


- 1 School of Pharmacy, Shandong University, Jinan, Shandong, China
- 2 School of Public Health, Shandong University, Jinan, Shandong, China
Bestatin, a small molecular weight dipeptide, is a potent inhibitor of various aminopeptidases as well as LTA4 hydrolase. Various physiological functions of Bestatin have been identified, viz.: (1) an immunomodifier for enhancing the proliferation of normal human bone marrow granulocyte–macrophage progenitor cells to form CFU-GM colonies; Bestatin exerts a direct stimulating effect on lymphocytes via its fixation on the cell surface and an indirect effect on monocytes via aminopeptidase B inhibition of tuftsin catabolism; (2) an immunorestorator and curative or preventive agent for spontaneous tumor; Bestatin alone or its combination with chemicals can prolongate the disease-free interval and survival period in adult acute or chronic leukemia, therefore, it was primarily marketed in 1987 in Japan as an anticancer drug and servers as the only marketed inhibitor of Aminopeptidase N (APN/CD13) to cure leukemia to date; (3) a pan-hematopoietic stimulator and restorator; Bestatin promotes granulocytopoiesis and thrombocytopoiesis in vitro and restores them in myelo-hypoplastic men; (4) an inhibitor of several natural opioid peptides. Based on the knowledge that APN can cleave several bioactive neuropeptides such as Met-enkaphalins, Leu-enkaphalins, β-Endorphin, and so on, the anti-aminopeptidase action of Bestatin also allows it to protect endopeptides against their catabolism, exhibiting analgesic activity. Although many scientific studies and great accomplishments have been achieved in this field, a large amount of problems are unsolved. This article reviews the promising results obtained for future development of the analgesic activity of Bestatin that can be of vital interest in a number of severe and chronic pain syndromes.
Introduction
The endogenous opioids comprising Met-Enkaphalins (ME), Leu-Enkaphalins (LE), β-Endorphin (β-EP), Dynorphin (Dyn), Kyotorphin (KTP), Endomorphin, (EM) and numerous others all belong to the family of pain-killing opioid peptides, which potently agitate opioid receptors to attain the antinociceptive effect (Frenk et al., 1978 ; Schwartz et al., 1981 ; Lee et al., 2004 ). The relevant opioid receptors belong to the family of G-protein coupled receptors with at least four subtypes [κ-opioid receptor (KOR), µ-opioid receptor (MOR), δ-opioid receptor (DOR), and σ-opioid receptor] being found in the human central nervous system contributing to the analgesia. These endogenous opioids, which work as “natural pain relievers” by decreasing the perception of pain, have a homogenous structural relationship as they usually possess tyrosine as an N-terminal amino acid residue, which is essential for opioid activity (Ramamoorthy and Balasubramanian, 1992 ). In most cases, these peptides tend to share multiple opiate receptors to mediate analgesic actions, even though they actually show distinctive preferences towards different opioid receptors. For example, the Enkaphalin has a higher affinity for the δ receptor (Chaillet et al., 1984 ), whereas the Dyn more readily acts as the ligand of the κ subtype (Han and Xie, 1982 ).
Despite their marked functions, the majority of opioid peptides undergo rapid enzymatic degradation (Egleton et al., 1998 ). Most of the extracellular peptide-degrading enzymes are membrane-bound exo- and endopeptidases and aminopeptidase N is one of these crucial enzymes. APN is a transmembrane protease present in a wide range of human tissues and cell types (endothelial, epithelial, fibroblast, leukocyte), which is able to modulate bioactive peptide responses and to influence immune functions. APN is responsible for the degradation of several biologically active peptide substrates including Enkaphalins, neurokinin A and β-EP (Miller et al., 1994 ; Lucius et al., 1995 ; Bauvois and Dauzonne, 2006 ; Luan and Xu, 2007 ), which, therefore, can be considered as a useful clinical marker. It can be expected that opiate peptides-operated neural activity is potentiated by peptidase inhibitors.
Bestatin, N-(2S, 3R)-3-amino-2-hydroxy-4-phenyl butanoyl) L-leucine (Figure 1 ), was extracted from Streptomyces olivoreticuli (Umezawa et al., 1976 ) and totally synthesized by Suda et al. (1976) for the first time adopting the following synthetic routine (Figure 2 ). As a potent APN inhibitor, its wide application in the immune system has been searched for a long history, affirming its functions in immunomodification (Umezawa et al., 1976 ; Mathe, 1991 ), immunorecovery (Bruley-Rosset et al., 1979 ; Ota and Ogawa, 1990 ), hematopoiesis (Talmadge et al., 1990 ), antinociception (Mathe, 1991 ; Miller et al., 1994 ). Among these, its marked inhibition of the degradation of these endogenous peptides attracts considerable attention due to its non-morphine-like addiction. Hence, we have briefly summarized the development of this specific effect of peptidase inhibitors, exclusively Bestatin, on the enzymatic degradation of various natural analgesic substances in the human body in sequence to further estimate the possibility and potential role for Bestatin to serve in the nervous system.
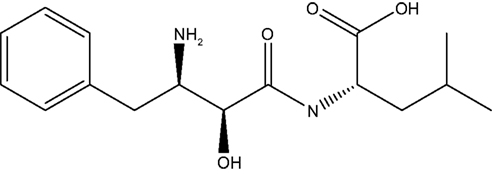
Figure 1. Bestatin.
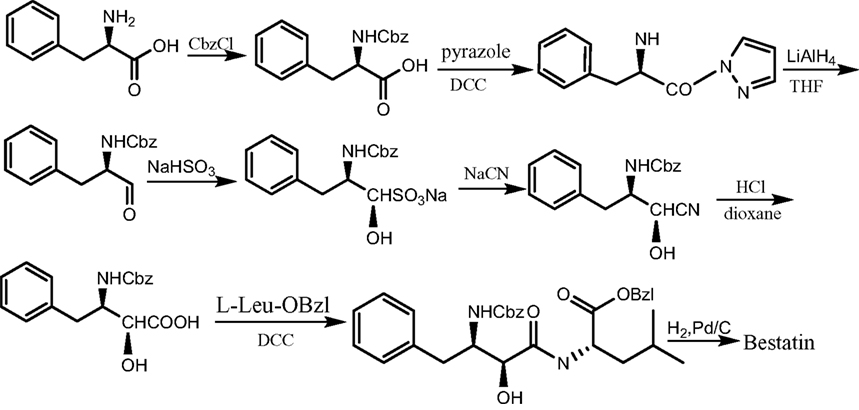
Figure 2. The synthetic route of Bestatin.
Defining the inactivation pathway of opioid peptides allows us to understand the possibility that opioid catabolism inhibitors elicit typical opioid responses by increasing the amount of endopeptides in vivo. Therefore, the increased analgesic effects of endogenous ligands by Bestatin may act on the opioid receptors, however, Bestatin and morphine have different mechanisms of action in the opioid system. It’s already known that chronic administration of morphine can induce tolerance and dependence, exhibiting a characteristic abstinence syndrome during the challenge of naloxone (NLX), an opioid antagonist, which can antagonize the antinociception of all the endogenous opioid ligands. Ozaki et al. (1994) firstly discovered that Bestatin could increase the ileal twitch inhibitory potency caused by ME as well as transient inhibition of twitch contraction after tetanic stimulation. These results suggested that Bestatin-sensitive aminopeptidase participated in the post-tetanic twitch inhibition. After a further study, the quantitatively different mechanisms of action in the opioid system between Bestatin and morphine were elucidated based on the fact that when challenged with NLX after a long exposure to the Bestatin and morphine respectively, the former didn’t induce any NLX-induced contraction which morphine did although they had similar effects on the post-tetanic contraction, which suggested the possibility that Bestatin had a smaller dependence liability (Ozaki, 2002 ).
Variously natural opioid peptides could act as substrates of APN including ME, LE, Dyn1–6, and Dyn1–7, which were commonly catalyzed by the N-terminal hydrolysis of tyrosine (Griffin et al., 1992 ). It was also proved that ME hydrolyzing enzymes were involved in the degradation of not only ME but also β-EP and Dyns in the rat central nervous system (Kishioka et al., 1994 ). Though ME and β-EP acted on both supraspinal and spinal sites, Dyns acted only on the spinal site. So it can be concluded that Bestatin can affect several opioid peptides concurrently. Given the considerably complex substances in the human body relevant to the antinociception responses, in order to organize and summarize more logically, we, followingly, deliberately state the analgesic effect of Bestatin in accordance with the list of each possibly related natural substance.
Functional Mechanisms
Enkaphalins
An enkaphalin is a pentapeptide involved in regulating nociception in the body of which two forms are determined; they are Met-enkaphalin (Tyr–Gly–Gly–Phe–Met) and Leu-enkaphalin (Tyr–Gly–Gly–Phe–Leu). Both are products of the proenkaphalin gene and it has been identified that MOR, rather than DOR, appears to be involved in enkaphalin-induced analgesia (Chaillet et al., 1984 ) based on the observed highly affinity between various enkaphalin analogs for the central or peripheral MOR, not DOR.
The inactivation of endogenous enkaphalins in neutral endopeptidase (NEP) and APN has been proven by several experiments (Dua et al., 1985 ; Leung et al., 1992 ; Miller et al., 1994 ; Kanai et al., 2002 ; Mosnaim Aron et al., 2004 ). Chaillet et al. (1983) found that not only the co-administration of Bestatin and Thiorphan, an enkaphalin dipeptidyl carboxypeptidase inhibitor, significantly protected [3H]ME-administered intracerebroventricular (i.c.v.) to mice from hydrolysis, but also both the two drugs alone caused a strong reduction in the accumulation of ME hydrolysis products in the brain. Furthermore, the antinociceptive activity of ME was additively potentiated by Bestatin and Thiorphan on either the hot-plate jump test or the phenyl-benzoquinone writhing test, respectively. Also, the hydrolysis of [3H]LE by slices from rat striatum was observed to be inhibited in the presence of Bestatin and Thiorphan.
Carenzi et al. (1983) provided another evidence in vivo to the degradatative effects by APN and NEP through detecting the pain threshold changes of mice and rats in vivo when Bestatin and Thiorphan were administrated as selective peptidase inhibitors. The results indicated that both enzymes were responsible for the enkaphalins’ degradation. However, it was argued that only concomitant intracerebral treatment with both inhibitors led to an increase in the threshold of animal pain, besides, in the presence of exogenous peptides, the co-administration of two inhibitors in mice elicited a synergetic analgesic activity and this response could be seen only after acute trauma. Later, opioid peptides contributing to the stress-induced antinociception with inflamed peripheral tissues of rats were studied (Parsons and Herz, 1990 ). Cohen et al. (1983) further affirmed the critical role of APN for terminating the pharmacological actions of enkaphalins in intact guinea pig ileal preparations. From their experiments, Bestatin enhanced the efficacy of enkaphalin in the guinea pig ileum and ileal longitudinal muscle in a dose-dependent fashion. Bestatin also decreased the formation of [3H] tyrosine and increased [3H]LE content. Later, Dickenson et al. (1987) stated that the enkaphalins may be degraded by at least three peptidases in the superficial dorsal horn of rabbit based on the study of three inhibitors of different peptidases by comparing the inhibitory effects mediated by endogenous enkaphalins. These inhibitors were Bestatin, a non-specific aminopeptidase inhibitor, Thiorphan, an inhibitor of NEP and Kelatorphan, a mixed inhibitor of several aminopeptidases, all of which enhanced the antinociception under the experiment with Kelatorphan to be the most efficient one. By observing the intense change of the voltage, the analgesic effects of peptidase inhibitors were also approved (Piguet and North, 1993 ). According to the experiment, the hyperpolarization of dentate granule cells of rat hippocampal slices by ME was potentiated by Bestatin and Thiorphan and blocked by antagonists selective for both MOR and DOR. Suzuki et al. (1994 , 1997) firstly tested the effects of peptidase inhibitors on physiological activities involving enkaphalins in the isolated spinal cord of the neonatal rat. Electrical stimulation of the saphenous nerve induced a slow ventral root potential (VRP), which was depressed by enkaphalins and augmented by NLX, suggesting that an opioid peptide-operated inhibitory mechanism was involved in the slow VRP. This effect of ME was markedly potentiated by addition of the mixture of peptidase inhibitors owing to the inhibition of the degradation of ME. Bestatin alone could potentiate the depressant effect of ME, which was less effective compared with Thiorphan, but still significantly inhibited the degradation by 65%. The important role of peptidases related to the hydrolysis of enkaphalins was recently studied with the NEP-knockout mice. It’s already known that in the brain, pharmacological inhibition of NEP-activity caused analgesia resulting from enhanced extracellular enkaphalin concentration. However, Fischer et al. (2002) was surprised to find that the NEP-knockout mice suffered from bradykinin-induced hyperalgesia rather than enkaphalin-mediated analgesia, what’s more, Thiorphan, the NEP-inhibitor couldn’t reduce writhing in NEP-deficient mice. Fortunately, Bestatin did exert remarkably inhibitory effects in both the NEP-knockout mice and the wild-type mice. This result further indicates the crucial role of APN in enkaphalin degradation.
On the other hand, the different metabolisms between ME and LE in the brain were investigated (Belyaev et al., 1990 ). Direct confirmation was obtained by determining changes in brain enkaphalin levels under the influence of Bestatin. It was surprised to note that Bestatin, which controlled the ME level in the midbrain, had no effect on the concentration of this peptide in the striatum, nor on the LE concentration in both parts of the brain studied. The selective action of Bestatin might indicate the regional differences of ME catabolism in the brain and also differences in the contribution of APN to the inactivation of ME and LE. A similar result was obtained by Jin et al. (1986) who reported that microinjection of Bestatin or Thiorphan into the nucleus accumbens of the rabbit produced a dose-dependent analgesic effect, which was totally reversed by NLX or by antibodies against ME administered to the same site, whereas antibodies against LE were not effective. Interestingly, Chaillet et al. (1983) and Mosnaim Aron et al. (2004) both successfully observed the inhibitory effect of the co-administration of Bestatin and Thiorphan on the hydrolysis of [3H]LE by slices from rat striatum. Besides, they also proved that the two inhibitors used together could potently protect ME administered i.c.v. to potentiate its antinociceptive properties in mice. Also, Bestatin itself displayed antinociceptive properties. Thus, a Bestatin-sensitive enkaphalin aminopeptidase activity together with the enkaphalin dipeptidyl carboxypeptidase activity apparently was mainly responsible for the inactivation of both exogenous and endogenous enkaphalins. In addition to the above-mentioned regional difference of the LE effect in the brain, some experiments showed the distinctive administration techniques might also lead to distinguished responses of LE (Mizuma et al., 1997 ). In accordance with the experiment, no analgesic effect was observed in the acetic acid writhing assay of mice after s.c. administration (for peripheral effect) of LE or cellobiose-coupled LE (CcpLE) without peptidase inhibitors, whereas CcpLE as well as LE had analgesic activity by subcutaneous administration (s.c.) to mice pretreated with peptidase inhibitors, On the other hand, hot-plate assay (for central effect) did not indicate obvious analgesic effect after s.c. administration of CcpLE or EcpLE with pretreatment of peptidase inhibitors. Therefore, it was concluded that CcpLE was a peripherally active analgesic. Also, the preference to different DORs was discovered in the mouse tail flick assay (Takemori and Portoghese, 1993 ). When LE or ME administered by i.c.v. together with the peptidase inhibitors, Bestatin and Thiorphan, the antinociceptive effects were significantly reversed by a selective DOR1 antagonist but not by a selective DOR2 antagonist. On the other hand, when given intrathecally (i.t.), the antinociceptive ED50 values of both enkaphalins were significantly raised by the opposite receptor subtypes respectively.
A large amount of work has been done to determine the precise active sites of APN to further understand the functional mechanism of Bestatin. The specific effects of Bestatin on exogenous and endogenous ME-induced antinociception were examined by a hot-plate method in mice (Matsuoka et al., 1988 ), which showed the elevation of pain threshold caused by an intracisternal administration of ME was enhanced by Bestatin and its analogs. Moreover, microinjection of Bestatin into the nucleus accumbens of rabbit resulted in a marked potentiation of the aftereffect of electroacupuncture-produced analgesia, as well as the analgesia induced by a small dose of morphine, both effects of which were mediated, at least in part, by a ME-like immunoreactive substance (Jin et al., 1986 ). These results not only indicated that the inhibition of APN might augment the exogenous ME antinociception but also suggested that Bestatin and its analogs might cause the enhancement of the endogenous ME-mediated antinociception. Berg and Marks (1984) primarily illuminated the specific acting sites of an Bestatin-sensitive aminopeptidase purified to homogeneity from the cytosol of rat brain, which cleaved Dyns and selected proenkaphalins at the Tyr–Gly bond proved by the release of only Tyr and the des Tyr fragments. Later on, Anderson et al. (1998) also supported the theory employing different experimental systems, in which ME was proven to be most readily degraded by the oligopeptidases contained in the venom of the Taiwan cobra, releasing a steady accumulation of Tyr, Gly–Gly and Phe–Met over time. The peptidase inhibitors, Bestatin and Thiorphan, were tested for their ability to inhibit the actions of the oligopeptidases. It was again verified that with ME as substrate, Bestatin inhibited the cleavage of the Tyr–Gly peptide bond depending on the observation of the reduced collection of Gly–Gly–Phe–Met, while there was no fall in the production of Tyr–Gly–Gly and Phe–Met and also an increase of Tyr–Gly–Gly. Meanwhile, Thiorphan was affirmed to accumulate the Gly–Phe peptide bond. Bestatin and Thiorphan also inhibited subsequent cleavage of the fragments Tyr–Gly–Gly and Gly–Gly–Phe–Met, respectively. LE was also found readily hydrolyzed to free tyrosine and Gly–Gly–Phe–Leu in rat microglia compared with other glial cells (Lucius et al., 1995 ) and inhibition experiments and immunostaining revealed that APN on the surface of microglia was responsible for enkaphalin cleavage. Later, Mosnaim et al. (2000) affirmed the inhibitory role of Bestatin in humans of various characters. They found that the [3H]LE degradation rate was pH and temperature dependent incubated in platelet-poor human plasma, which was independent of gender, age, or even the physical condition based on the observation that LE degradation was all rapid and complete in any case. More importantly, they pointed out that LE degradation was strongly and similarly inhibited by Bestatin with a lesser extent of various L-tyrosine-containing Leu fragments.
The antinociceptive effects of several enkaphalin analogues were also studied, among them, the heptapeptide, ME–Arg6–Phe7 (MEAP) was the exemplary one. As a precursor of ME, MEAP was indicated as an independent neuromodulator exhibiting the analgesic effect in vivo. Tang et al. (1983) claimed that APN might be important in the degradation of MEAP in vivo by using a preparation for the perfusion of the subarachnoidal spaces of the spinal cord of rats. Reggiani et al. (1984) reported that Bestatin could drastically potentiate the analgesic effect induced by the intracerebral injection of MEAP in cannulated rats, but Thiorphan was totally ineffective. These observations indicated that the predominant inactivating mechanism for MEAP was through the action of APN. Recently, MEAP-induced antinociception in chronic treatment was assumed to be caused by interaction with all three opioid receptors (µ, δ, and κ) in rats and differently regulating their expression (Vats et al., 2009 ). The analgesic activity could be potentiated by i.t. of Bestatin and Captoril owing to the accumulated recovery of MEAP.
β-Endorphine
β-Endorphin is released into blood from the pituitary gland and into the spinal cord and brain from hypothalamic neurons. Compared with other opioid peptides, β-EP is more resistant to the degrading-enzyme system thanks to its bigger molecular weight; nevertheless, once released into the blood it can’t enter the brain in large quantities because of the brain–blood barrier (BBB), so the physiological importance of the β-EP that can be measured in the blood is far from clear. β-EP has the highest affinity for the MOR1, a slightly lower affinity for MOR2 and DOR and low affinity for KOR. In common with ME, β-EP also plays an essential part in the antinociceptive system; actually, the β-EP may act as a releaser of ME. The effect of the intrathecal injection of Bestatin on the antinociception induced by β-EP administered i.c.v. was studied in male mice (Suh and Tseng, 1990b ). In the experiment, antinociceptive effects were valued by the tail-flick and hot-plate tests which showed Bestatin could potentiate inhibition of the tail-flick response induced by β-EP, but not antagonize inhibition of the hot-plate response. The results indicated that inhibition of the tail-flick response, induced by β-EP administered i.c.v. was mediated by the opioid system rather than by noradrenergic or serotonergic systems in the spinal cord, whereas the hot-plate response might be involved in other responsive mechanisms.
Further, Suh and Tseng (1990a) hypothesized that the antinociception induced by β-EP was mediated by release of ME. The theory was based on the fact that β-EP injected into the fourth ventricle could increase immunoreactive ME in the spinal perfusate in urethane-anesthetized rats; furthermore, Bestatin increased the amount of immunoreactive ME released by β-EP in a dose- dependent manner. This was also identified by Takemori and Portoghese (1993) , who found that β-EP-induced (i.c.v.) antinociception was antagonized by a selective DOR2 antagonist and potentiated by Bestatin, which seemed to have the same response with the enkaphalins administered i.t.. So it was assumed that the antinociception produced by i.c.v. administered β-EP was attributed to the release of ME in the spinal cord. However, some differences between the bioactive effects of β-EP and ME were also investigated based on the different reactions to the Bestatin. Compared to ME, β-EP also decreased the contractile responses to electrical field stimulation on airway smooth muscle (Horii et al., 1990 ) which was dose-dependent and reversed by NLX, however, was not augmented by the peptidase inhibitor, Bestatin, which is different from that of ME.
Dynorphins
Dynorphins are a class of opioid peptides that arise from the precursor protein prodynorphin. When prodynorphin is cleaved by proprotein convertase 2, multiple active peptides are released: Dyn A, Dyn B, and α/β-neoendorphin. Dyns exert their effects primarily through KOR, including KOR1 and KOR2. Besides, the peptides also have some affinities for MOR and DOR, meanwhile, different Dyns show different selectivities and potencies towards receptors. Calculating on a molar basis, Dyns can be 6–10 times more potent than morphine and 65–100 times more potent than morphiceptin, which was partially reversed by NLX (Han and Xie, 1982 ). Berg and Marks (1984) discovered an aminopeptidase, consisting of a single polypeptide chain when purified to homogeneity from the cytosol of rat brain, that cleaved Dyns having 5–17 residues and selected proenkaphalins at the Tyr–Gly bond to release Tyr and the desTyr fragments, which was inhibited by Bestatin in the micromolar range. Nakazawa et al. (1989) found that intracerebrally administered Dyn-B produced both an analgesia through opioid receptors and a motor dysfunction through non-opioid receptors in mice with the same dosage. More notably, Bestatin markedly potentiated the antinociceptive response induced by Dyn-B but not the motor dysfunction, and the potentiation could be antagonized by NLX. Another experimental result implied that some special conditions might be necessary for Bestatin to inhibit Dyns’ degradation. [3H]Dyn A 1–8 and [3H]Dyn A 1–9 with suspensions of guinea pig brain membrane could be both degraded at 25 and at 0°C (Gillan et al., 1985 ). Bestatin reduced degradation at 0°C and exhibited a similar degree of protection at 25°C. Additionally, Bestatin exerted less effect when the Dyns were bound with opioid receptor. Muller et al. (1995) illustrated the metabolism of Dyn A1–13 in human blood and plasma through the combination of enzyme inhibition experiments and non-compartmental kinetic analysis. They claimed that Dyn A1–13 was mainly metabolized by aminopeptidases and by carboxypeptidases, Bestatin-sensitive endopeptidases metabolized 30% of the subsequent product, Dyn A2–12. Based on the inhibitory roles of two types of neuropeptidases: NEP and APN, the combination of Bestatin plus Thiorphan could produce an increase in the level of both enkaphalins and Dyns family peptides. Cannabinoids elicited antinociception both at spinal and supraspinal levels, which can be readily potentiated by simultaneous activation of the opioid system and therefore, was therapeutically promising. Δ9-tetrahydrocannabinol (Δ9-THC), the most psychoactive natural cannabinoid, elicited antinociception in rodents through the central CB1 cannabinoid receptor subtype (Reche et al., 1998 ). In addition, Δ9-THC stimulated the release of Dyn-related peptides leading to κ-opioid spinal antinociception. Based on the existing knowledge that morphine was able to enhance cannabinoid antinociception, Bestatin was studied to identify its possible effect coupling with Δ9-THC, and the result showed its enhancement of the antinociceptive response of Δ9-THC, which was blocked by the CB1 cannabinoid receptor antagonist, as well as by NLX. This offered some encouragement to develop a new type of analgesic drug that might simultaneously activate both cannabinoid and opioid receptors.
Conversely, other studies have shown Dyn A is an endogenous opioid peptide that produces non-opioid receptor-mediated neural excitation identifying a role for Dyn and KOR stimulation in neuropathic pain (Lai et al., 2006 ; Mei et al., 2007 ). Furthermore, this group also reported that the Dyn-KOR system mediated astrocyte proliferation through the activation of p38 MAPK that was required for the effects of neuropathic pain on analgesic responses. Taken together, these results suggest that Dyn can elicit multiple effects on both KOR and non-opioid pathways to modulate pain-related responses.
Like the previously mentioned two types of peptides, Dyns similarly depressed the cholinergic twitch contractions of circular muscle strips from the guinea pig proximal colon induced by single pulse electrical field stimulation, which was abolished by atropine (Giuliani et al., 1996 ). This result totally contradicted the point that acetylcholine-induced contractions were not affected by Dyns as demonstrated before (Horii et al., 1990 ). Dyn A exhibited concentration-dependent inhibition of twitches, which could be enhanced by Bestatin and other peptidase inhibitors. Therefore, cholinergic twitches were considered to be modulated by multiple opioid receptors, among the KORs, activated by Dyn A1–13, mediating a powerful inhibition of acetylcholine release.
Kyotorphin
Kyotorphin, an endogenous ME-releasing antinociceptive dipeptide (l-Tyr–l-Arg), primarily obtained from the bovine rain, was formed by KTP synthetase from its constituent amino acids: L-Tyr and L-Arg, in an ATP-Mg2+-dependent manner. KTP produces analgesic effects possibly through ME release, by activating opioid receptors, especially the DOR (Takagi et al., 1979 ; Kawabata et al., 1996a ). Subsequently, Akasaki and Tsuji (1991) and Akasaki et al. (1991) ensured the degradating role of Bestatin-sensitive aminopeptidase, which could also catalyze the hydrolysis of LE and ME. Consequently, KTP was degraded by the aminopeptidase with a compatible Km value to those of LE and ME, and this conclusion was further identified using immunological analysis. Later on, a newly synaptic membrane-bound peptidase was supposed as a novel KTP-degrading enzyme which was inhibited by Bestatin exclusively in rat brain synaptosomes (Orawski and Simmons, 1992 ). Bestatin could also degrade other dipeptides including Phe–Arg, a synaptic membrane-generated metabolite of bradykinin. The deactivation of KTP in rats was studied by employing an amino acid analyzer, showing that Bestatin strongly inhibited the hydrolysis of KTP (Matsubayashi et al., 1984 ; Ueda et al., 1985a ,b ). According to the experiment, a dose of 50 µg of Bestatin administered intracisternally potentiated the analgesic activity of KTP by 4.8 times or so, and this effect was abolished by pretreatment with NLX. These facts suggested that the potentiation of analgesia by Bestatin might be due to the protection against the degradation of KTP and the release of enkaphalin by a membrane-bound aminopeptidase. Moreover, Satoh et al. (1985) claimed that the protection of Bestatin differed among regional areas based on the observation that co- administration of Bestatin with KTP enhanced the analgesic effect in the periaqueductal gray and lumbosacral subarachnoid space but not in the nucleus reticularis paragigantocellularis.
Later on, KTP synthetase was reported to also exist in the adrenal gland, in addition to the central nervous system (Kawabata et al., 1996b ), therefore, the peripheral contribution of KTP in pain regulation was studied (Inoue et al., 1997 ). Bradykinin given into the plantar region of the mouse hind-limb produced a flexor response, and KTP produced a dose-dependent reduction of the BK-induced nociceptive responses, which was not reversed by NLX, taking it into consideration that KTP-induced analgesia by central administration was completely blocked by NLX; therefore, the findings indicated that KTP produced analgesia through direct inhibition on Bradykinin responses without opioid mechanisms in peripheral tissues. It was also noteworthy to mention that the prerequisite of the effective antinociception of KTP was the presence of Bestatin given prior to the BK injection, When KTP was administered alone, the ED50 was disastrously higher and there was a complete recovery later. This further argues the significant protection of KTP from aminopeptidase by Bestatin.
Endomorphins
Endomorphins, isolated from bovine and human brain (Zadina et al., 1997 ), comprise two endogenous opioid peptides: endomorphin-1 (EM-1) (Tyr–Pro–Trp–Phe–NH2) and endomorphin-2 (EM-2) (Tyr–Pro–Phe–Phe–NH2), both of which are tetrapeptides with the highest affinity and specificity for the MOR to exert efficient antinociception (Zadina et al., 1999 ), which can be blocked by NLX. The physiological action of EMs as neurotransmitters or neuromodulators in the central nervous system is closely related to the degradation of EMs by peptidases. APN, due to the peptide bond type of its degradation, was assumed to play a crucial role in EM-degradation to tripeptides (Dua et al., 1985 ; Harbeck and Mentlein, 1991 ). Nevertheless, compared with the active sites of Bestatin on either enkaphalins or Dyns, some distinctive inhibitory mechanism was found as to that of EMs. Sakurada et al. (2003) and Sakurada (2004) reported that NEP and APN would be mainly responsible for the degradation of EM-2 at the synapse. In accordance with their experiment, NEP, as the first enzyme, cleaved EM-2 at the Pro2–Phe3 bond to generate Tyr–Pro and Phe–Phe–NH2, from which free phenylalanine and Phe–NH2 were produced by APN. This was proved by the fact that Bestatin could markedly enhance the generation of Phe–Phe–NH2, whereas free Phe and Phe–NH2 were decreased. These results might imply the involvement of APN in secondary cleavage of the initially formed cleavage products by the action of NEP; therefore, Bestatin might have relatively less effect on the degradation of EM-2 at supraspinal level. However, it was also noteworthy that Bestatin produced a large amount of Pro–Phe–Phe–NH2 whose peak was not detected in the absence of Bestatin, indicating the additional protection of Pro–Phe bond. In the behavioral studies, i.c.v. or i.t. administered EMs significantly increased nociceptive thresholds, which was blocked by NLX, implying that EMs might play a physiological role in modulating pain at the spinal and supraspinal levels (Fichna et al., 2007 ). Further research implied that the antinociception induced by EM-2 might be mediated by the release of Dyn A1–17 and ME, which subsequently acted on KORs and DORs to produce antinociception (Tseng et al., 2000 ; Sakurada et al., 2001 ).
The peripheral effect of EM-1 was primarily studied by Li et al. (2001) . In the experiment, EM-1 raised the pain threshold dose-dependently in tail stimulation-vocalization test in rats and inhibited the acetic acid writhing responses of mice, although the analgesic effect induced by peripheral intraperitoneal (i.p.) administration was obviously slower and weaker than that of the central (i.c.v. and i.t.) administration. However, both analgesic effects of EM-1 were reversed by NLX, which may predict the possible inhibitory effects of Bestatin, although no direct research has been done up to now.
Neurotension and Neuromedin U
Neurotension (NT) is a 13 amino acid neuropeptide (Glu–Leu–Tyr–Glu–Asn–Lys–Pro–Arg–Arg–Pro–Tyr–Ile–Leu–OH), which is synthesized as part of a 169–170 amino acid precursor protein that also contains the related neuropeptide neuromedin U (NM U), NM U is a highly conserved neuropeptide present in many species, existing as multiple isoforms; in humans, it is a 25 amino acid peptide. Both NT and its congener, NM, administrated i.c.v. in mice induce antinociceptive activity. Structure–activity studies reveal a close correlation between the analgesic potency of NT analogs and their affinity for the NT receptor 2 (NTR2) and discloses potent and selective agonists of this receptor, which confirms the fact that NTR2 mediates NT-induced analgesia.
The analgesic effects of both NT and NM were tested by the writhing and the tail-flick test (Coquerel et al., 1988 ). Compared to NT, the effect of NM was poorer. Bestatin alone promoted antinociceptive effects of NM with subthreshold dose in the tail-flick test though maximal antinociception was obtained by combining other inhibitors. More notably, NLX did not reduce the antinociceptive effect of either NT or NM indicating that NT and NM elicited analgesia in mice through an opiate-independent mechanism. Despite its peculiarity, Bestatin-sensitive aminopeptidase can degrade NM and has comparatively little effect on the NT. Fernandez and Alba (1996) further affirmed the crucial role of endogenous peptidases implicated in the inactivation of NT and NM in the prefrontal cortex by observing the inhibition of intracranial self-stimulation (ICSS) produced by injections of NT and NM into the medial prefrontal cortex of the rat. They studied the inhibitory change on ICSS after the administration of Bestatin and Thiorphan, two specific inhibitors of the peptidases that inactivate NM and NT, respectively, obtaining the result that microinjections of Bestatin and Thiorphan potentiated the inhibition of ICSS produced by the intracortical administration of NM and NT, correspondingly. The potentiation affected both the amplitude and the duration of the inhibition of ICSS produced by the neuropeptides.
Other Natural Substances
In addition to the above-mentioned natural peptides which are closely related with the analgesic activity of Bestatin, a large number of other relevant substances have been discovered.
Tuftsin (Thr–Lys–Pro–Arg)
Produced by enzymatic cleavage of the Fc-domain of the immunoglobulin G (Herman et al., 1981 , 1983 ; Ramamoorthy and Balasubramanian, 1992 ) and by acting as a naturally occurring immunopotentiating factor, also had marked analgesic activity. A cytosolic monkey brain aminopeptidase was responsible for cleaving tuftsin, which was inhibited by Bestatin.
The proenkephalin A-processing product peptide E (PE)
An opioid peptide originally isolated from the bovine adrenal medulla, was known to possess very low analgesic potency when injected by i.c.v. in mice alone (Westphal et al., 1985 ). However, the co-injection with Bestatin showed that PE-induced analgesia was increased remarkably. Inconsistently, Condamine et al. (1999) later rejected the potential PE-induced analgesia with the evidence that concomitant administration of PE with Bestatin did not produce analgesic responses. Therefore, further research on the structural and pharmacological characteristics of PE is urgently required.
I-menthol
An effective counterirritant has been widely used in the treatment of mild pains and itches by topical application. Taniguchi et al. (1994) found that I-Menthol-induced analgesia in the hot-plate test and hind paw pressure test of rats was both significantly potentiated by Bestatin and blocked by NLX. However, I-Menthol did not inhibit either carrageenin-induced paw edema of rats or the synthesis of prostaglandin E2 in vitro. All these observations suggested that I-menthol produced antinociceptive effects by activating the endogenous opioid system.
L-Arginine produced central antinociception by acting as a precursor of KTP
L-Ornithine, a metabolite of L-arginine, was assumed to produce L-arginine-like antinociception via KTP receptors. Kawabata et al. (1996a) identified this hypothesis depending on the evidence both L-Ornithine and L-arginine, when given by i.c.v. to mouse, elicited antinociception on the carrageenin-induced hyperalgesia even in intact mice, the effects being potentiated by Bestatin and abolished by NLX.
Substance P1–7
As one of the major metabolites of Substance P, administered by i.t. could increase tail-flick latency in a dose-dependent manner in the mouse tail-flick test. As endopeptidase played a major role in Substance P degradation in the mouse spinal cord, Bestatin potentiated the antinociception having only a mild effect (Sakurada et al., 2004 ).
More interestingly, Bestatin was discovered to be a powerful inducer of wound-response genes in tomato through specifically activating jasmonic acid signaling in plants, which can enhance the plant resistant system to various exotic nociceptive pain (Zheng et al., 2006 ).
Problems and Strategies
After a brief summary of the analgesic activity of Bestatin and its concerned biological substances, it can be safely concluded that the co-administration of Bestatin and the various peptides will offer us a newly promising strategy for the development of a creative class of analgesic. It is, however, quite necessary to point out that there are numerous drawbacks concerned about both of the two aspects.
Bestatin
The first issue is Bestatin’s marked toxicity and non-specificity (Giros et al., 1986 ; Bauvois and Dauzonne, 2006 ) by inhibiting other membrane-bound metalloproteases such as Leucyl-aminopeptidase, aminopeptidase B and aminopeptidase W or matrix metalloproteinases, suggesting that some of the observed chemotherapeutic actions of Bestatin may be due to inhibition of other cell surface peptidases. Together, these observations emphasize the need for more specific and targeted APN inhibitors.
Bestatin analogs
In order to counteract the negative problems with Bestatin, some analogs of Bestatin have been designed, synthesized and evaluated by i.c.v. injection in mice, several compounds, especially Kelatorphan, leading to NLX-reversible analgesic responses, that were slightly better than those produced by Bestatin and were also more efficient in potentiating the analgesia induced by a subanalgesic dose of ME (Fournie-Zaluski et al., 1985 ). More analogues of Bestatin were found to inhibit the hydrolysis of β-EP in different assay systems (Shang et al., 2008 ).
Co-administration
Another more advisable method to ensure the best use of Bestatin is the co-administration of the other drugs in a synchronic manner, which often involves several peptidases. Typical combinations included Bestatin or amastatin to inhibit APN, Thiorphan, or Phosphoramidon to inhibit NEP, and Captopril to inhibit angiotensin-convertion enzyme are effective in preventing the metabolic breakdown of a variety of opioid peptides (Hiranuma et al., 1998 ). The antinociceptive effect of i.t. administered Phosphoramidon and Bestatin was observed against the capsaicin-induced paw licking and biting response in mice (Tan-No et al., 1998 ), which was significantly antagonized by naltrindole, a selective DOR antagonist. Thus, Phosphoramidon coupling with Bestatin may block the degradation of enkaphalin. Cathinone-induced analgesia depended upon stimulation of α-adrenoceptors, which was counteracted by phenoxybenzamine. However, Bestatin strongly potentiated Cathinone-induced analgesia and this effect was sensitive to both NLX and Phenoxybenzamine blockade (Della Bella et al., 1985 ). Romero et al. (2009) reported that Bestatin remarkably potentiated the peripheral antinociceptive effect of Xylazine, an α2-adrenoceptor agonist by activating opioid receptors via endogenous opioid release. Also, Bestatin was found to enhance the analgesic effect of Celecoxib, a selective inhibitor of cyclooxygenase-2 in a model of peripherally induced inflammatory pain in rats (Rezende et al., 2009 ).
Dual inhibitors
Another powerful strategy as to develop ‘physiological’ analgesics devoid of the side effects of morphine is the application of the dual inhibitors, which can be metabolized once inside the brain to form two selective enzyme inhibitors and blocking both types of the zinc-metallopeptidase and enkaphalinase enzymes, resulting in the increase concentration of enkaphalins (Roques and Noble, 1995 ; Le Guen et al., 2003 ; Noble and Roques, 2007 ). These findings may constitute promising data for future development of a new class of analgesics that could be of major interest in severe and persistent pain syndromes.
Neuropeptides
The non-addictive morphine-like activity indicates that neuropeptides could potentially replace the morphine in pain management (Otero et al., 1993 ). However, as mentioned at the beginning, the therapeutic applications of these peptides for various disease conditions are restricted by their susceptibility to rapid degradation in biological systems. Furthermore, the BBB generally prevents the entry of peptides into the brain either by enzyme degradation or by specific properties of the BBB. Up to now, several tactics have been taken to prevent the transient degradation as well as the transportation barrier concerned with natural peptides.
Derivatives
Firstly, a great deal of derivatives of opioid peptides have been designed and synthesized, some of which exhibit significant improvements in both the blood–brain barrier penetration and half-life time in blood compared with the parent compounds, which predict the potential product of protein drugs arriving at the central nervous system; some stable peptide analogues of both ME and endomorphin-2 (Egleton et al., 1998 ), that lead to analgesia without side effects were successfully synthesized (Fichna et al., 2005 ). An engineered endomorphin-2 gene for use in neuropathic pain therapy was already obtained to randomly design peptides that acted at opioid receptors for analgesia (Wolfe et al., 2007 ) and to reduce the unwanted side effects associated with morphine, such as addiction and inhibition of gastric motility. Dyn1–13 efficiently traversed the BBB by a modification of the Oldendorf technique in cats with experimentally induced focal cerebral ischemia (Turner et al., 1998 ).
Chimeric peptides
The strategy of chimeric peptides linked by spacers was also investigated to enhance the antinociceptive property (Gupta et al., 2001 ; Kawano et al., 2007 ). An enzymatically stable analog of a chimeric peptide of ME and MEAP was synthesized, whose antinociceptive effect administered i.c.v. was investigated using the mouse radiant-heat tail-flick test and the result was encouraging.
Transportation
Also, some improvements have been achieved based on the study of transportation mechanisms. The enhancement of absorption and systemic delivery of enkaphalins via the human nose was realized through the co-administration of the peptide with protease inhibitors and absorption enhancers, resulting in drastically increased antinociception (Gwak et al., 2003 ; Agu et al., 2004 ). This was also applied to the improvement of EM-1, with the N- and C-termini of EM-1 modified by lipoamino acids.
Conclusion
The ubiquity of the APN in the body qualifies it to be a multifunctional enzyme, with its function mainly depending on its location. As it is already widely accepted, APN accounts for several biologically active peptides’ degradation, such as ME, LE, β-EM, Dyns and so on. Bestatin, as a potent APN inhibitor, therefore, may serve as a new and effective painkiller in alleviating severe and acute pain conditions with little or no morphine-related addition. Despite the obvious drawbacks of either the Bestatin, including the unavoidable toxicity and the poor selectivity or the quick degradation and the penetration difficulty concerned with its interacted substances, a commodious application space of Bestatin in the analgesic area is still conceivable. With scholars’ concerned, continuous efforts and the better understanding of the antinociceptive mechanisms occurring in animals as well as in the human body, we firmly believe that, someday, a completely new and effective class of analgesics derived from Bestatin and neuropeptides will emerge as therapies for several states of diseases. Besides, all of these available findings broaden our view on the role of Bestatin, which pave the way for Bestatin to be used as a potential composition in the treatment of different types of pharmacological nociception.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agu, R., Vu Dang, H., Jorissen, M., Kinget, R., and Verbeke, N. (2004). Metabolism and absorption enhancement of methionine enkephalin in human nasal epithelium. Peptides 25, 563–569.
Akasaki, K., Nakamura, A., Shiomi, H., and Tsuji, H. (1991). Identification and characterization of two distinct kyotorphin-hydrolyzing enzymes in rat brain. Neuropeptides 20, 103–107.
Akasaki, K., and Tsuji, H. (1991). An enkephalin-degrading aminopeptidase from rat brain catalyzes the hydrolysis of a neuropeptide, kyotorphin (L-Tyr-L-Arg). Chem. Pharm. Bull. (Tokyo) 39, 1883–1885.
Anderson, L., Bussler, B., Martins, H., and Dufton, M. (1998). Enkephalin-processing oligopeptidases in cobra venom: inhibition by thiorphan and bestatin reveals co-operative actions. Toxicon 36, 719–728.
Bauvois, B., and Dauzonne, D. (2006). Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: chemistry, biological evaluations, and therapeutic prospects. Med. Res. Rev. 26, 88–130.
Belyaev, N. A., Kolesanova, E. F., Kelesheva, L. F., Rotanova, T. V., and Panchenko, L. F. (1990). Effect of the aminopeptidase inhibitor bestatin on rat brain enkephalin levels. Bull. Exp. Biol. Med. 110, 1483–1485.
Berg, M. J., and Marks, N. (1984). Formation of desTyr dynorphins 5-17 by a purified cytosolic aminopeptidase of rat brain. J. Neurosci. Res. 11, 313–321.
Bruley-Rosset, M., Florentin, I., Kiger, N., Schulz, J., and Mathé, G. (1979). Restoration of impaired immune functions of aged animals by chronic bestatin treatment. Immunology 38, 75–83.
Carenzi, A., Frigeni, V., Reggiani, A., and Della Bella, D. (1983). Effect of inhibition of neuropeptidases on the pain threshold of mice and rats. Neuropharmacology 22, 1315–1319.
Chaillet, P., Coulaud, A., Zajac, J. M., Fournie-Zaluski, M. C., Costentin, J., and Roques, B. P. (1984). The mu rather than the delta subtype of opioid receptors appears to be involved in enkephalin-induced analgesia. Eur. J. Pharmacol. 101, 83–90.
Chaillet, P., Marcais-Collado, H., Costentin, J., Yi, C. C., De La Baume, S., and Schwartz, J. C. (1983). Inhibition of enkephalin metabolism by, and antinociceptive activity of, bestatin, an aminopeptidase inhibitor. Eur. J. Pharmacol. 86, 329–336.
Cohen, M. L., Geary, L. E., and Wiley, K. S. (1983). Enkephalin degradation in the guinea pig ileum: effect of aminopeptidase inhibitors, puromycin and bestatin. J. Pharmacol. Exp. Therapeut. 224, 379–385.
Condamine, E., Leprince, J., Suaudeau, C., Mayer, C., Davoust, D., Costentin, J., and Vaudry, H. (1999). The proenkephalin A-processing product peptide E, which encompasses two enkephalin sequences, has a much lower opioid activity than beta- endorphin. Peptides 20, 865–871.
Coquerel, A., Dubuc, I., Kitabgi, P., and Costentin, J. (1988). Potentiation by thiorphan and bestatin of the naloxone-insensitive analgesic effects of neurotensin and neuromedin N. Neurochem. Int. 12, 361–366.
Della Bella, D., Carenzi, A., Frigeni, V., Reggiani, A., and Zambon, A. (1985). Involvement of monoaminergic and peptidergic components in cathinone-induced analgesia. Eur. J. Pharmacol. 114, 231–234.
Dickenson, A. H., Sullivan, A. F., Fournie-Zaluski, M. C., and Roques, B. P. (1987). Prevention of degradation of endogenous enkephalins produces inhibition of nociceptive neurones in rat spinal cord. Brain Res. 408, 185–191.
Dua, A. K., Pinsky, C., and LaBella, F. S. (1985). Peptidases that terminate the action of enkephalins. Consideration of physiological importance for amino-, carboxy-, endo-, and pseudoenkephalinase. Life Sci. 37, 985–992.
Egleton, R. D., Abbruscato, T. J., Thomas, S. A., and Davis, T. P. (1998). Transport of opioid peptides into the central nervous system. J. Pharmaceut. Sci. 87, 1433–1439.
Fernandez, R., and Alba, F. (1996). Peptidase inhibitors potentiate the effects of neurotensin and neuromedin IM on self-stimulation of the medial prefrontal cortex. Neuroreport 7, 798.
Fichna, J., do-Rego, J. C., Kosson, P., Costentin, J., and Janecka, A. (2005). Characterization of antinociceptive activity of novel endomorphin-2 and morphiceptin analogs modified in the third position. Biochem. Pharmacol. 69, 179–185.
Fichna, J., Janecka, A., Costentin, J., and Do Rego, J. C. (2007). The endomorphin system and its evolving neurophysiological role. Pharmacol. Rev. 59, 88–123.
Fischer, H. S., Zernig, G., Hauser, K. F., Gerard, C., Hersh, L. B., and Saria, A. (2002). Neutral endopeptidase knockout induces hyperalgesia in a model of visceral pain, an effect related to bradykinin and nitric oxide. J. Mol. Neurosci. 18, 129–134.
Fournie-Zaluski, M. C., Coulaud, A., Bouboutou, R., Chaillet, P., Devin, J., Waksman, G., Costentin, J., and Roques, B. P. (1985). New bidentates as full inhibitors of enkephalin-degrading enzymes: synthesis and analgesic properties. J. Med. Chem. 28, 1158–1169.
Frenk, H., McCarty, B. C., and Liebeskind, J. C. (1978). Different brain areas mediate the analgesic and epileptic properties of enkephalin. Science 200, 335–337.
Gillan, M. G., Robson, L. E., McKnight, A. T., and Kosterlitz, H. W. (1985). Kappa-binding and degradation of [3H]dynorphin A (1-8) and [3H]dynorphin A (1-9) in suspensions of guinea pig brain membranes. J. Neurochem. 45, 1034–1042.
Giros, B., Gros, C., Solhonne, B., and Schwartz, J. C. (1986). Characterization of aminopeptidases responsible for inactivating endogenous (Met5)enkephalin in brain slices using peptidase inhibitors and anti- aminopeptidase M antibodies. Mol. Pharmacol. 29, 281–287.
Giuliani, S., Lecci, A., Tramontana, M., and Maggi, C. A. (1996). Role of kappa opioid receptors in modulating cholinergic twitches in the circular muscle of guinea-pig colon. Br. J. Pharmacol. 119, 985–989.
Griffin, K. J., Gierse, J., Krivi, G., and Fitzpatrick, F. A. (1992). Opioid peptides are substrates for the bifunctional enzyme LTA4 hydrolase/aminopeptidase. Prostaglandins 44, 251–257.
Gupta, S., Pasha, S., Gupta, Y., and Bhardwaj, D. (2001). Effects of intracerebroventricularly administered chimeric peptide of metenkephalin and FMRFa¡a[D-Ala2] YFa¡aon antinociception and its modulation in mice. Brain Res. Bull. 55, 51–57.
Gwak, H. S., Cho, Y. M., and Chun, I. K. (2003). Analgesic effects of intra-nasal enkephalins. J. Pharm. Pharmacol. 55, 1207–1212.
Han, J. S., and Xie, C. W. (1982). Dynorphin: potent analgesic effect in spinal cord of the rat. Life Sci. 31, 1781–1784.
Harbeck, H. T., and Mentlein, R. (1991). Aminopeptidase P from rat brain. Purification and action on bioactive peptides. Eur. J. Biochem. 198, 451–458.
Herman, Z. S., Stachura, Z., Krzemiski, T., Plech, A., Siemion, I. Z., and Nawrocka, E. (1983). Central effects of Tuftsin. Ann. N.Y. Acad. Sci. 419, 156–163.
Herman, Z. S., Stachura, Z., Opielka, L., Siemion, I. Z., and Nawrocka, E. (1981). Tuftsin and D-Arg3-tuftsin possess analgesic action. Experientia 37, 76–77.
Hiranuma, T., Kitamura, K., Taniguchi, T., Kanai, M., Arai, Y., Iwao, K., and Oka, T. (1998). Protection against Dynorphin-(1-8) hydrolysis in membrane preparations by the combination of Amastatin, Captopril and Phosphoramidon. Pharmocology 286, 863–869.
Horii, S., Tamaoki, J., Kanemura, T., Yamawaki, I., and Takizawa, T. (1990). Effects of beta-endorphin and dynorphin A on cholinergic neurotransmission in canine airway smooth muscle. Eur. J. Pharmacol. 182, 497–502.
Inoue, M., Nakayamada, H., Tokuyama, S., and Ueda, H. (1997). Peripheral non-opioid analgesic effects of kyotorphin in mice. Neurosci. Lett. 236, 60–62.
Jin, W. Q., Zhou, Z. F., and Han, J. S. (1986). Electroacupuncture and morphine analgesia potentiated by bestatin and thiorphan administered to the nucleus accumbens of the rabbit. Brain Res. 380, 317–324.
Kanai, M., Takahashi, S., Kosaka, K., Iwao, K., Kobayashi, H., and Oka, T. (2002). [Met5]enkephalin-Arg-Gly-Leu-induced antinociception is greatly increased by peptidase inhibitors. Eur. J. Pharmacol. 453, 53–58.
Kawabata, A., Iwatsubo, K., Takaya, S., and Takagi, H. (1996a). Central antinociceptive effect of l-ornithine, a metabolite of l-arginine, in rats and mice. Eur. J. Pharmacol. 296, 23–31.
Kawabata, A., Muguruma, H., Tanaka, M., and Takagi, H. (1996b). Kyotorphin synthetase activity in rat adrenal glands and spinal cord. Peptides 17, 407–411.
Kawano, S., Ito, R., Nishiyama, M., Kubo, M., Matsushima, T., Minamisawa, M., Ambo, A., and Sasaki, Y. (2007). Receptor binding properties and antinociceptive effects of chimeric peptides consisting of a i-opioid receptor agonist and an ORL1 receptor antagonist. Biol. Pharmaceut. Bull. 30, 1260–1264.
Kishioka, S., Miyamoto, Y., Fukunaga, Y., Nishida, S., and Yamamoto, H. (1994). Effects of a mixture of peptidase inhibitors (Amastatin, Captopril and Phosphoramidon) on Met-Enkephalin-, Â-Endorphin-, Dynorphin-(1-13) and electroacupuncture-induced antinociception in rats. Jpn. J. Pharmacol. 66, 337–345.
Lai, J., Luo, M. C., Chen, Q., Ma, S., Gardell, L. R., Ossipov, M. H., and Porreca, F. (2006). Dynorphin A activates bradykinin receptors to maintain neuropathic pain. Nat. Neurosci. 9, 1534–1540.
Le Guen, S., Mas Nieto, M., Canestrelli, C., Chen, H., Fournie-Zaluski, M. C., Cupo, A., Maldonado, R., Roques, B. P., and Noble, F. (2003). Pain management by a new series of dual inhibitors of enkephalin degrading enzymes: long lasting antinociceptive properties and potentiation by CCK2 antagonist or methadone. Pain 104, 139–148.
Lee, J. J., Hahm, E. T., Min, B. I., and Cho, Y. W. (2004). Activation of protein kinase C antagonizes the opioid inhibition of calcium current in rat spinal dorsal horn neurons. Brain Res. 1017, 108–119.
Leung, M. K., Le, S., Houston, S., and Stefano, G. B. (1992). Degradation of Met-enkephalin by hemolymph peptidases in Mytilus edulis. Cell Mol. Neurobiol. 12, 367–378.
Li, Z. H., Shan, L. D., Jiang, X. H., Guo, S. Y., Yu, G. D., Hisamitsu, T., and Yin, Q. Z. (2001). Analgesic effect of endomorphin-1. Acta Pharmacol. Sin. 22, 976–980.
Luan, Y., and Xu, W. (2007). The structure and main functions of aminopeptidase N. Curr. Med. Chem. 14, 639–647.
Lucius, R., Sievers, J., and Mentlein, R. (1995). Enkephalin metabolism by microglial aminopeptidase N (CD13). J. Neurochem. 64, 1841–1847.
Mathe, G. (1991). Bestatin, an aminopeptidase inhibitor with a multi-pharmacological function. Biomed. Pharmacother. 45, 49–54.
Matsubayashi, K., Kojima, C., Kawajiri, S., Ono, K., Takegoshi, T., Ueda, H., and Takagi, H. (1984). Hydrolytic deactivation of kyotorphin by the rodent brain homogenates and sera. J. Pharmacobiodyn. 7, 479–484.
Matsuoka, Y., Satoh, S., Uruno, T., and Kubota, K. (1988). 2S,3R 3-amino-2-hydroxy-4-phenylbutanoic acid derivatives, enkephalinase inhibitors, augment met5-enkephalin-induced antinociception. Jpn. J. Pharmacol. 46, 205–210.
Mei, X., Bruchas, M. R., Ippolito, D. L., Gendron, L., and Chavkin, C. (2007). Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by κ opioid activation of p38 mitogen-activated protein kinase. J. Neurosci. 27, 2570–2581.
Miller, B. C., Thiele, D. L., Hersh, L. B., and Cottam, G. L. (1994). Methionine enkephalin is hydrolyzed by aminopeptidase N on CD4+ and CD8+ spleen T cells. Arch. Biochem. Biophys. 311, 174–179.
Mizuma, T., Ohta, K., and Awazu, S. (1997). Peripherally active analgesia of aminopeptidase-resistant sugar-coupled leucine enkephalin. Res. Commun. Mol. Pathol. Pharmacol. 95, 83–91.
Mosnaim, A. D., Wolf, M. E., Nguyen, T. D., Puente, J., Freitag, F., and Diamond, S. (2000). Degradation kinetics of leucine5-enkephalin by plasma samples from healthy controls and various patient populations: in vitro drug effects. Am. J. Therapeut. 7, 185–194.
Mosnaim Aron, D., Javier, P., Vasant, R., Catherine, H., and Wolf, M. E. (2004). Inhibition of human plasma leucine5-enkephalin aminopeptidase hydrolysis by various endogenous peptides and a select number of clinically used drugs. Am. J. Therapeut. 11, 459–465.
Muller, S., and Hochhaus, G. (1995). Metabolism of dynorphin A 1-13 in human blood and plasma. Pharm Res 12, 1165–1170.
Nakazawa, T., Ikeda, M., Kaneko, T., Yamatsu, K., Kitagawa, K., and Kiso, Y. (1989). Bestatin potentiates the antinociception but not the motor dysfunction induced by intracerebrally administered dynorphin-B in mice. Neuropeptides 13, 277–283.
Noble, F., and Roques, B. P. (2007). Protection of endogenous enkephalin catabolism as natural approach to novel analgesic and antidepressant drugs. Expert Opin. Therapeut. Targets 11, 145–159.
Orawski, A. T., and Simmons, W. H. (1992). Dipeptidase activities in rat brain synaptosomes can be distinguished on the basis of inhibition by bestatin and amastatin: identification of a kyotorphin (Tyr-Arg)-degrading enzyme. Neurochem. Res. 17, 817–820.
Ota, K., and Ogawa, N. (1990). Randomized controlled study of chemoimmunotherapy with bestatin of acute nonlymphocytic leukemia in adults. Biomed. Pharmacother. 44, 93–101.
Otero, M. J., Iglesias, T., and Fuentes, J. A. (1993). Hypoalgesic action of bestatin analogues that inhibit central aminopeptidases, but not neutral endopeptidase. Neuropeptides 25, 175–182.
Ozaki, M. (2002). Effects of naloxone and post-tetanic stimulation on isolated guinea-pig ileum followed by long exposure to morphine and bestatin. J. Toxicol. Sci. 27, 173–182.
Ozaki, M., Miyamoto, Y., Kishioka, S., Masuda, Y., and Yamamoto, H. (1994). Effect of some peptidase inhibitors on exogenous and endogenous opioid actions in guinea-pig ileum. Biol. Pharm. Bull. 17, 62–69.
Parsons, C. G., and Herz, A. (1990). Peripheral opioid receptors mediating antinociception in inflammation. Evidence for activation by enkephalin-like opioid peptides after cold water swim stress. J. Pharmacol. Exp. Ther. 255, 795–802.
Piguet, P., and North, R. A. (1993). Opioid actions at mu and delta receptors in the rat dentate gyrus in vitro. J. Pharmacol. Exp. Ther. 266, 1139–1149.
Ramamoorthy, S., and Balasubramanian, A. S. (1992). Chemical modification of the monkey brain cytosolic aminopeptidase that cleaves enkephalin to its constituent amino acids. Evidence for essential tyrosine residue (residues). Biochem. Int. 27, 861–868.
Reche, I., Ruiz-Gayo, M., and Fuentes, J. A. (1998). Inhibition of opioid-degrading enzymes potentiates delta9- tetrahydrocannabinol-induced antinociception in mice. Neuropharmacology 37, 215–222.
Reggiani, A., Carenzi, A., Frigeni, V., and Della Bella, D. (1984). Effect of bestatin and thiorphan on [Met5]enkephalin-Arg6-Phe7-induced analgesia. Eur. J. Pharmacol. 105, 361–364.
Rezende, R. M., Dos Reis, W. G. P., Duarte, I. D. G., Lima, P. P., Bakhle, Y. S., and de Francischi, J. N. (2009). The analgesic actions of centrally administered celecoxib are mediated by endogenous opioids. Pain 142, 94–100.
Romero, T. R. L., de Castro Perez, A., de Francischi, J. N., and Gama Duarte, I. D. (2009). Probable involvement of alpha(2C)-adrenoceptor subtype and endogenous opioid peptides in the peripheral antinociceptive effect induced by xylazine. Eur. J. Pharmacol. 608, 23–27.
Roques, B. P., and Noble, F. (1995). Dual inhibitors of enkephalin-degrading enzymes (neutral endopeptidase 24.11 and aminopeptidase N) as potential new medications in the management of pain and opioid addiction. NIDA Res. Monogr. 147, 104–145.
Sakurada, C. (2004). [Development of a new analgesic based on metabolism of endomorphin, an endogenous opioid peptide]. Yakugaku Zasshi 124, 549–554.
Sakurada, C., Sakurada, S., Hayashi, T., Katsuyama, S., Tan-No, K., and Sakurada, T. (2003). Degradation of endomorphin-2 at the supraspinal level in mice is initiated by dipeptidyl peptidase IV: an in vitro and in vivo study. Biochem. Pharmacol. 66, 653–661.
Sakurada, C., Watanabe, C., and Sakurada, T. (2004). Occurrence of substance P(1-7) in the metabolism of substance P and its antinociceptive activity at the mouse spinal cord level. Methods Find Exp. Clin. Pharmacol. 26, 171–176.
Sakurada, S., Hayashi, T., Yuhki, M., Orito, T., Zadina, J. E., Kastin, A. J., Fujimura, T., Murayama, K., Sakurada, C., Sakurada, T., Narita, M., Suzuki, T., Tan-No, K., and Tseng, L. F. (2001). Differential antinociceptive effects induced by intrathecally administered endomorphin-1 and endomorphin-2 in the mouse. Eur. J. Pharmacol. 427, 203–210.
Satoh, M., Wada, T., Iwama, T., and Takagi, H. (1985). Sites of analgesic actions of kyotorphin and D-kyotorphin in the central nervous system of rats. Neuropeptides 5, 415–418.
Schwartz, J.-C., Malfroy, B., and De La Baume, S. (1981). Biological inactivation of enkephalins and the role of enkephalin-dipeptidyl-carboxypeptidase (“enkephalinase”) as neuropeptidase. Life Sci. 29, 1715–1740.
Shang, L. Q., Maeda, T., Xu, W. F., and Kishioka, S. (2008). Evaluation of a novel aminopeptidase N inhibitor, in vitro, using two assay systems. J. Enzyme Inhib. Med. Chem. 23, 198–205.
Suda, H., Takita, T., Aoyagi, T., and Umezawa, H. (1976). The chemical synthesis of bestatin. J. Antibiot. 29, 600–601.
Suh, H. H., and Tseng, L. L. (1990a). Intrathecal administration of thiorphan and bestatin enhances the antinociception and release of Met-enkephalin induced by beta- endorphin intraventricularly in anesthetized rats. Neuropeptides 16, 91–96.
Suh, H. H., and Tseng, L. L. (1990b). Intrathecal administration of thiorphan, bestatin, desipramine and fluoxetine differentially potentiate the antinociceptive effects induced by beta-endorphin and morphine, administered intracerebroventricularly. Neuropharmacology 29, 207–214.
Suzuki, H., Yanagisawa, M., Yoshioka, K., Hosoki, R., and Otsuka, M. (1997). Enzymic inactivation of enkephalin neurotransmitters in the spinal cord of the neonatal rat. Neurosci. Res. 28, 261–267.
Suzuki, H., Yoshioka, K., Yanagisawa, M., Urayama, O., Kurihara, T., Hosoki, R., Saito, K., and Otsuka, M. (1994). Involvement of enzymatic degradation in the inactivation of tachykinin neurotransmitters in neonatal rat spinal cord. Br. J. Pharmacol. 133, 310–316.
Takagi, H., Shiomi, H., Ueda, H., and Amano, H. (1979). A novel analgesic dipeptide from bovine brain is a possible Met-enkephalin releaser. Nature 282, 410–412.
Takemori, A. E., and Portoghese, P. S. (1993). Enkephalin antinociception in mice is mediated by delta 1- and delta 2-opioid receptors in the brain and spinal cord, respectively. Eur. J. Pharmacol. 242, 145–150.
Talmadge, J. E., Black, P. L., Pelus, L. M., and Abe, F. (1990). Hematopoietic and hematologic properties of bestatin in normal and cyclophosphamide myelosuppressed mice. Biomed. Pharmacother. 44, 85–91.
Tan-No, K., Taira, A., Inoue, M., Ohshima, K., Sakurada, T., Sakurada, C., Nylander, I., Demuth, H., Silberring, J., and Terenius, L. (1998). Intrathecal administration of p- hydroxymercuribenzoate or phosphoramidon/bestatin-combined induces antinociceptive effects through different opioid mechanisms. Neuropeptides 32, 411–415.
Tang, J., Chou, J., Yang, H. Y., and Costa, E. (1983). The effect of peptidase inhibitors on the release of Met5-Enk-Arg6-Phe7 (YGGFMRF) and Met5-enkephalin (YGGFM) from spinal cord induced by substance P in vivo. Life Sci. 33(Suppl. 1), 121–124.
Taniguchi, Y., Deguchi, Y., Saita, M., and Noda, K. (1994). Antinociceptive effects of counterirritants. Nippon Yakurigaku Zasshi 104, 433–446.
Tseng, L. F., Narita, M., Suganuma, C., Mizoguchi, H., Ohsawa, M., Nagase, H., and Kampine, J. P. (2000). Differential antinociceptive effects of endomorphin-1 and endomorphin-2 in the mouse. J. Pharmacol. Exp. Therapeut. 292, 576–583.
Turner, T. D., Browning, J. L., Widmayer, M. A., and Baskin, D. S. (1998). Penetration of dynorphin 1-13 across the blood-brain barrier. Neuropeptides 32, 141–149.
Ueda, H., Ming, G., Hazato, T., Katayama, T., and Takagi, H. (1985a). Degradation of kyotorphin by a purified membrane-bound-aminopeptidase from monkey brain: potentiation of kyotorphin-induced analgesia by a highly effective inhibitor, bestatin. Life Sci. 36, 1865–1871.
Ueda, H., Yoshihara, Y., Nakamura, A., Shiomi, H., Satoh, M., and Takagi, H. (1985b). How is kyotorphin (Tyr-Arg) generated in the brain? Neuropeptides 5, 525–528.
Umezawa, H., Aoyagi, T., Suda, H., Hamada, M., and Takeuchi, T. (1976). bestatin, an inhibitor of aminopeptidaseB, produced by actinomycetes. J. Antibiot. 29, 97–99.
Vats, I., Chaudhary, S., Karar, J., Nath, M., Pasha, Q., and Pasha, S. (2009). Endogenous peptide: Met-enkephalin-Arg-Phe, differently regulate expression of opioid receptors on chronic treatment. Neuropeptides 43, 355–362.
Westphal, M., Hammonds, R. G., Jr., and Li, C. H. (1985). Protease inhibition increases the analgesic potency of peptide E. Neuropharmacology 24, 173–175.
Wolfe, D., Hao, S., Hu, J., Srinivasan, R., Goss, J., Mata, M., Fink, D. J., and Glorioso, J. C. (2007). Engineering an endomorphin-2 gene for use in neuropathic pain therapy. Pain 133, 29–38.
Zadina, J. E., Hackler, L., Ge, L. J., and Kastin, A. J. (1997). A potent and selective endogenous agonist for the mu-opiate receptor. Nature 386, 499–502.
Zadina, J. E., Martin-Schild, S., Gerall, A. A., Kastin, A. J., Hackler, L., Ge, L. J., and Zhang, X. (1999). Endomorphins: novel endogenous mu-opiate receptor agonists in regions of high mu-opiate receptor density. Ann. N.Y. Acad. Sci. 897, 136–144.
Keywords: Bestatin, analgesic activity, mechanism, neuropeptides, application
Citation: Jia MR, Wei T and Xu WF (2010) The analgesic activity of Bestatin as a potent APN inhibitor. Front. Neurosci. 4:50. doi: 10.3389/fnins.2010.00050
This article was submitted to Frontiers in Neuropharmacology, a specialty of Frontiers in Neuroscience.
Received: 09 April 2010;
Paper pending published: 08 June 2010;
Accepted: 10 June 2010;
Published online: 28 June 2010
Edited by:
Xiao-Ming Ou, University of Mississippi Medical Center, USAReviewed by:
Junming Wang, University of Mississippi Medical Center, USAXiao-Ming Ou, University of Mississippi Medical Center, USA
Copyright: © 2010 Jia, Wei and Xu. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Wen-Fang Xu, Department of Medicinal Chemistry, School of Pharmacy, Shandong University, 44 West Culture Road, Jinan, 250012, People’s Republic of China. e-mail:d2Z4dUB5YWhvby5jbg==
Abbreviations: APN, Aminopeptidase N; ME, Met-enkaphalin; LE, Leu- enkaphalin; β-EP, β-Endorphin; Dyns, Dynorphins; EM, Endomorphin; KOR, κ-opioid receptor; MOR, µ-opioid receptor; DOR, δ-opioid receptor; NLX, Naloxone; NEP, Neutral endopeptidase; i.c.v., Intracerebroventricular; VRP, ventral root potential; s.c, subcutaneously; i.t., intrathecal; Δ9- THC, Δ9-Tetrahydrocannabinol; i.p., Intraperitoneally; NT, Neurotension; NMU, Neuromedin U; NTR2, NT receptor 2; ICSS, inhibition of intracranial self-stimulation; PE, peptide E.