 Bruno P. Meloni
Bruno P. Meloni Frank L. Mastaglia2,3
Frank L. Mastaglia2,3- 1Department of Neurosurgery, QEII Medical Centre, Sir Charles Gairdner Hospital, Nedlands, WA, Australia
- 2Perron Institute for Neurological and Translational Science, Nedlands, WA, Australia
- 3Centre for Neuromuscular and Neurological Disorders, The University of Western Australia, Nedlands, WA, Australia
There are virtually no clinically available neuroprotective drugs for the treatment of acute and chronic neurological disorders, hence there is an urgent need for the development of new neuroprotective molecules. Cationic arginine-rich peptides (CARPs) are an expanding and relatively novel class of compounds, which possess intrinsic neuroprotective properties. Intriguingly, CARPs possess a combination of biological properties unprecedented for a neuroprotective agent including the ability to traverse cell membranes and enter the CNS, antagonize calcium influx, target mitochondria, stabilize proteins, inhibit proteolytic enzymes, induce pro-survival signaling, scavenge toxic molecules, and reduce oxidative stress as well as, having a range of anti-inflammatory, analgesic, anti-microbial, and anti-cancer actions. CARPs have also been used as carrier molecules for the delivery of other putative neuroprotective agents across the blood-brain barrier and blood-spinal cord barrier. However, there is increasing evidence that the neuroprotective efficacy of many, if not all these other agents delivered using a cationic arginine-rich cell-penetrating peptide (CCPPs) carrier (e.g., TAT) may actually be mediated largely by the properties of the carrier molecule, with overall efficacy further enhanced according to the amino acid composition of the cargo peptide, in particular its arginine content. Therefore, in reviewing the neuroprotective mechanisms of action of CARPs we also consider studies using CCPPs fused to a putative neuroprotective peptide. We review the history of CARPs in neuroprotection and discuss in detail the intrinsic biological properties that may contribute to their cytoprotective effects and their usefulness as a broad-acting class of neuroprotective drugs.
Introduction
Despite the enormous global impact of neurological disorders and the extensive research over many decades, there is still a lack of proven clinically effective pharmacological neuroprotective therapies capable of reducing the severity of brain or spinal cord tissue injury in acute (e.g., stroke, traumatic brain injury and spinal cord injury, and hypoxic-ischemic encephalopathy) or chronic (Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis) neurological disorders. The few neuroprotective treatments that are available, such as riluzole for amyotrophic lateral sclerosis and memantine for Alzheimer's disease provide only modest benefits. While hypothermia is used as a neuroprotective therapy for neonatal encephalopathy and for comatose survivors of cardiac arrest, it is difficult to implement due to the need for specialized equipment and intensive patient monitoring, and its efficacy is also limited.
Hence, the development of effective neuroprotective drugs for the treatment of a variety of neurological disorders remains an urgent priority. To make matters worse, due to past clinical failures, some researchers, physicians, and pharmaceutical companies are reluctant to continue research focused on the development of neuroprotective agents. However, most impartial observers would agree that the benefits of continuing to pursue the discovery of neuroprotective therapies far outweigh the risks. With this in mind, it is also intuitive that in order to increase the chances of achieving translational success at the clinical level, it is preferable that any new neuroprotective drug should have a multimodal mechanism of action. To this end, cationic arginine-rich peptides (CARPs) represent a relatively novel and expanding class of compounds, which possess an array of intrinsic neuroprotective properties, and are thus ideal molecules for development as therapies for a broad range of neurological disorders.
General Aspects of CARPs
As the name suggests, critical factors for CARP neuroprotection are their positive charge and arginine content as well as, the ability to traverse membrane lipid bilayers. Whereas, cationic charge can be imparted by the presence of the positively charged amino acids arginine and lysine (Figure 1A), which have a net charge of +1 at pH 7, arginine is the amino acid essential for neuroprotection. Histidine, the other positively charged amino acid only provides a modest contribution to peptide charge with a net charge of +0.1 at pH 7. Furthermore, CARPs represent a broader class of bioactive peptides with a number of other properties that may contribute to their neuroprotective actions, including the ability to reduce intracellular calcium influx, antagonize cell surface receptor function, target mitochondria, scavenge reactive molecules, induce cell signaling, stabilize proteins, inhibit proteolytic enzymes, and reduce inflammation, and in addition to being neuroprotective also have anti-nociceptive, cardioprotective, anti-microbial and anti-cancer properties.
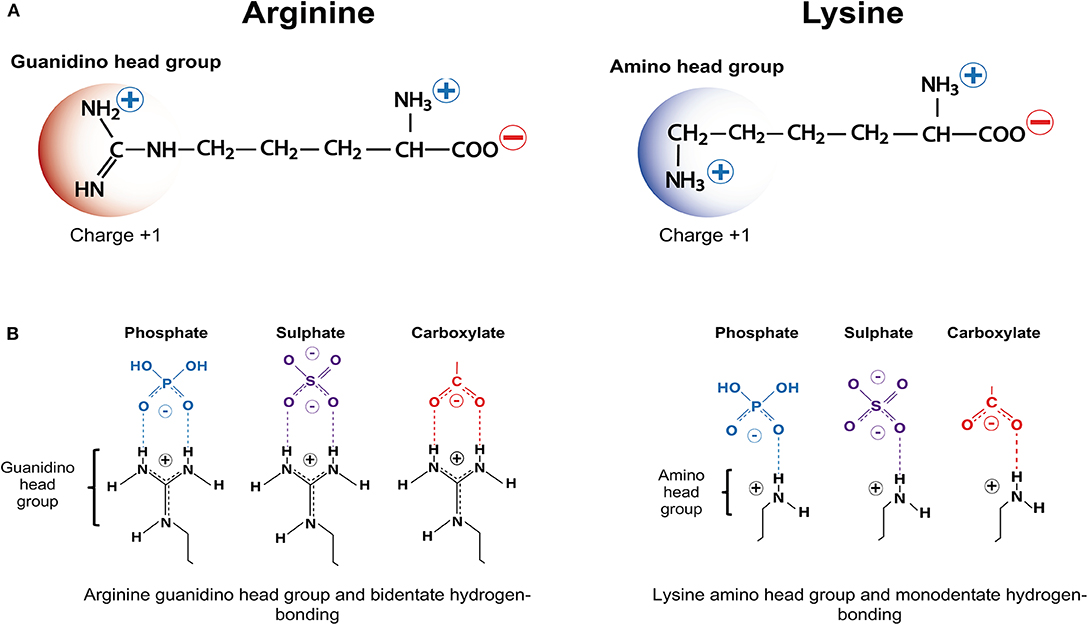
Figure 1. Positively charged amino acids arginine and lysine, and hydrogen bonding. (A) Arginine and lysine depicting positively charged guanindino head group and amino head group, respectively. (B) Arginine guanindino head groups and lysine amino head groups forming bidentate hydrogen-bonding and monodentate hydrogen-bonding, respectively, with phosphate, sulfate and carboxylate anionic moieties.
CARPs have demonstrated neuroprotection in in vitro neuronal injury models (e.g., excitotoxicity, oxygen-glucose deprivation), in in vivo models of acute central nervous system (CNS) injury (e.g., stroke, traumatic brain injury, perinatal hypoxia-ischemia, traumatic brain injury, spinal cord injury, and epilepsy) and in models of chronic neurodegenerative disorders (e.g., Parkinson's and Alzheimer's disease) and neuropathic pain (Tables 1–3). Furthermore, it is important to acknowledge that neuroprotective CARPs can be categorized into three main groups; (i) poly-arginine peptides, cationic arginine-rich cell-penetrating peptides (CCPPs) or peptides derived from proteins (Table 1); (ii) putative neuroprotective peptides fused to CCPPs (Table 2); and (iii) endogenous peptides (Table 3).
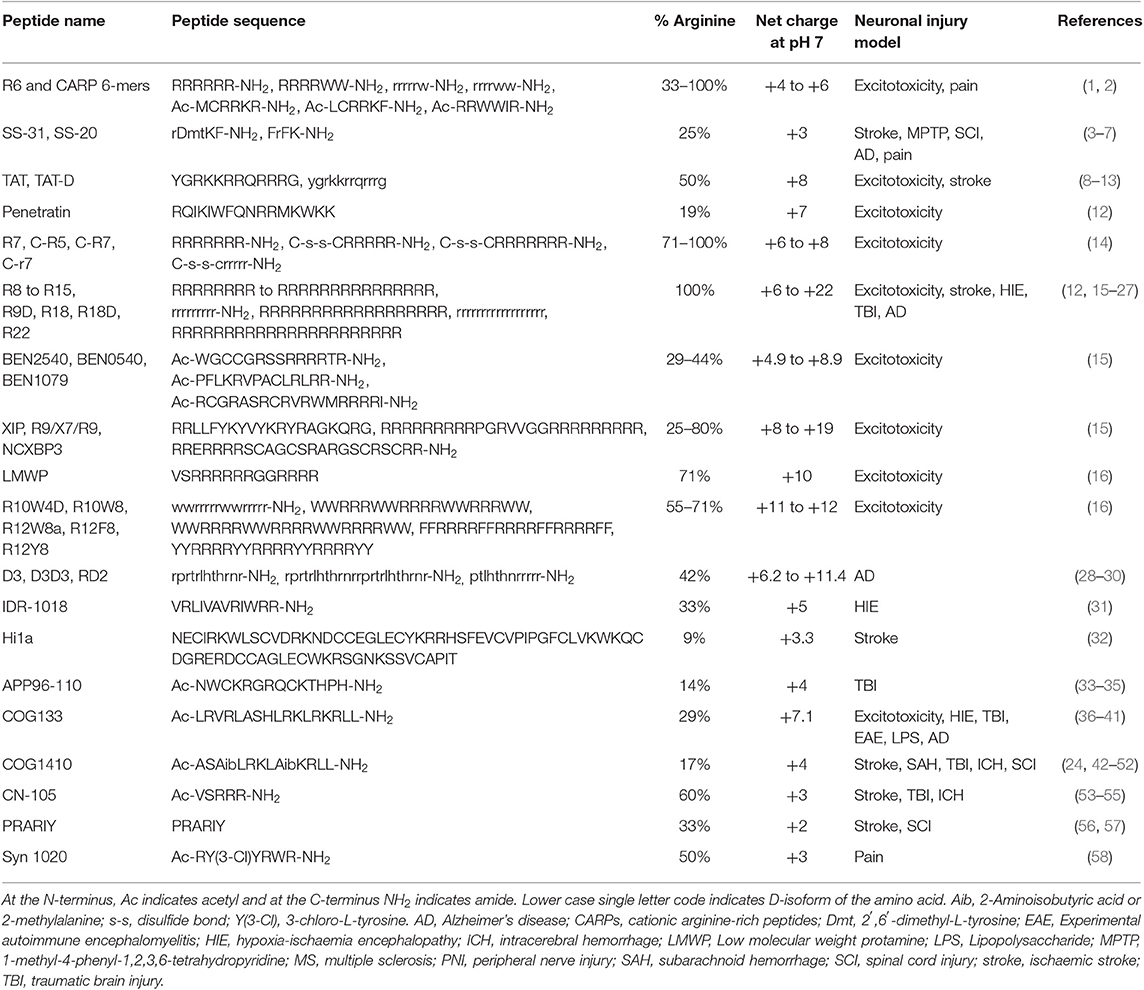
Table 1. CARPs with neuroprotective and other neuroactive properties.
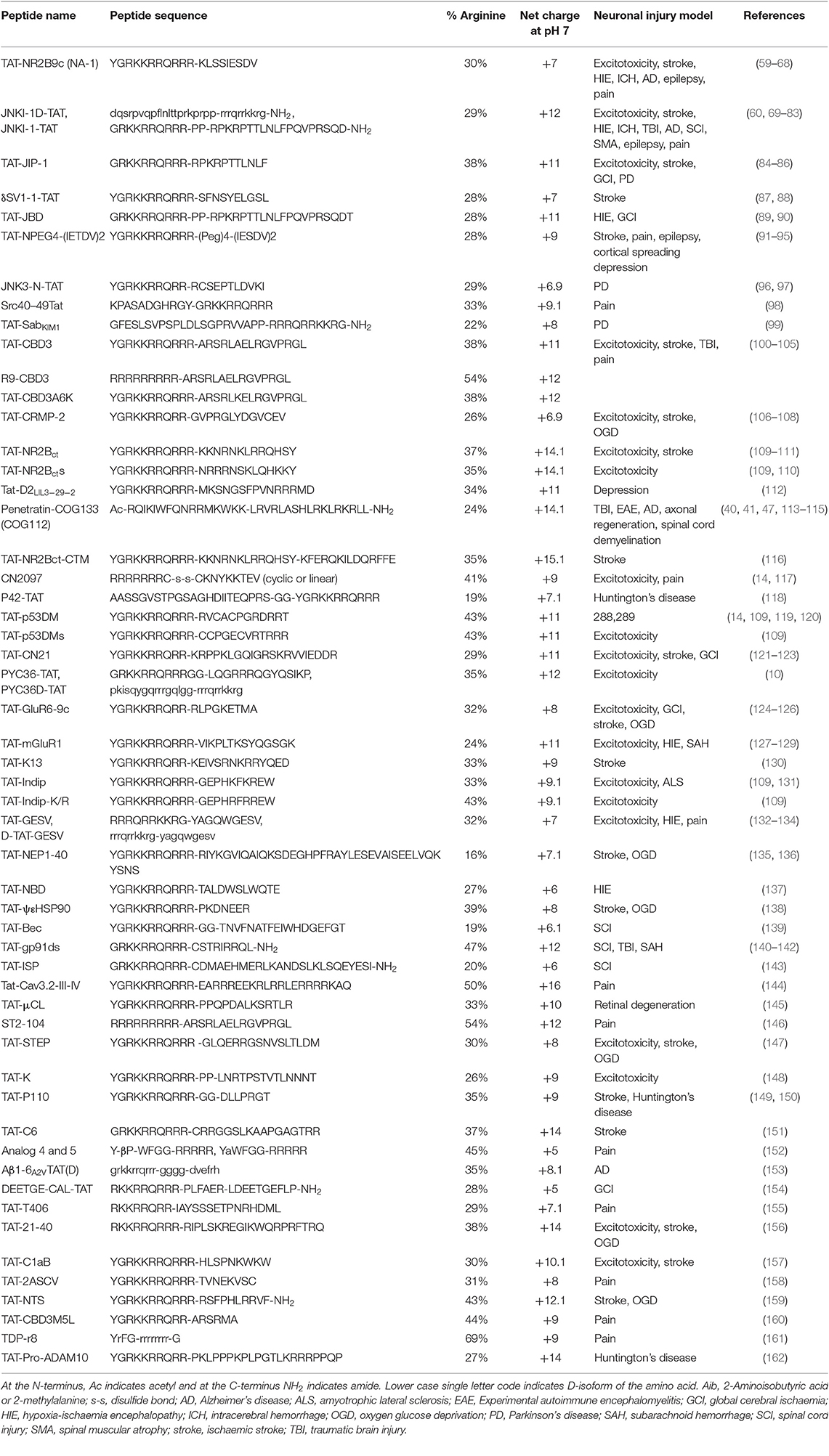
Table 2. Studies demonstrating neuroprotective and other neuroactive properties of peptides fused to TAT and other cell penetrating peptides.
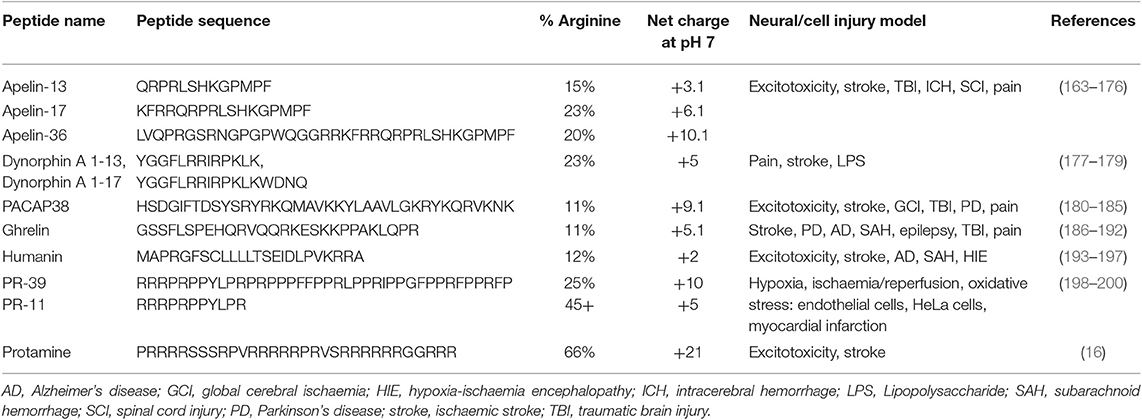
Table 3. Endogenous CARPs with neuroprotective and cytoprotective properties.
The aim of this review is to highlight the recognition of CARPs as a novel class of peptide with great promise for the treatment of acute and chronic neurological disorders, and in so doing summarize their known neuroprotective mechanisms of action, as well as other potential actions whereby they may exert beneficial effects in injured or affected cells. Within this group of compounds are included many putative neuroprotective peptides fused to CCPPs (e.g., TAT, R9, penetratin) that have been developed (Table 2). In this review, such peptides are also classified as CARPs, and we propose that in many, if not all instances their putative neuroprotective effects may actually be mediated by the arginine content and positive charge of the carrier and/or cargo peptide, rather than the cargo peptide itself.
Generic Features of Neuroprotective CARPs
In general terms, neuroprotective CARPs typically possess the following properties: (i) range in size from 4 to 40 amino acids; (ii) positive net charge ≥ +2 to +20; (iii) one or more positively charged arginine residues that comprise between 20 and 100% of the peptide; (iv) other positively charged amino acids namely lysine and histidine; (v) amphiphilicity due to the presence of both hydrophilic (e.g., arginine, lysine) and hydrophobic (e.g., tryptophan, phenylalanine, tyrosine) amino acids; and (vi) endocytic and/or non-endocytic cell membrane traversing properties, including the ability to cross the blood-brain and blood-spinal cord barriers (BBB/BSCB). Invariably, CARPs are commercially or chemically synthesized using solid-phase peptide synthesis. One exception is the CARP, protamine (Table 3), which is purified from salmon milt or generated recombinantly. Due to the capacity of CARPs to traverse cellular membranes and localize to different organs within the body, they have been the subject of several experimental and review articles examining their bioavailability (201–203) and therefore this subject is not covered in this review.
Historical Overview of CARPs and Neuroprotection Studies
Key historical events in the recognition and application of CARPs as neuroprotective agents are summarized in Figure 2. The first study to identify the neuroprotective properties of CARPs was in 1998 when Ferrer-Montiel et al. (1) screened a 6-mer peptide library containing over 49,000 different peptides for their ability to block glutamate-evoked ionic currents in Xenopus oocytes expressing the NR1 and NR2A NMDA receptor subunits. Hexapeptides containing at least two arginine (R) residues at any position as well as one or more lysine (K), tryptophan (W), and cysteine (C) residues displayed ionic current blocking activity. Further analysis revealed that C-carboxyl amidated (-NH2; note C-carboxyl amidation removes the negatively charged COO− C-terminus thereby increasing peptide net charge by +1) dipeptides RR-NH2 (net charge +3) and RW-NH2 (net charge +2) were also capable of blocking NMDA receptor activity. Similarly, certain amino acid residues within arginine-rich hexapeptides inhibited the NMDA receptor blocking ability of the peptide (e.g., RFMRNR-NH2; net charge +4, was ineffective; M, methionine; N, asparagine). In addition, increasing oligo-arginine peptide length from 2 to 6 resides (e.g., R2-NH2 vs. R3-NH2 vs. R6-NH2) increased blocking activity. In a NMDA excitotoxicity model (NMDA: 200 μM/20 min) using cultured hippocampal neurons, arginine-rich hexapeptides (Table 1), especially those also containing one or two tryptophan residues displayed high-levels of neuroprotection, and the neuroprotective action of the peptides was not stereo-selective with L- and D-isoform peptides showing similar efficacy. The ability of tryptophan to improve peptide neuroprotective efficacy is of particular interest as tryptophan residues also increase the uptake efficacy of CCPPs (204–208).
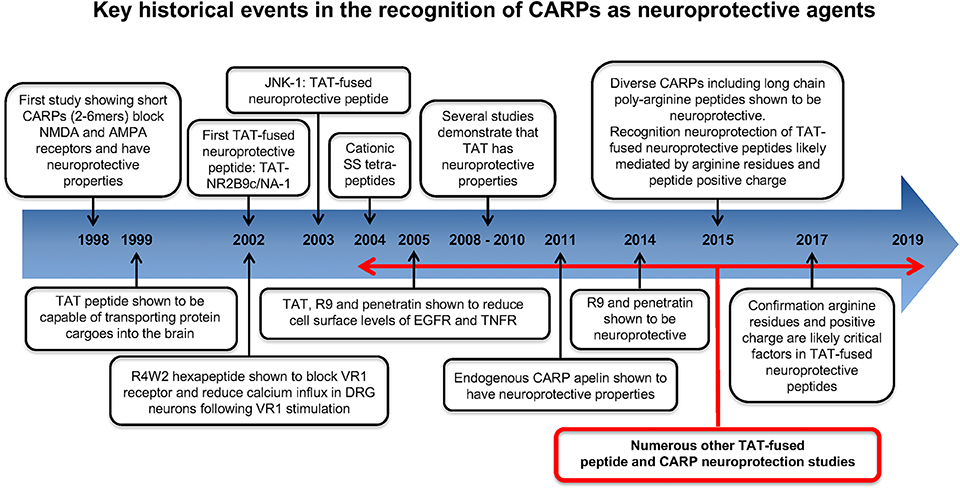
Figure 2. Historical time-line for the recognition of CARPs as neuroprotective agents.
Other observations concluded that: (i) whereas cationic arginine-rich hexapeptides were highly efficient at blocking NMDA receptor evoked ionic currents (80–100%), some peptides (e.g., RRRCWW-NH2 and RYYRRW-NH2) also blocked AMPA receptor currents by over 60%. In subsequent studies, the peptide RRRRWW-NH2 was demonstrated to antagonize the vanilloid receptor 1 (VR1; also known as the transient receptor potential cation channel subfamily V member 1; TRPV1) mediated currents in a Xenopus expression system and reduce calcium influx in rat dorsal root ganglion neurons following capsaicin or resiniferatoxin VR1 receptor stimulation (2, 209).
Neuroprotective Properties of Cationic Arginine-Rich Cell-Penetrating Peptides TAT, R9, and Penetratin
Shortly after the work of Ferrer-Montiel et al. (1), the CCPP TAT (see Table 1 for sequence and net charge) was demonstrated to have the capacity to transport large protein cargos across the BBB (210). Subsequently, the TAT peptide became increasingly utilized as a carrier molecule to deliver various cargos into the brain, including putative neuroprotective peptides and proteins (Figure 2). To date over fifty different TAT-fused neuroprotective peptides have been shown to have positive effects in different in vitro and/or animal CNS injury models (Table 2). However, not surprisingly in light of the Ferrer-Montiel et al. (1) findings, experiments in other laboratories demonstrated that the TAT peptide itself possesses modest neuroprotective actions in in vitro excitotoxicity and in vivo ischemic injury models (8–11). Subsequently, it was reported that the CCPPs, R9 (Table 1), and penetratin (Table 1) were 17- and 4.6-fold, respectively more neuroprotective than TAT in a severe cortical neuronal glutamic acid excitotoxicity cell death model (glutamic acid: 100 μM/5 min; Figure 3) (12). These findings also raised the first clues that the neuroprotective actions of putative neuroprotective peptides fused to CCPPs may in fact be mediated by the carrier peptide.
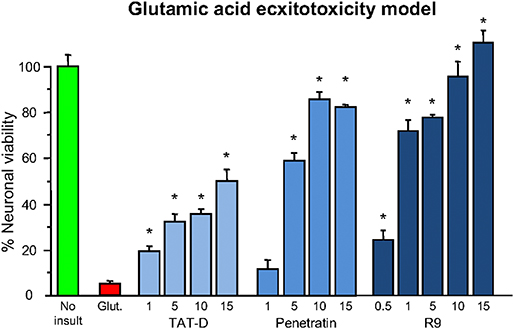
Figure 3. Neuroprotective efficacy of cationic arginine-rich cell-penetrating peptides in glutamic acid excitotoxicity model. Peptides present in neuronal cultures for 10 min before and during (half concentration) 5-min glutamic acid exposure. Neuronal viability measured 24 h following glutamic acid exposure. Concentration of peptide in μM. MTS assay data were expressed as percentage neuronal viability with no insult control taken as 100% viability and glutamic acid control (Glut.) taken as 5% (mean ± SE; n = 4; *P < 0.05). Adapted from Meloni et al. (12).
Further Validation and Characterization of CARPs as Neuroprotective Agents
Later, in vitro studies confirmed that other CARPs (e.g., protamine, LMWP, XIP; Tables 1, 3) and long-chain poly-arginine peptides (Table 1) were also highly neuroprotective, with efficacy increasing with increasing arginine content and peptide positive charge, plateauing at around 15–18 arginine residues for arginine polymers (15, 16). Furthermore, the requirement for arginine residues, rather than lysine residues, was demonstrated to be critical for neuroprotection, with the K10 peptide (10-mer of lysine; net charge +10) displaying limited efficacy in a neuronal glutamic acid excitotoxic model (15). In addition, the importance of peptide charge was confirmed by the finding that the glutamic acid containing neutrally charged R9/E9 peptide (RRRRRRRRREEEEEEEEE; net charge 0; E = glutamic acid) displayed no neuroprotection in the excitotoxic model (15).
Based on the above findings, it was hypothesized that CARP neuroprotection is largely mediated by the positively charged guanidinium head-group, which is unique to arginine (Figure 1A) (note: lysine possesses a positively charged amide group; Figure 1A) (15, 109). These findings also support the notion that peptide neuroprotective efficacy appears to be correlated with the same features that are critical for the endocytic and/or non-endocytic membrane traversing properties of CCPPs (14, 15, 109, 211). It was also demonstrated in an in vitro glutamic acid excitotoxicity model that the hydrophobic aromatic amino acids tryptophan, and to a lesser extent phenylalanine and tyrosine can significantly improve CARP neuroprotective efficacy. In contrast, alanine and glycine resides reduce peptide neuroprotective efficacy (15, 16). Importantly, tryptophan residues are also known to increase the cell-penetrating properties of CCPPs, providing further evidence that neuroprotection is closely linked to the peptide membrane traversing capacity of the peptides.
Studies have also demonstrated that a 10-min pre-treatment of neuronal cultures with CARPs induces a pre-conditioning neuroprotective response lasting up to 2–5 h post-treatment (15–17). Similar to the findings of Ferrer-Montiel et al. (1), it was also observed that there was no stereo-selectivity in terms of neuroprotective efficacy of L- and D-enantiomer CARPs, which suggests that with respect to neuroprotection, peptide electrostatic interactions are more important than peptide structural interactions of the peptide with specific biological targets. Importantly, CARPs have the capacity to significantly inhibit neuronal intracellular calcium influx in the glutamic acid excitotoxicity model (15–17, 109).
Consistent with in vitro findings, CARPs (e.g., R9D, R12, R18, R18D, protamine; Tables 1, 3) were also demonstrated to provide significant neuroprotection and improve functional outcomes in rat models of permanent and/or transient middle cerebral artery occlusion (MCAO), perinatal hypoxia-ischemia and traumatic brain injury (15, 16, 18–24, 212) and a non-human primate MCAO stroke model (26). Positive neuroprotective effects with R9D and R18D, which are the D-enantiomers of R9 and R18, also confirmed the lack of stereo-specificity for CARP efficacy in vivo.
In 2015, Marshall et al. (14) also confirmed the in vivo neuroprotective properties of CARPs including poly-arginine R7 (Table 1), as well as the TAT and TAT-NR2B9c (also known as NA-1; Table 2) peptides in rat retinal ganglion cells exposed to NMDA (20 nmol; 3 μL intravitreal injection). The study also demonstrated that CARPs containing a terminal cysteine residue improved neuroprotective efficacy; this could be due to the cysteine residue improving peptide stability and/or enhancing anti-oxidant properties. Marshall et al. (14) also considered that it was likely that the cell-penetrating properties of the CARPs along with the guanidinium head group of arginine and peptide positive charge were the “driving force” for neuroprotection. Furthermore, and as proposed by Meloni et al. (211), Marshall et al. (14) also suggested that cargo peptides designed to inhibit cell death following NMDA excitotoxicity (e.g., peptides CN2097; CKNYKKTEV and NR2B9c; KLSSIESDV) and fused to a CCPP (e.g., R7 for CN2097 and TAT for NR2B9c) were unlikely to be the active component mediating neuroprotection in the retinal ganglion cell NMDA excitotoxic injury model.
In 2017, McQueen et al. (110) re-evaluated the neuroprotective mechanism of action of the death-associated protein kinase 1 protein (DAPK1) blocking peptide TAT-NR2Bct (Table 2) and its scrambled control TAT-NR2Bcts (Table 2). DAPK1 is a calcium-calmodulin regulated protein activated in neurons following NMDA receptor over-stimulation as occurs in ischemia mediated excitotoxicity. TAT-NR2Bct was designed to competitively inhibit activated DAPK1 binding to the NR2B subunit protein, and thereby block subsequent downstream damaging cellular events caused by NMDA receptor over-activation. Interestingly, Meloni et al. (109) had earlier examined the TAT-NR2Bct and TAT-NR2Bcts peptides and demonstrated high neuroprotective efficacy for both peptides in the glutamic acid excitotoxicity model. Therefore, it was not surprising that McQueen et al. (110) also found that both TAT-NR2Bct and TAT-NR2Bcts, along with a randomly designed CARP (RRRTQNRRNRRTSRQNRRRSRRRR; net charge +15) were neuroprotective in a neuronal NMDA excitotoxicity model. On the basis of their findings they concluded that neuroprotection was dependent on peptide positive charge and independent of peptide sequence and DAPK1 signaling.
Taken together, the above studies provide irrefutable evidence of the neuroprotective properties of CARPs in various experimental situations and in doing so, raise two important issues in regard their application in neuroprotection: (i) what are the precise neuroprotective mechanisms operating; and (ii) the need to re-evaluate studies using CARPs and CCPPs for the delivery of neuroactive cargos into the CNS, particularly putative neuroprotective peptides. Both these topics are discussed below. Also, because it is likely that CARPs interact with negatively charged cell membrane structures, an interaction that appears to be critical for neuroprotection, the mechanisms associated with the affinity of CARPs to cell membranes will also be discussed. Interestingly, it is the interaction of CARPs with negatively charged bacterial and cancer cell cytoplasmic membrane structures that is considered to be one of the mechanisms responsible for their anti-bacterial and anti-cancer properties (213, 214).
Putative Neuroactive Peptides Fused to Cationic Arginine-Rich Cell-Penetrating Peptides and Neuroprotection
Given that CARPs possess intrinsic neuroprotective properties raises questions regarding the mode of action of other putative neuroprotective peptides when they are fused to a carrier CCPP (Table 2). As alluded to above, it is likely that the neuroprotection provided by such putative neuroprotective peptides fused to CCPPs, is mediated not by the actions of the cargo molecule per se, but by the carrier itself with potency being further enhanced by the amino acid content (e.g., arginine, lysine, cysteine, and tryptophan resides) and/or stability provided by the cargo peptide. In essence, a putative neuroprotective peptide fused to an arginine-rich cell-penetrating carrier peptide will possess the properties of a CARP; the only exception being if a negatively charged cargo peptide neutralizes the positive charge of the carrier peptide.
In 2015 we published a review article (109) highlighting the likelihood of the neuroprotective mechanism of action of putative neuroprotective peptide fused to cell-penetrating carrier peptides being mediated by the carrier molecule. Three of the most commonly used TAT-fused neuroprotective peptides TAT-NR2B9c, TAT-JNKI-1 and TAT-CBD3, as well as several other less characterized TAT-fused peptides (e.g., TAT-p53DM, TAT-s-p53DM, TAT-NR2Bct, TAT-NR2Bcts, Indip/IndipK-R) were analyzed based on theoretical grounds, and on our own and other previous experimental studies in relation to neuroprotective mechanism of action. Following this analysis, we provided several lines of evidence to support the view that TAT-fused neuroprotective peptides are behaving as neuroprotective CARPs, and not by the proposed intended mechanism of action of the cargo peptide. This evidence included: (1) the ability of the peptides to reduce intracellular calcium influx, even though this was never an intended mechanism of action of the cargo peptide; (2) despite targeting intracellular proteins, the peptides often reduced surface expression or interfered with plasma membrane ion channel receptors; (3) lack of efficacy and inability of the peptide to reduce neuronal calcium influx when introduced directly into the cell; (4) improved peptide efficacy when TAT was replaced with R9 (increasing peptide positive charge and arginine content) or replacing neutral or negatively charged amino acids with positively charged arginine or lysine; (5) decreased peptide efficacy when replacing amino acids with alanine, which is known to reduce membrane traversing properties of cell-penetrating peptides; (6) demonstrating neuroprotective properties of CCPP-fused scrambled cargo control peptides; and (7) due to endosomal entrapment and/or peptide degradation it is possible cargo peptides have a limited capacity to interact with their intended intracellular target. Importantly, the subsequent studies of Marshall et al. (14) and McQueen et al. (110) (described above) further validate the view that the mechanism of action of TAT-fused neuroprotective peptides is likely to be mediated by the carrier peptide, and by extension the arginine content and positive charge of the peptide.
In order to confirm the specific action of a neuroactive peptide cargo fused to a carrier CCPP, we recommend that the neuroprotective or other intended neuroactive actions of the peptide should be reassessed after the introduction of arginine substitutions into the cargo peptide. The introduction of arginine residues into the cargo peptide should abolish the proposed/intended neuroprotective action of the cargo peptide. However, if the action of the carrier-cargo peptide is maintained or enhanced it is likely that the neuroprotective action of the peptide was mediated by the cationic and arginine-rich properties of the peptide. Alternatively, the peptide could be synthesized in the same amino acid sequence (as opposed to retro-inversely) with D-isoform amino acids, which would drastically alter the peptide's steric structure and binding specificity/affinity to its intended target, whereas its electro-physiochemical properties would be similar. Finally, the CCPP carrier molecule could be replaced with a non-arginine containing cell-penetrating peptide (e.g., TP10 or MAP).
CARPs and Their Interaction With Cellular Membranes
CARPs have the capacity to form electrostatic interactions with anionic phosphate, sulfate and carboxylate moieties (Figure 1B) present on structures found in the plasma membrane and in membranes of cellular organelles (e.g., mitochondria, nucleus, endoplasmic reticulum, golgi, endosomes). These anionic chemical moieties are located within membrane proteoglycans (heparin sulfate proteoglycans: HSPGs; chondroitin sulfate proteoglycans: CSPGs; dermatan sulfate proteoglycans: DSPGs; keratin sulfate proteoglycans: KSPGs), glycoproteins, glycosphingolipids, and phospholipids as well as negatively charged aspartate and glutamate residues within protein receptors and other protein structures embedded in cellular membranes (Supplementary Table 1).
Negatively charged phosphate groups are a component of phospholipids that make-up cellular membrane bilayers (e.g., plasma membrane, inner and outer mitochondrial membrane, nuclear membrane, and endoplasmic reticulum membrane). There are at least five negatively charged membrane phospholipids including the mitochondrial membrane specific phospholipid cardiolipin, which possess a net charge of between −1 to −4 at pH 7 (Supplementary Table 1).
Proteoglycans a type of glycoprotein found on the surface of most cells and consist of a protein core and glycosaminoglycans (GAGs), which are long un-branched polysaccharides consisting of a repeating disaccharide subunit. Negatively charged sulfate groups are located on the polysaccharide repeating disaccharide subunits. In addition, the monosaccharide sialic acid is located at the end of the sugar chains attached to glycoproteins and has a negatively charged carboxyl group. Glycoproteins have important cellular functions, such as cell surface ligand receptor binding, cell signaling, cell adhesion, endocytosis, and binding extracellular matrix molecules (e.g., growth factors, enzymes, protease inhibitors, chemokines).
Glycolipids consist of a membrane lipid moiety covalently attached to a monosaccharide or polysaccharide. Glycolipids, namely glycosphingolipids, are negatively charged due to the presence of sialic acid. A glycosphingolipid containing one or more sialic acid residues is also known as a ganglioside. Gangliosides are expressed on most cells, but are more abundantly expressed on the cell surface of neurons, and are found ubiquitously throughout the CNS (215). They play a key role in modulating ion channel function, receptor signaling, cell-to-cell recognition and adhesion and regulation of neuronal excitability (216, 217). Membrane protein receptors rich in the acidic amino acids aspartate and glutamate also possess a negatively charged carboxylic moiety on their side chain.
With respect to the interaction of CARPs with anionic moieties, the positively charged arginine guanidinium head group forms bidentate hydrogen bonds with sulfates, carboxylates and phosphates, whereas the positively charged lysine amide head group forms weaker monodentate hydrogen bonds (Figure 1B). In addition, arginine and lysine cationic side chains can form salt bridges with the negatively charged aspartate and glutamate carboxylate C-termini, and cation-π interactions with the aromatic amino acids tryptophan, phenylalanine and tyrosine in proteins (218, 219). Interestingly, many neurotransmitters and drug-receptor interactions involve cation-π interactions (219). Together, the different electrostatic interaction between CARPs and plasma membrane structures can induce cellular uptake of the peptide by endocytic and non-endocytic pathways (220–224). Furthermore, peptide charge, arginine content and arginine distribution within the peptide, and the extent and density of the negatively charged moieties present on the cell surface play a significant role in terms of uptake efficacy (225–227). Peptide positive charge and arginine guanidinium head groups are also critical elements responsible for the ability of CARPs to target organelle membranes, such as the outer and inner mitochondrial membranes, which contain the negatively charged phospholipids cardiolipin (charge −2) and phosphatidylinositol 4, 5-bisphosphate (PIP2; charge −4) (Supplementary Table 1).
Importantly, studies have demonstrated that peptide characteristics that are known to increase the cell membrane traversing properties of CARPs, such as arginine content, peptide charge and presence of the aromatic amino acid tryptophan are also linked to increased peptide neuroprotective potency (1, 15, 16, 211).
CARPs Have Multimodal Neuroprotective Mechanisms Of Action
Data obtained in our laboratory and others using neuronal and non-neuronal cells indicate that CARPs have multimodal mechanisms of action targeting cell surface ion channel receptors and other receptors, mitochondria, proteolytic enzymes, oxidative stress/free radical molecules, protein stability, and pro-survival signaling, as well as having anti-inflammatory and immune regulatory actions (Figure 4). Evidence supporting these different neuroprotective mechanisms is provided below.
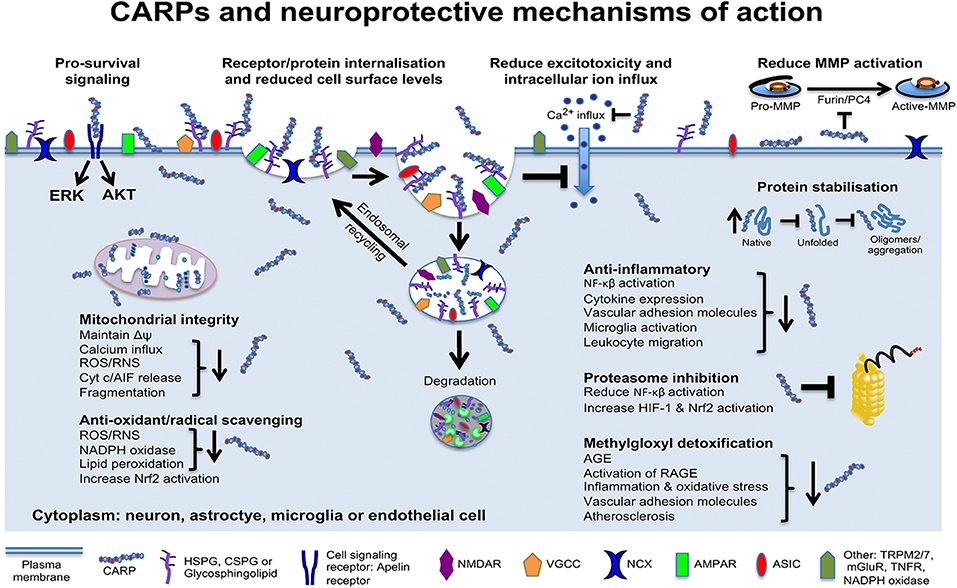
Figure 4. Schematic representation of CARP neuroprotective mechanisms of action. Model applies to neurons and potentially astrocytes, brain endothelial cells, oligodendrocytes, pericytes, and microglia. AGE, advanced glycation end products; RAGE, AGE receptors; AIF, apoptosis inducing factor; AKT, protein kinase B; Cyt c, cytochrome c; ERK, extracellular signal–regulated kinase; HIF-1, hypoxia-inducible factor-1; MMPs, matrix metalloproteinases; Δψ, mitochondrial transmembrane potential; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; Nrf2, nuclear factor erythroid 2-related factor 2; RNS, reactive nitrogen species; ROS, reactive oxygen species. NMDAR, N-methyl-D-aspartate receptor; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; NCX, sodium calcium exchanger; VGCC, voltage-gated calcium channels; ASIC, acid-sensing ion channels; TRPM2/7, transient receptor potential cation channels 2 and 7; mGluR, metabotropic glutamate receptor; TNFR, tumor necrosis factor receptor.
Inhibition of Excitotoxic Neuronal Death and Excitotoxic Neuronal Calcium Influx
Our laboratory has established that CARPs are highly effective at reducing excitotoxic neuronal death and that they have the capacity to reduce glutamic acid induced neuronal calcium influx (9, 10, 15, 19, 109). These findings provide a mechanism in which CARPs inhibit glutamate-evoked ionic currents in Xenopus oocytes expressing NMDA receptors (1), and NMDA excitotoxic neuronal death in neuronal cultures in vitro and retinal ganglion cells in vivo (14, 59, 69, 110). In addition, other CARPs reduce potassium depolarization-induced calcium-influx (e.g., R9-CBD3-A6K, TAT-L1, TAT-ct-dis) and sodium currents and sodium influx (e.g., t-CSM) in cultured dorsal root ganglion neurons (see Supplementary Table 2 for details).
The ability of CARPs to reduce glutamate receptor and other receptor mediated intracellular neuronal calcium influx is likely to be a primary mechanism accounting for their neuroprotective efficacy in protecting neurons in injury models associated with excitotoxicity and excessive neuronal intracellular calcium influx. As a mechanism whereby CARPs act to reduce the intracellular influx of calcium and potentially other ions, we hypothesized (109) that CARPs have the capacity to induce the endocytic internalization of cell surface ion channel receptors (Figure 4). In support of this hypothesis we subsequently showed that R12, as well as the TAT-fused neuroprotective peptide TAT-NR2B9c, reduces neuronal cell surface expression of the glutamate receptor subunit protein, NR2B (228). Importantly, several CCPPs (e.g., TAT, penetratin, R9) have also been demonstrated to reduce TNF (tumor necrosis factor) and EGF (epidermal growth factor) receptors in non-neuronal cells via an endocytic internalization mechanism (Supplementary Table 2) (229).
Interaction With Membrane Ion Receptors/Channels/Transporters
Many other studies have described the ability of CARPs to reduce neuronal and non-neuronal cell surface levels and/or activity of NMDA receptors, and other ion and non-ion channel receptors (see Supplementary Table 2). While these studies provide ample evidence for the ability of CARPs to perturb cell surface receptors it raises the question how different peptides with diverse amino acid sequences possess the ability to reduce cell surface levels and/or antagonize receptor function. As mentioned above, one mechanism involves CARP induced internalization of cell surface receptors. However, it is also possible CARPs antagonize ion channel receptor function by electrostatic interactions. For example, CARP electrostatic interactions with receptor anionic moieties may alter receptor function or interfere with ion transport within the receptor pore. In support of this, the guanidine moiety in arginine residues and in other molecules play a critical role in voltage-gated and ligand-gated ion channel function (see section Compounds Containing the Guanidinium Moiety and Neuroprotection) (230–237). For example, the guanidino moiety in agmatine, a molecule with neuroprotective properties (Supplementary Table 3), has been identified as being capable of interacting with a site within the NMDA receptor channel and calcium voltage channels and blocking their function (238). Interestingly, polyamines (e.g., putrescine and spermine) a class of compounds that also contain positively charged amino groups also have the capacity to block ion channels, including glutamate receptor and potassium channels (239). Given that positive charge is a critical factor for CARP neuroprotection and charge is independent of peptide amino acid sequence, provides additional support for a mechanism involving an electrostatic interaction perturbing ion channel function. Hence, there is good evidence to indicate that the structure and charge of the guanidine moieties in CARPs have the capacity to block ion channel receptor function, providing an additional mechanism whereby the peptides can reduce the toxic effects of intracellular ion influx associated with excitotoxicity and ion channel over-stimulation.
Mitochondrial Targeting and Maintenance of Mitochondrial Integrity
CARPs have the capacity to target mitochondria and exert positive effects on the organelle, with potential neuroprotective outcomes. This topic has been the subject of several reviews by the developers of the mitochondrial targeting SS cationic arginine-containing tetrapeptides (240), and more recently by our own laboratory (241), and therefore will be discussed only briefly here.
After entering cells, CARPs target and enter mitochondria due to the presence in the outer and inner mitochondrial membranes of negatively charged phospholipids (e.g., cardiolipin, PIP2), and because of the mitochondrial transmembrane potential (ΔΨm). It is also possible that electrostatic interactions of CARPs with negatively charged free mitochondrial DNA contributes to the retention of the peptides in the organelle. At the site of the outer mitochondrial membrane, CARPs can inhibit the toxic influx of calcium into mitochondria, possibly by perturbing ion channel receptors (e.g., MCU, VDAC, NCX) and other membrane proteins (e.g., mitochondrial permeability transition pore proteins) responsible for the movement of calcium ions into mitochondria.
CARPs can also perturb other outer membrane proteins that are detrimental to mitochondrial function and cell survival. For example, CARPs interfere with BAX, the mitochondrial permeability transition pore and other pro-apoptotic proteins that localize to the outer mitochondrial membrane during cell death or interfere with proteins that promote mitochondrial fission and mitophagy. Inhibition of mitochondrial fission enables maintenance of mitochondria as filamentous structures, which enables toxic products generated by dysfunctional mitochondria to be distributed over a large organelle volume and thereby minimizing any detrimental effects. Furthermore, due to their interactions with cardiolipin, CARPs assist in stabilizing and preserving cristae architecture and the electron transport chain with positive effects on ATP maintenance, reduced reactive nitrogen species/reactive oxygen species (ROS/RNS) generation, as well as maintenance of cytochrome c native tertiary structure, function, oxidation state and location within the inner mitochondrial membrane. In addition, the anti-oxidant and free radical scavenging properties of CARPs (discussed below) would also have positive influences in reducing the toxic effects of excessive ROS/RNS generation by mitochondria during cellular stress.
Anti-oxidant and Free Radical Scavenging Properties
Due to the amino acid arginine, CARPs are likely to act as anti-oxidant and/or free radical scavenging molecules in their own right. Although L-arginine is utilized by nitric oxide synthase as a substrate for nitric oxide generation, which is a key regulator of endothelial cell function and blood flow, the amino acid has other properties. Arginine is unique in possessing a N-terminal guanidinium head group (Figure 1A) and guanidinium containing small molecules are known to possess properties that mitigate the effects of oxidative stress. For example, L- and D-arginine, along with aminoguanidine, methylguanidine, guanidine, and creatine, which are all structurally related to arginine have the ability to scavenge one or more of the following reactive molecules: superoxide, peroxynitrate, hydroxyl radicals, hydrogen peroxide, hypochlorous acid, and breakdown products of lipid peroxidation (e.g., reactive aldehydes: malondialdehyde and 4-hydroxynonenal) (242–249).
The anti-oxidant properties of aminoguanidine, which also possesses neuroprotective actions (Supplementary Table 3), were demonstrated in vitro with the agent reducing rat retinal Muller cell hydrogen peroxide oxidant induced apoptosis, ROS production and lipid peroxidation, and in vivo by reducing the level of lipid peroxides in the vitreous of diabetic rats (246). Furthermore, both L- and D-arginine reduced oxidative impairment to myocardial contractility of perfused rat hearts subjected to oxygen radical generation (249). L- and D-arginine and L- and D-arginine polymers (e.g., poly-arginine R9) have beneficial effects on vascular endothelial cell and cardiovascular function and have anti-atherosclerotic properties (242, 250–252). These positive effects have also been observed with D-arginine containing peptides and therefore are likely to be independent of the nitric oxide pathway, as D-arginine is not readily metabolized by nitric oxide synthase. In support of this, in Caenorhabditis elegans, which lacks nitric oxide synthase (NOS), exposure to exogenous L-arginine prolongs worm lifespan under oxidative stress growth conditions (253).
Szeto-Schiller (SS) peptides are short tetrapeptides with alternating basic (e.g., arginine, lysine, or ornithine) and aromatic (e.g., tyrosine, dimethyltyrosine, tryptophan, or phenylalanine) amino acids, but usually containing at least one arginine and one tyrosine or dimethyltyrosine residue (240). Several SS peptides (e.g., SS-31, SS-20, mCPP-1; Table 1) have demonstrated anti-oxidant properties by way of reducing ROS levels in cells grown under normal or oxidative stress conditions. Whereas, the anti-oxidant action of SS peptides has been attributable to the tyrosine and dimethyltyrosine residues, based on the free radial scavenging properties of guanidinium containing molecules it is likely that the arginine residue also contributes to the anti-oxidant property of SS peptides.
Larger CARP's also have anti-oxidant and lipid peroxidation reducing properties. A lactoferrin derived peptide f8 (GRRRRSVQWCAVSQPEATKCFQWQRNMRKVRGPPVSCIKRDSPIQCIQ; net charge +8.7) and a casein derived peptide f12 YPYYGTNLYQRRPAIAINNPYVPRTYYANPAVVRPHAQIPQRQYLPNSHPPTVVRRP; net charge +7.2) have demonstrated anti-oxidant activity in an in vitro free radical scavenging assay (254). While both f8 and f12 are large peptides with interspersed arginine residues, molecular modeling revealed the peptides display a configuration with a highly cationic electrostatic surface, with arginine residues facing on the outside of the peptide. In addition, the human cathelicidin anti-microbial peptide LL-27 (LGDFFRKSKEKIGKEFKRIVQRIKDFL; net charge +5) inhibits the oxidation of low density (LDL) and high density (HDL) lipoproteins and can reduce fatty acid hydroperoxides in in vitro oxidation models (255). As demonstrated with protamine, CARPs also bind negatively charged oxidized-LDLs, inhibit their engagement to the lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), which can stimulate intracellular signaling cascades detrimental in ischemia-reperfusion cerebral injury (256, 257). Similarly, the cathelicidin PR-39 (RRRPRPPYLPRPRPPPFFPPRLPPRIPPGFPPRFPPRFP; net charge +10) protects HeLa cells from apoptotic cell death induced by the oxidizing agent tert-butyl hydroperoxide (198), and inhibits hypoxia induced cell death of endothelial cells (199).
Another mechanism how CARPs can reduce oxidative stress is by inhibiting the activity of the plasma membrane superoxide generating enzyme complex nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), with one study indicating inhibition is associated with the presence of poly basic amino acid consisting of arginine, lysine or histidine motifs (258). The CARPs PR-39, PR-26 (amino acids 1–26 of PR-39; net charge +8), gp91ds-tat (Table 2), and TAT-NR2B9c all inhibit NADPH oxidase function or superoxide generation in cell free systems and/or in different cells both in vitro and in vivo (259–261). The NADPH oxidase complex consists of 5 subunits with PR-39 and PR-26 binding to the SH3 (SRC homology 3) domain within the p47phox subunit, which disrupts binding to the p22phox subunit (261). The gp91ds-tat is derived from the NADPH oxidase Nox2 cytosolic B loop (mouse Nox2; amino acids 86–94) subunit and was designed to inhibit Nox2 interacting with p47phox (260). TAT-NR2B9c inhibits the generation of superoxide and phosphorylation of p47phox in cultured neurons exposed to NMDA (259). It was concluded that inhibition of NMDA receptor-PDS-95 mediated signaling by TAT-NR2B9c prevented phosphorylation of p47phox and activation of NADPH oxidase. However, for both the gp91ds-tat and TAT-NR2B9c peptides a direct inhibitory action on NADPH oxidase associated with the arginine content and positive charge of the peptides cannot be ruled out. In addition, given all four peptides have cell-penetrating properties, it is possible that these and other CARPs disrupt the membrane assembly of NADPH oxidase units within the plasma membrane. Since superoxide generation is also associated with inflammatory responses, inhibition of NADPH oxidase activity would also contribute to the anti-inflammatory properties of CARPs.
While the anti-oxidant properties of CARPs need to be further investigated, available evidence suggests that arginine residues within the peptide have the potential to exert anti-oxidant and/or free radical scavenging actions. Furthermore, it could be hypothesized that the multiple arginine resides within CARPs will act as a multivalent anti-oxidant compound, which depending on the number and arrangement of the guanidinium moieties would provide considerably more potency per molecule than arginine alone or a molecule containing a single guanidine moiety.
Methylglyoxal Scavenging and Glycation End-Products
Glyoxal compounds are highly reactive cell permeable dicarbonyls produced predominantly as a by-product of glycolysis, and are precursors in the formation of advanced glycation end-products (AGEs). An important dicarbonyl with respect to cellular toxicity is methylglyoxal, which reacts irreversibly with arginine and lysine residues and reversibly with cysteine resides on proteins causing functional impairment (262). Methylgyloxal also reacts with nucleic acids and lipids, and its production is associated with oxidative stress and ROS generation. The glycation of arginine and lysine by methylglyoxal forms the AGEs hydro-imidazolone, methylglyoxal-hydroimdazolone 1 (MG-H1), argpyrimidine, and MG-derived lysine dimer. In addition, to altering protein function, AGE-modified proteins interact with AGE receptors (RAGE), which stimulate the expression of inflammatory genes and ROS generation. AGEs are increased in diabetes, vascular disease, cerebral ischemia, renal failure, aging and chronic disorders, such as Alzheimer's disease, Parkinson's disease and liver cirrhosis. Methylgyloxal can affect mitochondrial function, up-regulate vascular adhesion molecules (e.g., P-selectin and E-selectin) that contribute to leucocyte adhesion (263), and cause glycation of the BBB vascular tight junction protein occluding (264) and the basement membrane extracellular protein fibronectin (265) resulting in altered endothelial function.
Given that methylglyoxal readily targets basic amino acids, it is likely that arginine (and lysine/cysteine) residues within CARPs react with and act as methylglyoxal scavengers, thereby reducing their toxic effects on intra- and extra-cellular proteins, and RAGE activation. Methylgloxal is normally detoxified by the glyoxalase system (glyoxalase-1 and glyoxalase-2), which utilizes glutathione as a co-factor, however neurons are particularly susceptible to methylgloxal due to the high glycolytic activity of the brain and the reduced capacity of the glyoxalase system in neurons. The capacity of the glyoxalase system in the brain deceases with age, especially after the fifth decade of life, and an increase in MG-H1 modified mitochondrial proteins is linked to aging and increased ROS production (266); increasing glyoxalase capacity in C. elegan increases life span in this organism (267). Furthermore, any additional stress within the brain as occurs in cerebral ischemia/reperfusion, as well as in chronic neurological disorders is likely to lead to an excessive production of methylgloxal and/or reduced capacity to detoxify methylgloxal.
Importantly, arginine and other guanidinium containing molecules (e.g., aminoguanidine, metformin) have the capacity to scavenge methylglyoxal and prevent AGEs (268, 269). For example, at one stage aminoguanidine was being developed as a therapeutic agent for the prevention of AGEs in diabetes, and both L- and D-arginine can effectively scavenge and attenuate the harmful effects of methylgloxal on cultured endothelial cells (270). However, it is considered that small molecule methylglyoxal scavengers are not sufficiently potent and/or suffer from short half-lives to be effective in vivo. In contrast, arginine-containing penta-peptides peptides (CycK[Myr]RRRRE; Cyc, cyclic peptide; Myr, myristic acid; and myr-KRRRRE; net charge +4) also possess methylglyoxal scavenging activity (271), and CycK(Myr)RRRRE prevents methylglyoxal induced pain in mice, and is being considered as a therapy for pain and other diabetic complications associated with methylglyoxal toxicity (271).
Inhibition of Matrix Metalloproteinase Activation and the Proteasome
Another mechanism whereby CARPs may exert a neuroprotective effect is by their ability to indirectly prevent the activation of matrix metalloproteinases (MMPs) by inhibiting proprotein convertase (PC) activation. Proprotein convertase consists of a family of proteolytic enzymes that cleave inactive proteins, including MMPs into an active state. Poly-arginine peptides and other CARPs are potent inhibitors of convertases, such as furin, PC1, PC4, PC5/6, and PC7 (272–276).
Furin is a ubiquitously expressed convertase that is regulated by hypoxia-inducible factor-1 (HIF-1) (277) and up-regulated in the ischemic brain (278, 279), and can activate MMP2, MMP3, and MMP14 (280). Furthermore, MMP3 can activate MMP1, MMP7 and MMP9, and MMP14 can activate MMP2 and MMP2 can activate MMP9 (281, 282). Significantly, following ischemic stroke MMP2, MMP3, MMP7, MMP9, and MMP14 are either up-regulated or activated in the brain. Moreover, MMP activation is associated with degradation of the neurovascular unit and BBB disruption, which in turn can result in cerebral edema, leukocyte infiltration and secondary hemorrhage after ischemia (279, 281, 282). It is also possible that proprotein convertases have other protein substrates, which when activated are potentially neuro-damaging, however this is an area that has as yet not been explored.
The ability of poly-arginine peptides to inhibit proprotein convertases, similar to peptide neuroprotection (15), increases with increasing polymer length (e.g., R9 > R8 > R7 > R6) (272). In addition, convertase inhibition is not stereospecific with both to L- and D-isoform peptides having the capacity to inhibit enzyme activity. The electrostatic interaction between CARPs and the negatively charged surface of convertases is believed to be the mechanism responsible for the inhibitory actions of the peptides. Interestingly, a penetratin-fused peptide (P-IQACRH: RQIKIWFQNRRMKWKK-IQACRG; net charge +7.9) that mimics the active site of caspases 1, 2, 3, 6, 7 and 14 and acts as a competitive inhibitor for these enzymes, inhibited caspase and MMP9 activation following NMDA-induced excitotoxicity in an in vivo retinal ganglion cell injury model (283). However, the peptide also reduced NMDA-induced retinal ganglion cell death in culture and in vivo, and hence it is possible that the anti-excitotoxic properties of P-IQACRH, rather than a direct down-stream inhibition of caspases and MMP9 was responsible for blocking the activation of the enzymes. The P-IQACRH study highlights the caution that is needed when analyzing the neuroprotective actions of CARPs.
CARPs can also inhibit other proteolytic enzymes, such as cathepsin C (284) as well as, the activity of the proteasome (285–289). Importantly, treatments known to inhibit the proteasome, which is responsible for the degradation of short-lived cytosolic proteins, is known to reduce the severity of brain injury after stroke (290–293).
With respect to proteasomal inhibition, the CARP PR-39 (see above) can reversibly bind to the α7 subunit of the 26S proteasome and block degradation of the nuclear factor-κB (NF-κB) inhibitor protein IκBα. Interestingly, studies utilizing PR-39 indicate that proteasomal inhibition occurs via a unique allosteric, reversible and substrate selective mechanism without inhibiting overall-proteasome proteolytic activity, which in itself could be deleterious by interfering with normal cellular processes. In contrast, mild levels of proteasome inhibition can induce a protective pre-conditioning response that can protect cells from oxidative stress (294). Similarly, ischemic pre-conditioning, which can reduce brain injury following stroke is associated with proteasomal inhibition (293). PR-39 abolished NF-κB-dependent gene expression in cultured endothelial cells exposed to TNF-α, and in the pancreases and hearts of mice following induction of acute pancreatitis and myocardial infarction, including the up-regulation of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) (285). Other studies have also demonstrated that PR-39 can reduce infarct size and have beneficial effects on microvascular cells in myocardial reperfusion injury models by blocking proteasome-mediated degradation of IκBα (200).
In contrast to blocking activation of the NF-κB, inhibition of the proteasome is likely to enhance activation of the transcription factor HIF-1, which is considered one of the most critical adaptive gene expression responses to low oxygen concentrations. Hypoxia-inducible factor-1 consists of the HIF-α and HIF-β subunits, the former undergoing proteasomal degradation during normoxia, and the latter being constitutively expressed. Therefore, inhibition of the proteasome will enhance and/or prolong HIF-1 activation in the brain during and following cerebral ischemia, thereby enhancing any neuroprotective actions of the transcription factor. For example, PR-39 was demonstrated to inhibit the proteasome-dependent degradation of HIF-α and stimulate angiogenesis by accelerating the formation of vascular structures in cultured endothelial cells and in vivo in the myocardium (295). A similar effect was demonstrated in the brain after stroke with a small molecule proteasome inhibitor resulting in accumulation of HIF-α and enhanced angio-neurogenesis (291).
Inhibition of the proteasome can also enhance the activity of the transcription factor nuclear factor E2-related factor 2 (Nrf2), which regulates the expression of multiple cytoprotective genes, particularly those involved in mitigating oxidative stress (e.g., HO-1, SOD1, NAD[P]H dehydrogenase, glutathione S-transferase) (296). Under normal conditions, cytoplasmic Nrf2 is bound to kelch-like ECH-associated protein 1 (Keap1), in which it is subject to proteasomal degradation, however oxidative stress disables keap1, allowing Nrf2 to accumulate, translocate to the nucleus and activate gene expression. Interestingly, a Nrf2 amino acid derived sequence (LQLDEETGEFLPIQ) has been developed that disrupts the Nrf2-Keap1 interaction, and when fused to TAT (TAT-14: YGRKKRRQRRR-LQLDEETGEFLPIQ; charge +4) or R7 (7R-ETGE: RRRRRRRR-LQLDEETGEFLPIQ; net charge +4) has been demonstrated to activate Nrf2 and cytoprotective gene expression in THP-1 monocyte and RAW 264.7 macrophage cell lines (297, 298). Furthermore, the TAT-14 peptide modified to contain a calpain cleavage sequence (TAT-CAL-DEETGE: Table 2) increased Nrf2-regulated gene expression in the brain (299) and is beneficial when administered to rodents after global cerebral ischemia and TBI (154, 299). It remains to be determined if the Nrf2 peptides are activating Nrf2 by directly disrupting the Nrf2-Keap1 interaction or by inhibiting the proteasome. Also of interest is the demonstration that in rats, oral treatment with arginine, resulted in the up-regulation of proteins associated with the Nrf2 pathway in liver and plasma (300).
Reducing the Inflammatory Response
Whereas, few studies have specifically examined neuroprotective CARPs in the setting of neuro-inflammation, this class of peptide has well-established anti-inflammatory properties that are potentially beneficial in neurodegenerative disorders. It is likely CARPs exert differential effects on the immune response by targeting both the CNS and peripheral immune responses by several mechanisms. As explained above, the ability of CARPs to inhibit the proteasome will reduce NF-κB activation and the expression of genes involved in pro-inflammatory pathways. Interestingly, the CARP AIP6 (RLRWR; net charge +3) can inhibit NF-κB activity by an alternative mechanism, by binding to and blocking NF-κβ p65 sub-unit binding to DNA and inhibiting its transcriptional activity (301). The p65 subunit is a negatively charged protein, and hence it is possible that CARPs have the capacity to interfere with this NF-κβ sub-unit through an electrostatic interaction. Similarly, because CARPs can interfere with cell surface receptors levels and/or function, it is also possible they reduce the inflammatory response associated with ligands (e.g., cytokines, chemokines, intracellular molecules) binding to receptors on immune cells.
Proteins regulated by NF-κB and involved in the inflammatory response include cytokines (e.g., IL-1, TNF-α), chemokines (e.g., MCP-1, CXCL1) and vascular adhesion molecules (e.g., ICAM-1, VAM-1) (302). To this end, NF-κB is responsible for up-regulating cerebral vascular adhesion molecules VCAM-1 and ICAM-1, which during cerebral reperfusion promotes macrophage and neutrophil infiltration into the brain. Although the ability of CARPs to reduce vascular adhesion molecule expression in the cerebral vasculature has not been examined, PR-39 can reduce VCAM-1 and ICAM-1 protein levels in heart tissue following myocardial infarction in mice and in cultured vascular endothelial cells following exposure to TNF-α (285), and leukocyte adhesion to rat mesenteric venules after ischemia and reperfusion (303). In addition, the TAT peptide reduces the production of multiple cytokines (e.g., G-CSF, IL-6, MIP1α, TNF-α, and IFN-γ) in cultured human lung epithelial cells following protein kinase C stimulation by phorbol 12, 13-dibutyrate, a stimulus associated with NF-κβ activation (304). In line with the ability of CARPs to inhibit the proteasome, TAT reduced degradation of the NF-κβ inhibitory subunit IK-κβ in lung epithelial cells following protein kinase C activation. Similarly, AIP6 demonstrated anti-inflammatory effects in cultured activated macrophages by decreasing TNF-α and prostaglandin-E secretion and in a mouse model of paw inflammation reduced levels of TNF-α, IL-1β, and IL-6 protein in affected tissue (301).
CARPs can also bind to oxidized phospholipids (e.g., ox-LDLs) which are known pro-inflammatory molecules and play an important role in atherosclerosis and other inflammatory disorders. Due to the high lipid content of the brain, any conditions that increase oxidative stress will generate oxidized phospholipids. Binding of CARPs to oxidized phospholipids is believed to enhance their clearance, as well as reduce their inflammatory potential and inhibitory effects on anti-oxidant enzymes associated with lipoproteins and the cell membrane (255, 305). The CARP E5 (Ac-SHLRKLRKRLLRDADDKRLA-NH2; net charge +6) was demonstrated to bind oxidized phospholipids and inhibit their pro-inflammatory function in human blood (305). In addition, pre-treatment of macrophage (RAW264.7) and endothelial (HUVEC) cell lines with the LL-27 reduces pro-inflammatory gene expression, whereas pre-incubation of oxidized phospholipid with the peptide prior to administration to mice reduces serum IL-6 and TNF-α levels (255). Similarly, the Apolipoprotein E (ApoE) protein derived CARP Ac-hE18A-NH2 (Ac-RKLRKRLLRDWLKAFYDKVAEKLKEAF-NH2; net charge +6) can bind bacterial lipopolysaccharides (LPS) and reduce its inflammatory (e.g., TNF-α, IL-6 production) inducing properties in human blood and primary leukocytes and a monocyte cell line (306). Ac-hE18A-NH2 can also inhibit LPS-induced VCAM-1 expression, and reduce monocyte adhesion in HUVECs, as well as the secretion of IL-6 and monocyte chemoattractant protein-1 (MCP-1) from THP-1 monocyte cells exposed to LPS (307). Also, the CARP TAT-14 (see above), which while developed to activate Nrf-2, reduces TNF-α production in THP-1 monocyte cells following LPS stimulation (297).
Other ApoE derived peptides have also demonstrated anti-inflammatory properties. The two almost identical ApoE derived peptides, ApoE-133–150 (ApoE-133–150: Ac-LRVRLASHLRKLRKRLLR-NH2; net charge +8.1) and COG-133 (Table 1) suppress cytokine expression (IL-8 or TNF-α) and other inflammatory mediators (e.g., COX or NO) in THP-1 monocytes or BV-2 microglia cells stimulated with LPS (308, 309). COG-133 treatment can suppress systemic and brain levels of TNF-α and IL-6 in mice after LPS administration (36). The COG112 peptide, which comprises COG133 fused to the CCPP penetratin (Table 2) inhibits the inflammatory response in mouse models of pathogen or injury induced colitis by reducing several pro-inflammatory mediators. For example, COG112 attenuated cytokine and chemokine expression, iNOS expression and nitric oxide production in mouse colon epithelial cell cultures and in colon tissue in mice following exposure to Citrobacter rodentium (310, 311). It was also demonstrated that COG112 inhibited NF-κB activation in colon cells and tissue following bacterial stimulation (310, 311).
The CARP PACAP38 (Table 3), which is derived from the neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) binds the adenylate-cyclase-activating receptor stimulating adenylate cyclase and subsequently increases intracellular cAMP, which is a signaling molecule important in many biological processes. PACAP38 is a predominant cleavage product of PACAP, which exerts several functions within the CNS, including acting as a neurotransmitter and neuromodulator and modulating inflammatory responses. The peptide has cell-penetrating properties (312), is widely distributed within the CNS and has neuroprotective actions in excitotoxicity, retinal ischemia, stroke and traumatic brain injury models (313–318). With respect to its anti-inflammatory actions, PACAP38 can reduce the activation of cultured primary microglia to hypoxia by inhibiting induction of nitric oxide, iNOS, and p38 as well as reducing TNF-α secretion (315). Furthermore, following traumatic brain injury, PACAP38 treatment reduces cerebral inflammation by reducing toll-like receptor-4 (TLR-4) up-regulation, and its downstream mediators. For example, treatment reduced TNF-α and IL-1β levels, reduced NF-κβ p65 sub-unit levels in nuclei, and increased levels of the NF-κβ inhibitory subunit IκB-α in the brain (318). Additionally, PACAP38 ablated TLR-4 up-regulation in the brain and in BV-2 microglia following exposure to the TLR-4 agonist LPS (319). Similarly, the LL-37 anti-microbial CARP (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES; net charge +6) attenuates activation of cultured dendritic cells to different TLR ligands (320).
The CARP dRK (rrkrrr; net charge +6; lower case indicates D-isoform amino acids) was identified based on its ability to block the interaction between VEGF and the VEGF receptor. The dRK peptide can reduce TNF-α and IL-6 production in normal peripheral blood monocytes and synovial fluid mononuclear cells of rheumatoid arthritis patients following VEGF stimulation (321). In a mouse model of collagen-induced arthritis, dKR reduced paw inflammation and serum levels of IL-6 (321). While it was believed that dKR was directly inhibiting the pro-inflammatory effects of VEGF and its receptor, it is possible the peptide was inhibiting VEGF induced activation of NF-κβ. In a different collagen-induced arthritis mouse model, the CARPs IG-19 (IGKEFKRIVQRIKDFLRNL-NH2; net charge +5) and IDR-1018 (Table 1) both reduced the number of limbs affected, and IG-19 reduced overall disease severity (322). Subsequent examination of IG-19 treated mice revealed reduced serum levels of the pro-inflammatory cytokines TNF-α and IFN-γ and reduced cellular infiltration and cartilage degradation in arthritic joints. Interestingly, in a more recent study, IDR-108 prolonged anti-inflammatory TGFβ gene expression and suppressed early pro-inflammatory IL-1β gene expression levels in a human endothelial cell line (EA.hy926) cultivated in a high glucose environment to induce cell stress (323). In another study, treatment of arthritic mice with a peptide developed to mimic the action of Bcl-2 homology 3 (BH3) domain-only proteins (TAT-BH3: Ac-RKKRR-O-RRR-EIWIAQELRRIGDEFNAYYAR; net charge +6) ameliorated arthritis development and reduced the number of myeloid cells in the affected joint (324).
The CARP R9-SOCS1-KIR (RRRRRRRRR-DTHFRTFRSHSDYRRI; net charge +11.2) was developed to inhibit suppressor of cytokine signaling 1 (SOCS1) signaling, which can result in JAK/STAT or NF-κβ activation. This peptide blocked the activation and nuclear translocation of STAT1α, STAT3, and NF-κB p65 and inflammatory effects induced by IFN-γ, TNF-α, and IL-17A in the ARPE-19 human retinal pigment epithelial cell line (325). Topical delivery of R9-SOCS1-KIR also reduced inflammatory cell infiltration into the eye in a mouse model of experimental autoimmune uveitis. Furthermore, in a mouse model of Pseudomonas aeruginosa induced keratitis, R9D (Table 1) treatment reduced disease severity and concentrations of corneal TNF-α, IFN-γ, IL-10, and GM-CSF (326).
The cationic arginine-rich human beta-defensin derived peptide hBD3-3 (GKCSTRGRKCCRRKK; net charge +8) has demonstrated in vitro and in vivo anti-inflammatory actions. In a macrophage cell line (RAW264.7) pre-treatment with hBD3-3 reduced iNOS, TNF-α, and IL-6 protein expression (327). In addition, mice treated with hBD3-3 and injected with LPS had reduced plasma levels of TNF-α and IL-1β and reduced neutrophil infiltration into lung regions affected by LPS induced inflammation. Finally, as NF-κβ activation is involved in iNOS, TNF-α and IL-6 expression, studies in RAW264.7 cells revealed that hBD3-3 significantly inhibited degradation of the NF-κβ inhibitory subunit IκB-α, as well as the translocation of the NF-κβ p65 subunit to the nucleus.
CARPs may also inhibit inflammation by reducing activation of components of the complement system. Protamine (Table 1) and large poly-L-arginine peptides antagonize complement protein C5a binding to its receptor C5aR1 (or CD88) in leukocytes (328). C5aR is a transmembrane G-protein-coupled receptor expressed on neutrophils, monocytes, eosinophils, and non-myeloid cells, including liver cells and alveolar and kidney tubular epithelial cells, some classes of neurons and microglia and astrocytes. Importantly activation of the complement system following stroke/cerebral ischemia and other neurological conditions is associated with unfavorable outcomes and inhibition of C5a improves outcomes (329). Antagonism of the C5aR is thought to be due to an electrostatic interaction between the CARP and anionic sites within the receptor (328).
In some situations, CARPs may induce pro-inflammatory responses. For example, a large poly-arginine peptide (R100; 100-mer, 12.5–13.5 kDa) can bind to TLR-4 and induce cytokine and interferon gene expression in mouse splenocytes comprising mostly of B-cells, but also T-cells and monocytes (330). The peptides ApoE-133–150 and IDR-1018 increase secretion of the cytokine MCP-1 in human blood mononuclear cells (309). In one study, IDR-1018 also increased neutrophil adherence to EA.hy926 endothelial cells and promoted neutrophil migration and cytokine production (e.g., IL-8, MCP-1, MCP-3) (331).
Pro-survival Signaling
Due to the ability of CARPs to interact with cell surface receptors it appears they also have the capacity to stimulate receptor mediated pro-survival signaling pathways. The best example of CARP pro-survival signaling has been demonstrated with apelin peptides.
Apelin is a highly conserved arginine-rich peptide first identified in 1998 following its isolation in bovine stomach extracts. The peptide is expressed as a 77 amino acid preprotein, which can be processed into at least three bioactive carboxy-terminal fragments including apelin-36, apelin-17, and apelin-13 (Table 3). All apelin peptides can bind the G-protein coupled apelin receptor (originally named APJ) and induce cell signaling (332), with positively charged arginine and lysine resides in apelin, and negatively charged aspartate and glutamate resides in the extracellular N-terminal region of the apelin receptor important for receptor binding and internalization (333, 334). Apelin peptides and the apelin receptor are widely expressed throughout the body including brain, heart, adipose, skeletal muscle, kidney, and lung. The apelin/apelin receptor system regulates cardiac and vascular function, glucose metabolism, fluid homeostasis, cell survival, and angiogenesis. Other CARPs including poly-arginine peptide R9D and protamine can bind to the apelin receptor (333, 335). Interestingly, pre-treatment of cells with R9D and protamine appears to inhibit subsequent apelin receptor signaling. However, this is likely due to the R9D and protamine peptides desensitizing the receptor or inducing receptor internalization because pre-treatment of cells with apelin peptides also decreases apelin receptor cell signaling (336). The apelin receptor can dimerise with the κ-opioid G-protein-coupled receptor (KOR) and bind both apelin and dynorphin A peptides (Table 3) and activate extracellular signal–regulated kinase 1/2 (ERK1/2) signaling (337).
Established signaling events activated by the apelin receptor are the AMP-activated protein kinase (AMPK), ERK1/2 and phosphatidyl inositol 3-kinase/protein kinase B (PI3K/AKT) pathways (338–340). The AMPK pathway is a major energy sensing system that monitors for low levels of energy molecules, such as ATP and AMP to induce cellular adaptive metabolic changes to preserve and better utilize remaining energy substrates and maintain mitochondrial function. In addition, AMPK can activate the transcription factor Nrf2, resulting in the expression of anti-oxidant proteins. The ERK pathway has diverse actions including cell survival mediated by the expression of pro-survival proteins (e.g., BCL2) and inhibition of pro-apoptotic proteins (e.g., BAD). Similarly, the PI3K/AKT pathway promotes cell survival by targeting and phosphorylating proteins that regulate cell death and survival, cell migration and metabolism and angiogenesis.
With respect to neuroprotection, apelin peptides reduce intracellular calcium influx and neuronal death following NMDA receptor mediated excitotoxicity (163–166, 341), and improve outcomes in animal stroke, perinatal hypoxia-ischemia, traumatic brain injury, intracerebral hemorrhage and Alzheimer's disease models (see Table 3). The neuroprotective mechanism of action of apelin peptides have been attributed to AMPK, ERK, and/or AKT mediated signaling by inhibiting apoptosis, suppressing inflammation, reducing ER stress, preserving BBB integrity and stimulating angiogenesis (163, 166–170, 340–342), as well as mechanisms independent of apelin receptor signaling (164, 165).
The endogenous CARP toddler (also known as elabela/apela; QRPVNLTMRRKLRKHNCLQRRCMPLHSRVPFP; net charge +9.1) can also bind the apelin receptor and induce ERK signaling (343). Whereas, an anti-microbial CARP SR-0379 (MLKLIFLHRLKRMRKRLKRK; net charge +11) can stimulate ERK and AKT phosphorylation in dermal fibroblasts via a cell surface integrin receptor (344). In addition, the anti-microbial LL-37 peptide can bind to cell surface receptors in different cells, and activate downstream ERK, AKT, or P38 signaling (345, 346). Finally, protamine and polycationic arginine and lysine peptides interact with and enhance the EGF receptor tyrosine kinase activity and thereby enhance cell signaling activated by the receptor (347, 348).
Inhibiting Protein Aggregation in Neurodegenerative Disorders
Protein misfolding can lead to protein aggregation, and the accumulation of specific protein oligomers, aggregates and fibrils is the hallmark of several chronic neurodegenerative disorders, such as Alzheimer's disease (e.g., Aβ peptide, tau), Parkinson's disease (e.g., α-synuclein), Huntington's disease (e.g., Huntingtin), and amyotrophic lateral sclerosis (e.g., SOD1). Arginine is a common additive to protein solutions to facilitate protein folding, and to help maintain protein stability and inhibit protein aggregation (349, 350). Since arginine can stabilize proteins and inhibit self-aggregation, it is possible CARPs can also reduce protein misfolding and aggregation, and is a mechanism through which CARPs may be beneficial in animal models of human neurodegenerative disorders associated with proteinopathies.
The guanidinium head group of arginine and the ability of arginine to self-associate to form clusters (n-mers; n > 2) is considered a critical mechanism responsible for suppressing protein aggregation (349). Arginine clusters associate with the surface of proteins, namely aromatic (e.g., tryptophan) and negatively charged (e.g., glutamate) amino acid residues via cation-π interactions and hydrogen bonding, respectively. The interaction of arginine clusters with hydrophobic residues not normally exposed in the native state stabilizes partially unfolded proteins and act to “crowd around” proteins to prevent aggregation (350). Hence, it is conceivable that CARPs, such as poly-arginine molecules due to their multivalent arginine arrangement behave in a similar fashion to arginine clusters to prevent protein aggregation. For example, CARPs that inhibit Aβ oligomer formation, which is considered neurotoxic include KLVFFRRRRRR (net charge +7) and R5 (RRRRRR: net charge +5) (351), 15M (Ac-VITNPNRRNRTPQMLKR-NH2: net charge +5) (352) SRPGLRR (net charge +3) (353), RR-7-animo-4-trifluromethylcoumarin (net charge +3) (354) RI-OR2-TAT (Ac-rGffvlkGrrrrkkrGy-NH2: charge +9) (355) R8-Aβ (25–35) (rrrrrrrr-gsnkgaiiglm: net charge +10) (356), and the related D3 and RD2 peptides (Table 1) (28, 29). In a mouse model of Alzheimer's disease, R9 administered subcutaneously over 4 weeks, decreased brain Aβ deposits by 15%, albeit the reduction was not statistically significant (357). In addition, poly-arginine (5–15 kDa; 32–96-mers) can inhibit the aggregation of a tau mutant protein (P301L) commonly associated with tauopathy (358), and the CARPs P42-TAT (Table 2) (118) and TAT-P110 (Table 2) (149) inhibit aggregation of mutant Huntingtin protein. Interestingly, several of the key proteins that accumulate in the CNS in chronic neurodegenerative disorders are negatively-charged (e.g., Aβ 1–42: −2.7; tau: −6.2; α-synuclein: −8.9; Huntington: −59.7; SOD1: −5.5), which would increase their electrostatic affinity to positively-charged molecules, and make them ideal therapeutic targets for CARPs.
Endogenous CARPs and Neuroprotection
The endogenous PACAP38 peptide is a member of the secretin/glucagon/growth hormone-releasing hormone superfamily, and its neuroprotective properties have been discussed above. Dynorphins are widely distributed in the CNS and consist of two main peptides dynorphin A (Table 3) and dynorphin B (Table 3) that bind the κ-opioid receptor to induce analgesia (359). Interestingly, many other synthetic CARPs also have analgesic properties (see Tables 1, 2). Dynorphin A and dynorphin B are synthesized as the precursor protein predynorphin, which is then proteolytically cleaved to the smaller peptides. The dynorphin A peptide has also been shown to be neuroprotective in a rat stroke model (177).
Similarly, different classes of endogenous anti-microbial peptides (e.g., defensins, cathelicidins, bactenecin) have been derived from mammals, many of which are cationic and arginine-rich. Anti-microbial peptides are mainly produced by leukocytes and act as a defense against bacteria, fungi and viruses, and act either directly or by modulating inflammatory responses. Interestingly, several cationic arginine-rich anti-microbial peptides have also been shown to have neuroprotective properties in stroke, perinatal hypoxia-ischemia and traumatic brain injury animal models (Table 2).
Compounds Containing the Guanidinium Moiety and Neuroprotection
As mentioned above, arginine is unique in possessing a guanidinium head group, and most likely the critical element imparting the neuroprotective properties of CARPs. It is therefore not surprising that compounds containing the guanidinium moiety including arginine, arginine-based NOS inhibitors (e.g., L-NNA, L-NAME), the drugs metformin, phenformin, amiloride, and aminoguanidine, the toxin tetrodotoxin and the endogenous neuroactive molecule agmatine have neuroprotective properties in in vitro neuronal injury models (e.g., excitotoxicity, oxygen-glucose deprivation) and in animal models of stroke, perinatal hypoxia-ischemia, spinal cord injury, traumatic brain injury Parkinson's disease and Alzheimer's disease (Supplementary Table 3). It is thus conceivable that CARPs and other guanidinium moiety containing small molecules, at least in part, share the same neuroprotective mechanism of actions including anti-excitotoxic properties. In support of their anti-excitotoxic properties different guanidinium moiety containing molecules have been demonstrated to inhibit voltage gated and ligand-gated ion channels (230–237). However, because CARPs are multivalent guanidinium-agents they are likely to possess greater potency at the molar level than molecules that contain only one or several guanidine moieties, and have a greater capacity to traverse cell membranes.
Metformin and phenformin are biguanindine anti-hyperglycemic agents, which have been used for the treatment of diabetes for over 50 years. Like CARPs, metformin can activate AMPK signaling, target and suppress mitochondrial ROS production, limit calcium induced intracellular toxicity, scavenge methylglyoxal and reduce neuroinflammation (269). Aminoguanidine can also scavenge methylglyoxal and other dicarbynols (25). Agmatine is an endogenous divalent cationic guanidine. In the brain it is considered a putative neurotransmitter, in which it can be released from synaptic vesicles following membrane depolarization. It binds to various receptors (e.g., α2 adrenergic receptor) and can block NMDA receptors and other cation ligand-gated channels, with studies indicating that agmatine binds to the receptor near the channel pore and that the guanidinium group is critical for binding (360).
Arginine-based nitric oxide inhibitors, such as L-NNA and L-NAME are commonly used in in vitro excitotoxic and animal stroke studies to determine the neurodamaging role of nitric oxide over-production in neuronal death and ischemic brain tissue injury. However, given the potential anti-excitotoxic action of the guanidine moiety, it is possible that arginine-based NOS inhibitors in the setting of excitotoxicity are actually suppressing the activation of NMDA receptors and ion voltage gated channels, thereby indirectly rather than directly inhibiting NOS activation. There are several lines of evidence that support this hypothesis. Following excitotoxicity, it is difficult to imagine that by blocking neuronal nitric oxide production, but not the toxic intracellular influx of calcium is able to provide high level neuroprotection (361). In addition, arginine-based NOS inhibitors are not readily taken up by cells and possess slow NOS binding kinetics (362), but are neuroprotective when added at the same time as the excitotoxic agent, which favors an extracellular (i.e., cell surface) rather than an intracellular (i.e., NOS) mechanism of action. In addition, L-NAME is a weak NOS inhibitor, which is hydrolyzed by ubiquitous esterases to the more potent L-NNA, thus requiring additional time for the inhibitor to exert its NOS inhibitory effects.
Concluding Remarks
There is now overwhelming evidence from experimental studies that CARPs represent a novel class of neuroprotective agent with great potential for the treatment of neurological disorders. However, only two CARPs with neuroprotective properties (TAT-NR2B9c/NA-1 and CN-105) have so far progressed to clinical trials for a neurological condition (363–365). Further studies are required to obtain a more complete understanding of the neuroprotective mechanisms of action of CARPs in acute CNS injury and chronic neurodegenerative disease models. Despite this, based on experimental studies to date it appears that CARPs have the potential to be developed as therapeutics for the treatment of a diverse range of neurological disorders including stroke, perinatal hypoxia-ischemia, traumatic brain injury and spinal cord injury as well as, epilepsy and pain, and potentially even chronic degenerative neurological disorders, such as Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and Huntington's disease. Importantly, CARPs have properties that greatly enhance the likelihood of translational success at the clinical level including possessing a pluripotent mechanism of action, the capacity to enter the CNS, and the ability to exert a broad range of beneficial extracellular, intracellular and intra-organelle effects. Based on human studies with TAT-fused peptides, such as TAT-NR2B9c/NA-1 (366), the poly-arginine peptide R9/ALX40-4C (367), and arginine-rich peptides CN-105 (365), protamine (368) and RD2 (369), it appears this class of peptide has a favorable safety profile. Moreover, our recent experimental neuroprotection study with the poly-arginine peptide R18 in a non-human primate stroke model (26), has not disclosed any neurological or other toxic effects, which also augurs well for the translational potential of other CARPs to the clinical arena.
With respect to previous studies using cationic and arginine-rich peptides including those fused to a CCPP in neuroprotective, neuroactive or cytoprotective studies we believe that due to the cofounding effects of peptide positive charge and arginine residues, the mechanisms of action of theses peptides need to be critically re-evaluated.
Finally, given that CARPs with different amino acid sequences or modifications will have different physio-chemical and biological properties, future studies should focus on examining if new CARPs with more targeted molecular mechanisms of actions can be designed to improve therapeutic efficacy for specific neurological disorders.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
BM conceptualized and wrote the review. FM and NK provided the additional input into the content of the review and in drafting the manuscript. BM, FM, and NK contributed to the writing of the manuscript. BM prepared the figures and tables provided in the manuscript.
Conflict of Interest
BM and NK are named inventors of several patent applications regarding the use of CARPs as neuroprotective agents.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study has been supported by the Perron Institute and the Department of Neurosurgery, Sir Charles Gairdner Hospital. We thank Raphe Patmore for assistance with illustrations.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.00108/full#supplementary-material
References
1. Ferrer-Montiel AV, Merino JM, Blondelle SE, Perez-Payà E, Houghten RA, Montal M. Selected peptides targeted to the NMDA receptor channel protect neurons from excitotoxic death. Nat Biotechnol. (1998) 16:286–91. doi: 10.1038/nbt0398-286
2. Planells-Cases R, Aracil A, Merino JM, Gallar J, Pérez-Payá E, Belmonte C, et al. Arginine-rich peptides are blockers of VR-1 channels with analgesic activity. FEBS Lett. (2000) 481:131–36. doi: 10.1016/S0014-5793(00)01982-7
3. Yang L, Zhao K, Calingasan NY, Luo G, Szeto HH, Beal MF. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid Redox Signal. (2009) 11:2095–104. doi: 10.1089/ars.2009.2445
4. Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, et al. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer's disease neurons. J Alzheimers Dis. (2010) 20:609–31. doi: 10.3233/JAD-2010-100564
5. Kim EH, Tolhurst AT, Szeto HH, Cho SH. Targeting CD36-mediated inflammation reduces acute brain injury in transient but not permanent ischemic stroke. CNS Neurosci Ther. (2015) 21:385–91. doi: 10.1111/cns.12326
6. Zhu LL, Li MQ, He F, Zhou SB, Jiang W. Mitochondria targeted peptide attenuates mitochondrial dysfunction controls inflammation and protects against spinal cord injury-induced lung injury. Cell Physiol Biochem. (2017) 44:388–400. doi: 10.1159/000484919
7. Toyama S, Shimoyama N, Szeto HH, Schiller PW, Shimoyama M. Protective effect of a mitochondria-targeted peptide against the development of chemotherapy-induced peripheral neuropathy in mice. ACS Chem Neurosci. (2018) 9:1566–71. doi: 10.1021/acschemneuro.8b00013
8. Xu W, Zhou M, Baudry M. Neuroprotection by cell permeable TAT-mGluR1 peptide in ischemia: synergy between carrier and cargo sequences. Neuroscientist. (2008) 14:409–14. doi: 10.1177/1073858407309762
9. Vaslin A, Rummel C, Clarke PG. Unconjugated TAT carrier peptide protects against excitotoxicity. Neurotox Res. (2009) 15:123–6. doi: 10.1007/s12640-009-9012-6
10. Meade AJ, Meloni BP, Mastaglia FL, Watt PM, Knuckey NW. AP-1 inhibitory peptides attenuate in vitro cortical neuronal cell death induced by kainic acid. Brain Res. (2010) 1360:8–16. doi: 10.1016/j.brainres.2010.09.007
11. Meade AJ, Meloni BP, Cross JL, Bakker AJ, Fear MW, Mastaglia FL, et al. AP-1 inhibitory peptides are neuroprotective following acute glutamate excitotoxicity in primary cortical neuronal cultures. J Neurochem. (2010) 112:258–70. doi: 10.1111/j.1471-4159.2009.06459.x
12. Meloni BP, Craig AJ, Milech N, Hopkins RM, Watt PM, Knuckey NW. The neuroprotective efficacy of cell-penetrating peptides TAT penetratin Arg-9 and Pep-1 in glutamic acid kainic acid and in vitro ischemia injury models using primary cortical neuronal cultures. Cell Mol Neurobiol. (2014) 34:173–81. doi: 10.1007/s10571-013-9999-3
13. Craig AJ, Meloni BP, Watt PM, Knuckey NW. Attenuation of neuronal death by peptide inhibitors of AP-1 activation in acute and delayed in vitro ischaemia (oxygen/glucose deprivation) models. Int J Pept Res Ther. (2011) 17:1–6. doi: 10.1007/s10989-010-9234-8
14. Marshall J, Wong KY, Rupasinghe CN, Tiwari R, Zhao X, Berberoglu ED, Sinkler C, et al. Inhibition of N-methyl-D-aspartate-induced retinal neuronal death by polyarginine peptides is linked to the attenuation of stress-induced hyperpolarization of the inner mitochondrial membrane potential. J Biol Chem. (2015) 290:22030–48. doi: 10.1074/jbc.M115.662791
15. Meloni BP, Brookes LM, Clark VW, Cross JL, Edwards AB, Anderton RS, et al. Poly-arginine and arginine-rich peptides are neuroprotective in stroke models. J Cereb Blood Flow Metab. (2015) 35:993–1004. doi: 10.1038/jcbfm.2015.11
16. Meloni BP, Milani D, Cross JL, Clark VW, Edwards AB, Anderton RS, et al. Assessment of the neuroprotective effects of arginine-rich protamine peptides poly-arginine peptides (R12-cylic R22) and arginine-tryptophan containing peptides following in vitro excitotoxicity and/or permanent middle cerebral artery occlusion in rats. Neuromol Med. (2017) 19:271–85. doi: 10.1007/s12017-017-8441-2
17. Edwards AB, Cross JL, Anderton RS, Knuckey NW, Meloni BP. Characterisation of neuroprotective efficacy of modified poly-arginine-9 (R9) peptides in neuronal glutamic acid excitotoxicity model. Mol Cell Biochem. (2017) 426:75–85. doi: 10.1007/s11010-016-2882-z
18. Milani D, Clark VW, Cross JL, Anderton RS, Knuckey NW, Meloni BP. Poly-arginine peptides reduce infarct volume in a permanent middle cerebral artery rat stroke model. BMC Neurosci. (2016) 17:19. doi: 10.1186/s12868-016-0253-z
19. Milani D, Knuckey NW, Cross JL, Anderton RS, Meloni BP. The R18 polyarginine peptide is more effective than the TAT-NR2B9c (NA-1) peptide when administered 60 minutes after permanent middle cerebral artery occlusion in the rat. Stroke Res Treat. (2016) 2016:2372710. doi: 10.1155/2016/2372710
20. Milani D, Cross JL, Anderton RS, Blacker DJ, Knuckey NW, Meloni BP. Neuroprotective efficacy of R18 poly-arginine and NA-1 (TAT-NR2B9c) peptides following transient middle cerebral artery occlusion in the rat. Neurosci Res. (2017) 114:9–15. doi: 10.1016/j.neures.2016.09.002
21. Milani D, Bakeberg MC, Cross JL, Clark VW, Anderton RS, Blacker DJ, et al. Comparison of neuroprotective efficacy of poly-arginine R18 and R18D (D-enantiomer) peptides following permanent middle cerebral artery occlusion in the Wistar rat and in vitro toxicity studies. PLoS ONE. (2018) 13:e0193884. doi: 10.1371/journal.pone.0193884
22. Edwards AB, Cross JL, Anderton RS, Knuckey NW, Meloni BP. Poly-arginine R18 and R18D (D-enantiomer) peptides reduce infarct volume and improves behavioural outcomes following perinatal hypoxic-ischaemic encephalopathy in the P7 rat. Mol Brain. (2018) 11:1–12. doi: 10.1186/s13041-018-0352-0
23. Edwards AB, Anderton RS, Knuckey NW, Meloni BP. Assessment of therapeutic window for poly-arginine-18D (R18D) in a P7 rat model of perinatal hypoxic-ischaemic encephalopathy. J Neurosci Res. (2018) 96:1816–26. doi: 10.1002/jnr.24315
24. Chiu LS, Anderton RS, Cross JL, Clark VW, Edwards AB, Knuckey NW, et al. Assessment of neuroprotective peptides poly-arginine R18 COG1410 and APP96–110 experimental traumatic brain injury and in vitro neuronal excitotoxicity. Transl Neurosci. (2017) 15:147–57. doi: 10.1515/tnsci-2017-0021
25. Kikuchi S, Shinpo K, Moriwaka F, Makita Z, Miyata T, Tashiro K. Neurotoxicity of methylglyoxal and 3-deoxyglucosone on cultured cortical neurons: synergism between glycation and oxidative stress possibly involved in neurodegenerative diseases. J Neurosci Res. (1999) 57:280–9. doi: 10.1002/(SICI)1097-4547(19990715)57:2<280::AID-JNR14>3.0.CO;2-U
26. Meloni BP, Chen Y, Harrison KA, Nashed JY, Blacker DJ, South SM, et al. Poly-arginine peptide-18 (R18) reduces brain injury and improves functional outcomes in a nonhuman primate stroke model. Neurotherapeutics. (2019). doi: 10.1007/s13311-019-00809-1. [Epub ahead of print].
27. Batulu H, Du G-J, Li D-Z, Sailike D, Fan Y-H, Geng D. Effect of poly-arginine R18 on neurocyte cell growth via autophagy in traumatic brain injury. Exp Ther Med. (2019) 17:4109–15. doi: 10.3892/etm.2019.7423
28. van Groen T, Wiesehan K, Funke SA, Kadish I, Nagel-Steger L, Willbold D. Reduction of Alzheimer's disease amyloid plaque load in transgenic mice by D3, a D-enantiomeric peptide identified by mirror image phage display. ChemMedChem. (2008) 3:1848–52. doi: 10.1002/cmdc.200800273
29. van Groen T, Schemmert S, Brener O, Gremer L, Ziehm T, Tusche M, et al. The Aβ oligomer eliminating D-enantiomeric peptide RD2 improves cognition without changing plaque pathology. Sci Rep. (2017) 7:16275. doi: 10.1038/s41598-017-16565-1
30. Dunkelmann T, Teichmann K, Ziehm T, Schemmert S, Frenzel D, Tusche M, et al. Aβ oligomer eliminating compounds interfere successfully with pEAβ(3–42) induced motor neurodegenerative phenotype in transgenic mice. Neuropeptides. (2018) 67:27–35. doi: 10.1016/j.npep.2017.11.011
31. Bolouri H, Sävman K, Wang W, Thomas A, Maurer N, Dullaghan E, et al. Innate defense regulator peptide 1018 protects against perinatal brain injury. Ann Neurol. (2014) 75:395–41. doi: 10.1002/ana.24087
32. Chassagnon IR, McCarthy CA, Chin YK, Pineda SS, Keramidas A, Mobli M, et al. Potent neuroprotection after stroke afforded by a double-knot spider-venom peptide that inhibits acid-sensing ion channel 1a. Proc Natl Acad Sci USA. (2017) 114:3750–5. doi: 10.1073/pnas.1614728114
33. Corrigan F, Pham CL, Vink R, Blumbergs PC, Masters CL, van den Heuvel C, et al. The neuroprotective domains of the amyloid precursor protein in traumatic brain injury are located in the two growth factor domains. Brain Res. (2011) 1378:137–43. doi: 10.1016/j.brainres.2010.12.077
34. Corrigan F, Thornton E, Roisman LC, Leonard AV, Vink R, Blumbergs PC, et al. The neuroprotective activity of the amyloid precursor protein against traumatic brain injury is mediated via the heparin binding site in residues 96–110. J Neurochem. (2014) 128:196–204. doi: 10.1111/jnc.12391
35. Plummer SL, Corrigan F, Thornton E, Woenig JA, Vink R, Cappai R, et al. The amyloid precursor protein derivative APP96–110 is efficacious following intravenous administration after traumatic brain injury. PLoS ONE. (2018) 13:e0190449. doi: 10.1371/journal.pone.0190449
36. Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, et al. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. (2003) 278:48529–33. doi: 10.1074/jbc.M306923200
37. Aono M, Bennett ER, Kim KS, Lynch JR, Myers J, Pearlstein RD, et al. Protective effect of apolipoprotein E-mimetic peptides on N-methyl-D-aspartate excitotoxicity in primary rat neuronal-glial cell cultures. Neuroscience. (2003) 116:437–45. doi: 10.1016/S0306-4522(02)00709-1
38. Lynch JR, Wang H, Mace B, Leinenweber S, Warner DS, Bennett ER, et al. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp Neurol. (2005) 192:109–16. doi: 10.1016/j.expneurol.2004.11.014
39. McAdoo JD, Warner DS, Goldberg RN, Vitek MP, Pearlstein R, Laskowitz DT. Intrathecal administration of a novel apoE-derived therapeutic peptide improves outcome following perinatal hypoxic-ischemic injury. Neurosci Lett. (2005) 381:305–8. doi: 10.1016/j.neulet.2005.02.036
40. Li FQ, Sempowski GD, McKenna SE, Laskowitz DT, Colton CA, Vitek MP. Apolipoprotein E-derived peptides ameliorate clinical disability and inflammatory infiltrates into the spinal cord in a murine model of multiple sclerosis. J Pharmacol Exp Ther. (2006) 318:956–65. doi: 10.1124/jpet.106.103671
41. Sarantseva S, Timoshenko S, Bolshakova O, Karaseva E, Rodin D, Schwarzman AL, et al. Apolipoprotein E-mimetics inhibit neurodegeneration and restore cognitive functions in a transgenic Drosophila model of Alzheimer's disease. PLoS ONE. (2009) 4:e8191. doi: 10.1371/journal.pone.0008191
42. Gao J, Wang H, Sheng H, Lynch JR, Warner DS, Durham L, Vitek MP, et al. A novel apoE-derived therapeutic reduces vasospasm and improves outcome in a murine model of subarachnoid haemorrhage. Neurocrit Care. (2006) 4:25–31. doi: 10.1385/NCC:4:1:025
43. Hoane MR, Pierce JL, Holland MA, Birky ND, Dang T, Vitek MP, et al. The novel apolipoprotein E-based peptide COG1410 improves sensorimotor performance and reduces injury magnitude following cortical contusion injury. J Neurotrauma. (2007) 24:1108–18. doi: 10.1089/neu.2006.0254
44. Hoane MR, Kaufman N, Vitek MP, McKenna SE. COG1410 improves cognitive performance and reduces cortical neuronal loss in the traumatically injured brain. J Neurotrauma. (2009) 26:121–9. doi: 10.1089/neu.2008.0565
45. Jiang Y, Brody DL. Administration of COG1410 reduces axonal amyloid precursor protein immunoreactivity and microglial activation after controlled cortical impact in mice. J Neurotrauma. (2012) 29:2332–41. doi: 10.1089/neu.2012.2362
46. Kaufman NA, Beare JE, Tan AA, Vitek M P, McKenna SE, Hoane MR. COG1410 an apolipoprotein E-based peptide improves cognitive performance and reduces cortical loss following moderate fluid percussion injury in the rat. Behav Brain Res. (2010) 214:395–401. doi: 10.1016/j.bbr.2010.06.017
47. Laskowitz DT, McKenna SE, Song P, Wang H, Durham L, Yeung N, et al. COG1410 a novel apolipoprotein E-based peptide improves functional recovery in a murine model of traumatic brain injury. J Neurotrauma. (2007) 24:1093–97. doi: 10.1089/neu.2006.0192
48. Laskowitz DT, Lei B, Dawson HN, Wang H, Bellows ST, Christensen DJ, et al. The apoE-mimetic peptide COG1410 improves functional recovery in a murine model of intracerebral haemorrhage. Neurocrit Care. (2012) 16:316–26. doi: 10.1007/s12028-011-9641-5
49. James ML, Sullivan PM, Lascola CD, Vitek MP, Laskowitz DT. Pharmacogenomic effects of apolipoprotein E on intracerebral haemorrhage. Stroke. (2009) 40:632–9. doi: 10.1161/STROKEAHA.108.530402
50. Tukhovskaya EA, Yukin AY, Khokhlova ON, Murashev AN, Vitek MP. COG1410 a novel apolipoprotein-E mimetic improves functional and morphological recovery in a rat model of focal brain ischemia. J Neurosci Res. (2009) 87:677–82. doi: 10.1002/jnr.21874
51. Wang R, Hong J, Lu M, Neil JE, Vitek MP, Liu X, et al. ApoE mimetic ameliorates motor deficit and tissue damage in rat spinal cord injury. J Neurosci Res. (2014) 92:884–92. doi: 10.1002/jnr.23371
52. Wu Y, Pang J, Peng J, Cao F, Vitek MP, Li F, Jiang Y, et al. An apoE-derived mimic peptide COG1410 alleviates early brain injury via reducing apoptosis and neuroinflammation in a mouse model of subarachnoid haemorrhage. Neurosci Lett. (2016) 627:92–9. doi: 10.1016/j.neulet.2016.05.058
53. Lei B, James ML, Liu J, Zhou G, Venkatraman TN, Lascola CD, et al. Neuroprotective pentapeptide CN-105 improves functional and histological outcomes in a murine model of intracerebral haemorrhage. Sci Rep. (2016) 6:34834. doi: 10.1038/srep34834
54. Laskowitz DT, Wang H, Chen T, Lubkin DT, Cantillana V, Tu TM, et al. Neuroprotective pentapeptide CN-105 is associated with reduced sterile inflammation and improved functional outcomes in a traumatic brain injury murine model. Sci Rep. (2017) 7:46461. doi: 10.1038/srep46461
55. Tu TM, Kolls BJ, Soderblom EJ, Cantillana V, Ferrell PD, Moseley MA, et al. Apolipoprotein E mimetic peptide CN-105 improves outcomes in ischemic stroke. Ann Clin Transl Neurol. (2017) 4:246–65. doi: 10.1002/acn3.399
56. Zhao LR, Spellman S, Kim J, Duan WM, McCarthy JB, Low WC. Synthetic fibronectin peptide exerts neuroprotective effects on transient focal brain ischemia in rats. Brain Res. (2005) 1054:1–8. doi: 10.1016/j.brainres.2005.04.056
57. King VR, Hewazy D, Alovskaya A, Phillips JB, Brown RA, Priestley JV. The neuroprotective effects of fibronectin mats and fibronectin peptides following spinal cord injury in the rat. Neuroscience. (2010) 168:523–30. doi: 10.1016/j.neuroscience.2010.03.040
58. Khroyan TV, Polgar WE, Orduna J, Zaveri NT, Judd AK, Tuttle DJ, et al. Anti-nociceptive and anti-allodynic effects of a high affinity NOP hexapeptide [Ac-RY(3-Cl)YRWR-NH2] (Syn 1020) in rodents. Eur J Pharmacol. (2007) 560:29–35. doi: 10.1016/j.ejphar.2006.12.015
59. Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, et al. Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science. (2002) 298:846–50. doi: 10.1126/science.1072873
60. Soriano FX, Martel MA, Papadia S, Vaslin A, Baxter P, Rickman C, et al. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J Neurosci. (2008) 28:10696–710. doi: 10.1523/JNEUROSCI.1207-08.2008
61. Sun HS, Doucette TA, Liu Y, Fang Y, Teves L, Aarts M, et al. Effectiveness of PSD95 inhibitors in permanent and transient focal ischemia in the rat. Stroke. (2008) 39:2544–53. doi: 10.1161/STROKEAHA.107.506048
62. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. (2010) 142:387–97. doi: 10.1016/j.cell.2010.06.036
63. Bråtane BT, Cui H, Cook DJ, Bouley J, Tymianski M, Fisher M. Neuroprotection by freezing ischemic penumbra evolution without cerebral blood flow augmentation with a postsynaptic density-95 protein inhibitor. Stroke. (2011) 42:3265–70. doi: 10.1161/STROKEAHA.111.618801
64. D'Mello R, Marchand F, Pezet S, McMahon SB, Dickenson AH. Perturbing PSD-95 interactions with NR2B-subtype receptors attenuates spinal nociceptive plasticity and neuropathic pain. Mol Ther. (2012) 19:1780–92. doi: 10.1038/mt.2011.42
65. Cook DJ, Teves L, Tymianski M. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature. (2012) 483:213–7. doi: 10.1038/nature10841
66. Srejic LR, Hutchison WD, Aarts MM. Uncoupling PSD-95 interactions leads to rapid recovery of cortical function after focal stroke. J Cereb Blood Flow Metab. (2013) 33:1937–43. doi: 10.1038/jcbfm.2013.153
67. Xu B, Xiao AJ, Chen W, Turlova E, Liu R, Barszczyk A, et al. Neuroprotective effects of a PSD-95 inhibitor in neonatal hypoxic-ischemic brain injury. Mol Neurobiol. (2016) 53:5962–70. doi: 10.1007/s12035-015-9488-4
68. Wang Z, Chen Z, Yang J, Yang Z, Yin J, Duan X, et al. Treatment of secondary brain injury by perturbing postsynaptic density protein-95-NMDA receptor interaction after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab. (2019) 39:1588–601. doi: 10.1177/0271678X18762637
69. Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. (2003) 9:1180–86. doi: 10.1038/nm911
70. Hirt L, Badaut J, Thevenet J, Granziera C, Regli L, Maurer F, et al. D-JNKI1 a cell-penetrating c-Jun-N-terminal kinase inhibitor protects against cell death in severe cerebral ischemia. Stroke. (2004) 35:1738–43. doi: 10.1161/01.STR.0000131480.03994.b1
71. Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, et al. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. (2006) 26:3551–60. doi: 10.1523/JNEUROSCI.5290-05.2006
72. Centeno C, Repici M, Chatton JY, Riederer BM, Bonny C, Nicod P, et al. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ. (2007) 14:240–53. doi: 10.1038/sj.cdd.4401988
73. Esneault E, Castagne V, Moser P, Bonny C, Bernaudin M. D-JNKi a peptide inhibitor of c-Jun N-terminal kinase promotes functional recovery after transient focal cerebral ischemia in rats. Neuroscience. (2008) 152:308–20. doi: 10.1016/j.neuroscience.2007.12.036
74. Wiegler K, Bonny C, Coquoz D, Hirt L. The JNK inhibitor XG-102 protects from ischemic damage with delayed intravenous administration also in the presence of recombinant tissue plasminogen activator. Cerebrovasc Dis. (2008) 26:360–6. doi: 10.1159/000151639
75. Dykstra CM, Ratnam M, Gurd JW. Neuroprotection after status epilepticus by targeting protein interactions with postsynaptic density protein 95. J Neuropathol Exp Neurol. (2009) 68:823–31. doi: 10.1097/NEN.0b013e3181ac6b70
76. Ginet V, Puyal J, Magnin G, Clarke PG, Truttmann AC. Limited role of the c-Jun N-terminal kinase pathway in a neonatal rat model of cerebral hypoxia-ischemia. J Neurochem. (2009) 108:552–62. doi: 10.1111/j.1471-4159.2008.05797.x
77. Ortolano F, Colombo A, Zanier ER, Sclip A, Longhi L, Perego C, et al. c-Jun N-terminal kinase pathway activation in human and experimental cerebral contusion. J Neuropathol Exp Neurol. (2009) 68:964–71. doi: 10.1097/NEN.0b013e3181b20670
78. Bessero AC, Chiodini F, Rungger-Brändle E, Bonny C, Clarke PG. Role of the c-Jun N-terminal kinase pathway in retinal excitotoxicity and neuroprotection by its inhibition. J Neurochem. (2010) 113:1307–18. doi: 10.1111/j.1471-4159.2010.06705.x
79. Michel-Monigadon D, Bonny C, Hirt L. c-Jun N-terminal kinase pathway inhibition in intracerebral haemorrhage. Cerebrovasc Dis. (2010) 29:564–70. doi: 10.1159/000306643
80. Spigolon G, Veronesi C, Bonny C, Vercelli A. c-Jun N-terminal kinase signaling pathway in excitotoxic cell death following kainic acid-induced status epilepticus. Eur J Neurosci. (2010) 31:1261–72. doi: 10.1111/j.1460-9568.2010.07158.x
81. Sclip A, Antoniou X, Colombo A, Camici GG, Pozzi L, Cardinetti D, et al. c-Jun N-terminal kinase regulates soluble Aβ oligomers and cognitive impairment in AD mouse model. J Biol Chem. (2011) 286:43871–80. doi: 10.1074/jbc.M111.297515
82. Repici M, Chen X, Morel MP, Doulazmi M, Sclip A, Cannaya V, et al. Specific inhibition of the JNK pathway promotes locomotor recovery and neuroprotection after mouse spinal cord injury. Neurobiol Dis. (2012) 46:710–21. doi: 10.1016/j.nbd.2012.03.014
83. Schellino R, Boido M, Borsello T, Vercelli A. Pharmacological c-Jun NH2-terminal kinase (JNK) pathway inhibition reduces severity of spinal muscular atrophy disease in mice. Front Mol Neurosci. (2018) 11:308. doi: 10.3389/fnmol.2018.00308
84. Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. (2005) 25:694–712. doi: 10.1038/sj.jcbfm.9600062
85. Guan QH, Pei DS, Zong YY, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience. (2006) 139:609–27. doi: 10.1016/j.neuroscience.2005.11.067
86. Arthur PG, Matich GP, Pang WW, Yu DY, Bogoyevitch MA. Necrotic death of neurons following an excitotoxic insult is prevented by a peptide inhibitor of c-jun N-terminal kinase. J Neurochem. (2007) 102:65–76. doi: 10.1111/j.1471-4159.2007.04618.x
87. Bright R, Raval AP, Dembner JM, Pérez-Pinzón MA, Steinberg GK, Yenari MA, et al. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J Neurosci. (2004) 24:6880–8. doi: 10.1523/JNEUROSCI.4474-03.2004
88. Bright R, Steinberg GK, Mochly-Rosen D. DeltaPKC mediates microcerebrovascular dysfunction in acute ischemia and in chronic hypertensive stress in vivo. Brain Res. (2007) 1144:146–55. doi: 10.1016/j.brainres.2007.01.113
89. Nijboer CH, van der Kooij MA, Van Bel F, Ohl F, Heijnen CJ, Kavelaars A. Inhibition of the JNK/AP-1 pathway reduces neuronal death and improves behavioral outcome after neonatal hypoxic-ischemic brain injury. Brain Behav Immun. (2010) 24:812–21. doi: 10.1016/j.bbi.2009.09.008
90. Nijboer CH, Bonestroo HJ, Zijlstra J, Kavelaars A, Heijnen CJ. Mitochondrial JNK phosphorylation as a novel therapeutic target to inhibit neuroinflammation and apoptosis after neonatal ischemic brain damage. Neurobiol Dis. (2013) 54:432–44. doi: 10.1016/j.nbd.2013.01.017
91. Bach A, Clausen BH, Kristensen LK, Andersen MG, Ellman DG, Hansen PBL, et al. Selectivity efficacy and toxicity studies of UCCB01–144 a dimeric neuroprotective PSD-95 inhibitor. Neuropharmacology. (2019) 150:100–11. doi: 10.1016/j.neuropharm.2019.02.035
92. Bach A, Clausen BH, Møller M, Vestergaard B, Chi CN, Round A, et al. A high-affinity dimeric inhibitor of PSD-95 bivalently interacts with PDZ1–2 and protects against ischemic brain damage. Proc Natl Acad Sci USA. (2012) 109:3317–22. doi: 10.1073/pnas.1113761109
93. Andreasen JT, Nasser A, Caballero-Puntiverio M, Sahlholt M, Bach A, Gynther M, et al. Effects of the dimeric PSD-95 inhibitor UCCB01–144 in mouse models of pain cognition and motor function. Eur J Pharmacol. (2016) 780:166–73. doi: 10.1016/j.ejphar.2016.03.045
94. Kucharz K, Søndergaard Rasmussen I, Bach A, Strømgaard K, Lauritzen M. PSD-95 uncoupling from NMDA receptors by Tat-N-dimer ameliorates neuronal depolarization in cortical spreading depression. J Cereb Blood Flow Metab. (2017) 37:1820–8. doi: 10.1177/0271678X16645595
95. Colciaghi F, Nobili P, Cipelletti B, Cagnoli C, Zambon S, Locatelli D, et al. Targeting PSD95-nNOS interaction by Tat-N-dimer peptide during status epilepticus is neuroprotective in MAM-pilocarpine rat model. Neuropharmacology. (2019) 153:82–97. doi: 10.1016/j.neuropharm.2019.04.028
96. Pan J, Qian J, Zhang Y, Ma J, Wang G, Xiao Q, et al. Small peptide inhibitor of JNKs protects against MPTP-induced nigral dopaminergic injury via inhibiting the JNK-signaling pathway. Lab Invest. (2010) 90:156–67. doi: 10.1038/labinvest.2009.124
97. Pan J, Li H, Zhang B, Xiong R, Zhang Y, Kang WY, et al. Small peptide inhibitor of JNK3 protects dopaminergic neurons from MPTP induced injury via inhibiting the ASK1-JNK3 signaling pathway. PLoS ONE. (2015) 10:e0119204. doi: 10.1371/journal.pone.0119204
98. Liu XJ, Gingrich JR, Vargas-Caballero M, Dong YN, Sengar A, Beggs S, et al. Treatment of inflammatory and neuropathic pain by uncoupling Src from the NMDA receptor complex. Nat Med. (2008) 14:1325–32. doi: 10.1038/nm.1883
99. Chambers JW, Howard S, LoGrasso PV. Blocking c-Jun N-terminal kinase (JNK) translocation to the mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro and in vivo. J Biol Chem. (2013) 288:1079–87. doi: 10.1074/jbc.M112.421354
100. Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, et al. Neuroprotection against traumatic brain injury by a peptide derived from the collapsin response mediator protein 2 (CRMP2). J Biol Chem. (2011) 286:37778–92. doi: 10.1074/jbc.M111.255455
101. Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat Med. (2011) 17:822–9. doi: 10.1038/nm.2345
102. Brittain JM, Pan R, You H, Brustovetsky T, Brustovetsky N, Zamponi GW, et al. Disruption of NMDAR-CRMP-2 signaling protects against focal cerebral ischemic damage in the rat middle cerebral artery occlusion model. Channels. (2012) 6:52–9. doi: 10.4161/chan.18919
103. Piekarz AD, Due MR, Khanna M, Wang B, Ripsch MS, Wang R, et al. CRMP-2 peptide mediated decrease of high and low voltage-activated calcium channels attenuation of nociceptor excitability and anti-nociception in a model of AIDS therapy-induced painful peripheral neuropathy. Mol Pain. (2012) 8:2–19. doi: 10.1186/1744-8069-8-54
104. Ripsch MS, Ballard CJ, Khanna M, Hurley JH, White FA, Khanna R. A peptide uncoupling CRMP-2 from the presynaptic Ca2+ channel complex demonstrates efficacy in animal models of migraine and aids therapy-induced neuropathy. Transl Neurosci. (2012) 3:1–8. doi: 10.2478/s13380-012-0002-4
105. Moutal A, François-Moutal L, Brittain JM, Khanna M, Khanna R. Differential neuroprotective potential of CRMP2 peptide aptamers conjugated to cationic hydrophobic and amphipathic cell penetrating peptides. Front Cell Neurosci. (2015) 8:471. doi: 10.3389/fncel.2014.00471
106. Bu X, Nan Zhang N, Yang X, Liu Y, Du J, Liang J, et al. Proteomic analysis of cPKCβII-interacting proteins involved in HPC-induced neuroprotection against cerebral ischemia of mice. J Neurochem. (2011) 117:346–56. doi: 10.1111/j.1471-4159.2011.07209.x
107. Yin Y, Wang Y, Chen L, Han S, Zhao L, Luo Y, et al. Tat-collapsin response mediator protein 2 (CRMP2) increases the survival of neurons after NMDA excitotoxity by reducing the cleavage of CRMP2. Neurochem Res. (2013) 38:2095–104. doi: 10.1007/s11064-013-1118-9
108. Yang X, Zhang X, Li Y, Han S, Howells DW, Li S, et al. Conventional protein kinase Cβ-mediated phosphorylation inhibits collapsin response-mediated protein 2 proteolysis and alleviates ischemic injury in cultured cortical neurons and ischemic stroke-induced mice. J Neurochem. (2016) 137:446–59. doi: 10.1111/jnc.13538
109. Meloni BP, Milani D, Edwards AB, Anderton RS, O'Hare Doig RL, Fitzgerald M, et al. Neuroprotective peptides fused to arginine-rich cell penetrating peptides: neuroprotective mechanism likely mediated by peptide endocytic properties. Pharmacol Ther. (2015) 153:36–54. doi: 10.1016/j.pharmthera.2015.06.002
110. McQueen J, Ryan TJ, McKay S, Marwick K, Baxter P, Carpanini SM, et al. Pro-death NMDA receptor signaling is promoted by the GluN2B C-terminus independently of Dapk1. Elife. (2017) 6:e17161. doi: 10.7554/eLife.17161.024
111. Tu W, Xu X, Peng L, Zhong X, Zhang W, Soundarapandian MM, et al. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell. (2010) 140:222–34. doi: 10.1016/j.cell.2009.12.055
112. Pei L, Li S, Wang M, Diwan M, Anisman H, Fletcher PJ, et al. Uncoupling the dopamine D1-D2 receptor complex exerts antidepressant-like effects. Nat Med. (2010) 16:1393–5. doi: 10.1038/nm.2263
113. Li FQ, Fowler KA, Neil JE, Colton CA, Vitek MP. An apolipoprotein E-mimetic stimulates axonal regeneration and remyelination after peripheral nerve injury. J Pharmacol Exp Ther. (2010) 334:106–15. doi: 10.1124/jpet.110.167882
114. Ghosal K, Stathopoulos A, Thomas D, Phenis D, Vitek MP, Pimplikar SW. The apolipoprotein-E-mimetic COG112 protects amyloid precursor protein intracellular domain-overexpressing animals from Alzheimer's disease-like pathological features. Neurodegener Dis. (2013) 12:51–8. doi: 10.1159/000341299
115. Gu Z, Li F, Zhang YP, Shields LB, Hu X, Zheng Y, et al. Apolipoprotein E mimetic promotes functional and histological recovery in lysolecithin-induced spinal cord demyelination in mice. J Neurol Neurophysiol. (2013) 2014:10. doi: 10.4172/2155-9562.S12-010
116. Fan X, Jin W Y, Lu J, Wang J, Wang YT. Rapid and reversible knockdown of endogenous proteins by peptide-directed lysosomal degradation. Nat Neurosci. (2014) 17:471–80. doi: 10.1038/nn.3637
117. LeBlanc BW, Iwata M, Mallon AP, Rupasinghe CN, Goebel DJ, Marshall J, et al. A cyclic peptide targeted against PSD-95 blocks central sensitization and attenuates thermal hyperalgesia. Neuroscience. (2010) 167:490–500. doi: 10.1016/j.neuroscience.2010.02.031
118. Arribat Y, Talmat-Amar Y, Paucard A, Lesport P, Bonneaud N, Bauer C, et al. Systemic delivery of P42 peptide: a new weapon to fight Huntington's disease. Acta Neuropathol Commun. (2014) 2:86. doi: 10.1186/s40478-014-0086-x
119. Pei L, Shang Y, Jin H, Wang S, Wei N, Yan H, et al. DAPK1-p53 interaction converges necrotic and apoptotic pathways of ischemic neuronal death. J Neurosci. (2014) 34:6546–56. doi: 10.1523/JNEUROSCI.5119-13.2014
120. Wang X, Pei L, Yan H, Wang Z, Wei N, Wang S, et al. Intervention of death-associated protein kinase 1-p53 interaction exerts the therapeutic effects against stroke. Stroke. (2014) 45:3089–91. doi: 10.1161/STROKEAHA.114.006348
121. Vest RS, O'Leary H, Coultrap SJ, Kindy MS, Bayer KU. Effective post-insult neuroprotection by a novel Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor. J Biol Chem. (2010) 285:20675–82. doi: 10.1074/jbc.M109.088617
122. Ashpole NM, Hudmon A. Excitotoxic neuroprotection and vulnerability with CaMKII inhibition. Mol Cell Neurosci. (2011) 46:720–30. doi: 10.1016/j.mcn.2011.02.003
123. Ahmed ME, Dong Y, Lu Y, Tucker D, Wang R, Zhang Q. Beneficial effects of a CaMKIIα inhibitor TatCN21 peptide in global cerebral ischemia. J Mol Neurosci. (2017) 61:42–51. doi: 10.1007/s12031-016-0830-8
124. Pei DS, Wang XT, Liu Y, Sun YF, Guan QH, Wang W, et al. Neuroprotection against ischaemic brain injury by a GluR6–9c peptide containing the TAT protein transduction sequence. Brain. (2006) 129:465–79. doi: 10.1093/brain/awh700
125. Liu XM, Pei DS, Guan QH, Sun YF, Wang XT, Zhang QX, et al. Neuroprotection of Tat-GluR6–9c against neuronal death induced by kainate in rat hippocampus via nuclear and non-nuclear pathways. J Biol Chem. (2006) 281:17432–45. doi: 10.1074/jbc.M513490200
126. Yu CZ, Li C, Pei DS, Zong YY, Shi Q, Wen XR, et al. Neuroprotection against transient focal cerebral ischemia and oxygen-glucose deprivation by interference with GluR6-PSD95 protein interaction. Neurochem Res. (2009) 34:2008–21. doi: 10.1007/s11064-009-9990-z
127. Xu W, Wong TP, Chery N, Gaertner T, Wang YT, Baudry M. Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. (2007) 53:399–412. doi: 10.1016/j.neuron.2006.12.020
128. Zhou M, Xu W, Liao G, Bi X, Baudry M. Neuroprotection against neonatal hypoxia/ischemia-induced cerebral cell death by prevention of calpain-mediated mGluR1alpha truncation. Exp Neurol. (2009) 218:75–82. doi: 10.1016/j.expneurol.2009.04.006
129. Wang W, Han P, Xie R, Yang M, Zhang C, Mi Q, et al. TAT-mGluR1 attenuation of neuronal apoptosis through prevention of mGluR1α truncation after experimental subarachnoid hemorrhage. ACS Chem Neurosci. (2018) 10:746–56. doi: 10.1021/acschemneuro.8b00531
130. Zhang S, Taghibiglou C, Girling K, Dong Z, Lin SZ, Lee W, et al. Critical role of increased PTEN nuclear translocation in excitotoxic and ischemic neuronal injuries. J Neurosci. (2013) 33:7997–8008 doi: 10.1523/JNEUROSCI.5661-12.2013
131. Taghibiglou C, Martin HG, Lai TW, Cho T, Prasad S, Kojic L, et al. Role of NMDA receptor-dependent activation of SREBP1 in excitotoxic and ischemic neuronal injuries. Nat Med. (2009) 15:1399–406. doi: 10.1038/nm.2064
132. Li LL, Ginet V, Liu X, Vergun O, Tuittila M, Mathieu M, et al. The nNOS-p38MAPK pathway is mediated by NOS1AP during neuronal death. J Neurosci. (2013) 33:8185–201. doi: 10.1523/JNEUROSCI.4578-12.2013
133. Li LL, Melero-Fernandez de Mera RM, Chen J, Ba W, Kasri NN, Zhang M, et al. Unexpected heterodivalent recruitment of NOS1AP to nNOS reveals multiple sites for pharmacological intervention in neuronal disease models. J Neurosci. (2015) 35:7349–64. doi: 10.1523/JNEUROSCI.0037-15.2015
134. Lee WH, Li LL, Chawla A, Hudmon A, Lai YY, Courtney MJ, et al. Disruption of nNOS-NOS1AP protein-protein interactions suppresses neuropathic pain in mice. Pain. (2018) 159:849–63. doi: 10.1097/j.pain.0000000000001152
135. Wang Q, Gou X, Xiong L, Jin W, Chen S, Hou L, et al. Trans-activator of transcription-mediated delivery of NEP1–40 protein into brain has a neuroprotective effect against focal cerebral ischemic injury via inhibition of neuronal apoptosis. Anesthesiology. (2008) 108:1071–80. doi: 10.1097/ALN.0b013e318173f66b
136. Wang Q, Gou X, Jin W, Xiong L, Hou L, Chen S, et al. TAT-mediated protein transduction of Nogo extracellular peptide 1–40 and its biological activity. Cell Mol Neurobiol. (2009) 29:97–108. doi: 10.1007/s10571-008-9301-2
137. van der Kooij MA, Nijboer CH, Ohl F, Groenendaal F, Heijnen CJ, van Bel F, et al. NF-kappaB inhibition after neonatal cerebral hypoxia-ischemia improves long-term motor and cognitive outcome in rats. Neurobiol Dis. (2010) 38:266–72. doi: 10.1016/j.nbd.2010.01.016
138. Sun X, Budas GR, Xu L, Barreto GE, Mochly-Rosen D, Giffard RG. Selective activation of protein kinase Cϵ in mitochondria is neuroprotective in vitro and reduces focal ischemic brain injury in mice. J Neurosci Res. (2013) 91:799–807. doi: 10.1002/jnr.23186
139. He M, Ding Y, Chu C, Tang J, Xiao Q, Luo ZG. Autophagy induction stabilizes microtubules and promotes axon regeneration after spinal cord injury. Proc Natl Acad Sci USA. (2016) 113:11324–9. doi: 10.1073/pnas.1611282113
140. Zhang L, Li Z, Feng D, Shen H, Tian X, Li H, et al. Involvement of Nox2 and Nox4 NADPH oxidases in early brain injury after subarachnoid haemorrhage. Free Radic Res. (2017) 51:316–28. doi: 10.1080/10715762.2017.1311015
141. Khayrullina G, Bermudez S, Byrnes KR. Inhibition of NOX2 reduces locomotor impairment inflammation and oxidative stress after spinal cord injury. J Neuroinflammation. (2015) 12:172. doi: 10.1186/s12974-015-0391-8
142. Zhang QG, Laird MD, Han D, Nguyen K, Scott E, Dong Y, et al. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS ONE. (2012) 7:e34504. doi: 10.1371/journal.pone.0034504
143. Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, et al. Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature. (2015) 518:404–8. doi: 10.1038/nature13974
144. García-Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav32 channel activity. Neuron. (2014) 83:1144–58. doi: 10.1016/j.neuron.2014.07.036
145. Ozaki T, Ishiguro S, Hirano S, Baba A, Yamashita T, Tomita H, et al. Inhibitory peptide of mitochondrial μ-calpain protects against photoreceptor degeneration in rhodopsin transgenic S334ter and P23H rats. PLoS ONE. (2013) 8:e71650. doi: 10.1371/annotation/7a8aaf1d-e968-4b39-abb0-867d6078b2af
146. Ju W, Li Q, Allette YM, Ripsch MS, White FA, Khanna R. Suppression of pain-related behavior in two distinct rodent models of peripheral neuropathy by a homopolyarginine-conjugated CRMP2 peptide. J Neurochem. (2013) 124:869–79. doi: 10.1111/jnc.12070
147. Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, et al. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. (2009) 29:9330–43. doi: 10.1523/JNEUROSCI.2212-09.2009
148. Gamir-Morralla A, López-Menéndez C, Ayuso-Dolado S, Tejeda GS, Montaner J, Rosell A, et al. Development of a neuroprotective peptide that preserves survival pathways by preventing Kidins220/ARMS calpain processing induced by excitotoxicity. Cell Death Dis. (2015) 6:e1939. doi: 10.1038/cddis.2015.307
149. Disatnik MH, Joshi AU, Saw NL, Shamloo M, Leavitt BR, Qi X, et al. Potential biomarkers to follow the progression and treatment response of Huntington's disease. J Exp Med. (2016) 213:2655–69. doi: 10.1084/jem.20160776
150. Guo X, Sesaki H, Qi X. Drp1 stabilizes p53 on the mitochondria to trigger necrosis under oxidative stress conditions in vitro and in vivo. Biochem J. (2014) 461:137–46. doi: 10.1042/BJ20131438
151. Du W, Huang J, Yao H, Zhou K, Duan B, Wang Y. Inhibition of TRPC6 degradation suppresses ischemic brain damage in rats. J Clin Invest. (2010) 120:3480–92. doi: 10.1172/JCI43165
152. Wang CL, Qiu TT, Diao YX, Zhang Y, Gu N. Novel endomorphin-1 analogs with C-terminal oligoarginine-conjugation display systemic antinociceptive activity with less gastrointestinal side effects. Biochimie. (2015) 116:24–33. doi: 10.1016/j.biochi.2015.06.008
153. Cimini S, Sclip A, Mancini S, Colombo L, Messa M, Cagnotto A, et al. The cell-permeable Aβ1–6A2VTAT(D) peptide reverts synaptopathy induced by Aβ1–42wt. Neurobiol Dis. (2016) 89:101–11. doi: 10.1016/j.nbd.2015.12.013
154. Tu J, Zhang X, Zhu Y, Dai Y, Li N, Yang F, et al. Cell-permeable peptide targeting the Nrf2-Keap1 interaction: a potential novel therapy for global cerebral ischemia. J Neurosci. (2015) 35:14727–39. doi: 10.1523/JNEUROSCI.1304-15.2015
155. Liu J, Du J, Yang Y, Wang Y. Phosphorylation of TRPV1 by cyclin-dependent kinase 5 promotes TRPV1 surface localization leading to inflammatory thermal hyperalgesia. Exp Neurol. (2015) 273:253–62. doi: 10.1016/j.expneurol.2015.09.005
156. Zhang Y, Su P, Liang P, Liu T, Liu X, Liu XY, et al. The DREAM protein negatively regulates the NMDA receptor through interaction with the NR1 subunit. J Neurosci. (2010) 30:7575–86. doi: 10.1523/JNEUROSCI.1312-10.2010
157. Yeh CY, Bulas AM, Moutal A, Saloman JL, Hartnett KA, Anderson CT, et al. Targeting a potassium channel/syntaxin interaction ameliorates cell death in ischemic stroke. J Neurosci. (2017) 37 5648–58. doi: 10.1523/JNEUROSCI.3811-16.2017
158. Wattiez AS, Dupuis A, Privat AM, Chalus M, Chapuy E, Aissouni Y, et al. Disruption of 5-HT2A-PDZ protein interaction differently affects the analgesic efficacy of SSRI SNRI and TCA in the treatment of traumatic neuropathic pain in rats. Neuropharmacology. (2017) 125:308–18. doi: 10.1016/j.neuropharm.2017.07.034
159. Li X, Zheng L, Xia Q, Liu L, Mao M, Zhou H, et al. A novel cell-penetrating peptide protects against neuron apoptosis after cerebral ischemia by inhibiting the nuclear translocation of annexin A1. Cell Death Differ. (2019) 26:260–75. doi: 10.1038/s41418-018-0116-5
160. Moutal A, Li W, Wang Y, Ju W, Luo S, Cai S, et al. Homology-guided mutational analysis reveals the functional requirements for antinociceptive specificity of collapsin response mediator protein 2-derived peptides. Br J Pharmacol. (2018) 175:2244–60. doi: 10.1111/bph.13737
161. Kashkin VA, Shekunova EV, Titov MI, Eliseev II, Gureev MA, Porozov YB, et al. A new tridecapeptide with an octaarginine vector has analgesic therapeutic potential and prevents morphine-induced tolerance. Peptides. (2018) 99:61–9. doi: 10.1016/j.peptides.2017.11.011
162. Vezzoli E, Caron I, Talpo F, Besusso D, Conforti P, Battaglia E, et al. Inhibiting pathologically active ADAM10 rescues synaptic and cognitive decline in Huntington's disease. J Clin Invest. (2019) 130:120616. doi: 10.1172/JCI120616
163. O'Donnell LA, Agrawal A, Sabnekar P, Dichter MA, Lynch DR, Kolson DL. Apelin an endogenous neuronal peptide protects hippocampal neurons against excitotoxic injury. J Neurochem. (2007) 102:1905–17. doi: 10.1111/j.1471-4159.2007.04645.x
164. Cook DR, Gleichman AJ, Cross SA, Doshi S, Ho W, Jordan-Sciutto KL, et al. NMDA receptor modulation by the neuropeptide apelin: implications for excitotoxic injury. J Neurochem. (2011) 118:1113–23. doi: 10.1111/j.1471-4159.2011.07383.x
165. Sakamoto K, Murakami Y, Sawada S, Ushikubo H, Mori A, Nakahara T, et al. Apelin-36 is protective against N-methyl-D-aspartic-acid-induced retinal ganglion cell death in the mice. Eur J Pharmacol. (2016) 791:213–20. doi: 10.1016/j.ejphar.2016.08.036
166. Ishimaru Y, Sumino A, Kajioka D, Shibagaki F, Yamamuro A, Yoshioka Y, et al. Apelin protects against NMDA-induced retinal neuronal death via an APJ receptor by activating Akt and ERK1/2 and suppressing TNF-α expression in mice. J Pharmacol Sci. (2017) 133:34–41. doi: 10.1016/j.jphs.2016.12.002
167. Gu Q, Zhai L, Feng X, Chen J, Miao Z, Ren L, et al. Apelin-36 a potent peptide protects against ischemic brain injury by activating the PI3K/Akt pathway. Neurochem Int. (2013) 63:535–40. doi: 10.1016/j.neuint.2013.09.017
168. Chen D, Lee J, Gu X, Wei L, Yu SP. Intranasal delivery of apelin-13 is neuroprotective and oromotes angiogenesis after ischemic stroke in mice. ASN Neuro. (2015) 7:1–15. doi: 10.1177/1759091415605114
169. Yang Y, Zhang XJ, Li LT, Cui HY, Zhang C, Zhu CH, et al. Apelin-13 protects against apoptosis by activating AMP-activated protein kinase pathway in ischemia stroke. Peptides. (2016) 75:96–100. doi: 10.1016/j.peptides.2015.11.002
170. Qiu J, Wang X, Wu F, Wan L, Cheng B, Wu Y, et al. Low dose of apelin-36 attenuates ER stress-associated apoptosis in rats with ischemic stroke. Front Neurol. (2017) 8:556. doi: 10.2174/97816810853261170401
171. Xu N, Wang H, Fan L, Chen Q. Supraspinal administration of apelin-13 induces antinociception via the opioid receptor in mice. Peptides. (2009) 30:1153–7. doi: 10.1016/j.peptides.2009.02.011
172. Khaksari M, Aboutaleb N, Nasirinezhad F, Vakili A, Madjd Z. Apelin-13 protects the brain against ischemic reperfusion injury and cerebral edema in a transient model of focal cerebral ischemia. J Mol Neurosci. (2012) 48:201–8. doi: 10.1007/s12031-012-9808-3
173. Bao H, Yang X, Huang Y, Qiu H, Huang G, Xiao H, et al. The neuroprotective effect of apelin-13 in a mouse model of intracerebral haemorrhage. Neurosci Lett. (2016) 628:219–24. doi: 10.1016/j.neulet.2016.06.046
174. Bao HJ, Zhang L, Han WC, Dai DK. Apelin-13 attenuates traumatic brain injury-induced damage by suppressing autophagy. Neurochem Res. (2015) 40:89–97. doi: 10.1007/s11064-014-1469-x
175. Xin Q, Cheng B, Pan Y, Liu H, Yang C, Chen J, et al. Neuroprotective effects of apelin-13 on experimental ischemic stroke through suppression of inflammation. Peptides. (2015) 63:55–62. doi: 10.1016/j.peptides.2014.09.016
176. Hajimashhadi Z, Aboutaleb N, Nasirinezhad F. Chronic administration of [Pyr1] apelin-13 attenuates neuropathic pain after compression spinal cord injury in rats. Neuropeptides. (2017) 61:15–22. doi: 10.1016/j.npep.2016.08.010
177. Kao TK, Ou YC, Liao SL, Chen WY, Wang CC, Chen SY, et al. Opioids modulate post-ischemic progression in a rat model of stroke. Neurochem Int. (2008) 52:1256–65. doi: 10.1016/j.neuint.2008.01.007
178. Liu B, Qin L, Yang SN, Wilson BC, Liu Y, Hong JS. Femtomolar concentrations of dynorphins protect rat mesencephalic dopaminergic neurons against inflammatory damage. J Pharmacol Exp Ther. (2001) 298:1133–41.
179. Hall SM, Lee YS, Hruby VJ. Dynorphin A analogs for the treatment of chronic neuropathic pain. Future Med Chem. (2016) 8:165–77. doi: 10.4155/fmc.15.164
180. Yamamoto T, Tatsuno I. Antinociceptive effect of intrathecally administered pituitary adenylate cyclase activating polypeptide (PACAP) on the rat formalin test. Neurosci Lett. (1995) 184:32–5. doi: 10.1016/0304-3940(94)11161-B
181. Uchida D, Arimura A, Somogyvári-Vigh A, Shioda S, Banks WA. Prevention of ischemia-induced death of hippocampal neurons by pituitary adenylate cyclase activating polypeptide. Brain Res. (1996) 736:280–6. doi: 10.1016/0006-8993(96)00716-0
182. Shoge K, Mishima HK, Saitoh T, Ishihara K, Tamura Y, Shiomi H, et al. Attenuation by PACAP of glutamate-induced neurotoxicity in cultured retinal neurons. Brain Res. (1999) 839:66–73. doi: 10.1016/S0006-8993(99)01690-X
183. Reglodi D, Borzsei R, Bagoly T, Boronkai A, Racz B, Tamas A, et al. Agonistic behavior of PACAP6–38 on sensory nerve terminals and cytotrophoblast cells. J Mol Neurosci. (2008) 36:270–8. doi: 10.1007/s12031-008-9089-z
184. Reglodi D, Lubics A, Tamás A, Szalontay L, Lengvári I. Pituitary adenylate cyclase activating polypeptide protects dopaminergic neurons and improves behavioral deficits in a rat model of Parkinson's disease. Behav Brain Res. (2004) 151:303–12. doi: 10.1016/j.bbr.2003.09.007
185. Reglodi D, Tamás A, Somogyvári-Vigh A, Szántó Z, Kertes E, Lénárd L, et al. Effects of pretreatment with PACAP on the infarct size and functional outcome in rat permanent focal cerebral ischemia. Peptides. (2002) 23:2227–34. doi: 10.1016/S0196-9781(02)00262-0
186. Sibilia V, Lattuada N, Rapetti D, Pagani F, Vincenza D, Bulgarelli I, et al. Ghrelin inhibits inflammatory pain in rats: involvement of the opioid system. Neuropharmacology. (2006) 51:497–505. doi: 10.1016/j.neuropharm.2006.04.009
187. Erşahin M, Toklu HZ, Erzik C, Cetinel S, Akakin D, Velioglu-Ogünç A, et al. The anti-inflammatory and neuroprotective effects of ghrelin in subarachnoid hemorrhage-induced oxidative brain damage in rats. J Neurotrauma. (2010) 27:1143–55. doi: 10.1089/neu.2009.1210
188. Andrews ZB, Erion D, Beiler R, Liu ZW, Abizaid A, Zigman J, et al. Ghrelin promotes and protects nigrostriatal dopamine function via a UCP2-dependent mitochondrial mechanism. J Neurosci. (2009) 29:14057–65. doi: 10.1523/JNEUROSCI.3890-09.2009
189. Hwang S, Moon M, Kim S, Hwang L, Ahn KJ, Park S. Neuroprotective effect of ghrelin is associated with decreased expression of prostate apoptosis response-4. Endocr J. (2009) 56:609–17. doi: 10.1507/endocrj.K09E-072
190. Lopez NE, Gaston L, Lopez KR, Coimbra RC, Hageny A, Putnam J, et al. Early ghrelin treatment attenuates disruption of the blood brain barrier and apoptosis after traumatic brain injury through a UCP-2 mechanism. Brain Res. (2012) 1489:140–8. doi: 10.1016/j.brainres.2012.10.031
191. Ge T, Yang W, Fan J, Li B. Preclinical evidence of ghrelin as a therapeutic target in epilepsy. Oncotarget. (2017) 8:59929–39. doi: 10.18632/oncotarget.18349
192. Eslami M, Sadeghi B, Goshadrou F. Chronic ghrelin administration restores hippocampal long-term potentiation and ameliorates memory impairment in rat model of Alzheimer's disease. Hippocampus. (2018) 28:724–34. doi: 10.1002/hipo.23002
193. Xu X, Chua CC, Gao J, Hamdy RC, Chua BH. Humanin is a novel neuroprotective agent against stroke. Stroke. (2006) 37:2613–19. doi: 10.1161/01.STR.0000242772.94277.1f
194. Niikura T, Sidahmed E, Hirata-Fukae C, Aisen PS, Matsuoka Y. A humanin derivative reduces amyloid beta accumulation and ameliorates memory deficit in triple transgenic mice. PLoS ONE. (2011) 6:e16259. doi: 10.1371/journal.pone.0016259
195. Wang T, Huang Y, Zhang M, Wang L, Wang Y, Zhang L, et al. [Gly14]-Humanin offers neuroprotection through glycogen synthase kinase-3β inhibition in a mouse model of intracerebral hemorrhage. Behav Brain Res. (2013) 247:132–39. doi: 10.1016/j.bbr.2013.03.023
196. Chen J, Sun M, Zhang X, Miao Z, Chua BH, Hamdy RC, et al. Increased oligodendrogenesis by humanin promotes axonal remyelination and neurological recovery in hypoxic/ischemic brains. Hippocampus. (2015) 25:62–71. doi: 10.1002/hipo.22350
197. Yang X, Zhang H, Wu J, Yin L, Yan LJ, Zhang C. Humanin attenuates NMDA-induced excitotoxicity by inhibiting ROS-dependent JNK/p38 MAPK pathway. Int J Mol Sci. (2018) 19:E2982. doi: 10.3390/ijms19102982
198. Ross CR, Ricevuti G, Scovassi AI. The antimicrobial peptide PR-39 has a protective effect against HeLa cell apoptosis. Chem Biol Drug Des. (2007) 70:154–57. doi: 10.1111/j.1747-0285.2007.00540.x
199. Wu J, Parungo C, Wu G, Kang PM, Laham RJ, Sellke FW, et al. PR39 inhibits apoptosis in hypoxic endothelial cells: role of inhibitor apoptosis protein-2. Circulation. (2004) 109:1660–7. doi: 10.1161/01.CIR.0000124067.35915.E0
200. Bao J, Sato K, Li M, Gao Y, Abid R, Aird W, et al. PR-39 and PR-11 peptides inhibit ischemia-reperfusion injury by blocking proteasome-mediated I kappa B alpha degradation. Am J Physiol Heart Circ Physiol. (2001) 281:2612–18. doi: 10.1152/ajpheart.2001.281.6.H2612
201. Sarko D, Beijer B, Garcia Boy R, Nothelfer EM, Leotta K, Eisenhut M, et al. The pharmacokinetics of cell-penetrating peptides. Mol Pharm. (2010) 7:2224–31. doi: 10.1021/mp100223d
202. Stalmans S, Bracke N, Wynendaele E, Gevaert B, Peremans K, Burvenich C, et al. Cell-penetrating peptides selectively cross the blood-brain barrier in vivo. PLoS ONE. (2015) 10:e0139652. doi: 10.1371/journal.pone.0139652
203. Collado Camps E, Brock R. An opportunistic route to success: towards a change of paradigm to fully exploit the potential of cell-penetrating peptides. Bioorg Med Chem. (2018) 26:2780–87. doi: 10.1016/j.bmc.2017.11.004
204. Bechara C, Pallerla M, Zaltsman Y, Burlina F, Alves ID, Lequin O, et al. Tryptophan within basic peptide sequences triggers glycosaminoglycan-dependent endocytosis. FASEB J. (2013) 27:738–49. doi: 10.1096/fj.12-216176
205. Bechara C, Pallerla M, Burlina F, Illien F, Cribier S, Sagan S. Massive glycosaminoglycan-dependent entry of Trp-containing cell-penetrating peptides induced by exogenous sphingomyelinase or cholesterol depletion. Cell Mol Life Sci. (2015) 72:809–20. doi: 10.1007/s00018-014-1696-y
206. Jobin ML, Blanchet M, Henry S, Chaignepain S, Manigand C, Castano S, et al. The role of tryptophans on the cellular uptake and membrane interaction of arginine-rich cell penetrating peptides. Biochim Biophys Acta. (2015) 1848:593–602. doi: 10.1016/j.bbamem.2014.11.013
207. Rydberg HA, Matson M, Amand HL, Esbjörner EK, Nordén B. Effects of tryptophan content and backbone spacing on the uptake efficiency of cell-penetrating peptides. Biochemistry. (2012) 51:5531–39. doi: 10.1021/bi300454k
208. Walrant A, Vogel A, Correia I, Lequin O, Olausson BE, Desbat B, et al. Membrane interactions of two arginine-rich peptides with different cell internalization capacities. Biochim Biophys Acta. (2012) 1818:1755–63. doi: 10.1016/j.bbamem.2012.02.024
209. Himmel HM, Kiss T, Borvendeg SJ, Gillen C, Illes P. The arginine-rich hexapeptide R4W2 is a stereoselective antagonist at the vanilloid receptor 1: a Ca2+ imaging study in adult rat dorsal root ganglion neurons. J Pharmacol Exp Ther. (2002) 301:981–6. doi: 10.1124/jpet.301.3.981
210. Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. (1999) 285:1569–72. doi: 10.1126/science.285.5433.1569
211. Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. (2000) 56:318–25. doi: 10.1034/j.1399-3011.2000.00723.x
212. Meloni BP, South SM, Gill DA, Marriott AL, Déziel RA, Jacques A, et al. Poly-arginine peptides R18 and R18D improve functional outcomes after endothelin-1 (ET-1)-induced stroke in the Sprague Dawley rat. J Neuropathol Exp Neurol. (2019) 78:426–35. doi: 10.1093/jnen/nlz014
213. Bechinger B, Gorr SU. Antimicrobial peptides: mechanisms of action and resistance. J Dent Res. (2017) 96:254–60. doi: 10.1177/0022034516679973
214. Leite ML, da Cunha NB, Costa FF. Antimicrobial peptides nanotechnology and natural metabolites as novel approaches for cancer treatment. Pharmacol Ther. (2018) 183:160–76. doi: 10.1016/j.pharmthera.2017.10.010
215. Schnaar RL. Gangliosides of the vertebrate nervous system. J Mol Biol. (2016) 428:3325–36. doi: 10.1016/j.jmb.2016.05.020
216. Ravindran MS, Tanner LB, Wenk MR. Sialic acid linkage in glycosphingolipids is a molecular correlate for trafficking and delivery of extracellular cargo. Traffic. (2013) 14:1182–91. doi: 10.1111/tra.12100
217. Yu RK, Tsai YT, Ariga T, Yanagisawa M. Structures biosynthesis and functions of gangliosides-an overview. J Oleo Sci. (2011) 60:537–44. doi: 10.5650/jos.60.537
218. Aliste MP, MacCallum JL, Tieleman DP. Molecular dynamics simulations of pentapeptides at interfaces: salt bridge and cation-pi interactions. Biochemistry. (2003) 42:8976–87. doi: 10.1021/bi027001j
220. Chauhan A, Tikoo A, Kapur AK, Singh M. The taming of the cell penetrating domain of the HIV Tat: myths and realities. J Control Release. (2007) 117:148–62. doi: 10.1016/j.jconrel.2006.10.031
221. Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The design of guanidinium-rich transporters and their internalization mechanisms. Adv Drug Deliv Rev. (2008) 60:452–72. doi: 10.1016/j.addr.2007.10.016
222. Tyagi M, Rusnati M, Presta M, Giacca M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem. (2001) 276:3254–61. doi: 10.1074/jbc.M006701200
223. Suzuki T, Futaki S, Niwa M, Tanaka S, Ueda K, Sugiura Y. Possible existence of common internalization mechanisms among arginine-rich peptides. J Biol Chem. (2002) 277:2437–43. doi: 10.1074/jbc.M110017200
224. Takeuchi T, Futaki S. Current understanding of direct translocation of arginine-rich cell-penetrating peptides and its internalization mechanisms. Chem Pharm Bull (Tokyo). (2016) 64:1431–37. doi: 10.1248/cpb.c16-00505
225. Rothbard JB, Kreider E, VanDeusen CL, Wright L, Wylie BL, Wender PA. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J Med Chem. (2002) 45:3612–18. doi: 10.1021/jm0105676
226. Amand HL, Rydberg HA, Fornander LH, Lincoln P, Nordén B, Esbjörner EK. Cell surface binding and uptake of arginine- and lysine-rich penetratin peptides in absence and presence of proteoglycans. Biochim Biophys Acta. (2012) 1818:2669–78. doi: 10.1016/j.bbamem.2012.06.006
227. Rullo A, Qian J, Nitz M. Peptide-glycosaminoglycan cluster formation involving cell penetrating peptides. Biopolymers. (2011) 95:722–31. doi: 10.1002/bip.21641
228. MacDougall G, Anderton RS, Edwards AB, Knuckey NW, Meloni BP. The Neuroprotective peptide poly-arginine-12 (R12) reduces cell surface levels of NMDA NR2B receptor subunit in cortical neurons; investigation into the involvement of endocytic mechanisms. J Mol Neurosci. (2017) 61:235–46. doi: 10.1007/s12031-016-0861-1
229. Fotin-Mleczek M, Welte S, Mader O, Duchardt F, Fischer R, Hufnagel H, et al. Cationic cell-penetrating peptides interfere with TNF signalling by induction of TNF receptor internalization. J Cell Sci. (2005) 118:3339–51. doi: 10.1242/jcs.02460
230. Weng XC, Gai XD, Zheng JQ, Li J. Agmatine blocked voltage-gated calcium channel in cultured rat hippocampal neurons. Acta Pharmacol Sin. (2003) 24:746–50.
231. Garcia ML, King VF, Shevell JL, Slaughter RS, Suarez-Kurtz G, Winquist RJ, et al. Amiloride analogs inhibit L-type calcium channels and display calcium entry blocker activity. J Biol Chem. (1990) 265:3763–71.
232. Keana JF, McBurney RN, Scherz MW, Fischer JB, Hamilton PN, Smith SM, et al. Synthesis and characterization of a series of diarylguanidines that are noncompetitive N-methyl-D-aspartate receptor antagonists with neuroprotective properties. Proc Natl Acad Sci USA. (1989) 86:5631–35. doi: 10.1073/pnas.86.14.5631
233. Goldin SM, Subbarao K, Sharma R, Knapp AG, Fischer JB, Daly D, et al. Neuroprotective use-dependent blockers of Na+ and Ca2+ channels controlling presynaptic release of glutamate. Ann N Y Acad Sci. (1995) 765:210–29. doi: 10.1111/j.1749-6632.1995.tb16578.x
234. Kalia J, Swartz KJ. Elucidating the molecular basis of action of a classic drug: guanidine compounds as inhibitors of voltage-gated potassium channels. Mol Pharmacol. (2011) 80:1085–95. doi: 10.1124/mol.111.074989
235. Hong L, Kim IH, Tombola F. Molecular determinants of Hv1 proton channel inhibition by guanidine derivatives. Proc Natl Acad Sci USA. (2014) 111:9971–6. doi: 10.1073/pnas.1324012111
236. Armstrong CT, Mason PE, Anderson JL, Dempsey CE. Arginine side chain interactions and the role of arginine as a gating charge carrier in voltage sensitive ion channels. Sci Rep. (2016) 6:21759. doi: 10.1038/srep21759
237. Durán-Riveroll LM, Cembella AD. Guanidinium toxins and their interactions with voltage-gated sodium ion channels. Mar Drugs. (2017) 15:303. doi: 10.3390/md15100303
238. Yang XC, Reis DJ. Agmatine selectively blocks the N-methyl-D-aspartate subclass of glutamate receptor channels in rat hippocampal neurons. J Pharmacol Exp Ther. (1999) 288:544–9.
239. Williams K. Interactions of polyamines with ion channels. Biochem J. (1997) 325:289–97. doi: 10.1042/bj3250289
240. Szeto HH. Cell-permeable mitochondrial-targeted peptide antioxidants. AAPS J. (2006) 8:277–83. doi: 10.1007/BF02854898
241. MacDougall G, Anderton RS, Mastaglia FL, Knuckey NW, Meloni BP. Mitochondria and neuroprotection in stroke: cationic arginine-rich peptides (CARPs) as a novel class of mitochondria-targeted neuroprotective therapeutics. Neurobiol Dis. (2019) 121:17–33. doi: 10.1016/j.nbd.2018.09.010
242. Wascher TC, Posch K, Wallner S, Hermetter A, Kostner GM, Graier WF. Vascular effects of L-arginine: anything beyond a substrate for the NO-synthase? Biochem Biophys Res Commun. (1997) 234:35–8. doi: 10.1006/bbrc.1997.9994
243. Haklar G, Ulukaya-Durakbaşa C, Yüksel M, Dagli T, Yalçin AS. Oxygen radicals and nitric oxide in rat mesenteric ischaemia-reperfusion: modulation by L-arginine and NG-nitro-L-arginine methyl ester. Clin Exp Pharmacol Physiol. (1998) 25:908–12. doi: 10.1111/j.1440-1681.1998.tb02342.x
244. Yildiz G, Demiryürek AT, Sahin-Erdemli I, Kanzik I. Comparison of antioxidant activities of aminoguanidine methylguanidine and guanidine by luminol-enhanced chemiluminescence. Br J Pharmacol. (1998) 124:905–10. doi: 10.1038/sj.bjp.0701924
245. Courderot-Masuyer C, Dalloz F, Maupoil V, Rochette L. Antioxidant properties of aminoguanidine. Fundam Clin Pharmacol. (1999) 13:535–40. doi: 10.1111/j.1472-8206.1999.tb00358.x
246. Giardino I, Fard AK, Hatchell DL, Brownlee M. Aminoguanidine inhibits reactive oxygen species formation lipid peroxidation and oxidant-induced apoptosis. Diabetes. (1998) 47:1114–20. doi: 10.2337/diabetes.47.7.1114
247. Philis-Tsimikas A, Parthasarathy S, Picard S, Palinski W, Witztum JL. Aminoguanidine has both pro-oxidant and antioxidant activity toward LDL. Arterioscler Thromb Vasc Biol. (1995) 15:367–76. doi: 10.1161/01.ATV.15.3.367
248. Lawler JM, Barnes WS, Wu G, Song W, Demaree S. Direct antioxidant properties of creatine. Biochem Biophys Res Commun. (2002) 290:47–52. doi: 10.1006/bbrc.2001.6164
249. Lass A, Suessenbacher A, Wölkart G, Mayer B, Brunner F. Functional and analytical evidence for scavenging of oxygen radicals by L-arginine. Mol Pharmacol. (2002) 61:1081–88. doi: 10.1124/mol.61.5.1081
250. Uemura S, Fathman CG, Rothbard JB, Cooke JP. Rapid and efficient vascular transport of arginine polymers inhibits myointimal hyperplasia. Circulation. (2000) 102:2629–35. doi: 10.1161/01.CIR.102.21.2629
251. Suessenbacher A, Lass A, Mayer B, Brunner F. Antioxidative and myocardial protective effects of L-arginine in oxygen radical-induced injury of isolated perfused rat hearts. Naunyn Schmiedebergs Arch Pharmacol. (2002) 365:269–76. doi: 10.1007/s00210-001-0523-9
252. Kown MH, Lijkwan MA, Jahncke CL, Murata S, Rothbard JB, Robbins RC. L-Arginine polymers enhance coronary flow and reduce oxidative stress following cardiac transplantation in rats. J Thorac Cardiovasc Surg. (2003) 126:1065–70. doi: 10.1016/S0022-5223(03)00354-4
253. Ma H, Ma Y, Zhang Z, Zhao Z, Lin R, Zhu J, et al. l-Arginine enhances resistance against oxidative stress and heat stress in Caenorhabditis elegans. Int J Environ Res Public Health. (2016) 13:E969. doi: 10.3390/ijerph13100969
254. Mandal SM, Bharti R, Porto WF, Gauri SS, Mandal M, Franco OL, et al. Identification of multifunctional peptides from human milk. Peptides. (2014) 56:84–93. doi: 10.1016/j.peptides.2014.03.017
255. Aluganti Narasimhulu C, Selvarajan K, Brown M, Parthasarathy S. Cationic peptides neutralize Ox-LDL prevent its uptake by macrophages and attenuate inflammatory response. Atherosclerosis. (2014) 236:133–41. doi: 10.1016/j.atherosclerosis.2014.06.020
256. Akhmedov A, Bonetti NR, Reiner MF, Spescha RD, Amstalden H, Merlini M, et al. Deleterious role of endothelial lectin-like oxidized low-density lipoprotein receptor-1 in ischaemia/reperfusion cerebral injury. J Cereb Blood Flow Metab. (2019) 39:2233–45. doi: 10.1177/0271678X18793266
257. Takemura Y, Okamoto M, Hasegawa M, Hatanaka K, Kubota S. Protamine may have anti-atherogenic potential by inhibiting the binding of oxidized-low density lipoprotein to LOX-1. Biosci Biotechnol Biochem. (2019) 15:1–8. doi: 10.1080/09168451.2019.1588096
258. Joseph G, Gorzalczany Y, Koshkin V, Pick E. Inhibition of NADPH oxidase activation by synthetic peptides mapping within the carboxyl-terminal domain of small GTP-binding proteins: lack of amino acid sequence specificity and importance of polybasic motif. J Biol Chem. (1994) 269:29024–31.
259. Chen Y, Brennan-Minnella AM, Sheth S, El-Benna J, Swanson RA. Tat-NR2B9c prevents excitotoxic neuronal superoxide production. J Cereb Blood Flow Metab. (2015) 35:739–42. doi: 10.1038/jcbfm.2015.16
260. Cifuentes-Pagano E, Csanyi G, Pagano PJ. NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol Life Sci. (2012) 69:2315–25. doi: 10.1007/s00018-012-1009-2
261. Shi J, Ross CR, Leto TL, Blecha F. PR-39 a proline-rich antibacterial peptide that inhibits phagocyte NADPH oxidase activity by binding to Src homology 3 domains of p47 phox. Proc Natl Acad Sci USA. (1996) 93:6014–18. doi: 10.1073/pnas.93.12.6014
262. Allaman I, Bélanger M, Magistretti PJ. Methylglyoxal the dark side of glycolysis. Front Neurosci. (2015) 9:23. doi: 10.3389/fnins.2015.00023
263. Su Y, Lei X, Wu L, Liu L. The role of endothelial cell adhesion molecules P-selectin E-selectin and intercellular adhesion molecule-1 in leucocyte recruitment induced by exogenous methylglyoxal. Immunology. (2012) 137:65–79. doi: 10.1111/j.1365-2567.2012.03608.x
264. Wang B, Aw TY, Stokes KY. The protection conferred against ischemia-reperfusion injury in the diabetic brain by N-acetylcysteine is associated with decreased dicarbonyl stress. Free Radic Biol Med. (2016) 96:89–98. doi: 10.1016/j.freeradbiomed.2016.03.038
265. McDonald DM, Coleman G, Bhatwadekar A, Gardiner TA, Stitt AW. Advanced glycation of the Arg-Gly-Asp (RGD) tripeptide motif modulates retinal microvascular endothelial cell dysfunction. Mol Vis. (2009) 15:1509–20.
266. Rabbani N, Thornalley PJ. Dicarbonyl proteome and genome damage in metabolic and vascular disease. Biochem Soc Trans. (2014) 42:425–32. doi: 10.1042/BST20140018
267. Morcos M, Du X, Pfisterer F, Hutter H, Sayed AA, Thornalley P, et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. (2008) 7:260–9. doi: 10.1111/j.1474-9726.2008.00371.x
268. Thornalley PJ. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys. (2003) 419:31–40. doi: 10.1016/j.abb.2003.08.013
269. Kinsky OR, Hargraves TL, Anumol T, Jacobsen NE, Dai J, Snyder SA, et al. Metformin scavenges methylglyoxal to form a novel imidazolinone metabolite in humans. Chem Res Toxicol. (2016) 29:227–34. doi: 10.1021/acs.chemrestox.5b00497
270. Dhar I, Dhar A, Wu L, Desai K. Arginine attenuates methylglyoxal- and high glucose-induced endothelial dysfunction and oxidative stress by an endothelial nitric-oxide synthase-independent mechanism. J Pharmacol Exp Ther. (2012) 342:196–204. doi: 10.1124/jpet.112.192112
271. Brings S, Fleming T, De Buhr S, Beijer B, Lindner T, Wischnjow A, et al. A scavenger peptide prevents methylglyoxal induced pain in mice. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:654–62. doi: 10.1016/j.bbadis.2016.12.001
272. Cameron A, Appel J, Houghten RA, Lindberg I. Polyarginines are potent furin inhibitors. J Biol Chem. (2000) 275:36741–9. doi: 10.1074/jbc.M003848200
273. Kacprzak MM, Peinado JR, Than ME, Appel J, Henrich S, Lipkind G, et al. Inhibition of furin by polyarginine-containing peptides: nanomolar inhibition by nona-D-arginine. J Biol Chem. (2004) 279:36788–94. doi: 10.1074/jbc.M400484200
274. Peinado JR, Kacprzak MM, Leppla SH, Lindberg I. Cross-inhibition between furin and lethal factor inhibitors. Biochem Biophys Res Commun. (2004) 321:601–5. doi: 10.1016/j.bbrc.2004.07.012
275. Fugere M, Appel J, Houghten RA, Lindberg I, Day R. Short polybasic peptide sequences are potent inhibitors of PC5/6 and PC7: use of positional scanning-synthetic peptide combinatorial libraries as a tool for the optimization of inhibitory sequences. Mol Pharmacol. (2007) 71:323–32. doi: 10.1124/mol.106.027946
276. Ramos-Molina B, Lick AN, Nasrolahi Shirazi A, Oh D, Tiwari R, El-Sayed NS, et al. Cationic cell-penetrating peptides are potent furin inhibitors. PLoS ONE. (2015) 10:e0130417. doi: 10.1371/journal.pone.0130417
277. McMahon S, Grondin F, McDonald PP, Richard DE, Dubois CM. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: impact on the bioactivation of proproteins. J Biol Chem. (2005) 280:6561–9. doi: 10.1074/jbc.M413248200
278. Yokota N, Uchijima M, Nishizawa S, Namba H, Koide Y. Identification of differentially expressed genes in rat hippocampus after transient global cerebral ischemia using subtractive cDNA cloning based on polymerase chain reaction. Stroke. (2001) 32:168–74. doi: 10.1161/01.STR.32.1.168
279. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. (2007) 27:697–709. doi: 10.1038/sj.jcbfm.9600375
280. Tian S, Huang Q, Fang Y, Wu J. FurinDB: a database of 20-residue furin cleavage site motifs substrates and their associated drugs. Int J Mol Sci. (2011) 12:1060–5. doi: 10.3390/ijms12021060
281. Yang Y, Rosenberg GA. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. (2015) 1623:30–8. doi: 10.1016/j.brainres.2015.04.024
282. Turner RJ, Sharp FR. Implications of MMP9 for blood brain barrier disruption and hemorrhagic transformation following ischemic stroke. Front Cell Neurosci. (2016) 10:56. doi: 10.3389/fncel.2016.00056
283. Seki M, Soussou W, Manabe S, Lipton SA. Protection of retinal ganglion cells by caspase substrate-binding peptide IQACRG from N-methyl-D-aspartate receptor-mediated excitotoxicity. Invest Ophthalmol Vis Sci. (2010) 51:1198–207. doi: 10.1167/iovs.09-4102
284. Horn M, Pavlík M, Dolecková L, Baudys M, Mares M. Arginine-based structures are specific inhibitors of cathepsin C application of peptide combinatorial libraries. Eur J Biochem. (2000) 267:3330–6. doi: 10.1046/j.1432-1327.2000.01364.x
285. Gao Y, Lecker S, Post MJ, Hietaranta AJ, Li J, Volk R, et al. Inhibition of ubiquitin-proteasome pathway-mediated I kappa B alpha degradation by a naturally occurring antibacterial peptide. J Clin Invest. (2000) 106:439–48. doi: 10.1172/JCI9826
286. Gaczynska M, Osmulski PA, Gao Y, Post MJ, Simons M. Proline- and CARPs constitute a novel class of allosteric inhibitors of proteasome activity. Biochemistry. (2003) 42:8663–70. doi: 10.1021/bi034784f
287. Kloss A, Henklein P, Siele D, Schmolke M, Apcher S, Kuehn L, et al. The cell-penetrating peptide octa-arginine is a potent inhibitor of proteasome activities. Eur J Pharm Biopharm. (2009) 72:219–25. doi: 10.1016/j.ejpb.2008.10.016
288. Anbanandam A, Albarado DC, Tirziu DC, Simons M, Veeraraghavan S. Molecular basis for proline- and arginine-rich peptide inhibition of proteasome. J Mol Biol. (2008) 384:219–27. doi: 10.1016/j.jmb.2008.09.021
289. Karpowicz P, Osmulski PA, Witkowska J, Sikorska E, Gizynska M, Belczyk-Ciesielska A, et al. Interplay between structure and charge as a key to allosteric modulation of human 20S proteasome by the basic fragment of HIV-1 tat protein. PLoS ONE. (2015) 10:e0143038. doi: 10.1371/journal.pone.0143038
290. Wojcik C, Di Napoli M. Ubiquitin-proteasome system and proteasome inhibition: new strategies in stroke therapy. Stroke. (2004) 35:1506–18. doi: 10.1161/01.STR.0000126891.93919.4e
291. Doeppner TR, Mlynarczuk-Bialy I, Kuckelkorn U, Kaltwasser B, Herz J, Hasan MR, et al. The novel proteasome inhibitor BSc2118 protects against cerebral ischaemia through HIF1A accumulation and enhanced angioneurogenesis. Brain. (2012) 135:3282–97. doi: 10.1093/brain/aws269
292. Doeppner TR, Kaltwasser B, Schlechter J, Jaschke J, Kilic E, Bähr M, et al. Systemic proteasome inhibition induces sustained post-stroke neurological recovery and neuroprotection via mechanisms involving reversal of peripheral immunosuppression and preservation of blood-brain-barrier integrity. Mol Neurobiol. (2016) 53:6332–41. doi: 10.1007/s12035-015-9533-3
293. Doeppner TR, Doehring M, Kaltwasser B, Majid A, Lin F, Bähr M, et al. Ischemic post-conditioning induces post-stroke neuroprotection via Hsp70-mediated proteasome inhibition and facilitates neural progenitor cell transplantation. Mol Neurobiol. (2017) 54:6061–73. doi: 10.1007/s12035-016-0137-3
294. Bieler S, Meiners S, Stangl V, Pohl T, Stangl K. Comprehensive proteomic and transcriptomic analysis reveals early induction of a protective anti-oxidative stress response by low-dose proteasome inhibition. Proteomics. (2009) 9:3257–67. doi: 10.1002/pmic.200800927
295. Li J, Post M, Volk R, Gao Y, Li M, Metais C, et al. PR39 a peptide regulator of angiogenesis. Nat Med. (2000) 6:49–55. doi: 10.1038/71527
296. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. (2016) 73:3221–7. doi: 10.1007/s00018-016-2223-0
297. Steel R, Cowan J, Payerne E, O'Connell MA, Searcey M. Anti-inflammatory effect of a cell-penetrating peptide targeting the Nrf2/Keap1 interaction. ACS Med Chem Lett. (2012) 3:407–10. doi: 10.1021/ml300041g
298. Kanzaki H, Shinohara F, Kajiya M, Fukaya S, Miyamoto Y, Nakamura Y. Nuclear Nrf2 induction by protein transduction attenuates osteoclastogenesis. Free Radic Biol Med. (2014) 77:239–48. doi: 10.1016/j.freeradbiomed.2014.09.006
299. Zhao J, Redell JB, Moore AN, Dash PK. A novel strategy to activate cytoprotective genes in the injured brain. Biochem Biophys Res Commun. (2011) 407:501–6. doi: 10.1016/j.bbrc.2011.03.046
300. Liang M, Wang Z, Li H, Cai L, Pan J, He H, et al. L-Arginine induces antioxidant response to prevent oxidative stress via stimulation of glutathione synthesis and activation of Nrf2 pathway. Food Chem Toxicol. (2018) 115:315–28. doi: 10.1016/j.fct.2018.03.029
301. Wang YF, Xu X, Fan X, Zhang C, Wei Q, Wang X, et al. A cell-penetrating peptide suppresses inflammation by inhibiting NF-κB signaling. Mol Ther. (2011) 19:1849–57. doi: 10.1038/mt.2011.82
302. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. (2009) 1:a001651. doi: 10.1101/cshperspect.a001651
303. Korthuis RJ, Gute DC, Blecha F, Ross CR. PR-39 a proline/arginine-rich antimicrobial peptide prevents postischemic microvascular dysfunction. Am J Physiol. (1999) 277:1007–13. doi: 10.1152/ajpheart.1999.277.3.H1007
304. Kim H, Moodley S, Liu M. TAT cell-penetrating peptide modulates inflammatory response and apoptosis in human lung epithelial cells. Drug Deliv Transl Res. (2015) 5:275–8. doi: 10.1007/s13346-015-0230-6
305. Nankar SA, Pande AH. Properties of apolipoprotein E derived peptide modulate their lipid-binding capacity and influence their anti-inflammatory function. Biochim Biophys Acta. (2014) 184:620–9. doi: 10.1016/j.bbalip.2014.01.006
306. Sharifov OF, Xu X, Gaggar A, Tabengwa EM, White CR, Palgunachari MN, et al. L-4F inhibits lipopolysaccharide-mediated activation of primary human neutrophils. Inflammation. (2014) 37:1401–12. doi: 10.1007/s10753-014-9864-7
307. Datta G, White CR, Dashti N, Chaddha M, Palgunachari MN, Gupta H, et al. Anti-inflammatory and recycling properties of an apolipoprotein mimetic peptide Ac-hE18A-NH(2). Atherosclerosis. (2010) 208:134–41. doi: 10.1016/j.atherosclerosis.2009.07.019
308. Laskowitz DT, Thekdi AD, Thekdi SD, Han SK, Myers JK, Pizzo SV, et al. Downregulation of microglial activation by apolipoprotein E and apoE-mimetic peptides. Exp Neurol. (2001) 167:74–85. doi: 10.1006/exnr.2001.7541
309. Pane K, Sgambati V, Zanfardino A, Smaldone G, Cafaro V, Angrisano T, et al. A new cryptic cationic antimicrobial peptide from human apolipoprotein E with antibacterial activity and immunomodulatory effects on human cells. FEBS J. (2016) 283:2115–31. doi: 10.1111/febs.13725
310. Singh K, Chaturvedi R, Asim M, Barry DP, Lewis ND, Vitek MP, et al. The apolipoprotein E-mimetic peptide COG112 inhibits the inflammatory response to Citrobacter rodentium in colonic epithelial cells by preventing NF-kappaB activation. J Biol Chem. (2008) 283:16752–161. doi: 10.1074/jbc.M710530200
311. Singh K, Chaturvedi R, Barry DP, Coburn LA, Asim M, Lewis ND, et al. The apolipoprotein E-mimetic peptide COG112 inhibits NF-kappaB signaling proinflammatory cytokine expression and disease activity in murine models of colitis. J Biol Chem. (2011) 286:3839–50. doi: 10.1074/jbc.M110.176719
312. Neree AT, Nguyen PT, Bourgault S. Cell-penetrating ability of peptide hormones: key role of glycosaminoglycans clustering. Int J Mol Sci. (2015) 16:27391–400. doi: 10.3390/ijms161126025
313. Morio H, Tatsuno I, Hirai A, Tamura Y, Saito Y. Pituitary adenylate cyclase-activating polypeptide protects rat-cultured cortical neurons from glutamate-induced cytotoxicity. Brain Res. (1996) 741:82–8. doi: 10.1016/S0006-8993(96)00920-1
314. Seki T, Nakatani M, Taki C, Shinohara Y, Ozawa M, Nishimura S, et al. Neuroprotective effect of PACAP against kainic acid-induced neurotoxicity in rat retina. Ann N Y Acad Sci. (2006) 1070:531–4. doi: 10.1196/annals.1317.074
315. Werling D, Banks WA, Salameh TS, Kvarik T, Kovacs LA, Vaczy A, et al. Passage through the ocular barriers and beneficial effects in retinal ischemia of topical application of PACAP1–38 in rodents. Int J Mol Sci. (2017) 18:E675. doi: 10.3390/ijms18030675
316. Reglodi D, Somogyvari-Vigh A, Vigh S, Kozicz T, Arimura A. Delayed systemic administration of PACAP38 is neuroprotective in transient middle cerebral artery occlusion in the rat. Stroke. (2000) 31:1411–7. doi: 10.1161/01.STR.31.6.1411
317. Lazarovici P, Cohen G, Arien-Zakay H, Chen J, Zhang C, Chopp M, et al. Multimodal neuroprotection induced by PACAP38 in oxygen-glucose deprivation and middle cerebral artery occlusion stroke models. J Mol Neurosci. (2012) 48:526–40. doi: 10.1007/s12031-012-9818-1
318. Mao SS, Hua R, Zhao XP, Qin X, Sun ZQ, Zhang Y, et al. Exogenous administration of PACAP alleviates traumatic brain injury in rats through a mechanism involving the TLR4/MyD88/NF-κB pathway. J Neurotrauma. (2012) 29:1941–59. doi: 10.1089/neu.2011.2244
319. Suk K, Park JH, Lee WH. Neuropeptide PACAP inhibits hypoxic activation of brain microglia: a protective mechanism against microglial neurotoxicity in ischemia. Brain Res. (2004) 1026:151–6. doi: 10.1016/j.brainres.2004.08.017
320. Kandler K, Shaykhiev R, Kleemann P, Klescz F, Lohoff M, Vogelmeier C, et al. The anti-microbial peptide LL-37 inhibits the activation of dendritic cells by TLR ligands. Int Immunol. (2006) 18:1729–36. doi: 10.1093/intimm/dxl107
321. Yoo SA, Bae DG, Ryoo JW, Kim HR, Park GS, Cho CS, et al. Arginine-rich anti-vascular endothelial growth factor (anti-VEGF) hexapeptide inhibits collagen-induced arthritis and VEGF-stimulated productions of TNF-alpha and IL-6 by human monocytes. J Immunol. (2005) 174:5846–55. doi: 10.4049/jimmunol.174.9.5846
322. Chow LN, Choi KY, Piyadasa H, Bossert M, Uzonna J, Klonisch T, et al. Human cathelicidin LL-37-derived peptide IG-19 confers protection in a murine model of collagen-induced arthritis. Mol Immunol. (2014) 57:86–92. doi: 10.1016/j.molimm.2013.08.011
323. Marin-Luevano P, Trujillo V, Rodriguez-Carlos A, González-Curiel I, Enciso-Moreno JA, Hancock REW, et al. Induction by innate defence regulator peptide 1018 of pro-angiogenic molecules and endothelial cell migration in a high glucose environment. Peptides. (2018) 101:135–44. doi: 10.1016/j.peptides.2018.01.010
324. Scatizzi JC, Hutcheson J, Pope RM, Firestein GS, Koch AE, Mavers M, et al. Bim-Bcl-2 homology 3 mimetic therapy is effective at suppressing inflammatory arthritis through the activation of myeloid cell apoptosis. Arthritis Rheum. (2010) 62:441–51. doi: 10.1002/art.27198
325. Ahmed CM, Massengill MT, Brown EE, Ildefonso CJ, Johnson HM, Lewin AS. A cell penetrating peptide from SOCS-1 prevents ocular damage in experimental autoimmune uveitis. Exp Eye Res. (2018) 177:12–22. doi: 10.1016/j.exer.2018.07.020
326. Karicherla P, Aras S, Aiyar A, Hobden JA. Nona-D-arginine amide suppresses corneal cytokines in Pseudomonas aeruginosa keratitis. Cornea. (2010) 29:1308–14. doi: 10.1097/ICO.0b013e3181ca3a69
327. Lee JY, Suh JS, Kim JM, Kim JH, Park HJ, Park YJ, et al. Identification of a cell-penetrating peptide domain from human beta-defensin 3 and characterization of its anti-inflammatory activity. Int J Nanomedicine. (2015) 10:5423–34. doi: 10.2147/IJN.S90014
328. Olsen UB, Selmer J, Kahl JU. Complement C5a receptor antagonism by protamine and poly-L-Arg on human leukocytes. Complement. (1988) 5:153–62. doi: 10.1159/000463049
329. Zhang T, Garstka MA, Li K. The controversial C5a receptor C5aR2: its role in health and disease. J Immunol Res. (2017) 2017:8193932. doi: 10.1155/2017/8193932
330. Yang Y, Wolfram J, Fang X, Shen H, Ferrari M. Polyarginine induces an antitumor immune response through binding to toll-like receptor 4. Small. (2014) 10:1250–4. doi: 10.1002/smll.201302887
331. Niyonsaba F, Madera L, Afacan N, Okumura K, Ogawa H, Hancock RE. The innate defense regulator peptides IDR-HH2 IDR-1002 and IDR-1018 modulate human neutrophil functions. J Leukoc Biol. (2013) 94:159–70. doi: 10.1189/jlb.1012497
332. Shin K, Chapman NA, Sarker M, Kenward C, Huang SK, Weatherbee-Martin N, et al. Bioactivity of the putative apelin proprotein expands the repertoire of apelin receptor ligands. Biochim Biophys Acta Gen Subj. (2017) 1861:1901–12. doi: 10.1016/j.bbagen.2017.05.017
333. Zhou N, Fang J, Acheampong E, Mukhtar M, Pomerantz RJ. Binding of ALX40–4C to APJ a CNS-based receptor inhibits its utilization as a co-receptor by HIV-1. Virology. (2003) 312:196–203. doi: 10.1016/S0042-6822(03)00185-5
334. Zhou N, Zhang X, Fan X, Argyris E, Fang J, Acheampong E, et al. The N-terminal domain of APJ a CNS-based coreceptor for HIV-1 is essential for its receptor function and coreceptor activity. Virology. (2003) 317:84–94. doi: 10.1016/S0042-6822(03)00638-X
335. Le Gonidec S, Chaves-Almagro C, Bai Y, Kang HJ, Smith A, Wanecq E, et al. Protamine is an antagonist of apelin receptor and its activity is reversed by heparin. FASEB J. (2017) 31:2507–19. doi: 10.1096/fj.201601074R
336. Masri B, Morin N, Pedebernade L, Knibiehler B, Audigier Y. The apelin receptor is coupled to Gi1 or Gi2 protein and is differentially desensitized by apelin fragments. J Biol Chem. (2006) 281:18317–26. doi: 10.1074/jbc.M600606200
337. Li Y, Chen J, Bai B, Du H, Liu Y, Liu H. Heterodimerization of human apelin and kappa opioid receptors: roles in signal transduction. Cell Signal. (2012) 24:991–1001. doi: 10.1016/j.cellsig.2011.12.012
338. Liu J, Liu M, Chen L. Novel pathogenesis: regulation of apoptosis by Apelin/APJ system. Acta Biochim Biophys Sin (Shanghai). (2017) 49:471–8. doi: 10.1093/abbs/gmx035
339. O'Carroll AM, Lolait SJ, Harris LE, Pope GR. The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis. J Endocrinol. (2013) 219:13–35. doi: 10.1530/JOE-13-0227
340. Bertrand C, Valet P, Castan-Laurell I. Apelin and energy metabolism. Front Physiol. (2015) 6:115. doi: 10.3389/fphys.2015.00115
341. Zeng XJ, Yu SP, Zhang L, Wei L. Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Exp Cell Res. (2010) 316:1773–83. doi: 10.1016/j.yexcr.2010.02.005
342. Wu Y, Wang X, Zhou X, Cheng B, Li G, Bai B. Temporal expression of apelin/apelin receptor in ischemic stroke and its therapeutic potential. Front Mol Neurosci. (2017) 10:1. doi: 10.3389/fnmol.2017.00001
343. Perjés Á, Kilpiö T, Ulvila J, Magga J, Alakoski T, Szabó Z, et al. Characterization of apela, a novel endogenous ligand of apelin receptor, in the adult heart. Basic Res Cardiol. (2016) 111:2. doi: 10.1007/s00395-015-0521-6
344. Tomioka H, Nakagami H, Tenma A, Saito Y, Kaga T, Kanamori T, et al. Novel anti-microbial peptide SR-0379 accelerates wound healing via the PI3 kinase/Akt/mTOR pathway. PLoS ONE. (2014) 9:e92597. doi: 10.1371/journal.pone.0092597
345. Bowdish DM, Davidson DJ, Speert DP, Hancock RE. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J Immunol. (2004) 172:3758–65. doi: 10.4049/jimmunol.172.6.3758
346. Vandamme D, Landuyt B, Luyten W, Schoofs L. A comprehensive summary of LL-37 the factotum human cathelicidin peptide. Cell Immunol. (2012) 280:22–35. doi: 10.1016/j.cellimm.2012.11.009
347. Lokeshwar VB, Huang SS, Huang JS. Protamine enhances epidermal growth factor (EGF)-stimulated mitogenesis by increasing cell surface EGF receptor number. Implications for existence of cryptic EGF receptors. J Biol Chem. (1989) 264:19318–26.
348. Hubler L, Leventhal PS, Bertics PJ. Alteration of the kinetic properties of the epidermal growth factor receptor tyrosine kinase by basic proteins. Biochem J. (1992) 281:107–14. doi: 10.1042/bj2810107
349. Baynes BM, Wang DI, Trout BL. Role of arginine in the stabilization of proteins against aggregation. Biochemistry. (2005) 44:4919–25. doi: 10.1021/bi047528r
350. Shukla D, Trout BL. Interaction of arginine with proteins and the mechanism by which it inhibits aggregation. J Phys Chem B. (2010) 114:13426–38. doi: 10.1021/jp108399g
351. Gibson TJ, Murphy RM. Design of peptidyl compounds that affect beta-amyloid aggregation: importance of surface tension and context. Biochemistry. (2005) 44:8898–907. doi: 10.1021/bi050225s
352. Taddei K, Laws SM, Verdile G, Munns S, D'Costa K, Harvey AR, et al. Novel phage peptides attenuate beta amyloid-42 catalysed hydrogen peroxide production and associated neurotoxicity. Neurobiol Aging. (2010) 31:203–14. doi: 10.1016/j.neurobiolaging.2008.03.023
353. Kawasaki T, Onodera K, Kamijo S. Identification of novel short peptide inhibitors of soluble 37/48 kDa oligomers of amyloid β42. Biosci Biotechnol Biochem. (2011) 75:1496–501. doi: 10.1271/bbb.110198
354. Kawasaki T, Kamijo S. Inhibition of aggregation of amyloid β42 by arginine-containing small compounds. Biosci Biotechnol Biochem. (2012) 76:762–6. doi: 10.1271/bbb.110879
355. Parthsarathy V, McClean PL, Hölscher C, Taylor M, Tinker C, Jones G, et al. A novel retro-inverso peptide inhibitor reduces amyloid deposition oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer's disease. PLoS ONE. (2013) 8:e54769. doi: 10.1371/annotation/57e0a947-8600-4658-b04c-cf7a45c8bd8d
356. Cheng YS, Chen ZT, Liao TY, Lin C, Shen HC, Wang YH, et al. An intranasally delivered peptide drug ameliorates cognitive decline in Alzheimer transgenic mice. EMBO Mol Med. (2017) 9:703–15. doi: 10.15252/emmm.201606666
357. Fonar G, Polis B, Meirson T, Maltsev A, Samson AO. Subcutaneous sustained-release of poly-arginine ameliorates cognitive impairment in a transgenic mouse model of Alzheimer's disease. Adv Alzheimer's Dis. (2018) 7:153–82. doi: 10.4236/aad.2018.74011
358. Nadimidla K, Ismail T, Kanapathipillai M. Tau peptides and tau mutant protein aggregation inhibition by cationic polyethyleneimine and polyarginine. Biopolymers. (2017) 107:e23024. doi: 10.1002/bip.23024
359. Podvin S, Yaksh T, Hook V. The emerging role of spinal dynorphin in chronic pain: a therapeutic perspective. Annu Rev Pharmacol Toxicol. (2016) 56:511–33. doi: 10.1146/annurev-pharmtox-010715-103042
360. Reis DJ, Yang XC, Milner TA. Agmatine containing axon terminals in rat hippocampus form synapses on pyramidal cells. Neurosci Lett. (1998) 250:185–88. doi: 10.1016/S0304-3940(98)00466-2
361. Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. (1991) 88:6368–71. doi: 10.1073/pnas.88.14.6368
362. Víteček J, Lojek A, Valacchi G, Kubala L. Arginine-based inhibitors of nitric oxide synthase: therapeutic potential and challenges. Mediators Inflamm. (2012) 2012:318087. doi: 10.1155/2012/318087
363. Field Randomization of NA-1 Therapy in Early Responders (FRONTIER). Available online at: https://clinicaltrials.gov/ct2/show/NCT02315443?term=NA-1&draw=2&rank=2
364. Safety and Efficacy of NA-1 in Subjects Undergoing Endovascular Thrombectomy for Stroke (ESCAPE-NA1). Available online at: https://clinicaltrials.gov/ct2/show/NCT02930018?term=NA-1&draw=2&rank=1
365. Guptill JT, Raja SM, Boakye-Agyeman F, Noveck R, Ramey S, Tu TM, et al. Phase 1 randomized, double-blind, placebo-controlled study to determine the safety, tolerability, and pharmacokinetics of a single escalating dose and repeated doses of CN-105 in healthy adult subjects. J Clin Pharmacol. (2017) 57:770–6. doi: 10.1002/jcph.853
366. Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, Terbrugge KG, et al. ENACT trial investigators. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. (2012) 11:942–50. doi: 10.1016/S1474-4422(12)70225-9
367. Doranz BJ, Grovit-Ferbas K, Sharron MP, Mao SH, Goetz MB, Daar ES, et al. Safe use of the CXCR4 inhibitor ALX40–4C in humans. AIDS Res Hum Retroviruses. (2001) 17:475–86. doi: 10.1089/08892220151126508
368. Sokolowska E, Kalaska B, Miklosz J, Mogielnicki A. The toxicology of heparin reversal with protamine: past, present and future. Expert Opin Drug Metab Toxicol. (2016) 12:897–909. doi: 10.1080/17425255.2016.1194395
Keywords: cationic arginine-rich peptides, neuroprotection, cell-penetrating peptides, arginine, guanidinium head group, TAT
Citation: Meloni BP, Mastaglia FL and Knuckey NW (2020) Cationic Arginine-Rich Peptides (CARPs): A Novel Class of Neuroprotective Agents With a Multimodal Mechanism of Action. Front. Neurol. 11:108. doi: 10.3389/fneur.2020.00108
Received: 10 December 2019; Accepted: 30 January 2020;
Published: 25 February 2020.
Edited by:
Heike Wulff, University of California, Davis, United StatesReviewed by:
Rajesh Khanna, University of Arizona, United StatesMaria Calvo-Rodriguez, Massachusetts General Hospital, Harvard Medical School, United States
Copyright © 2020 Meloni, Mastaglia and Knuckey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno P. Meloni, YnJ1bm8ubWVsb25pQHBlcnJvbi51d2EuZWR1LmF1