 Lidia Garcia-Bonilla
Lidia Garcia-Bonilla Corinne Benakis
Corinne Benakis Jamie Moore
Jamie Moore Costantino Iadecola
Costantino Iadecola Josef Anrather
Josef Anrather- Brain and Mind Research Institute, Weill Cornell Medical College, New York, NY, USA
Stressor-induced tolerance is a central mechanism in the response of bacteria, plants, and animals to potentially harmful environmental challenges. This response is characterized by immediate changes in cellular metabolism and by the delayed transcriptional activation or inhibition of genetic programs that are not generally stressor specific (cross-tolerance). These programs are aimed at countering the deleterious effects of the stressor. While induction of this response (preconditioning) can be established at the cellular level, activation of systemic networks is essential for the protection to occur throughout the organs of the body. This is best signified by the phenomenon of remote ischemic preconditioning, whereby application of ischemic stress to one tissue or organ induces ischemic tolerance (IT) in remote organs through humoral, cellular and neural signaling. The immune system is an essential component in cerebral IT acting simultaneously both as mediator and target. This dichotomy is based on the fact that activation of inflammatory pathways is necessary to establish IT and that IT can be, in part, attributed to a subdued immune activation after index ischemia. Here we describe the components of the immune system required for induction of IT and review the mechanisms by which a reprogrammed immune response contributes to the neuroprotection observed after preconditioning. Learning how local and systemic immune factors participate in endogenous neuroprotection could lead to the development of new stroke therapies.
Introduction
Inflammation is a central component in the pathophysiology of cerebral ischemia. Brain ischemia triggers both a local and systemic inflammatory response. These responses play contradictory roles: contributing to progression of the ischemic lesion on one hand, and to processes of tissue repair in the injured brain on the other (Iadecola and Anrather, 2011a; Macrez et al., 2011). Since post-ischemic inflammation shows detrimental and beneficial aspects, anti-inflammatory therapies that indiscriminately target both arms of this immune response have not been successful (Iadecola and Anrather, 2011a; Macrez et al., 2011). Therefore, a deeper understanding of post-ischemic inflammation is needed in order to harness its beneficial effects for therapeutic purposes.
In addition to its beneficial role in the repair and regeneration of ischemic tissue, inflammatory pathways are also involved in evoking neuroprotective mechanisms that lead to ischemic tolerance (IT). The brain is equipped with a remarkable capacity to mount self-protective programs that are tuned to limit the deleterious effects of ischemia, commonly referred to as endogenous neuroprotection (Iadecola and Anrather, 2011b). These protective programs can be evoked by preconditioning (PC) stimuli (Table 1), such as sublethal stressors, resulting in IT (Kirino, 2002; Narayanan et al., 2013). Thus, understanding the endogenous inflammatory pathways involved in brain IT may lead to novel therapeutic strategies for the prevention and repair of neuronal damage in stroke patients (Shpargel et al., 2008; Macrez et al., 2011).
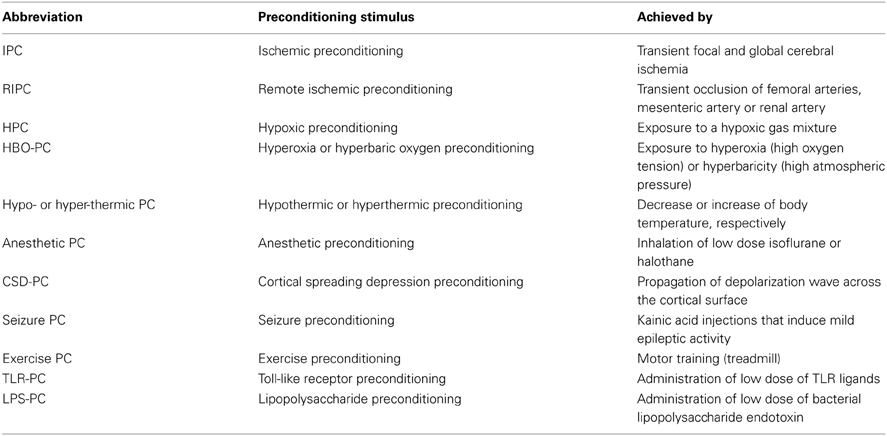
Table 1. List of preconditioning stimuli inducing ischemic tolerance.
Cerebral IT can be acquired by several stimuli including short episodes of transient focal and global cerebral ischemia, remote organ ischemia, hypoxia, hypothermia, hyperthermia, exposure to inhalation anesthetics, cortical spreading depression (CSD), brief episodes of seizures or by exposure to low-dose bacterial lipopolysaccharide (LPS) prior to cerebral ischemia (Gidday, 2006; Kunz et al., 2007; Shpargel et al., 2008). IT is characterized by the activation of evolutionary conserved programs that serve to increase the resistance of the brain to ischemia. In the case of early preconditioning, these programs can be activated through preexisting signaling modules that converge in the mitochondria to improved mitochondrial function and energy metabolism during the ischemic event (Dirnagl and Meisel, 2008). In contrast, delayed PC involves gene transcription and protein synthesis (Kitagawa et al., 1990). Because activation of transcription is the main mechanism by which many pro-inflammatory signaling cascades induce a specific cellular response, inflammation is thought to be a significant component of delayed PC. Table 2 summarizes the inflammatory pathways involved in IT achieved by different PC stimuli (Table 1). Inflammation is activated by innate immune receptors such as Toll-like receptors (TLRs) or cytokines receptors such as tumor necrosis factor receptor 1 (TNFR1) and interleukin-1 receptor (IL-1R). Activation of these pathways results in the induction of inflammatory genes that are mediators or effectors of the PC stimulus. In addition to conferring cytoprotection directly, the acquired IT protects the brain by suppressing post-ischemic proinflammatory gene expression, microglial and endothelial activation and leukocyte infiltration (Huang et al., 2006; Iadecola and Anrather, 2011b).
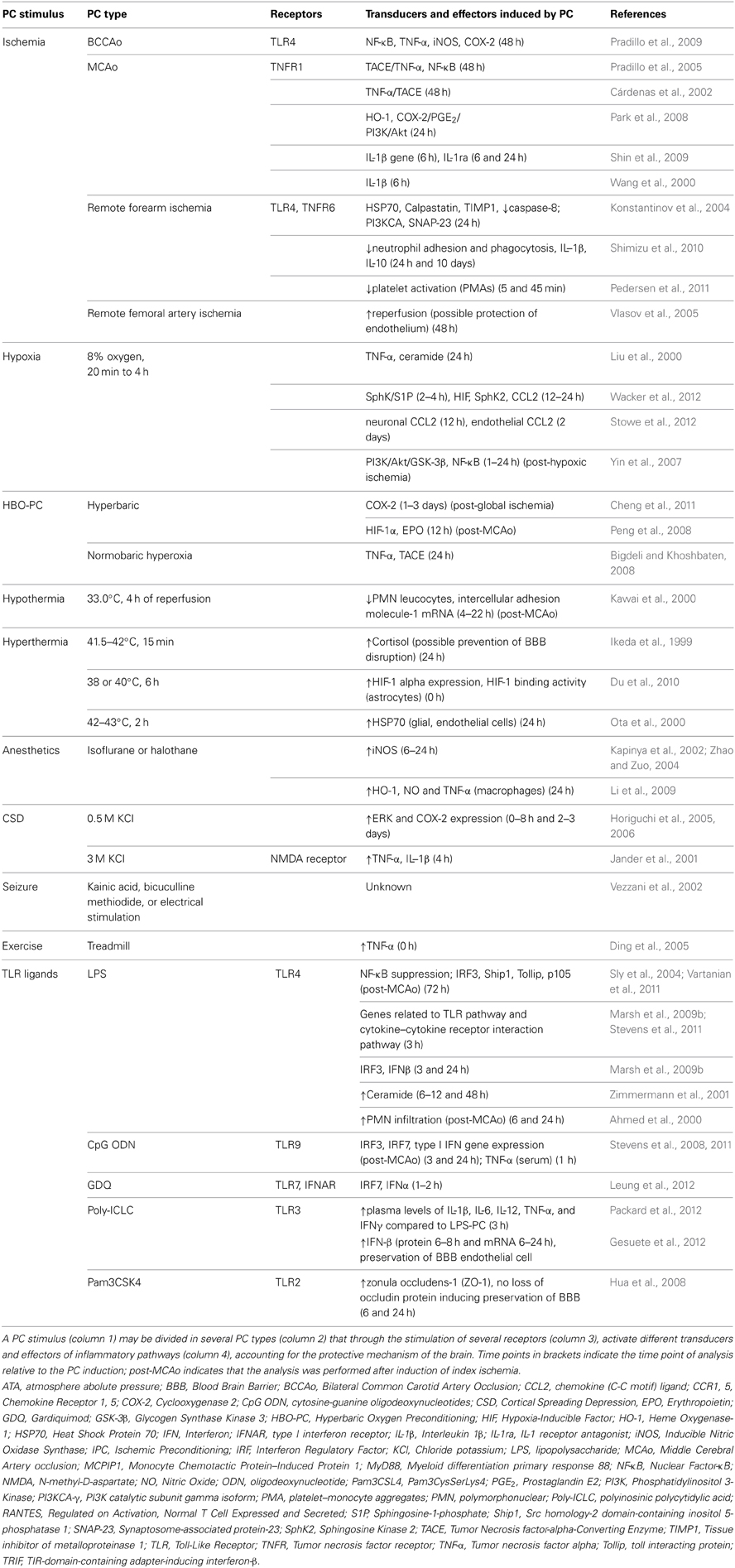
Table 2. Molecular inflammatory mechanisms of brain preconditioning.
In this review we will focus on inflammatory pathways leading to cerebral IT. We will review the signaling cascades involved in immune activation in different PC modalities and highlight the molecular components involved. Lastly, this review will highlight the importance of epigenetic modifications and microRNAs in PC-induced reprogramming of the immune system.
Evidence for a Role of Inflammatory Pathways in Different PC Paradigms
A wide variety of preconditioning stimuli trigger the activation of inflammatory pathways that lead to brain IT (Table 2). The receptors, transducers and effector elements of these pathways are shared among different preconditioning paradigms. For example, TLRs are potent mediators of both ischemic (Pradillo et al., 2009; Wang et al., 2010) and endotoxic PC (acquired by systemic administration of LPS) (Tasaki et al., 1997; Ahmed et al., 2000; Vartanian et al., 2011). Likewise, the TNF pathway is involved in ischemic, hypoxic, hyperthermic and exercise-induced PC. Although inflammatory pathways are not independently sufficient for the induction of IT, there is evidence that several PC modalities are dependent upon the induction of such pathways.
Ischemic, Hypoxic, and Hyperoxic PC
Short non-damaging episodes of ischemia prior to a lethal ischemic episode leads to IT (Kirino, 2002). Cerebral IT can be acquired by local PC, mediated either by global (Kitagawa et al., 1990) or focal cerebral ischemia (Barone et al., 1998), or by remote ischemic PC (RIPC), typically achieved by limb ischemia (Tapuria et al., 2008). Although both TLR2 and TLR4 have been implicated in the post-ischemic inflammatory response (Cao et al., 2007; Abe et al., 2010; Brea et al., 2011; Shichita et al., 2012), only TLR4 has been shown to participate in ischemic preconditioning (IPC) (Konstantinov et al., 2004; Pradillo et al., 2009). TLR4 signaling activates transcription nuclear factor-κB (NF-κB) leading to the expression of TNF, inducible nitric oxide (iNOS) and cyclooxygenase-2 (COX-2), which are required to evoke a PC effect (see Table 2). Similarly, TNFR1, another central receptor of the innate immune system and activator of NF-κB, is essential for the induction of IPC (Konstantinov et al., 2004; Pradillo et al., 2005; Figure 1).
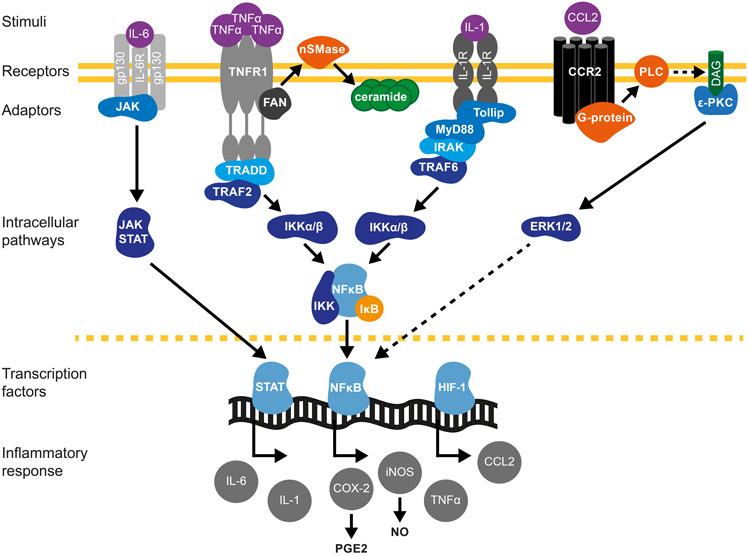
Figure 1. Cytokine signaling pathways in brain preconditioning. Following preconditioning (PC), the increase in tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 and chemokine (C-C motif) ligand 2 (CCL2) leads to the activation of inflammatory cascades and induction of gene expression such as inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2, mediators of delayed PC. IL-6/IL-6R activates Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway. Phosphorylation and nuclear translocation of STAT induces iNOS and COX-2 up-regulation (Dawn et al., 2004). TNF-α activates TNF receptor 1(TNFR1)/Tumor necrosis factor receptor type 1-associated DEATH domain protein (TRADD)/TNF-α receptor–associated factor 2 (TRAF2) pathway (Hallenbeck, 2002; Pradillo et al., 2005). Furthermore, TNF-α activation promotes ceramide synthesis that also mediates TNFα-preconditioning (Liu et al., 2000; Ginis et al., 2002). The neutral sphingomyelinase domain (NSD) of TNFR1 recruits factor associated with neutral sphingomyelinase activation (FAN). FAN activates neutral sphingomyelinase (nSMase) in the outer leaflet of the plasma membrane to produce ceramide (Hallenbeck, 2002). IL-1R activates toll interacting protein (tollip)/myeloid differentiation primary response 88 (MyD88)-IL-1R-associated kinase (IRAK)/TRAF6 signaling. TRAF mediates the activation of inhibitor of κB kinase (IKK), which triggers the phosphorylation-induced proteasomal degradation of the IκB, NF-κB activation and subsequent iNOS and COX-2 expression. CCL2/CCR2 activation promotes phospholipase-C (PLC)- diacylglycerol (DAG)-protein kinase C (PKC) pathway that leads to extracellular signal-regulated kinases (ERK1/2) activation and ultimately causes NF-κB activation (Rehni and Singh, 2012). In addition, HIF-1-induced expression of CCL-2 confers cerebral ischemic tolerance (Stowe et al., 2012). COX-2-derived prostaglandin E2 (PGE2) (Horiguchi et al., 2006) and iNOS-derived nitric oxide (NO) (Cho et al., 2005) are molecular mediators of PC.
IT is not limited to the organ to which a stressor is applied. A PC stimulus applied to one organ also leads to IT in other organs, referred to remote PC (for reviews see Tapuria et al., 2008; Anrather and Hallenbeck, 2013; Hess et al., 2013). Remote ischemia can be achieved in rodents by transient occlusion of one or both femoral arteries, the mesenteric artery or the renal artery prior to cerebral ischemia. RIPC results in a decrease of the infarct volume following cerebral ischemia [for a comprehensive list in animal model see (Hess et al., 2013) and (Anrather and Hallenbeck, 2013)]. The effect of RIPC on circulating immune cells has been investigated in humans following forearm transient ischemia (Table 2). These studies showed that the expression of pro-inflammatory genes is down-regulated in circulating leukocytes (Konstantinov et al., 2004) and neutrophil function is altered (Shimizu et al., 2010).
Hypoxic-ischemic brain damage is mediated by TLRs (Stridh et al., 2011). Therefore, TLRs might also trigger inflammatory signaling in hypoxic PC (HPC), although no report to date has confirmed their involvement. To induce HPC in rodents, animals are placed in chambers and exposed to a hypoxic gas mixture, usually 8% oxygen and 92% nitrogen, for 3–4 h (Yin et al., 2007; Wacker et al., 2012). Hypoxia-inducible factor 1 (HIF-1) and NF-κB are activated in the brain after HPC (Yin et al., 2007; Wacker et al., 2012) leading to the expression of iNOS (Jung et al., 2000), TNF-α (Liu et al., 2000) and chemokine (C-C motif) ligand 2 (CCL2) (Stowe et al., 2012), which have been implicated in establishing IT after HPC (see below section Molecular Components of the Immune System Involved in PC).
The immune system is also involved in hyperoxic PC, which can be achieved by either normobaric hyperoxia or hyperbaric oxygen PC (HBO-PC). Delivering a gas mixture containing 90–95% oxygen induces normobaric hyperoxia. HBO-PC consists of the inspiration of an oxygen-containing gas mixture at higher than one atmospheric absolute pressure (ATA), usually 2–2.5 ATA. Bigdeli et al. showed that rats subjected to 16 h of normobaric hyperoxia had reduced infarct volume after Middle Cerebral Artery occlusion (MCAo) and that the TNF-α converting enzyme (TACE)/TNF-α/NF-κB pathway is associated with the protective effect (Bigdeli and Khoshbaten, 2008). Moreover, COX-2 has also been reported to be a mediator of HBO-PC (Cheng et al., 2011). Administration of the specific COX-2 inhibitor, NS-398, before HBO exposure reversed the protective effect of HBO-PC. Cheng et al. further identified COX-2-dependent upregulation of HIF-1α as a possible down-stream mediator of HBO-PC.
Hypothermic and Hyperthermic PC
Hypothermia induced during and/or after ischemia has a well-established protective effect in acute ischemic stroke (Wu and Grotta, 2013) and may be partly due the inhibition of the inflammatory response occurring during stroke (Ceulemans et al., 2010). Brief exposure to hyperthermia represents an additional method of PC (Table 2). In animal model of stroke, transient hypothermia prior an ischemic insult (Nishio et al., 2000; Yunoki et al., 2003) or during reperfusion (Kawai et al., 2000) reduces infarct volume. The latter study has reported a decrease number of polymorphonuclear leucocytes during reperfusion—shown by a reduced myeloperoxidase activity in rats subjected to hypothermia (Kawai et al., 2000). Hyperthermic PC reduces brain damage in newborn rats as well (Ota et al., 2000). The protective mechanism of hyperthermic PC has been linked to the induction of the heat shock protein 70 (HSP70) within glial and endothelial cells (Ota et al., 2000). In addition to its role as an intracellular chaperon, HSP70 acts as an endogenous ligand of TLR4. Extracellular HSP70 is released from damaged cells and serves as a damage-associated molecular pattern (DAMPs) molecule (Bianchi, 2007). It has been shown that HSP70 mediates endotoxin tolerance by signaling through the TLR4/CD14 pathway (Vabulas et al., 2002; Aneja et al., 2006), similar to the canonical TLR4 ligand LPS. Thus, induction of HSP70 following hyperthermia could be a factor in the establishment of IT.
Anesthetic PC
Inhalation anesthetics are strong inducers of IT and can directly modulate immune functions in neutrophils, monocytes and lymphocytes (Clarkson, 2007; Wang et al., 2008). Rats pretreated with low dose isoflurane or halothane were protected after permanent MCAo (Kapinya et al., 2002). The protective effect was attributed to increased iNOS expression after anesthetic PC, and was reversed by treating the animals with the iNOS inhibitor aminoguanidine. Volatile anesthetics might directly alter the response of immune cells to inflammatory stimuli. Isoflurane PC of mouse macrophages induced the expression of heme oxygenase-1 (HO-1) and decreased NO and TNF-α release after subsequent LPS exposure (Li et al., 2009).
Cortical Spreading Depression, Brain Stimulation, and Seizures
CSD is an electrophysiological phenomenon characterized by a slowly propagating depolarization wave across the cortical surface that confers delayed IT (Yanamoto et al., 1998; Horiguchi et al., 2006; Shpargel et al., 2008). In animal models, CSD PC is elicited by either topical application or superfusion of the cortical surface with a 0.5–5 M KCl solution that induces IT lasting 1–15 days (Kawahara et al., 1995; Yanamoto et al., 1998; Horiguchi et al., 2006). The delayed tolerance induced by CSD is protein synthesis dependent and has been linked to the up-regulation of trophic factors and glial cell activation (Kawahara et al., 1999). On the other hand, spreading depression in organotypic hippocampal slice cultures increases the expression of several pro-inflammatory cytokines including TNF-α, IL-1β, IL-1α, and IL-6 (Kunkler et al., 2004). Indeed, in vivo experiments found an early induction of both TNF-α and IL-1β after CSD (Jander et al., 2001). Pre-treatment with the non-competitive N-methyl-D-aspartate (NMDA) receptor antagonist MK-801 completely abolished the upregulation of these cytokines, implicating NMDA receptors as a critical element driving their production. Additionally, Horiguchi et al. reported that CSD-induced neuroprotection against ischemic injury resulting from MCAo is dependent upon increased COX-2 expression (Horiguchi et al., 2005, 2006), supporting the involvement of the inflammatory response in CSD PC.
Electrical stimulation of the cerebellar fastigial nucleus, but not other cerebellar nuclei, can induce potent and long-lasting protection from focal or global cerebral ischemic injury (Reis et al., 1991, 1998; Golanov et al., 1998). At the same time, fastigial nucleus stimulation evokes a strong anti-inflammatory response and suppresses post-ischemic iNOS expression and brain inflammation after cortical IL-1β injection (Galea et al., 1998a,b). The effect is mediated, at least in part, by increasing the tolerance of mitochondria to calcium overload, and suppressing the release of mitochondrial pro-apoptototic factors induced by cerebral ischemia (Zhou et al., 2005). The molecular mechanisms of the protective effects on mitochondria involve upregulation of prohibitin, an integral protein of the inner mitochondrial membrane, which protects mitochondrial structure and function during cell stress (Zhou et al., 2012). Consistent with its role in PC, overexpression of prohibitin renders neurons more resistant to injury in a wide variety of models (Zhou et al., 2012).
Neuroprotection against cerebral damage induced by lethal ischemic/hypoxia or global ischemia can also be acquired through induction of mild epileptic activity elicited by kainic acid injections (Plamondon et al., 1999; Towfighi et al., 1999). Although the mechanisms are unknown, synthesis and release of TNF-α, IL-1β, IL-1α, and IL-6 by glia might contribute to this tolerance modality (Vezzani et al., 2002).
Exercise PC
Physical exercise prevents stroke and forced exercise training affords neuroprotection against ischemic injury (Endres et al., 2003). Experimentally, exercise PC can be achieved by training rodents on a motor driven treadmill for 1–3 weeks (Wang et al., 2001; Ding et al., 2005; Curry et al., 2010). Among other mechanisms, such as protection against the blood-brain barrier (BBB) disruption, promotion of angiogenesis and inhibition of apoptosis, exercise PC involves activation of the immune system (Zhang et al., 2011). The skeletal muscle is an important source of several cytokines, often referred to as myokines, including IL-6, IL-8, IL-15, BDNF, LIF, and FGF21 (Pedersen, 2011). The release of these myokines into circulation might be responsible for the systemic effects of exercise, including its neuroprotective potential (Iadecola and Anrather, 2011b). Downstream mediators of exercise PC may include TNF-α (Ding et al., 2005) and extracellular signal-regulated kinases 1 and 2 (ERK1/2) (Guo et al., 2008; Curry et al., 2010). In one study, exercised rats submitted to MCAo showed reduced infarct volume when compared to non-exercised rats and this protection was associated with a gradual increase in the level of TNF-α in the brain over the 3 week course of treadmill exercise (Ding et al., 2005). Pre-ischemic blockage of TNF-α signaling with an anti-TNF-α antibody or inhibition of ERK1/2 activation abolished the protective effect (Guo et al., 2008). Exercise PC can also change the expression of innate immunity receptors. Treadmill exercise decreased cerebral TLR4 receptor expression in rats, although the importance of this finding as a mechanism for cerebral IT has not been investigated to date (Zwagerman et al., 2010).
Molecular Components of the Immune System Involved in PC
Inflammatory signaling is governed by a complex array of molecules that not only modulate the response of the target cell, but also change the local microenvironment and cellular tissue composition by recruiting cell types that are not normally present in that tissue. This inflammatory cascade is driven by cytokines, chemokines and their receptors and is regulated by immunomodulatory molecules. Several of these components have been implicated in PC. Evidence for direct involvement of some of these immune factors in cerebral IT is summarized in Table 3.
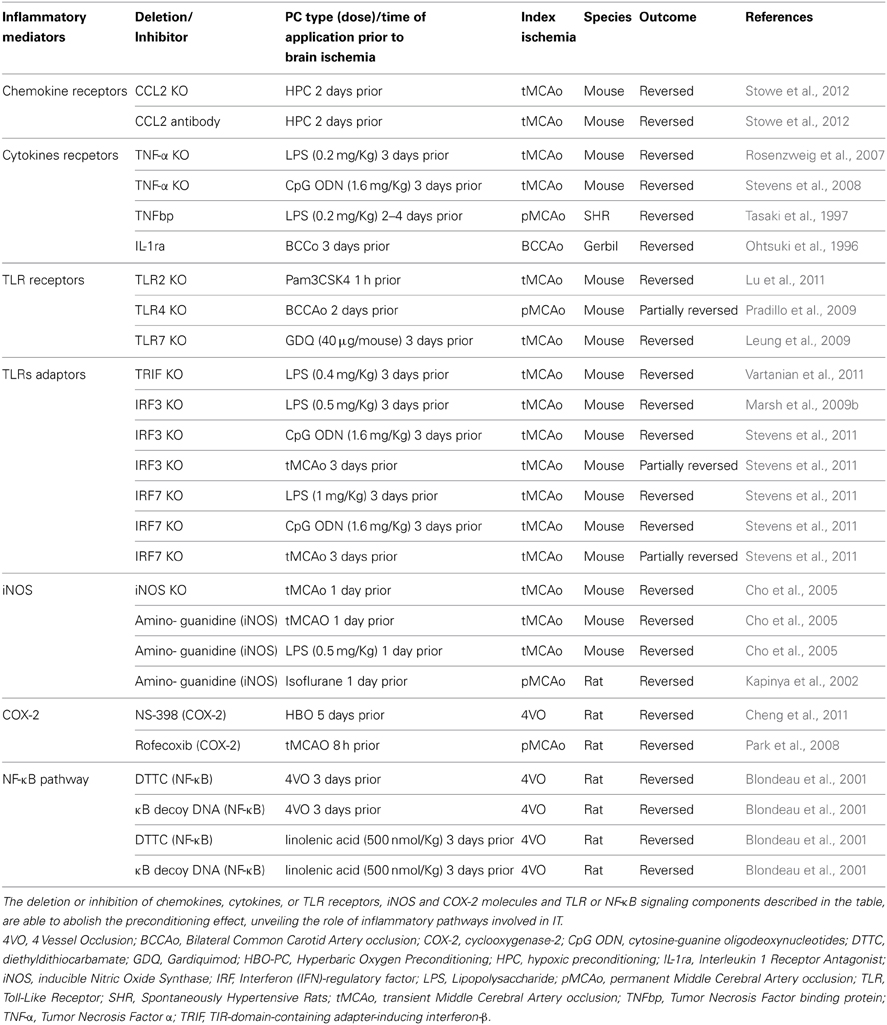
Table 3. Evidence for the involvement of inflammatory mediators in PC.
TLRs
The Toll receptor was first described in the context of Drosophila embryogenesis (Anderson et al., 1985). Subsequently, it was discovered that the Toll pathway is activated upon Drosophila fungal infection revealing a role of Toll receptors in the immune response (Lemaitre et al., 1996). Exposure of immune cells to certain pathogens activates these receptors resulting, in the activation of intracellular signaling pathways that lead to the expression of genes involved in the inflammatory response. In vertebrates, TLRs, named for their homology to the toll gene in Drosophila, are transmembrane proteins containing domains that are able to recognize viral or microbial components known as pathogen-associated molecular patterns (PAMPs) (Wang et al., 2011). Peptidoglycans (PGN), lipoproteins, lipoteichoic acids (LTA), lipopolysaccharides (LPS), as well as viral and bacterial nucleic acids, serve as PAMPs recognized by individual TLRs (Akira, 2009). To date 13 members of the TLR family have been identified in mammals. Among the most extensively studied TLRs are TLR2 which binds to LTA component of Gram-positive bacteria and TLR4, which recognizes LPS on the cell wall of Gram-negative bacteria (Verstak et al., 2007). Upon ligand binding, TLRs further signal by recruiting intracellular Toll/IL-1R (TIR)-homology domain containing adaptor proteins, which selectively activate signaling cascades that lead to immune responses. Several TLR adapter molecules that are associated with functionally different signaling cascades, such as myeloid differentiation factor-88 (MyD88) and TIR domain containing adaptor protein inducing interferon β (TRIF), are known to date. With the exception of TLR3, TLRs signal through MyD88-dependent pathways, while TLR4 can activate both MyD88-dependent and—independent pathways (Wang et al., 2011; Mallard, 2012).
MyD88 mobilizes members of the IL-1R-associated kinase family (IRAK) leading to nuclear translocation of the transcription factor NF-κB. NF-κB is a major regulator of the inflammatory response (Li and Verma, 2002) and is responsible for the induction of various inflammatory genes including cytokines, chemokines, adhesion molecules, proinflammatory enzymes, and growth factors (Pahl, 1999). NF-κB is commonly found as a heterodimer consisting of p50 (NFKB1) and p65 (RelA) subunits. In resting cells, the inactive form of NF-κB is located in the cytoplasm by association with inhibitor proteins, IκBα and IκBβ (Baeuerle and Henkel, 1994). Upon stimulation, IκB proteins are phosphorylated and proteolytically degraded, and NF-κB dimers are able to translocate to the nucleus and bind to the promoter region of target genes initiating their transcription (Baldwin, 1996). NF-κB is activated in many cell types including cells of the central nervous system (Kaltschmidt et al., 1994). In the rodent brain, NF-κB can be activated by numerous factors known to be induced after ischemia-reperfusion, such as glutamate, increased intracellular Ca2+, reactive oxygen species (ROS) and inflammatory cytokines (Harari and Liao, 2010). Furthermore, target genes of NF-κB have been implicated in the pathogenesis of cerebral ischemia, such as IL-1, IL-6, TNF-α, ICAM-1, MMP9, COX-2, and iNOS (Baeuerle and Henkel, 1994; Allan et al., 2005; Kaltschmidt et al., 2005; Harari and Liao, 2010). Thus, it has been postulated that stimulation of signaling pathways that lead to NF-κB activation are associated with detrimental outcome in cerebral ischemia (Stephenson et al., 2000). Supporting this notion, inhibition of NF-κB using pharmacologic agents results in reduced infarct size after stroke in rodents (Clemens et al., 1998; Nurmi et al., 2004). It has also been shown that mice lacking the p50 NF-κB subunit have reduced ischemic damage (Schwaninger et al., 1999; Nurmi, 2004). However, subsequent studies have also revealed a deleterious effect of NF-κB inhibition in neonatal hypoxia (Nijboer et al., 2008) and an increase of neuronal death after transient ischemia in p50 deficient mice (Duckworth et al., 2006) raising the possibility for a dual role of NF-κB in stroke.
TLR4 stimulation also leads to the activation of MyD88-independent pathways. In this case, binding of the adaptor molecule TRIF activates the transcription factors interferon regulatory factor 3 (IRF3) and 7 (IRF7), resulting in expression of IFNα and IFNβ type I IFN genes (Marsh et al., 2009a; Wang et al., 2011; Mallard, 2012) (Figure 2). Type I IFNs are released in the extracellular space and signal in an auto- or paracrine manner through a single heterodimeric receptor, IFNAR, to activate the JAK/STAT pathway, leading to the expression of several chemokines such as CCL2, CCL7, and CXCL10, while inhibiting CXCL1 and CXCL2 expression. Depending on the concentration of IFN and type of target cell, the resulting immune response can be pro- or anti-inflammatory (Trinchieri, 2010).
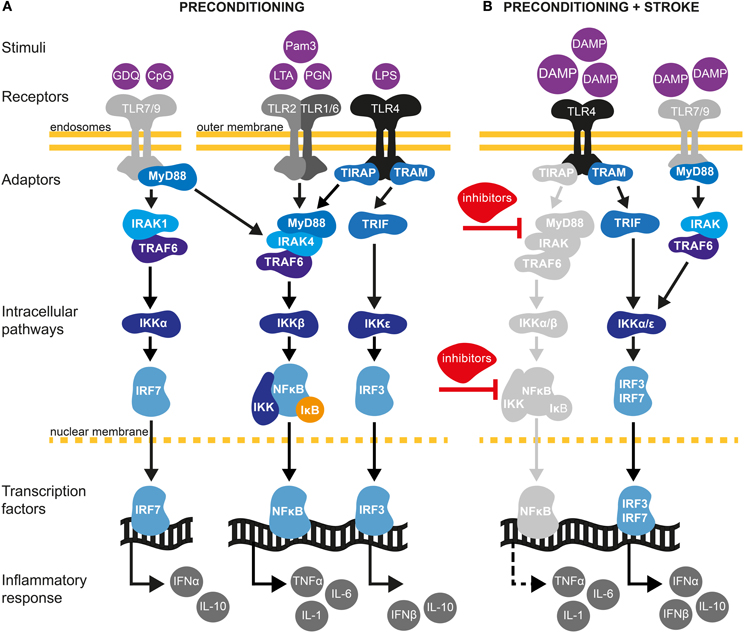
Figure 2. TLR signaling pathways in cerebral IT. (A) Preconditioning through TLRs may be afford by exposure with low dose of pathogen-associated molecular pattern (PAMPs) molecules (Mallard, 2012). TLRs are localized either at the outer cell surface (TLR1, TLR2, TLR4, and TLR6) or within endosomes (TLR7 and TLR9) (Marsh et al., 2009a). TLR2 dimerizes with TLR1 or TLR6 and is activated upon binding of PAMPs such as the synthetic lipopeptide Pam3CysSerLys4 (Pam3CSK4), the non-peptide ligand lipoteichoic acid (LTA) or peptidoglycans (PGN). TLR4 predominantly recognizes lipopolysaccharides (LPS) from Gram-negative bacteria. TLR7 and TRL9 can be activated by the synthetic imidazoquinoline Gardiquimod (GDQ) or small DNA such as non-methylated cytosine-guanosine (CpG), respectively (Wang et al., 2011). Preconditioning of TLRs will favor the activation of the myeloid differentiation factor-88 (MyD88)-dependent pathway. Upon binding of ligands, MyD88 is activated either directly (TLR7/9) or via the toll-interleukin 1 receptor (TIR) domain containing adaptor protein TIRAP (TLR2/4). The latter further mobilizes members of the IL-1R-associated kinase family (IRAK) leading to the subsequent binding/activation of TNF receptor associated factor-6 (TRAF6), the inhibitor of κB kinase (IKK) complex (composed of IKKα and IKKβ) and IκB. Once IκB is phosphorylated and degraded by the proteasome, nuclear factor-κB (NF-κB) translocates to the nucleus leading to low level of pro-inflammatory cytokine synthesis, such as TNF-α. TLR PC also induces the production of several negative inhibitors of TLR signaling, targeting mainly MyD88 and NF-κB pathways. Induction of proinflammatory cytokines and activation of such inhibitors are required to induce a state of ischemic tolerance (Marsh et al., 2009a; Vartanian and Stenzel-Poore, 2010; Wang et al., 2011). In addition to activating the MyD88-dependent pathway, TLR4 signals through the MyD88-independent pathway to activate IRF3 by sequential recruiting of the adaptor molecule TIR inducing interferon β (TRIF) and IKKε resulting in expression of anti-inflammatory type I interferon (IFN) genes, including IFNβ (Marsh et al., 2009b). Type I IFNγ genes are also induced by TLR7/9 activation through the TRAF6/IKKα pathway (Hoshino et al., 2006). (B) Damage-associated molecular pattern (DAMPs) molecules are endogenous ligands of TLRs produced in response to ischemic injury (Vabulas et al., 2002). Following preconditioning, activation of TLR4/7/9 by DAMPs will fail to activate MyD88 and the subsequent signaling molecules, such as IRAK and NF-κB. Indeed, several inhibitors produced during the first exposition of TLRs to exogenous stimuli will block the signaling of TLRs to NF-κB. Whether pro-inflammatory cytokines are suppressed or up-regulated is controversial and might be model specific (Marsh et al., 2009a; Vartanian and Stenzel-Poore, 2010). On one hand, TLR preconditioning induces a down-regulation of NF-κB. On the other hand, it has been shown that neuroprotection following cerebral ischemia is achieved by an up-regulation of the TLR-IRF axis generating an increase of TGF-β, IL-10, IFNα, and IFNβ anti-inflammatory cytokines (Stevens et al., 2011; Leung et al., 2012). Thus, TLR preconditioning induces a reorganization of the TLR signaling pathways after cerebral ischemia toward TRIF/IRF signaling that may confer protection of the brain against ischemic damage.
Aside from binding to PAMPs, TLRs also recognize endogenous molecules known as DAMPs, which are released during ischemic cellular injury (Chen and Nuñez, 2010). Prototypical DAMPs released from injured cells include high-mobility group box 1 (HMGb1) protein that activates TLR2, 4, and 9; heat shock proteins (HSPs; TLR2, 4); RNA (TLR3); mitochondrial DNA (TLR9); hyaluronic acid (TLR2, TLR4) and peroxiredoxins (TLR2, TLR4) (Chen and Nuñez, 2010; Patel et al., 2012; Shichita et al., 2012). HMGb1 and peroxiredoxins have been reported to increase cerebral ischemic injury by augmenting the post-ischemic inflammatory response (Kim, 2006; Yang et al., 2011; Shichita et al., 2012). While the role of DAMPs in cerebral IT has not been addressed, it has been shown that systemic administration of HMGb1 protects against myocardial ischemia–reperfusion injury and liver ischemia-reperfusion injury, a process that involves TLR4 (Izuishi et al., 2006; Hu et al., 2010). Additionally, HSP70 mediates endotoxin tolerance by signaling through TLR4 receptors (Aneja et al., 2006). Therefore, these findings support DAMPs to participate in the PC effect through TLRs activation. In contrast to the detrimental effect of TLR activation in response to ischemia (Wang et al., 2011), stimulation of some TLRs prior to ischemia provides robust neuroprotection (Marsh et al., 2009a; Wang et al., 2011). The ability of TLR ligands to induce cerebral IT was first demonstrated after systemic administration of low-dose LPS, the major TLR4 ligand, causing spontaneously hypertensive rats to become tolerant to subsequent ischemic brain damage induced by MCAo (Tasaki et al., 1997). Subsequently, LPS-induced tolerance to brain ischemia has been demonstrated in several experimental stroke models in mice, gerbils and pigs (Rosenzweig et al., 2004; Hickey et al., 2007; Yu et al., 2010). A dose below 1 mg/Kg of LPS administered 1–3 days prior to cerebral ischemia is generally used in rodents to induce IT. Furthermore, TLR4-mutant mice were refractory to IPC, indicating that TLR4 plays an universal role in establishing IT (Pradillo et al., 2009). Comparable to TLR4, pre-treatment with the TLR2 lipopetide ligand Pam3CysSerLys4 (Pam3CSK4) resulted in significantly decreased cerebral infarct volume after focal cerebral ischemia/reperfusion in mice (Hua et al., 2008). Preservation of the BBB has been suggested to play a role in this PC mechanism. Similarly, stimulation of TLR7 by administration of its agonist gardiquimod, an imidazoquinoline compound, provides neuroprotection against ischemic injury and was associated with the up-regulation of the IFN pathway and IFNα production (Leung et al., 2012). Interestingly and in contrast to TLR4 and TLR9 induced PC, TLR7-mediated PC was abolished in IFNAR mutant mice. Finally, activation of TLR9 by unmethylated CpG oligodeoxynucleotides induced IT after transient MCAo in mice (Stevens et al., 2008). Similar to LPS-PC, the protective effect after TLR9 stimulation lasted for up to a week and was dependent on the expression of TNF-α (Figure 2A) (Stevens et al., 2008; Marsh et al., 2009a). Accordingly, CpG oligodeoxynucleotides administration to TNF-α null mice failed to induce IT.
IT induced by TLR activation involves the reprogramming of inflammatory pathways. This is achieved by at least two different mechanisms. Concomitantly with induction of pro-inflammatory mediators, activation of the MyD88-dependent pathway will result in the upregulation of negative regulators, which prevent the interaction of several adaptors along the TLR4-MyD88 pathway (Figure 2A) (Sly et al., 2004). At the same time, activating the MyD88-independent pathway evokes a type I IFN response. Indeed, recent studies have found a neuroprotective role of the MyD88-independent pathway in TLR-induced IT after stroke (Marsh et al., 2009a). It has been postulated that pretreatment with LPS induces a switch in the transcriptional response to subsequent TLR4 stimulation by increasing the expression of the IRF3-induced cytokine IFNβ (Veldhuis et al., 2003) (Figure 2B). Consistent with this hypothesis, gene expression analysis following LPS-PC revealed upregulation of inhibitors of the Myd88-dependent and NF-κB pathways (Vartanian et al., 2011). Interestingly, the expression of pro-inflammatory genes related to the NF-κB signaling pathway was similar in both mice undergoing LPS-PC and in control mice following cerebral ischemia (Figure 2B). However, the study showed that LPS-PC increased IRF3 activity following ischemia and evoked the upregulation of anti-inflammatory genes including TGF-β and IL-10 (Vartanian et al., 2011). The importance of the MyD88-independent pathway in establishing IT is further supported by the fact that LPS-PC was unable to induce IT in TRIF- but not in Myd88-deficient mice (Vartanian et al., 2011).
Cytokines
Cytokines are secreted proteins with growth, differentiation, and activation functions that shape the nature of the immune response. Among the more than 70 candidate cytokines, TNF-α, IL-1, IL-6, and IL-17 play major roles in initiating and amplifying the post-ischemic inflammatory response, while IL-10 and TGF-β are the main anti-inflammatory factors (Iadecola and Anrather, 2011a). Since immune cells, including monocytes/macrophages, in the periphery and microglia in the brain express TLR receptors (Downes and Crack, 2010), they are capable of responding to systemic TLR agonists used as a preconditioning stimulus and modify cytokine expression or secretion (Rosenzweig et al., 2004; Ransohoff and Brown, 2012). Accordingly, several cytokines secreted by cells of the innate immune system have been implicated in PC. For example, TNF-α has been identified as an essential mediator of LPS-PC. It has been shown that LPS-preconditioned TNF-α null mice were not protected from ischemic brain injury (Rosenzweig et al., 2007). This study confirmed previous findings demonstrating that administration of a specific TNF antagonist reversed the protective effect of LPS PC in a model of permanent focal ischemia in mice (Tasaki et al., 1997). Similarly, upregulation of IL-1α and IL-1β after bilateral common carotid artery occlusion (BCCAo) triggered tolerance to global ischemia (Ohtsuki et al., 1996; Shin et al., 2009). Ohtsuki et al. also showed that systemic delivery of either IL-1α or IL-1β was sufficient to induce IT to a subsequent episode of lethal global ischemia and that intraperitoneal injection of recombinant human IL-1 receptor antagonist prior to IPC abolished the neuroprotective effect.
In addition to systemic administration of cytokines, neuroprotection has also been reported with direct delivery of cytokines to the brain before ischemia. Previous intracerebrovascular injection of IL-6 in rats subjected to permanent ischemia protects from cell death (Loddick et al., 1998). Likewise, intracisternal administration of TNF-α in mice before distal MCAo is neuroprotective (Nawashiro et al., 1997). However, in the same study, both intravenous and intraperitoneal routes of TNF-α administration failed to exert neuroprotection against cerebral ischemia. While differences in modality of administration and stroke models may account for the observed disparities in outcome, it remains unclear how systemic cytokines exert their effect within the brain. TNF-α, IL-1β, and IL-6 are transported unidirectionally across the BBB (Banks, 2005). Thus, systemic induction of cytokines can lead to an increase of them in the brain. For instance, IL-1 expression in the brain has been documented after an increase of circulating IL-1 induced by LPS PC (Gabellec et al., 1995). On the other hand, inducers of PC, such as LPS and cytokines, can act through interaction with receptors in circumventricular organs that lack a BBB, triggering upregulation of cytokine levels in the brain. Accordingly, it has been shown that perivascular cells and neurons in the circumventricular organs produce TNF-α in response to systemically administered LPS (Breder et al., 1994).
Chemokines
Chemokines are small molecules involved in the recruitment of immune cells to sites of inflammation or injury (Charo and Ransohoff, 2006). In the central nervous system, chemokines are highly upregulated in neuropathologic conditions. Neurons, astrocytes, microglia, endothelial cells as well as circulating leukocytes are potential sources of chemokines. Several chemokines have been linked to the pathogenesis of ischemic brain injury (Jaerve and Müller, 2012). Upon release, they exacerbate tissue injury by increasing leukocyte and monocytes/macrophages infiltration, potentiating neuronal injury. However, chemokines also possess neuroprotective and neurotrophic functions (Semple et al., 2009). The prevalence of beneficial or detrimental effects may depend on various factors, including the type of the chemokine, its concentration, the time course of production relative to the time of injury and the target cell type (Semple et al., 2009). The contribution of chemokines to PC has been investigated in several models of IT. For instance, HPC or BCCAo prior to transient or global MCAo resulted in reduced infarct size attributed to CCL2 (MCP-1), a leukocyte-derived pro-inflammatory chemokine (Rehni and Singh, 2012; Stowe et al., 2012). Following HPC, expression of CCL2 increased in both neurons and cerebral endothelial cells prior to the induction of the ischemic injury. Moreover, the induction of IT was blocked by CCL2 neutralizing antibodies and was not observed in CCL2-knockout mice (Stowe et al., 2012). Although the protection conferred by HPC is associated with the number of CCR2+ monocytes in blood and leaving the circulation, their direct protective role in brain IT needs to be further characterized. Rehni et al. have shown that IT induced by BCCAo PC was lost after treatment with a selective antagonist of CCR2 (Rehni and Singh, 2012), corroborating the role of CCL2 chemokines and its receptor in IT.
Cannabinoids
The endocannabinoid system consists of the cannabinoid type 1 (CB1) and type 2 (CB2) receptors and their ligands (Howlett et al., 2002). The expression of these two receptor subtypes varies among tissues. CB1 receptors are abundant in the CNS and are also present in several peripheral tissues, albeit to a lesser extent (Matsuda et al., 1990; Galiègue et al., 1995). Although also present in the brain (Gong et al., 2006), CB2 is mostly expressed in immune cells (Galiègue et al., 1995). Cannabinoid receptors may be activated upon binding of their endogenous ligands arachidonoyl ethanolamide or anandamide (AEA), 2-arachidonoylglycerol (2-AG), and 2-arachidonyl glyceryl ether (noladin ether), or synthetic analogs which are derivatives of herbal cannabinoids, such as the terpenoid Δ9-tetrahydrocannabinol (THC), the active compound of the cannabis plant (Di Marzo et al., 1998). Both CB1 and CB2 receptors are members of the seven-transmembrane G-protein coupled receptor superfamily. They may initiate downstream signaling pathways that activate potassium channels, phosphatidylinositol-3-kinase and mitogen-activated protein kinases (Di Marzo et al., 1998). The CNS endocannabinoid system has a variety of physiological roles, which are mainly mediated by CB1 receptors. These include, but are not limited to: psychotropic effect, pain inhibition, memory function and increased appetite (Ameri, 1999; Pacher and Haskó, 2008). Modulation of immune responses and the release of inflammatory mediators have been primarily attributed to CB2 receptors (Pacher et al., 2006; Ullrich et al., 2007; Elliott et al., 2011).
The endocannabinoid system has been implicated in ischemic injury (Pacher and Haskó, 2008). Several studies have shown a neuroprotective effect of CB activation in brain ischemia (Nagayama et al., 1999; Panikashvili et al., 2001) by decreasing intracellular Ca2+ (Zhuang et al., 2005), modulating brain temperature (Leker et al., 2003), inhibiting pro-inflammatory signaling cascades (Panikashvili et al., 2005) and by preventing endothelial cell activation and leukocyte adhesion (Zhang et al., 2007, 2009). There is emerging evidence that cannabinoids also play a role in PC. Induction of IT by electroacupuncture PC improves neuronal survival in a rat model of focal ischemia (Wang et al., 2005, 2009; Ma et al., 2011). While early PC in this model was dependent on the activation of CB1 receptors as demonstrated by the reversal of the protective effect after administration of the CB1 receptor antagonist AM251 or CB1 siRNA (Wang et al., 2009), delayed PC was dependent upon the activation of CB2 receptors (Ma et al., 2011). It was shown that IT was partially reversed when animals were treated with the specific CB2 antagonist AM630. Inhibition of the CB1 receptor, however, did not block the induction of delayed IT. Although the cellular components of this response have not been investigated, the nearly exclusively expression of CB2 receptors on immune cells makes it possible that modulation of the immune response accounts for the observed neuroprotection.
iNOS
Nitric oxide (NO) is produced by nitric oxidase synthase (NOS) through oxidation of the guanidino nitrogen of L-arginine. Endothelial (eNOS), neuronal (nNOS) and inducible or inflammatory (iNOS) isoforms are found in the brain, and play important roles in IT (for recent review see (Iadecola et al., 2011). iNOS is specifically expressed under pathological conditions, typically those associated with inflammation (Nathan, 1997). Data from our laboratory have shown that iNOS mediates IT following either IPC or LPS-PC in a mouse model of transient MCAo (Cho et al., 2005). PC-mediated neuroprotection was abolished when the selective inhibitor, aminoguanidine, was delivered prior to PC, or when iNOS null mice were used. iNOS expression and accumulation of peroxynitrite in cerebral blood vessels was observed 24 h after IPC (Cho et al., 2005), whereas LPS-PC was associated with accumulation of peroxynitrite in neurons and vessels (Kunz et al., 2007). The role of iNOS in PC has also been reported with anesthetic preconditioning (Kapinya et al., 2002). Prior treatment with isoflurane or halothane, which reduces infarct volume after permanent MCAo, induced iNOS expression in the cortex 18–24 h after PC. Analogous to the studies mentioned above, anesthetic PC was blocked when animals were treated with the iNOS inhibitor aminoguanidine.
COX-2
Similar to iNOS, brain cycloxygenase-2 (COX-2) is markedly upregulated during post-ischemic inflammation and its reaction products contribute to the evolution of ischemic damage (Iadecola et al., 2001). Likewise, the role of COX-2 has been investigated in several PC paradigms. Increased brain levels of COX-2 following PC by CSD, was associated with the development of cerebral IT in a rat model of MCAo (Horiguchi et al., 2006). IPC by transient MCAo induced COX-2 and HO-1 expression, and significantly reduced infarct volume after index MCAo. Administration of the COX-2-selective inhibitor rofecoxib abolished the neuroprotective effect of IPC, indicating a key role of COX-2 in establishing IT (Park et al., 2008). More recently, the role of COX-2 in PC induced by HBO has also been reported (Cheng et al., 2011). COX-2 induction is associated with protection against subsequent global cerebral occlusion in rats and IT was abolished by treatment with COX-2 selective inhibitor NS-398. Contrary to IPC, no role of COX-2 in LPS PC was found in a excitotoxic model of brain injury induced by NMDA (Kawano et al., 2007).
ROS
ROS play a key role in the pathogenesis of the ischemic cascade. ROS are mainly generated after cerebral ischemia by the injured tissue during the acute phase (minutes-hours) and by infiltrating leukocytes during the sub-acute ischemic phase (Chan, 2001; Kahles et al., 2007). For example, neutrophils that infiltrate the ischemic brain produce excessive superoxide via NADPH oxidase, contributing to the exacerbation of ischemic injury (Kunz et al., 2006; Chen et al., 2009; Pun et al., 2009). In preconditioning, the role of ROS via inflammatory mechanisms has also been reported. Kunz et al. showed improvement of cerebrovascular function after transient MCAo in LPS preconditioned mice, an effect associated with peroxynitrite formation (Kunz et al., 2007). Interestingly, peroxynitrite was specifically formed after PC from the reaction of iNOS-derived NO and Nox2 (NADPH oxidase)-derived superoxide, and was not observed in iNOS or Nox2-null mice. Moreover, peroxynitrite was also found to be beneficial in the tolerance induced by LPS-PC to brain injury resulting from cortical injection with NMDA (Kawano et al., 2007).
Epigenetic Mechanisms
In delayed PC, the time window during which IT is observed can last for days or even weeks. This contrasts with the limited duration of inflammatory signaling where responses are often short lived, and several negative feedback loops are in place to restrict the duration of the inflammatory response. This is achieved by the expression of a series of cellular inhibitors that are concomitantly produced along with pro-inflammatory molecules, leading to a timely termination of gene expression after the stimulus subsides. These inhibitors can directly target transcription factors, as is the case with NF-κB, which is sequestered in the cytoplasm by newly synthetized IκB proteins—or, by inactivating key proteins within the pro-inflammatory signaling cascade. For example, A20, an ubiquitin editing enzyme that is highly inducible by pro-inflammatory stimuli, targets several proteins in the TNFR signaling cascades by removing K63-linked ubiquitin chains from TRAF2, TRAF6 and NEMO. It results in suppression of NF-κB signaling, and by replacing K63- with K48-linked ubiquitin chains in RIP1, thus targeting it for proteasomal degradation (Verstrepen et al., 2010). Similarly, suppressor of cytokine signaling (SOCS), a family of 40 related proteins, and PIAS (protein inhibitor of activated STAT) target primarily the JAK (Janus kinase)/STAT (signal transducer and activator of transcription) pathway, which is the main cellular transducer of cytokine signals (Yoshimura et al., 2007). Accordingly, short-acting inflammatory signals, as applied during PC protocols, are transient, and not suitable to alter the cellular state for an extended period of time. This view is supported by experimental data in ischemic and LPS preconditioned animals, that show transient global alterations of gene expression profiles after PC, which have largely dissipated by the time of index ischemia (Stenzel-Poore et al., 2003; Marsh et al., 2009b). This raises the question of how the “memory” of the PC event is preserved if the attendant mRNA response has already subsided. An essential feature of such a mechanism, is its persistence beyond the initial stimulation and its decay over time, thus, equipping the cell or organ with a relatively short lived memory of a previous exposure to a potentially lethal stressor. One possibility is that the protein products of the subsided transcriptional activity are still present at the time of induction of ischemia, which could steer the response from injury toward protection. Another possibility is that the transcriptional response to PC induces longer lasting epigenetic changes that shape the genetic response when injurious ischemia occurs. Epigenetic mechanisms include all processes that regulate gene expression without alteration of the underlying DNA sequence, and include, for example, histone modifications, DNA methylation, and non-coding RNAs.
Histone and DNA Modifications in Preconditioning of the Immune System and the Brain
Histone modifications by methylation, acetylation, phosphorylation, ubiquitination, and sumoylation are key events in regulating chromatin structure and gene expression. The mechanism by which these modifications alter gene expression is not entirely understood, but may involve tethering of chromatin remodeling machinery, transcriptional repressors and activators. While acetylation of N-terminal histone tails are a hallmark of actively transcribed genes, it is less likely that histone acetylation plays a major role in imprinting a lasting memory from a previous exposure. Supporting this view is the fact that histone deacetylase (HDAC) is recruited early to the promoter of induced genes, in most cases during the gene activation process itself, resulting in a high turnover rate of acetylation marks in the order of minutes and fast disposal of this modification once transcription of the particular gene subsides (Hazzalin and Mahadevan, 2005). On the other hand, DNA methylation is a very thermostable epigenetic mark and, with the possible exception of 5-hydroxymethylcytosine, refractory to environmental stimuli. Therefore, DNA methylation is unsuited to act as an on-off switch for epigenetic memory (Qureshi and Mehler, 2010). Histone H3 tri-methylation at lysine 4 (H3K4me3), in contrast, has been shown to mark active or poised enhancers for an extended period of time. In a model of Candida albicans infected macrophages, persistent H3K4me3 modifications in the enhancer regions of inflammatory genes was associated with a more robust inflammatory response and reduced re-infection rate upon re-exposure (Quintin et al., 2012). Notably, the K4 trimethylation was observed for up to a week after deposition. The importance of H3K4me3 in changing transcriptional responses has been shown in LPS-preconditioned macrophages (Foster et al., 2007). Macrophages pre-exposed to LPS responded to a second LPS exposure with a transcriptional program distinct from that of naïve macrophages exposed to LPS. Genes could be divided into “tolerizeable” genes (T), which were suppressed in tolerant macrophages, and “non-tolerizeable” genes (NT), expressed at higher levels in tolerant than non-tolerant macrophages. It was found that promoter regions of T-class genes were refractory to histone H4 acetylation and H3K4 trimethylation, two modifications positively correlated with transcriptional activity, upon the second LPS challenge (Foster et al., 2007). Depending on the chromatin status in the resting state, genes can be divided in primary and secondary response genes (Ramirez-Carrozzi et al., 2006). Primary genes show a chromatin structure that is suited for immediate transcription and their activation is independent of new protein synthesis. Secondary genes show histone modifications characteristic of inactivated genes and the removal of this block and gene induction is protein synthesis dependent. Interestingly, this study found that LPS-PC changed many genes from secondary to primary response genes upon LPS re-stimulation, further implicating that epigenetic memory has been established in this model.
Several histone modifiers have been implicated in the PC response of the brain, including sirtuin family HDACs (Morris et al., 2011) and polycomb group proteins (PcG), a multimeric protein complex that mediates gene silencing and PC (Stapels et al., 2010). In addition to their role in neuronal physiology, both protein families have been implicated in the regulation of immune function (Swigut and Wysocka, 2007; Gallí et al., 2011). Whether they also regulate the immune signaling during PC, however, has not been directly addressed thus far.
microRNAs and Long Non-Coding RNAs
microRNAs (miRNAs) are small non-coding RNAs (18- to 24- nucleotides) that repress translation of mRNAs. Mature miRNAs are excised from a precursor RNA by the activity of Dicer and are guided to the 3′-untranslated region (3′-UTR) of mRNAs targets by the RNA-induced silencing complex (RISC). This leads to the down-regulation of gene expression via degradation or translational inhibition (Bartel, 2009). miRNAs have an essential quality that makes them compatible with the maintenance of preconditioning memory, namely their stability (Rüegger and Großhans, 2012). In addition, they are inducible by environmental stimuli, and are regulated at the levels of transcription, biogenesis, stability and decay. Supportive for a role of some miRNAs in establishing epigenetic memory, is the fact that they are extremely long lived, more than 12 days in the case of miR-208 in the heart (van Rooij et al., 2007). Furthermore, ablation of many miRNAs is well tolerated in the mouse, with these mutant animals only developing substantially altered phenotypes after being subjected to some kind of stress. These observations suggest that the persistence of inducible miRNAs due to their long half-lives after initial stimulation, may enable the maintenance of gene-expression programs that enhance the resistance of cells to repeated stress-exposure. When such “memory” miRNAs are absent (such as in animals depleted of the genes encoding the miRNAs), aberrant phenotypes become apparent (Leung and Sharp, 2010). The findings that miRNA can be actively secreted and transferred from cell-to-cell via microvesicles makes it possible that miRNA can work at a distance by transferring epigenetic signatures from a stimulus-exposed cell to a cell that has not encountered the particular environmental stressor (Valadi et al., 2007).
It is estimated that 500–1000 different miRNAs are expressed in mammalian cells and several miRNAs have been implicated in the regulation of the immune response and inflammation (O'Connell et al., 2012). Certain miRNAs, for instance, miR-9, -21, -146a, -147, -203, are induced in response to TLR activation, and serve as negative feedback regulators of the MyD88-dependent TLR signaling pathway and NF-κB activity (O'Neill et al., 2011). Conversely, miR-155 enhances the inflammatory response by down-regulating negative regulators Src Homology-2 domain-containing inositol-5′-phosphatase 1 (SHIP1) and SOCS1 leading to prolonged activation of AKT and IFN signaling pathways (O'Connell et al., 2009).
Cerebral ischemia-reperfusion injury alters the expression of several miRNAs (Dharap et al., 2009; Wang et al., 2013). Interestingly, IPC and HPC have been shown to rapidly alter miRNA levels in the brain (Dharap and Vemuganti, 2010; Lee et al., 2010; Liu et al., 2012). Although it is unknown if these alterations directly affect immune pathways participating in PC, the finding that expression of miRNAs involved in inflammatory signaling is altered by IPC in mice, is suggestive for such a role. For example, miR-17-5p, an inhibitor of monocyte maturation, and the TLR4-targeting miRNA let-7e were down-regulated in the cerebral cortex of mice after IPC (Lusardi et al., 2010). Furthermore, the peroxisome proliferator-activated receptor gamma (PPARγ)-targeting miR-27b and miR-146a, which regulates Traf6, IRAK1, and IRAK2 expression, were up-regulated after PC by transient MCAo in rats (Dharap and Vemuganti, 2010). The same study showed down-regulation of miR-145, which targets the essential TLR4-associated signaling molecule MyD88-adapter-like (MAL). Tellingly, monocyte chemotactic protein-induced protein 1 (MCPIP1), a multifunctional protein with RNAse activity that antagonizes Dicer, and thereby inhibits miRNA biogenesis (Suzuki et al., 2011), has been shown to abrogate the effects of LPS-PC in a mouse model of transient cerebral ischemia (Liang et al., 2011). Taken together, although PC-induced regulation of immune components by miRNA has not been conclusively shown to contribute to cerebral IT, the correlative data gathered so far are supportive for a role of miRNAs in regulation of immune pathways during PC.
Long non-coding RNAs range from 200 bp to several kb, and are expressed in a cell type specific manner (Mercer et al., 2009). Although most of these molecules have not been functionally characterized, they may have diverse roles ranging from regulation of chromosome inactivation to control of gene expression (Mercer et al., 2009). Because several long non-coding RNAs have been shown to be inducible and are involved in regulating immune cell activity and inflammatory signaling, they constitute another potential player by which epigenetic processes can establish a cellular memory. Their role in PC has yet to be investigated.
Conclusions
Although post-ischemic inflammation is considered a major pathogenic factor in stroke, emerging evidence supports a beneficial role of selected inflammatory pathways. One such mechanism is the induction of endogenous neuroprotection through ischemic PC (Figure 3). The evidence summarized in this review indicates that the immune system plays a central role in the mechanisms eliciting neuroprotective responses after PC. While activation of IT-inducing immune pathways shows robust neuroprotection in animal models, the clinical translation of such approaches is complicated by side effects including systemic inflammation and immunodepression (Meisel et al., 2005; McColl et al., 2009). The identification of specific pathways and effector molecules involved in establishing inflammatory PC will help to design targeted PC protocols that can be potentially translated into clinical practice (Keep et al., 2010; Anrather and Hallenbeck, 2013; Narayanan et al., 2013). Notably, immune pathways are not only involved in PC induced by inflammatory mediators, but have also been implicated in several PC paradigms that are not triggered by inflammatory signals, such as ischemic and anesthetic PC, attesting a pivotal role for the immune system in mediating and/or establishing IT. Because of the involvement of inflammatory pathways in a wide variety of diseases, many of the immune components involved in PC are prime therapeutic targets and subjects of extensive drug development. Several proteins involved in PC, such as type I IFNs, are currently in clinical use, and re-purposing them for stroke therapy or prevention could provide a fast track for clinical translation. However, much remains to be learned. For example, few studies have investigated the role of specific immune cells in IT, an area of research that could result in cell-specific therapies and would therefore mitigate the likelihood of adverse effects associated with administration of inflammatory cytokines. In addition, a better understanding of the epigenetic mechanisms involved in IT could lead to therapies that preserve PC-induced epigenetic changes, thereby substantially extending the time frame in which IT is observed.
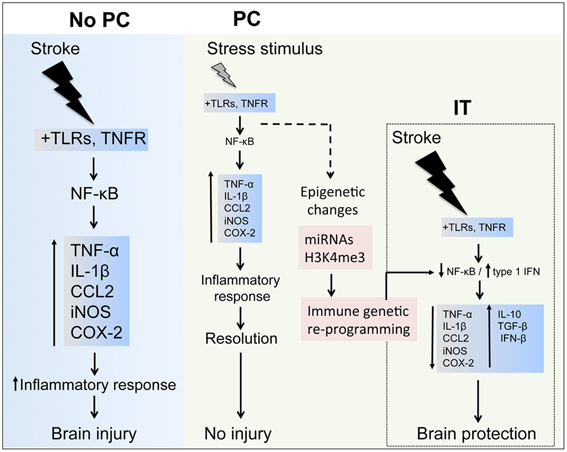
Figure 3. Inflammatory components in stroke and preconditioning. Stroke induces a major inflammatory response through toll-like receptors (TLRs) and tumor necrosis factor receptor (TNFR) signaling, which activate nuclear factor-κB (NF-κB) resulting in upregulation of inflammatory molecules, such as TNF-α, interleukin (IL)-1β, CCL2, inducible nitric oxidase synthase (iNOS) and cyclooxygenase (COX)-2, contributing to the ischemic brain injury. In preconditioning (PC), the exposure to a wide range of stressors activates inflammatory pathways and leads to upregulation of inflammatory molecules similarly to those induced by stroke. After the effects of the stressor have subsided, the inflammatory response is resolved during the early phase of PC and gene expression returns to basal levels. However, longer lasting epigenetic changes induced in the immune system components by micro-RNAs (miRNAs) and histone methylation (i.e., histone H3 trimethylation at lysine 4, H3K4me3) may reprogram inflammatory pathways to respond differently after an episode of severe ischemia, for instance favoring the expression of anti-inflammatory cytokines (IL-10, tumor growth factor-(TGF)-β and interferon-(IFN)-β that induces ischemic tolerance (IT).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grants NS34179 (Costantino Iadecola) and NS081179 (Josef Anrather). Corinne Benakis was supported by grant P3SMP3 148367 from the Swiss National Science Foundation and the Swiss Foundation for Grants in Biology and Medicine (SFGBM).
References
Abe, T., Shimamura, M., Jackman, K., Kurinami, H., Anrather, J., Zhou, P., et al. (2010). Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke 41, 898–904. doi: 10.1161/STROKEAHA.109.572552
Ahmed, S. H., He, Y. Y., Nassief, A., Xu, J., Xu, X. M., Hsu, C. Y., et al. (2000). Effects of lipopolysaccharide priming on acute ischemic brain injury. Stroke 31, 193–199. doi: 10.1161/01.STR.31.1.193
Akira, S. (2009). Pathogen recognition by innate immunity and its signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 85, 143–156. doi: 10.2183/pjab.85.143
Allan, S. M., Tyrrell, P. J., and Rothwell, N. J. (2005). Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 5, 629–640. doi: 10.1038/nri1664
Ameri, A. (1999). The effects of cannabinoids on the brain. Prog. Neurobiol. 58, 315–348. doi: 10.1016/S0301-0082(98)00087-2
Anderson, K. V., Bokla, L., and Nüsslein-Volhard, C. (1985). Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell 42, 791–798. doi: 10.1016/0092-8674(85)90275-2
Aneja, R., Odoms, K., Dunsmore, K., Shanley, T. P., and Wong, H. R. (2006). Extracellular heat shock protein-70 induces endotoxin tolerance in THP-1 cells. J. Immunol. 177, 7184–7192.
Anrather, J., and Hallenbeck, J. M. (2013). Biological networks in ischemic tolerance—rethinking the approach to clinical conditioning. Transl. Stroke Res. 4, 114–129. doi: 10.1007/s12975-012-0244-z
Baeuerle, P. A., and Henkel, T. (1994). Function and activation of NF-kappaB in the immune system. Annu. Rev. Immunol. 12, 141–179. doi: 10.1146/annurev.iy.12.040194.001041
Baldwin, A. S. (1996). The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 14, 649–683. doi: 10.1146/annurev.immunol.14.1.649
Banks, W. A. (2005). Blood-brain barrier transport of cytokines: a mechanism for neuropathology. Curr. Pharm. Des. 11, 973–984. doi: 10.2174/1381612053381684
Barone, F. C., White, R. F., Spera, P. A., Ellison, J., Currie, R. W., Wang, X., et al. (1998). Ischemic preconditioning and brain tolerance: temporal histological and functional outcomes, protein synthesis requirement, and interleukin-1 receptor antagonist and early gene expression. Stroke 29, 1937–1950. discussion: 1950–1951.
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukocyte Biol. 81, 1–5. doi: 10.1189/jlb.0306164
Bigdeli, M. R., and Khoshbaten, A. (2008). In vivo preconditioning with normobaric hyperoxia induces ischemic tolerance partly by triggering tumor necrosis factor-alpha converting enzyme/tumor necrosis factor-alpha/nuclear factor-kappaB. Neuroscience 153, 671–678. doi: 10.1016/j.neuroscience.2008.02.064
Blondeau, N., Widmann, C., Lazdunski, M., and Heurteaux, C. (2001). Activation of the nuclear factor-kappaB is a key event in brain tolerance. J. Neurosci. 21, 4668–4677.
Brea, D., Blanco, M., Ramos-Cabrer, P., Moldes, O., Arias, S., Pérez-Mato, M., et al. (2011). Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. J. Cereb. Blood Flow Metab. 31, 1424–1431. doi: 10.1038/jcbfm.2010.231
Breder, C. D., Hazuka, C., Ghayur, T., Klug, C., Huginin, M., Yasuda, K., et al. (1994). Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration. Proc. Natl. Acad. Sci. U.S.A. 91, 11393–11397. doi: 10.1073/pnas.91.24.11393
Cao, C.-X., Yang, Q.-W., Lv, F.-L., Cui, J., Fu, H.-B., and Wang, J.-Z. (2007). Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochem. Biophys. Res. Commun. 353, 509–514. doi: 10.1016/j.bbrc.2006.12.057
Cárdenas, A., Moro, M. A., Leza, J. C., O'Shea, E., Dávalos, A., Castillo, J., et al. (2002). Upregulation of TACE/ADAM17 after ischemic preconditioning is involved in brain tolerance. J. Cereb. Blood Flow Metab. 22, 1297–1302. doi: 10.1097/00004647-200211000-00002
Ceulemans, A.-G. A., Zgavc, T. T., Kooijman, R. R., Hachimi-Idrissi, S. S., Sarre, S. S., and Michotte, Y. Y. (2010). The dual role of the neuroinflammatory response after ischemic stroke: modulatory effects of hypothermia. J. Neuroinflammation 7, 74–74. doi: 10.1186/1742-2094-7-74
Chan, P. H. (2001). Reactive oxygen radicals in signaling and damage in the ischemic brain. J. Cereb. Blood Flow Metab. 21, 2–14. doi: 10.1097/00004647-200101000-00002
Charo, I. F., and Ransohoff, R. M. (2006). The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621. doi: 10.1056/NEJMra052723
Chen, G. Y., and Nuñez, G. (2010). Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837. doi: 10.1038/nri2873
Chen, H., Song, Y. S., and Chan, P. H. (2009). Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J. Cereb. Blood Flow Metab. 29, 1262–1272. doi: 10.1038/jcbfm.2009.47
Cheng, O., Ostrowski, R. P., Wu, B., Liu, W., Chen, C., and Zhang, J. H. (2011). Cyclooxygenase-2 mediates hyperbaric oxygen preconditioning in the rat model of transient global cerebral ischemia. Stroke 42, 484–490. doi: 10.1161/STROKEAHA.110.604421
Cho, S., Park, E.-M., Zhou, P., Frys, K., Ross, M. E., and Iadecola, C. (2005). Obligatory role of inducible nitric oxide synthase in ischemic preconditioning. J. Cereb. Blood Flow Metab. 25, 493–501. doi: 10.1038/sj.jcbfm.9600058
Clarkson, A. N. (2007). Anesthetic-mediated protection/preconditioning during cerebral ischemia. Life Sci. 80, 1157–1175. doi: 10.1016/j.lfs.2006.12.022
Clemens, J. A., Stephenson, D. T., Yin, T., Smalstig, E. B., Panetta, J. A., Little, S. P., et al. (1998). Drug-induced neuroprotection from global ischemia is associated with prevention of persistent but not transient activation of nuclear factor-B in rats editorial comment. Stroke 29, 677–682. doi: 10.1161/01.STR.29.3.677
Curry, A., Guo, M., Patel, R., Liebelt, B., Sprague, S., Lai, Q., et al. (2010). Exercise pre-conditioning reduces brain inflammation in stroke via tumor necrosis factor-α, extracellular signal-regulated kinase 1/2 and matrix metalloproteinase-9 activity. Neurol. Res. 32, 756–762. doi: 10.1179/174313209X459101
Dawn, B., Xuan, Y.-T., Guo, Y., Rezazadeh, A., Stein, A. B., Hunt, G., et al. (2004). IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc. Res. 64, 61–71. doi: 10.1016/j.cardiores.2004.05.011
Dharap, A., Bowen, K., Place, R., Li, L.-C., and Vemuganti, R. (2009). Transient focal ischemia induces extensive temporal changes in rat cerebral MicroRNAome. J. Cereb. Blood Flow Metab. 29, 675–687. doi: 10.1038/jcbfm.2008.157
Dharap, A., and Vemuganti, R. (2010). Ischemic pre-conditioning alters cerebral microRNAs that are upstream to neuroprotective signaling pathways. J. Neurochem. 113, 1685–1691. doi: 10.1111/j.1471-4159.2010.06735.x
Di Marzo, V., Melck, D., Bisogno, T., and De Petrocellis, L. (1998). Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 21, 521–528. doi: 10.1016/S0166-2236(98)01283-1
Ding, Y.-H., Young, C. N., Luan, X., Li, J., Rafols, J. A., Clark, J. C., et al. (2005). Exercise preconditioning ameliorates inflammatory injury in ischemic rats during reperfusion. Acta Neuropathol. 109, 237–246. doi: 10.1007/s00401-004-0943-y
Dirnagl, U., and Meisel, A. (2008). Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology 55, 334–344. doi: 10.1016/j.neuropharm.2008.02.017
Downes, C. E., and Crack, P. J. (2010). Neural injury following stroke: are Toll-like receptors the link between the immune system and the CNS? Br. J. Pharmacol. 160, 1872–1888. doi: 10.1111/j.1476-5381.2010.00864.x
Du, F., Zhu, L., Qian, Z.-M., Wu, X.-M., Yung, W.-H., and Ke, Y. (2010). Hyperthermic preconditioning protects astrocytes from ischemia/reperfusion injury by up-regulation of HIF-1 alpha expression and binding activity. Biochim. Biophys. Acta 1802, 1048–1053. doi: 10.1016/j.bbadis.2010.06.013
Duckworth, E. A. M., Butler, T., Collier, L., Collier, S., and Pennypacker, K. R. (2006). NF-κB protects neurons from ischemic injury after middle cerebral artery occlusion in mice. Brain Res. 1088, 167–175. doi: 10.1016/j.brainres.2006.02.103
Elliott, M. B., Tuma, R. F., Amenta, P. S., Barbe, M. F., and Jallo, J. I. (2011). Acute effects of a selective cannabinoid-2 receptor agonist on neuroinflammation in a model of traumatic brain injury. J. Neurotrauma 28, 973–981. doi: 10.1089/neu.2010.1672
Endres, M., Gertz, K., Lindauer, U., Katchanov, J., Schultze, J., Schröck, H., et al. (2003). Mechanisms of stroke protection by physical activity. Ann. Neurol. 54, 582–590. doi: 10.1002/ana.10722
Foster, S. L., Hargreaves, D. C., and Medzhitov, R. (2007). Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447, 972–978. doi: 10.1038/nature05836
Gabellec, M. M., Griffais, R., Fillion, G., and Haour, F. (1995). Expression of interleukin 1 alpha, interleukin 1 beta and interleukin 1 receptor antagonist mRNA in mouse brain: regulation by bacterial lipopolysaccharide (LPS) treatment. Brain Res. Mol. Brain Res. 31, 122–130. doi: 10.1016/0169-328X(95)00042-Q
Galea, E., Glickstein, S. B., Feinstein, D. L., Golanov, E. V., and Reis, D. J. (1998a). Stimulation of cerebellar fastigial nucleus inhibits interleukin-1beta-induced cerebrovascular inflammation. Am. J. Physiol. 275, H2053–H2063.
Galea, E., Golanov, E. V., Feinstein, D. L., Kobylarz, K. A., Glickstein, S. B., and Reis, D. J. (1998b). Cerebellar stimulation reduces inducible nitric oxide synthase expression and protects brain from ischemia. Am. J. Physiol. 274, H2035–H2045.
Galiègue, S., Mary, S., Marchand, J., Dussossoy, D., Carrière, D., Carayon, P., et al. (1995). Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 232, 54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x
Gallí, M., Van Gool, F., and Leo, O. (2011). Sirtuins and inflammation: friends or foes? Biochem. Pharmacol. 81, 569–576. doi: 10.1016/j.bcp.2010.12.010
Gesuete, R., Packard, A. E. B., Vartanian, K. B., Conrad, V. K., Stevens, S. L., Bahjat, F. R., et al. (2012). Poly-ICLC preconditioning protects the blood-brain barrier against ischemic injury in vitro through type I interferon signaling. J. Neurochem. 123(Suppl. 2), 75–85. doi: 10.1111/j.1471-4159.2012.07946.x
Gidday, J. M. (2006). Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 7, 437–448. doi: 10.1038/nrn1927
Ginis, I., Jaiswal, R., Klimanis, D., Liu, J., Greenspon, J., and Hallenbeck, J. M. (2002). TNF-alpha-induced tolerance to ischemic injury involves differential control of NF-kappaB transactivation: the role of NF-kappaB association with p300 adaptor. J. Cereb. Blood Flow Metab. 22, 142–152. doi: 10.1097/00004647-200202000-00002
Golanov, E. V., Liu, F., and Reis, D. J. (1998). Stimulation of cerebellum protects hippocampal neurons from global ischemia. Neuroreport 9, 819–824. doi: 10.1097/00001756-199803300-00010
Gong, J.-P., Onaivi, E. S., Ishiguro, H., Liu, Q.-R., Tagliaferro, P. A., Brusco, A., et al. (2006). Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 1071, 10–23. doi: 10.1016/j.brainres.2005.11.035
Guo, M., Lin, V., Davis, W., Huang, T., Carranza, A., Sprague, S., et al. (2008). Preischemic induction of TNF-alpha by physical exercise reduces blood-brain barrier dysfunction in stroke. J. Cereb. Blood Flow Metab. 28, 1422–1430. doi: 10.1038/jcbfm.2008.29
Hallenbeck, J. M. (2002). The many faces of tumor necrosis factor in stroke. Nat. Med. 8, 1363–1368. doi: 10.1038/nm1202-1363
Harari, O. A., and Liao, J. K. (2010). NF-κB and innate immunity in ischemic stroke. Ann. N.Y. Acad. Sci. 1207, 32–40. doi: 10.1111/j.1749-6632.2010.05735.x
Hazzalin, C. A., and Mahadevan, L. C. (2005). Dynamic acetylation of all lysine 4-methylated histone H3 in the mouse nucleus: analysis at c-fos and c-jun. PLoS Biol. 3:e393. doi: 10.1371/journal.pbio.0030393
Hess, D. C., Hoda, M. N., and Bhatia, K. (2013). Remote limb perconditioning and postconditioning: will it translate into a promising treatment for acute stroke? Stroke 44, 1191–1196. doi: 10.1161/STROKEAHA.112.678482
Hickey, E. J., You, X., Kaimaktchiev, V., Stenzel-Poore, M., and Ungerleider, R. M. (2007). Lipopolysaccharide preconditioning induces robust protection against brain injury resulting from deep hypothermic circulatory arrest. J. Thorac. Cardiovasc. Surg. 133, 1588–1596. doi: 10.1016/j.jtcvs.2006.12.056
Horiguchi, T., Snipes, J. A., Kis, B., Shimizu, K., and Busija, D. W. (2005). The role of nitric oxide in the development of cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats. Brain Res. 1039, 84–89. doi: 10.1016/j.brainres.2005.01.047
Horiguchi, T., Snipes, J. A., Kis, B., Shimizu, K., and Busija, D. W. (2006). Cyclooxygenase-2 mediates the development of cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats. Neuroscience 140, 723–730. doi: 10.1016/j.neuroscience.2006.02.025
Hoshino, K., Sugiyama, T., Matsumoto, M., Tanaka, T., Saito, M., Hemmi, H., et al. (2006). IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440, 949–953. doi: 10.1038/nature04641
Howlett, A. C. A., Barth, F. F., Bonner, T. I. T., Cabral, G. G., Casellas, P. P., Devane, W. A. W., et al. (2002). International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 54, 161–202. doi: 10.1124/pr.54.2.161
Hu, X., Jiang, H., Cui, B., Xu, C., Lu, Z., and He, B. (2010). Preconditioning with high mobility group box 1 protein protects against myocardial ischemia-reperfusion injury. Int. J. Cardiol. 145, 111–112. doi: 10.1016/j.ijcard.2009.05.057
Hua, F., Ma, J., Ha, T., Kelley, J., Williams, D. L., Kao, R. L., et al. (2008). Preconditioning with a TLR2 specific ligand increases resistance to cerebral ischemia/reperfusion injury. J. Neuroimmunol. 199, 75–82. doi: 10.1016/j.jneuroim.2008.05.009
Huang, J., Upadhyay, U. M., and Tamargo, R. J. (2006). Inflammation in stroke and focal cerebral ischemia. Surg. Neurol. 66, 232–245. doi: 10.1016/j.surneu.2005.12.028
Iadecola, C., and Anrather, J. (2011a). The immunology of stroke: from mechanisms to translation. Nat. Med. 17, 796–808. doi: 10.1038/nm.2399
Iadecola, C., and Anrather, J. (2011b). Stroke research at a crossroad: asking the brain for directions. Nat. Neurosci. 14, 1363–1368. doi: 10.1038/nn.2953
Iadecola, C., Kahles, T., Gallo, E. F., and Anrather, J. (2011). Neurovascular protection by ischaemic tolerance: role of nitric oxide. J. Physiol. (Lond.) 589, 4137–4145. doi: 10.1113/jphysiol.2011.210831
Iadecola, C., Niwa, K., Nogawa, S., Zhao, X., Nagayama, M., Araki, E., et al. (2001). Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 1294–1299. doi: 10.1073/pnas.98.3.1294
Ikeda, T., Xia, X. Y., Xia, Y. X., and Ikenoue, T. (1999). Hyperthermic preconditioning prevents blood-brain barrier disruption produced by hypoxia-ischemia in newborn rat. Brain Res. Dev. Brain Res. 117, 53–58. doi: 10.1016/S0165-3806(99)00097-8
Izuishi, K., Tsung, A., Jeyabalan, G., Critchlow, N. D., Li, J., Tracey, K. J., et al. (2006). Cutting edge: high-mobility group box 1 preconditioning protects against liver ischemia-reperfusion injury. J. Immunol. 176, 7154–7158.
Jaerve, A., and Müller, H. W. (2012). Chemokines in CNS injury and repair. Cell Tissue Res. 349, 229–248. doi: 10.1007/s00441-012-1427-3
Jander, S., Schroeter, M., Peters, O., Witte, O. W., and Stoll, G. (2001). Cortical spreading depression induces proinflammatory cytokine gene expression in the rat brain. J. Cereb. Blood Flow Metab. 21, 218–225. doi: 10.1097/00004647-200103000-00005
Jung, F., Palmer, L. A., Zhou, N., and Johns, R. A. (2000). Hypoxic regulation of inducible nitric oxide synthase via hypoxia inducible factor-1 in cardiac myocytes. Circ. Res. 86, 319–325. doi: 10.1161/01.RES.86.3.319
Kahles, T., Luedike, P., Endres, M., Galla, H.-J., Steinmetz, H., Busse, R., et al. (2007). NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke 38, 3000–3006. doi: 10.1161/STROKEAHA.107.489765
Kaltschmidt, B., Widera, D., and Kaltschmidt, C. (2005). Signaling via NF-kappaB in the nervous system. Biochim. Biophys. Acta 1745, 287–299. doi: 10.1016/j.bbamcr.2005.05.009
Kaltschmidt, C., Kaltschmidt, B., Neumann, H., Wekerle, H., and Baeuerle, P. A. (1994). Constitutive NF-kappa B activity in neurons. Mol. Cell Biol. 14, 3981–3992.
Kapinya, K. J., Löwl, D., Fütterer, C., Maurer, M., Waschke, K. F., Isaev, N. K., et al. (2002). Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke 33, 1889–1898. doi: 10.1161/01.STR.0000020092.41820.58
Kawahara, N., Ruetzler, C. A., and Klatzo, I. (1995). Protective effect of spreading depression against neuronal damage following cardiac arrest cerebral ischaemia. Neurol. Res. 17, 9–16.
Kawahara, N., Ruetzler, C. A., Mies, G., and Klatzo, I. (1999). Cortical spreading depression increases protein synthesis and upregulates basic fibroblast growth factor. Exp. Neurol. 158, 27–36. doi: 10.1006/exnr.1999.7091
Kawai, N. N., Okauchi, M. M., Morisaki, K. K., and Nagao, S. S. (2000). Effects of delayed intraischemic and postischemic hypothermia on a focal model of transient cerebral ischemia in rats. Stroke 31, 1982–1989. doi: 10.1161/01.STR.31.8.1982
Kawano, T., Kunz, A., Abe, T., Girouard, H., Anrather, J., Zhou, P., et al. (2007). iNOS-derived NO and nox2-derived superoxide confer tolerance to excitotoxic brain injury through peroxynitrite. J. Cereb. Blood Flow Metab. 27, 1453–1462. doi: 10.1038/sj.jcbfm.9600449
Keep, R. F., Wang, M. M., Xiang, J., Hua, Y., and Xi, G. (2010). Is there a place for cerebral preconditioning in the clinic? Transl. Stroke Res. 1, 4–18. doi: 10.1007/s12975-009-0007-7
Kim, J. B. (2006). HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 26, 6413–6421. doi: 10.1523/JNEUROSCI.3815-05.2006
Kirino, T. (2002). Ischemic tolerance. J. Cereb. Blood Flow Metab. 22, 1283–1296. doi: 10.1097/00004647-200211000-00001
Kitagawa, K., Matsumoto, M., Tagaya, M., Hata, R., Ueda, H., Niinobe, M., et al. (1990). “Ischemic tolerance” phenomenon found in the brain. Brain Res. 528, 21–24. doi: 10.1016/0006-8993(90)90189-I
Konstantinov, I. E., Arab, S., Kharbanda, R. K., Li, J., Cheung, M. M. H., Cherepanov, V., et al. (2004). The remote ischemic preconditioning stimulus modifies inflammatory gene expression in humans. Physiol. Genomics 19, 143–150. doi: 10.1152/physiolgenomics.00046.2004
Kunkler, P. E., Hulse, R. E., and Kraig, R. P. (2004). Multiplexed cytokine protein expression profiles from spreading depression in hippocampal organotypic cultures. J. Cereb. Blood Flow Metab. 24, 829–839. doi: 10.1097/01.WCB.0000126566.34753.30
Kunz, A., Anrather, J., Zhou, P., Orio, M., and Iadecola, C. (2006). Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J. Cereb. Blood Flow Metab. 27, 545–551. doi: 10.1038/sj.jcbfm.9600369
Kunz, A., Park, L., Abe, T., Gallo, E. F., Anrather, J., Zhou, P., et al. (2007). Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J. Neurosci. 27, 7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007
Lee, S.-T., Chu, K., Jung, K.-H., Yoon, H.-J., Jeon, D., Kang, K.-M., et al. (2010). MicroRNAs induced during ischemic preconditioning. Stroke 41, 1646–1651. doi: 10.1161/STROKEAHA.110.579649
Leker, R. R., Gai, N., Mechoulam, R., and Ovadia, H. (2003). Drug-induced hypothermia reduces ischemic damage: effects of the cannabinoid HU-210. Stroke 34, 2000–2006. doi: 10.1161/01.STR.0000079817.68944.1E
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M., and Hoffmann, J. A. (1996). The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983. doi: 10.1016/S0092-8674(00)80172-5
Leung, A. K. L., and Sharp, P. A. (2010). MicroRNA functions in stress responses. Mol. Cell 40, 205–215. doi: 10.1016/j.molcel.2010.09.027
Leung, P. Y., Packard, A. E., and Stenzel-Poore, M. P. (2009). It's all in the family: multiple Toll-like receptors offer promise as novel therapeutic targets for stroke neuroprotection. Future Neurol. 4, 201–208. doi: 10.2217/14796708.4.2.201
Leung, P. Y., Stevens, S. L., Packard, A. E. B., Lessov, N. S., Yang, T., Conrad, V. K., et al. (2012). Toll-like receptor 7 preconditioning induces robust neuroprotection against stroke by a novel type I interferon-mediated mechanism. Stroke 43, 1383–1389. doi: 10.1161/STROKEAHA.111.641522
Li, Q., and Verma, I. M. (2002). NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2, 725–734. doi: 10.1038/nri910
Li, Q.-F., Zhu, Y.-S., Jiang, H., Xu, H., and Sun, Y. (2009). Heme oxygenase-1 mediates the anti-inflammatory effect of isoflurane preconditioning in LPS-stimulated macrophages. Acta Pharmacol. Sin. 30, 228–234. doi: 10.1038/aps.2008.19
Liang, J., Wang, J., Saad, Y., Warble, L., Becerra, E., and Kolattukudy, P. E. (2011). Participation of MCP-induced protein 1 in lipopolysaccharide preconditioning-induced ischemic stroke tolerance by regulating the expression of proinflammatory cytokines. J. Neuroinflammation 8:182. doi: 10.1186/1742-2094-8-182
Liu, C., Peng, Z., Zhang, N., Yu, L., Han, S., Li, D., et al. (2012). Identification of differentially expressed microRNAs and their PKC-isoform specific gene network prediction during hypoxic pre-conditioning and focal cerebral ischemia of mice. J. Neurochem. 120, 830–841. doi: 10.1111/j.1471-4159.2011.07624.x
Liu, J., Ginis, I., Spatz, M., and Hallenbeck, J. M. (2000). Hypoxic preconditioning protects cultured neurons against hypoxic stress via TNF-alpha and ceramide. Am. J. Physiol. Cell Physiol. 278, C144–C153.
Loddick, S. A., Turnbull, A. V., and Rothwell, N. J. (1998). Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 18, 176–179. doi: 10.1097/00004647-199802000-00008
Lu, C., Liu, L., Chen, Y., Ha, T., Kelley, J., Schweitzer, J., et al. (2011). TLR2 ligand induces protection against cerebral ischemia/reperfusion injury via activation of phosphoinositide 3-kinase/Akt signaling. J. Immunol. 187, 1458–1466. doi: 10.4049/jimmunol.1003428
Lusardi, T. A., Farr, C. D., Faulkner, C. L., Pignataro, G., Yang, T., Lan, J., et al. (2010). Ischemic preconditioning regulates expression of microRNAs and a predicted target, MeCP2, in mouse cortex. J. Cereb. Blood Flow Metab. 30, 744–756. doi: 10.1038/jcbfm.2009.253
Ma, L., Zhu, Z., Zhao, Y., Hou, L., Wang, Q., Xiong, L., et al. (2011). Cannabinoid receptor type 2 activation yields delayed tolerance to focal cerebral ischemia. Curr. Neurovasc. Res. 8, 145–152. doi: 10.2174/156720211795495394
Macrez, R., Ali, C., Toutirais, O., Le Mauff, B., Defer, G., Dirnagl, U., et al. (2011). Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol. 10, 471–480. doi: 10.1016/S1474-4422(11)70066-7
Mallard, C. (2012). Innate immune regulation by toll-like receptors in the brain. ISRN Neurol. 2012:701950. doi: 10.5402/2012/701950
Marsh, B., Stevens, S. L., Packard, A. E. B., Gopalan, B., Hunter, B., Leung, P. Y., et al. (2009b). Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J. Neurosci. 29, 9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009
Marsh, B. J., Williams-Karnesky, R. L., and Stenzel-Poore, M. P. (2009a). Toll-like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 158, 1007–1020. doi: 10.1016/j.neuroscience.2008.07.067
Matsuda, L. A., Lolait, S. J., Brownstein, M. J., Young, A. C., and Bonner, T. I. (1990). Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346, 561–564. doi: 10.1038/346561a0
McColl, B. W., Allan, S. M., and Rothwell, N. J. (2009). Systemic infection, inflammation and acute ischemic stroke. Neuroscience 158, 1049–1061. doi: 10.1016/j.neuroscience.2008.08.019
Meisel, C., Schwab, J. M., Prass, K., Meisel, A., and Dirnagl, U. (2005). Central nervous system injury-induced immune deficiency syndrome. Nat. Rev. Neurosci. 6, 775–786. doi: 10.1038/nrn1765
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. doi: 10.1038/nrg2521
Morris, K. C., Lin, H. W., Thompson, J. W., and Perez-Pinzon, M. A. (2011). Pathways for ischemic cytoprotection: role of sirtuins in caloric restriction, resveratrol, and ischemic preconditioning. J. Cereb. Blood Flow Metab. 31, 1003–1019. doi: 10.1038/jcbfm.2010.229
Nagayama, T. T., Sinor, A. D. A., Simon, R. P. R., Chen, J. J., Graham, S. H. S., Jin, K. K., et al. (1999). Cannabinoids and neuroprotection in global and focal cerebral ischemia and in neuronal cultures. J. Neurosci. 19, 2987–2995.
Narayanan, S. V., Dave, K. R., and Perez-Pinzon, M. A. (2013). Ischemic preconditioning and clinical scenarios. Curr. Opin. Neurol. 26, 1–7. doi: 10.1097/WCO.0b013e32835bf200
Nathan, C. (1997). Inducible nitric oxide synthase: what difference does it make? J. Clin. Invest. 100, 2417–2423. doi: 10.1172/JCI119782
Nawashiro, H., Tasaki, K., Ruetzler, C. A., and Hallenbeck, J. M. (1997). TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 17, 483–490. doi: 10.1097/00004647-199705000-00001
Nijboer, C. H., Heijnen, C. J., Groenendaal, F., May, M. J., van Bel, F., and Kavelaars, A. (2008). A dual role of the NF-kappaB pathway in neonatal hypoxic-ischemic brain damage. Stroke 39, 2578–2586. doi: 10.1161/STROKEAHA.108.516401
Nishio, S., Yunoki, M., Chen, Z. F., Anzivino, M. J., and Lee, K. S. (2000). Ischemic tolerance in the rat neocortex following hypothermic preconditioning. J. Neurosurg. 93, 845–851. doi: 10.3171/jns.2000.93.5.0845
Nurmi, A. (2004). Nuclear factor-B contributes to infarction after permanent focal ischemia. Stroke 35, 987–991. doi: 10.1161/01.STR.0000120732.45951.26
Nurmi, A., Vartiainen, N., Pihlaja, R., Goldsteins, G., Yrjanheikki, J., and Koistinaho, J. (2004). Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J. Neurochem. 91, 755–765. doi: 10.1111/j.1471-4159.2004.02756.x
O'Connell, R. M., Chaudhuri, A. A., Rao, D. S., and Baltimore, D. (2009). Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. U.S.A. 106, 7113–7118. doi: 10.1073/pnas.0902636106
O'Connell, R. M., Rao, D. S., and Baltimore, D. (2012). microRNA regulation of inflammatory responses. Annu. Rev. Immunol. 30, 295–312. doi: 10.1146/annurev-immunol-020711-075013
Ohtsuki, T., Ruetzler, C. A., Tasaki, K., and Hallenbeck, J. M. (1996). Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal CA1 neurons. J. Cereb. Blood Flow Metab. 16, 1137–1142. doi: 10.1097/00004647-199611000-00007
O'Neill, L. A., Sheedy, F. J., and McCoy, C. E. (2011). MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat. Rev. Immunol. 11, 163–175. doi: 10.1038/nri2957
Ota, A., Ikeda, T., Xia, X. Y., Xia, Y. X., and Ikenoue, T. (2000). Hypoxic-ischemic tolerance induced by hyperthermic pretreatment in newborn rats. J. Soc. Gynecol. Investig. 7, 102–105. doi: 10.1016/S1071-5576(00)00038-1
Pacher, P., Bátkai, S., and Kunos, G. (2006). The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 58, 389–462. doi: 10.1124/pr.58.3.2
Pacher, P., and Haskó, G. (2008). Endocannabinoids and cannabinoid receptors in ischaemia-reperfusion injury and preconditioning. Br. J. Pharmacol. 153, 252–262. doi: 10.1038/sj.bjp.0707582
Packard, A. E. B., Hedges, J. C., Bahjat, F. R., Stevens, S. L., Conlin, M. J., Salazar, A. M., et al. (2012). Poly-IC preconditioning protects against cerebral and renal ischemia-reperfusion injury. J. Cereb. Blood Flow Metab. 32, 242–247. doi: 10.1038/jcbfm.2011.160
Pahl, H. L. (1999). Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18, 6853–6866. doi: 10.1038/sj.onc.1203239
Panikashvili, D., Mechoulam, R., Beni, S. M., Alexandrovich, A., and Shohami, E. (2005). CB1 cannabinoid receptors are involved in neuroprotection via NF-κB inhibition. J. Cereb. Blood Flow Metab. 25, 477–484. doi: 10.1038/sj.jcbfm.9600047
Panikashvili, D., Simeonidou, C., Ben-Shabat, S., Hanus, L., Breuer, A., Mechoulam, R., et al. (2001). An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413, 527–531. doi: 10.1038/35097089
Park, M. K., Kang, Y. J., Lee, H. S., Kim, H. J., Seo, H. G., Lee, J. H., et al. (2008). The obligatory role of COX-2 expression for induction of HO-1 in ischemic preconditioned rat brain. Biochem. Biophys. Res. Commun. 377, 1191–1194. doi: 10.1016/j.bbrc.2008.10.149
Patel, H., Shaw, S. G., Shi-Wen, X., Abraham, D., Baker, D. M., and Tsui, J. C. S. (2012). Toll-like receptors in ischaemia and its potential role in the pathophysiology of muscle damage in critical limb ischaemia. Cardiol. Res. Pract. 2012:121237. doi: 10.1155/2012/121237
Pedersen, B. K. (2011). Exercise-induced myokines and their role in chronic diseases. Brain Behav. Immun. 25, 811–816. doi: 10.1016/j.bbi.2011.02.010
Pedersen, C. M., Cruden, N. L., Schmidt, M. R., Lau, C., Bøtker, H. E., Kharbanda, R. K., et al. (2011). Remote ischemic preconditioning prevents systemic platelet activation associated with ischemia-reperfusion injury in humans. J. Thromb. Haemost. 9, 404–407. doi: 10.1111/j.1538-7836.2010.04142.x
Peng, Z., Ren, P., Kang, Z., Du, J., Lian, Q., Liu, Y., et al. (2008). Up-regulated HIF-1α is involved in the hypoxic tolerance induced by hyperbaric oxygen preconditioning. Brain Res. 1212, 71–78. doi: 10.1016/j.brainres.2008.03.027
Plamondon, H., Blondeau, N., Heurteaux, C., and Lazdunski, M. (1999). Mutually protective actions of kainic acid epileptic preconditioning and sublethal global ischemia on hippocampal neuronal death: involvement of adenosine A1 receptors and K(ATP) channels. J. Cereb. Blood Flow Metab. 19, 1296–1308. doi: 10.1097/00004647-199912000-00002
Pradillo, J. M., Fernández-López, D., García-Yébenes, I., Sobrado, M., Hurtado, O., Moro, M. A., et al. (2009). Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J. Neurochem. 109, 287–294. doi: 10.1111/j.1471-4159.2009.05972.x
Pradillo, J. M., Romera, C., Hurtado, O., Cárdenas, A., Moro, M. A., Leza, J. C., et al. (2005). TNFR1 upregulation mediates tolerance after brain ischemic preconditioning. J. Cereb. Blood Flow Metab. 25, 193–203. doi: 10.1038/sj.jcbfm.9600019
Pun, P. B. L., Lu, J., and Moochhala, S. (2009). Involvement of ROS in BBB dysfunction. Free Radic. Res. 43, 348–364. doi: 10.1080/10715760902751902
Quintin, J., Saeed, S., Martens, J. H. A., Giamarellos-Bourboulis, E. J., Ifrim, D. C., Logie, C., et al. (2012). Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 12, 223–232. doi: 10.1016/j.chom.2012.06.006
Qureshi, I. A., and Mehler, M. F. (2010). Emerging role of epigenetics in stroke: part 1: DNA methylation and chromatin modifications. Arch. Neurol. 67, 1316–1322. doi: 10.1001/archneurol.2010.275
Ramirez-Carrozzi, V. R., Nazarian, A. A., Li, C. C., Gore, S. L., Sridharan, R., Imbalzano, A. N., et al. (2006). Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 20, 282–296. doi: 10.1101/gad.1383206
Ransohoff, R. M., and Brown, M. A. (2012). Innate immunity in the central nervous system. J. Clin.l Invest. 122, 1164–1171. doi: 10.1172/JCI58644
Rehni, A. K., and Singh, T. G. (2012). Involvement of CCR-2 chemokine receptor activation in ischemic preconditioning and postconditioning of brain in mice. Cytokine 60, 83–89. doi: 10.1016/j.cyto.2012.05.009
Reis, D. J., Berger, S. B., Underwood, M. D., and Khayata, M. (1991). Electrical stimulation of cerebellar fastigial nucleus reduces ischemic infarction elicited by middle cerebral artery occlusion in rat. J. Cereb. Blood Flow Metab. 11, 810–818. doi: 10.1038/jcbfm.1991.139
Reis, D. J., Kobylarz, K., Yamamoto, S., and Golanov, E. V. (1998). Brief electrical stimulation of cerebellar fastigial nucleus conditions long-lasting salvage from focal cerebral ischemia: conditioned central neurogenic neuroprotection. Brain Res. 780, 161–165. doi: 10.1016/S0006-8993(97)01017-2
Rosenzweig, H. L., Lessov, N. S., Henshall, D. C., Minami, M., Simon, R. P., and Stenzel-Poore, M. P. (2004). Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 35, 2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae
Rosenzweig, H. L., Minami, M., Lessov, N. S., Coste, S. C., Stevens, S. L., Henshall, D. C., et al. (2007). Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J. Cereb. Blood Flow Metab. 27, 1663–1674. doi: 10.1038/sj.jcbfm.9600464
Rüegger, S., and Großhans, H. (2012). MicroRNA turnover: when, how, and why. Trends Biochem. Sci. 37, 436–446. doi: 10.1016/j.tibs.2012.07.002
Schwaninger, M., Schneider, A., Martin-Villalba, A., Weih, F., Vogel, J., and Wirth, T. (1999). NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 5, 554–559. doi: 10.1038/8432
Semple, B. D., Kossmann, T., and Morganti-Kossmann, M. C. (2009). Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 30, 459–473. doi: 10.1038/jcbfm.2009.240
Shichita, T., Hasegawa, E., Kimura, A., Morita, R., Sakaguchi, R., Takada, I., et al. (2012). Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 18, 911–917. doi: 10.1038/nm.2749
Shimizu, M., Saxena, P., Konstantinov, I. E., Cherepanov, V., Cheung, M. M. H., Wearden, P., et al. (2010). Remote ischemic preconditioning decreases adhesion and selectively modifies functional responses of human neutrophils. J. Surg. Res. 158, 155–161. doi: 10.1016/j.jss.2008.08.010
Shin, J. A., Park, E.-M., Choi, J.-S., Seo, S.-M., Kang, J. L., Lee, K.-E., et al. (2009). Ischemic preconditioning-induced neuroprotection is associated with differential expression of IL-1beta and IL-1 receptor antagonist in the ischemic cortex. J. Neuroimmunol. 217, 14–19. doi: 10.1016/j.jneuroim.2009.06.001
Shpargel, K. B., Jalabi, W., Jin, Y., Dadabayev, A., Penn, M. S., and Trapp, B. D. (2008). Preconditioning paradigms and pathways in the brain. Cleve. Clin. J. Med. 75(Suppl. 2), S77–S82. doi: 10.3949/ccjm.75.Suppl_2.S77
Sly, L. M., Rauh, M. J., Kalesnikoff, J., Song, C. H., and Krystal, G. (2004). LPS-Induced Upregulation of SHIP is essential for endotoxin tolerance. Immunity 21, 227–239. doi: 10.1016/j.immuni.2004.07.010
Stapels, M., Piper, C., Yang, T., Li, M., Stowell, C., Xiong, Z. G., et al. (2010). Polycomb group proteins as epigenetic mediators of neuroprotection in ischemic tolerance. Sci. Signal. 3, ra15–ra15. doi: 10.1126/scisignal.2000502
Stenzel-Poore, M. P., Stevens, S. L., Xiong, Z., Lessov, N. S., Harrington, C. A., Mori, M., et al. (2003). Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 362, 1028–1037. doi: 10.1016/S0140-6736(03)14412-1
Stephenson, D., Yin, T., Smalstig, E. B., Hsu, M. A., Panetta, J., Little, S., et al. (2000). Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 20, 592–603. doi: 10.1097/00004647-200003000-00017
Stevens, S. L., Ciesielski, T. M. P., Marsh, B. J., Yang, T., Homen, D. S., Boule, J.-L., et al. (2008). Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J. Cereb. Blood Flow Metab. 28, 1040–1047. doi: 10.1038/sj.jcbfm.9600606
Stevens, S. L., Leung, P. Y., Vartanian, K. B., Gopalan, B., Yang, T., Simon, R. P., et al. (2011). Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J. Neurosci. 31, 8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011
Stowe, A. M., Wacker, B. K., Cravens, P. D., Perfater, J. L., Li, M. K., Hu, R., et al. (2012). CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J. Neuroinflammation 9:33. doi: 10.1186/1742-2094-9-33
Stridh, L., Smith, P. L. P., Naylor, A. S., Wang, X., and Mallard, C. (2011). Regulation of toll-like receptor 1 and -2 in neonatal mice brains after hypoxia-ischemia. J. Neuroinflammation 8:45. doi: 10.1186/1742-2094-8-45
Suzuki, H. I., Arase, M., Matsuyama, H., Choi, Y. L., Ueno, T., Mano, H., et al. (2011). MCPIP1 ribonuclease antagonizes dicer and terminates microRNA biogenesis through precursor microRNA degradation. Mol. Cell 44, 424–436. doi: 10.1016/j.molcel.2011.09.012
Swigut, T., and Wysocka, J. (2007). H3K27 demethylases, at long last. Cell 131, 29–32. doi: 10.1016/j.cell.2007.09.026
Tapuria, N., Kumar, Y., Habib, M. M., Abu-Amara, M., Seifalian, A. M., and Davidson, B. R. (2008). Remote ischemic preconditioning: a novel protective method from ischemia reperfusion injury—a review. J. Surg. Res. 150, 304–330. doi: 10.1016/j.jss.2007.12.747
Tasaki, K., Ruetzler, C. A., Ohtsuki, T., Martin, D., Nawashiro, H., and Hallenbeck, J. M. (1997). Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 748, 267–270. doi: 10.1016/S0006-8993(96)01383-2
Towfighi, J., Housman, C., Mauger, D., and Vannucci, R. C. (1999). Effect of seizures on cerebral hypoxic-ischemic lesions in immature rats. Brain Res. Dev. Brain Res. 113, 83–95. doi: 10.1016/S0165-3806(99)00004-8
Trinchieri, G. (2010). Type I interferon: friend or foe? J. Exp. Med. 207, 2053–2063. doi: 10.1084/jem.20101664
Ullrich, O., Merker, K., Timm, J., and Tauber, S. (2007). Immune control by endocannabinoids—new mechanisms of neuroprotection? J. Neuroimmunol. 184, 127–135. doi: 10.1016/j.jneuroim.2006.11.018
Vabulas, R. M., Ahmad-Nejad, P., Ghose, S., Kirschning, C. J., Issels, R. D., and Wagner, H. (2002). HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J. Biol. Chem. 277, 15107–15112. doi: 10.1074/jbc.M111204200
Valadi, H., Ekström, K., Bossios, A., Sjöstrand, M., Lee, J. J., and Lötvall, J. O. (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659. doi: 10.1038/ncb1596
van Rooij, E., Sutherland, L. B., Qi, X., Richardson, J. A., Hill, J., and Olson, E. N. (2007). Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316, 575–579. doi: 10.1126/science.1139089
Vartanian, K., and Stenzel-Poore, M. (2010). Toll-like receptor tolerance as a mechanism for neuroprotection. Transl. Stroke Res. 1, 252–260. doi: 10.1007/s12975-010-0033-5
Vartanian, K. B., Stevens, S. L., Marsh, B. J., Williams-Karnesky, R., Lessov, N. S., and Stenzel-Poore, M. P. (2011). LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J. Neuroinflammation 8:140. doi: 10.1186/1742-2094-8-140
Veldhuis, W. B., Derksen, J. W., Floris, S., van der Meide, P. H., de Vries, H. E., Schepers, J., et al. (2003). Interferon-beta blocks infiltration of inflammatory cells and reduces infarct volume after ischemic stroke in the rat. J. Cereb. Blood Flow Metab. 1029–1039. doi: 10.1097/01.WCB.0000080703.47016.B6
Verstak, B., Hertzog, P., and Mansell, A. (2007). Toll-like receptor signalling and the clinical benefits that lie within. Inflamm. Res. 56, 1–10. doi: 10.1007/s00011-007-6093-7
Verstrepen, L., Verhelst, K., van Loo, G., Carpentier, I., Ley, S. C., and Beyaert, R. (2010). Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem. Pharmacol. 80, 2009–2020. doi: 10.1016/j.bcp.2010.06.044
Vezzani, A., Moneta, D., Richichi, C., Aliprandi, M., Burrows, S. J., Ravizza, T., et al. (2002). Functional role of inflammatory cytokines and antiinflammatory molecules in seizures and epileptogenesis. Epilepsia 43(Suppl. 5), 30–35. doi: 10.1046/j.1528-1157.43.s.5.14.x
Vlasov, T. D., Korzhevskii, D. E., and Polyakova, E. A. (2005). Ischemic preconditioning of the rat brain as a method of endothelial protection from ischemic/repercussion injury. Neurosci. Behav. Physiol. 35, 567–572. doi: 10.1007/s11055-005-0095-0
Wacker, B. K., Perfater, J. L., and Gidday, J. M. (2012). Hypoxic preconditioning induces stroke tolerance in mice via a cascading HIF, sphingosine kinase, and CCL2 signaling pathway. J. Neurochem. 123, 954–962. doi: 10.1111/jnc.12047
Wang, F., Birch, S. E., He, R., Tawadros, P., Szaszi, K., Kapus, A., et al. (2010). Remote ischemic preconditioning by hindlimb occlusion prevents liver ischemic/reperfusion injury. Ann. Surg. 251, 292–299. doi: 10.1097/SLA.0b013e3181bfda8c
Wang, L., Traystman, R. J., and Murphy, S. J. (2008). Inhalational anesthetics as preconditioning agents in ischemic brain. Curr. Opin. Pharmacol. 8, 104–110. doi: 10.1016/j.coph.2007.09.005
Wang, Q., Peng, Y., Chen, S., Gou, X., Hu, B., Du, J., et al. (2009). Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke 40, 2157–2164. doi: 10.1161/STROKEAHA.108.541490
Wang, Q., Xiong, L., Chen, S., Liu, Y., and Zhu, X. (2005). Rapid tolerance to focal cerebral ischemia in rats is induced by preconditioning with electroacupuncture: window of protection and the role of adenosine. Neurosci. Lett. 381, 158–162. doi: 10.1016/j.neulet.2005.02.019
Wang, R. Y., Yang, Y. R., and Yu, S. M. (2001). Protective effects of treadmill training on infarction in rats. Brain Res. 922, 140–143. doi: 10.1016/S0006-8993(01)03154-7
Wang, X., Li, X., Currie, R. W., Willette, R. N., Barone, F. C., and Feuerstein, G. Z. (2000). Application of real-time polymerase chain reaction to quantitate induced expression of interleukin-1beta mRNA in ischemic brain tolerance. J. Neurosci. Res. 59, 238–246. doi: 10.1016/S1385-299X(00)00015-5
Wang, Y., Wang, Y., and Yang, G.-Y. (2013). MicroRNAs in cerebral ischemia. Stroke Res. Treat. 2013:276540. doi: 10.1155/2013/276540
Wang, Y.-C., Lin, S., and Yang, Q.-W. (2011). Toll-like receptors in cerebral ischemic inflammatory injury. J. Neuroinflammation 8:134. doi: 10.1186/1742-2094-8-134
Wu, T.-C., and Grotta, J. C. (2013). Hypothermia for acute ischaemic stroke. Lancet Neurol. 12, 275–284. doi: 10.1016/S1474-4422(13)70013-9
Yanamoto, H., Hashimoto, N., Nagata, I., and Kikuchi, H. (1998). Infarct tolerance against temporary focal ischemia following spreading depression in rat brain. Brain Res. 784, 239–249. doi: 10.1016/S0006-8993(97)01344-9
Yang, Q.-W., Lu, F.-L., Zhou, Y., Wang, L., Zhong, Q., Lin, S., et al. (2011). HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. J. Cereb. Blood Flow Metab. 31, 593–605. doi: 10.1038/jcbfm.2010.129
Yin, W., Signore, A. P., Iwai, M., Cao, G., Gao, Y., Johnnides, M. J., et al. (2007). Preconditioning suppresses inflammation in neonatal hypoxic ischemia via Akt activation. Stroke 38, 1017–1024. doi: 10.1161/01.STR.0000258102.18836.ca
Yoshimura, A., Naka, T., and Kubo, M. (2007). SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 7, 454–465. doi: 10.1038/nri2093
Yu, J. T., Lee, C. H., Yoo, K.-Y., Choi, J. H., Li, H., Park, O. K., et al. (2010). Maintenance of anti-inflammatory cytokines and reduction of glial activation in the ischemic hippocampal CA1 region preconditioned with lipopolysaccharide. J. Neurol. Sci. 296, 69–78. doi: 10.1016/j.jns.2010.06.004
Yunoki, M., Nishio, S., Ukita, N., Anzivino, M. J., and Lee, K. S. (2003). Hypothermic preconditioning induces rapid tolerance to focal ischemic injury in the rat. Exp. Neurol. 181, 291–300. doi: 10.1016/S0014-4886(03)00056-6
Zhang, F., Wu, Y., and Jia, J. (2011). Exercise preconditioning and brain ischemic tolerance. Neuroscience 177, 170–176. doi: 10.1016/j.neuroscience.2011.01.018
Zhang, M., Adler, M. W., Abood, M. E., Ganea, D., Jallo, J., and Tuma, R. F. (2009). CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc. Res. 78, 86–94. doi: 10.1016/j.mvr.2009.03.005
Zhang, M., Martin, B. R., Adler, M. W., Razdan, R. K., Jallo, J. I., and Tuma, R. F. (2007). Cannabinoid CB2 receptor activation decreases cerebral infarction in a mouse focal ischemia/reperfusion model. J. Cereb. Blood Flow Metab. 27, 1387–1396. doi: 10.1038/sj.jcbfm.9600447
Zhao, P., and Zuo, Z. (2004). Isoflurane preconditioning induces neuroprotection that is inducible nitric oxide synthase-dependent in neonatal rats. Anesthesiology 101, 695–703. doi: 10.1097/00000542-200409000-00018
Zhou, P., Qian, L., D'Aurelio, M., Cho, S., Wang, G., Manfredi, G., et al. (2012). Prohibitin reduces mitochondrial free radical production and protects brain cells from different injury modalities. J. Neurosci. 32, 583–592. doi: 10.1523/JNEUROSCI.2849-11.2012
Zhou, P., Qian, L., Zhou, T., and Iadecola, C. (2005). Mitochondria are involved in the neurogenic neuroprotection conferred by stimulation of cerebellar fastigial nucleus. J. Neurochem. 95, 221–229. doi: 10.1111/j.1471-4159.2005.03358.x
Zhuang, S.-Y., Bridges, D., Grigorenko, E., McCloud, S., Boon, A., Hampson, R. E., et al. (2005). Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine-sensitive stores. Neuropharmacology 48, 1086–1096. doi: 10.1016/j.neuropharm.2005.01.005
Zimmermann, C., Ginis, I., Furuya, K., Klimanis, D., Ruetzler, C., Spatz, M., et al. (2001). Lipopolysaccharide-induced ischemic tolerance is associated with increased levels of ceramide in brain and in plasma. Brain Res. 895, 59–65. doi: 10.1016/S0006-8993(01)02028-5
Keywords: preconditioning, ischemic tolerance, stroke, TLR, epigenetics, microRNAs, TNF, inflammation
Citation: Garcia-Bonilla L, Benakis C, Moore J, Iadecola C and Anrather J (2014) Immune mechanisms in cerebral ischemic tolerance. Front. Neurosci. 8:44. doi: 10.3389/fnins.2014.000
Received: 29 September 2013; Accepted: 17 February 2014;
Published online: 04 March 2014.
Edited by:
Adam Denes, University of Manchester, UKReviewed by:
Sophie Layé, Université Bordeaux, FranceCraig J. Smith, Salford Royal NHS Foundation Trust, UK
Copyright © 2014 Garcia-Bonilla, Benakis, Moore, Iadecola and Anrather. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Josef Anrather, Brain and Mind Research Institute, Weill Cornell Medical College, 407 East 61st Street, RR-409, New York, NY 10065, USA e-mail:am9hMjAwNkBtZWQuY29ybmVsbC5lZHU=