



- Department of Pharmacology and Toxicology, School of Medical Sciences, University of Otago, Dunedin, New Zealand
Some toxins and drugs can trigger lasting neuroprotective mechanisms that enable neurons to resist a subsequent severe insult. This “pharmacological preconditioning” has far-reaching implications for conditions in which blood flow to the brain is interrupted. We have previously shown that in vitro preconditioning with the AMPA receptor antagonist GYKI 52466 induces tolerance to kainic acid (KA) toxicity in hippocampus. This effect persists well after washout of the drug and may be mediated via inverse agonism of G-protein coupled receptors (GPCRs). Given the amplifying nature of metabotropic modulation, we hypothesized that GYKI 52466 may be effective in reducing seizure severity at doses well below those normally associated with adverse side effects. Here we report that pharmacological preconditioning with low-dose GYKI imparts a significant protection against KA-induced seizures in vivo. GYKI (3 mg/kg, s.c.), 90–180 min prior to high-dose KA, markedly reduced seizure scores, virtually abolished all level 3 and level 4 seizures, and completely suppressed KA-induced hippocampal c-FOS expression. In addition, preconditioned animals exhibited significant reductions in high frequency/high amplitude spiking and ECoG power in the delta, theta, alpha, and beta bands during KA. Adverse behaviors often associated with higher doses of GYKI were not evident during preconditioning. The fact that GYKI is effective at doses well-below, and at pre-administration intervals well-beyond previous studies, suggests that a classical blockade of ionotropic AMPA receptors does not underlie anticonvulsant effects. Low-dose GYKI preconditioning may represent a novel, prophylactic strategy for neuroprotection in a field almost completely devoid of effective pharmaceuticals.
Introduction
GYKI 52466 (1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy- 5H-2,3-benzodiazepine hydrochloride) is a non-competitive AMPA receptor negative allosteric modulator (Donevan and Rogawski, 1993; Zorumski et al., 1993) which has been examined in a number of seizure models including audiogenic seizures (Smith et al., 1991; De Sarro et al., 1998a,b, 2003; Szabados et al., 2001), maximal electroshock seizures (Donevan et al., 1994; Borowicz et al., 1995; Szabados et al., 2001; Barton et al., 2003; De Sarro et al., 2003; Gressens et al., 2005), amygdala-kindled seizures (Borowicz et al., 2001), and chemoconvulsant-induced seizures (Donevan et al., 1994; De Sarro et al., 2003). Most of these studies found GYKI 52466 to be an effective anticonvulsant, exhibiting ED50′s of approximately 10–25 mg/kg when administered 15–30 min prior to seizure induction. GYKI 52466’s effects have been shown to last from 60 to 90 min (De Sarro et al., 1998a, 2003), with plasma concentrations peaking within 15 min and dropping to less than 5% of peak levels within 90 min of i.p injection (De Sarro et al., 1998b). Unfortunately, debilitating motor and cognitive side effects have been routinely documented at doses as low as 10–15 mg/kg (Donevan et al., 1994; Borowicz et al., 1995, 2001). Given the short duration of action and magnitude of adverse effects seen at effective doses, the therapeutic utility of GYKI 52466 has remained limited.
Seizures and epilepsy often arise following stroke or traumatic brain injury (Karhunen et al., 2007). At the same time, uncontrolled seizure activity can lead to CNS damage (Sloviter, 1987; Pitkänen and Sutula, 2002). It is well-established that neuroprotectants are often ineffective when administration is significantly delayed, as is often the case in human stroke (Chauhan et al., 2003). Ideal neuroprotectants would be those that trigger endogenous neuroprotective pathways when administered to at-risk patients before an ischemic insult occurs, and could be of real benefit in cases of cardiac or brain surgery in which global or focal CNS ischemia is likely. A number of studies in recent years have shown that CNS neurons do in fact possess inducible neuroprotective mechanisms and can be preconditioned in a variety of ways to resist hypoxic or excitotoxic insults. In addition, evidence indicates that preconditioning procedures can impart a rapid tolerance to subsequent seizurogenic or excitotoxic insults, which may be observed within minutes of preconditioning.
In the course of assessing pharmacological preconditioning mechanisms, we found that in vitro preconditioning with GYKI 52466 induces a lasting tolerance versus kainic acid (KA) toxicity in hippocampal CA1, and that this effect may be metabotropic in nature (Hesp et al., 2004). In those studies we found that a number of ionotropic AMPA and KA antagonists (including GYKI 52466) also acted as inverse agonists of G-protein coupled receptor (GPCR) function, significantly reducing constitutive GTPase activity in hippocampal membranes from young but not aged rats. In addition, in rat hippocampal CA1, low concentrations of GYKI 52466, and NBQX-induced tolerance to the adverse electrophysiological effects of high-dose KA, even after the antagonists had been washed from the slice preparation. Taken together, our findings suggest that tolerance is triggered by a selective reduction in constitutive GPCR activity, and this metabotropic neuroprotective mechanism is lost in aged hippocampus (Hesp et al., 2004). Given the lasting and amplifying nature of metabotropic modulation, we hypothesized that GYKI 52466 may be effective in reducing seizure severity at doses well below those normally associated with adverse side effects. In the present study we examined the anticonvulsant effects of GYKI 52466 versus KA-induced seizures across a range of low doses and extended time points. Parts of this study have appeared in abstract form (Kerr, 2007).
Materials and Methods
Animals
All procedures were approved by the University of Otago Animal Ethics Committee and conducted in accordance with the New Zealand Animal Welfare Act. Male Sprague–Dawley rats, 2–3 months old and weighing 200–280 g, were obtained from the University of Otago Hercus-Taieri animal colony and housed for at least one week in the departmental holding facility on a 12-h light–dark cycle, with free access to food and water.
Drugs and Reagents
Kainic acid and GYKI 52466 hydrochloride were obtained from Tocris Bioscience (UK), dissolved in sterile saline (with small pH adjustment as necessary) and administered at rates of 1 mL/kg body weight. Immunohistochemistry reagents were sourced as described below. All other chemicals and reagents were acquired from BDH Chemicals (Poole, England).
Behavioral Observation and Analysis
Prior to behavioral observation animals were allowed to acclimate for 30 min in a custom-made Perspex™ observation chamber (Aburn Glass, Dunedin, NZ). For the initial parametric analysis of GYKI preconditioning, rats were injected with GYKI 52466 (0.3, 1, or 3 mg/kg or saline vehicle; s.c.) 10, 90, or 180 min prior to a single injection of KA (10 mg/kg; a dose which reliably produces clonic–tonic convulsions within 120 min of administration). For the subsequent ECoG study, rats were injected with saline or GYKI 52466 (1 mg/kg or 3 mg/kg, s.c.) at selected time points (10, 90, 180 min) prior to a single injection of KA (10 mg/kg, s.c.). Treatment groups and Ns are shown in Table 1.
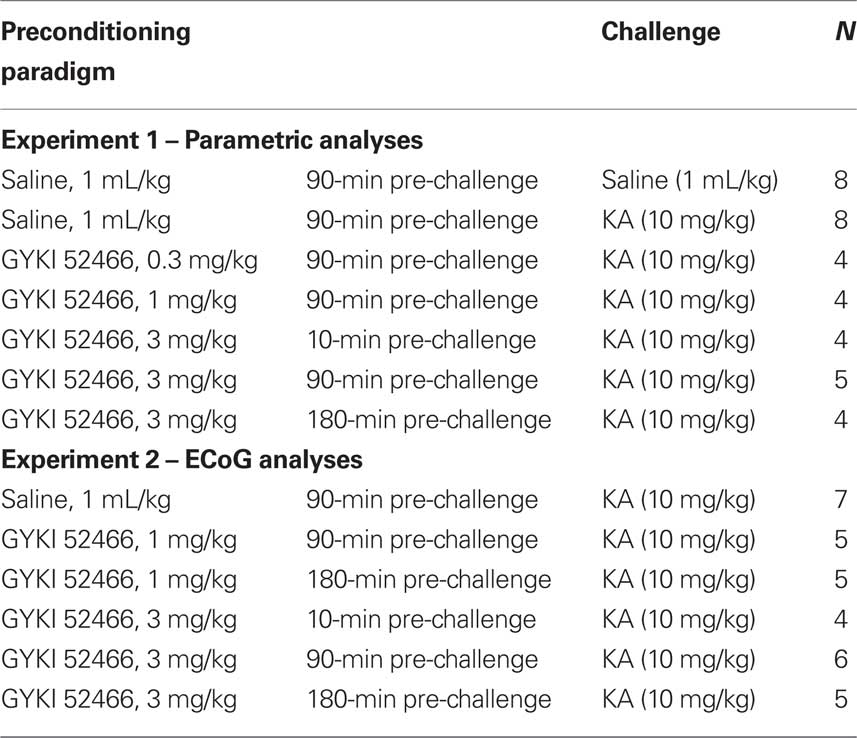
Table 1. Drug treatment conditions.
Immediately following KA injection, animals were returned to the observation chamber and behaviors recorded in a 1-min-on and 1-min-off cycle for 2 h, with all behaviors during each observation period coded and logged to a laptop computer, using a spreadsheet based program developed in our lab. To ensure that observations were recorded systematically, when a particular behavior (e.g., sleeping, walking, etc.) was continuously expressed, observations were only logged once every 15 s. Discrete changes in behavioral state were additionally logged as they occurred. Behaviors were rated using a seizure scale previously employed in our lab (Hesp et al., 2007; Sawant et al., 2010b) in which level 0 consists of normal resting or exploratory behaviors, level 1 – discomfort behaviors, level 2 – stereotypical behaviors confined to the head and neck region, level 3 – moderate seizure behaviors associated with stereotypical movements of the limbs or trunk, and level 4 – severe generalized seizure behaviors leading to level 5 clonic–tonic convulsions (CTC; for more detail, see Hesp et al., 2007). If an animal exhibited two periods of prolonged CTC, the animal was immediately sacrificed and all subsequent observation periods were recorded as level 5. In the ECoG study, the entire experiment was videotaped and coded for subsequent blind behavioral analysis.
Cumulative seizure scores were defined as the sum of the highest scores observed during each 1-min period during the 2 h following KA injection. In addition, average scores during each 1-min observation period were summed and assessed for all drug conditions. Cumulative averages were found to be tightly correlated to cumulative maxima, and consequently, only cumulative maxima are reported here. Furthermore, the total number of wet dog shakes (WDS), level 3, and level 4 behaviors were discretely quantified over the 2-h observation period. At the conclusion of behavioral observations, rats were rendered unconscious by CO2 and decapitated. Brains were then removed, frozen on dry ice and stored for subsequent immunohistochemical staining.
Dual Channel Digital Transmitter Implantation
Carprofen 5 mg/kg and strepcin 250 IU s.c. were administered to animals 30 min prior to surgery and for 3 days post-operatively. Animals were anesthetized with ketamine hydrochloride 75 mg/kg, Domitor® (medetomidine) 0.4 mg/kg, and atropine 0.05 mg/kg. Body temperature was maintained at 37ºC throughout the procedure. A 2-cm transverse incision was made in the skin over the lower abdomen and a two-channel digital transmitter (Telemetry Research, Auckland, NZ) was implanted within a small subcutaneous pocket. An ECG reference lead was sutured to the xiphoid process and other leads were subcutaneously tunneled to the rostral thorax. A small incision exposed the tracheal muscle, and the cardiac recording lead was formed into a U shape, then positioned close to the right atrium for ECG recording. The two ECoG electrode leads were tunneled subcutaneously to a point just posterior to the foramen magnum. The rat was placed into a stereotaxic frame, an incision made lengthwise along the skull, and the skin retracted. Two stainless steel anchoring screws were drilled bilaterally into the skull. The stereotaxic co-ordinates for recording the electrode were 5.2 mm posterior to bregma, 5 mm left of the midline, and 2-mm depth (from skull surface). The reference electrode was placed 5 mm posterior to bregma, 2 mm right of the midline and 1-mm depth. Electrodes were inserted slowly to reduce tissue damage, and were immediately fixed in place using superglue to prevent movement. The leads were secured by suturing to muscle on the neck, so that the vigorous body movements commonly occurring during seizures would not displace the electrodes. Finally, electrodes and screws were fixed to the skull using cranioplastic cement and the scalp sutured. Animals were allowed 8–10 days to recover from implantation surgery before experimentation.
ECoG Recordings and Spectral Analysis
Data were recorded with a Powerlab 2/25 signal conditioner and Chart 6 analysis software (AD Instruments, Sydney). ECoG activity was acquired continuously for the duration of the experiment at 2 kHz, with receivers set to 0.1 Hz high pass and 1000 Hz low pass filters. Fast Fourier transformation was used to quantify the following frequency bands: delta (1.25–4.50 Hz), theta (4.75–6.75 Hz), alpha (7.00–12.50 Hz), beta (12.75–35 Hz), and gamma (35.5–100 Hz). Simultaneous ECG recording allowed the removal of high frequency/high amplitude movement artifacts which invariably occurred during (WDS). ECoG recordings were examined in 10-min periods. These sampling periods were analyzed by dividing them into half overlapping half-second epochs, using a weighted Cosine bell window. Power spectral density (PSD) for each period was calculated as the sum of all epochs. Baseline PSD for each animal was determined as the mean of the three 10-min periods during acclimatization. PSD values for subsequent periods were expressed as a percentage of baseline and averaged within treatment groups. High frequency/high amplitude (HF/HA) spiking was also quantified as a measure of seizure activity. These were defined as spikes of at least three times the average spike amplitude during baseline.
c-Fos Immunohistochemistry
A large body of research indicates that c-Fos protein expression increases with seizure severity, peaks in hippocampus 1–2 h following a seizure, and is particularly robust in the dentate gyrus. Immunohistochemical c-Fos staining was carried out using the protocol described by Sundquist and Nisenbaum (2005), with slightly longer incubations to ensure strong immunoreactivity. Frozen brains were cryosectioned (10 μ), mounted on poly-l-lysine coated slides and post-fixed in 4% paraformaldehyde. Endogenous peroxidases were quenched with 0.3% H2O2 (Merck) and non-specific antibody binding was blocked by incubating sections in 0.015% normal goat serum (Vectorlabs, Burlingame, CA, USA). Sections were incubated over night in 1:2000 rabbit anti-c-Fos primary antibody (Calbiochem, San Diego, CA, USA) prior to incubation with biotinylated goat anti-rabbit secondary antibody (Vectorlabs). Antibody binding was visualized using the ABC complex (Vectorlabs) and a Sigma Fast™ 3,′3 diaminobenzidine (DAB) peroxidase substrate tablet set (Sigma Chemicals, St. Louis, MO, USA) in distilled H2O. Sections were then dehydrated in ascending concentrations of methanol and cleared in xylene before coverslipping.
Seizure-related c-Fos immunoreactivity was observed in all sub-regions of the hippocampus. As expression was strongest in the granule cells of the dentate gyrus (DG), only DG granule cell layer c-Fos expression is reported here. Immunoreactivity was assessed relative to non-seizure (saline only) controls using blind observer scoring (Kapoor et al., 2004; Clarkson et al., 2005). Micrographs of sections from experimental conditions were paired with micrographs of control tissue processed on the same slide. Five blind observers were presented with pairs and asked to compare the right image to the left image (control and experimental micrographs were randomly alternated between left and right). Observers rated the relative degree of c-Fos immunoreactivity (right vs. left) on a scale of −3 to +3, where +3 is more immunoreactive, −3 is less immunoreactive and a score of 0 represents no difference.
Statistics
For behavioral analysis and immunohistochemical grading, Kruskal–Wallis ANOVAs with Dunn’s post hoc tests were conducted for each data set. For post hoc purposes, the saline/saline control and saline/KA seizure groups were compared either to the 0.3, 1, and 3 mg/kg 90 min GYKI 52466 pre-treatment groups, or to the 3 mg/kg 10, 90, or 180 min GYKI 524466 pre-treatment groups, to differentiate dose- and time-dependent effects. p < 0.05 was considered to be a significant difference. A two-tailed Spearman’s non-parametric correlation was used to determine the relationship between c-Fos staining and cumulative maximum seizure scores. For analysis of electrocortigraphic data, HF/HA spiking data was transformed using a natural log, and analyzed using one way ANOVA with Bonferroni post hoc. ECoG power spectral density data was found to violate assumptions of equal variance, so was likewise transformed using a natural log.
Results
Seizure Induction Following Saline or GYKI Preconditioning
Behavioral analysis was undertaken for low-dose GYKI preconditioning, and all periods were compared to 60-min saline preconditioning controls. The pre-treatment dose for GYKI 52466 was set at 3 mg/kg or lower, as previous studies have shown that these doses cause no adverse behavioral effects (Rorick-Kehn et al., 2005). Our observations confirmed that low-dose GYKI 52466 does not adversely affect animal behaviors (data not shown); during GYKI preconditioning rats exhibited normal behaviors such as exploring, grooming and periodic resting, and these behaviors were not significantly different from those behaviors observed during the baseline acclimation period.
Following saline preconditioning, rats were injected with either saline (saline controls) or KA (10 mg/kg, s.c.; seizure controls). In saline controls, a second injection of saline caused no observable changes in animal behaviors. In contrast, in saline preconditioned seizure controls, high-dose KA produced seizures which steadily increased during the 2-h observation period (cumulative seizure scores: seizure controls vs saline controls; p < 0.01; Figure 1).
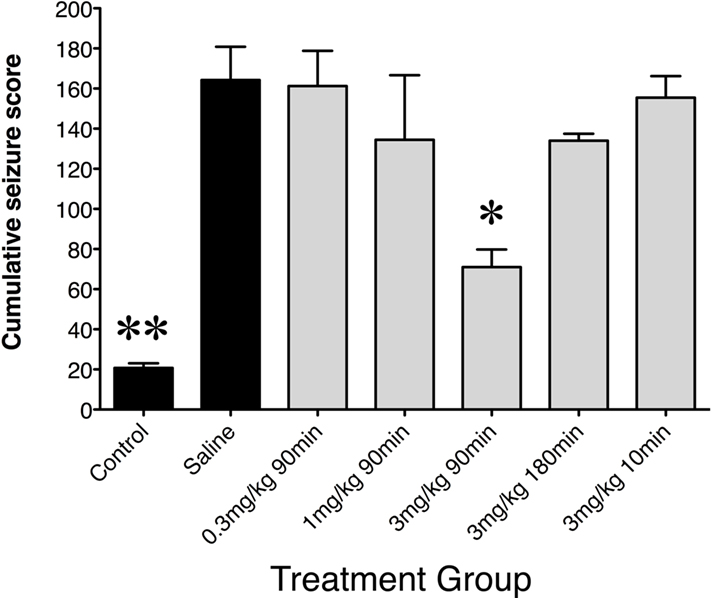
Figure 1. Cumulative seizure scores following saline or low-dose GYKI 52466 preconditioning. Histograms show mean ± SEM cumulative seizure scores. Labels along the x axis indicate preconditioning paradigm. All groups received KA (10 mg/kg) except control animals, which received a second saline injection. Scores were calculated from the 2-h monitoring period following KA injection. *p < 0.05 vs. Saline, **p < 0.01 vs. Saline.
When GYKI 52466 (3 mg/kg) was administered only 10 min prior to KA, cumulative seizure scores were comparable to those seen in saline preconditioned rats (N.S.). The same was seen when GYKI 52466 (3 mg/kg) was administered 180 min prior to KA. However, when GYKI (3 mg/kg) was administered 90 min prior to KA, animals exhibited a significant reduction in cumulative scores in the 2-h period following high-dose KA (p < 0.05). These rats only exhibited level 1 and 2 behaviors, and infrequent WDS. Lower preconditioning doses of GYKI (0.3 and 1 mg/kg; 90 min pre-challenge) did not significantly reduce seizure scores (Figure 1).
To further assess differences between rats pre-treated with saline or GYKI 52466, the number of WDS (a pronounced level 3 behavior), the net occurrence of level 3 or above behaviors, and the net occurrence of level 4 behaviors were quantified (Figure 2). GYKI 52466 (3 mg/kg), 90 min prior to high-dose KA, virtually abolished all level 3 and level 4 seizures (p < 0.05). GYKI 52466 (3 mg/kg) preconditioning 180 min prior to seizure induction also markedly reduced level 4 behaviors (Figure 2). In contrast, lower doses of GYKI or a shorter pre-administration period (10 min) prior to KA, was ineffective.
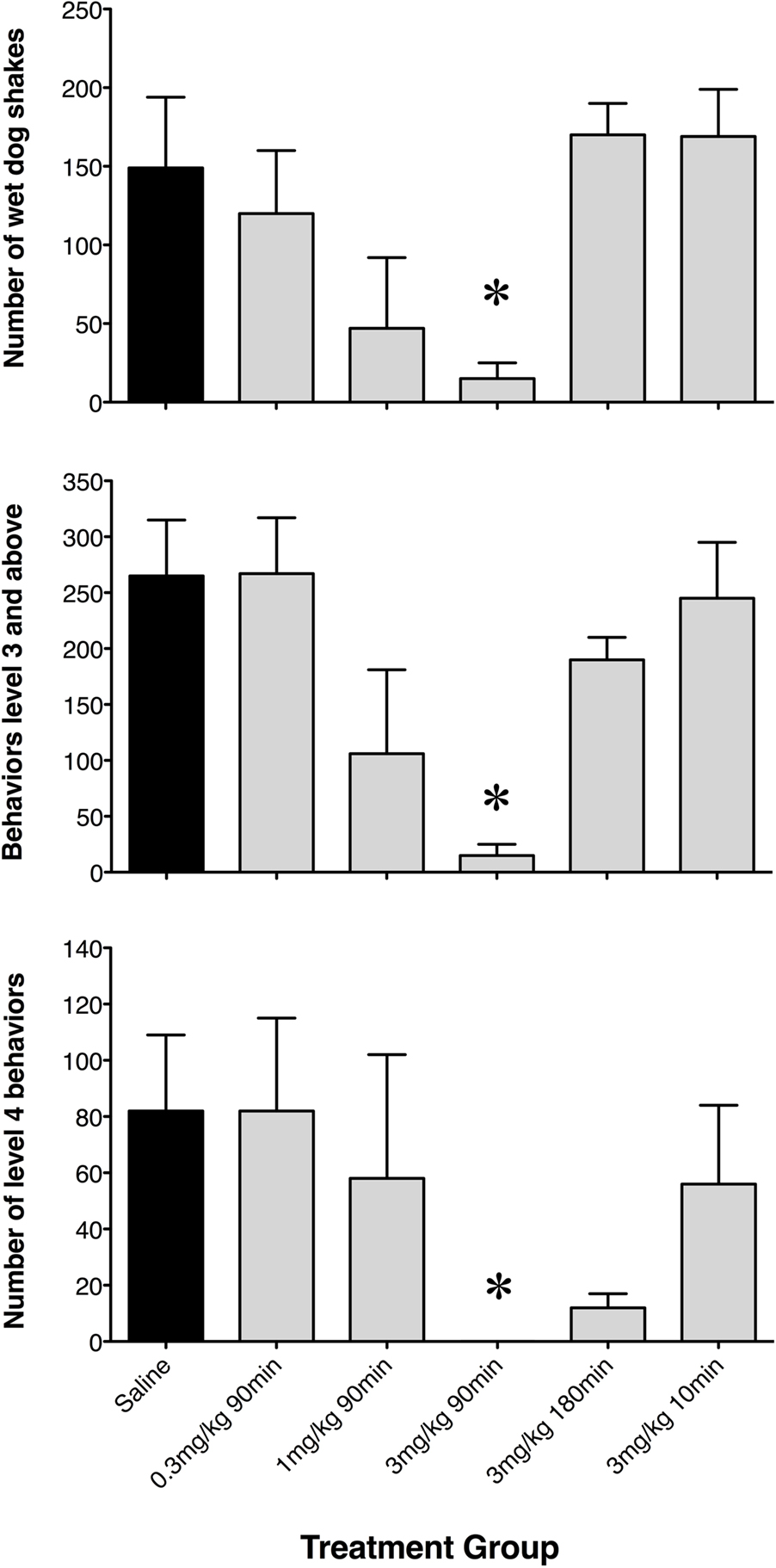
Figure 2. Net occurrence of wet dog shakes, level 3 and level 4 seizure behaviors exhibited following saline or low-dose GYKI 52466 preconditioning. Histograms show mean ± SEM number of discrete events during the 2-h seizure induction period. Labels along the x axis indicate preconditioning paradigm.*p < 0.05 vs. saline.
c-Fos Immunohistochemistry
Minimal c-Fos labeling was seen in tissue from saline control rats. This tissue was used as a comparison for the estimation of c-Fos immunoreactivity in experimental animals. Seizure control rats displayed intense c-Fos immunoreactivity in the dentate gyrus of the hippocampus. Seizure-induced c-Fos expression was significantly attenuated by administration of 3 mg/kg GYKI 52466 90 min prior to KA administration (Figure 3A; p < 0.05). Comparable reductions in c-Fos immunoreactivity were not evident when seizure animals were pre-treated with 1 mg/kg GYKI 52466 given 90 min or 3 mg/kg GYKI 52466 given 10 or 180 min before KA (Figure 3B). The relationship between behavioral measures and c-Fos expression was examined using individual data from each animal and revealed a positive, significant correlation between cumulative maximum seizure score and c-Fos expression in the dentate gyrus (Figure 3C).
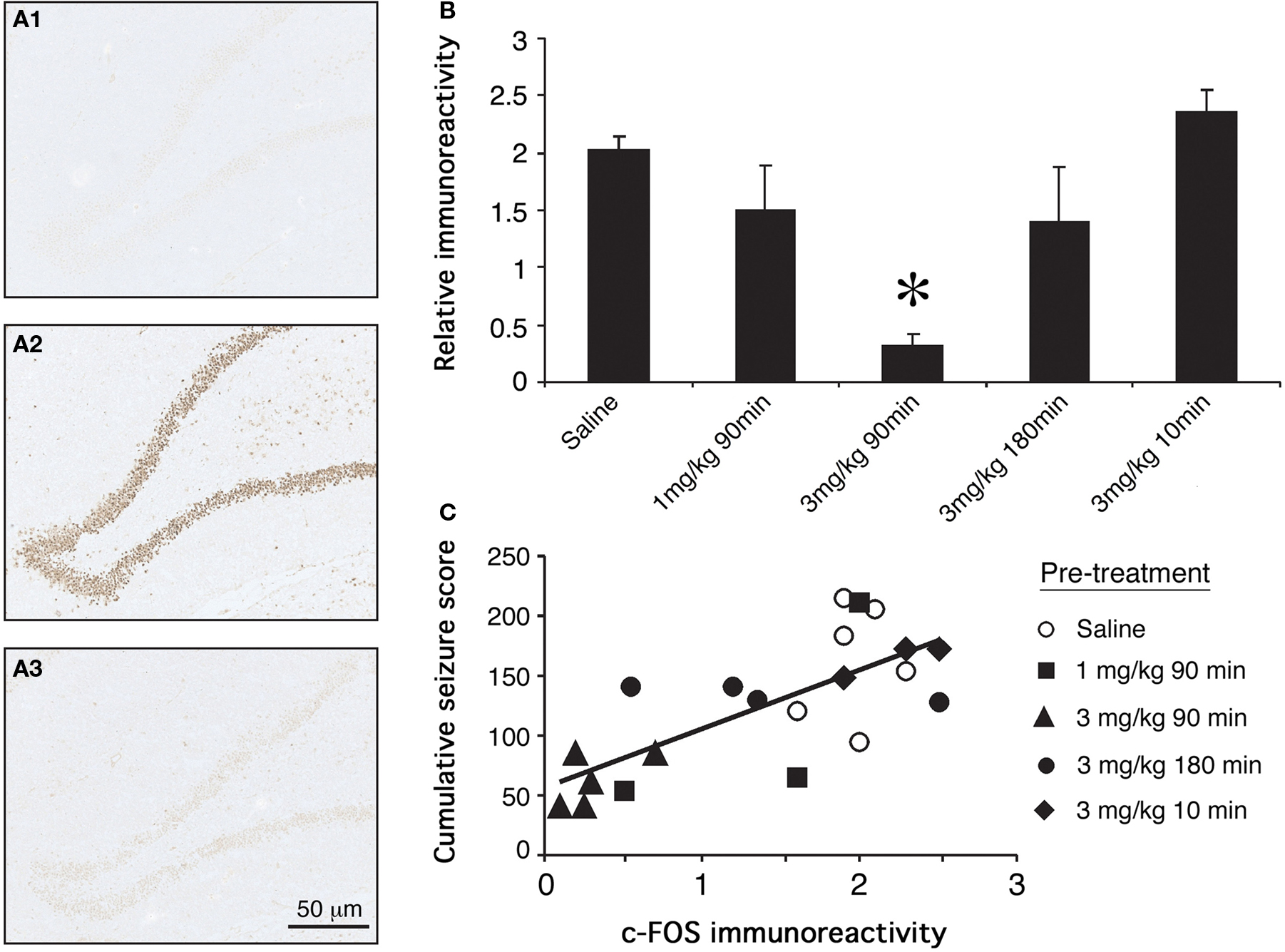
Figure 3. Effects of GYKI 52466 preconditioning on KA-induced c-Fos expression in the dentate gyrus. (A) Representative micrographs of c-Fos immunoreactivity in the dentate gyri of (A1) saline treated controls; (A2) KA treated animals that received saline pre-treatment; (A3) KA treated animals that received 3 mg/kg GYKI 52466 90 min beforehand. (B) Histograms of varying GYKI 52466 dose and pre-treatment period on KA-induced c-Fos expression in the dentate gyrus relative to saline controls (mean ± SEM; *p <0.05). (C) Spearman’s non-parametric correlation between c-Fos immunoreactivity and cumulative maximum seizure score; p < 0.05.
ECoG Activity During Pharmacological Preconditioning and Seizure Induction
Based on results obtained during the parametric analyses described above, a second set of experiments were conducted to determine whether changes in electrocorticographic activity parallel behavioral changes observed following saline and GYKI preconditioning. During saline preconditioning, cortical activity was not significantly different from that observed during the baseline (acclimation) recording period. ECoG traces routinely exhibited extended periods of low power activity interrupted by occasional spikes or brief flurries of activity associated with exploring and grooming (Figure 4). Following the high-dose KA challenge, saline preconditioned animals exhibited several minutes of hyperactivity and exploratory behaviors followed by a short (approx. 15 min) period of hypoactivity. Within 20 min of KA administration, head tremors, mastication, freezing and WDS occurred with increasing frequency, and these were associated with the appearance of regular high frequency/high amplitude (HF/HA) electrical activity in the cortex. High level convulsive seizures (level 3 and 4 behaviors) of variable intensity occurred approximately 40 min after KA in saline preconditioned animals (Figures 4 and 5), and in all cases were associated with robust increases in ECoG power and electrographic spiking. ECoG recording revealed strong seizure activity during the kainic acid challenge in 100% of saline treated rats.
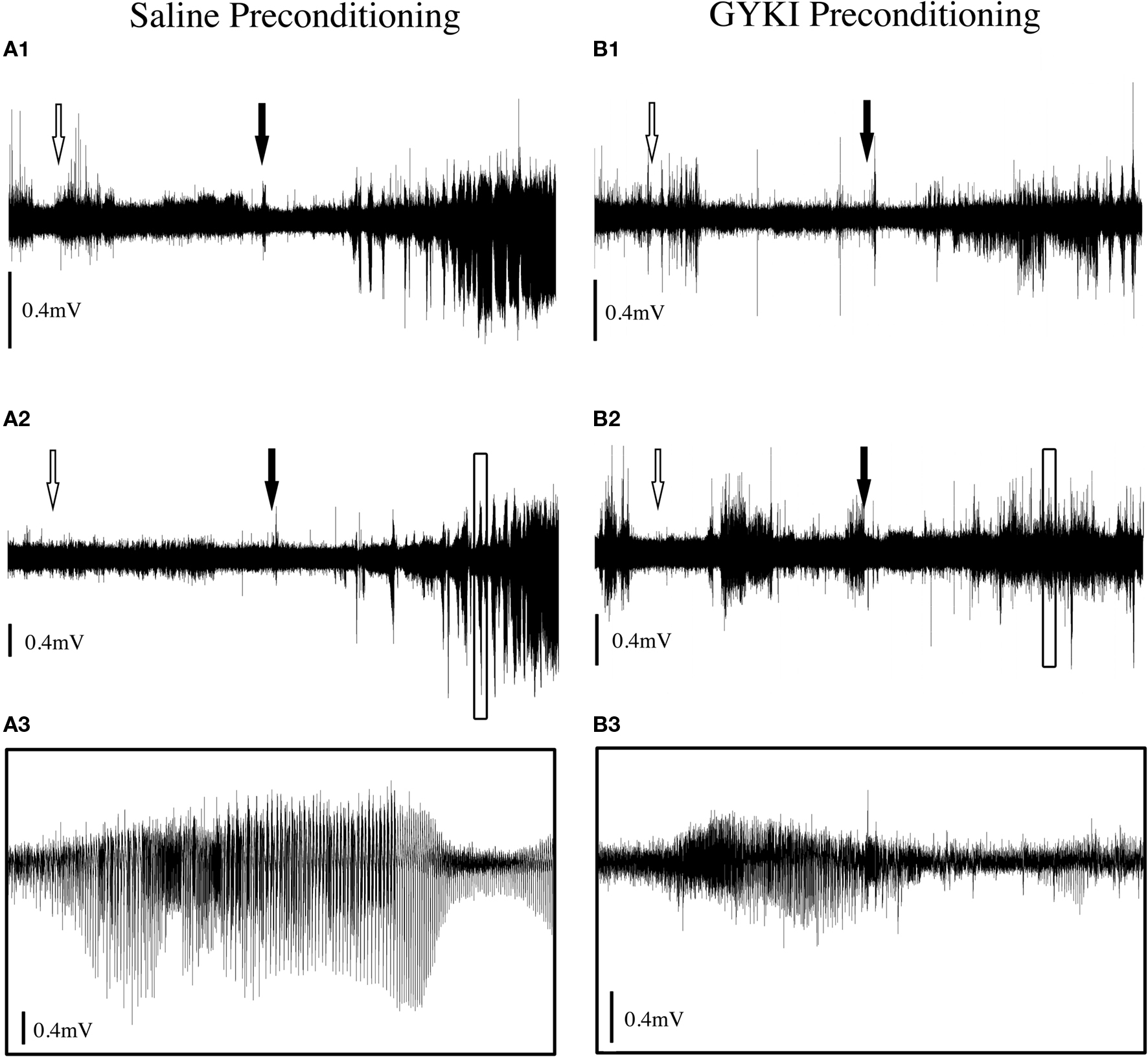
Figure 4. Typical ECoG traces from rats preconditioned with (A) saline or (B) GYKI 52466 (3 mg/kg 90 min) followed by KA (10 mg/kg) seizure challenge. (A1, A2): Example traces from two saline preconditioned rats. (A3) Magnification of boxed section in (A2). (B1, B2) Example traces from two rats preconditioned with GYKI (3 mg/kg), 90 min prior to KA administration. (B3) Magnification of boxed section in (B2). White arrows indicate time of preconditioning injection. Filled arrows indicate time of KA injection. Complete traces are 2-h duration; magnified traces are 2 min duration. Traces adjusted vertically for visual comparison as signal strength varied between experiments.
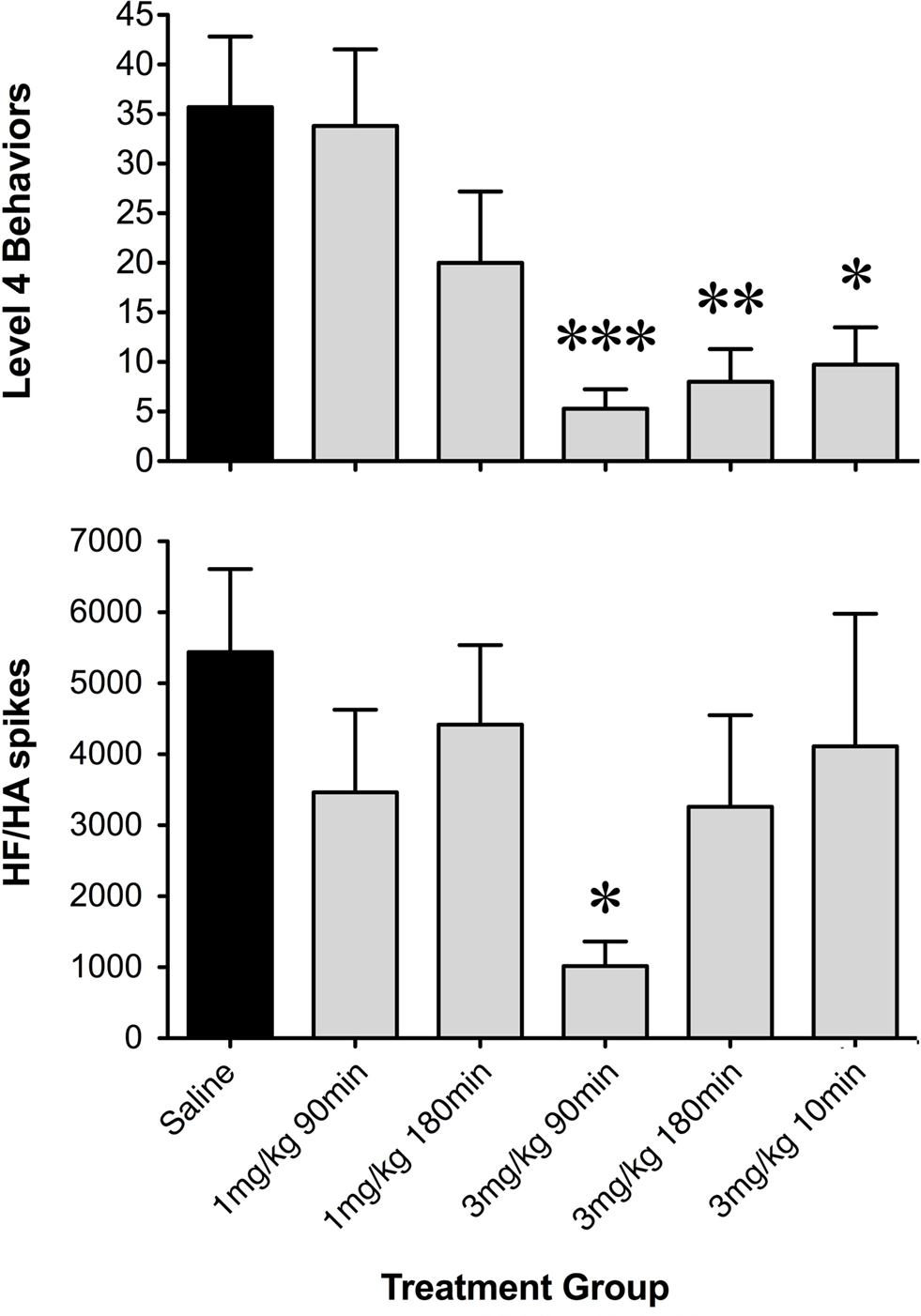
Figure 5. Histograms of net occurrence of level 4 seizure behaviors and high frequency/high amplitude (HF/HA) electrographic spikes during the 2-h seizure induction period following saline or GYKI 52466 preconditioning. Totals were averaged within treatment groups (mean ± SEM; *p < 0.05, **p < 0.01, ***p = 0.001).
During low-dose GYKI preconditioning, animals exhibited ECoGs not significantly different from those obtained during the baseline recording period. Following the KA challenge, GYKI treated animals exhibited low-level exploratory behaviors that were sometimes interrupted by wet-dog shakes and other level 2 and 3 seizure behaviors. These behavioral manifestations were associated with moderate increases in ECoG power and generally persisted for less than 1 h. Following this, GYKI preconditioned animals settled into sleeping and resting behaviors with occasional periods of exploring and grooming; ECoG power during this latter period was not different from baseline (Figures 4 and 5). Consistent with our earlier observations, the 3 mg/kg 90-min GYKI preconditioning group exhibited a small but significant reduction in cumulative seizure scores (p = 0.027; data not shown). Non-significant decreases were also observed in the 1 mg/kg 180 min, 3 mg/kg 10 min, and 3 mg/kg 180 min groups. There were no significant differences in the average number of level 3 behaviors. However, consistent with our previous work, significant decreases in Level 4 behaviors were observed for several treatment groups (Figure 5). The greatest reduction was observed for the 3 mg/kg 90 min group which exhibited only 5.3 ± 2.0 discrete level 4 seizure behaviors over the entire 2-h observation period (versus saline controls; 35.7 ± 7.1; p = 0.001; Figure 5). Significant reductions in level 4 seizure behaviors were also observed in the 3 mg/kg 180 min group (p = 0.007) and 3 mg/kg 10-min group (p = 0.019). The number of HF/HA spikes observed during the 2-h observation periods were analyzed (Figure 5). Observation of ECoG traces showed no evidence of HF/HA spiking during baseline or during saline or GYKI preconditioning for any of the animals in either group. However, relative to saline, GYKI preconditioned animals exhibited reductions in HF/HA spiking during the high-dose KA challenge. In particular, HF/HA spiking during KA was markedly reduced in the 3 mg/kg GYKI 90 min preconditioning group (1067 ± 324), and this reduction was significantly different from that observed in saline controls (5553 ± 1114; p = 0.031; Figure 5).
ECoG Power Spectral Density
Consistent with current and prior behavioral observations, we generally observed changes in ECoG signal intensity as early as 10–15 min, and peak changes from 90 to 120 min following KA injection (Figure 6). Fast Fourier transform was used to assess ECoG power spectral density (PSD) over the entire 2-h recording period, and analysis of variance indicated a main effect of drug treatment in seizure expression following KA (F = 4.86; p = 0.004); GYKI preconditioned animals exhibited significant reductions in ECoG PSD across frequency bands relative to saline controls and these reductions were most pronounced at 3 mg/kg either 90 or 180 min prior to seizure induction (delta: p < 0.01; theta: p = 0.003; alpha: p < 0.01; beta: p < 0.01; gamma: N.S; Figure 6).
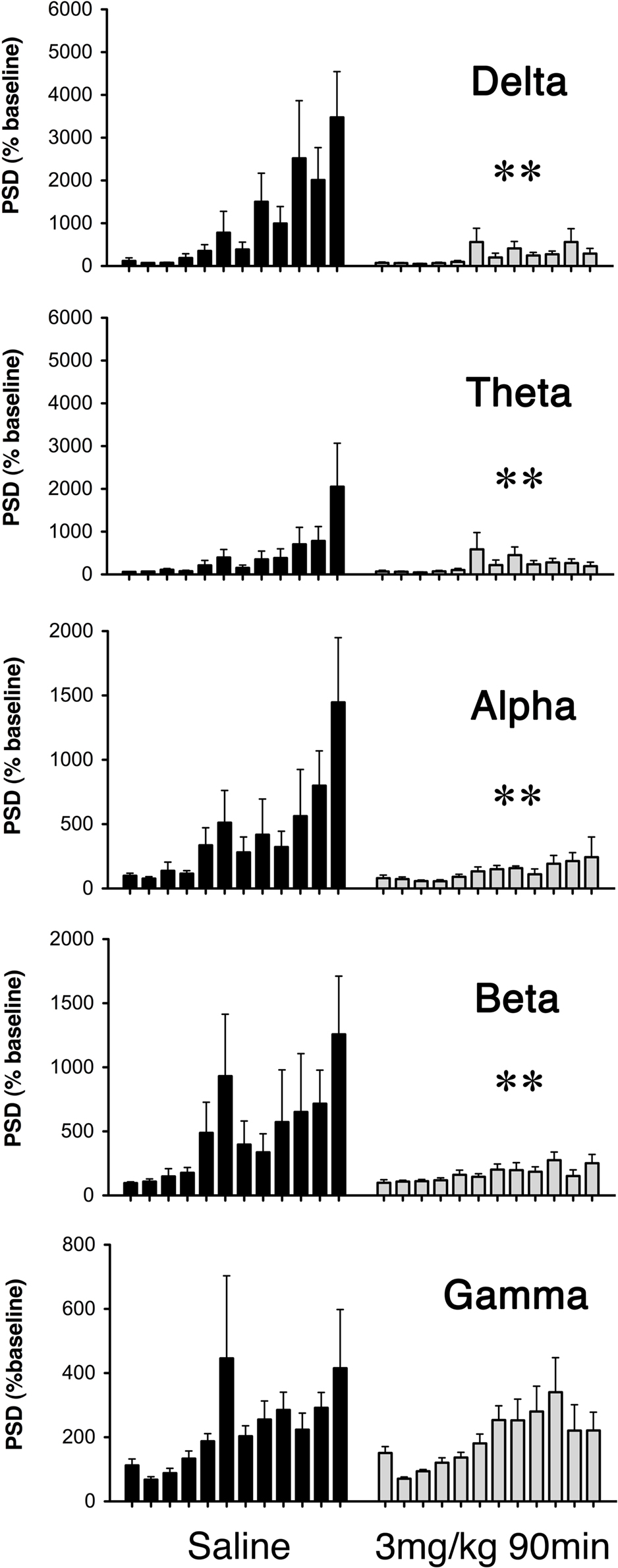
Figure 6. Average power spectral density (PSD) of ECoG recordings during seizure induction for saline (dark bars) and GYKI 52466 (3 mg/kg 90 min; light bars) preconditioned rats. Histograms show average PSD (mean ± SEM) for each 10-min epoch during the 2-h KA challenge. Preconditioning effects were analyzed separately for each frequency band (Delta (1.25–4.5 Hz); Theta (4.75–6.75 Hz); Alpha (7.00–12.5 Hz); Beta (12.75–35 Hz); Gamma (35.5–100 Hz). **p < 0.01.
Discussion
Anticonvulsant and Neuroprotective Actions of GYKI 52466
GYKI 52466 has been assessed as a potential anticonvulsant in a number of animal models of seizure, including audiogenic seizures, maximal electroshock seizures, amygdala-kindled seizures, and chemically induced seizures (for an extensive review see De Sarro et al., 2005). In most of these studies ED50s ranged from 13 to 25 mg/kg when the drug was administered 15–30 min prior to seizure induction. Worryingly, debilitating motor and cognitive effects have been seen at 10–15 mg/kg and most investigators have concluded that the “therapeutic utility of GYKI 52466 is limited” (Barton et al., 2003; De Sarro et al., 2005; Weiczner et al., 2008).
Here, we obtained in vivo evidence that pharmacological preconditioning with low-dose GYKI 52466 is sufficient to impart a significant protection against KA-induced seizures. In these studies young adult rats were preconditioned with saline or GYKI 52466 at doses 5- to 20-fold lower than published ED50s for anticonvulsant activity, and seizures were induced 10–180 min later. Low-dose GYKI (3 mg/kg, s.c.), 90 min prior to high-dose KA, markedly reduced cumulative seizure scores, virtually abolished all level 3 and level 4 seizures, and completely suppressed KA-induced hippocampal c-FOS expression. In the present study we further assessed functional CNS protection by low-dose GYKI preconditioning using telemetric electrocorticographic (ECoG) recording coupled with behavioral analysis and found that preconditioned animals exhibited significant reductions in high frequency/high amplitude spiking and ECoG power in the delta, theta, alpha, and beta bands during high-dose KA. This anticonvulsant effect was not observed when GYKI was administered 10 or 180 min before seizure induction or at lower doses (≤1 mg/kg). In addition, adverse behaviors (loss of motor control, sedation, etc.) were not evident during the preconditioning period, and our observation that both HF/HA spiking and c-FOS expression were dramatically reduced by GYKI preconditioning confirms that the reduction of high level (motor) seizure behaviors were not simply due to GYKI-induced paralysis or sedation. Although we cannot directly confirm that GYKI 52466 suppresses seizures via KA-sensitive GPCRs, the fact that the compound was effective at doses well-below, and at time intervals well-beyond previous studies, suggests that a classical blockade of ionotropic AMPA receptors does not underlie the anticonvulsant effect.
A Novel, Metabotropic Mechanism of Action?
GYKI 54266 has been classed as a selective non-competitive allosteric AMPA receptor antagonist. GYKI binding presumably causes a conformational change in the receptor that inhibits the binding of glutamate and related agonists such as KA. As such, negative allosteric modulation has been assumed to account for GYKI’s anticonvulsant activity versus excitotoxicity and seizures (Donevan et al., 1994; De Sarro et al., 2003). However prior work by our group suggests that this compound may also act via a metabotropic mechanism to prevent seizures (Hesp et al., 2004).
Using the hippocampus as a model system, we have shown that low dose preconditioning with either kainate or the neurotoxin domoic acid (Kerr et al., 2002; Hesp et al., 2004, 2007; Sawant et al., 2007, 2008, 2010a) induces tolerance against the hyperexcitatory actions of much higher doses of these toxins, both in vitro and in vivo. Interestingly, these effects are generally less robust in aged animals. Given the lasting nature of the tolerance effect, we examined G-protein coupled receptor function. Results of these studies (Hesp et al., 2004) suggest that tolerance induction likely occurs via recently described KA-sensitive G-protein-linked receptors (Rozas et al., 2003; Lerma, 2006; Rodriguez-Moreno and Sihra, 2007). We found that a number of ionotropic KA receptor agonists (KA, DOM, ATPA, and SYM-2081) significantly reduce constitutive GTPase activity in hippocampal membranes from young but not aged rats. Furthermore, in the course of searching for other ligands which reliably induce tolerance to excitotoxins we found, quite unexpectedly, that a number of ionotropic AMPA and KA antagonists (such as CNQX, NBQX, GYKI 52466, NS-102, and GAMS) also act as strong inverse agonists of G-protein receptor function in young but not aged rats, and some of these act as strong preconditioning agents in vitro (Hesp et al., 2004). Notably, in young rat hippocampal CA1, low concentrations of the AMPA/KA blockers GYKI 52466 and NBQX-induced tolerance to high-dose KA administered well after the antagonists had been washed from the slice preparation. These findings suggest that tolerance is triggered by a selective reduction in constitutive KA-sensitive GPCR activity, and this metabotropic neuroprotective mechanism may be lost in aged hippocampus.
In the present study we found that low-dose GYKI 52466, administered 90–180 min prior to systemic administration of kainic acid, produces significant reductions in behavioral, electrographic, and immunohistological indices of seizures. Our parametric analysis indicates that preconditioning protection is optimal at 3 mg/kg, 90 min prior to seizure induction. No prior studies have shown a protective effect with GYKI 52466 at doses as low as 3 mg/kg (Donevan et al., 1994; Borowicz et al., 2001; De Sarro et al., 2003). Of particular interest is the fact that low-dose GYKI 52466 markedly reduced KA-induced seizures when injected 90 min prior to KA. This lasting anticonvulsant efficacy is difficult to explain in terms of classical AMPA receptor antagonism by GYKI. As an ionotropic AMPA receptor antagonist, 3 mg/kg GYKI 52466 should have little effect 90 min after administration when very little GYKI is remaining in the plasma (De Sarro et al., 1998b). We cannot rule out the possibility that GYKI crosses the blood–brain barrier and lingers in the CNS even after it has been cleared from the plasma. However it remains clear that low-dose GYKI preconditioning offers clinical efficacy without dosing at toxic levels. Taken together, our findings support the possibility of a long-lasting preconditioning effect mediated by a metabotropic mechanism, and suggest that GYKI 52466 and related compounds may be used prophylactically to protect the CNS.
While the exact location and function of the G-protein that GYKI 52466 may influence is not known, the findings of the current study coupled with prior data (Hesp et al., 2004) are compatible with the proposal that excitotoxic preconditioning is mediated via inverse agonism of a metabotropic pathway. This as yet unidentified receptor may bind KA, DOM, and GYKI 52466 in a similar manner, lowering the constitutive activity of a G-protein complex leading to downstream effects that protect the cell from subsequent insult. The G-protein complex may be a separate membrane bound GPCR or may be linked to a currently known (but misunderstood) ionotropic receptor (Lerma, 2006). Indeed, recent studies have challenged the dogma that non-NMDA glutamate receptors are purely ionotropic in action, suggesting that KA and AMPA receptor complexes play metabotropic roles in regulating neuronal activity. It has been demonstrated that KA receptors can have a G-protein linked function (Cunha et al., 1999; Melyan et al., 2002; Fernandes et al., 2009; Segerstråle et al., 2010) that may involve protein kinase C (PKC; Rodriguez-Moreno et al., 2000; Kurkinen et al., 2001; Jung et al., 2005), protein kinase A- (PKA; Rodriguez-Moreno and Sihra, 2004) or mitogen-activated protein kinase (MAPK; DeCoster et al., 1998). AMPA receptors have also been linked to second messenger cascades (Wang and Durkin, 1995; Perkinton et al., 1999). Our data suggest that GYKI 52466 may be equally efficacious via metabotropic receptors (relative to its classical effects as a negative allosteric modulator of the AMPA receptor), with second messenger amplification accounting for the effects of GYKI at the unusually low doses and extended time points used in this study.
The long-lasting anticonvulsant efficacy of low-dose GYKI preconditioning seen here fits well with existing literature regarding early and delayed windows of preconditioning protection. A large number of studies in recent years have shown that CNS neurons do in fact possess inducible neuroprotective mechanisms, and can be preconditioned in a variety of ways to resist hypoxic or excitotoxic insults. In general most preconditioning paradigms have involved moderate to severe insults many hours to days apart, and in many cases resulted in lasting neuroprotection (Pohle and Rauca, 1994; Best et al., 1996; El Bahh et al., 1997; Najm et al., 1998; Lere et al., 2002; Tandon et al., 2002; Chang et al., 2005; Wang et al., 2005). However, a growing body of evidence indicates that preconditioning procedures can impart a rapid tolerance to subsequent excitotoxic insults which may be observed within minutes of conditioning. For example, early work showed that anoxic preconditioning or brief exposure to depolarizing agents imparts a robust, Ca2+-dependent tolerance to subsequent anoxic depolarization (Centeno et al., 1999; Perez-Pinzon et al., 1999), and evidence of rapid “cross conditioning” in vitro has also emerged (Schurr et al., 2001; Youssef et al., 2001). Importantly, in studies of in vitro ischemic preconditioning, an “early window” of neuroprotection has been identified which may involve rapid shifts in glutamate and GABA release (Dave et al., 2005; for review, see Perez-Pinzon, 2007).
Clinical Implications
It is well-established that stroke and seizure share overlapping mechanisms of damage (Meldrum, 1993; Pitkänen and Sutula, 2002). Ischemia-induced neuronal death, anatomical remodeling, and pathological shifts in glutamate and GABA signaling appear to underlie epileptogenesis (Epsztein et al., 2008). The incidence of stroke, seizures, and refractory epilepsy is markedly increased with advancing age, and a significant percentage of age-related epilepsies occur following the onset of cerebrovascular pathology in the elderly (Cloyd et al., 2006). GYKI 52466 and related compounds (e.g., GYKI 53773, GYKI 53405, and EGIS 8332) have been examined in animal models of stroke. In general, most of these compounds have proven effective in reduction of infarct volume when administered during, or up to 2 or 3 h following stroke induction (Elger et al., 2005; Erdo et al., 2005; Gressens et al., 2005; Denes et al., 2006; Matucz et al., 2006; Gigler et al., 2007) at doses ranging from 10 to 30 mg/kg i.v. or i.p. Unfortunately, almost all of the studies to date have been limited to anatomical–morphological outcomes with only one recent study assessing recovery of cognitive and motor functions 30 days post-stroke (Erdo et al., 2006). In addition, as previously noted, the therapeutic utility of high-dose GYKI 52466 in cases of human stroke has been questioned.
Studies are currently underway in our lab to assess the potential of low-dose GYKI as a prophylactic neuroprotectant in a rat model of neonatal hypoxia/ischemia. The prophylactic induction of tolerance in the brain has far-reaching implications for clinical procedures such as cardiac and brain surgery, in which blood flow to the brain is interrupted. Ischemia induced under these conditions frequently leads to excitotoxic damage (Doyle et al., 2008), transhemispheric diaschisis (Clarkson et al., 2005), seizures and lasting neurological deficits (Newman et al., 2001; Hazell, 2007; Naegele, 2007). At present there are few pharmacological tools available to the clinician to protect the brain from the effects of ischemic or excitotoxic insults. Developing drugs which can induce cross-tolerance in at-risk patients before an excitotoxic insult occurs offers a valuable opportunity to improve long-term survival rates and functional outcomes for surgical patients and stroke victims. As such, prophylactic neuroprotection by metabotropic induction of endogenous neuroprotective pathways may provide clinicians with a new and much needed basis of treatment.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge financial support provided by the Community Trust of Otago (AG-279) and administrative support provided by the Otago Medical Research Foundation.
References
Barton, M. E., Peters, S. C., and Shannon, H. E. (2003). Comparison of the effect of glutamate receptor modulators in the 6 Hz and maximal electroshock seizure models. Epilepsy Res. 56, 17–26.
Borowicz, K. K., Duda, A. M., Kleinrok, Z., and Czucwar, S. J. (2001). Interaction of GYKI 52466, a selective non- competitive antagonist of AMPA/kainate receptors, with conventional antiepileptic drugs in amygdala-kindled seizures in rats. Polish J. Pharmacol. 53, 101–108.
Borowicz, K. K., Gasior, M., Kleinrok, Z., and Czucwar, S. J. (1995). The noncompetitive Ampa/Kainate receptor antagonist, GYKI 52466, potentiates the activity of conventional antiepileptics. Eur. J. Pharmacol. 281, 319–326.
Best, N., Sundstrom, L. E., Mitchell, J., and Wheal, H. V. (1996). Pre-exposure to subtoxic levels prevents kainic acid lesions in organotypic hippocampal slice cultures: effects of kainic acid on parvalbumin-immunoreactive neurons and expression of heat shock protein 72 following the induction of tolerance. Eur. J. Neuorsci. 8, 1209–1219.
Centeno, J. M., Orti, M., Salom, J. B., Sick, T. J., and Pérez-Pinzon, M. A. (1999). Nitric oxide is involved in anoxic preconditioning neuroprotection in rat hippocampal slices. Brain Res. 836, 62–69.
Chang, A. Y., Wang, C. H., Chiu, T. H., Chi, J. W., Chen, C. F., Ho, L. T., and Lin, A. M. (2005). Hypoxic preconditioning attenuated in kainic acid-induced neurotoxicity in rat hippocampus. Exp. Neurol. 195, 40–48.
Chauhan, N., Zhao, Z., Barber, P. A., and Buchan, A. M. (2003). Lessons in experimental ischemia for clinical stroke medicine. Curr. Opin. Neurol. 16, 65–71.
Clarkson, A., Liu, H., Rahman, R., Appleton, I., Jackson, D., and Kerr, D. S. (2005). Clomethiazole: mechanisms underlying lasting neuroprotection following hypoxia-ischemia. FASEB J. 19, 1036–1038.
Cloyd, J., Hauser, W., Towne, A., Ramsay, R., Mattson, R., Gilliam, F., and Walczak, T. (2006). Epidemiological and medical aspects of epilepsy in the elderly. Epilepsy Res. 68S, S39–S48.
Cunha, R. A., Malva, J. O., and Ribeiro, J. A. (1999). Kainate receptors coupled to G(i)/G(o) proteins in the rat hippocampus. Mol. Pharmacol. 56, 429–433.
Dave, K. R., Lange-Asschenfeldt, C., Raval, A. P., Prado, R., Busto, R., Saul, I., and Perez-Pinzon, M. A. (2005). Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/gamma-aminobutyric acid release and biosynthesis. J. Neurosci. Res. 82, 665–673.
DeCoster, M. A., Mukherjee, P. K., Davis, R. J., and Bazan, N. G. (1998). Platelet-activating factor is a downstream messenger of kainate-induced activation of mitogen-activated protein kinases in primary hippocampal neurons. J. Neurosci. Res. 53, 297–303.
De Sarro, A., De Sarro, G., Gitto, R., Grasso, S., Micale, N., Quartaone, S., and Zappala, M. (1998a). 7,8- methylenedioxy-4H-2,3-benzodiazepin-4-ones as novel AMPA receptor antagonists. Bioorg. Med. Chem. Lett. 8, 971–976.
De Sarro, G., Rizzo, M., Sinopoli, V. A., Gitto, R., De Sarro, A., Zappala, M., and Chimirri, A. (1998b). Relationship between anticonvulsant activity and plasma level of some 2,3- benzodiazepines in genetically epilepsy-prone rats. Pharmacol. Biochem. Behav. 61, 215–220.
De Sarro, G., Ferreri, G., Gareri, P., Russo, E., De Sarro, A., and Gitto, R., and Chimirri, A. (2003). Comparative anticonvulsant activity of some 2,3-benzodiazepine derivatives in rodents. Pharmacol. Biochem. Behav. 74, 595–602.
De Sarro, G., Gitto, R., Russo, E., Ibbadu, G.F., Barreca, M.L., De Luca, L., and Chimirri, A. (2005). AMPA receptor antagonists as potential anticonvulsant drugs. Curr. Top. Med. Chem. 5, 31–42.
Denes, L., Szilagyi, G., Gal, A., and Nagy, Z. (2006). Talampanel a non-competitive AMPA-antagonist attenuates caspase-3 dependent apoptosis in mouse brain after transient focal cerebral ischemia. Brain Res. Bull. 70, 260–262.
Donevan, S. D., and Rogawski, M. A. (1993). Gyki 52466, a 2,3- benzodiazepine, is a highly selective, noncompetitive antagonist of Ampa/Kainate receptor responses. Neuron 10, 51–59.
Donevan, S. D., Yamaguchi, S., and Rogawski, M. A. (1994). Non-N-Methyl-d-Aspartate receptor antagonism by 3-N-substituted 2,3-benzodiazepines – Relationship to anticonvulsant activity. J. Pharmacol. Exp. Therap. 271, 25–29.
Doyle, K. P., Simon, R. P., and Stenzel-Poore, M. P. (2008). Mechanisms of ischemic brain damage. Neuropharmacology 55, 310–318.
Elger, B., Huth, A., Neuhaus, R., Ottow, E., Schneider, H., Seilheimer, B., and Turski, L. (2005). Novel alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptor antagonists of 2,3-benzodiazepine type: chemical synthesis, in vitro characterization, and in vivo prevention of acute neurodegeneration. J. Med. Chem. 48, 4618–4627.
El Bahh, B., Lurton, D., Sundstrom, L. E., and Rougier, A. (1997). Induction of tolerance and mossy fibre neuropeptide-Y expression in the contralateral hippocampus following a unilateral intrahippocampal kainic acid injection in the rat. Neurosci. Lett. 227, 135–139.
Epsztein, J., Ben-Ari, Y., Represa, A., and Crépel, V. (2008). Late-onset epileptogenesis and seizure genesis: lessons from models of cerebral ischemia. Neuroscientist 14, 78–90.
Erdo, F., Berzsenyi, P., and Andrasi, F. (2005). The AMPA-antagonist talampanel is neuroprotective in rodent models of focal cerebral ischemia. Brain Res. Bull. 66, 43–49.
Erdo, F., Berzsenyi, P., Nemet, L., and Andrasi, F. (2006). Talampanel improves the functional deficit after transient focal cerebral ischemia in rats. A 30-day follow up study. Brain Res. Bull. 68, 269–276.
Fernandes, H. B., Catches, J. S., Petralia, R. S., Copits, B. A., Xu, J., Russell, T. A., Swanson, G. T., and Contractor, A. (2009). High-affinity kainate receptor subunits are necessary for ionotropic but not metabotropic signaling. Neuron 63, 818–829.
Gigler, G., Moricz, K., Agoston, M., Simo, A., Albert, M., Benedek, A., Kapus, G., Kertesz, S., Vegh, M., Barkoczy, J., Marko, B., Szabo, G., Matucz, E., Gacsalyi, I., Levay, G., Harsing, L. G. Jr., and Szenasi, G. (2007). Neuroprotective and anticonvulsant effects of EGIS-8332, a non- competitive AMPA receptor antagonist, in a range of animal models. Br. J. Pharmacol. 152, 151–160.
Gressens, P., Spedding, M., Gigler, G., Kertesz, S., Villa, P., Medja, F., Williamson, T., Kapus, G., Levay, G., Szenasi, G., Barkoczy, J., and Harsing, L. G. Jr. (2005). The effects of AMPA receptor antagonists in models of stroke and neurodegeneration. Eur. J. Pharmacol. 519, 58–67.
Hazell, A. S. (2007). Excitotoxic mechanisms in stroke: an update of concepts and treatment strategies. Neurochem. Int. 50, 941–953.
Hesp, B. R., Clarkson, A., Sawant, P. M., and Kerr, D. S. (2007). Domoic acid pre-conditioning and seizure induction in young and aged rats. Epilepsy Res. 76, 103–112.
Hesp, B. R., Wrightson, T., Mullaney, I. M., and Kerr, D. S. (2004). Kainate receptors mediate tolerance to kainic acid and reduce high-affinity GTPase activity in young but not aged rat hippocampus. J. Neurochem. 90, 70–79.
Jung, Y. S., Lee, B. K., Park, H. S., Shim, J. K., Kim, S. U., Lee, S. H., Baik, E. J., and Moon, C. H. (2005). Activation of protein kinase C-delta attenuates kainate-induced cell death of cortical neurons. Neuroreport 16, 741–744.
Kapoor, M., Howard, R., Hall, I., and Appleton, I. (2004). Effects of epicatechin gallate on wound healing and scar formation in a full thickness incisional wound healing model in rats. Am. J. Pathol. 165, 299–307.
Karhunen, H., Bezvenyuk, Z., Nissinen, J., Sivenius, J., Jolkkonen, J., and Pitkänen, A. (2007). Epileptogenesis after cortical photothrombotic brain lesion in rats. Neuroscience 148, 314–324.
Kerr, D. S. (2007). In vivo pharmacological preconditioning reduces domoic acid- and kainic acid-induced seizures in rats: a role of G protein coupled receptors? ASCEPT Intern. Confer. on Neuropharmacology of Glutamate; Hunter Valley, Newcastle, Australia, July 8–11, 2007.
Kerr, D. S., Razak, A., and Crawford, N. (2002). Age-related changes in tolerance to the marine algal excitotoxin domoic acid. Neuropharmacol. 43, 357–366.
Kurkinen, K., Busto, R., Goldsteins, G., Koistinaho, J., and Perez-Pinzon, M. A. (2001). Isoform-specific membrane translocation of protein kinase C after ischemic preconditioning. Neurochem. Res. 26, 1139–1144.
Lere, C., El Bahh, B., Le Gal La Salle, G., and Rougier, A. (2002). A model of ‘epileptic tolerance’ for investigating neuroprotection, epileptic susceptibility and gene expression-related plastic changes. Brain Res. Brain Res. Protoc. 9, 49–56.
Matucz, E., Moricz, K., Gigler, G., Benedek, A., Barkoczy, J., Levay, G., Harsing, L. G. Jr., and Szenasi, G. (2006). Therapeutic time window of neuroprotection by non-competitive AMPA antagonists in transient and permanent focal cerebral ischemia in rats. Brain Res. 1123, 60–67.
Meldrum, B. S. (1993). Excitotoxicity and selective neuronal loss in epilepsy. Brain Pathol. 3, 405–412.
Melyan, Z., Wheal, H. V., and Lancaster, B. (2002). Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron 34, 107–114.
Naegele, J. R. (2007). Neuroprotective strategies to avert seizure-induced neurodegeneration in epilepsy. Epilepsia 48(Suppl. 2), 107–117.
Najm, I. M., Hadam, J., Chakraverty, D., Mikuni, N., Penrod, C., Sopa, C., Markarian, G., Luders, H. O., Babb, T., and Baudry, M. (1998). A short episode of seizure activity protects from status epilepticus-induced neuronal damage in rat brain. Brain Res. 810, 72–75.
Newman, M. F., Kirchner, J. L., Phillips-Bute, B., Gaver, V., Grocott, H., Jones, R. H., Mark, D. B., Reves, J. G., and Blumenthal, J. A. (2001). Longitudinal assessment of neurocognitive function after coronary-artery bypass surgery. New England J. Med. 344, 395–402.
Perez-Pinzon, M. A. (2007). Mechanisms of neuroprotection during ischemic preconditioning: lessons from anoxic tolerance. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 147, 291–299.
Perez-Pinzon, M. A., Born, J. G., and Centeno, J. M. (1999). Calcium and increased excitability promote tolerance against anoxia in hippocampal slices. Brain Res. 833, 20–26.
Perkinton, M. S., Sihra, T. S., and Williams, R. J. (1999). Ca2+-permeable AMPA receptors induce phosphorylation of cAMP response element-binding protein through a phosphatidylinositol 3-kinase- dependent stimulation of the mitogen-activated protein kinase signaling cascade in neurons. J. Neurosci. 19, 5861–5874.
Pitkänen, A., and Sutula, T. P. (2002). Is epilepsy a progressive disorder? Prospects fornew therapeutic approaches in temporal-lobeepilepsy. Lancet Neurol. 1, 173–181.
Pohle, W., and Rauca C. (1994). Hypoxia protects against the neurotoxicity of kainic acid. Brain Res. 644, 297–304.
Rorick-Kehn, L. M., Hart, J. C., and McKinzie, D. L. (2005). Pharmacological characterization of stress-induced hyperthermia in DBA/2 mice using metabotropic and ionotropic glutamate receptor ligands. Psychopharmacology 183, 226–240.
Rodriguez-Moreno, A., Lopez-Garcia, J. C., and Lerma, J. (2000). Two populations of kainate receptors with separate signaling mechanisms in hippocampal interneurons. Proc. Natl. Acad. Sci. U.S.A. 97, 1293–1298.
Rodriguez-Moreno, A., and Sihra, T. S. (2004). Presynaptic kainate receptor facilitation of glutamate release involves protein kinase A in the rat hippocampus. J. Physiol. 557, 733–745.
Rodriguez-Moreno, A., and Sihra, T. S. (2007). Kainate receptors with a metabotropic modus operandi. Trends Neurosci. 30, 630–637.
Rozas, J. L., Paternain, A. V., and Lerma, J. (2003). Noncanonical signaling by ionotropic kainate receptors. Neuron 39, 543–553.
Sawant, P. M., Munday, R., Holland, P. T., Mountfort, D. O., and Kerr, D. S. (2008). In vivo studies of seizure induction and pharmacological preconditioning by domoic acid and isodomoic acids A, B and C. Neuropharmacology 55, 1412–1418.
Sawant, P. M., Munday, R., Holland, P. T., Mountfort, D. O., and Kerr, D. S. (2010a). Spectral analysis of electrocorticographic activity during pharmacological preconditioning and seizure induction by intrahippocampal domoic acid. Hippocampus 20, 994–1002.
Sawant, P. M., Tyndall J. D. A., Holland, P. T., Mountfort, D. O., and Kerr, D. S. (2010b). In vivo seizure induction and affinity studies of domoic acid and isodomoic acids D, E and F. Neuropharmacology 59, 129–138.
Sawant, P. M., Weare, B. A., Holland, P. T., Selwood, A. I., King, K. L., Mikulski, C. M., Doucette, G. J., Mountfort, D. O., and Kerr, D. S. (2007). Isodomoic acids A and C exhibit low KA receptor affinity and reduced in vitro potencyin hippocampal region CA1. Toxicon 50, 627–638.
Segerstråle, M., Juuri, J., Lanore, F., Piepponen, P., Lauri, S. E., Mulle, C., and Taira, T. (2010). High firing rate of neonatal hippocampal interneurons is caused by attenuation of after hyperpolarizing potassium currents by tonically active kainate receptors. J. Neurosci. 30, 6507–6514.
Schurr, A., Payne, R. S., Tseng, M. T., Gozal, E., and Gozal, D. (2001). Excitotoxic preconditioning elicited by both glutamate and hypoxia and abolished by lactate transport inhibition in rat hippocampal slices. Neurosci. Lett. 307, 151–154.
Sloviter, R. S. (1987). Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science 235, 73–76.
Smith, S. E., Dürmüller, N., and Meldrum, B. S. (1991). The non-N-methyl-d-aspartate receptor antagonists, GYKI 52466 and NBQX are anticonvulsant in two animal models of reflex epilepsy. Eur. J. Pharmacol. 201, 179–183.
Sundquist, S. J., and Nisenbaum, L. K. (2005). Fast Fos: rapid protocols for single- and double-labeling c-Fos immunohistochemistry in fresh frozen brain sections. J. Neurosci. Meth. 141, 9–20.
Szabados, T., Gigler, G., Gacsályi, I., Gyertyán, I., and Lévay, G. (2001). Comparison of anticonvulsive and acute neuroprotective activity of three 2,3-benzodiazepine compounds, GYKI 52466, GYKI 53405, and GYKI 53655. Brain Res. Bull. 55, 387–391.
Tandon, P., Yang, Y., Stafstrom, C. E., and Holmes, G. L. (2002). Downregulation of kainate receptors in the hippocampus following repeated seizures in immature rats. Dev. Brain Res. 136, 145–150.
Wang, C. H., Chang, A., Tsai, M. J., Cheng, H., Liao, L. P., and Lin, A. M. (2005). Kainic acid-induced oxidative injury is attenuated by hypoxic preconditioning. Ann. N.Y. Acad. Sci. 1042, 314–324.
Wang, Y. Z., and Durkin, J. P. (1995). alpha-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, but not N-methyl-d-aspartate, activates mitogen-activated protein kinase through G-protein beta gamma subunits in rat cortical neurons.. J. Biol. Chem. 270, 22783–22787.
Weiczner, R., Krisztin-Péva, B., and Mihály, A. (2008). Blockade of AMPA-receptors attenuates 4- aminopyridine seizures, decreases the activation of inhibitory neurons but is ineffective against seizure-related astrocytic swelling. Epilepsy Res. 78, 22–32.
Youssef, F. F., Addae, J. I., McRae, A., and Stone. T. W. (2001). Long-term potentiation protects rat hippocampal slices from the effects of acute hypoxia. Brain Res. 907, 144–150.
Keywords: GYKI 52466, protection, seizures, EEG, preconditioning, metabotropic
Citation: Goulton CS, Patten AR, Kerr JR and Kerr DS(2010) Pharmacological preconditioning with GYKI 52466: a prophylactic approach to neuroprotection. Front. Neurosci. 4:54. doi: 10.3389/fnins.2010.00054
Received: 15 April 2010;
Paper pending published: 10 May 2010;
Accepted: 02 July 2010;
Published online: 03 August 2010
Edited by:
Xiao-Ming Ou, University of Mississippi Medical Center, USAReviewed by:
Roland Weiczner, University of Szeged, HungaryCopyright: © 2010 Goulton, Patten, Kerr and Kerr. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: D. Steven Kerr, Department of Pharmacology and Toxicology, School of Medical Sciences, University of Otago, P.O. Box 913, Dunedin, New Zealand. e-mail:c3RldmUua2VyckBvdGFnby5hYy5ueg==