

- Neuroscience Program, Department of Psychology, Trinity College, Hartford, CT, USA
A link between metabolism and brain function is clear. Since ancient times, epileptic seizures were noted as treatable with fasting, and historical observations of the therapeutic benefits of fasting on epilepsy were confirmed nearly 100 years ago. Shortly thereafter a high fat, low-carbohydrate ketogenic diet (KD) debuted as a therapy to reduce seizures. This strict regimen could mimic the metabolic effects of fasting while allowing adequate caloric intake for ongoing energy demands. Today, KD therapy, which forces predominantly ketone-based rather than glucose-based metabolism, is now well-established as highly successful in reducing seizures. Cellular metabolic dysfunction in the nervous system has been recognized as existing side-by-side with nervous system disorders – although often with much less obvious cause-and-effect as the relationship between fasting and seizures. Rekindled interest in metabolic and dietary therapies for brain disorders complements new insight into their mechanisms and broader implications. Here we describe the emerging relationship between a KD and adenosine as a way to reset brain metabolism and neuronal activity and disrupt a cycle of dysfunction. We also provide an overview of the effects of a KD on cognition and recent data on the effects of a KD on pain, and explore the relative time course quantified among hallmark metabolic changes, altered neuron function and altered animal behavior assessed after diet administration. We predict continued applications of metabolic therapies in treating dysfunction including and beyond the nervous system.
The Ketogenic Diet and Ketone-Based Metabolism
Metabolism influences brain activity, and metabolic dysfunction is associated with a wide variety of neurological disorders. The cause-and-effect relationship between metabolic and neuronal dysfunction is often unclear, though not in the case of epilepsy and diet. Historical observations noted the therapeutic benefits of fasting on epilepsy, but fasting is necessarily a time-limited practice. Therapeutic benefits of the metabolic condition of fasting were confirmed over 90 years ago when the high fat, low-carbohydrate ketogenic diet (KD) was described as alternative to fasting which still reduced epileptic seizures (Wilder, 1921). In turn, anticonvulsant drugs debuted over the next two decades, such that since then the KD has been used mostly for inoperable and medication-resistant epilepsy, which has been estimated to be 15% up to 45% of cases (Picot et al., 2008; Dong et al., 2011).
Although used clinically for many decades, prescribed most often to children, and increasing in popularity over the last two decades, the KD’s mechanism of action remains controversial. The KD was designed to produce ketosis without fasting by strictly limiting carbohydrate intake (Wilder, 1921). To make up for lost calories and augment ketosis, fat content is increased dramatically. When carbohydrate intake is strongly limited (as during the KD or fasting), the liver increases production of the ketone bodies β-hydroxybutyrate, acetoacetate, and acetone from circulating fatty acids (Aoki, 1981). Because of the β-hydroxyl substitution, β-hydroxybutyrate is not actually a ketone, although by convention it is grouped with the other two ketone bodies.
Ketone bodies are released into the circulation as an alternative energy source to generate ATP (“ketolytic” metabolism) within tissues, including the brain and spinal cord. Hallmark changes in blood chemistry are produced reliably in rodents (Figure 1). Formulation of the KD is calculated using a ratio of fat content to combined protein and carbohydrate content, varying in the clinic from 5:1 to 1:1 depending on a patient’s individual needs (Swink et al., 1997; Vining, 1999). We found that a KD-fed ad libitum at ratio of 7:1 or 3:1 to rats produced similar changes in blood chemistry (Figure 1). Clinically, the trend has been to decrease the ratio where possible and thus make the diet more palatable (including the more liberal modified Atkins diet; Kang et al., 2007; Kossoff et al., 2008b) but more systematic research is needed. Regarding different food types, the KD has now been adapted for widely varying cultures and cuisines in different countries around the world (e.g., India, Korea, United Kingdom, Saudi Arabia, Republic of Georgia; Kang et al., 2007; Neal et al., 2008a; Sharma et al., 2009; B. Zupec-Kania, personal communication). Understanding the mechanisms by which a diet controls seizures, along with broader opportunities for metabolic therapies, remains an active research topic because of accessibility, efficacy, and economics.
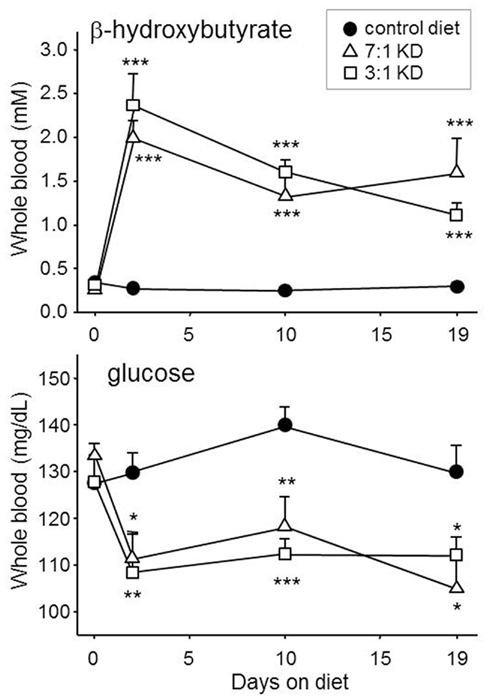
Figure 1. Ketogenic diets can produce prompt and sustained ketosis and mild hypoglycemia in experimental rodents. Here, young male Sprague-Dawley rats were fed with one of two ketogenic diets for 19 days, or remained fed with normal rodent chow. Both KDs, with strengths of 3:1 and 7:1 (BioServ 5140 and 3666, respectively), produced similar and significantly increased blood ketones and reduced blood glucose within 2 days and lasting until the last test day. Number of subjects was 12–14. *p < 0.05, **p < 0.01, ***p < 0.001, comparisons to control diet. Authors’ unpublished data.
Metabolism, Plasticity, and Synaptic Activity
The KD might alleviate seizures and other pathological states partially by providing elevated levels of high-energy molecules (e.g., ATP, phosphocreatine) and increased capacity for energy generation (increased mitochondrial number; Seyfried and Mukherjee, 2005; Bough and Rho, 2007; Masino and Geiger, 2008). Yet, numerous other changes due to the KD have been hypothesized to underlie increased inhibition and/or decreased excitation in brain, and thus to an anticonvulsant/neuroprotective state. In normal humans fed a KD, electroencephalography and transcranial magnetic stimulation demonstrated increased inhibition in the cerebral cortex, with a magnitude similar to that seen after benzodiazepine administration (Cantello et al., 2007). With the more extensive investigation possible in experimental animals, a KD was shown to enhance paired-pulse depression, shift the input/output relationship rightward, elevate the threshold for maximal electrical activation, and to block spreading depression-style events in the hippocampus in vivo (Bough et al., 2003). There have been surprisingly few detailed studies on detailed synaptic effects, likely because of the difficulty in performing such studies in vivo, coupled with the typical glucose-based incubation protocol for in vitro slices; to date, a “KD” incubation protocol has not been standardized, although recent work sampling cerebrospinal fluid in KD-fed animals might provide a starting point (Samala et al., 2011). Currently, the major proposed mechanisms for such increased inhibition and/or decreased excitation include increased levels of adenosine, a major inhibitory neuromodulator (Masino and Geiger, 2008); increased levels of γ-aminobutyric acid (GABA), a major inhibitory neurotransmitter (Yudkoff et al., 2007; Omote et al., 2011); decreased glutamate, a major excitatory neurotransmitter (Lund et al., 2009; Juge et al., 2010) and direct effects of elevated ketone bodies on ion channels (Ma et al., 2007).
Increased inhibition or decreased excitability, if sufficiently strong, might not only suppress seizures but also influence normal brain function. Many types of normal brain function, as well as recovery from injury, are thought to depend on synaptic plasticity, i.e., the malleability, either temporary or long-lasting, of the strength of neuronal communication (Davis et al., 1992; Goosens and Maren, 2002). Long-term potentiation (LTP) is a sustained increase in synaptic efficacy which can be observed in a number of brain regions including its original discovery site, the hippocampus (Bliss and Lømo, 1973; Bramham and Srebro, 1989; Clugnet and LeDoux, 1990; Bonci and Malenka, 1999; Mahon et al., 2004). Studies have linked metabolism and LTP (Potter et al., 2010); we and our collaborators characterized the effects of a KD on hippocampal LTP with the hypothesis that KD-related inhibition or reduced excitation might affect brain plasticity (Koranda et al., 2011). We recorded hippocampal signals through chronically implanted electrodes in freely moving rats. After 3 weeks on a 7:1 KD, baseline synaptic measurements were taken in the perforant path-dentate gyrus pathway and LTP was induced with tetanic stimulation and the response measured over the next 2 days. The KD had no significant effects on measures of short-term plasticity (paired-pulse depression, paired-pulse facilitation), and did not prevent LTP induction, whereas the magnitude of the potentiation was significantly smaller in KD-fed rats. The LTP magnitude remained lower in these rats out to the longest tested time point (48 h). As discussed below, cognitive effects of the diet are mixed in animals and overall positive in humans. In addition, it is important to note that 7:1 is a stronger diet ratio than that used clinically, animals used had never had seizures, and another paper looking at the KD on LTP in vivo in anesthetized animals did not find any differences (Thio et al., 2010).
To test the role of adenosine in the KD’s ability to reduce seizures, we and our collaborators recently tested the effectiveness of a KD in a transgenic mouse with spontaneous hippocampal electrographic seizures due to adenosine deficiency. These mice overexpress the adenosine-metabolizing enzyme adenosine kinase (ADK) in brain (Fedele et al., 2005), and tonic levels of the endogenous inhibitor adenosine are therefore lower than normal. At baseline, seizures recorded with chronically implanted electrodes occur five times per hour, on average (Masino et al., 2011). After being fed on a 7:1 KD for 3 weeks, seizure frequency dropped almost 90%. This antiseizure effect depended on low glucose (seizures were restored by a peripheral injection of glucose), and activation of the adenosine A1 receptor subtype (A1R; seizure activity was restored by injection of a selective A1R antagonist). Together, this evidence suggests that the KD exerts antiseizure effects by restoring adenosine levels and A1R activation via a mechanism related to low glucose.
Further support for this idea is provided by transgenic mice lacking A1Rs. These mice also have spontaneous electrographic seizures in the hippocampus, but the KD has no effect on seizure frequency in A1R knockout mice, and is partially effective in mice heterozygous for the A1R (Masino et al., 2011). Although these models all involve seizures induced by a lack of adenosinergic modulation, the results are likely generalizable: adenosine has been found to be anticonvulsive/antiseizure in virtually every seizure model in which it has been tested (excepting A1R knockout mice – providing further evidence for the primary anticonvulsant role of A1Rs). Adenosine in particular, and a KD in general, might offer more homeostatic “upstream” bioenergetic regulation of neuronal activity, and possibly long-term benefits on brain homeostasis, than highly specific drug therapies (Boison et al., 2011). Regarding LTP, previous results consistent with the involvement of adenosine in KD effects have shown that adenosine reduces LTP magnitude when present during induction (Mitchell et al., 1993; Costenla et al., 1999; de Mendonca and Ribeiro, 2000; Fujii et al., 2000a,b; Tabata et al., 2001; Zhang et al., 2004; Rex et al., 2005; but see Pascual et al., 2005) and, when applied after induction, promotes reversal of existing LTP (Huang et al., 1999; Fujii et al., 2000a). Yet, the lack of effects of the KD on input–output relationships and short-term plasticity seem to argue against the tonic involvement of adenosine (Koranda et al., 2011). Mechanism aside, the KD can limit excessive neuronal activity (a class into which the neuronal activity during an LTP induction burst certainly applies) and perhaps reset baseline activity.
Ketogenic Diet for a Brain Slice: Relaxing in Reduced Glucose?
Compared to in vivo, in vitro paradigms can provide tighter control over experimental variables, allowing for a more thorough characterization of mechanisms. Effects of KD feeding on baseline excitability are inconsistent in vitro, however (Stafstrom et al., 1999; Thio et al., 2000; Bough et al., 2006; Nylen et al., 2008). Certainly, the metabolic state established by a KD might be disrupted during tissue preparation for in vitro work. As introduced briefly above, one of the biochemical effects associated with a KD is an abundance of high-energy molecules (DeVivo et al., 1978; Nakazawa et al., 1983; Pan et al., 1999; Masino et al., 2007), as well as increased mitochondrial biogenesis, respiration, and expression of ATP synthesis-related proteins (Noh et al., 2004; Sullivan et al., 2004; Bough et al., 2006; Nylen et al., 2009; Balietti et al., 2010). Several lines of evidence suggest that reduced glucose is critical for antiseizure effects.
We modeled key aspects of the KD in vitro by maintaining or increasing intracellular ATP while decreasing extracellular glucose in individual CA3 pyramidal neurons in acute hippocampal slices. We varied ATP (0.5–5.0 mM; 2 mM is standard) in the patch pipet and changed glucose concentration of the bathing solution from 11 mM (standard) to either 7 or 3 mM (Kawamura et al., 2010). Note that 3 mM glucose is still a physiological level: in vivo brain concentrations are near 3 mM (Hu and Wilson, 1997; Shram et al., 1997). Moderately lowered extracellular glucose has been reported to attenuate epileptiform activity in brain slices (Kirchner et al., 2006), whereas experimental studies of pathological hypoglycemia often remove glucose completely from the bathing medium (aglycemia; Tromba et al., 1992; Zhu and Krnjevic, 1993).
We found that when intracellular ATP levels were adequate or high (1.0–5.0 mM), reducing extracellular glucose provoked an outward (inhibitory) current, with a larger current found with a reduction to 3 mM versus to 7 mM (Figure 2). This outward current was fully reversible on return to 11 mM glucose and had a reversal potential near the equilibrium potential for K+, and was blocked by the non-selective K+ channel antagonist Ba2+ (Kawamura et al., 2010). If intracellular ATP levels were low (0.5 mM), reducing glucose produced a transient inward (excitatory) current instead (Figure 2). Therefore, moderately low extracellular glucose can inhibit hippocampal neurons that have sufficient or abundant energy stores. Furthermore, this inhibition was completely blocked by application of an A1R antagonist and was not present in neurons from A1R knockout mice (Figure 2; similar to observations in vivo: Masino et al., 2011) implying increased adenosine levels produced the inhibition (conversely, diabetic hyperglycemia seems to be related to reduced signaling through A1Rs (Duarte et al., 2006). A similar consistent mechanism was reflected presynaptically (measured as decreased spontaneous postsynaptic current frequency); an A1R-dependent presynaptic inhibition was produced by adequate/high postsynaptic intracellular ATP combined with low extracellular glucose (Kawamura et al., 2010). Together, this study and Masino et al. (2011) suggest that a KD can limit seizures (at least those involving the hippocampus) through a mechanism dependent on low glucose and abundant high-energy molecules and involving augmentation of adenosine levels.
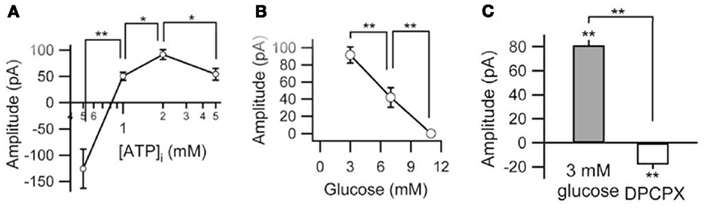
Figure 2. Relationship among membrane current, intracellular ATP, and extracellular glucose. (A) Peak currents produced by lowering extracellular glucose from 11 to 3 mM depend on intracellular ATP concentration (0.5–5 mM). Current is outward, except at 0.5 mM ATP. (B) Concentration-dependence of glucose-related outward current (with 2 mM intracellular ATP). (C) Outward current produced in low-glucose buffer reversed completely (and became slightly inward) with the A1R antagonist DPCPX. *p < 0.05, **p < 0.01. Adapted with permission from Kawamura et al. (2010).
In our in vitro study, we manipulated ATP only in the patched neuron, suggesting an autocrine mechanism to increase adenosine. How might this autoinhibition occur? ATP might be metabolized intracellularly to adenosine, which would then be released. Loading pyramidal neurons with adenosine + ATP versus ATP alone, however, suggested that the current was not mediated by direct adenosine release (Kawamura et al., 2010). Alternatively, ATP might be released and then metabolized to adenosine. Cells can release ATP by several mechanisms (Dubyak, 2009), and extracellular ATP is metabolized rapidly to adenosine (Dunwiddie et al., 1997). One prominent non-exocytotic ATP release mechanism in neurons and glia is ATP passage through channels composed of connexins or pannexins (Stout et al., 2002; Schock et al., 2008; Iwabuchi and Kawahara, 2011). Through a series of physiological and pharmacological experiments, we determined that pannexin channels were the source of extracellular ATP. Taken together, our data are consistent with a process by which lowered extracellular glucose promotes release of ATP via pannexins. ATP is then converted extracellularly to adenosine, which activates A1Rs coupled, under these conditions, to KATP channels (Kawamura et al., 2010). This pathway is likely to underlie the A1R-mediated anticonvulsant effect produced by the KD in vivo. Certainly, mild hypoglycemia and enhanced adenosine tone can underlie its anticonvulsant effect (Masino et al., 2011), whereas the in vivo involvement of pannexin channels and ATP release remains to be demonstrated directly.
Ketogenic Diet’s Effect on Cognition and Mood: Negative, then Positive?
Altered cognition and affect in children with seizure disorders has always been a concern. Regarding pharmacological therapies, several authors have shown that children with epilepsy – even those whose seizures were well-controlled with antiepileptic drugs – had decreased cognitive function compared to their peers (Devinsky, 1995; Thompson et al., 2000; Drane and Meador, 2002). The exact mechanism of cognitive decline is unknown: traditional antiepileptic drugs decrease membrane excitability, increase postsynaptic inhibition, or reduce network synchronization to decrease excessive excitability associated with seizure development (Loring, 2005). These neurophysiological mechanisms, if sufficiently strong, will not only suppress seizures but also impair normal brain function. The incidence of cognitive side effects is increased at higher dosing and with polypharmacy which might be necessary for significant seizure control (Loring and Kimford, 2001). Thus, the cognitive and affective state of a medicated epileptic patient results from a balance of forces including the negative effects of the disease state (seizures, abnormal interictal brain activity, abnormal sleep), the positive effects of the anticonvulsive medication (seizure control), and the negative side effects of the anticonvulsive mediation (which can include sedation and/or abnormal sleep).
The KD might offer fewer chronic negative side effects than medication, and given that it has been in use for over 90 years, serious or systematic negative consequences would likely have surfaced by now. In research studies, KDs (albeit at a much stronger ratio than used clinically) reduced brain mass in juvenile rodents (Cheng et al., 2004; Zhao et al., 2004) and KDs can affect body growth in children (who are typically on the diet temporarily; Liu et al., 2003; Peterson et al., 2005; Neal et al., 2008b) but to our knowledge negative KD effects on human brain development and growth have not been quantified. Notably, recurrent clinical hypoglycemia can lead to a cumulative cognitive impairment (Langan et al., 1991; Deary et al., 1993) – although this effect might not be directly applicable because the hypoglycemia in these studies was episodic and much more severe than the chronic reduced (but not abnormal) glucose levels associated with the KD. Overall, positive and negative short- and long-term effects of this strict diet on cognition and mood remain under-examined clinically, particularly in pediatric patients.
It is worthwhile to consider that any assessment of cognitive or affective state associated with a KD should occur at multiple time points, as effects of the KD (including anticonvulsive effects) clearly evolve. There are limitations to combining data from different laboratories due to differing methodologies, different KDs, etc. Yet in surveying the research literature, it seems fairly clear that there is a biphasic effect on locomotor behavior: reduced activity characterizes KD onset, whereas increased activity predominate after a few weeks. Effects of a KD on locomotion in rodents (compiled informally from the literature) are shown in Figure 3. Notably, a biphasic pattern over time after diet initiation is found in clinical literature relating to cognition, mood, and vitality. Soon after beginning a KD, subjects often complain of lethargy (Vining et al., 1998; Lefevre and Aronson, 2000); in children, intolerable drowsiness is a reported side-effect that sometimes leads to cessation of KD treatment (Neal et al., 2008a). Yet, after weeks on the diet, subjects report heightened vitality, physical functioning, and alertness (Hallböök et al., 2007; Mosek et al., 2009; Yancy et al., 2009). In some cases these positive effects may be at least partially due to reduced seizure frequency, but similar positive effects are also described in non-epileptic subjects. This delay in beneficial effects is reminiscent of the delay often observed in anticonvulsant effects (Kossoff et al., 2008a).
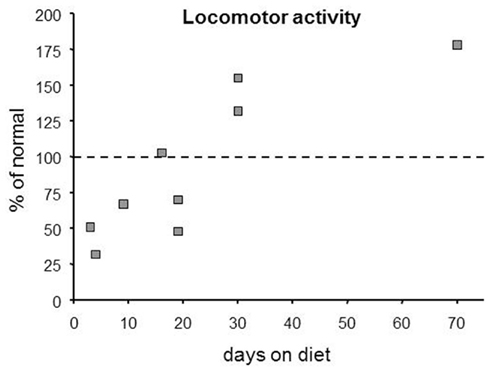
Figure 3. Over time, KDs produce a biphasic effect on locomotor activity based on this compilation of published rodent data. Note that hypoactivity predominated in studies with short diet treatments (≤20 day), whereas hyperactivity predominated with longer treatments (≥30 day). Points were estimated from published graphs and tables in multiple references (Zhao et al., 2004; Murphy et al., 2005; Ziegler et al., 2005; Murphy and Burnham, 2006; Mantis et al., 2009; Oishi et al., 2010; Thio et al., 2010).
Studies of the KD in epileptic patients rarely characterize mood, which might understandably be poor during the initial lethargic/drowsy stage. Several weight-loss studies, however, included affective measures and found positive effects of KD on mood in overweight subjects as early as 2 weeks into diet treatment, and lasting many weeks (Halyburton et al., 2007; McClernon et al., 2007; Brinkworth et al., 2009; Yancy et al., 2009). Two of these studies provide some evidence against this result simply being a psychological effect of weight loss (Brinkworth et al., 2009; Yancy et al., 2009). Thus, beneficial effects on mood (as well as weight loss) await those who conquer the early stage after KD initiation. Studies of patients with epilepsy on the KD, including children, have either reported improved cognition anecdotally (Sirven et al., 1999) or reported improvements in more general measures such as attention and social functioning (Kinsman et al., 1992; Pulsifer et al., 2001). It is difficult to determine if these effects are due to reduced seizures, to concomitantly reduced medications, or a direct action on cognition/attention. Investigations in non-epileptic adult subjects (thus without confounding antiepileptic medications) have more specifically addressed cognition and the KD. One study found a transient, moderate impairment in one cognitive task (but not two other tasks) at 1 week of diet treatment but found no impairments at later time points (Wing et al., 1995); two studies examining chronic KD treatments reported improved processing speed and working memory lasting up to 1 year (Halyburton et al., 2007; Brinkworth et al., 2009). This pattern seems to parallel the biphasic effect on activity and vitality noted above.
A minority of animal studies have reported impairments in learning and memory, specifically in a task of spatial reference memory (Su et al., 2000; Zhao et al., 2004). Other studies, however, have failed to find any detrimental effect of the KD on learning and memory in rodents in various mazes or in fear conditioning (Hori et al., 1997; Todorova et al., 2000; Silva et al., 2005; Appelberg et al., 2009; Thio et al., 2010). We tested normal mice of both sexes in a simple working memory task after feeding on a 7:1 KD at a number of time points, up to 10 weeks, and found no effect of the KD (though hyperactivity did appear beginning at 2 weeks (Ruskin et al., 2011a). It is worth noting that a KD not only does not impair but in fact reverses age-related deficits in learning and other cognitive measures in aged, but otherwise healthy, dogs and rodents (Pan et al., 2010; Xu et al., 2010). Taken together, these results largely support the beneficial nature of KD feeding on mood and cognition in patients.
Nociception and Inflammation: Multiple Mechanisms Likely
Converging lines of evidence suggest the utility of a KD for pain relief. First, it has long been known that reducing glucose metabolism influences pain. There is an overall increase in pain thresholds (and thus reduced pain) when glycolytic enzymes are inhibited by exogenous 2-deoxy-D-glucose (Bodnar et al., 1979). This effect is mediated centrally (Bodnar et al., 1981), and might involve increased brain/spinal cord inhibition by adenosine, the release of which is stimulated by 2-deoxy-D-glucose (Zhao et al., 1997; Minor et al., 2001). 2-Deoxy-D-glucose is also anticonvulsant (Garriga-Canut et al., 2006), and while the mechanisms might not overlap entirely with the KD (Stafstrom et al., 2009; Gasior et al., 2010) there might be some common pathways. Second, anticonvulsant drugs such as gabapentin, felbamate, and valproate are useful in treating pain, particularly neuropathic pain and migraine (Johannessen Landmark, 2008). These drugs typically act by decreasing neuronal activity or excitability, and it is clear that reducing central activity with adenosine or GABA agonists alleviates pain (Karlsten et al., 1992; Malmberg and Yaksh, 1993; Belfrage et al., 1995; Malan et al., 2002; Gwak et al., 2006). Thus, we predicted that the KD, which reduces glucose metabolism and is anticonvulsant, would reduce pain.
We fed rats a 7:1 KD in order to test the effects in the hotplate test. In this test, the latency to withdraw a hindpaw from the warm surface indicates the animal’s sensitivity to painful heat. In young rats, we found that KD feeding for 3–4 weeks increased paw withdrawal latency (i.e., decreased the sensitivity) to plate temperatures from 48 to 51°C (Ruskin et al., 2009). In adult rats, the effect seemed to be smaller in magnitude, and was significant only at 49 and 50°C. We recently found similar results with a less stringent 3:1 KD (Ruskin et al., 2011b). Curiously, another study reported increased thermal pain sensitivity (tail flick) after 12 weeks of KD feeding in young rats (Ziegler et al., 2005); methodological differences such as rat strain, body part (paw vs. tail), diet composition, and stimulus strength might be factors. The difference in diet treatment length (3 vs. 12 weeks) does not seem to explain the disparity, as subsequently we have found decreased thermal pain sensitivity present after 10–11 weeks of feeding with a 3:1 KD (Ruskin et al., 2011b). Thus far the specific mechanism of altered thermal nociception in KD-fed rats is unknown, and could involve hypoglycemia, ketosis, fatty acids, and/or adenosine.
One recently published clinical report on KD effects on “quality of life” reported that beneficial effects on self-reported general bodily pain were at the threshold of statistical significance (Yancy et al., 2009), suggesting that KD effects on overall pain might be positive. This report, however, was not a dedicated study of pain, but rather a study of overall quality of life; as such, there was no underlying painful condition to treat. In the same study, a low-fat diet also alleviated bodily pain. Overall, an assessment of pain in KD-treated patients is warranted.
A better understanding of the relationship between metabolism and pain could help multiple and comorbid conditions, and the KD might prove uniquely useful against diabetes and diabetes-related neuropathy. Although work with rodents has produced mixed results (Al-Khalifa et al., 2009, 2011; Garbow et al., 2011; Park et al., 2011; Poplawski et al., 2011), clinical studies have found exclusively positive outcomes: after KD treatment, patients with type I or II diabetes had improved control of blood glucose, and many could have their medications reduced or eliminated (Gumbiner et al., 1996; Yancy et al., 2005; Westman et al., 2008; Dressler et al., 2010). In addition, type I diabetic patients (and, based on one report, children with epilepsy) prefer foods that are high in fat and low in carbohydrates (Amari et al., 2007; Snell-Bergeon et al., 2009), which might be attempted self-medication. The mixed animal results might result from the use of very strict KDs (Garbow et al., 2011; Park et al., 2011), or from the diabetic propensity of many laboratory rodent strains. Thus, the KD might benefit diabetic patients both by alleviating neuropathic pain and treating the underlying glycemic control dysfunction.
Finally, the KD would be predicted to be effective against inflammatory pain. Chronic inflammation is typically accompanied by pain due to the release of prostaglandins and the consequent sensitization of sensory neurons (Mense, 1983). Some of the most common sources of inflammatory pain are rheumatoid arthritis, chronic inflammatory bowel disease, pancreatitis, back pain, and some cancers. We found that a KD reduced experimental inflammation-induced swelling and plasma extravasation (Ruskin et al., 2009), and clinical studies describe positive effects of a KD on liver inflammation in non-alcoholic fatty liver disease (Tendler et al., 2007; Pérez-Guisado and Muñoz-Serrano, 2011). Regarding mechanisms linking metabolism to inflammatory pain, reactive oxygen species are a major component of inflammation, and limiting reactive oxygen species should contribute to limiting inflammation. Accordingly, ketone-based metabolism should produce fewer free radicals and reactive oxygen species through affecting the mitochondrial co-enzyme Q couple and the cytoplasmic glutathione couple (Veech, 2004). Indeed, as expected, treatment with ketones reduces the level of reactive oxygen species (Noh et al., 2006a; Kim et al., 2007, 2010; Maalouf et al., 2007; Haces et al., 2008; Maalouf and Rho, 2008), as does KD feeding (Sullivan et al., 2004).
Regarding inflammatory pain, by virtue of their high-fat content KDs should also activate peroxisome proliferator-activated receptors (PPARs). These nuclear receptors bind long-chain polyunsaturated fatty acids, and consequently induce transcriptional changes that culminate in enhanced lipid metabolism (Moya-Camarena et al., 1999; Diradourian et al., 2005; Michalik et al., 2006). Genetic knockout of a major PPAR (the α subtype) augments inflammatory reactions (Cuzzocrea et al., 2006), whereas synthetic PPAR agonists reduce experimentally induced inflammation (Cuzzocrea et al., 2003; LoVerme et al., 2005). This latter effect appears to involve reduced transcription of pro-inflammatory genes (Blanquart et al., 2003) and seems to be invoked by the KD (Jeong et al., 2011). Synthetic PPAR agonists are analgesic against inflammatory pain (LoVerme et al., 2006). In addition to these effects, PPAR activation augments expression of the enzymes involved in ketogenesis (Cullingford et al., 2002), promoting the shift to a ketone-based metabolism, in agreement with findings of stronger ketosis with a high-polyunsaturated fat KD (Fuehrlein et al., 2004). Although polyunsaturated fatty acid content of the KD seems not to be important in the diet’s anticonvulsant effect (Dell et al., 2001; Dahlin et al., 2007), it might be a crucial characteristic for KD influence on inflammation.
It might seem ironic that the KD is discussed here as reducing inflammation, given that other high-fat diets and obesity are definitely linked to chronic inflammation (Thaler and Schwartz, 2010; Ding and Lund, 2011; Laugerette et al., 2011). Those high-fat diets that lead to obesity, including the so-called Western diet, include a high amount of fat along with normal amounts of carbohydrate, a crucial difference from the very low-carbohydrate KD which typically leads to weight loss (Gumbiner et al., 1996; Halyburton et al., 2007; Tendler et al., 2007; Westman et al., 2008). Thus, the high-fat-plus-carbohydrate diet promotes fat storage whereas the high fat, low-carbohydrate diet promotes fat metabolism. Nevertheless, more clinical work with the KD and inflammation is warranted, particularly regarding long-term effects. It will be crucial to determine which of the mechanisms described above is most important for the KD’s alleviation of inflammation. Future work on the relationship between the KD’s hallmark changes in blood chemistry, ketosis and mild hypoglycemia, and its anti-inflammatory and anti-nociceptive effects should help characterize the pertinent mechanisms.
Attenuating Brain Injury and Neurodegeneration
Animal studies have found find that the KD protects against seizure-induced neurodegeneration and related sequelae (such as aberrant neurite sprouting; Muller-Schwarze et al., 1999; Noh et al., 2003, 2005, 2006b; Linard et al., 2010). The KD is also neuroprotective against ischemic damage (Tai et al., 2008, 2009), hypoglycemic damage (Yamada et al., 2005), and traumatic brain and spinal injury (Prins et al., 2005; Appelberg et al., 2009; Hu et al., 2009a,b; Prins and Hovda, 2009; Schwartzkroin et al., 2010; Streijger et al., 2011), and improves injury-related deficits in cognition and movement after traumatic brain and spinal injury, respectively (Appelberg et al., 2009; Streijger et al., 2011). Ketosis is apparently crucial to these effects as direct application of ketones to in vitro tissue is also protective against hypoglycemia and ischemia (Samoilova et al., 2010), oxidative stress (Kim et al., 2007), and excitotoxicity (Massieu et al., 2003; Noh et al., 2006b; Maalouf et al., 2007; Samoilova et al., 2010). The mechanisms are likely to involve reduced reactive oxygen species, reduced tissue excitability, and enhanced production of high-energy molecules.
Based on evidence for neuroprotection against acute insults, and recognition that metabolic dysfunction accompanies chronic neurological disease, researchers are expanding into animal models of more slowly-acting neurodegenerative diseases. Positive effects of KD feeding have been found in models of amyotrophic lateral sclerosis (Zhao et al., 2006), Parkinson’s disease (Cheng et al., 2009; Yang and Cheng, 2010), and Alzheimer’s disease (Van der Auwera et al., 2005; Mohamed et al., 2010). In addition, KD feeding reverses aging-related impairments in brain biochemistry in animals (Studzinski et al., 2008; Balietti et al., 2010). Direct application of ketones is also beneficial in models of Parkinson’s disease (Kashiwaya et al., 2000; Tieu et al., 2003) and Alzheimer’s disease (Kashiwaya et al., 2000).
Huntington’s disease, which involves the death of neurons in the caudate and putamen, is thought to involve excitotoxicity and mitochondrial dysfunction (Estráda-Sanchez et al., 2008; Damiano et al., 2010). Based on findings reviewed above, we characterized the effects of a strict (7:1) KD in a rapidly progressing Huntington’s disease model, the R6/2 mouse (lifespan less than 16 weeks). KD feeding began at 6 weeks of age, when motor impairments are still minor (Ruskin et al., 2011a). The KD did not increase lifespan or alleviate motor impairments, but, importantly, it did not negatively affect either. However, the KD did delay significantly the onset of progressive weight loss, which is a major problem in patients (Sanberg et al., 1981; Lanska et al., 1988). In addition, the KD reversed a modest working memory impairment in female mice, and working memory is known to be affected in patients with Huntington’s disease (Lange et al., 1995; Lawrence et al., 1996) as well as other neurological disorders and aging.
The lack of effect on lifespan or locomotor activity may signal that beneficial effects of a KD might not be similar across neurodegenerative disorders, might depend on the severity or rate of progression, or might differ in different animal models of a disorder; alternatively, the KD might need to be optimized for strength and composition for different conditions. Although it seems paradoxical that the KD, normally associated with weight loss, might maintain, or increase body weight under particular conditions, our data suggest that KD feeding could alleviate Huntington’s disease-associated cachexia, and, as noted above, in patients a higher body mass is associated with slower disease progression (Myers et al., 1991). Based on this finding, the KD might also deserve consideration for treatment of other cachexias; for instance, that associated with cancer (Colomer et al., 2007). Indeed, the KD is beginning to be used as an anti-tumorigenic treatment (Klement and Kammerer, 2011; Seyfried et al., 2012) and so could provide dual benefits. If the anti-neurodegenerative effects found in animal models of Parkinson’s disease, Alzheimer’s disease, and aging are successfully extended to humans, the KD could also have dual benefits, delaying the primary degenerative condition and alleviating the working memory problems common to these conditions (Halyburton et al., 2007; Brinkworth et al., 2009; Ruskin et al., 2011a).
Looking Ahead
A KD offers known benefits for epilepsy, and it is apparent that the relationship between metabolism and brain function offers primary therapeutic opportunities. Basic and clinical research is acutely aware that metabolic dysfunction and comorbidities promulgate lifelong impacts on nervous system function. Particularly promising unrealized opportunities for intervention and restoration of metabolic homeostasis occur during development, after injury, and during disease progression – all windows with high levels of plasticity and remodeling. New insight into mechanisms could accelerate development of treatments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported by the National Institutes of Health (P20RR017699, R15NS065446, R15NS066392, R01NS065957), the National Science Foundation (IOS0843585), the CHDI Foundation, and Trinity College.
References
Al-Khalifa, A., Mathew, T. C., Al-Zaid, N. S., Mathew, E., and Dashti, H. (2011). Low carbohydrate ketogenic diet prevents the induction of diabetes using streptozotocin in rats. Exp. Toxicol. Pathol. 63, 663–669.
Al-Khalifa, A., Mathew, T. C., Al-Zaid, N. S., Mathew, E., and Dashti, H. M. (2009). Therapeutic role of low-carbohydrate ketogenic diet in diabetes. Nutrition 25, 1177–1185.
Amari, A., Dahlquist, L., Kossoff, E. H., Vining, E. P. G., Trescher, W. H., and Slifer, K. J. (2007). Children with seizures exhibit preferences for foods compatible with the ketogenic diet. Epilepsy Behav. 11, 98–104.
Aoki, T. T. (1981). Metabolic adaptations to starvation, semistarvation, and carbohydrate restriction. Prog. Clin. Biol. Res. 67, 161–177.
Appelberg, K. S., Hovda, D. A., and Prins, M. L. (2009). The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J. Neurotrauma 26, 497–506.
Balietti, M., Giorgetti, B., Di Stefano, G., Casoli, T., Platano, D., Solazzi, M., Bertoni-Freddari, C., Aicardi, G., Lattanzio, F., and Fattoretti, P. (2010). A ketogenic diet increases succinic dehydrogenase (SDH) activity and recovers age-related decrease in numeric density of SDH-positive mitochondria in cerebellar Purkinje cells of late-adult rats. Micron 41, 143–148.
Belfrage, M., Sollevi, A., Segerdahl, M., Sjölund, K. F., and Hansson, P. (1995). Systemic adenosine infusion alleviates spontaneous and stimulus evoked pain in patients with peripheral neuropathic pain. Anesth. Analg. 81, 713–717.
Blanquart, C., Barbier, O., Fruchart, J. C., Staels, B., and Glineur, C. (2003). Peroxisome proliferator-activated receptors: regulation of transcriptional activities and roles in inflammation. J. Steroid Biochem. Mol. Biol. 85, 267–273.
Bliss, T. V. P., and Lømo, T. (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. (Lond.) 232, 331–356.
Bodnar, R. J., Kelly, D. D., and Glusman, M. (1979). 2-Deoxy-D-glucose analgesia: influences of opiate and non-opiate factors. Pharmacol. Biochem. Behav. 11, 297–301.
Bodnar, R. J., Merrigan, K. P., and Wallace, M. M. (1981). Analgesia following intraventricular administration of 2-deoxy-D-glucose. Pharmacol. Biochem. Behav. 14, 579–581.
Boison, D., Masino, S. A., and Geiger, J. D. (2011). Homeostatic bioenergetic network regulation: a novel concept to avoid pharmacoresistance in epilepsy. Expert Opin. Drug Discov. 6, 713–724.
Bonci, A., and Malenka, R. C. (1999). Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. J. Neurosci. 19, 3723–3730.
Bough, K. J., and Rho, J. M. (2007). Anticonvulsant mechanisms of the ketogenic diet. Epilepsia 48, 43–58.
Bough, K. J., Schwartzkroin, P. A., and Rho, J. M. (2003). Caloric restriction and ketogenic diet diminish neuronal excitability in rat dentate gyrus in vivo. Epilepsia 44, 752–760.
Bough, K. J., Wetherington, J., Hassel, B., Pare, J. F., Gawryluk, J. W., Greene, J. G., Shaw, R., Smith, Y., Geiger, J. D., and Dingledine, R. J. (2006). Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 60, 223–235.
Bramham, C. R., and Srebro, B. (1989). Synaptic plasticity in the hippocampus is modulated by behavioral state. Brain Res. 493, 74–86.
Brinkworth, G. D., Buckley, J. D., Noakes, M., Clifton, P. M., and Wilson, C. J. (2009). Long-term effects of a very low-carbohydrate diet and a low-fat diet on mood and cognitive function. Arch. Intern. Med. 169, 1873–1880.
Cantello, R., Varrasi, C., Tarletti, R., Cecchin, M., D’Andrea, F., Veggiotti, P., Bellomo, G., and Monaco, F. (2007). Ketogenic diet: electrophysiological effects on the normal human cortex. Epilepsia 48, 1756–1763.
Cheng, B., Yang, X., An, L., Gao, B., Liu, X., and Liu, S. (2009). Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease. Brain Res. 1286, 25–31.
Cheng, C. M., Hicks, K., Wang, J., Eagles, D. A., and Bondy, C. A. (2004). Caloric restriction augments brain glutamic acid decarboxylase-65 and -67 expression. J. Neurosci. Res. 77, 270–276.
Clugnet, M.-C., and LeDoux, J. E. (1990). Synaptic plasticity in fear conditioning circuits: induction of LTP in the lateral nucleus of the amygdala by stimulation of the medial geniculate body. J. Neurosci. 10, 2818–2824.
Colomer, R., Moreno-Nogueira, J. M., García-Luna, P. P., García-Peris, P., García-De-Lorenzo, A., Zarazaga, A., Quecedo, L., Del Llano, J., Usán, L., and Casimiro, C. (2007). N-3 fatty acids, cancer and cachexia: a systematic review of the literature. Br. J. Nutr. 97, 823–831.
Costenla, A. R., De Mendonca, A., and Ribeiro, J. A. (1999). Adenosine modulates synaptic plasticity in hippocampal slices from aged rats. Brain Res. 851, 228–234.
Cullingford, T. E., Dolphin, C. T., and Sato, H. (2002). The peroxisome proliferator-activated receptor α-selective activator ciprofibrate upregulates expression of genes encoding fatty acid oxidation and ketogenesis enzymes in rat brain. Neuropharmacology 42, 724–730.
Cuzzocrea, S., Mazzon, E., Di Paolo, R., Peli, A., Bonato, A., Britti, D., Genovese, T., Muia, C., Crisafulli, C., and Caputi, A. P. (2006). The role of the peroxisome proliferator-activated receptor-alpha (PPAR-alpha) in the regulation of acute inflammation. J. Leukoc. Biol. 79, 999–1010.
Cuzzocrea, S., Mazzon, E., Dugo, L., Patel, N. S., Serraino, I., Di Paolo, R., Genovese, T., Britti, D., De Maio, M., Caputi, A. P., and Thiemermann, C. (2003). Reduction in the evolution of murine type II collagen-induced arthritis by treatment with rosiglitazone, a ligand of the peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 48, 3544–3556.
Dahlin, M., Hjelte, L., Nilsson, S., and Åmark, P. (2007). Plasma phospholipid fatty acids are influenced by a ketogenic diet enriched with n-3 fatty acids in children with epilepsy. Epilepsy Res. 73, 199–207.
Damiano, M., Galvan, L., Déglon, N., and Brouillet, E. (2010). Mitochondria in Huntington’s disease. Biochim. Biophys. Acta 1802, 52–61.
Davis, S., Butcher, S. P., and Morris, R. G. M. (1992). The NMDA receptor antagonist D-2-amino-5-phosphonopentanoate (D-AP5) impairs spatial learning and LTP in vivo at intracerebral concentrations comparable to those that block LTP in vitro. J. Neurosci. 12, 21–34.
de Mendonca, A., and Ribeiro, J. A. (2000). Long-term potentiation observed upon blockade of adenosine A1 receptors in rat hippocampus is N-methyl-D-aspartate receptor-dependent. Neurosci. Lett. 291, 81–84.
Deary, I. J., Crawford, J. R., Hepburn, D. A., Langan, S. J., Blackmore, L. M., and Frier, B. M. (1993). Severe hypoglycemia and intelligence in adult patients with insulin-treated diabetes. Diabetes 42, 341–344.
Dell, C. A., Likhodii, S. S., Musa, K., Ryan, M. A., Burnham, W. C., and Cunnane, S. C. (2001). Lipid and fatty acid profiles in rats consuming different high-fat ketogenic diets. Lipids 36, 373–378.
Devinsky, O. (1995). Cognitive and behavioral effects of antiepileptic drugs. Epilepsia 36, S46–S65.
DeVivo, D. C., Leckie, M. P., Ferrendelli, J. S., and McDougal, D. B. Jr. (1978). Chronic ketosis and cerebral metabolism. Ann. Neurol. 3, 331–337.
Ding, S., and Lund, P. K. (2011). Role of intestinal inflammation as an early event in obesity and insulin resistance. Curr. Opin. Clin. Nutr. Metab. Care 14, 328–333.
Diradourian, C., Girard, J., and Pegorier, J.-P. (2005). Phosphorylation of PPARs: from molecular characterization to physiological relevance. Biochimie 87, 33–38.
Dong, L., Luo, R., Tong, Y., Cai, X., Mao, M., and Yu, D. (2011). Lack of association between ABCB1 gene polymorphisms and pharmacoresistant epilepsy: an analysis in a western Chinese pediatric population. Brain Res. 1391, 114–124.
Drane, D. L., and Meador, K. J. (2002). Cognitive and behavioral effects of antiepileptic drugs. Epilepsy Behav. 3, S49–S53.
Dressler, A., Reithofer, E., Trimmel-Schwahofer, P., Klebermasz, K., Prayer, D., Kasprian, G., Rami, B., Schober, E., and Feucht, M. (2010). Type 1 diabetes and epilepsy: efficacy and safety of the ketogenic diet. Epilepsia 51, 1086–1089.
Duarte, J. M. N., Oliveira, C. R., Ambrosio, A. F., and Cunha, R. A. (2006). Modification of adenosine A1 and A2A receptor density in the hippocampus of streptozotocin-induced diabetic rats. Neurochem. Int. 48, 144–150.
Dubyak, G. R. (2009). Both sides now: multiple interactions of ATP with pannexin-1 hemichannels. Am. J. Physiol. Cell Physiol. 296, C235–C241.
Dunwiddie, T. V., Diao, L., and Proctor, W. R. (1997). Adenine nucleotides undergo rapid, quantitative conversion to adenosine in the extracellular space in rat hippocampus. J. Neurosci. 17, 7673–7682.
Estráda-Sanchez, A. M., Mejía-Tober, J., and Massieu, L. (2008). Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch. Med. Res. 39, 265–276.
Fedele, D. E., Gouder, N., Guttinger, M., Gabernet, L., Scheurer, L., Rulicke, T., Crestani, F., and Boison, D. (2005). Astrogliosis in epilepsy leads to overexpression of adenosine kinase, resulting in seizure aggravation. Brain 128, 2383–2395.
Fuehrlein, B. S., Rutenberg, M. S., Silver, J. N., Warren, M. W., Theriaque, D. W., Duncan, G. E., Stacpoole, P. W., and Brantly, M. L. (2004). Differential metabolic effects of saturated versus polyunsaturated fats in ketogenic diets. J. Clin. Endocrinol. Metab. 89, 1641–1645.
Fujii, S., Kato, H., Ito, K.-I., Itoh, S., Yamazaki, Y., Sasaki, H., and Kuroda, Y. (2000a). Effects of A1 and A2 adenosine receptor antagonists on the induction and reversal of long-term potentiation in guinea pig hippocampal slices of CA1 neurons. Cell. Mol. Neurobiol. 20, 331–350.
Fujii, S., Kuroda, Y., Ito, K.-I., Yoshioka, M., Kaneko, K., Yamazaki, Y., Sasaki, H., and Kato, H. (2000b). Endogenous adenosine regulates the effects of low-frequency stimulation on the induction of long-term potentiation in CA1 neurons of guinea pig hippocampal slices. Neurosci. Lett. 279, 121–124.
Garbow, J. R., Doherty, J. M., Schugar, R. C., Travers, S., Weber, M. L., Wentz, A. E., Ezenwajiaku, N., Cotter, D. G., Brunt, E. M., and Crawford, P. A. (2011). Hepatic steatosis, inflammation, and ER stress in mice maintained long term on a very low-carbohydrate ketogenic diet. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G956–G967.
Garriga-Canut, M., Schoenike, B., Qazi, R., Bergendahl, K., Daley, T. J., Pfender, R. M., Morrison, J. F., Ockuly, J., Stafstrom, C., Sutula, T., and Roopra, A. (2006). 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nat. Neurosci. 9, 1382–1387.
Gasior, M., Yankura, J., Hartman, A. L., French, A., and Rogawski, M. A. (2010). Anticonvulsant and proconvulsant actions of 2-deoxy-D-glucose. Epilepsia 51, 1385–1394.
Goosens, K. A., and Maren, S. (2002). Long-term potentiation as a substrate for memory: evidence from studies of amygdaloid plasticity and Pavlovian fear conditioning. Hippocampus 12, 592–599.
Gumbiner, B., Wendel, J. A., and McDermott, M. P. (1996). Effects of diet composition and ketosis on glycemia during very-low-energy-diet therapy in obese patients with non-insulin-dependent diabetes mellitus. Am. J. Clin. Nutr. 63, 110–115.
Gwak, Y. S., Tan, H. Y., Nam, T. S., Paik, K. S., Hulsebosch, C. E., and Leem, J. W. (2006). Activation of spinal GABA receptors attenuates chronic central neuropathic pain after spinal cord injury. J. Neurotrauma 23, 1111–1124.
Haces, M. L., Hernandez-Fonseca, K., Medina-Campos, O. N., Montiel, T., Pedraza-Chaverri, J., and Massieu, L. (2008). Antioxidant capacity contributes to protection of ketone bodies against oxidative damage induced during hypoglycemic conditions. Exp. Neurol. 211, 85–96.
Hallböök, T., Lundgren, J., and Rosén, I. (2007). Ketogenic diet improves sleep quality in children with therapy-resistant epilepsy. Epilepsia 48, 59–65.
Halyburton, A. K., Brinkworth, G. D., Wilson, C. J., Noakes, M., Buckley, J. D., Keogh, J. B., and Clifton, P. M. (2007). Low- and high-carbohydrate weight-loss diets have similar effects on mood but not cognitive performance. Am. J. Clin. Nutr. 86, 580–587.
Hori, A., Tandon, P., Holmes, G. L., and Stafstrom, C. E. (1997). Ketogenic diet: effects on expression of kindled seizures and behavior in adult rats. Epilepsia 38, 750–758.
Hu, Y., and Wilson, G. S. (1997). Rapid changes in local extracellular rat brain glucose observed with an in vivo glucose sensor. J. Neurochem. 68, 1745–1752.
Hu, Z.-G., Wang, H.-D., Qiao, L., Yan, W., Tan, Q.-F., and Yin, H.-X. (2009a). The protective effect of the ketogenic diet on traumatic brain injury-induced cell death in juvenile rats. Brain Inj. 23, 459–465.
Hu, Z. G., Wang, H. D., Jin, W., and Yin, H. X. (2009b). Ketogenic diet reduces cytochrome c release and cellular apoptosis following traumatic brain injury in juvenile rats. Ann. Clin. Lab. Sci. 39, 76–83.
Huang, C.-C., Liang, Y.-C., and Hsu, K.-S. (1999). A role for extracellular adenosine in the time-dependent reversal of long-term potentiation by low-frequency stimulation at hippocampal CA1 synapses. J. Neurosci. 19, 9728–9738.
Iwabuchi, S., and Kawahara, K. (2011). Functional significance of the negative-feedback regulation of ATP release via pannexin-1 hemichannels under ischemic stress in astrocytes. Neurochem. Int. 58, 376–384.
Jeong, E. A., Jeon, B. T., Shin, H. J., Kim, N., Lee, D. H., Kim, H. J., Kang, S. S., Cho, G. J., Choi, W. S., and Roh, G. S. (2011). Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 232, 195–202.
Johannessen Landmark, C. (2008). Antiepileptic drugs in non-epileptic disorders: relations between mechanisms of action and clinical efficacy. CNS Drugs 22, 27–47.
Juge, N., Gray, J. A., Omote, H., Miyaji, T., Inoue, T., Hara, C., Uneyama, H., Edwards, R. H., Nicoll, R. A., and Moriyama, Y. (2010). Metabolic control of vesicular glutamate transport and release. Neuron 68, 99–112.
Kang, H.-C., Lee, H. S., You, S. J., Kang, D. C., Ko, T.-S., and Kim, H. D. (2007). Use of a modified Atkins diet in intractable childhood epilepsy. Epilepsia 48, 182–186.
Karlsten, R., Gordh, T., and Post, C. (1992). Local antinociceptive and hyperalgesic effects in the formalin test after peripheral administration of adenosine analogues in mice. Pharmacol. Toxicol. 70, 434–438.
Kashiwaya, Y., Takeshima, T., Mori, N., Nakashima, K., Clarke, K., and Veech, R. L. (2000). D-β-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 97, 5440–5444.
Kawamura, M. Jr., Ruskin, D. N., and Masino, S. A. (2010). Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors and KATP channels. J. Neurosci. 30, 3886–3895.
Kim, D. Y., Davis, L. M., Sullivan, P. G., Maalouf, M., Simeone, T. A., Van Brederode, J., and Rho, J. M. (2007). Ketone bodies are protective against oxidative stress in neocortical neurons. J. Neurochem. 101, 1316–1326.
Kim, D. Y., Vallejo, J., and Rho, J. M. (2010). Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J. Neurochem. 114, 130–141.
Kinsman, S. L., Vining, E. P., Quaskey, S. A., Mellits, D., and Freeman, J. M. (1992). Efficacy of the ketogenic diet for intractable seizure disorders: review of 58 cases. Epilepsia 33, 1132–1136.
Kirchner, A., Veliskova, J., and Velisek, L. (2006). Differential effects of low glucose concentrations on seizures and epileptiform activity in vivo and in vitro. Eur. J. Neurosci. 23, 1512–1522.
Klement, R. J., and Kammerer, U. (2011). Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr. Metab. 8, 75.
Koranda, J. L., Ruskin, D. N., Masino, S. A., and Blaise, J. H. (2011). A ketogenic diet reduces long-term potentiation in the dentate gyrus of freely-behaving rats. J. Neurophysiol. 106, 662–666.
Kossoff, E. H., Laux, L. C., Blackford, R., Morrison, P. F., Pyzik, P. L., Hamdy, R. M., Turner, Z., and Nordli, D. R. Jr. (2008a). When do seizures usually improve with the ketogenic diet? Epilepsia 49, 329–333.
Kossoff, E. H., Rowley, H., Sinha, S. R., and Vining, E. P. G. (2008b). A prospective study of the modified Atkins diet for intractable epilepsy in adults. Epilepsia 49, 316–319.
Langan, S. J., Deary, I. J., Hepburn, D. A., and Frier, B. M. (1991). Cumulative cognitive impairment following recurrent severe hypoglycaemia in adult patients with insulin-treated diabetes mellitus. Diabetologia 34, 337–344.
Lange, K. W., Sahakian, B. J., Quinn, N. P., Marsden, C. D., and Robbins, T. W. (1995). Comparison of executive and visuospatial memory function in Huntington’s disease and dementia of Alzheimer type matched for degree of dementia. J. Neurol. Neurosurg. Psychiatr. 58, 598–606.
Lanska, D. J., Lanska, M. J., Lavine, L., and Schoenberg, B. S. (1988). Conditions associated with Huntington’s disease at death: a case-control study. Arch. Neurol. 45, 878–880.
Laugerette, F., Vors, C., Peretti, N., and Michalski, M.-C. (2011). Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie 93, 39–45.
Lawrence, A. D., Sahakian, B. J., Hodges, J. R., Rosser, A. E., Lange, K. W., and Robbins, T. W. (1996). Executive and mnemonic functions in early Huntington’s disease. Brain 119, 1633–1645.
Lefevre, F., and Aronson, N. (2000). Ketogenic diet for the treatment of refractory epilepsy in children: a systematic review of efficacy. Pediatrics 105, e46.
Linard, B., Ferrandon, A., Koning, E., Nehlig, A., and Raffo, E. (2010). Ketogenic diet exhibits neuroprotective effects in hippocampus but fails to prevent epileptogenesis in the lithium-pilocarpine model of mesial temporal lobe epilepsy in adult rats. Epilepsia 51, 1829–1836.
Liu, Y.-M. C., Williams, S., Basualdo-Hammond, C., Stephens, D., and Curtis, R. (2003). A prospective study: growth and nutritional status of children treated with the ketogenic diet. J. Am. Diet. Assoc. 103, 707–712.
Loring, D. W. (2005). Cognitive side effects of antiepileptic drugs in children. Psychiatr. Times 22, 41.
Loring, D. W., and Kimford, J. (2001). Cognitive and behavioral effects of epilepsy treatment. Epilepsia 42, 24–32.
LoVerme, J., Fu, J., Astarita, G., La Rana, G., Russo, R., Calignano, A., and Piomelli, D. (2005). The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 67, 15–19.
LoVerme, J., Russo, R., La Rana, G., Fu, J., Farthing, J., Mattace-Raso, G., Meli, R., Hohmann, A., Calignano, A., and Piomelli, D. (2006). Rapid broad-spectrum analgesia through activation of peroxisome-proliferator activated receptor-α. J. Pharmacol. Exp. Ther. 319, 1051–1061.
Lund, T. M., Risa, Ø., Sonnewald, U., Schousboe, A., and Waagepetersen, H. S. (2009). Availability of neurotransmitter glutamate is diminished when β-hydroxybutyrate replaces glucose in cultured neurons. J. Neurochem. 110, 80–91.
Ma, W., Berg, J., and Yellen, G. (2007). Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J. Neurosci. 27, 3618–3625.
Maalouf, M., and Rho, J. M. (2008). Oxidative impairment of hippocampal long-term potentiation involves activation of protein phosphatase 2A and is prevented by ketone bodies. J. Neurosci. Res. 86, 3322–3330.
Maalouf, M., Sullivan, P. G., Davis, L., Kim, D. Y., and Rho, J. M. (2007). Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 145, 256–264.
Mahon, S., Deniau, J.-M., and Charpier, S. (2004). Corticostriatal plasticity: life after the depression. Trends Neurosci. 27, 460–467.
Malan, T. P. Jr., Mata, H. P., and Porreca, F. (2002). Spinal GABAA and GABAB receptor pharmacology in a rat model of neuropathic pain. Anesthesiology 96, 1161–1167.
Malmberg, A. B., and Yaksh, T. L. (1993). Pharmacology of the spinal action of ketorolac, morphine, ST-91, U50488H, and L-PIA on the formalin test and an isobolographic analysis of the NSAID interaction. Anesthesiology 79, 270–281.
Mantis, J. G., Fritz, C. L., Marsh, J., Heinrichs, S. C., and Seyfried, T. N. (2009). Improvement in motor and exploratory behavior in Rett syndrome mice with restricted ketogenic and standard diets. Epilepsy Behav. 15, 133–141.
Masino, S. A., and Geiger, J. D. (2008). Are purines mediators of the anticonvulsant/neuroprotective effects of ketogenic diets? Trends Neurosci. 31, 273–278.
Masino, S. A., Gockel, J. A., Wasser, C. D., Pomeroy, L. T., Wagener, J. F., Gawryluk, J. W., and Geiger, J. D. (2007). The relationship among ATP, adenosine and a ketogenic diet. Soc. Neurosci. Abstr. 595, 512.
Masino, S. A., Li, T., Theofilas, P., Ruskin, D. N., Fredholm, B. B., Geiger, J. D., Aronica, E., and Boison, D. (2011). A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J. Clin. Invest. 121, 2679–2683.
Massieu, L., Haces, M. L., Montiel, T., and Hernández-Fonseca, K. (2003). Acetoacetate protects hippocampal neurons against glutamate-mediated neuronal damage during glycolysis inhibition. Neuroscience 120, 365–378.
McClernon, F. J., Yancy, W. S. Jr., Eberstein, J. A., Atkins, R. C., and Westman, E. C. (2007). The effects of a low-carbohydrate ketogenic diet and a low-fat diet on mood, hunger, and other self-reported symptoms. Obesity (Silver Spring). 15, 182–187.
Michalik, L., Auwerx, J., Berger, J. P., Chatterjee, V. K., Glass, C. K., Gonzalez, F. J., Grimaldi, P. A., Kadowaki, T., Lazar, M. A., O’Rahilly, S., Palmer, C. N. A., Plutzky, J., Reddy, J. K., Spiegelman, B. M., Staels, B., and Wahli, W. (2006). International Union of Pharmacology. LXI. Perixosome proliferator-activated receptors. Pharmacol. Rev. 58, 726–741.
Minor, T. R., Rowe, M. K., Soames-Job, R. F., and Ferguson, E. C. (2001). Escape deficits induced by inescapable shock and metabolic stress are reversed by adenosine receptor antagonists. Behav. Brain Res. 120, 203–212.
Mitchell, J. B., Miller, K., and Dunwiddie, T. V. (1993). Adenosine-induced suppression of synaptic responses and the initiation and expression of long-term potentiation in the CA1 region of the hippocampus. Hippocampus 3, 77–86.
Mohamed, H. E., El-Swefy, S. E., Rashed, L. A., and El-Latif, S. K. A. (2010). Biochemical effect of a ketogenic diet on the brains of obese adult rats. J. Clin. Neurosci. 17, 899–904.
Mosek, A., Natour, H., Neufeld, M. Y., Shiff, Y., and Vaisman, N. (2009). Ketogenic diet treatment in adults with refractory epilepsy: a prospective pilot study. Seizure 18, 30–33.
Moya-Camarena, S. Y., Vanden Heuvel, J. P., Blanchard, S. G., Leesnitzer, L. A., and Belury, M. A. (1999). Conjugated linoleic acid is a potent naturally occurring ligand and activator of PPARα. J. Lipid Res. 40, 1426–1433.
Muller-Schwarze, A. B., Tandon, P., Liu, Z., Lang, Y., Holmes, G. L., and Stafstrom, C. E. (1999). Ketogenic diet reduces spontaneous seizures and mossy fiber sprouting in the kainic acid model. Neuroreport 10, 1517–1522.
Murphy, P., and Burnham, W. M. (2006). The ketogenic diet causes a reversible decrease in activity level in Long-Evans rats. Exp. Neurol. 201, 84–89.
Murphy, P., Likhodii, S. S., Hatamian, M., and Burnham, W. M. (2005). Effect of the ketogenic diet on the activity level of Wistar rats. Pediatr. Res. 57, 353–357.
Myers, R. H., Sax, D. S., Koroshetz, W. J., Mastromauro, C., Cupples, L. A., Kiely, D. K., Pettengill, F. K., and Bird, E. D. (1991). Factors associated with slow progression in Huntington’s disease. Arch. Neurol. 48, 800–804.
Nakazawa, M., Kodama, S., and Matsuo, T. (1983). Effects of ketogenic diet on electroconvulsive threshold and brain contents of adenosine nucleotides. Brain Dev. 5, 375–380.
Neal, E. G., Chaffe, H., Schwartz, R. H., Lawson, M. S., Edwards, N., Fitzsimmons, G., Whitney, A., and Cross, J. H. (2008a). The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol. 7, 500–506.
Neal, E. G., Chaffe, H. M., Edwards, N., Lawson, M. S., Schwartz, R. H., and Cross, J. H. (2008b). Growth of children on classical and medium-chain triglyceride ketogenic diets. Pediatrics 122, e334–e340.
Noh, H. S., Hah, Y.-S., Nilufar, R., Han, J., Bong, J.-H., Kang, S. S., Cho, G. J., and Choi, W. S. (2006a). Acetoacetate protects neuronal cells from oxidative glutamate toxicity. J. Neurosci. Res. 83, 702–709.
Noh, H. S., Kim, Y. S., Kim, Y. H., Han, J. Y., Park, C. H., Kang, A. K., Shin, H. S., Kang, S. S., Cho, G. J., and Choi, W. S. (2006b). Ketogenic diet protects the hippocampus from kainic acid toxicity by inhibiting the dissociation of Bad from 14-3-3. J. Neurosci. Res. 84, 1829–1836.
Noh, H. S., Kang, S. S., Kim, D. W., Kim, Y. H., Park, C. H., Han, J. Y., Cho, G. J., and Choi, W. S. (2005). Ketogenic diet increases calbindin-D28k in the hippocampi of male ICR mice with kainic acid seizures. Epilepsy Res. 65, 153–159.
Noh, H. S., Kim, Y. S., Lee, H. P., Chung, K. M., Kim, D. W., Kang, S. S., Cho, G. J., and Choi, W. S. (2003). The protective effect of a ketogenic diet on kainic acid-induced hippocampal cell death in the male ICR mice. Epilepsy Res. 53, 119–128.
Noh, H. S., Lee, H. P., Kim, D. W., Kang, S. S., Cho, G. J., Rho, J. M., and Choi, W. S. (2004). A cDNA microarray analysis of gene expression profiles in rat hippocampus following a ketogenic diet. Brain Res. Mol. Brain Res. 129, 80–87.
Nylen, K., Velazquez, J. L. P., Likhodii, S. S., Cortez, M. A., Shen, L., Leshchenko, Y., Adeli, K., Gibson, K. M., Burnham, W. M., and Snead, O. C. III. (2008). A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp. Neurol. 210, 449–457.
Nylen, K., Velazquez, J. L. P., Sayed, V., Gibson, K. M., Burnham, W. M., and Snead, O. C. III. (2009). The effects of a ketogenic diet on ATP concentrations and the number of hippocampal mitochondria in Aldh5a1-/- mice. Biochim. Biophys. Acta 1790, 208–212.
Oishi, K., Sakamoto, K., Konishi, M., Murata, Y., Itoh, N., and Sei, H. (2010). FGF21 is dispensable for hypothermia induced by fasting in mice. Neuro Endocrinol. Lett. 31, 101–105.
Omote, H., Miyaji, T., Juge, N., and Moriyama, Y. (2011). Vesicular neurotransmitter transporter: bioenergetics and regulation of glutamate transport. Biochemistry 50, 5558–5565.
Pan, J. W., Bebin, E. M., Chu, W. J., and Hetherington, H. P. (1999). Ketosis and epilepsy: 31P spectroscopic imaging at 4.1 T. Epilepsia 40, 703–707.
Pan, Y., Larson, B., Araujo, J. A., Lau, W., De Rivera, C., Santana, R., Gore, A., and Milgram, N. W. (2010). Dietary supplementation with medium-chain TAG has long-lasting cognition-enhancing effects in aged dogs. Br. J. Nutr. 103, 1746–1754.
Park, S., Kim, D. S., Kang, S., and Daily, J. W. III. (2011). A ketogenic diet impairs energy and glucose homeostasis by the attenuation of hypothalamic leptin signaling and hepatic insulin signaling in a rat model of non-obese type 2 diabetes. Exp. Biol. Med. 236, 194–204.
Pascual, O., Casper, K. B., Kubera, C., Zhang, J., Revilla-Sanchez, R., Sul, J.-Y., Takano, H., Moss, S. J., McCarthy, K., and Haydon, P. G. (2005). Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116.
Pérez-Guisado, J., and Muñoz-Serrano, A. (2011). The effect of the Spanish Ketogenic Mediterranean Diet on nonalcoholic fatty liver disease: a pilot study. J. Med. Food 14, 677–680.
Peterson, S. J., Tangney, C. C., Pimentel-Zablah, E. M., Hjelmgren, B., Booth, G., and Berry-Kravis, E. (2005). Changes in growth and seizure reduction in children on the ketogenic diet as a treatment for intractable epilepsy. J. Am. Diet. Assoc. 105, 718–725.
Picot, M. C., Baldy-Moulinier, M., Daurès, J. P., Dujols, P., and Crespel, A. (2008). The prevalence of epilepsy and pharmacoresistant epilepsy in adults: a population-based study in a Western European country. Epilepsia 49, 1230–1238.
Poplawski, M. M., Mastaitis, J. W., Isoda, F., Grosjean, F., Zheng, F., and Mobbs, C. V. (2011). Reversal of diabetic nephropathy by a ketogenic diet. PLoS ONE 6, e18604. doi:10.1371/journal.pone.0018604
Potter, W. B., O’Riordan, K. J., Barnett, D., Osting, S. M. K., Wagoner, M., Burger, C., and Roopra, A. (2010). Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS ONE 5, e8996. doi:10.1371/journal.pone.0008996
Prins, M. L., Fujima, L. S., and Hovda, D. A. (2005). Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J. Neurosci. Res. 82, 413–420.
Prins, M. L., and Hovda, D. A. (2009). The effects of age and ketogenic diet on local cerebral metabolic rates of glucose after controlled cortical impact injury in rats. J. Neurotrauma 26, 1083–1093.
Pulsifer, M. B., Gordon, J. M., Brandt, J., Vining, E. P. G., and Freeman, J. M. (2001). Effects of ketogenic diet on development and behavior: preliminary report of a prospective study. Dev. Med. Child Neurol. 43, 301–306.
Rex, C. S., Kramar, E. A., Colgin, L. L., Lin, B., Gall, C. M., and Lynch, G. (2005). Long-term potentiation is impaired in middle-aged rats: regional specificity and reversal by adenosine receptor antagonists. J. Neurosci. 25, 5956–5966.
Ruskin, D. N., Kawamura, M. Jr., and Masino, S. A. (2009). Reduced pain and inflammation in juvenile and adult rats fed a ketogenic diet. PLoS ONE 4, e8349. doi:10.1371/journal.pone.0008349
Ruskin, D. N., Ross, J. L., Kawamura, M. Jr., Ruiz, T. L., Geiger, J. D., and Masino, S. A. (2011a). A ketogenic diet delays weight loss and does not impair working memory or motor function in the R6/2 1J mouse model of Huntington’s disease. Physiol. Behav. 103, 501–507.
Ruskin, D. N., Suter, T. A. C. S., and Masino, S. A. (2011b). Dissociation of hypoalgesia, ketosis, and hypoglycemia with two ketogenic diets in the rat. Soc. Neurosci. Abstr. 383.04.
Samala, R., Klein, J., and Borges, K. (2011). The ketogenic diet changes metabolite levels in hippocampal extracellular fluid. Neurochem. Int. 58, 5–8.
Samoilova, M., Weisspapir, M., Abdelmalik, P., Velumian, A. A., and Carlen, P. L. (2010). Chronic in vitro ketosis is neuroprotective but not anticonvulsant. J. Neurochem. 113, 826–835.
Sanberg, P. R., Fibiger, H. C., and Mark, R. F. (1981). Body weight and dietary factors in Huntington’s disease patients compared with matched controls. Med. J. Aust. 1, 407–409.
Schock, S. C., LeBlanc, D., Hakim, A. M., and Thompson, C. S. (2008). ATP release by way of connexin 36 hemichannels mediates ischemic tolerance in vitro. Biochem. Biophys. Res. Commun. 368, 138–144.
Schwartzkroin, P. A., Wenzel, H. J., Lyeth, B. G., Poon, C. C., DeLance, A., Van, K. C., Campos, L., and Nguyen, D. V. (2010). Does ketogenic diet alter seizure sensitivity and cell loss following fluid percussion injury? Epilepsy Res. 82, 74–84.
Seyfried, T. N., Marsh, J., Shelton, L. M., Huysentruyt, L. C., and Mukherjee, P. (2012). Is the restricted ketogenic diet a viable alternative to the standard of care for managing malignant brain cancer? Epilepsy Res. PMID:21885251. [Epub ahead of print].
Seyfried, T. N., and Mukherjee, P. (2005). Targeting energy metabolism in brain cancer: review and hypothesis. Nutr. Metab. 2, 30.
Sharma, S., Gulati, S., Kalra, V., Agarwala, A., and Kabra, M. (2009). Seizure control and biochemical profile on the ketogenic diet in young children with refractory epilepsy – Indian experience. Seizure 18, 446–449.
Shram, N. F., Netchiporouk, L. I., Martelet, C., Jaffrezic-Renault, N., and Cespuglio, R. (1997). Brain glucose: voltammetric determination in normal and hyperglycaemic rats using a glucose microsensor. Neuroreport 8, 1109–1112.
Silva, M. C., Rocha, J., Pires, C. S., Ribeiro, L. C., Brolese, G., Leite, M. C., Almeida, L. M. V., Tramontina, F., Ziegler, D. R., and Gonçalves, C. A. (2005). Transitory gliosis in the CA3 hippocampal region in rats fed on a ketogenic diet. Nutr. Neurosci. 8, 259–264.
Sirven, J., Whedon, B., Caplan, D., Liporace, J., Glosser, D., O’Dwyer, J., and Sperling, M. R. (1999). The ketogenic diet for intractable epilepsy in adults: preliminary results. Epilepsia 40, 1721–1726.
Snell-Bergeon, J. K., Chartier-Logan, C., Maahs, D. M., Ogden, L. G., Hokanson, J. E., Kinney, G. L., Eckel, R. H., Ehrlich, J., and Rewers, M. (2009). Adults with type 1 diabetes eat a high fat, atherogenic diet which is associated with coronary artery calcium. Diabetologia 52, 801–809.
Stafstrom, C. E., Ockuly, J. C., Murphree, L., Valley, M. T., Roopra, A., and Sutula, T. P. (2009). Anticonvulsant and antiepileptic actions of 2-deoxy-d-glucose in epilepsy models. Ann. Neurol. 65, 435–447.
Stafstrom, C. E., Wang, C., and Jensen, F. E. (1999). Electrophysiological observations in hippocampal slices from rats treated with the ketogenic diet. Dev. Neurosci. 21, 393–399.
Stout, C. E., Costantin, J. L., Naus, C. C., and Charles, A. C. (2002). Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 277, 10482–10488.
Streijger, F., Plunet, W. T., Lee, H. T., Liu, J., Lam, C. K., Park, S., Hilton, B. J., Fransen, B. L., Matheson, K. A., Assinck, P., Kwon, B. K., and Tetzlaff, W. (2011). Ketogenic diet improves forelimb motor function after spinal cord injury. Soc. Neurosci. Abstr. 255.09.
Studzinski, C. M., Mackay, W. A., Beckett, T. L., Henderson, S. T., Murphy, M. P., Sullivan, P. G., and Burnham, W. M. (2008). Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-β precursor protein (APP) levels in the aged dog. Brain Res. 1226, 209–217.
Su, S. W., Sogawa, M. R. C. Y., Silveira, D. C., Holmes, G. L., and Stafstrom, C. E. (2000). Timing of ketogenic diet initiation in an experimental epilepsy model. Brain Res. Dev. Brain Res. 125, 131–138.
Sullivan, P. G., Rippy, N. A., Dorenbos, K., Concepcion, R. C., Agarwal, A. K., and Rho, J. M. (2004). The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann. Neurol. 55, 576–580.
Swink, T. D., Vining, E. P., and Freeman, J. M. (1997). The ketogenic diet: 1997. Adv. Pediatr. 44, 297–329.
Tabata, K., Matsumoto, K., Murakami, Y., and Watanabe, H. (2001). Ameliorative effects of paeoniflorin, a major constituent of peony root, on adenosine A1 receptor-mediated impairment of passive avoidance performance and long-term potentiation in the hippocampus. Biol. Pharm. Bull. 24, 496–500.
Tai, K.-K., Nguyen, N., Pham, L., and Truong, D. D. (2008). Ketogenic diet prevents cardiac arrest-induced cerebral ischemic neurodegeneration. J. Neural Transm. 115, 1011–1017.
Tai, K.-K., Pham, L., and Truong, D. D. (2009). Intracisternal administration of glibenclamide or 5-hydroxydecanoate does not reverse the neuroprotective effect of ketogenic diet against ischemic brain injury-induced neurodegeneration. Brain Inj. 23, 1081–1088.
Tendler, D., Lin, S., Yancy, W. S. Jr., Mavropoulos, J., Sylvestre, P., Rockey, D. C., and Westman, E. C. (2007). The effect of a low-carbohydrate, ketogenic diet on nonalcoholic fatty liver disease: a pilot study. Dig. Dis. Sci. 52, 589–593.
Thaler, J. P., and Schwartz, M. W. (2010). Minireview: inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology 151, 4109–4115.
Thio, L. L., Rensing, N., Maloney, S., Wozniak, D. F., Xiong, C., and Yamada, K. A. (2010). A ketogenic diet does not impair rat behavior or long-term potentiation. Epilepsia 51, 1619–1623.
Thio, L. L., Wong, M., and Yamada, K. A. (2000). Ketone bodies do not directly alter excitatory or inhibitory hippocampal synaptic transmission. Neurology 54, 325–331.
Thompson, P. J., Baxendale, S. A., Duncan, J. S., and Sander, J. W. (2000). Effects of topiramate on cognitive function. J. Neurol. Neurosurg. Psychiatr. 69, 636–641.
Tieu, K., Perier, C., Caspersen, C., Teismann, P., Wu, D.-C., Yan, S.-D., Niani, A., Vila, M., Jackson-Lewis, V., Ramasamy, R., and Przedborsky, S. (2003). d-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Invest. 112, 892–901.
Todorova, M. T., Tandon, P., Madore, R. A., Stafstrom, C. E., and Seyfried, T. N. (2000). The ketogenic diet inhibits epileptogenesis in EL mice: a genetic model for idiopathic epilepsy. Epilepsia 41, 933–940.
Tromba, C., Salvaggio, A., Racagni, G., and Volterra, A. (1992). Hypoglycemia-activated K+ channels in hippocampal neurons. Neurosci. Lett. 143, 185–189.
Van der Auwera, I., Wera, S., Van Leuven, F., and Henderson, S. T. (2005). A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2, 28.
Veech, R. L. (2004). The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fatty Acids 70, 309–319.
Vining, E. P., Freeman, J. M., Ballaban-Gil, K., Camfield, C. S., Camfield, P. R., Holmes, G. L., Shinnar, S., Shuman, R., Trevathan, E., and Wheless, J. W. (1998). A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 55, 1433–1437.
Westman, E. C., Yancy, W. S. Jr., Mavropoulos, J. C., Marquart, M., and McDuffie, J. R. (2008). The effect of a low-carbohydrate, ketogenic diet versus a low-glycemic index diet on glycemic control in type 2 diabetes mellitus. Nutr. Metab. (Lond.) 5, 36.
Wilder, R. M. (1921). The effects of ketonemia on the course of epilepsy. Mayo Clin. Bull. 2, 307–308.
Wing, R. R., Vazquez, J. A., and Ryan, C. M. (1995). Cognitive effects of ketogenic weight-reducing diets. Int. J. Obes. Relat. Metab. Disord. 19, 811–816.
Xu, K., Sun, X., Eroku, B. O., Tsipis, C. P., Puchowicz, M. A., and LaManna, J. C. (2010). Diet-induced ketosis improves cognitive performance in aged rats. Adv. Exp. Med. Biol. 662, 71–75.
Yamada, K. A., Rensing, N., and Thio, L. L. (2005). Ketogenic diet reduces hypoglycemia-induced neuronal death in young rats. Neurosci. Lett. 385, 210–214.
Yancy, W. S. Jr., Almirall, D., Maciejewski, M. L., Kolotkin, R. L., McDuffie, J. R., and Westman, E. C. (2009). Effects of two weight-loss diets on health-related quality of life. Qual. Life Res. 18, 281–289.
Yancy, W. S. Jr., Foy, M., Chalecki, A. M., Vernon, M. C., and Westman, E. C. (2005). A low-carbohydrate, ketogenic diet to treat type 2 diabetes. Nutr. Metab. 2, 34.
Yang, X., and Cheng, B. (2010). Neuroprotective and anti-inflammatory activities of ketogenic diet on MPTP-induced neurotoxicity. J. Mol. Neurosci. 42, 145–153.
Yudkoff, M., Daikhin, Y., Melø, T. M., Nissim, I., Sonnewald, U., and Nissim, I. (2007). The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu. Rev. Nutr. 27, 415–430.
Zhang, D. S., Ren, L. M., and Zhang, L. (2004). Relation between adenosine A1 receptor and NMDA receptor on synaptic transmission in dentate gyrus of hippocampus. Yao Xue Xue Bao 39, 245–249.
Zhao, Q., Stafstrom, C. E., Fu, D. D., Hu, Y., and Holmes, G. L. (2004). Detrimental effects of the ketogenic diet on cognitive function in rats. Pediatr. Res. 55, 498–506.
Zhao, Y. T., Tekkök, S., and Krnjevic, K. (1997). 2-Deoxy-d-glucose-induced changes in membrane potential, input resistance, and excitatory postsynaptic potentials of CA1 hippocampal neurons. Can. J. Physiol. Pharmacol. 75, 368–374.
Zhao, Z., Lange, D. J., Voustianiouk, A., MacGrogan, D., Ho, L., Suh, J., Humala, N., Thiyagarajan, M., Wang, J., and Pasinetti, G. M. (2006). A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 7, 29. doi:10.1186/1471-2202-7-29
Zhu, P. J., and Krnjevic, K. (1993). Adenosine release is a major cause of failure of synaptic transmission during hypoglycaemia in rat hippocampal slices. Neurosci. Lett. 155, 128–131.
Keywords: adenosine, epilepsy, glucose, inflammation, long-term potentiation, metabolism, pain, seizure
Citation: Ruskin DN and Masino SA (2012) The nervous system and metabolic dysregulation: emerging evidence converges on ketogenic diet therapy. Front. Neurosci. 6:33. doi: 10.3389/fnins.2012.00033
Received: 23 December 2011; Paper pending published: 16 January 2012;
Accepted: 23 February 2012; Published online: 26 March 2012.
Edited by:
Yuri Zilberter, INSERM U751, FranceReviewed by:
Jason B. Wu, Cedars-Sinai Medical Center, USARobert W. Greene, University of Texas Southwestern Medical Center and Veterans Affairs Medical Center, USA
Copyright: © 2012 Ruskin and Masino. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Susan A. Masino, Neuroscience Program, Department of Psychology, Trinity College, Life Sciences Center, 300 Summit Street, Hartford, CT 06106, USA. e-mail:c3VzYW4ubWFzaW5vQHRyaW5jb2xsLmVkdQ==