 Xinran Ma
Xinran Ma Ji He
Ji He Xiaoxuan Liu
Xiaoxuan Liu Dongsheng Fan
Dongsheng Fan- 1Department of Neurology, Peking University Third Hospital, Beijing, China
- 2Beijing Municipal Key Laboratory of Biomarker and Translational Research in Neurodegenerative Diseases, Beijing, China
- 3Key Laboratory for Neuroscience, National Health Commission/Ministry of Education, Peking University, Beijing, China
Background: REEP1 is a common cause of autosomal dominant hereditary spastic paraplegia (HSP) but is rare in China. The pathological mechanism of REEP1 is not fully understood.
Methods: We screened for REEP1 mutations in 31 unrelated probands from Chinese HSP families using next-generation sequencing targeting pathogenic genes for HSP and other related diseases. All variants were validated by Sanger sequencing. The proband family members were also screened for variants for the segregation analysis. All previously reported REEP1 mutations and cases were reviewed to clarify the genetic and clinical features of REEP1-related HSP.
Results: A pathogenic mutation, REEP1c. 125G>A (p.Trp42*), was detected in a pure HSP family from North China out of 31 HSP families (1/31). This locus, which is located in the second hydrophobic domain of REEP1, is detected in both Caucasian patients with complicated HSP phenotypes and Chinese pure HSP families.
Conclusion: REEP1-related HSP can be found in the Chinese population. The 42nd residue is a novel transethnic mutation hotspot. Mutations in this spot can lead to both complicated and pure form of HSP. Identification of transethnic hotspot will contribute to clarify the underlying pathological mechanisms.
Introduction
Hereditary spastic paraplegia (HSP) comprises a group of neurodegenerative diseases characterized by spastic paraplegia of the lower limbs (1). Hereditary spastic paraplegia is classified as pure or complicated HSP based on whether impairment is restricted to the pyramidal system (2). Approximately 79 pathogenic genes for HSPs have been found (3). These diseases can be inherited in various ways, including autosomal dominant (AD), recessive, X-linked, mitochondrial, and other mechanisms (4). The only treatment to date for HSP is symptomatic treatment. Because HSPs are monogenic diseases, gene therapies, and precision medicine may be appropriate (3).
Loss-of-function mutations of REEP1 (receptor expression enhancing protein 1), a mediator of endoplasmic reticulum (ER)–mitochondrial interactions, can lead to AD HSP (5–7). In a previous study of HSP cohorts, REEP1 mutations were found to be rare in the Chinese population (8). Here, we screened for REEP1 mutations using next-generation sequencing (NGS) in 31 Chinese HSP families and performed a general review of REEP1-related HSP, which helped to elucidate the genetic and clinical features of this disease.
Methods
Subjects
From January 2012 to September 2019, 31 Chinese families clinically diagnosed with HSP according to Harding's criteria (2) in Peking University Third Hospital were enrolled in this study. All the probands and their relatives received detailed clinical examinations. All participants provided written informed consent. The study was approved by the Peking University Third Hospital ethics committee.
Genetic Test and Mutation Analysis
Peripheral blood was obtained from all the participants, and DNA was isolated. Next-generation sequencing targeting ~160 genes related to Charcot-Marie-Tooth disease, HSP, and amyotrophic lateral sclerosis, including REEP1(NM_022912.2), was conducted (the gene list and detailed sequencing and mutation analysis procedure are shown in Supplementary File and Supplementary Table 1). All identified variants were validated by Sanger sequencing. The relatives of the probands were also screened for these variants via Sanger sequencing for the segregation analysis. The detailed Sanger sequencing procedure for the identified REEP1 variants is shown in Supplementary Table 2.
Results
Thirty-one unrelated HSP probands and their relatives from mainland China were recruited for the study (Table 1). Twenty-one probands were male, and 10 were female. The average age at onset was 33.8 ± 13.3 years. Ten families presented with a complicated phenotype. The accompanying symptoms included neuropathy (5/10), extrapyramidal impairments (parkinsonism 1/10, dystonia 1/10), white matter lesions (1/10), dysphagia (1/10), deafness (1/10), nystagmus (1/10), and cognitive impairment (1/10).

Table 1. Clinical features of the HSP cohort in this study.
Genetic Results
Genetic variants in pathogenic genes of HSP were identified in eight probands, with a diagnostic rate of 25.8%. Three of them were known causative mutations for HSP (Table 2) (9–11). Two known pathogenic mutations and a novel mutation of SPAST were detected in three probands (9.7%). The possible damaging variants were listed in Supplementary Table 3, including KIAA0196 (1/31), AP5Z1 (1/31), DDHD1 (1/31), and SPG7 (1/31). A previously reported (9) pathogenic non-sense mutation of REEP1 c. 125G>A (p.Trp42*) (RefSeq NM_022912) in exon 3 was detected in a pure HSP proband via NGS and then validated by Sanger sequencing (Figure 1).

Table 2. Pathogenic and likely pathogenic mutations of REEP1 and SPAST detected in the HSP cohort.
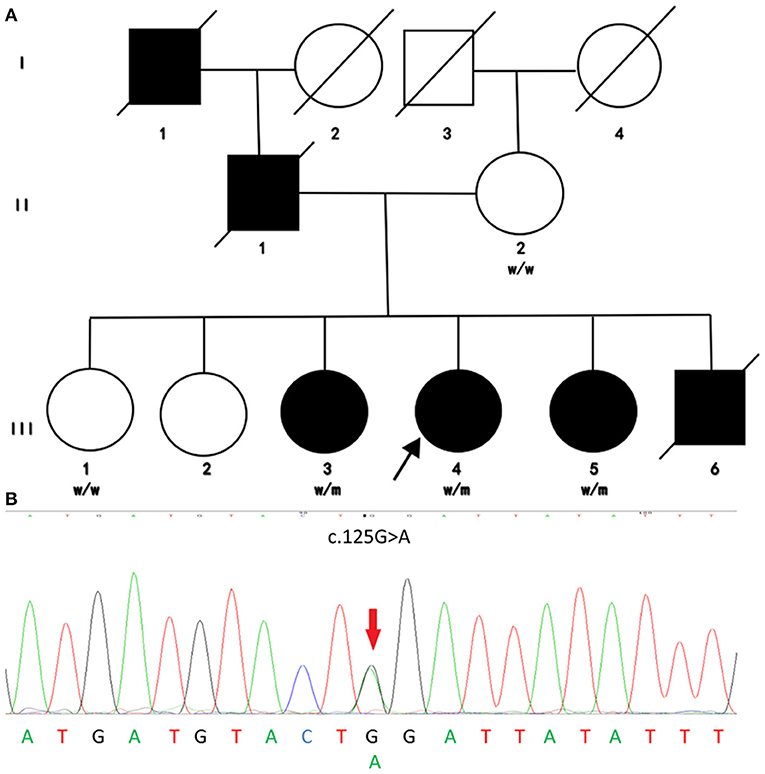
Figure 1. Family pedigree and genomic sequencing electropherograms of the investigated HSP family. (A) Pedigree of the investigated family. Males and females are represented by squares and circles, respectively, and filled and unfilled symbols represent affected and unaffected individuals, respectively. The crossed symbols indicate deceased individuals. W, wild type; M, mutated. (B) Genomic sequencing electropherograms. The c.125G>A(p.Trp42*) heterozygous non-sense mutation of REEP1 was detected in this HSP family. This mutation cosegregated with an early-onset pure HSP phenotype, supporting the notion that this mutation is pathogenic for HSP.
This mutation was detected in all affected members but not the unaffected ones via Sanger sequencing, which is consistent with an AD model of inheritance. Moreover, the mutation was not found in population databases such as ExAC and 1,000 Genomes. Obvious cosegregation was found in the examined family. Next-generation sequencing showed no pathogenic or likely pathogenic mutations in other causative genes of spastic paraplegia such as SPAST and so on. The phenotype of these individuals was also consistent with a previous case. Therefore, we concluded that REEP1 c.125G>A (p.Trp42*) is a pathogenic mutation in this family.
Clinical Manifestation of the HSP Family With REEP1 Mutation
All the cases in the family from North China with the mutation were consistent with pure HSP (Figure 1B). The proband (III-4) was a 51-year-old woman who complained of walking difficulty and lower limb stiffness starting at ~20 years of age. Recently, she had also experienced urgency of urination without urinary incontinence. Her symptoms progressed slowly during subsequent years. Her family members (I-1, II-1, III-3, III-5, and III-6) had similar symptoms that were limited to lower limb stiffness and urgency of urination. They all received detailed clinical examinations. Other systems were normal. The age at onset for III-3, III-4, III-5, and III-6 ranged from 10 to 30 years. Patient III-6 died of a traffic accident when he was 31 years old.
Discussion
In this study, genetic variants in HSP genes were detected in eight probands. The frequencies of rare HSP genes in our study are similar to those in a previous study (12). Although SPAST was the most common cause for ADHSP (12), accounting for ~50% of ADHSP families in China (13), known pathogenic SPAST mutations were detected in only two probands in our cohort (the detailed information are shown in Supplementary File). That may be due to different sequencing methods and small sample size. (We used NGS-based method to detect copy number variation in Supplementary File). Our study may indicate the advantages and disadvantages of NGS. Other sequencing methods could not be substituted.
A known pathogenic mutation, p.W42*, in the second hydrophobic domain (HD) of REEP1, was detected, which was previously detected in a pure HSP patient in Norway (9). W42R is a missense mutation in the same amino acid that was found to cause complicated HSP with neuropathy in French Caucasians (14). Both non-sense and missense mutations at the W42 locus have been found to be pathogenic in different ethnicities, indicating that this locus is a transethnic hotspot that plays an important role in the pathogenesis of HSP. Mutations in this locus can lead to both pure and complicated phenotypes, indicating substantial heterogeneity of this transethnic hotspot.
REEP1 is a causative gene of HSP and distal hereditary motor neuropathy type 5B (15), and REEP1-related diseases also include 2p11.2-2p12 deletion syndrome (16). The extension of the REEP1 protein and mislocalized REEP1 can lead to “toxic gain of function” and result in dHMN (15, 17), whereas loss of function may lead to HSP (5–7).
The REEP1 protein is located in the mitochondria and ER and participates in the functional activities of organelles, such as the interaction between the tubular ER and microtubules and peripheral ER shaping (5–7). To date, ~60 pathogenic mutations of REEP1 have been reported, including missense mutations, non-sense mutations, exon deletions, splicing site mutations, and miRNA binding site mutations (Figure 2). The REEP1 protein has a conserved TB2/DP1/HVA22 domain that may have a chaperone-like function (7, 18). Additionally, it contains a mitochondria-localization domain (6) (between aa116 and aa157 in NP_075063.1) and a cytoplasmic C-terminus that is in contact with microtubules (19). There is also a highly conserved miRNA binding site in the 3′ UTR of REEP1 mRNA, and pathogenic mutations in this region influence its post-transcriptional regulation (20). Many missense mutations of REEP1 are located near the N terminus (20, 21), indicating that it is a hotspot region. Mutations in the N terminus (before the 55th amino acid) influence the localization of REEP1 in the ER (21). This region contains two HDs, HD1 in the N terminus, and HD2 near the middle, which is located in the conserved domain. HD2 forms a hairpin-like structure to interact with SPAST and ALT1 in the ER (19, 21). Their interactions mediate ER shaping and are very important for the ER network between the cell body and axon in motor neurons (22). Disruption of the hairpin domain harms the ER organization in distal axons, which may explain the length-dependent degeneration of upper motor neurons in HSP (22). The novel transethnic hotspot W42 is located in the hairpin domain. Thus, it can disturb the normal function of this domain and lead to pathogenesis.
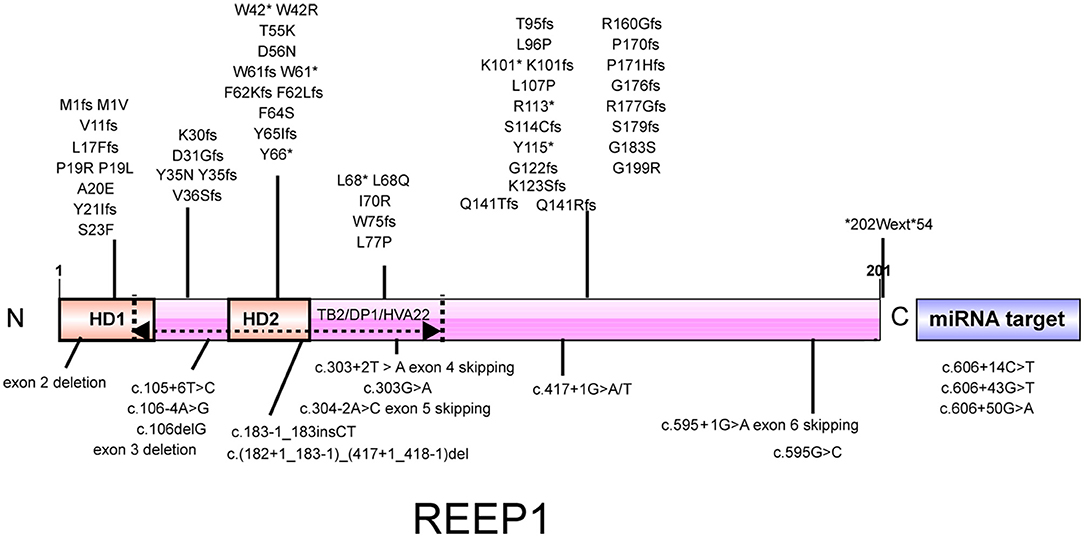
Figure 2. Illustration of the REEP1 protein. REEP1 contains 2 hydrophobic domains and a conserved TB2/DP1/HVA22 domain. There is also a miRNA target region in the 3′ UTR of REEP1 mRNA. All reported pathogenic mutations of REEP1 are shown except for 2p11.2-2p12 deletions. HD, hydrophobic domain.
More than 70 REEP1-related HSP pedigrees have been reported (7, 9, 14, 15, 17, 20, 23–35), and their genotypes and phenotypes are summarized in Supplementary Table 4. There is generally an early age at onset, commonly 0–20 and 30–35 years of age (14). The mutation of REEP1 typically results in AD pure HSP but can cause complicated HSP. The accompanying symptoms include neuropathy (23), tremor, and cognitive impairment (14). Few mutations can lead to both complicated and pure HSP phenotypes. The clinical manifestations can also vary among different ethnicities.
The mutation rate of REEP1 in HSP varies in different regions (7, 13, 14, 24, 26–29, 31, 36) (Table 3). Although REEP1 was reported to be the third most common cause of HSP in some countries (20), previous screening studies in Chinese patients did not find pathogenic REEP1 variants (8, 13). In the present study, we found one family with pathogenic REEP1 mutation out of 31 HSP families, which is uncommon.
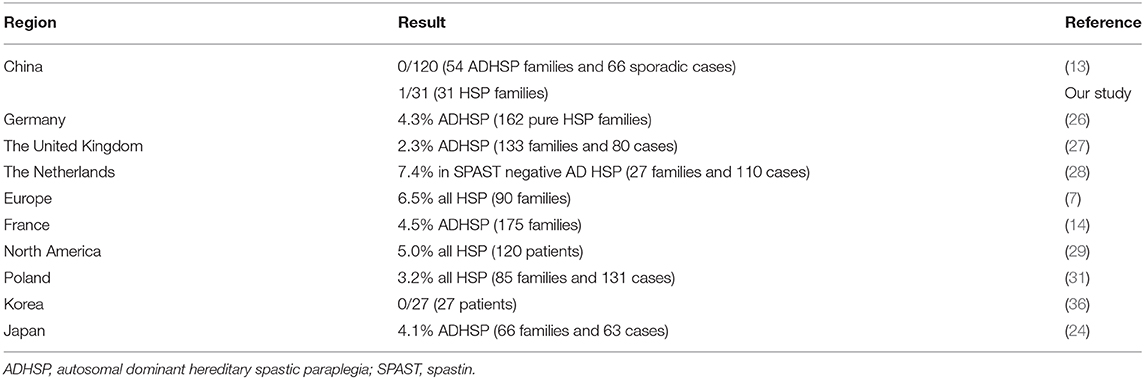
Table 3. REEP1 mutation rate in different regions.
Conclusion
REEP1-related HSP can be found in the Chinese population. The 42nd residue is a novel transethnic mutation hotspot. Mutations in this spot can lead to both complicated and pure form of HSP. Identification of transethnic hotspot will contribute to clarify the underlying pathological mechanisms.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the Peking University Third Hospital Ethics Committee. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
DF conceived this study and provide financial support. XM and JH performed the experiments, analyzed the data, and wrote the manuscript. XL provided supplementary data.
Funding
This work was funded by the National Natural Science Foundation of China (81601105, 81471184, 81974197).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fneur.2020.00499/full#supplementary-material
References
1. Harding AE. Hereditary spastic paraplegias. Semin Neurol. (1993) 13:333–6. doi: 10.1055/s-2008-1041143
2. Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. (1983) 1:1151–5. doi: 10.1016/S0140-6736(83)92879-9
3. Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. (2019) 18:1136–46. doi: 10.1016/S1474-4422(19)30235-2
4. Finsterer J, Loscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. (2012) 318:1–18. doi: 10.1016/j.jns.2012.03.025
5. Beetz C, Koch N, Khundadze M, Zimmer G, Nietzsche S, Hertel N, et al. A spastic paraplegia mouse model reveals REEP1-dependent ER shaping. J Clin Invest. (2013) 123:4273–82. doi: 10.1172/JCI65665
6. Lim Y, Cho IT, Schoel LJ, Cho G, Golden JA. Hereditary spastic paraplegia-linked REEP1 modulates endoplasmic reticulum/mitochondria contacts. Ann Neurol. (2015) 78:679–96. doi: 10.1002/ana.24488
7. Zuchner S, Wang G, Tran-Viet KN, Nance MA, Gaskell PC, Vance JM, et al. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet. (2006) 79:365–9. doi: 10.1086/505361
8. Du J, Shen L, Zhao GH, Wang YG, Liao SS, Chen C, et al. Receptor expression-enhancing protein 1 gene (SPG31) mutations are rare in Chinese Han patients with hereditary spastic paraplegia. Chin Med J. (2009) 122:2064–6. doi: 10.3760/cma.j.issn.0366-6999.2009.17.019
9. Iqbal Z, Rydning SL, Wedding IM, Koht J, Pihlstrom L, Rengmark AH, et al. Targeted high throughput sequencing in hereditary ataxia and spastic paraplegia. PLos ONE. (2017) 12:e0174667. doi: 10.1371/journal.pone.0174667
10. Shoukier M, Neesen J, Sauter SM, Argyriou L, Doerwald N, Pantakani DV, et al. Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. Eur J Hum Genet. (2009) 17:187–94. doi: 10.1038/ejhg.2008.147
11. Svenstrup K, Bross P, Koefoed P, Hjermind LE, Eiberg H, Born AP, et al. Sequence variants in SPAST, SPG3A and HSPD1 in hereditary spastic paraplegia. J Neurol Sci. (2009) 284:90–5. doi: 10.1016/j.jns.2009.04.024
12. Dong EL, Wang C, Wu S, Lu YQ, Lin XH, Su HZ, et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol Neurodegener. (2018) 13:36. doi: 10.1186/s13024-018-0269-1
13. Luo Y, Chen C, Zhan Z, Wang Y, Du J, Hu Z, et al. Mutation and clinical characteristics of autosomal-dominant hereditary spastic paraplegias in China. Neurodegener Dis. (2014) 14:176–83. doi: 10.1159/000365513
14. Goizet C, Depienne C, Benard G, Boukhris A, Mundwiller E, Sole G, et al. REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum Mutat. (2011) 32:1118–27. doi: 10.1002/humu.21542
15. Beetz C, Pieber TR, Hertel N, Schabhuttl M, Fischer C, Trajanoski S, et al. Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type V. Am J Hum Genet. (2012) 91:139–45. doi: 10.1016/j.ajhg.2012.05.007
16. Stevens SJC, Blom EW, Siegelaer ITJ, Smeets EEJGL. A recurrent deletion syndrome at chromosome bands 2p11.2–2p12 flanked by segmental duplications at the breakpoints and including REEP1. Eur J Hum Genet. (2015) 23:543–6. doi: 10.1038/ejhg.2014.124
17. Bock AS, Gunther S, Mohr J, Goldberg LV, Jahic A, Klisch C, et al. A nonstop variant in REEP1 causes peripheral neuropathy by unmasking a 3'UTR-encoded, aggregation-inducing motif. Hum Mutat. (2018) 39:193–6. doi: 10.1002/humu.23369
18. Chen CN, Chu CC, Zentella R, Pan SM, Ho TH. AtHVA22 gene family in Arabidopsis: phylogenetic relationship, ABA and stress regulation, and tissue-specific expression. Plant Mol Biol. (2002) 49:633–44. doi: 10.1023/A:1015593715144
19. Park SH, Zhu PP, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. (2010) 120:1097–110. doi: 10.1172/JCI40979
20. Beetz C, Schule R, Deconinck T, Tran-Viet KN, Zhu H, Kremer BP, et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain. (2008) 131(Pt 4):1078–86. doi: 10.1093/brain/awn026
21. Falk J, Rohde M, Bekhite MM, Neugebauer S, Hemmerich P, Kiehntopf M, et al. Functional mutation analysis provides evidence for a role of REEP1 in lipid droplet biology. Hum Mutat. (2014) 35:497–504. doi: 10.1002/humu.22521
22. Yalcin B, Zhao L, Stofanko M, O'Sullivan NC, Kang ZH, Roost A, et al. Modeling of axonal endoplasmic reticulum network by spastic paraplegia proteins. Elife. (2017) 6:e23882. doi: 10.7554/eLife.23882
23. Toft A, Birk S, Ballegaard M, Duno M, Hjermind LE, Nielsen JE, et al. Peripheral neuropathy in hereditary spastic paraplegia caused by REEP1 variants. J Neurol. (2019) 266:735–44. doi: 10.1007/s00415-019-09196-1
24. Ishiura H, Takahashi Y, Hayashi T, Saito K, Furuya H, Watanabe M, et al. Molecular epidemiology and clinical spectrum of hereditary spastic paraplegia in the Japanese population based on comprehensive mutational analyses. J Hum Genet. (2014) 59:163–72. doi: 10.1038/jhg.2013.139
25. Liu SG, Che FY, Heng XY, Li FF, Huang SZ, Lu DG, et al. Clinical and genetic study of a novel mutation in the REEP1 gene. Synapse. (2009) 63:201–5. doi: 10.1002/syn.20602
26. Schlang KJ, Arning L, Epplen JT, Stemmler S. Autosomal dominant hereditary spastic paraplegia: novel mutations in the REEP1 gene (SPG31). BMC Med Genet. (2008) 9:71. doi: 10.1186/1471-2350-9-71
27. Hewamadduma C, McDermott C, Kirby J, Grierson A, Panayi M, Dalton A, et al. New pedigrees and novel mutation expand the phenotype of REEP1-associated hereditary spastic paraplegia (HSP). Neurogenetics. (2009) 10:105–10. doi: 10.1007/s10048-008-0163-z
28. de Bot ST, Veldink JH, Vermeer S, Mensenkamp AR, Brugman F, Scheffer H, et al. ATL1 and REEP1 mutations in hereditary and sporadic upper motor neuron syndromes. J Neurol. (2013) 260:869–75. doi: 10.1007/s00415-012-6723-z
29. McCorquodale DS III, Ozomaro U, Huang J, Montenegro G, Kushman A, Citrigno L, et al. Mutation screening of spastin, atlastin, and REEP1 in hereditary spastic paraplegia. Clin Genet. (2011) 79:523–30. doi: 10.1111/j.1399-0004.2010.01501.x
30. Polymeris AA, Tessa A, Anagnostopoulou K, Rubegni A, Galatolo D, Dinopoulos A, et al. A series of Greek children with pure hereditary spastic paraplegia: clinical features and genetic findings. J Neurol. (2016) 263:1604–11. doi: 10.1007/s00415-016-8179-z
31. Elert-Dobkowska E, Stepniak I, Krysa W, Rajkiewicz M, Rakowicz M, Sobanska A, et al. Molecular spectrum of the SPAST, ATL1 and REEP1 gene mutations associated with the most common hereditary spastic paraplegias in a group of Polish patients. J Neurol Sci. (2015) 359:35–9. doi: 10.1016/j.jns.2015.10.030
32. Erro R, Cordivari C, Bhatia KP. SPG31 presenting with orthostatic tremor. Eur J Neurol. (2014) 21:e34–5. doi: 10.1111/ene.12360
33. Battini R, Fogli A, Borghetti D, Michelucci A, Perazza S, Baldinotti F, et al. Clinical and genetic findings in a series of Italian children with pure hereditary spastic paraplegia. Eur J Neurol. (2011) 18:150–7. doi: 10.1111/j.1468-1331.2010.03102.x
34. Kamada M, Kawarai T, Miyamoto R, Kawakita R, Tojima Y, Montecchiani C, et al. Spastic paraplegia type 31: a novel REEP1 splice site donor variant and expansion of the phenotype variability. Parkinsonism Relat Disord. (2018) 46:79–83. doi: 10.1016/j.parkreldis.2017.10.012
35. Richard S, Lavie J, Banneau G, Voirand N, Lavandier K, Debouverie M. Hereditary spastic paraplegia due to a novel mutation of the REEP1 gene: case report and literature review. Medicine. (2017) 96:e5911. doi: 10.1097/MD.0000000000005911
Keywords: hereditary spastic paraplegia, receptor expression-enhancing protein 1, mutation analysis, transethnic, hotspot
Citation: Ma X, He J, Liu X and Fan D (2020) Screening for REEP1 Mutations in 31 Chinese Hereditary Spastic Paraplegia Families. Front. Neurol. 11:499. doi: 10.3389/fneur.2020.00499
Received: 05 January 2020; Accepted: 06 May 2020;
Published: 23 June 2020.
Edited by:
Christos Proukakis, University College London, United KingdomReviewed by:
Jordi Clarimon, Sant Pau Institute for Biomedical Research, SpainCraig Blackstone, National Institute of Neurological Disorders and Stroke (NINDS), United States
Copyright © 2020 Ma, He, Liu and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongsheng Fan, ZHNmYW4yMDEwQGFsaXl1bi5jb20=
†These authors have contributed equally to this work