, hypothermia has been shown to produce an anti-inflammatory effect in the brain. Amongst other mechanisms, hypothermia suppresses microglial activation (47) and inhibits inflammatory transcription factor nuclear factor kappa B (48). Hypothermia also reduces excitotoxic insult by attenuating glutamate release following ischemia (49). NMDA receptor-mediated Ca2+ entry has been shown to play a major role in excitotoxic neuronal injury and cell death (11, 12, 50). We have shown that hypothermia reduces Ca2+ entry through NMDA receptors (26), and this could be an important mechanism for the strong neuroprotective effects observed after SE following hypothermia. Thus, the mechanisms underlying the neuroprotective effects of hypothermia are multifactorial. Although they have different inciting injuries, stroke, TBI, and SE share the common pathology of elevated [Ca2+]i, and subsequent neuronal loss (5). Therefore, it stands to reason that hypothermia would provide similar neuroprotection after SE as it does with stroke and TBI. Indeed, hypothermia has been shown to reduce neurological injuries associated with out-of-hospital cardiac arrests (17–19), stroke (22, 23), and TBI (20, 21).
Hypothermia could be a promising therapeutic alternative for modifying epileptogenesis and AE outcomes after SE. It has been shown to have anticonvulsant properties and has been used to treat refractory SE (51, 52). The mechanism by which hypothermia prevents the persistent rise in [Ca2+]i is most likely mediated by its ability to modulate Ca2+ influx into the cell via NMDA and ryanodine receptors. However, NMDA receptor activation is not responsible for Ca2+ influx post-SE. At this point, the influx of Ca2+ following SE recruits ryanodine receptors, resulting in enhanced Ca2+-induced Ca2+ release. Ryanodine receptor activation is believed to be involved in maintaining the Ca2+ plateau in SE (12, 27, 28). Interestingly, hypothermia is also effective at reducing Ca2+ entry via ryanodine receptors (26), thus preventing the formation of the Ca2+ plateau. Indeed, as shown in this study, hypothermia administered 30 min post-SE blocked the protracted hippocampal Ca2+ elevations, and extended significant neuroprotection in hippocampus following SE.
The results of this study have important clinical implications. The neuroprotective mechanisms of hypothermia are complex and not completely understood. These findings offer additional insight into how hypothermia protects against neurological injury. In addition, the results suggest that hypothermia may prevent epileptogenesis by blocking the formation of the Ca2+ plateau. Further research to investigate the long-term effects of hypothermia following SE are necessary to evaluate the therapeutic potential of hypothermia as an anti-epileptogenic intervention.
Author Contributions
RD conceptualized the study. KP and LD designed and conducted the experiments. KP, LD, and RD analyzed the data and drafted the manuscript the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the National Institute of Neurological Disorders and Stroke (NINDS), Grant Number R56NS092494-01 to RD.
References
1. Hauser WA, Hesdorffer DC. Epilepsy: Frequency, Causes and Consequences. New York, NY: Demos (1990).
2. DeLorenzo RJ, Hauser WA, Towne AR, Boggs JG, Pellock JM, Penberthy L, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology (1996) 46:1029–35. doi: 10.1212/WNL.46.4.1029
3. Hesdorffer DC, Logroscino G, Cascino G, Annegers JF, Hauser WA. Risk of unprovoked seizure after acute symptomatic seizure: effect of status epilepticus. Ann Neurol. (1998) 44:908–12. doi: 10.1002/ana.410440609
4. Thurman DJ, Beghi E, Begley CE, Berg AT, Buchhalter JR, Ding D, et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia (2011) 52(Suppl. 7):2–26. doi: 10.1111/j.1528-1167.2011.03121.x
5. DeLorenzo RJ, Sun DA, Deshpande LS. Cellular mechanisms underlying acquired epilepsy: the calcium hypothesis of the induction and maintainance of epilepsy. Pharmacol Ther. (2005) 105:229–66. doi: 10.1016/j.pharmthera.2004.10.004
6. Pitkanen A, Lukasiuk K, Dudek FE, Staley KJ. Epileptogenesis. Cold Spring Harb Perspect Med. (2015) 5:a022822. doi: 10.1101/cshperspect.a022822
7. McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE (2006) 2006:re12. doi: 10.1126/stke.3562006re12
8. Raza M, Blair RE, Sombati S, Carter DS, Deshpande LS, DeLorenzo RJ. Evidence that injury-induced changes in hippocampal neuronal calcium dynamics during epileptogenesis cause acquired epilepsy. Proc Natl Acad Sci USA. (2004) 101:17522–7. doi: 10.1073/pnas.0408155101
9. DeLorenzo RJ, Sun DA, Blair RE, Sombati S. An in vitro model of stroke-induced epilepsy: elucidation of the roles of glutamate and calcium in the induction and maintenance of stroke-induced epileptogenesis. Int Rev Neurobiol. (2007) 81:59–84. doi: 10.1016/S0074-7742(06)81005-6
10. Sun DA, Deshpande LS, Sombati S, Baranova A, Wilson MS, Hamm RJ, et al. Traumatic brain injury causes a long-lasting calcium-plateau of elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons surviving the brain injury. Eur J Neurosci. (2008) 27:1659–72. doi: 10.1111/j.1460-9568.2008.06156.x
11. Deshpande LS, Carter DS, Blair RE, DeLorenzo RJ. Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol Sci. (2010) 116:623–31. doi: 10.1093/toxsci/kfq157
12. Deshpande LS, Carter DS, Phillips KF, Blair RE, DeLorenzo RJ. Development of status epilepticus, sustained calcium elevations and neuronal injury in a rat survival model of lethal paraoxon intoxication. Neurotoxicology (2014) 44C:17–26. doi: 10.1016/j.neuro.2014.04.006
13. Nagarkatti N, Deshpande LS, DeLorenzo RJ. Development of the calcium plateau following status epilepticus: role of calcium in epileptogenesis. Expert Rev Neurother. (2009) 9:813–24. doi: 10.1586/ern.09.21
14. Berridge MJ. Neuronal calcium signaling. Neuron (1998) 21:13–26. doi: 10.1016/S0896-6273(00)80510-3
15. Deshpande LS, Blair RE, Phillips KF, DeLorenzo RJ. Role of the calcium plateau in neuronal injury and behavioral morbidities following organophosphate intoxication. Ann NY Acad Sci. (2016) 1374:176–83. doi: 10.1111/nyas.13122
16. Bengtson CP, Bading H. Nuclear calcium signaling. Adv Exp Med Biol. (2012) 970:377–405. doi: 10.1007/978-3-7091-0932-8_17
17. Scholefield B, Duncan H, Davies P, Gao Smith F, Khan K, Perkins GD, et al. Hypothermia for neuroprotection in children after cardiopulmonary arrest. Cochrane Database Syst Rev. (2013) 2:CD009442. doi: 10.1002/14651858.CD009442.pub2
18. Kim HJ, Kim GW, Oh SH, Park SH, Choi JH, Kim KH, et al. Therapeutic hypothermia after cardiac arrest caused by self-inflicted intoxication: a multicenter retrospective cohort study. Am J Emerg Med. (2014) 32:1378–81. doi: 10.1016/j.ajem.2014.08.045
19. Lascarrou JB, Meziani F, Le Gouge A, Boulain T, Bousser J, Belliard G, et al. Therapeutic hypothermia after nonshockable cardiac arrest: the HYPERION multicenter, randomized, controlled, assessor-blinded, superiority trial. Scand J Trauma Resusc Emerg Med. (2015) 23:26. doi: 10.1186/s13049-015-0103-5
20. McIntyre LA, Fergusson DA, Hebert PC, Moher D, Hutchison JS. Prolonged therapeutic hypothermia after traumatic brain injury in adults: a systematic review. JAMA (2003) 289:2992–9. doi: 10.1001/jama.289.22.2992
21. Madden LK, DeVon HA. A systematic review of the effects of body temperature on outcome after adult traumatic brain injury. J Neurosci Nurs. (2015) 47:190–203. doi: 10.1097/JNN.0000000000000142
22. Kuramatsu JB, Kollmar R, Gerner ST, Madzar D, Pisarcikova A, Staykov D, et al. Is hypothermia helpful in severe subarachnoid hemorrhage? An exploratory study on macro vascular spasm, delayed cerebral infarction and functional outcome after prolonged hypothermia. Cerebrovasc Dis. (2015) 40:228–35. doi: 10.1159/000439178
23. Yao Z, You C, He M. Effect and feasibility of therapeutic hypothermia in patients with hemorrhagic stroke: a systematic review and meta-analysis. World Neurosurg. (2018) 111:404–412.e2. doi: 10.1016/j.wneu.2018.01.020
24. Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. (2009) 37:S186–202. doi: 10.1097/CCM.0b013e3181aa5241
25. Karnatovskaia LV, Wartenberg KE, Freeman WD. Therapeutic hypothermia for neuroprotection: history, mechanisms, risks, and clinical applications. Neurohospitalist (2014) 4:153–63. doi: 10.1177/1941874413519802
26. Phillips KF, Deshpande LS, DeLorenzo RJ. Hypothermia reduces calcium entry via the N-methyl-D-aspartate and ryanodine receptors in cultured hippocampal neurons. Eur J Pharmacol (2013) 698:186–92. doi: 10.1016/j.ejphar.2012.10.010
27. Nagarkatti N, Deshpande LS, Carter DS, DeLorenzo RJ. Dantrolene inhibits the calcium plateau and prevents the development of spontaneous recurrent epileptiform discharges following in vitro status epilepticus. Eur J Neurosci. (2010) 32:80–8. doi: 10.1111/j.1460-9568.2010.07262.x
28. Deshpande LS, Blair RE, Huang BA, Phillips KF, DeLorenzo RJ. Pharmacological blockade of the calcium plateau provides neuroprotection following organophosphate paraoxon induced status epilepticus in rats. Neurotoxicol Teratol. (2016) 56:81–6. doi: 10.1016/j.ntt.2016.05.002
29. Falenski KW, Carter DS, Harrison AJ, Martin BR, Blair RE, DeLorenzo RJ. Temporal characterization of changes in hippocampal cannabinoid CB(1) receptor expression following pilocarpine-induced status epilepticus. Brain Res. (2009) 1262:64–72. doi: 10.1016/j.brainres.2009.01.036
30. Deshpande LS, Nagarkatti N, Ziobro JM, Sombati S, Delorenzo RJ. Carisbamate prevents the development and expression of spontaneous recurrent epileptiform discharges and is neuroprotective in cultured hippocampal neurons. Epilepsia (2008) 49:1795–802. doi: 10.1111/j.1528-1167.2008.01667.x
31. Nagarkatti N, Deshpande LS, DeLorenzo RJ. Levetiracetam inhibits both ryanodine and IP3 receptor activated calcium induced calcium release in hippocampal neurons in culture. Neurosci Lett. (2008) 436:289–93. doi: 10.1016/j.neulet.2008.02.076
32. Deshpande LS, Blair RE, Ziobro JM, Sombati S, Martin BR, DeLorenzo RJ. Endocannabinoids block status epilepticus in cultured hippocampal neurons. Eur J Pharmacol. (2007) 558:52–59. doi: 10.1016/j.ejphar.2006.11.030
33. Wallace MJ, Blair RE, Falenski KW, Martin BR, DeLorenzo RJ. The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J Pharmacol Exp Ther. (2003) 307:129–37. doi: 10.1124/jpet.103.051920
34. Phillips KF, Deshpande LS. Repeated low-dose organophosphate DFP exposure leads to the development of depression and cognitive impairment in a rat model of Gulf War Illness. Neurotoxicology (2016) 52:127–33. doi: 10.1016/j.neuro.2015.11.014
35. Scholefield BR, Lyttle MD, Berry K, Duncan HP, Morris KP. Survey of the use of therapeutic hypothermia after cardiac arrest in UK paediatric emergency departments. Emerg Med J. (2013) 30:24–7. doi: 10.1136/emermed-2011-200348
36. Sherman AL, Wang MY. Hypothermia as a clinical neuroprotectant. Phys Med Rehabil Clin N Am. (2014) 25:519–29. doi: 10.1016/j.pmr.2014.04.003
37. Raza M, Pal S, Rafiq A, DeLorenzo RJ. Long-term alteration of calcium homeostatic mechanisms in the pilocarpine model of temporal lobe epilepsy. Brain Res. (2001) 903:1–12. doi: 10.1016/S0006-8993(01)02127-8
38. Baker KD, Edwards TM, Rickard NS. The role of intracellular calcium stores in synaptic plasticity and memory consolidation. Neurosci Biobehav Rev. (2013) 37:1211–39. doi: 10.1016/j.neubiorev.2013.04.011
39. Adasme T, Haeger P, Paula-Lima AC, Espinoza I, Casas-Alarcon MM, Carrasco MA, et al. Involvement of ryanodine receptors in neurotrophin-induced hippocampal synaptic plasticity and spatial memory formation. Proc Natl Acad Sci USA. (2011) 108:3029–34. doi: 10.1073/pnas.1013580108
40. Bodalia A, Li H, Jackson MF. Loss of endoplasmic reticulum Ca2+ homeostasis: contribution to neuronal cell death during cerebral ischemia. Acta Pharmacol Sin. (2013) 34:49–59. doi: 10.1038/aps.2012.139
41. Chadwick W, Mitchell N, Martin B, Maudsley S. Therapeutic targeting of the endoplasmic reticulum in Alzheimer's disease. Curr Alzheimer Res. (2013) 9:110–9. doi: 10.2174/156720512799015055
42. Pitkanen A, Nehlig A, Brooks-Kayal AR, Dudek FE, Friedman D, Galanopoulou AS, et al. Issues related to development of antiepileptogenic therapies. Epilepsia (2013) 54(Suppl. 4):35–43. doi: 10.1111/epi.12297
43. Lanier WL. Cerebral metabolic rate and hypothermia: their relationship with ischemic neurologic injury. J Neurosurg Anesthesiol. (1995) 7:216–21. doi: 10.1097/00008506-199507000-00021
44. Nosaka N, Okada A, Tsukahara H. Effects of therapeutic hypothermia for neuroprotection from the viewpoint of redox regulation. Acta Med Okayama (2017) 71:1–9. doi: 10.18926/AMO/54819
45. Zhang Z, Sobel RA, Cheng D, Steinberg GK, Yenari MA. Mild hypothermia increases Bcl-2 protein expression following global cerebral ischemia. Brain Res Mol Brain Res. (2001) 95:75–85. doi: 10.1016/S0169-328X(01)00247-9
46. Yenari MA, Iwayama S, Cheng D, Sun GH, Fujimura M, Morita-Fujimura Y, et al. Mild hypothermia attenuates cytochrome c release but does not alter Bcl-2 expression or caspase activation after experimental stroke. J Cereb Blood Flow Metab. (2002) 22:29–38. doi: 10.1097/00004647-200201000-00004
47. Zheng Z, Yenari MA. Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res. (2004) 26:884–92. doi: 10.1179/016164104X2357
48. Han HS, Karabiyikoglu M, Kelly S, Sobel RA, Yenari MA. Mild hypothermia inhibits nuclear factor-kappaB translocation in experimental stroke. J Cereb Blood Flow Metab. (2003) 24:589–98. doi: 10.1097/01.WCB.0000059566.39780.8D
49. Busto R, Globus MY, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Effect of mild hypothermia on ischemia-induced release of neurotransmitters and free fatty acids in rat brain. Stroke (1989) 20:904–10.
50. de Araujo Furtado M, Lumley LA, Robison C, Tong LC, Lichtenstein S, Yourick DL. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague-Dawley rats. Epilepsia (2010) 51:1503–10. doi: 10.1111/j.1528-1167.2009.02478.x
51. Bennett AE, Hoesch RE, DeWitt LD, Afra P, Ansari SA. Therapeutic hypothermia for status epilepticus: a report, historical perspective, and review. Clin Neurol Neurosurg. (2014) 126:103–9. doi: 10.1016/j.clineuro.2014.08.032
Keywords: status epilepticus, pilocarpine, hypothermia, intra neuronal calcium levels, mortality, neuroprotection, Sprague-Dawley rats
Citation: Phillips KF, Deshpande LS and DeLorenzo RJ (2018) Hypothermia Reduces Mortality, Prevents the Calcium Plateau, and Is Neuroprotective Following Status Epilepticus in Rats. Front. Neurol. 9:438. doi: 10.3389/fneur.2018.00438
Received: 01 March 2018; Accepted: 24 May 2018;
Published: 11 June 2018.
Edited by:
Batool F. Kirmani, Epilepsy Center, Baylor Scott and White Health Neuroscience Institute, United StatesReviewed by:
Ashok K. Shetty, Institute for Regenerative Medicine, Texas A&M University College of Medicine, United StatesDinesh Upadhya, Manipal Academy of Higher Education, India
Olagide Wagner Castro, Federal University of Alagoas, Brazil
Copyright © 2018 Phillips, Deshpande and DeLorenzo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert J. DeLorenzo, cm9iZXJ0LmRlbG9yZW56b0B2Y3VoZWFsdGgub3Jn