 Jian Zou
Jian Zou Fulton T. Crews*
Fulton T. Crews*- Bowles Center for Alcohol Studies, The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Regulation of hippocampal neurogenesis is poorly understood, but appears to contribute to mood and cognition. Ethanol and neuroinflammation are known to reduce neurogenesis. We have found that ethanol induces neuroinflammation supporting the hypothesis that ethanol induction of neuroinflammation contributes to ethanol inhibition of neurogenesis. To identify the key proinflammatory molecule that may be responsible for ethanol-impaired neurogenesis we used an ex vivo model of organotypic hippocampal-entorhinal cortex brain slice cultures. Here, we demonstrated a key role of proinflammatory cytokine IL-1β signaling in mediating ethanol inhibition of neurogenesis. Ethanol inhibition of neurogenesis was reversed by neutralizing antibody to IL-1β or blockade of the IL-1β receptor with antagonist IL-1RIa. Ethanol-impaired neurogenesis is associated with strong induction of IL-1β and inflammasome proteins NALP1 and NALP3 in both neurons and astrocytes. Blockade of IL-1β synthesis with inflammasome inhibitors Parthenolide and Bay11708 significantly reversed ethanol inhibited neurogenesis. Furthermore, we also found that IL-1β and inflammasome proteins NALP1 and NALP3 are increased in hippocampal neurons and astrocytes in postmortem alcoholic human brain. Together, these novel findings demonstrate that targeting inflammasome-IL-1β signaling can normalize ethanol-impaired hippocampal neurogenesis, which may have therapeutic implications for treatment of cognitive impairment associated with hippocampal dysfunction in alcoholics.
Introduction
Acute and chronic binge ethanol exposure has been found to inhibit adult hippocampal neurogenesis in rat brain (Crews and Nixon, 2009). Further, chronic ethanol drinking by mice induces depression-like behavior and decreases neurogenesis, with both reversed by antidepressants (Stevenson et al., 2009). Exercise on a running wheel also reverses ethanol induced inhibition of neurogenesis in mice (Crews et al., 2004b). Adult hippocampal neurogenesis significantly contributes to hippocampal neuroplasticity that underlies learning and memory and modulates reward pathway in drug addiction. Adult born new neurons in hippocampus may also be involved in certain aspects of alcohol addiction. It can be hypothesized that the reduced hippocampal neurogenesis may result in a more robust and long-lasting memory of alcohol taking and seeking or decrease extinction learning (Mandyam and Koob, 2012). Therefore, it is clear that studying how to modulate hippocampal neurogenesis impaired by alcohol exposure has beneficial effects on the maintenance of hippocampal neuroplasticity and reducing the vulnerability to alcohol taking and relapse during abstinence.
Neuroinflammation has been implicated in regulation of neurogenesis. Adult hippocampal neurogenesis has also been suggested to be required for certain, but not all, antidepressant-induced behavioral responses in depression (Sahay and Hen, 2007). Recent studies have identified stress-induced proinflammatory cytokine IL-1β as a critical mediator of the loss of neurogenesis and the depression-like behavior caused by stress (Koo and Duman, 2008; Koo et al., 2010). Similar to alcohol, stress activates NF-κB resulting in induction of proinflammatory cytokines including IL-1β (Koo et al., 2010). Blockade of IL-1β signaling with anti-IL-1β neutralizing antibody or IL-1 surface receptor antagonist IL-1RIa abolishes stress-induced inhibition of neurogenesis and depression-like behavior (Koo and Duman, 2008, 2009a). Mice that lack IL-1 receptor are protected against stress reduced neurogenesis and depression-like behavior (Koo and Duman, 2009b). We have found that ethanol increases expression of proinflammatory genes such as TNFα, IL-1β, and MCP-1, through activation of NF-κB, a key transcription factor known to increase expression of proinflammatory genes, in animals, and brain slice cultures (Crews et al., 2006a; Zou and Crews, 2006, 2010). These results suggest induction of neuroinflammatory genes may contribute to ethanol inhibition of neurogenesis. However, the in vivo model limits in dissecting the primary proinflammatory molecules in mediating ethanol effects on hippocampal neurogenesis.
The mechanisms of regulating hippocampal neurogenesis are complex. Likely, adult hippocampal neurogenesis is regulated by multiple intrinsic and extrinsic factors including genetic background, age, sex, neurotransmitters, behavior, physical exercise, stress, hormones, and drugs (Deng et al., 2010; Ming and Song, 2011). Therefore, multiple signals that alter neurogenesis make it difficult to delineate the primary signals that regulate neurogenesis in vivo. Recent studies support a significant role of glial cells in regulating neurogenic niche signaling and neurogenesis (Ming and Song, 2011). These findings and others indicate hippocampal DG has unique cellular elements that regulate adult neurogenesis. To investigate the mechanisms of ethanol inhibition of neurogenesis we used an ex vivo model of organotypic hippocampal-entorhinal cortex (HEC) brain slice cultures that contain major cellular components of the adult neurogenic niche in hippocampal DG and cytoarchitecture of brain (Raineteau et al., 2004; Laskowski et al., 2005; Noraberg et al., 2005). We report here that adult hippocampal neurogenesis in HEC slice model is inhibited by ethanol similar to in vivo studies. Anti-IL-1β neutralizing antibody and IL-1 surface receptor antagonist IL-1RIa blunt ethanol inhibition of neurogenesis. Ethanol-impaired hippocampal neurogenesis is accompanied with strong induction of IL-1β and inflammasome proteins NALP1 and NALP3, a multiple protein complex that cleaves and releases mature IL-1β (Bryant and Fitzgerald, 2009). Blockade of IL-1β synthesis with inflammasome inhibitors as well as other anti-inflammatory drugs reverses ethanol inhibition of neurogenesis. Further, for the first time we reported that IL-1β and inflammasome proteins are strongly increased in postmortem human alcoholic brain. These novel findings suggest ethanol inhibits neurogenesis through induction of inflammasome-IL-1β signaling molecules.
Materials and Methods
Slice Culture
All protocols followed in this study were approved by the Institutional Animal Care Use Committee and were in accordance with National Institute of Health regulation for the care and use of animals in research. The preparation of organotypic HEC slice cultures has been described elsewhere (Zou and Crews, 2010). In the present study, we optimized culture condition by gradually reducing horse serum that kept HEC healthy while maintaining high levels of DCX expression and BrdU incorporation predominantly into hippocampal DG region that mimicked adult neurogenesis as described previously (Raineteau et al., 2004; Namba et al., 2007). Briefly, HEC slices were initially cultured for 4 days in vitro (DIV) with MEM medium containing 25% horse serum (HS), followed by three DIV in medium with 12.5% HS and then in serum-free medium supplemented with N2 until the end of experiment. The cultures after 14 DIV were used for experiments and drug treatments were done during final four to seven DIV in serum-free N2 medium.
5-Bromo-2-deoxyuridine Labeling and Immunostaining
BrdU labeling was used to follow NPC survival. Intrinsic newly generated cells were labeled by intraperitoneal injection of 5-bromo-2-deoxyuridine (BrdU; Sigma) dissolved in 0.9% NaCl (10 mg/kg bodyweight) at 24 h and 30 min prior to HEC slice preparation. HEC slices after 14 DIV were treated with ethanol for 4 DIV and BrdU+ cells determined. For some experiments, BrdU was applied to culture to label extrinsic proliferating cells in HEC slices, in which HEC slices after 10 DIV were incubated with medium containing BrdU (50 μM) for 24 h and then returned to BrdU-free medium for another 4 DIV or 11 DIV. At the end of experiments, all slices were fixed for BrdU immunohistochemistry to visualize BrdU+ cells. BrdU+ immunoreactivity (BrdU + IR) cells in DG were quantified with computer-assisted BioQuant imaging software (Zou et al., 2012).
Ethanol and Drug Treatments
Ethanol treatment with the indicated concentrations occurred in a dessiccator containing 300 ml water saturated with equal concentrations of ethanol to balance evaporation of ethanol from the media. All drug treatments were performed in serum-free N2 supplemented medium. At the end of experiments, slices are removed for analysis. For immunohistochemistry, slices are fixed with 4% paraformaldehyde + 5% sucrose in 0.1 M PBS for overnight at 4°C and stored in 0.1 M PBS. For mRNA using RT-PCR study, slices are rinsed with cold PBS, followed by total RNA purification using the RNeasy Mini Kit (see below). For protein determination using western blotting, slices are rinsed with cold PBS, and immediately stored at −80°C for later protein analysis (see below).
Immunohistochemistry and DoubleCortin Quantification
Slices were fixed at the end of experiment and removed for processing immunohistochemistry. Floating slices were washed with TBS and followed by incubation with 0.6% H2O2 to quench endogenous peroxidase activity. Slices were then blocked with 3% horse/goat/rabbit serum containing 0.25% Triton X100 for 1 h at room temperature and followed by incubation with primary antibodies for 48 h at 4°C. The detailed information regarding primary antibodies used in the present study is summarized in Table 1.
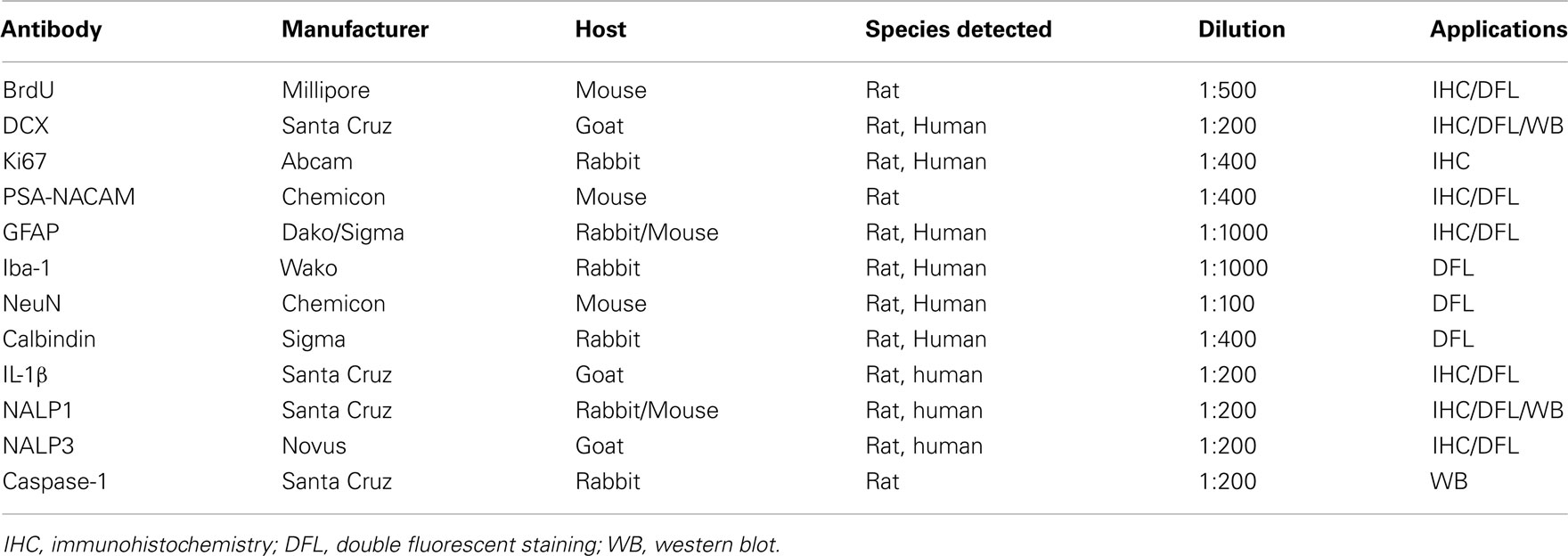
Table 1. Summary of antibodies used in the present study.
Immunoreactivity was detected using an ABC kit and followed by DAB method. DCX is a cytoskeleton marker of neuroprogenitors expressed during maturation of dendrites and other neuronal structures. DCX is expressed over the course of stem cell maturation, but not in mature dentate granule neurons making it a good index of neurogenesis (Couillard-Despres et al., 2005). For quantification of DCX IR, DCX+ cells, and processes in DG region of the hippocampus were imaged live at 8× magnification, background corrected, and DCX IR measured with BioQuant imaging software (Crews et al., 2006a).
Double Immunofluorescent Staining and Confocal Analysis
Free-floating slices were used for double immunofluorescent staining. Primary antibodies used are summarized in Table 1. Specific cellular markers used include anti-neuronal-specific nuclear protein (NeuN) for neurons, Iba-1 for microglia, and GFAP for astrocytes. All primary antibodies were incubated for 48 h at 4°C. Either Alexa Fluor 594 or Alexa Fluor 488 secondary antibodies (1:2000; Molecular Probes, Eugene, OR, USA) were used for immunofluorescent staining and incubated for 1 h at room temperature. For negative control immunofluorescent staining, the primary antibody was omitted to assess non-specific binding of the secondary antibody. The slices were coverslipped with anti-fade mounting medium (pro-long; Molecular Probes). Confocal analysis was performed using a Leica SP2 AOBS Upright Laser Scanning Confocal in Michael Hooker Microscopy Facility, UNC.
Western Blotting
Hippocampal-entorhinal cortex slices were incubated in lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, pH 7.9) plus protease inhibitor cocktail (Sigma) for 15 min and then disrupted with sonication (three time, 2 min apart in ice). After centrifugation of the slice homogenate, the supernatant were collected, and protein determined using a BioRad Bradford reagent kit. For western blotting, an equal amount of protein (100 μg), mixed with an equal volume of 2× RIPA loading buffer, was separated using a 4–15% Tris mini-gel (BioRad) and transferred onto nitrocellulose membranes. After blocking with Licor blocking buffer for 2 h, the membrane was probed with primary antibodies (Table 1) at 4°C overnight. After washing, membrane was incubated with second antibodies coupled with fluorescence (Licor) and then scanned with Odyssey machine. The densitometry of each band was quantified with BioQuant imaging software.
RNA Isolation, Reverse Transcription, and Real Time Quantitative RT-PCR
After treatments, the slices were removed, rinsed with cold PBS and immediately followed by total RNA purification using the RNeasy Mini Kit (Qiagen Inc., CA, USA). For reverse transcription, 2 μg of total RNA was used to synthesize the first strand cDNA using random primers (Invitrogen, CA, USA) and reverse transcriptase Moloney murine leukemia virus (Invitrogen, CA, USA). Total of 1 μl of the first strand cDNA solution was used for RT-PCR. SYBER Green Supermix (BioRad) was used as a RT-PCR solution. The following primers were used: for IL-1β, forward 5′-GAAACAGCAATGGTCGGGAC-3′ and reverse 5′-AAGACACGGGTTCCATGGTG-3′; for NALP1, forward 5′-GGACCAGAATCCTGAGCTGTGT-3′ and reverse 5′-GAAGCCTCAGGAAGGATGGAT-3′; for CREB, forward 5′-CATGGCACGTAATGGAGACTACCGCA-3′ and reverse 5′-CCGCCAGCATGCCTTC-3′; for BDNF, forward 5′-GTGACARTATTAGCGAGTGGG-3′ and reverse 5′-GGGTAGTTCGGCATTGC-3′ and; for β-Actin: forward 5′-CTACAATGAGCTGCGTGTGGC-3′ and reverse 5′-CAGGTCCAGACGCAGGATGGC-3′.
All experiments were run in triplicate. The real time PCR was run with initial activation for 10 min at 95°C and followed by 40–45 cycles of denaturation (95°C, 40 s), annealing (58°C, 45 s), and extension (72°C, 45 s). The threshold cycle (CT) of each target product was determined and normalized to internal standard beta-actin. Difference in CT values (CT) of two genes was calculated [difference = 2 − (CT of target genes − CT of beta-actin) = 2 − CT], and data are expressed the percentage of control.
Human Brain Immunohistochemistry and Quantification
Human postmortem brain tissue was obtained from the New South Wales Tissue Resource Center in Australia (ethics committee approval number: X11-0107). Paraffin sections of hippocampus at thickness of 10 μm were used in this study. The detailed patients’ medical and alcohol consumption history is presented in Table 2.
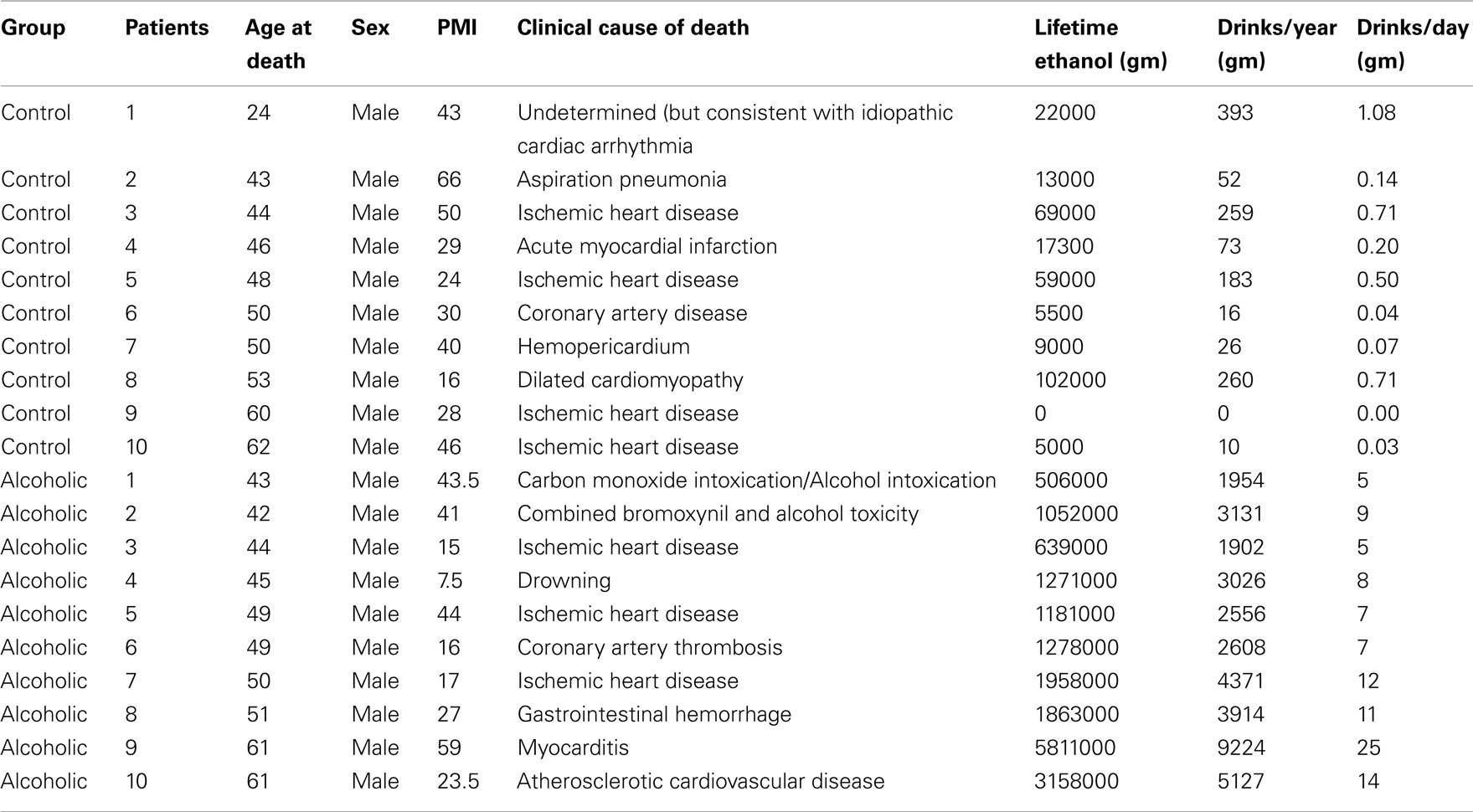
Table 2. Clinical data of patients with controls and alcoholics.
Hippocampal sections from 10 controls and 10 alcoholic patients were stained and histochemistry quantitated. Although the cause of death varied among subjects, 9 of 10 controls, and 6 of 10 alcoholics died from cardiovascular disease, and none of the subjects died from inflammation; it is possible that unknown factors other than ethanol contribute to changes in human hippocampus.
For immunohistochemistry, the sections were deparaffinized, followed by the procedure of antigen retrieval, in which the sections were treated in a microwave oven for 15 min in a citric acid solution (0.1 mol/l citric acid monohydrate and 0.1 mol/l trisodium citrate dihydrate), pH 6.0, and then allowed to cool down at room temperature for 30 min. After washing with PBS trice, the sections were incubated for 25 min in 1% hydrogen peroxide in PBS to quench endogenous peroxidases. Afterward, they were incubated overnight in a humid chamber with primary antibodies (see Table 1). Immunoreactivity was detected using an ABC kit and followed by DAB method. For quantification, positive cells in the hippocampal dentate gyrus and CA fields were imaged at 10× magnification, background corrected, and +IR cells quantitated using BioQuant imaging software.
For double fluorescent immunostaining, the sections after antigen retrieval were incubated with blocking serum for 2 h and then incubated with primary antibodies (see Table 1) overnight at 4°C. After washing, the sections were incubated with second antibodies for 1 h at room temperature and then coverslipped with anti-fade mounting medium (pro-long; Molecular Probes). Confocal analysis was performed using a Leica SP2 AOBS Upright Laser Scanning Confocal in Michael Hooker Microscopy Facility, UNC.
Statistical Analysis
Experimental results were expressed as mean ± SEM from the indicated number of experiments. Statistical comparisons were made with ANOVA and the difference between the experimental groups was further compared by using post hoc Fisher PLSD test. Variations were considered to be statistically significant at a p value of <0.05.
Results
Characterization of Neurogenesis in HEC Brain Slice Cultures
Organotypic hippocampal slice culture is an ex vivo model that contains the neurogenic niche of hippocampal DG and all the cellular elements and circuitry of brain including NPC that proliferate, migrate, and differentiate into mature integrated hippocampal DG granule neurons similar to in vivo NPC (Namba et al., 2007). To characterize NPC differentiation within our HEC model, we pre-labeled intrinsic NPCs by injecting BrdU into P6 neonates prior to slice preparation and then followed NPC differentiation in HEC slice cultures at 14 DIV. The majority of BrdU + IR cells were within hippocampal DG and subgranular regions (Figure 1A), although scattered BrdU+ cells could be found throughout the hippocampus. BrdU+ cell counts were stable from 10 to 21 DIV (data not shown) indicating mimicking maturation and survival of NPC similar to maturation in vivo (Nixon and Crews, 2002). To further compare HEC slice neurogenesis to in vivo neurogenesis, we determined Ki67 immunohistochemistry (Ki67 + IR), a marker of endogenous NPC proliferation in vivo (Nixon and Crews, 2002). As shown in Figure 1B, Ki67 + IR was found in proliferating cells in HEC DG similar to that found in rat hippocampal DG in vivo (Nixon and Crews, 2002). DCX, a neuroprogenitor microtubule-associated protein expressed specifically in maturing neurons, has been characterized as a reliable index of adult hippocampal neurogenesis (Gleeson et al., 1998; Couillard-Despres et al., 2005). In HEC slice DCX + IR was robust and localized to the DG of the slices over 2–4 weeks in vitro (Figure 1C). Double immunostaining and confocal analysis revealed that the majority of pre-labeled BrdU + IR cells develop into neuronal cells expressing DCX (Figure 1D) and NeuN, a marker for mature neurons (Figure 1E), but rarely associated with other cell specific markers for GFAP+ astrocytes (Figure 1F) or oligodendrocytes (data not shown). Approximately 70% of BrdU + IR in DG at 14 DIV co-labeled with NeuN indicating differentiation of NPC in HEC into DG neurons, mimicking the developmental course of adult NPC maturation within the DG similar to that found in vivo (Crews et al., 2006b). Furthermore, other markers of NPC, e.g., PSA-NACAM and calbindin, a marker for mature granule neurons and the newly formed cells that functionally integrated into the hippocampus (van Praag et al., 2002), also co-label DCX+ neurons mimicking NPC in vivo (Figures 1G,H). In vitro-labeled BrdU+ cells were occasionally seen to co-label with DCX under culture conditions used (Figure 1I). These results are consistent with HEC slice culture providing a model for investigating molecular mechanisms of regulation of neurogenesis.
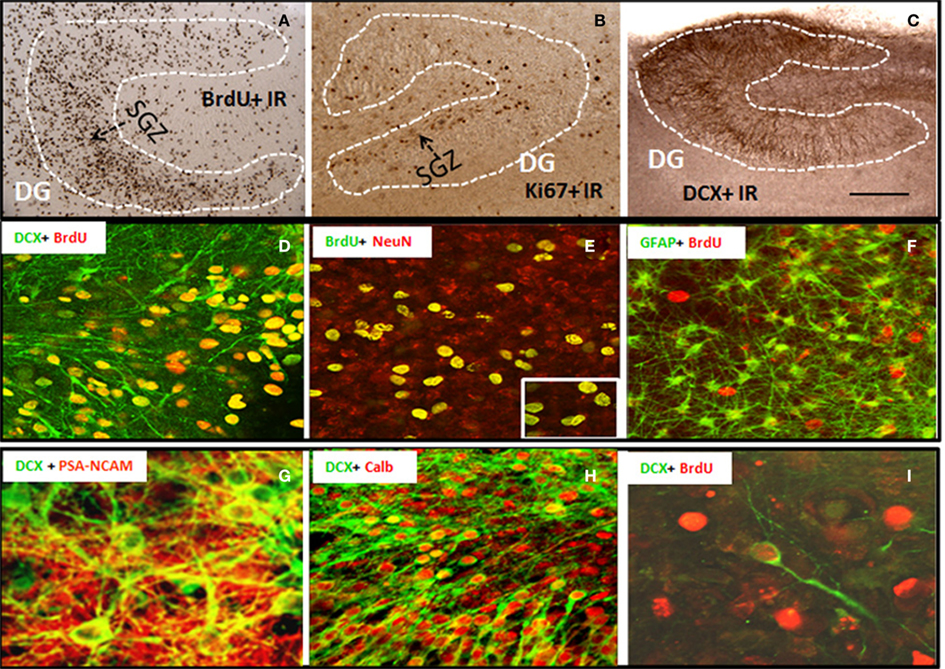
Figure 1. Characterization of neural progenitor cells and neurogenesis in HEC slice culture model. Shown in the top panel are representative microphotographs of BrdU+ IR cells (A) from 14 DIV slice pre-labeled with BrdU (see Materials and Methods); positive cells stained with Ki67 (B), a marker for endogenous progenitors, and DCX+ IR (C) confined in dentate gyrus of hippocampus (scale bar = 500 μm). Shown in the middle panel are (D) a merger image of double immunofluorescent staining with BrdU and DCX (green) indicating the majority of in vivo pre-labeled BrdU+ cells develop into DCX+ immature neurons; (E) a merger image of double immunofluorescent staining with BrdU (green) and NeuN (red) indicating that majority of BrdU+ cell in the subgranular layer of DG co-expressed neuronal marker NeuN at 14 DIV (original magnification 20×, insert-80×); and (F) a merger image of double immunofluorescent staining with BrdU (red) and astrocyte marker GFAP (green), indicating a rare colocalization of BrdU+ cells with GFAP+ astrocytes (original magnification 20×). Shown in the bottom panel are representative confocal images showing (G) DCX+ cells (green) coexpressing PSA-NCAM (red, a marker for young neurons, original magnification 120×); (H) a majority of DCX+ cells (green) coexpressing calbindin (red, a marker for mature neurons, original magnification 20×); and (I) in vitro-labeled BrdU+ cells (red) and DCX+ IR (green) indicating that in vitro-labeled BrdU+ cells occasionally become DCX+ neurons under the culture conditions used (original magnification 120×).
Ethanol Inhibition of NPC Proliferation, Neurogenesis, and Survival in HEC Slices
Ethanol treatment of rats in vivo can reduce neurogenesis (Nixon and Crews, 2002) and in mice after chronic ethanol consumption in parallel with depression-like behavior (Stevenson et al., 2009). To determine if ethanol exposure of HEC slices inhibits neurogenesis, HEC slices were treated with ethanol (100 mM, 4 days) and neurogenesis determined. Ethanol treatment following BrdU labeling in which majority of BrdU + IR cells develop into DCX + IR neurons (see Figure 1) found reduced BrdU + IR by about 30% (Figure 2, top panel). The reduced survival of BrdU + IR cells mimics that found with ethanol treatment of rats (Nixon and Crews, 2002). Endogenous proliferating cells labeled by Ki67 were also profoundly affected by ethanol exposure. As shown in Figure 2 (bottom panel), ethanol exposure significantly decreased the number of Ki67 + IR cells in DG by 80% (Ki67 cells/dentate gyrus: control 156.9 ± 24.2 and Ethanol 30.8 ± 5.3; Fisher post hoc: ***p < 0.001 compared with control, n = 6). To further investigate neurogenesis in HEC we used DCX + IR, a reliable index of neurogenesis (Couillard-Despres et al., 2005). DCX + IR was reduced by ethanol exposure by 50–70% (Figure 3, top panel). DCX protein levels analyzed with western blotting in slice show an ethanol dose dependent decrease similar to the decreased DCX + IR with 100 mM ethanol appearing close to a maximal concentration that inhibits DCX expression by about 70% (Figure 3, bottom panel). These concentrations of ethanol are high modeling binge drinking by chronic alcoholics (Crews et al., 2000; Obernier et al., 2002). These studies are consistent with ethanol reducing NPC survival (BrdU + IR), NPC proliferation (Ki67 + IR), and neurogenesis (DCX + IR) in HEC slices similar to in vivo binge drinking models.
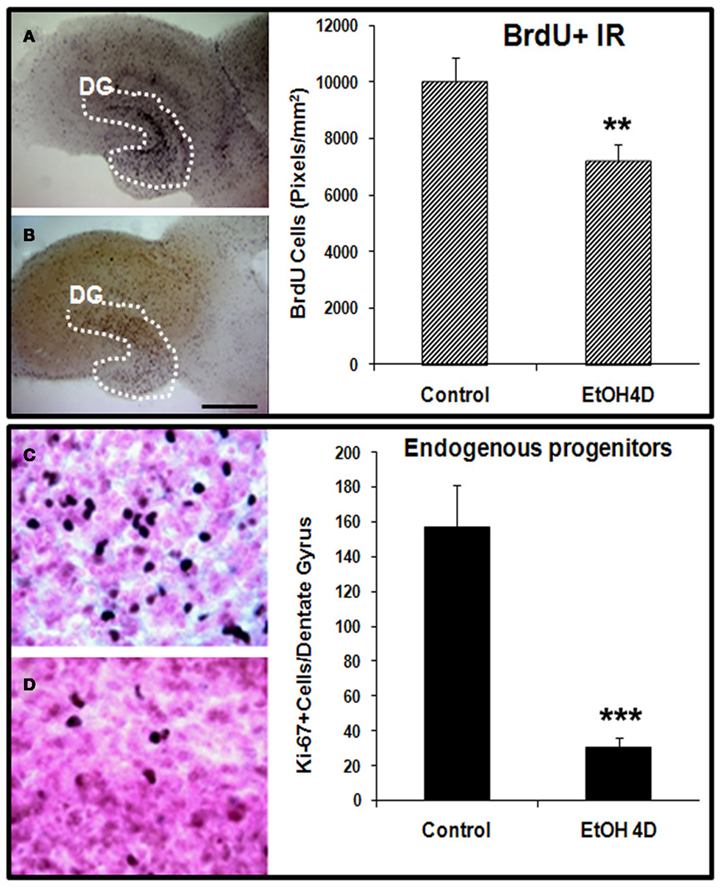
Figure 2. Ethanol inhibition of neural cell proliferation in HEC slices. Top panel: ethanol inhibition of BrdU+ cell proliferation with in vivo prelabeling protocol. Representative photographs of BrdU+ cell IR indicate fewer BrdU+ cells in ethanol treated (100 mM, 4 days) slice (B) compared with control (A) (Scale bar = 500 μm). Shown in bar graph are mean ± SEM of BrdU+ IR measured from DG regions of the control and ethanol treated slices with BioQuant imaging software (**p < 0.01 compared with control, n = 6–8). Bottom panel: ethanol inhibition of proliferation of endogenous progenitors. HEC slices at 10 DIV were treated with ethanol (100 mM) for 96 h and then stained with Ki67, a marker for endogenous progenitors. Ki67 positive cells in DG of the hippocampus were quantified as described in Methods. Shown in bar graph are mean ± SEM of Ki67+ cells counted in each dentate gyrus. Ethanol treatment for 4 days significantly reduced number of endogenous progenitors labeled by Ki67. ANOVA following Fisher post hoc: ***p < 0.001 compared with control, n = 6. The representative photographs of Ki67+ IR were shown in control [(C), 40×] and ethanol group [(D), 40×].
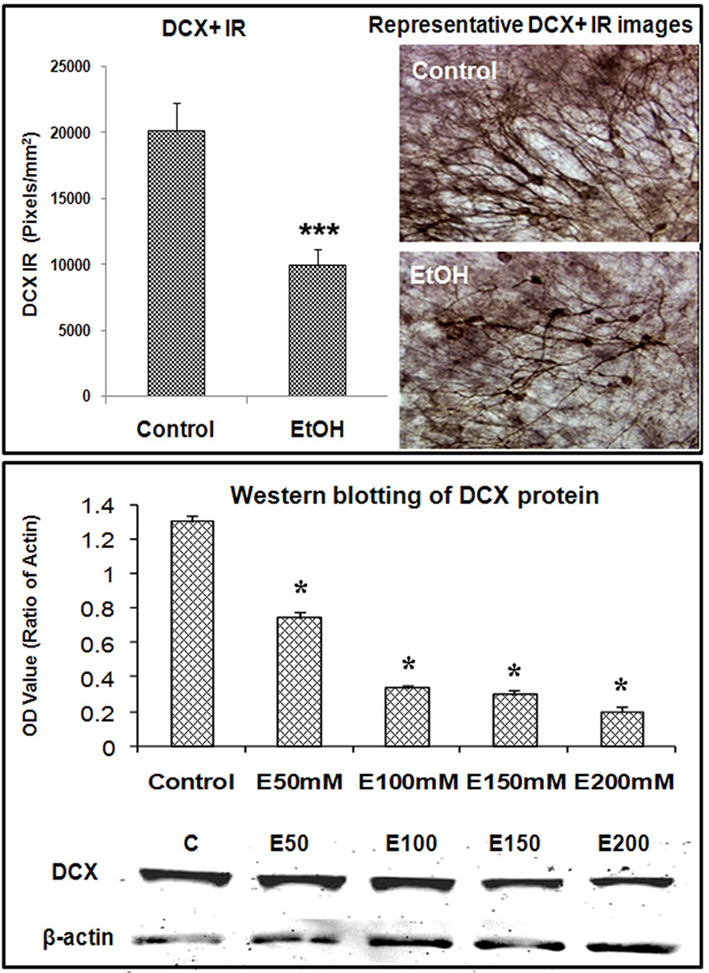
Figure 3. Ethanol inhibition of neurogenesis as indicated by DCX expression in HEC slices. Using DCX as a marker for neurogenesis, we investigated the effects of ethanol exposure on DCX expression in HEC slices. Top panel: shown in bar graph (left) are mean ± SEM of DCX+ IR measured with BioQuant imaging software from both control and ethanol groups. ANOVA following Fisher post hoc: ***p < 0.001 compared with control, n = 6–8. The representative images of DCX IR from control and ethanol treated cultures were shown in the right (original magnification 40×). Bottom panel: western blotting of DCX from control and ethanol treated slices. Shown in bar graph are mean ± SEM of DCX optical density values. ANOVA following Fisher post hoc: *p < 0.05 compared with control, n = 4 blots. The images of western blot were shown in the bottom. The experiments were repeated at least three times with similar designs and results.
Proinflammatory Cytokine IL-1β Mediates Ethanol Inhibition of Neurogenesis
In our previous studies we have found that ethanol treatment of HEC slice cultures induces NF-κB transcription and expression of proinflammatory innate immune genes including TNFα, IL-1β, MCP-1, and iNOS (Zou and Crews, 2010). Since proinflammatory molecules have been found to reduce neurogenesis (Monje et al., 2003; Das and Basu, 2008), we used neutralizing antibodies to different cytokines to gain insight into ethanol inhibition of neurogenesis. Ethanol reduced DCX + IR that was further decreased by exogenous IL-1β. Ethanol decreased DCX + IR was significantly blunted by inclusion of neutralizing antibodies to IL-1β and interleukin 1 surface receptor antagonist (IL-1RIa), but not by neutralizing antibodies to TNFα or MCP-1 (Figure 4). These results suggest a critical role of IL-1β in mediating ethanol inhibition of neurogenesis.
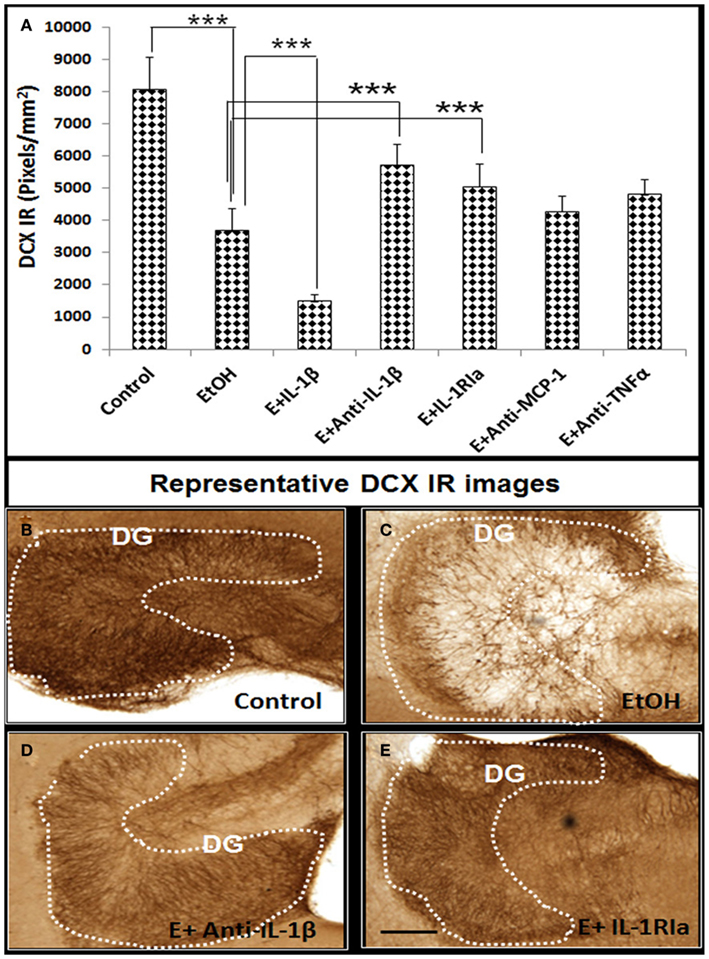
Figure 4. Ethanol-impaired neurogenesis is mediated by IL-1β. HEC slices at 10 DIV were treated with ethanol for 96 h in the presence or absence of IL-1β (50 ng/ml), neutralizing antibodies against IL-1β (E + Anti-IL-1β, 2 μg/ml), TNFα (E + Anti-TNFα, 2 μg/ml), and MCP-1 (E + Anti-MCP-1, 5 μg/ml) or IL-1 surface receptor antagonist (E + IL-1RIa, 2 μg/ml). Bar graph (A) shows mean ± SEM of DCX IR measured from each group. Ethanol-impaired neurogenesis is exacerbated by exogenous IL-1β and reversed by addition of neutralizing anti-IL-1β antibody or blockade of IL-1 surface receptors. ANOVA F(6, 38) = 11.27, p < 0.0001, and Fisher post hoc: ***p < 0.001 compared with corresponding group as indicated, n = 6–8. Representative DCX+ IR photographs from the groups as indicated were shown in (B–E). The experiments were repeated at least four times with similar designs and results (scale bar = 200 μm).
The reversal of ethanol inhibition of neurogenesis by IL-1β antagonists and neutralizing antibodies prompted experiments on IL-1β and induction of IL-1β by ethanol. Previous studies found that ethanol induces IL-1β and other proinflammatory cytokines in HEC, somewhat different in culture media (Zou and Crews, 2010). Under culture conditions used in the present study, in which culture medium was optimized for neurogenesis, ethanol induced IL-1β mRNA about sixfold (Figure 5A). In addition, ethanol reduced expression of CREB and BDNF mRNA in the same experiment, two genes decreased in vivo by ethanol (Crews et al., 2004a). The effects of ethanol treatment on cell type expression of IL-1β protein was further analyzed with double immunofluorescent staining and confocal analysis. IL-1β + IR, likely both mature IL-1β and pro-IL-1β, showed low expression in controls (Figures 5B,D), but markedly increased by ethanol exposure in dentate granule neurons (NeuN + IR) and astrocytes (GFAP + IR; Figures 5C,E respectively). Similar to ethanol, exogenous IL-1β shows a concentration dependent decrease in DCX + IR with maximal reduction of about 80% at 100 ng/ml (∼6 nM) IL-1β (Figure 6). Ethanol plus exogenous IL-1β (50 ng/ml, Figure 4) reduced DCX almost identically to that of IL-1β (50 ng/ml) alone (Figure 6), consistent with ethanol acting through IL-1β. Taken together, these results indicate that IL-1β is a potent and effective inhibitor of neurogenesis and that targeting IL-1β by using IL-1β neutralizing antibodies and receptor antagonists can reverse ethanol inhibition of neurogenesis, supporting the hypothesis that ethanol inhibits neurogenesis through induction of IL-1β.
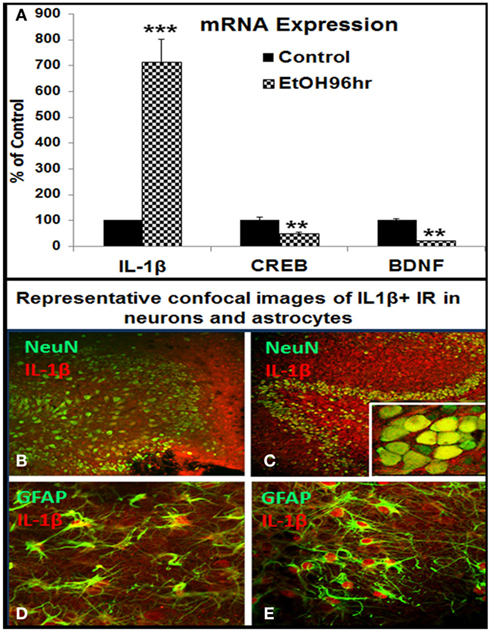
Figure 5. Ethanol induction of proinflammatory cytokine IL-1β in HEC slices. (A) Shown in bar graph are mRNA levels of IL-1β, CREB, and BDNF from a representative experiment. Ethanol treatment (100 mM, 4 days) upregulated IL-1β but downregulated CREB and BDNF mRNA expression. ANOVA following Fisher post hoc: ***p < 0.001 or **p < 0.01 compared with control, n = 3. Cellular expression of IL-1β IR was examined with double fluorescent immunostaining and confocal analysis. (B,C) Representative confocal photographs of double immunofluorescent staining with anti-IL-1β (red) and neuronal marker NeuN (green) show ethanol induction of IL-1β IR expression in neuronal cells of DG region [(B) control; (C) ethanol, original magnification 20×, insert in (B) 160×]; (D,E) representative confocal photographs of double immunofluorescent staining with anti-IL-1β (red) and astrocyte marker GFAP (green) show ethanol induced strong expression of IL-1β IR in astrocytes [(D) control; (E) ethanol, original magnification 40×].

Figure 6. Inhibition of exogenous IL-1β on neurogenesis in HEC slices. To test whether IL-1β may impair neurogenesis in HEC slices, we treated slices at 10 DIV with exogenous IL-1β (10, 50, and 100 ng/ml) for 4 days and then DCX IR was determined. Bar graph (A) indicates IL-1β dose-dependently inhibited neurogenesis as indicated by significant reduction of DCX+ IR. Data represent mean ± SEM. ANOVA F(3, 23) = 7.301, p < 0.0001, and Fisher post hoc: ***p < 0.001 compared with control, n = 6–8. The experiments were repeated with similar designs and results. Representative images of DCX+ IR from control and IL-1β-treated slices were shown in (B–E) (scale bar = 200 μm).
Ethanol Induction of Inflammasome Proteins
The synthesis of IL-1β, a 17.5-kDa protein, occurs through proteolytic processing of a larger precursor protein by a group of proteins that oligomerize into a common structure that include activated caspase-1, NALP proteins, and others that form a multi-protein complex called the “inflammasome” (Chakraborty et al., 2010). Therefore, we investigated the effect of ethanol on inflammasome proteins. Ethanol progressively increased inflammasome NALP1 mRNA during 4–24 h of treatment that stabilized at twofold to threefold higher levels during 24–96 h of ethanol exposure (Figure 7, bar graph). Across multiple experiments, NALP1 mRNA level was increased by 2.5- to 7-fold by ethanol after 96 h. We could not determine NALP3 mRNA due to no published primers for rat, although we did have antibodies for IHC. Protein levels assessed using immunohistochemistry indicated ethanol increased NALP1 and NALP3 expression (Figure 7). Specific cell marker immunostaining revealed that ethanol induced NALP1 in both DG granule neurons (NeuN + IR) and astrocytes (GFAP + IR; Figures 7B,D) and NALP 3 in astrocytes (Figure 7F). Western blot analysis further indicates that protein levels of short form NALP1 and caspase-1 were increased by ethanol treatment (see Figure 8). These findings provide the first evidence that ethanol induces inflammasome proteins in HEC slices that increase IL-1β levels.
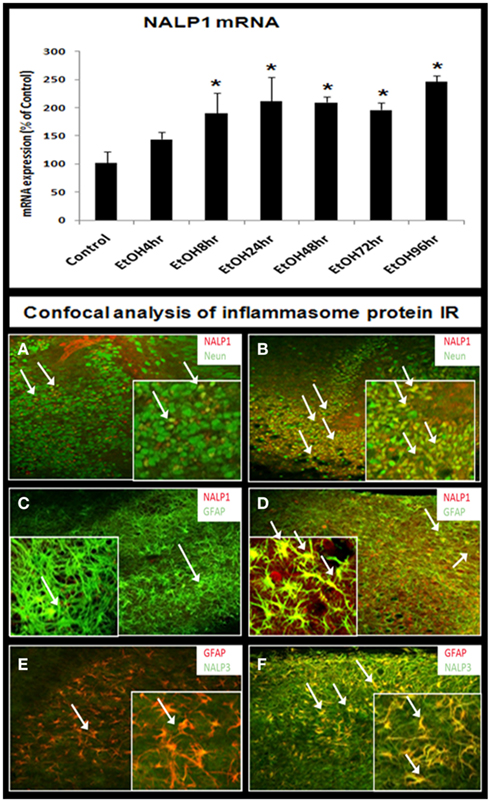
Figure 7. Ethanol induction of inflammasomes in HEC slices. RT-PCR analysis shows that ethanol exposure persistently up-regulates inflammasome NALP1 mRNA (bar graph, top). Representative confocal images are shown for cellular expression of inflammasome NALP1 and NALP3. (A,B) Double fluorescent immunostaining with NALP1 (red) and neuronal cell marker NeuN (green) show ethanol induction of NALP1 in DG granule neurons (arrows) [(A) control; (B) ethanol, original magnification 20×; insert-80×]. (C,D) Double fluorescent immunostaining with NALP1 (red) and astrocyte neuronal marker GFAP (green) show ethanol induced strong induction of NALP1 in astrocytes (yellow, arrows) [(C) control; (D) ethanol, original magnification 20×; insert-80×]. (E,F) Double fluorescent immunostaining with NALP3 (green) and astrocyte marker GFAP (red) show ethanol induction of inflammasome NALP3 in astrocytes (yellow, arrows) [(E) control; (F) ethanol, original magnification 20×; insert-80×].
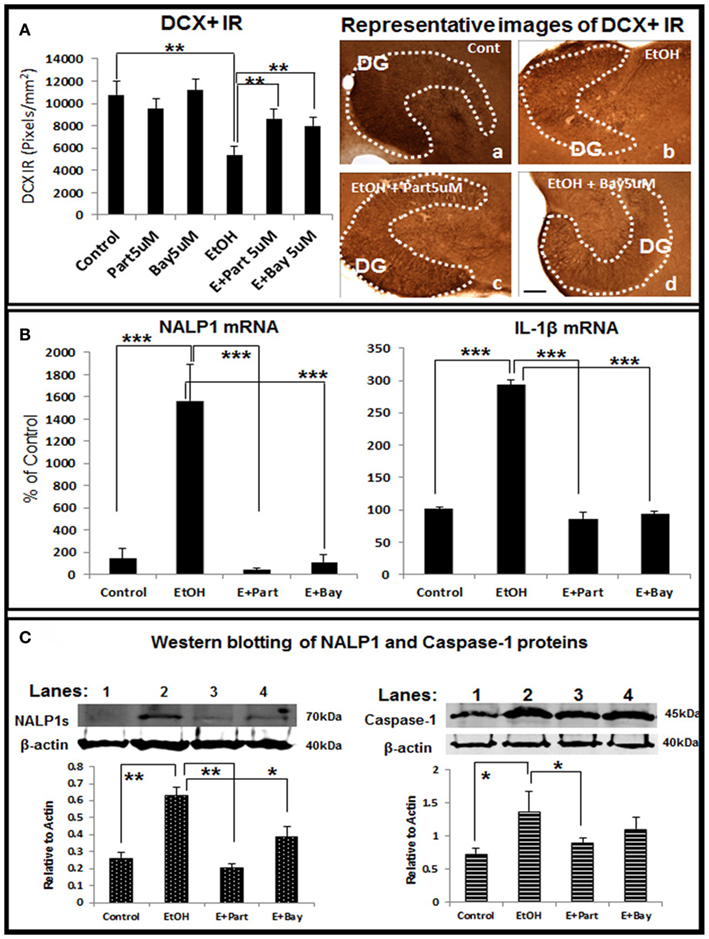
Figure 8. Direct inhibition of the inflammasomes protects ethanol-impaired neurogenesis. HEC slices after 10 DIV were treated with ethanol for 4 days in the absence or presence of anti-inflammatory compounds Parthenolide and Bay 11-7082 (5 μM, 2 h pretreatment), both compounds have been shown to directly inhibit the inflammasome activation (Juliana et al., 2010). The presence of both Parthenolide and Bay 11-7082 during ethanol treatment significantly reduces ethanol-impaired neurogenesis. (A) Shown in bar graph (left) are mean ± SEM of DCX IR density measured from DG region of hippocampus. ANOVA F(5, 20) = 10.38, p < 0.001) and Fisher post hoc: **p < 0.01 compared with Control or Ethanol; n = 4–6). The representative microphotographs (right) of DCX+ IR were taken from Control (A), Ethanol (B), Ethanol + Parthenolide (E + Part, c) and Ethanol + Bay 11-7082 (E + Bay, d) (scale bar = 200 μm). The experiments were repeated 3 times with the similar design and results. (B) Ethanol induction of NALP1 and IL-1β mRNA is blocked by inhibitors parthenolide (Part, 5 μM, pretreatment for 2 h) and Bay 11-7082 (Bay, 5 μM, pretreatment for 2 h). ANOVA following Fisher post hoc: ***p < 0.001 compared with Control or Ethanol, n = 3. (C) Western blot of NALP1 and pro-caspase-1 proteins. Consistent with mRNA data, ethanol exposure strongly induced short form (∼75 kDa) NALP1 and both inflammasome inhibitors blocked ethanol induction of NALP1 protein level (left). Western blot of caspase-1 also indicates increased expression of caspase-1 protein (45 kDa) by ethanol exposure (right). ANOVA following Fisher post hoc: **p < 0.01 compared with control; *p < 0.05 compared with EtOH, n = 3.
Anti-Inflammatory Agents Block Ethanol and IL-1β Inhibition of Neurogenesis
To further investigate the involvement of inflammasomes in ethanol inhibition of neurogenesis we studied the anti-inflammatory drugs Parthenolide and Bay 11708, both originally thought to block NF-κB activation, but more recently thought to be more specific for inflammasomes (Juliana et al., 2010). Parthenolide and Bay 11708 alone did not change the key marker of neurogenesis, e.g., DCX + IR, however, both restored DCX + IR in ethanol treated HEC slices (Figure 8A), indicating both inflammasome inhibitors significantly reversed ethanol inhibition of neurogenesis. RT-PCR analysis further indicates that ethanol induction of NALP1 and IL-1βmRNA was significantly blocked by both anti-inflammatory drugs (Figure 8B). Western blots of inflammasome proteins NALP 1 and caspase-1 proteins are increased by ethanol exposure and both inflammasome inhibitors blocked ethanol induction of these proteins (Figure 8C). Together, these results suggest that targeting inflammasome proteins that are involved in production of bioactive IL-1β can reverse ethanol inhibition of hippocampal neurogenesis.
We next investigated the effects of anti-inflammatory agents butylated hydroxytoluene (BHT) and rolipram, both found to protect against ethanol induced increased NF-κB transcription of proinflammatory cytokines and neurodegeneration (Crews et al., 2006a). BHT added to HEC slice cultures increased DCX + IR and reversed ethanol reduction of DCX + IR (Figure 9). Rolipram, a PDE IV inhibitor that increases cAMP, PKA activation, and pCREB that protect against ethanol neurotoxicity (Zou and Crews, 2006) also increased DCX + IR and reversed ethanol inhibition of DCX + IR (Figure 9). These studies suggest targeting IL-1β signaling pathway with anti-inflammatory inhibition can reverse ethanol inhibition of neurogenesis.
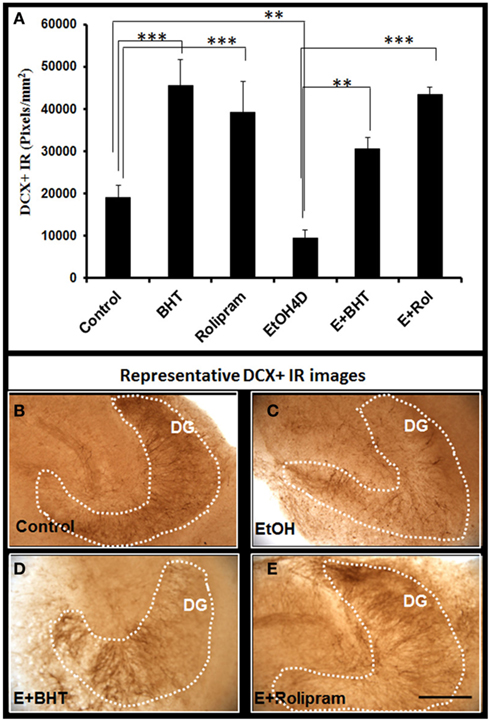
Figure 9. Rolipram and anti-oxidant BHT protect from ethanol-impaired neurogenesis. Shown in bar graph (A): PDE IV inhibitor rolipram and anti-oxidant BHT increase DCR+ IR and reverse ethanol inhibition of DCX+ IR. ANOVA F(5, 32) = 11.552, p < 0.001, and Fisher post hoc: ***p < 0.001 or **p < 0.01 compared with Control or EtOH4D, n = 6–8. The experiments were repeated with similar results. Representative photographs of DCX+ IR are shown in (B) (control); (C) (EtOH); (D) (Ethanol + BHT, 50 mM, pretreatment for 3 days); and (E) (Ethanol + Rolipram, 500 nM, pretreatment for 3 days; scale bar = 200 μm).
Increased Expression of IL-1β and Inflammasome Proteins in Human Alcoholic Brains
To determine if alteration of hippocampal neurogenesis and induction of proinflammatory cytokine IL-1β and inflammasome proteins could be found in human alcoholic brains we studied human postmortem brains from 10 paired controls and moderate drinking alcoholics (see Table 2). In both control and alcoholic hippocampus, some DCX + IR cells with neuronal morphology were found within the dense layer of granule cells, but variation between patients was too great to establish any patterns (data not shown). Similar to DCX staining, Ki67+ cells were scattered in the human hippocampus (data not shown). However, IL-1β and inflammasome proteins NALP1 and NALP3 were found to be consistently increased within alcoholic brains. IL-1β, NALP1, and NALP3 + IR cells were quantified in two regions (DG and CA) of hippocampus. As shown in Figure 10, IL-1β + IR cells were over twofold greater than controls (Figure 10 Top). NALP1 + IR was also increased in postmortem alcoholic brain relative to controls by at least twofold (Figure 10 middle). NALP3 + IR was also higher in postmortem alcoholic brain compared to controls (Control vs. Alcoholics: DG-662 ± 131 vs. 1876 ± 289 pixels/mm2, p < 0.001, n = 10; CA: 874 ± 156 vs. 2418 ± 242, p < 0.001, n = 10). Confocal analysis of double fluorescent immunostaining with cell markers was further performed to determine cell type expression of these molecules in human brain. IL-1β was primarily found in NeuN + IR cells in the DG granule layer and CA pyramidal cells of the hippocampus and increased IL-1β IR in the DG granule cells in alcoholic brain was depicted (Figure 10I). NALP1 was increased in alcoholic brains in hippocampal cells that co-localized with mature neuronal marker calbindin (Figure 10J) and in some GFAP+ cells (Figure 10K) but rarely co-expressed in Iba-1+ microglia (data not shown). NALP3 was co-localized with NeuN+ neuronal cells and Iba-1 + IR microglia in DG and CA region. The increased NALP3 IR in NeuN+ neurons and Iba-1+ microglia was obvious in alcoholic brain (Figures 10L–N). These findings in postmortem human brain are consistent with chronic alcohol consumption by human alcoholics inducing inflammasome-IL-1β signaling molecules.
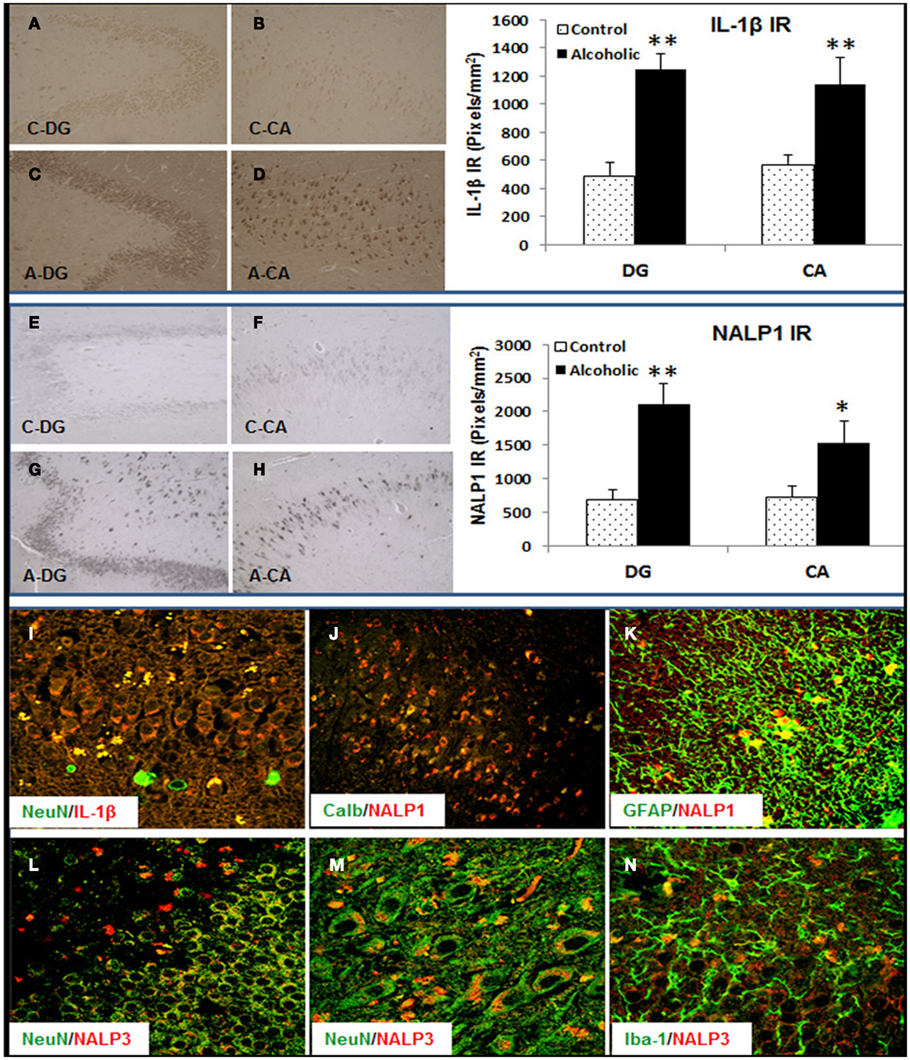
Figure 10. Increased expression of Inflammasome-IL-1β proteins in alcoholic human brains. Hippocampal sections from 10 controls and 10 alcoholic patients were stained and histochemistry quantitated. Although the cause of death varied among subjects, 9 of 10 controls and 6 of 10 alcoholics died from cardiovascular disease, and none of the subjects died from inflammation. Top panel: bar graph indicates average IL-1β+ IR measured from DG and CA in 10 paired controls and alcoholics. Data represent mean ± SEM. DG: ANOVA F(1,18) = 47.75, p < 0.0001, and Fisher post hoc: **p < 0.001 compared with Controls, n = 10; CA: ANOVA F(1,18) = 7.97, p < 0.011, and Fisher post hoc: **p < 0.001 compared with Controls, n = 10. Representative images of IL-1β+ IR are shown: (A) DG-Control, (B) CA-Control, (C) DG-Alcoholic, and (D) CA-Alcoholic. Middle panel: bar graph indicates average NALP1+ IR measured from DG and CA in 10 paired controls and alcoholics. Data represent mean ± SEM. DG: ANOVA F(1,18) = 18.35, p = 0.0004, and Fisher post hoc: **p < 0.001 compared with Controls, n = 10; CA: ANOVA F(1,18) = 4.7, p = 0.044, and Fisher post hoc: *p < 0.04 compared with Controls, n = 10. Representative images of NALP1+ IR are shown: (E) DG-Control, (F) CA-Control, (G) DG-Alcoholic, and (H) CA-Alcoholic. Bottom panel: confocal images of double fluorescent immunostaining show (I) NeuN (green) and IL-1β (red) from DG in alcoholic (original magnification 80×); (J) Calbindin (Calb, green) and NALP1 (red) from CA region of hippocampus in alcoholic (original magnification 20×); (K) GFAP (green) and NALP1 (red) from hippocampal DG of alcoholic (original magnification 80×); (L–N) NeuN (green) and NALP3 (red) from hippocampal DG (L) and CA (M) of alcoholic [original magnification 80× in (D) and 120× in (E)] and (N) microglial marker Iba-1 (green) and NALP3 (red) from hippocampal DG of alcoholic (original magnification 80×).
Discussion
We report here for the first time that in an ex vivo model of HEC slice culture ethanol treatment inhibits neurogenesis similar to in vivo studies. We identify the proinflammatory cytokine IL-1β as a key mediator for ethanol inhibition of neurogenesis. We also demonstrate for the first time that ethanol inhibition of neurogenesis is associated with induction of inflammasome proteins and that these proinflammatory molecules are also increased in postmortem alcoholic human brains. Our results find that IL-1β is induced by ethanol in our rat brain slice cultures consistent with our previous findings in mice (Qin et al., 2008). These findings continue to link neuroinflammation to ethanol disruption of hippocampal neurogenesis and neuroplasticity.
In rats, acute and chronic ethanol binge drinking reduces Ki67 + IR, and other indices of NPC proliferation (Nixon and Crews, 2002). In vivo BrdU prelabeling of NPC to follow survival of maturing NPC finds ethanol treatment reduces BrdU + IR in rats (Nixon and Crews, 2002) and mice (Crews et al., 2004b). Ethanol treatment of rats increases pyknotic nuclei in dentate gyrus consistent with increased NPC cell death as one mechanism of ethanol reduced neurogenesis (He et al., 2005). As reported in this study, treatment of HEC slices with concentrations of ethanol mimicking alcoholic binge drinking reduce Ki67 + IR in DG consistent with inhibition of NPC proliferation and reduce pre-labeled BrdU + IR cell number consistent with reduced NPC survival. In addition, ethanol treatment of rats (Crews et al., 2006a,b) and mice (He et al., 2005) reduces DCX + IR similar to our findings reported here in HEC slices. These data suggest ethanol inhibition of neurogenesis in HEC slice is similar to ethanol inhibition of hippocampal neurogenesis in in vivo rat and mouse brain.
Studies have found that neuroinflammation reduces neurogenesis (Monje et al., 2003; Mathieu et al., 2010) and that ethanol increases neuroinflammation (Crews et al., 2011). Ethanol activates and amplifies proinflammatory signals through NF-κB, a key proinflammatory transcription factor that induces multiple neuroimmune genes, including cytokines and their receptors, that further activate and amplify NF-κB transcription (Crews et al., 2011). Ethanol is known to increase brain NF-κB-DNA binding and activation in vivo (Ward et al., 1996; Crews et al., 2006a), in vitro in HEC slice culture (Zou and Crews, 2006, 2010), and astrocyte cultures (Valles et al., 2004). However, proinflammatory innate immune activation involves multiple molecules that induce each other complicating identification of key regulatory elements. Further confounds are that diverse cytokines have different opposing actions on adult hippocampal neurogenesis (Mathieu et al., 2010). We report here for the first time that neutralizing IL-1β and blockade of IL-1 surface receptor with antagonist IL-1RIa blunt ethanol inhibition of neurogenesis. Neutralizing antibodies to TNFα or MCP-1 did not reverse ethanol inhibition of neurogenesis. Addition of IL-1β reduced HEC neurogenesis and ethanol increased the synthesis of IL-1β and key proteins involved in IL-1β maturation, synthesis, and release, consistent with IL-1β as a primary molecule mediating ethanol inhibition of neurogenesis. These findings are consistent with elegant studies by Koo and Duman using chronic stress models that activate NF-κB, induce depression-like behavior and reduce neurogenesis (Koo et al., 2010). Similar to our findings reported here, stress-induced IL-1β was found to mediate stress-reduced neurogenesis (Koo and Duman, 2008). Consistent with reports that stress-reduced neurogenesis being associated with depression-like behavior (Koo et al., 2010) we previously reported that ethanol self-administration by mice reduces neurogenesis in association with increased depression-like behavior, with both reversed by antidepressants (Stevenson et al., 2009). Although neuroinflammation reduces neurogenesis, neuroinflammation particularly IL-1β, also increase glutamate excitability that can lead to seizures (Maroso et al., 2011) and seizures cause a delayed burst in neurogenesis similar to alcohol withdrawal (Crews and Nixon, 2009) even though the neuroimmune activation persists. Although the in vivo mechanisms of regulation of neurogenesis are complex, in the absence of seizures, IL-1β appears to be a key proinflammatory molecule mediating inhibition of neurogenesis by both chronic stress and ethanol.
IL-1β production involves the induction of proteins that form inflammasomes, a multi-protein complex that processes pro-IL-1β into mature bioactive IL-1β. A core group of proteins known as nucleotide oligomerization domain-like receptor with pyrin domain containing proteins (NLRP or NALP) binds together apoptotic speck-like protein with card (Valles et al., 2004) and pro-caspase-1 as well as other proteins that amplify proinflammatory responses and lead to proteolytic maturation and secretion of bioactive IL-1β (Bryant and Fitzgerald, 2009; Figure 11). NALP1 and NALP3 are core inflammasome proteins expressed in brain (Yin et al., 2009). We found ethanol increased expression of IL-1β as well as the inflammasome proteins NALP1 and NALP3. To our knowledge this is the first report showing increased induction of inflammasome proteins by ethanol in rat brain tissue as well as in alcoholic human brains. Interestingly, expression of inflammasome NALP1 and NALP3 in HEC slice appears to be cell type different, with NALP1 in both NeuN+ neurons and GFAP+ astrocytes and NALP3 only in GFAP+ astrocytes. Significance of NALP1 and NALP3 in different type of cells may underlie different mechanisms for activation of inflammasome and subsequent IL-1β release. For example, it has been reported that IL-1β production is differentially regulated in human monocytes and macrophage, in which a second signal is required for activation of inflammasome NALP3-caspase-1 and subsequent IL-1β processing and release (Netea et al., 2009). Although our human brain studies should be considered preliminary, we found that alcoholic brain had increased levels of IL-1β + IR in NeuN+ DG granule neurons. We also found increased inflammasome proteins NALP1 and NALP3 in postmortem alcoholic hippocampus. Consistent with findings in HEC slice, NALP1 is co-localized in both neuronal cells and GFAP+ astrocytes. Interestingly, NALP3 + IR is co-localized with NeuN+ neuronal cells as well as Iba-1+ microglia in postmortem alcoholic brain. Although many studies have found neuroimmune genes increased in diseased human brain, we are not aware of any other studies finding expression of inflammasome proteins within human hippocampus.
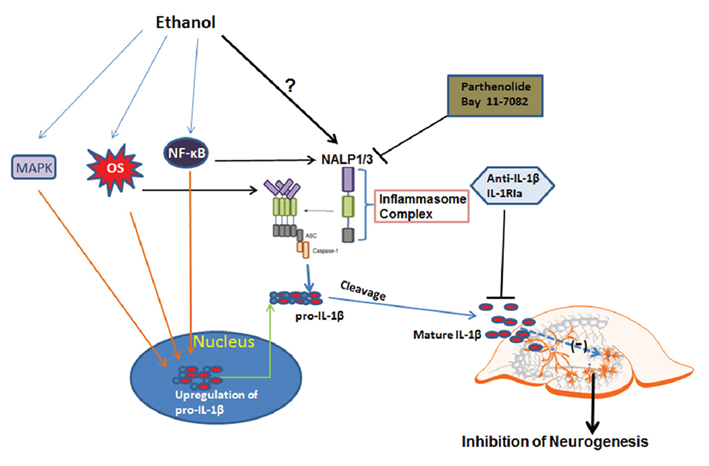
Figure 11. Schematic diagram for ethanol regulation of IL-1β and neurogenesis. The first step (the priming step): ethanol may up-regulate pro-IL-1β through NF-κB and MAPK signaling. The second step (the activation step): ethanol may directly regulate inflammasome activation through possible mechanisms such as oxidative stress, TLR4, or low potassium level, all of which have been shown to involve inactivation of inflammasome protein NALP1 or NALP3. Two events associated with ethanol exposure lead to increased production of active form IL-1β, contributing to inhibition of neurogenesis. Blockade of inflammasome activation with inhibitors to reduce IL-1β release or directly neutralizing IL-1β or blockade of IL-1 surface receptor can offer protection from ethanol-impaired neurogenesis.
To further investigate the role of neuroinflammatory activation and induction of inflammasome proteins in ethanol inhibition of neurogenesis we studied the actions of several anti-inflammatory drugs. Parthenolide and Bay 11-7082, anti-inflammatory drugs suggested to act as specific inflammasome inhibitors (Juliana et al., 2010), blocked ethanol, and IL-1β inhibition of neurogenesis, likely due to blocking inflammasome amplification of NF-κB proinflammatory responses. BHT, an anti-oxidant that blocks NF-κB induction of proinflammatory genes (Crews et al., 2006b; Zou and Crews, 2010), reversed ethanol inhibition of neurogenesis. The similar results were also observed in Rolipram, a PDE IV inhibitor that increases cAMP, Increased cAMP can increase PKA activation that inhibits proinflammatory NF-κB activation (Takahashi et al., 2002), key elements of proinflammatory gene induction. PKA may also increase BDNF in HEC that could contribute to the rolipram induced increase in neurogenesis (Zou and Crews, 2006). Thus, anti-inflammatory drugs reverse ethanol inhibition of neurogenesis consistent with IL-1β as a key proinflammatory molecule mediating the effects of ethanol.
In summary, ethanol inhibits neurogenesis and induces IL-1β and inflammasome proteins in neurons and glia. Targeting IL-1β signaling by using IL-1β neutralizing antibodies or IL-1 surface receptor antagonists as well as inflammasome inhibitors can normalize hippocampal neurogenesis impaired by ethanol and IL-1β (Figure 11). Inflammasome proteins are found in human hippocampus, and are increased in postmortem human alcoholic hippocampus. These findings support the hypothesis that IL-1β mediates ethanol inhibition of neurogenesis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The Authors wish to acknowledge support from the Bowles Center for Alcohol Studies, School of Medicine, University of North Carolina and the National Institutes of Health, National Institute on Alcoholism and Alcohol Abuse, AA020023, AA020024, AA020022, AA019767, AA11605. Thanks to Diana Lotito for assistance with manuscript preparation.
References
Bryant, C., and Fitzgerald, K. A. (2009). Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 19, 455–464.
Chakraborty, S., Kaushik, D. K., Gupta, M., and Basu, A. (2010). Inflammasome signaling at the heart of central nervous system pathology. J. Neurosci. Res. 88, 1615–1631.
Couillard-Despres, S., Winner, B., Schaubeck, S., Aigner, R., Vroemen, M., Weidner, N., Bogdahn, U., Winkler, J., Kuhn, H. G., and Aigner, L. (2005). Doublecortin expression levels in adult brain reflect neurogenesis. Eur. J. Neurosci. 21, 1–14.
Crews, F., Nixon, K., Kim, D., Joseph, J., Shukitt-Hale, B., Qin, L., and Zou, J. (2006a). BHT blocks NF-kappaB activation and ethanol-induced brain damage. Alcohol. Clin. Exp. Res. 30, 1938–1949.
Crews, F. T., Mdzinarishvili, A., Kim, D., He, J., and Nixon, K. (2006b). Neurogenesis in adolescent brain is potently inhibited by ethanol. Neuroscience 137, 437–445.
Crews, F. T., Braun, C. J., Hoplight, B., Switzer, R. C. III, and Knapp, D. J. (2000). Binge ethanol consumption causes differential brain damage in young adolescent rats compared with adult rats. Alcohol. Clin. Exp. Res. 24, 1712–1723.
Crews, F. T., Collins, M. A., Dlugos, C., Littleton, J., Wilkins, L., Neafsey, E. J., Pentney, R., Snell, L. D., Tabakoff, B., Zou, J., and Noronha, A. (2004a). Alcohol-induced neurodegeneration: when, where and why? Alcohol Clin. Exp. Res. 28, 350–364.
Crews, F. T., Nixon, K., and Wilkie, M. E. (2004b). Exercise reverses ethanol inhibition of neural stem cell proliferation. Alcohol 33, 63–71.
Crews, F. T., and Nixon, K. (2009). Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 44, 115–127.
Crews, F. T., Zou, J., and Qin, L. (2011). Induction of innate immune genes in brain create the neurobiology of addiction. Brain Behav. Immun. 25(Suppl. 1), S4–S12.
Das, S., and Basu, A. (2008). Inflammation: a new candidate in modulating adult neurogenesis. J. Neurosci. Res. 86, 1199–1208.
Deng, W., Aimone, J. B., and Gage, F. H. (2010). New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 11, 339–350.
Gleeson, J. G., Allen, K. M., Fox, J. W., Lamperti, E. D., Berkovic, S., Scheffer, I., Cooper, E. C., Dobyns, W. B., Minnerath, S. R., Ross, M. E., and Walsh, C. A. (1998). Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell 92, 63–72.
He, J., Nixon, K., Shetty, A. K., and Crews, F. T. (2005). Chronic alcohol exposure reduces hippocampal neurogenesis and dendritic growth of newborn neurons. Eur. J. Neurosci. 21, 2711–2720.
Juliana, C., Fernandes-Alnemri, T., Wu, J., Datta, P., Solorzano, L., Yu, J. W., Meng, R., Quong, A. A., Latz, E., Scott, C. P., and Alnemri, E. S. (2010). Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 285, 9792–9802.
Koo, J. W., and Duman, R. S. (2008). IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Natl. Acad. Sci. U.S.A. 105, 751–756.
Koo, J. W., and Duman, R. S. (2009a). Evidence for IL-1 receptor blockade as a therapeutic strategy for the treatment of depression. Curr. Opin. Investig. Drugs 10, 664–671.
Koo, J. W., and Duman, R. S. (2009b). Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci. Lett. 456, 39–43.
Koo, J. W., Russo, S. J., Ferguson, D., Nestler, E. J., and Duman, R. S. (2010). Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl. Acad. Sci. U.S.A. 107, 2669–2674.
Laskowski, A., Schmidt, W., Dinkel, K., Martínez-Sánchez, M., and Reymann, K. G. (2005). bFGF and EGF modulate trauma-induced proliferation and neurogenesis in juvenile organotypic hippocampal slice cultures. Brain Res. 1037, 78–89.
Mandyam, C. D., and Koob, G. F. (2012). The addicted brain craves new neurons: putative role for adult-born progenitors in promoting recovery. Trends Neurosci. 35, 250–260.
Maroso, M., Balosso, S., Ravizza, T., Liu, J., Bianchi, M. E., and Vezzani, A. (2011). Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: the importance of IL-1beta and high-mobility group box 1. J. Intern. Med. 270, 319–326.
Mathieu, P., Battista, D., Depino, A., Roca, V., Graciarena, M., and Pitossi, F. (2010). The more you have, the less you get: the functional role of inflammation on neuronal differentiation of endogenous, and transplanted neural stem cells in the adult brain. J. Neurochem. 112, 1368–1385.
Ming, G. L., and Song, H. (2011). Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70, 687–702.
Monje, M. L., Toda, H., and Palmer, T. D. (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765.
Namba, T., Mochizuki, H., Onodera, M., Namiki, H., and Seki, T. (2007). Postnatal neurogenesis in hippocampal slice cultures: early in vitro labeling of neural precursor cells leads to efficient neuronal production. J. Neurosci. Res. 85, 1704–1712.
Netea, M. G., Nold-Petry, C. A., Nold, M. F., Joosten, L. A., Opitz, B., van der Meer, J. H., van de Veerdonk, F. L., Ferwerda, G., Heinhuis, B., Devesa, I., Funk, C. J., Mason, R. J., Kullberg, B. J., Rubartelli, A., van der Meer, J. W., and Dinarello, C. A. (2009). Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113, 2324–2335.
Nixon, K., and Crews, F. T. (2002). Binge ethanol exposure decreases neurogenesis in adult rat hippocampus. J. Neurochem. 83, 1087–1093.
Noraberg, J., Poulsen, F. R., Blaabjerg, M., Kristensen, B. W., Bonde, C., Montero, M., Meyer, M., Gramsbergen, J. B., and Zimmer, J. (2005). Organotypic hippocampal slice cultures for studies of brain damage, neuroprotection and neurorepair. Curr. Drug Targets CNS Neurol. Disord. 4, 435–452.
Obernier, J. A., White, A. M., Swartzwelder, H. S., and Crews, F. T. (2002). Cognitive deficits and CNS damage after a 4-day binge ethanol exposure in rats. Pharmacol. Biochem. Behav. 72, 521–532.
Qin, L., He, J., Hanes, R. N., Pluzarev, O., Hong, J. S., and Crews, F. T. (2008). Increased systemic and brain cytokine production and neuroinflammation by endotoxin following ethanol treatment. J. Neuroinflammation 5, 10.
Raineteau, O., Rietschin, L., Gradwohl, G., Guillemot, F., and Gähwiler, B. H. (2004). Neurogenesis in hippocampal slice cultures. Mol. Cell. Neurosci. 26, 241–250.
Sahay, A., and Hen, R. (2007). Adult hippocampal neurogenesis in depression. Nat. Neurosci. 10, 1110–1115.
Stevenson, J. R., Schroeder, J. P., Nixon, K., Besheer, J., Crews, F. T., and Hodge, C. W. (2009). Abstinence following alcohol drinking produces depression-like behavior and reduced hippocampal neurogenesis in mice. Neuropsychopharmacology 34, 1209–1222.
Takahashi, N., Tetsuka, T., Uranishi, H., and Okamoto, T. (2002). Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur. J. Biochem. 269, 4559–4565.
Valles, S. L., Blanco, A. M., Pascual, M., and Guerri, C. (2004). Chronic ethanol treatment enhances inflammatory mediators and cell death in the brain and in astrocytes. Brain Pathol. 14, 365–371.
van Praag, H., Schinder, A. F., Christie, B. R., Toni, N., Palmer, T. D., and Gage, F. H. (2002). Functional neurogenesis in the adult hippocampus. Nature 415, 1030–1034.
Ward, R. J., Zhang, Y., Crichton, R. R., Piret, B., Piette, J., and de Witte, P. (1996). Identification of the nuclear transcription factor NFkappaB in rat after in vivo ethanol administration. FEBS Lett. 389, 119–122.
Yin, Y., Yan, Y., Jiang, X., Mai, J., Chen, N. C., Wang, H., and Yang, X. F. (2009). Inflammasomes are differentially expressed in cardiovascular and other tissues. Int. J. Immunopathol. Pharmacol. 22, 311–322.
Zou, J., and Crews, F. (2006). CREB and NF-kappaB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell. Mol. Neurobiol. 26, 385–405.
Zou, J., and Crews, F. (2010). Induction of innate immune gene expression cascades in brain slice cultures by ethanol: key role of NF-kappaB and proinflammatory cytokines. Alcohol. Clin. Exp. Res. 34, 777–789.
Keywords: IL-1β, neurogenesis, alcohol, inflammasomes, neuroprogenitors, brain slice culture
Citation: Zou J and Crews FT (2012) Inflammasome-IL-1β signaling mediates ethanol inhibition of hippocampal neurogenesis. Front. Neurosci. 6:77. doi: 10.3389/fnins.2012.00077
Received: 22 March 2012; Paper pending published: 13 April 2012;
Accepted: 09 May 2012; Published online: 30 May 2012.
Edited by:
Angelique Bordey, Yale University School of Medicine, USAReviewed by:
Ashok K. Shetty, Institute for Regenerative Medicine, USAGonzalo Alvarez-Bolado, University of Heidelberg, Germany
Copyright: © 2012 Zou and Crews. This is an openaccess article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Fulton T. Crews, Bowles Center for Alcohol Studies, University of North Carolina at Chapel Hill, CB#7178, Chapel Hill, NC 27599-7178, USA. e-mail:ZnRjcmV3c0BtZWQudW5jLmVkdQ==