 Mario Herrera-Marschitz1,2*
Mario Herrera-Marschitz1,2* Tanya Neira-Pena1,2,3
Tanya Neira-Pena1,2,3 Edgardo Rojas-Mancilla1Pablo Espina-Marchant1Daniela Esmar2Ronald Perez2Valentina Muñoz2Manuel Gutierrez-Hernandez1Benjamin Rivera2
Edgardo Rojas-Mancilla1Pablo Espina-Marchant1Daniela Esmar2Ronald Perez2Valentina Muñoz2Manuel Gutierrez-Hernandez1Benjamin Rivera2 Nicola Simola4
Nicola Simola4 Diego Bustamante2
Diego Bustamante2 Paola Morales2
Paola Morales2 Peter J. Gebicke-Haerter2,5
Peter J. Gebicke-Haerter2,5- 1Millenium Institute BNI-Chile, Santiago, Chile
- 2Department of Molecular and Clinical Pharmacology, ICBM, Medical Faculty, University of Chile, Santiago, Chile
- 3Department of Chemical-Biological Science, University Bernardo O'Higgins, Santiago, Chile
- 4Department of Biomedical Sciences, Section of Neuropsychopharmacology, Cagliari University, Cagliari, Italy
- 5Department of Psychopharmacology, Central Institute of Mental Health J5, Mannheim, Germany
Perinatal asphyxia constitutes a prototype of obstetric complications occurring when pulmonary oxygenation is delayed or interrupted. The primary insult relates to the duration of the period lacking oxygenation, leading to death if not re-established. Re-oxygenation leads to a secondary insult, related to a cascade of biochemical events required for restoring proper function. Perinatal asphyxia interferes with neonatal development, resulting in long-term deficits associated to mental and neurological diseases with delayed clinical onset, by mechanisms not yet clarified. In the experimental scenario, the effects observed long after perinatal asphyxia have been explained by overexpression of sentinel proteins, such as poly(ADP-ribose) polymerase-1 (PARP-1), competing for NAD+ during re-oxygenation, leading to the idea that sentinel protein inhibition constitutes a suitable therapeutic strategy. Asphyxia induces transcriptional activation of pro-inflammatory factors, in tandem with PARP-1 overactivation, and pharmacologically induced PARP-1 inhibition also down-regulates the expression of proinflammatory cytokines. Nicotinamide has been proposed as a suitable PARP-1 inhibitor. Its effect has been studied in an experimental model of global hypoxia in rats. In that model, the insult is induced by immersing rat fetus into a water bath for various periods of time. Following asphyxia, the pups are delivered, treated, and nursed by surrogate dams, pending further experiments. Nicotinamide rapidly distributes into the brain following systemic administration, reaching steady state concentrations sufficient to inhibit PARP-1 activity for several hours, preventing several of the long-term consequences of perinatal asphyxia, supporting the idea that nicotinamide constitutes a lead for exploring compounds with similar or better pharmacological profiles.
Introduction
It has been proposed that metabolic and environmental insults occurring during early development stages (i.e., pre- and perinatal) lead to psychiatric manifestations with an onset at adolescence and/or young adulthood stages (Basovich, 2010). Accordingly, epidemiologic studies have shown that pre- and perinatal factors, like maternal infections (Brown and Derkits, 2010) or hypoxia are related to schizophrenia (Cannon et al., 2002, 2008). Furthermore, high maternal anxiety during pregnancy has been associated to attention deficit and hyperactivity disorders, together with cognitive impairments and emotional symptoms (Van den Bergh et al., 2005, 2006; Loomans et al., 2011, 2012). In animal models, different risk factors have been identified occurring early in life, including maternal stress during pregnancy (Baier et al., 2012), early postnatal maternal separation (Zhu et al., 2010; Hulshof et al., 2011), neonatal bedding stress (Green et al., 2011), infections (Landreau et al., 2012) and asphyctic injury (Dell'Anna et al., 1991; Morales et al., 2011), promoting cognitive deficits, anxiety, and other behavioral disturbances at adulthood. Obstetric complications, in particular, are associated to psychiatric and neurological disorders (Cannon et al., 2002), hypoxia being a recurrent co-factor, adversely priming brain development by mechanisms not yet established (Low, 2004; Basovich, 2010; Herrera-Marschitz et al., 2011).
Delay in starting pulmonary ventilation at birth implies decrease of oxygen saturation in blood and reduced oxygen supply to the brain, which depends on aerobic metabolism for maintaining the respiratory chain and mitochondrial ATPase activity. Whenever hypoxia is sustained, there is a switch to glycolysis, which for neurons is a poor metabolic alternative, because of low stores of glucose in brain tissue and deficient ATP output by the glycolysis pathway. Glycolysis implies production of lactate, which is accumulated in extracellular compartments, causing acidosis, although it has also been suggested that lactate is an energy source for neurons (see Wyss et al., 2011). Prolonged hypoxia impairs gene expression, decreases transcription and translation, as well as activation of genes, such as hypoxia inducible factor (HIF) and its target molecules (Iyer et al., 1998).
Re-oxygenation is a requisite for survival, but it encompasses uneven metabolism, with metabolically privileged (e.g., heart, brain, and adrenal medulla) and less privileged (e.g., muscles, kidneys, and carcass) organs, including uneven distribution in brain regions. During the re-oxygenation period extracellular glutamate levels are increased, enhancing the activation of Na+/K+ ATPase, increasing further ATP consumption. Extracellular glutamate levels override the buffer capacity of astrocytes, resulting in sustained overactivation of glutamate receptors, mainly of the N-methyl-D-aspartate (NMDA) subtype, increasing Ca2+ conductance and further improper homeostasis. The metabolic crisis implies lactate accumulation and acidosis, differently reflected along development and brain regions. Indeed, we found that lactate levels remained increased (>2-fold) in neostriatum, but not in other regions of the basal ganglia of severely asphyxia-exposed, compared to control rats assayed 8 days after birth (Chen et al., 1997a).
All these changes are inherent biological reactions that result in partial recovery, but also in sustained overexpression of alternative or even antagonistic metabolic pathways, and molecular and cell cascades, prolonging the energy deficit and oxidative stress, associated with further cell damage and apoptotic or necrotic cell death (Figure 1). Molecular and cell cascades involved in removal of cells damaged by the reduced supply of oxygen are the ubiquitination, peroxisomal, and caspase pathways, whereas optional, compensatory cascades elicit multiple mechanisms of DNA repair, for prevention of cell loss or salvage of cells, respectively.
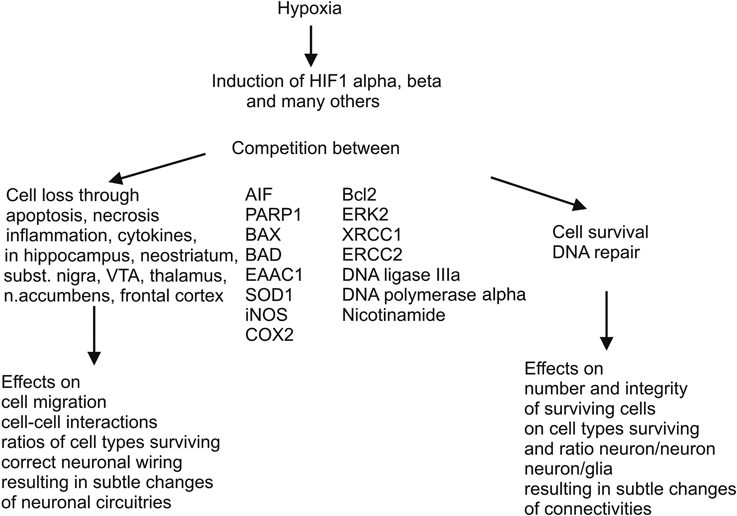
Figure 1. Antagonistic molecular and cell cascades elicited by perinatal asphyxia. There are two important molecular and cell cascades elicited by perinatal hypoxic insults, one leading to removal of cells damaged by the reduced supply of oxygen, implying activation of ubiquitination, peroxisomal, and caspase pathways, resulting in apoptosis or necrosis, the latter encompassing mild to severe inflammation. The other cascade is activated for compensating cell loss by multiple mechanisms of DNA repair. Naturally occurring nicotinamide with its various mechanisms of action could support these compensatory mechanisms if given systemically as a therapeutic means. In any case, both cell loss and cell rescue eventually entail more or less subtle consequences on brain development, neuronal wiring, and neuron glia interactions. These tentatively negative consequences are probably reinforced by additional negative impact during development (puberty and adolescence), resulting in psychiatric disorders if not aggravating neurological deficits.
Oxidative stress is inherent to re-oxygenation, resulting in overactivation, but also inactivation of a number of buffering enzymes; including those modulating the activity of mitochondria (see Gitto et al., 2002). In the clinical scenario, resuscitation may even imply hyperoxemia, leading to further production of free radicals and oxidative stress, worsening brain injury (Davis et al., 2004; Solberg et al., 2007; see Kapadia et al., 2013). Therefore, hypothermia has been proposed as a therapeutic intervention, targeting functional hyperoxemia and oxidative stress, decreasing the metabolic demands, attenuating the disturbances of metabolism elicited by re-oxygenation, preventing the short- and long-term consequences of perinatal asphyxia (Gunn et al., 1998; Engidawork et al., 2001). Indeed, hypothermia is now recommended as a routine intervention in the clinical practice (Azzopardi et al., 2009; Hagmann et al., 2011), although there is still concern about a narrow therapeutic window (Roelfsema et al., 2004; see also Van den Broek et al., 2010), and doubts about whether the treatment can really prevent the long-term consequences of perinatal asphyxia.
Cell Death and Regional Vulnerability: Endpoints for the Outcome of Perinatal Asphyxia
Delayed cell death is an important end-point of perinatal asphyxia, associated to caspase-dependent and caspase-independent mechanisms (Northington et al., 2001). Several pro-apoptotic proteins are increased following perinatal asphyxia, including B-cell lymphoma-2 (Bcl-2) associated X (BAX), and Bcl-2 associated death (BAD) factors, but also anti-apoptotic proteins, including Bcl-2, extracellular signal-regulated kinase 2 (ERK2), and basic fibroblast growth factor (bFGF) (Morales et al., 2008). Extensive and regionally selective nuclear fragmentation has been observed in control and asphyxia-exposed rat pups, depending upon the stage of development and the analyzed brain region (Dell'Anna et al., 1997). Consequently, signs of apoptosis have been found in normal and asphyxia-exposed animals, independently upon the severity of the insult. However, in neocortex, neostriatum and mesencephalon a significant increase of apoptotic cells is only observed in asphyxia-exposed animals, which documents regional vulnerability (Dell'Anna et al., 1997; Neira-Peña et al., 2014).
This regional vulnerability of the basal ganglia to anoxia/ischemia has been reported by several groups (Pasternak et al., 1991; Pastuzko, 1994; Cowan et al., 2003; Miller et al., 2005; Barkovich, 2006; Ferrari et al., 2011). It can be explained either by (1) the severity of the insult; (2) the local metabolic imbalance during the re-oxygenation period, and/or (3) the developmental stage of the affected regions. Immaturity of a particular brain region plays a role, because the insult affects the initial plastic changes required for establishing neurocircuitries and synaptogenesis. Furthermore, the re-establishment of homeostasis consumes extra energy that competes with the demands required for consolidating neurocircuitries and synapsis consolidation.
Development of neurocircuitries is based on a strict sequential programming. For instance, in terms of dopamine pathways, it is well-established that at P1 the brain of rats possesses a similar number of dopamine cell bodies as in adulthood, but, although dopamine fibers start to invade the neostriatum before birth (Seiger and Olson, 1973), dopamine-containing axon terminals reach a peak at the fourth postnatal week, and a mature targeting is only achieved after several postnatal weeks, when patches are replaced by a diffuse innervation pattern (Olson and Seiger, 1972; Seiger and Olson, 1973; Voorn et al., 1988; Antonopoulos et al., 2002; Loizou, 1972). From this time on, dopaminergic axons continue to grow at a slow rate during adulthood (Loizou, 1972; Voorn et al., 1988), followed by naturally occurring waves of dopamine cell death (Oo and Burke, 1997; Antonopoulos et al., 2002). The neocortex and hippocampus are also immature at postnatal stages. In the rat, neocortical pyramidal projections become physiologically viable only 1 week after birth (Li and Martin, 2000; Meng and Martin, 2003; Meng et al., 2004), and in humans the prefrontal cortex achieves a full mature stage long after postadolescent stages (Sowell et al., 1999; Segalowitz and Davies, 2004).
Henceforth, it is not surprising that mesostriatal, mesolimbic and mesocortical monoamine pathways are vulnerable to perinatal asphyxia. Long-lasting decreases of tyrosine hydroxylase (TH)-positive cell bodies have been observed in substantia nigra and ventral tegmental area (VTA) of rats exposed to severe perinatal asphyxia (Chen et al., 1995, 1997b), together with decreased dopamine utilization in neostriatum, accumbens, and olfactory tubercle (Ungethüm et al., 1996; Bustamante et al., 2003) 1 month after birth. A decrease in D-amphetamine stimulated dopamine release was also observed when asphyxia-exposed animals were evaluated using in vivo microdialysis 3 months after birth (Bustamante et al., 2007). In agreement, decreases in TH immunohistochemistry have been observed in neostriatum, hippocampus, thalamus, frontal cortex, and cerebellum of asphyxia-exposed rats evaluated 1–3 months after birth (Kohlhauser et al., 1999a,b). However, in the same animals, the excitatory amino acid carrier 1 (EAAC1) was increased in the frontal cortex (Kohlhauser et al., 1999b). The regional selectivity of the insult has been further investigated with triple organotypic cultures, finding a selective decrease in the number of dopamine neurons in cultures from asphyxia-exposed animals. In contrast, in the same cultures, nitric oxide synthase (NOS) positive neurons were increased in substantia nigra, decreased in neostriatum, and not changed in neocortex, again underlining the regionally different vulnerability (Klawitter et al., 2007). Neurite length and branching of neurons with dopamine and NOS phenotypes were also decreased in rats exposed to perinatal asphyxia (Morales et al., 2003; Klawitter et al., 2005, 2007). A similar effect has been observed in hippocampus. Neurite length and branching, as well as the expression of synaptophysin and postsynaptic density protein 95 (PSD95), pre- and postsynaptic markers, respectively, were found to be decreased at P30 in tissue from asphyxia-exposed animals (Rojas-Mancilla et al., 2013).
Sentinel Proteins
Suppression and/or overactivation of gene expression occur immediately or during the re-oxygenation period following perinatal asphyxia (Labudova et al., 1999; Mosgoeller et al., 2000; Seidl et al., 2000; Lubec et al., 2002). When DNA integrity is compromised, a number of sentinel proteins is activated, including poly(ADP-ribose) polymerases (PARPs) (Amé et al., 2004); X-Ray Cross Complementing Factor 1 (XRCC1) (Green et al., 1992); DNA ligase IIIα (Leppard et al., 2003); DNA polymerase β (Wilson, 1998; Mishra et al., 2003); Excision Repair Cross-Complementing Rodent Repair Group 2 (ERCC2) (Sung et al., 1993; Chiappe-Gutierrez et al., 1998; Lubec et al., 2002), and DNA-dependent protein kinases (De Murcia and Menissier de Murcia, 1994).
PARP proteins transfer adenosine diphosphate (ADP)-riboses from nicotinamide dinucleotide (NAD+) to glutamic and aspartic residues of the PARPs and their substrates. PARPs also catalyze the polymerization of ADP-riboses via glycosidic bonds, creating long and branched ADP-ribose polymers. PARP-1 is the most abundant and conserved member of a large superfamily comprising at least 18 PARP proteins, encoded by different genes, but displaying a conserved catalytic domain. PARP-1 is involved in DNA repair, but it also promotes cell death (see De Murcia and Menissier de Murcia, 1994; Kauppinen and Swanson, 2007; Cohen-Armon, 2008). When DNA damage is mild, PARP-1 is involved in the maintenance of chromatin integrity, by signaling cell-cycle arrest or activating DNA repairing molecular cascades. Furthermore, PARP-1 is involved in the regulation of cell proliferation and differentiation, modulating the transcription of several inflammatory signals, including nuclear factor κB (NF-κB) (Hassa and Hottinger, 1999). Excessive PARP-1 activation leads to NAD+ exhaustion and energy crisis (Berger, 1985), and to caspase-independent apoptosis, via translocation of the mitochondrial pro-apoptotic protein Apoptosis-Inducing Factor (AIF) to the nucleus, producing nuclear condensation (Jiang et al., 1996; Yu et al., 2002; Hong et al., 2004). PARP-1 has been involved in the long-term effects produced by perinatal asphyxia (Martin et al., 2005), interacting with XRCC1, DNA ligase IIIα, and DNA polymerase-β, working in tandem to repair single-strand breaks. DNA ligase IIIα has a N-terminal zinc finger interacting with the DNA binding domain of PARP-1 and DNA strand breaks. Further, DNA Ligase IIIα interacts with XRCC1, forming a DNA Ligase IIIα-XRCC1 complex (see Ellenberger and Tomkinson, 2008; Odell et al., 2011; Simsek et al., 2011), and DNA polymerase-β can couple to PARP-1, DNA Ligase IIIα, and XRCC1, providing the overall stability of the repair complex, promoting catalysis and fidelity (Sawaya et al., 1997). It has been shown that PARP-1, DNA polymerase-β, and XRCC1 expression are increased by hypoxia in newborn piglets (Mishra et al., 2003) and rats (Chiappe-Gutierrez et al., 1998). With an ischemic preconditioning model, Li et al. (2007) demonstrated a 5-fold increase of XRCC1 levels 30 min after ischemia, reaching a maximal expression after 4 h. DNA polymerase-β and DNA ligase IIIα levels were also increased, and co-expressed in neuron and glial cells. PARP-1 can be phosphorylated by ERK, probably via the isoform 2 (ERK2), as a requirement for maximal PARP-1 activation after DNA damage (Kauppinen et al., 2006). Nevertheless, Cohen-Armon et al. (2007) reported evidence for PARP-1 activation by phosphorylated ERK2, in the absence of DNA damage, via the signaling cascade of the transcription factor ETS domain-containing protein 1 (Elk1), increasing the expression of the immediate early gene c-fos, stimulating cell growth and differentiation. Thus, cell type specificity and regional distribution of sentinel proteins may provide regulatory mechanisms by which the long-term effects of metabolic insults occurring at birth are heterogeneous, targeting some, but leaving other brain regions apparently untouched.
PARP-1 Activity and Inflammatory Signaling
In the developing brain, re-oxygenation leads to oxidative stress, generating reactive oxygen species (ROS) and H2O2 accumulation, a dangerous outcome, because the immature brain expresses low activity of glutathione peroxidase (GPx) and catalase, the enzymes ameliorating the damaging effects of superoxide and hydrogen peroxide, respectively (Lafemina et al., 2006). While superoxide dismutase (SOD) is the only known enzymatic scavenger of extracellular superoxide levels, its overexpression in the cytosol (SOD1) of neonatal animals increases hypoxic-ischemic brain injury (Ditelberg et al., 1996), together with H2O2 accumulation (Lafemina et al., 2006). The metabolic insult enhances nitric oxide (NO) synthesis via inducible nitric oxide synthase (iNOS), producing large amounts of NO over extended periods, damaging cell constituents, promoting peroxynitrite formation, and lipid and protein peroxidation (Bonfocco et al., 1995), as well as activation of cyclooxygenase-2 (COX-2) (Nogawa et al., 1998), eliciting inflammatory cascades (see Hagberg et al., 1996).
DNA damage elicited by perinatal asphyxia implies PARP-1 overactivation, promoting NF-κB translocation and expression of proinflammatory cytokines (Ullrich et al., 2001), a phenomenon associated with microglial migration toward the site of neuronal injury (see Skaper, 2003; Gagne et al., 2008). Conversely, PARP-1 inhibition suppresses NF-κB dependent gene transcription in microglia (Kauppinen et al., 2008). In a model of bilateral carotid occlusion-reperfusion in rats, Kauppinen et al. (2009) demonstrated that treatment with the selective PARP-1 inhibitor PJ34 ([N-(6-Oxo-5, 6-dihydrophenanthridin-2-yl)-N, N-dimethylacetamide-HCl]), 48 h later, suppressed ischemia-induced microglial activation, enhancing long-term neuronal survival, neurogenesis, and spatial memory. Also, in a model of forebrain ischemia, PJ34 produced a near-complete inhibition of microglia activation and a significant reduction of neuronal death in the hippocampus (Hamby et al., 2007). Furthermore, it was reported that minocycline, a tetracycline derivative with anti-inflammatory properties (see Carty et al., 2008), protected neuronal cultures against genotoxic agents, such as N-methyl-N-nitro-N-nitrosoguanidine (MNNG) and 1, 3-morpholinosydonimine (SIN-1). Interestingly, minocycline also inhibited PARP-1 activity (Alano et al., 2006). It has been suggested that perinatal asphyxia induces transcriptional activation of pro-inflammatory factors, in tandem with PARP-1 overactivation (Neira-Peña et al., 2013). Indeed, there is an increase of mRNA levels of IL-1β and TNF-α in mesencephalon and hippocampus of asphyxia-exposed animals, in agreement with reports showing that TNF-α, IL-1β, as well as IL-6 modulate cell death, vascular permeability and recruitment of peripheral blood cells into the CNS (see Deverman and Patterson, 2009; Russo et al., 2011), supporting the idea that inflammasomes, a large intracellular multiprotein complex (see Schroder and Tschopp, 2010), plays a central role in the activation of IL-1β, via NACHT, LRR, and PYD domain-containing protein 3 (NALP3). NLRP3 recruits caspase-1, which cleaves pro-IL1β, generating the active form of IL-1β. Inflammasomes can also be activated by damage-associated molecular pattern molecules (DAMPs), known as danger-associated molecular pattern molecules, including extracellular ATP and molecules related to metabolic stress (see Schroder and Tschopp, 2010; Fann et al., 2013).
A relationship between serum levels of IL-1β, IL-6, and TNF-α and the outcome of infants that have suffered of perinatal asphyxia has been observed in humans (Foster-Barber et al., 2001), suggesting that proinflammatory cytokines have a predictive value. Indeed, a study of blood and CSF obtained during the first 24 h of life from infants who suffered hypoxic ischemic encephalopathy showed an increase in IL-1β, IL-6, and TNF-α levels, compared to control infants (Aly et al., 2006).
Targets for Neuroprotection
It is proposed that PARP-1 inhibition is a target for neuroprotection following hypoxia/ischemia. Several PARP inhibitors, with increasing degrees of potency, have been shown to decrease brain damage, improving the neurological outcome of perinatal brain injury (Zhang et al., 1995; Ducrocq et al., 2000; Sakakibara et al., 2000; see Virag and Szabo, 2002; Jagtap and Szabo, 2005; Kauppinen et al., 2009).
The idea that PARP-1 activation is beneficial has also been explored, depending upon the actual levels of cellular NAD+. While PARP inhibitors offer remarkable protection under conditions of NAD+ and ATP depletion, inhibition of PARP-1 in the presence of NAD+ sensitizes cells to DNA damage, and subsequent increase of cell death (Nagayama et al., 2000). Also, inhibition of PARP-1 induces apoptosis in rapidly dividing cells (Saldeen and Welsh, 1998), probably by blocking the access of repairing enzymes. Thus, PARP-1 acts as both a cell survival and cell death-inducing factor by regulation of DNA repair, chromatin remodeling, and regulation of transcription.
Nicotinamide is a reference compound regarding PARP-1 inhibition (Virag and Szabo, 2002). Because of its relative low potency, it is proposed to be useful when treating young animals, antagonizing the effects elicited by PARP-1 overactivation without impairing DNA repair or cell proliferation. Nicotinamide has also been proposed as an antioxidant compound (Wan et al., 1999; Yan et al., 1999), and its pharmacodynamics can provide advantages over more selective compounds (Griffin et al., 2013; Shetty et al., 2013; Turunc Bayrakdar et al., 2014). Apparently, it also helps to replenish the reduced pool of NAD+ by being metabolized through nicotinamide phosphoribosyltransferase; (NAMPT) (Pittelli et al., 2014).
An Experimental Working Model for Perinatal Asphyxia
While the short-and long-term clinical outcomes of perinatal asphyxia are well-established, pre-clinical research is still at an exploratory phase, mainly because of a lack of consensus on a reliable and predictable experimental model. A model for investigating the issue was proposed at the Karolinska Institutet, Stockholm, Sweden, at the beginning of the nineties (Bjelke et al., 1991; Andersson et al., 1992; Herrera-Marschitz et al., 1993). The model starts by evaluating the oestral cycle of young female rats for a programmed mating. A vaginal frottis is taken for evaluating the cycle, and the female is exposed to a male at the time of the pro-oestrous for 1 night. Thereafter the presence of a vaginal clot is evaluated, to predict the exact time of delivery (22 days after a vaginal clot has been recorded). At the time of delivery, a first spontaneous delivery can be observed before the dams are anaesthetized, neck dislocated, and subjected to a caesarean section and hysterectomy. The uterine horns containing the fetus are immediately immersed into a water bath at 37°C for various periods of time (0–22 min). Following asphyxia, the pups are removed from the uterine horns and resuscitated. Additional efforts and care are taken to induce and maintain pulmonary breathing. Several laboratories are running the model, although it has criticized that it works with on term pups and not with neonates at P7, arguing that the brain of neonate rats is premature when compared to the neonatal human brain, a statement mainly referring to the neocortex (see Romijn et al., 1991), and the pattern of oligodendrocyte lineage progression required for cerebral myelination (Craig et al., 2003). Our view is that the degree of maturity depends upon the tissue and functions selected for the comparisons, vulnerability relating to both the timing and the location of the insult (Craig et al., 2003, see also De Louw et al., 2002). In agreement, it has been reported that susceptibility to hypoxia-ischemia induced by common carotid artery ligation and hypoxemia is greater when performed at P7 rather than at earlier postnatal periods (Towfighi et al., 1997).
Our approach is rather pragmatic, inducing asphyxia at the time when rats are delivered. Thus, our model: (1) mimics well-some relevant aspects of human delivery; (2) it is largely non-invasive; (3) it allows studying short- and long-term consequences of the insult in the same preparation, and (4) it is highly reproducible among laboratories. Gert Lubec and co-workers in Vienna (Austria) (Lubec et al., 1997a,b; Seidl et al., 2000) have stressed the issue that the model is suitable for studying the early phase of perinatal asphyxia, as observed in the clinical setup, performed during delivery, acknowledging the fact that the model implies oxygen interruption, but not any additional lesions, including, by example, vessel occlusion. Moreover, it is followed by hypoxemia, acidosis and hypercapnia, mandatory criteria for a clinically relevant model of perinatal asphyxia (Seidl et al., 2000).
Pups exposed to zero or mild asphyxia start breathing with a gluttonous gasp, which is rapidly replaced by regular and synchronized breathing. For pups exposed to longer periods of asphyxia (19–21 min), resuscitation implies expert and skillful handling, taking a long time (4–6 min) for a first gasping, and even longer time for establishing a more or less regular breathing, always supported by gasping. Surrogate dams can nurture the pups, whether long-term experiments are desirable.
With that rat model (Bjelke et al., 1991; Andersson et al., 1992; Herrera-Marschitz et al., 1993), we reported that nicotinamide prevents several of the neuronal (Klawitter et al., 2006, 2007; Morales et al., 2010), neurochemical (Bustamante et al., 2003, 2007) and behavioral (Simola et al., 2008; Morales et al., 2010) effects produced by perinatal asphyxia. With microdialysis experiments performed 1 h after birth, it was found that after a single systemic dose (0.8 mmol/kg, i.p.), nicotinamide rapidly distributed along different compartments including the brain, achieving concentrations larger than 30 μ M for longer than 30 min, producing a long-lasting inhibition of PARP-1 activity in mesencephalon, telencephalon, and heart of asphyxia-exposed and control animals (Allende-Castro et al., 2012). Interestingly, PARP-1 inhibitors, which do not reach the brain at enough sustained concentrations, such as 1,3-dimethylxanthine (theophylline) (Geraets et al., 2006; Allende-Castro et al., 2012), do not protect against the long-term effects elicited by perinatal asphyxia, although it has been shown that theophylline reduces inflammation and innate immunity (Peters-Golden et al., 2005).
Cognitive Deficits
Motor and cognitive alterations of variable severity, including cerebral palsy, seizures, spasticity, attention deficit, hyperactivity, mental retardation, and other neuropsychiatric syndromes with delayed clinical onset have been associated to perinatal asphyxia (du Plessis and Volpe, 2002; Van Erp et al., 2002; Kaufman et al., 2003; Vannuci and Hagberg, 2004; De Hann et al., 2006; Odd et al., 2009). In the rat, several studies have investigated the behavioral effects associated with perinatal asphyxia, addressing motor function (Bjelke et al., 1991; Chen et al., 1995), emotional behavior (Dell'Anna et al., 1991; Hoeger et al., 2000; Venerosi et al., 2004, 2006; Morales et al., 2010), and spatial memory (Boksa et al., 1995; Iuvone et al., 1996; Hoeger et al., 2000, 2006; Loidl et al., 2000; Van de Berg et al., 2003; Venerosi et al., 2004).
We have investigated whether perinatal asphyxia produces long-term effects on cognitive performance, using an object recognition test (Ennaceur and Delacour, 1988), based on the ability of the rat to discriminate between objects differing in shape and color, without any association to rewarding or aversive stimuli. During a first session, two copies of the same object are presented to the rat for a short period, and then again, during a second session, when one of the previously presented copies is replaced by a novel object, similar in size, but different in shape and/or color. The idea is that the rat has to recognize the novel object, spending longer time exploring the novel than the previously presented object. The first and the second sessions can be separated by different time intervals, for evaluating learning consolidation. A good memory would be able to recognize a previously presented object after a long elapsing time between a first and a second session, implying that the animal concentrates on exploring the novel object. In our studies, we used 15 or 60 min intervals, and the animals were studied at 3 months of age (Simola et al., 2008). No differences were observed between animals exposed to severe asphyxia and control animals when a 15 min interval elapsed between the first and the second session. However, when 60 min elapsed between the first and second session, asphyxia-exposed animals spent less time exploring the novel object, indicating that asphyxia-exposed rats could not recognize its novelty.
Notably, the total exploration time was not affected by the insult, suggesting that asphyctic rats do not have non-specific deficits in object discrimination, but rather display a specific deficit in non-spatial working memory (Simola et al., 2008). This is a straightforward experiment showing a subtle consequence of a metabolic insult (hypoxia) occurring at birth, impairing a cognitive function that shows up only after a proper challenge. Results in line with these have also been obtained by subsequent studies in either rats or mice subjected to the same experimental model of perinatal asphyxia (Hutton et al., 2009; Strackx et al., 2010). This impairment in object recognition is very much reminiscent of the clinical experience revealing effects only when the child starts primary school (see Odd et al., 2009; Strackx et al., 2010).
Nicotinamide prevented the effect of perinatal asphyxia on novel object recognition evaluated after a 60 min elapse. Nicotinamide did not affect the pattern observed in the corresponding control rats (Morales et al., 2010). The effect of nicotinamide on novel object recognition deficit induced by perinatal asphyxia was related to that on hippocampus delayed cell death, suggesting an effect on a specific neuroanatomical substrate.
Conclusions
Perinatal asphyxia is still a health concern worldwide, a risk factor for several mental and neurological disorders showing a delayed clinical onset.
As shown by Figure 2, hypoxia implies a severe energetic crisis, leading to death if re-oxygenation is not promptly restored. Decrease of oxygen saturation implies a shift to anaerobic metabolism for preserving the fundamental demands of energy. Anaerobic metabolism is, however, inefficient for providing the energy required by a sophisticated, high consuming, but low energy-storing organ, such as the brain. A decrease of energy implies low metabolism, which can be enough for maintaining basic functioning, competing, however, with the high metabolism required for development of neuronal networks and neurocircuitries. Anaerobic metabolism implies also lactate accumulation and acidosis. Whether oxygen supply is not restored, the reserves are exhausted. Energy can still be compartmentalized among privileged and less privileged brain regions, without consideration, however, about the implications for long-term development. Re-oxygenation is a requisite for survival, but the kinetics of oxygen supply implies global and/or local hyperoxemia, free radicals accumulation, oxidative stress, cell damage, and overactivation of defense mechanisms, including sentinel proteins, but also inflammatory cascades and further cell stress.
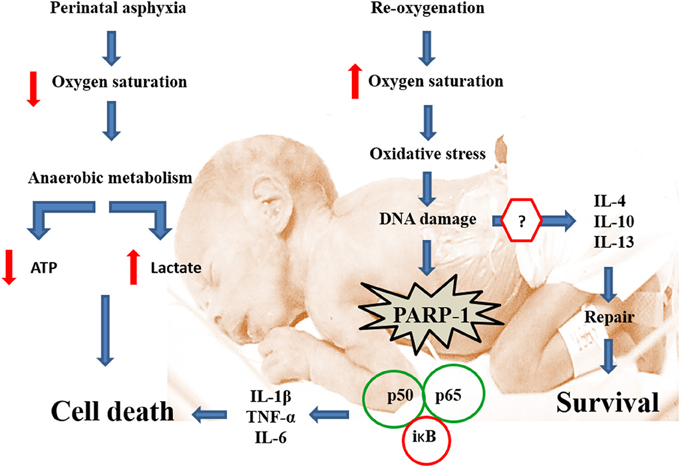
Figure 2. Transcriptional activation of pro-inflammatory signaling in tandem with PARP-1 overactivation. Perinatal asphyxia decreases oxygen saturation in blood, leading to a switch from aerobic to a less efficient anaerobic metabolism involving lactate accumulation, acidosis and cell death. Re-oxygenation is essential for survival. Nevertheless, re-oxygenation induces oxidative stress, damaging several biomolecules including DNA. In response to DNA damage, PARP-1 increases its activity, recruiting the DNA repair machine. PARP-1 overactivation modifies several target proteins via poly (ADP-ribose) polymers (pADPr). One of the targets of pADPr ribosylation is NF-κB, whose p65 subunit is translocated to the nucleus, activating the transcription of pro-inflammatory (e.g., TNF-α, IL-1β, and IL-6), but also anti-inflammatory (e.g., IL-4, IL-10; IL-13) cytokines. The balance between pro- and anti-inflammatory cytokines is crucial for determining cell survival or death.
Thus, the functional constraints produced by the lack of oxygen are exacerbated by and during the re-oxygenation period, implying oxidative stress, synthesis, and release of metabolic by-products delaying the onset of proper homeostasis and recovery. A number of sentinel proteins are rapidly activated whenever there is a risk of genome damage, stimulating base excision repair. PARP-1 plays a pivotal role for repairing damaged DNA, eliciting also caspase-independent cell death when repair is not viable. PARP-1 overactivation leads to NAD+ exhaustion, worsening the energy crisis, which is the basis for the hypothesis that PARP-1 is a suitable target for therapeutic interventions preventing the long-term effects of perinatal asphyxia, nicotinamide being a prototype for counteracting PARP-1 overactivation. Asphyxia also induces transcriptional activation of pro-inflammatory factors, in tandem with PARP-1 overactivation. In agreement, pharmacologically induced PARP-1 inhibition also down-regulates the expression of proinflammatory cytokines.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Contract grant sponsors: FONDECYT-Chile (contracts: 1120079; 1110263); Millenium Institute Initiative (BNI P09-015-F); BMBF (NGFN+ TP9), and DAAD (415/alechile)-Germany. Tanya Neira-Pena is a BNI fellow; Edgardo Rojas-Mancilla is CONICYT (#21090557) and ICBM fellow. Nicola Simola gratefully acknowledges Sardinia Regional Government for financial support (P.O.R. Sardegna F.S.E. Operational Programme of the Autonomous Region of Sardinia, European Social Fund 2007–2013—Axis IV Human Resources, Objective l.3, Line of Activity l.3.1 “Avviso di chiamata per il finanziamento di Assegni di Ricerca”).
Abbreviations
ADP, adenosine diphosphate; AIF, apoptosis-inducing factor; ATP, adenosine triphosphate; ATPase, ATP polymerase; BAD, Bcl-2-associated death factor; BAX, Bcl-2-associated X protein; BBB, blood-brain barrier; bFGF, basic fibroblast growth factor; Bcl-2, B-cell lymphoma-2; CA, cornus Ammonis (CA1, C2, C3); CAM, cell adhesion molecule; CNS, central nervous system; COX-2, cyclooxygenase-2; CS, caesarean-delivered saline treated animal; CSF, cerebral spinal fluid; DAMPs, damage-associated molecular pattern molecules; DG, dentate gyrus of the hippocampus; DNA, deoxyribonucleic acid; EAAC1, excitatory amino acid carrier 1; Elk1, ETS domain-containing protein 1; ERCC2, excision repair cross-complementing rodent repair group 2; ERK, extracellular signal-regulated kinases; FGFR, bFGF receptors; FLRT3, leucine-rich repeat transmembrane protein; GPx, glutathione peroxidase; HIF, hypoxia inducible factor; IκB, inhibitor of kappa B protein; iNOS, inducible NOS; IL-1β/-6, interleukin-1β/-6; ICAM-1, TNFα adhesion molecule-1; L1, L1 CAM; LPS, lipopolysaccharide; MAP-2, microtubule-associated protein-2; MAPK, mitogen-activated protein kinase; MNNG, N-methyl-N-nitro-N-nitrosoguanidine; NACHT, LRR and PYD domains-containing protein 3 (cryopyrin); NAD+, oxidized nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; NALP3, NACHT, LRR, and PYD domain-containing protein 3 (cryopyrin); NAMPT, nicotinamide phosphoribosyltransferase; NF-κB, nuclear factor κB; NMDA, N-methyl-D-aspartate; NO, nitric oxide; NOS, nitric oxide synthase; nNOS, neuronal NOS; p65/p50, protein subunits of 65/50 k Dalton MW; P1, postnatal day 1; PARP-1, poly(ADP-ribose) polymerase-1; PIP2, phosphatidylinositol-4,5-bisphosphate; PJ34, [N-(6-Oxo-5, 6-dihydrophenanthridin-2-yl)-N, N-dimethylacetamide.HCl]; PKC, protein kinase C; pADPr, poly (ADP-ribose) polymers; PSD95, postsynaptic density protein 95; RhoA, ras homolog gene family, member A, small GTPase protein; ROS, reactive oxygen species; Sef, similar expression fgf gene; SEM, standard error of the means; SIN, 1, 3-morpholinosydonimine; SIRT, sirtuin; SOD, superoxide dismutase; Spry, sprouty; SRY, sex determining region Y; SVZ, subventricular zone; TH, tyrosine hydroxylase; Thy1, thymocyte differentiation antigen 1; TNF-α, tumor necrosis factor-alpha cytokine; TUNEL, TdT-mediated dUTP nick-end labeling; VTA, ventral tegmental area; XRCC1, X-ray cross complementing F factor 1.
References
Alano, C. C., Kauppinen, T. M., Valls, A. V., and Swanson, R. A. (2006). Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc. Natl. Acad. Sci. U.S.A. 103, 9685–9690. doi: 10.1073/pnas.0600554103
Allende-Castro, C., Espina-Marchant, P., Bustamante, D., Rojas-Mancilla, E., Neira, T., Gutierrez-Hernandez, M. A., et al. (2012). Further studies on the hypothesis of PARP-1 inhibition as a strategy for lessening the long-term effects produced by perinatal asphyxia: effects of nicotinamide and theophylline on PARP-1 activity in brain and peripheral tissue. Neurotox. Res. 22, 79–90. doi: 10.1007/s12640-012-9310-2
Aly, H., Khashaba, M. T., El-Ayouty, M., El-Sayed, O., and Hasanein, B. M. (2006). IL-1beta, IL-6 and TNF-alpha and outcomes of neonatal hypoxic ischemic encephalopathy. Brain Dev. 28, 178–182. doi: 10.1016/j.braindev.2005.06.006
Amé, J.-C., Spenlehauer, C., and de Murcia, G. (2004). The PARP superfamily. Bioessays 26, 882–893. doi: 10.1002/bies.20085
Andersson, K., Bjelke, B., Bolme, P., and Ögren, S. O. (1992). “Asphyxia-induced lesion of the rat hippocampus (CA1, CA3) and the nigro-striatal dopamine system,” in Hypoxia and Ischemia. CNS, Vol. 41. ed J. Gross (Berlin: Wissenschafliche Publikationen der Humboldt-Universität zu Berlin, B. Medizin), 71–76.
Antonopoulos, J., Dori, I., Dinopoulos, A., Chiotelli, M., and Parnavelas, J. G. (2002). Postnatal development of the dopaminergic system of the striatum of the rat. Neuroscience 110, 245–256. doi: 10.1016/S0306-4522(01)00575-9
Azzopardi, D., Strohm, B., Edwards, A. D., Halliday, H., Juszczak, E., Kapellu, O., et al. (2009). Moderate hypothermia to treat perinatal asphyxia encephalopathy. N. Engl. J. Med. 361, 1349–1358. doi: 10.1056/NEJMoa0900854
Baier, C. J., Katunar, M. R., Adrover, E., Pallarés, M. E., and Antonelli, M. C. (2012). Gestational restraint stress and the developing dopaminergic system: an overview. Neurotox. Res. 22, 16–32. doi: 10.1007/s12640-011-9305-4
Barkovich, A. J. (2006). MR imagining of the neonatal. Neuroimaging Clin. N. Am. 16, 117–135. doi: 10.1016/j.nic.2005.10.003
Basovich, S. N. (2010). The role of hypoxia in mental development and in the treatment of mental disorders. Biosci. Trends 4, 288–296. doi: 10.1016/S0924-9338(12)75173-8
Berger, N. A. (1985). Poly (ADP-ribose) in the cellular response to DNA damage. Radiat. Res. 1001, 4–15. doi: 10.2307/3576299
Bjelke, B., Andersson, K., Ögren, S. O., and Bolme, P. (1991). Asphyctic lesion: proliferation of tyrosine hydroxylase immunoreactive nerve cell bodies in the rat substantia nigra and functional changes in dopamine transmission. Brain Res. 543, 1–9. doi: 10.1016/0006-8993(91)91041-X
Boksa, P., Krishnamurthy, A., and Brooks, W. (1995). Effects of a period of asphyxia during birth on spatial learning in the rat. Pediatr. Res. 37, 489–496. doi: 10.1203/00006450-199504000-00018
Bonfocco, E., Krainc, D., Ankarcrona, M., Nicotera, P., and Lipton, S. A. (1995). Apotosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. U.S.A. 92, 7162–7166. doi: 10.1073/pnas.92.16.7162
Brown, A. S., and Derkits, E. J. (2010). Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am. J. Psychiatry 167, 261–280. doi: 10.1176/appi.ajp.2009.09030361
Bustamante, D., Goiny, M., Åström, G., Gross, J., Andersson, K., and Herrera-Marschitz, M. (2003). Nicotinamide prevents the long-term effects of perinatal asphyxia on basal ganglia monoamine systems in the rat. Exp. Brain Res. 148, 227–232.
Bustamante, D., Morales, P., Torres-Pereyra, J., Goiny, M., and Herrera-Marschitz, M. (2007). Nicotinamide prevents the effect of perinatal asphyxia on dopamine release evaluated with in vivo microdialysis three months after birth. Exp. Brain Res. 177, 358–369. doi: 10.1007/s00221-006-0679-0
Cannon, M., Jones, P. B., and Murray, R. M. (2002). Obstetric complications and schizophrenia: historical and meta-analytic review. Am. J. Psychiatry 159, 1080–1092. doi: 10.1176/appi.ajp.159.7.1080
Cannon, T. D., Yolken, R., Buka, S., and Torrey, E. F. (2008). Collaborative study group on the perinatal origins of severe psychiatric disorders decreased neurotrophic response to birth hypoxia in the etiology of schizophrenia. Biol. Psychiatry 64, 797–802. doi: 10.1016/j.biopsych.2008.04.012
Carty, M. L., Wixey, J. H., Colditz, P. B., and Buller, K. M. (2008). Post-insult minocycline treatment attenuates hypoxia-ischemia-induced neuroinflammation and white matter injury in the neonatal rat: a comparison of two different dose regimes. Int. J. Dev. Neurosci. 26, 477–485. doi: 10.1016/j.ijdevneu.2008.02.005
Chen, Y., Engidawork, E., Loidl, F., Dell'Anna, E., Gony, M., Lubec, G., et al. (1997a). Short- and long-term effects of perinatal asphyxia on monoamine, amino acids and glycolysis product levels measured in the basal ganglia of the rat. Dev Brain Res 104, 19–30.
Chen, Y., Herrera-Marschitz, M., Bjelke, B., Blum, M., Gross, J., and Andersson, K. (1997b). Perinatal asphyxia-induced changes in rat brain tyrosine-hydroxylase-immunoreactive cell body number: effects of nicotine treatment. Neurosci. Lett. 221, 77–80. doi: 10.1016/S0304-3940(96)13293-6
Chen, Y., Ögren, S. O., Bjelke, B., Bolme, P., and Eneroth, P., Gross, J., et al. (1995). Nicotine treatment counteracts perinatal asphyxia-induced changes in the mesostriatal/limbic dopamine systems and in motor behaviour in the four-week old male rat. Neuroscience 68, 531–538. doi: 10.1016/0306-4522(95)00118-3
Chiappe-Gutierrez, M., Kitzmueller, E., Labudova, O., Fuerst, G., Hoeger, H., Hardmeier, R., et al. (1998). mRNA levels of the hypoxia inducible factor (HIF-1) and DNA repair genes in perinatal asphyxia of the rat. Life Sci. 63, 1157–1167. doi: 10.1016/S0024-3205(98)00377-4
Cohen-Armon, M. (2008). PARP-1 activation in the ERK signalling pathway. TIPs 28, 556–560. doi: 10.1016/j.tips.2007.08.005
Cohen-Armon, M., Visochek, L., Rozendal, D., Kalal, A., Geistrikh, I., Klein, R., et al. (2007). DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol. Cell 25, 297–308. doi: 10.1016/j.molcel.2006.12.012
Cowan, F., Rutherford, M., Groenendaal, F., Eken, P., Mercuri, E., Bydder, G. M., et al. (2003). Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet 361, 736–742. doi: 10.1016/S0140-6736(03)12658-X
Craig, A., Luo, N. L., Bearsley, D. J., Wingate-Pearse, N., Walker, D., Hohimer, A. R., et al. (2003). Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp. Neurol. 181, 231–240. doi: 10.1016/S0014-4886(03)00032-3
Davis, P. G., Tan, A., O'Donnell, C. P., and Schulze, A. (2004). Resuscitation of newborn infants with 100% oxygen or air: a systematic review and meta-analysis. Lancet 364, 1329–1333. doi: 10.1016/S0140-6736(04)17189-4
De Hann, M., Wyatt, J. S., Vargha-Khadem, F., Gadian, D., and Mishki, M. (2006). Brain and cognitive behavioural development after asphyxia at term birth. Dev. Sci. 9, 350–358. doi: 10.1111/j.1467-7687.2006.00499.x
Dell'Anna, E., Calzolari, S., Milinari, M., Luvone, L., and Calimici, R. (1991). Neonatal anoxia induces transitory hyperactivity, permanent spatial memory deficits and CA1 cell density reduction in the developing rats. Behav. Brain Res. 45, 125–134.
Dell'Anna, E., Chen, Y., Engidawork, E., Andersson, K., Lubec, G., Luthman, J., et al. (1997). Delayed neuronal death following perinatal asphyxia in rat. Exp. Brain Res. 115, 105–115.
De Louw, A. J., Van De Berg, W. D., De Vente, J., Blanco, C. E., Gavilanes, A. W., Steinbusch, H. P., et al. (2002). Developmental apoptosis in the spinal cord white matter in neonatal rats. Glia 37, 89–91. doi: 10.1002/glia.1130
De Murcia, G., and Menissier de Murcia, J. (1994). poly(ADP-ribose) polymerase: a molecular nick-sensor. TIBS 19, 172–176. doi: 10.1016/0968-0004(94)90280-1
du Plessis, A. J., and Volpe, J. J. (2002). Perinatal brain injury in the preterm and term newborn. Curr. Opin. Neurol. 15, 151–157. doi: 10.1097/00019052-200204000-00005
Deverman, B. E., and Patterson, P. H. (2009). Cytokines and CNS development. Neuron 64, 61–78. doi: 10.1016/j.neuron.2009.09.002
Ditelberg, J. S., Sheldon, R. A., Epstein, C. J., and Ferreiro, D. M. (1996). Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr. Res. 39, 204–208. doi: 10.1203/00006450-199602000-00003
Ducrocq, S., Benjelloun, N., Plotkine, M., Ben-Ari, Y., and Charriaut-Marlangue, C. (2000). Poly(ADP-ribose) synthase inhibition reduces ischemic injury and inflammation in neonatal rat brain. J. Neurochem. 74, 2504–2511. doi: 10.1046/j.1471-4159.2000.0742504.x
Ellenberger, T., and Tomkinson, A. E. (2008). Eukaryotic DNA ligases: structural and functional insights. Annu. Rev. Biochem. 77, 313–338. doi: 10.1146/annurev.biochem.77.061306.123941
Engidawork, E., Loidl, F., Chen, Y., Kohlhauser, C., Stoeckler, S., Dell'Anna, E., et al. (2001). Comparison between hypothermia and glutamate antagonism treatments on the immediate outcome of perinatal asphyxia. Exp. Brain Res. 138, 375–383. doi: 10.1007/s002210100710
Ennaceur, A., and Delacour, J. (1988). A new one-trial test for neurobiological studies of memory in rats. I: behavioral data. Behav Brain Res 31, 47–59. doi: 10.1016/0166-4328(88)90157-X
Fann, D. Y.-W., Lee, S.-Y., Manzanero, S., Chunduri, P., Sobey, C. G., and Arumugan, T. V. (2013). Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 12, 941–966. doi: 10.1016/j.arr.2013.09.004
Ferrari, F., Todeschini, A., Guidotti, I., Martinez-Biarge, M., Roversi, A., Cowan, F. M., et al. (2011). General movements in full-term infants with perinatal asphyxia are related to basal ganglia and thalamic lesions. J. Pediatr. 158, 904–911. doi: 10.1016/j.jpeds.2010.11.037
Foster-Barber, A., Dickens, B., and Ferriero, D. M. (2001). Human perinatal asphyxia: correlation of neonatal cytokines with MRI and outcome. Dev. Neurosci. 23, 213–218. doi: 10.1159/000046146
Gagne, J.-P., Isabelle, M., Lo, K. S., Bourassa, S., Hendzel, M. J., Dawson, V. L., et al. (2008). Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 36, 6959–6976. doi: 10.1093/nar/gkn771
Geraets, L., Moonen, H. J. J., Wouters, E. F. M., Bast, A., and Hageman, G. J. (2006). Caffeine metabolites are inhibitors of the nuclear enzyme poly(ADP-ribose)polymerase-1 at physiological concentrations. Biochem. Pharmacol. 72, 902–910. doi: 10.1016/j.bcp.2006.06.023
Gitto, E., Reiter, R. J., Karbownik, M., Tan, D.-X., Gitto, P., Barberi, S., et al. (2002). Causes of oxidative stress in the pre- and perinatal period. Biol. Neonate 81, 146–157. doi: 10.1159/000051527
Green, A., Prager, A., Stoudt, P. M., and Murray, D. (1992). Relationships between DNA damage and the survival of radiosensitive mutant Chinese hamster cell lines exposed to gamma-radiation. Part 1: Intrinsic radiosensitivity. Int. J. Radiat. Biol. 61, 465–472. doi: 10.1080/09553009214551221
Green, P. G., Chen, X., Alvarez, P., Ferrari, L. F., and Levine, J. D. (2011). Early-life stress produces muscle hyperalgesia and nociceptor sensitization in the adult rat. Pain 152, 2549–2556. doi: 10.1016/j.pain.2011.07.021
Griffin, S. M., Pickard, M. R., Orme, R. P., Hawkins, C. P., and Fricker, R. A. (2013). Nicotinamide promotes neuronal differentiation of mouse embryonic stem cells in vitro. Neuroreport 24, 1041–1046. doi: 10.1097/WNR.0000000000000071
Gunn, A. J., Gunn, T. R., Gunning, M. I., Williams, C. E., and Gluckman, P. D. (1998). Neuroprotection with prolonged head cooling started before postischemic seizures in fetal sheep. Pediatrics 102, 1098–1106. doi: 10.1542/peds.102.5.1098
Hagberg, H., Gilland, E., Bona, E., Hanson, L. A., Hahlin-Zoric, M., Biennow, M., et al. (1996). Enhanced expression of interleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia-ischemia in neonatal rats. Pediatr. Res. 40, 603–609. doi: 10.1203/00006450-199610000-00015
Hagmann, C. F., Brotschi, B., Bernet, V., Berger, T. M., and Robertson, N. J. (2011). Hypothermia for perinatal encephalopathy. Swiss Med. Wkly. 141, w13145. doi: 10.4414/smw.2011.13145
Hamby, A. M., Suh, S. W., Kauppinen, T. M., and Swanson, R. A. (2007). Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke 38, 632–636. doi: 10.1161/01.STR.0000250742.61241.79
Hassa, P. O., and Hottinger, M. O. (1999). A role of poly(ADP-ribose) polymerase in NF-κ B transcriptional activation. Biol. Chem. 380, 953–959. doi: 10.1515/BC.1999.118
Herrera-Marschitz, M., Loidl, C. F., Andersson, K., and Ungerstedt, U. (1993). Prevention of mortality induced by perinatal asphyxia: hypothermia or glutamate antagonism? Amino Acids 5, 413–419. doi: 10.1007/BF00806959
Herrera-Marschitz, M., Morales, P., Leyton, L., Bustamante, D., Klawitter, V., Espina-Marchant, P., et al. (2011). Perinatal asphyxia: current status and approaches towards neuroprotective strategies, focus on sentinel proteins. Neurotoxicity Res 19, 603–627. doi: 10.1007/s12640-010-9208-9
Hoeger, H., Engelmann, M., Bernet, G., Seidl, R., Bubna-Littitz, H., Mpsgoeller, W., et al. (2000). Long term neurological and behavioral effects of graded perinatal asphyxia in the rat. Life Sci. 66, 947–962. doi: 10.1016/S0024-3205(99)00678-5
Hoeger, H., Engidawork, E., Stolzlechner, D., Bubna-Littitz, H., and Lubec, B. (2006). Long-term effect of moderate and profound hyperthermia on morphology, neurological, cognitive and behavioural functions in a rat model of perinatal asphyxia. Amino Acids 31, 385–396. doi: 10.1007/s00726-006-0393-z
Hong, S. J., Dawson, T. M., and Dawson, V. L. (2004). Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signalling. TIPS 25, 259–264. doi: 10.1016/j.tips.2004.03.005
Hulshof, H. J., Novati, A., Sgoifo, A., Luiten, P. G., den Boer, J. A., and Meerlo, P. (2011). Maternal separation decreases adult hippocampal cell proliferation and impairs cognitive performance but has little effect on stress sensitivity and anxiety in adult Wistar rats. Behav. Brain Res. 216, 552–560. doi: 10.1016/j.bbr.2010.08.038
Hutton, L. C., Ratnayake, U., Shields, A., and Walker, D. W. (2009). Neuropathology and functional deficits in a model of birth asphyxia in the precocial spiny mouse (Acomys cahirinus). Dev. Neurosci. 31, 523–535. doi: 10.1159/000251907
Iuvone, L., Geloso, M. C., and Dell'Anna, E. (1996). Changes in open field behavior, spatial memory, and hippocampal parvalbumin immunoreactivity following enrichment in rats exposed to neonatal anoxia. Exp. Neurol. 139, 25–33.
Iyer, N. V., Kotch, L. E., Agani, F., Leung, S. W., Laugner, E., Wenger, R. H., et al. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 12, 149–162. doi: 10.1101/gad.12.2.149
Jagtap, P., and Szabo, C. (2005). Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. 4, 421–440. doi: 10.1038/nrd1718
Jiang, B.-H., Rue, E., Wang, G. L., Roe, R., and Semenza, G. L. (1996). Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J. Biol. Chem. 271, 17771–17778. doi: 10.1074/jbc.271.30.17771
Kapadia, V. S., Chalak, L. F., DuPont, T. L., Rollins, N. K., Brion, L. P., and Wyckoff, M. H. (2013). Perinatal asphyxia with hyperoxemia within the first hour of lfe is associated with moderate to severe hypoxic-ischemic encephalopathy. J. Pediatr. 163, 949–954. doi: 10.1016/j.jpeds.2013.04.043
Kaufman, S. A., Miller, S. P., Ferreiro, D. M., Glidden, D. H., Barkovich, A. J., and Partridge, J. C. (2003). Encephalopathy as a predictor of magnetic resonance imaging abnormalities in asphyxiated newborns. Pediatr. Neurol. 28, 342–346. doi: 10.1016/S0887-8994(03)00015-8
Kauppinen, T. M., Chan, W. Y., Suh, S. W., Wiggins, A. K., Huang, E. J., and Swanson, R. A. (2006). Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases1/2. Proc. Natl. Acad. Sci. U.S.A. 103, 7136–7141. doi: 10.1073/pnas.0508606103
Kauppinen, T. M., Higashi, Y., Suh, S. W., Escartin, C., Nagasawa, K., and Swanson, R. A. (2008). Zinc triggers microglial activation. J. Neurosci. 28, 5827–5835. doi: 10.1523/JNEUROSCI.1236-08.2008
Kauppinen, T. M., Suh, S. W., Berman, A. E., Hamby, A. M., and Swanson, R. A. (2009). Inhibition of poly(ADP-ribose) polymerase suppresses inflammation and promotes recovery after ischemic injury. J. Cereb. Blood Flow Metab. 29, 820–829. doi: 10.1038/jcbfm.2009.9
Kauppinen, T. M., and Swanson, R. A. (2007). The role of poly(ADP-ribose) polymerase-1 in CNS disease. Neuroscience 147, 1267–1272. doi: 10.1016/j.neuroscience.2006.09.034
Klawitter, V., Morales, P., Bustamante, D., Goiny, M., and Herrera-Marschitz, M. (2006). Plasticity of the central nervous system (CNS) following perinatal asphyxia: does nicotinamide provide neuroprotection? Amino Acids 31, 377–384. doi: 10.1007/s00726-006-0372-4
Klawitter, V., Morales, P., Bustamante, D., Gomez-Urquijo, S., Hökfelt, T., and Herrera-Marschitz, M. (2007). Neuronal plasticity of basal ganglia following perinatal asphyxia: neuroprotection by nicotinamide. Exp. Brain Res. 180, 139–152. doi: 10.1007/s00221-006-0842-7
Klawitter, V., Morales, P., Johansson, S., Bustamante, D., Goiny, M., Gross, J., et al. (2005). Effect of perinatal asphyxia on cell survival, neuronal phenotype and neurite growth evaluated with organotypic triple cultures. Amino Acids 28, 149–155. doi: 10.1007/s00726-005-0161-5
Kohlhauser, C., Kaehler, S., Mosgoeller, W., Singewald, N., Koulevas, D., Prast, H., et al. (1999a). Histological changes and neurotransmitter levels three months following perinatal asphyxia in the rat. Life Sci. 64, 2109–2124. doi: 10.1016/S0024-3205(99)00160-5
Kohlhauser, C., Mosgoeller, W., Hoeger, H., Lubec, G., and Lubec, B. (1999b). Cholinergic, monoaminergic and glutamatergic changes following perinatal asphyxia in the rat. Cell. Mol. Life Sci. 55, 1491–1501. doi: 10.1007/s000180050388
Labudova, O., Schuller, E., Yeghiazaryan, K., Kitzmueller, E., Hoeger, H., Lubec, G., et al. (1999). Genes involved in the pathophysiology of perinatal asphyxia. Life Sci. 64, 1831–1838. doi: 10.1016/S0024-3205(99)00125-3
Lafemina, M. J., Sheldon, A. A., and Ferreiro, D. M. (2006). Acute hypoxia-ischemia results in hydrogen peroxide accumulation in neonatal but not adult mouse brain. Pediatr. Res. 59, 680–683. doi: 10.1203/01.pdr.0000214891.35363.6a
Landreau, F., Galeano, P., Caltana, L. R., Masciotra, L., Chertcoff, A., Pontoriero, A., et al. (2012). Effects of two commonly found strains of influenza A virus on developing dopaminergic neurons, in relation to the pathophysiology of schizophrenia. PLoS ONE 7:e51068. doi: 10.1371/journal.pone.0051068
Leppard, J. B., Dong, Z., Mackey, Z. B., and Tomkinson, A. E. (2003). Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol. Cell. Biol. 23, 5919–5927. doi: 10.1128/MCB.23.16.5919-5927.2003
Li, N., Wu, H., Yang, S., and Chen, D. (2007). Ischemic preconditioning induces XRCC1, DNA polymerase-beta, and DNA ligase III and correlates with enhanced base excision repair. DNA Repair (Amst.) 6, 1297–1306. doi: 10.1016/j.dnarep.2007.02.027
Li, Q., and Martin, J. H. (2000). Postdevelopmental of differential projections from the caudal and rostral motor cortex subregions. Exp. Brain Res. 134, 187–198. doi: 10.1007/s002210000454
Loidl, F., Gavilanes, A. W., Van Dijk, E. H., Vreuls, W., Blokland, A., Vies, J. S., et al. (2000). Effects of hypothermia and gender on survival and behaviour after perinatal asphyxia in rats. Physiol. Behav. 68, 263–269. doi: 10.1016/S0031-9384(99)00125-0
Loizou, L. A. (1972). The postnatal ontogeny of monoamine-containing neurones in the central nervous system of the albino rat. Brain Res. 40, 395–418. doi: 10.1016/0006-8993(72)90142-4
Loomans, E. M., van der Stelt, O., van, E. M., Gemke, R. J., Vrijkotte, T., and den Bergh, B. R. (2011). Antenatal maternal anxiety is associated with problem behaviour at age five. Early Hum. Dev. 87, 565–570. doi: 10.1016/j.earlhumdev.2011.04.014
Loomans, E. M., van der Stelt, O., van, E. M., Gemke, R. J., Vrijkotte, T. G., and Van den Bergh, B. R. (2012). High levels of antenatal maternal anxiety are associated with altered cognitive control in five-year-old children. Dev. Psychobiol. 54, 441–450. doi: 10.1002/dev.20606
Low, J. A. (2004). Determining the contribution of asphyxia to brain damage in the neonate. J. Obstet. Gynaecol. Res. 30, 276–286. doi: 10.1111/j.1447-0756.2004.00194.x
Lubec, B., Dell'Anna, E., Fang-Kircher, S., Mark, M., Herrera-Marschitz, M., and Lubec, G. (1997b). Decrease of brain protein kinase C, protein kinase A, and cyclin-dependent kinase correlating with pH precedes neuronal death in neonatal asphyxia of the rat. J. Investig. Med. 45, 284–294.
Lubec, B., Labudova, O., Hoeger, H., Kirchner, L., and Lubec, G. (2002). Expression of transcription factors in the brain of rats with perinatal asphyxia. Biol. Neonate 81, 266–278. doi: 10.1159/000056758
Lubec, B., Mark, M., Herrera-Marschitz, M., Labudova, O., Hoeger, H., Gille, L., et al. (1997a). Decrease of heart protein kinase C and cyclin-dependent kinase precedes death in perinatal asphyxia of the rat. FAEB J. 11, 482–492.
Martin, S. S., Perez-Polo, J. R., Noppens, K. M., and Grafe, M. R. (2005). Biphasic changes in the levels of poly(ADPribose) polymerase-1 and caspase 3 in the immature brain following hypoxia-ischemia. Int. J. Dev. Neurosci. 23, 673–686. doi: 10.1016/j.ijdevneu.2005.08.002
Meng, Z., Li, Q., and Martin, J. H. (2004). The transition from development to motor control function in the corticospinal system. J. Neurosci. 24, 605–614. doi: 10.1523/JNEUROSCI.4313-03.2004
Meng, Z., and Martin, J. H. (2003). Postnatal development of corticospinal postsynaptic action. J. Neurophysiol. 90, 683–692. doi: 10.1152/jn.00152.2003
Miller, S. P., Ramaswamy, V., Michelson, D., Barkovich, J., Holshouser, B., Wycliff, N., et al. (2005). Patters of brain injury in term neonatal encephalopathy. J. Pediatr. 146, 453–460. doi: 10.1016/j.jpeds.2004.12.026
Mishra, O. P., Akhter, W., Ashraf, Q. M., and Delivoria-Papadopoulus, M. (2003). Hypoxia-induced modification of poly (ADP-Ribose) Polymerase and DNA polymerase β activity in cerebral cortical nuclei of newborn piglets: role of nitric oxide. Neuroscience 119, 1023–1032. doi: 10.1016/S0306-4522(03)00166-0
Morales, P., Bustamante, D., Espina-Marchant, P., Neira, T., Gutierrez, M., and Allende, C. (2011). Pathophysiology of perinatal asphyxia: can we predict and improve individual outcomes? EMPA J. 2, 211–230. doi: 10.1007/s13167-011-0100-3
Morales, P., Fiedler, J. L., Andres, S., Berrios, C., Huaiquin, P., Bustamante, D., et al. (2008). Plasticity of hippocampus following perinatal asphyxia: effects on postnatal apoptosis and neurogenesis. J. Neurosci. Res. 86, 2650–2662. doi: 10.1002/jnr.21715
Morales, P., Klawitter, V., Johansson, S., Huaiquin, P., Barros, V. G., Avalos, A. M., et al. (2003). Perinatal asphyxia impairs connectivity and dopamine branching in organotypic triple culture from rat substantia nigra. Neurosci. Lett. 348, 175–179. doi: 10.1016/S0304-3940(03)00507-X
Morales, P., Simola, N., Bustamante, D., Lisboa, F., Fiedler, J., Gebicke-Haerter, P., et al. (2010). Nicotinamide prevents the effect of perinatal asphyxia on apoptosis, non-spatial working memory and anxiety in rats. Exp. Brain Res. 202, 1–14. doi: 10.1007/s00221-009-2103-z
Mosgoeller, W., Kastner, P., Fang-Kircher, S., Kitzmueller, E., Hoeger, H., Sether, P., et al. (2000). Brain RNA polymerase and nucleolar structure in perinatal asphyxia of the rat. Exp. Neurol. 161, 174–182. doi: 10.1006/exnr.1999.7232
Nagayama, T., Simon, R. P., Chen, D., Henshall, D. C., Pei, W., Stetler, R. A., et al. (2000). Activation of Poly(ADP-Ribose) Polymerase in the rat hippocampus may contribute to cellular recovery following sublethal transient global ischemia. J. Neurochem. 74, 1636–1645. doi: 10.1046/j.1471-4159.2000.0741636.x
Neira-Peña, T., Espina-Marchant, P., Rojas-Mancilla, E., Esmar, D., Kraus, C., Munoz, V., et al. (2014). “Molecular, cellular, and behavioural effects produced by perinatal asphyxia: protection by poly(ADP-ribose) polymerase 1 (PARP-1) inhibition,” in Handbook of Neurotoxicity, ed R. M. Kostrzewa (New York, NY: Springer Science+Business Media). doi: 10.1007/978-1-4614-5836-4_115. (in press).
Neira-Peña, T., Rojas-Mancilla, E., Muñoz, V., Gutierrez-Hernández, M., Bustamante, D., Gebicke-Haerter, P., et al. (2013). Nicotinamide Decreases PARP-1 Activity, Inflammation and Cell Death Caused by Perinatal Asphyxia (Prague).
Nogawa, S., Forster, C., Zhang, F., Nsgayama, M., Ross, M. E., and Iadecola, C. (1998). Interaction between inducible nitric oxide synthase and cyclooxygenase-2 after cerebral ischemia. Proc. Natl. Acad. Sci. U.S.A. 95, 10966–10971. doi: 10.1073/pnas.95.18.10966
Northington, F. J., Ferreiro, D. M., Graham, E. M., Traystman, R. J., and Martin, L. J. (2001). Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol. Dis. 8, 207–219. doi: 10.1006/nbdi.2000.0371
Odd, D. E., Lewis, G., Whitelaw, A., and Gunnell, D. (2009). Resuscitation at birth and cognition at 8 years of age: a cohort study. Lancet 373, 1615–1622. doi: 10.1016/S0140-6736(09)60244-0
Odell, I. D., Barbour, J. E., Murphy, D. L., Della-Maria, J. A., Sweasy, J. B., Tomkinson, A. E., et al. (2011). Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell. Biol. 31, 4623–4632. doi: 10.1128/MCB.05715-11
Olson, L., and Seiger, Å. (1972). Early prenatal ontogeny of central monoamine neurons in the rat: fluorescence histochemical observations. Z. Anat. Entwicklungsgesch. 137, 301–316. doi: 10.1007/BF00519099
Oo, T. F., and Burke, R. E. (1997). The time course of the development cell death in phenotypically defined dopaminergic neurons of the substantia nigra. Dev. Brain Res. 98, 191–196. doi: 10.1016/S0165-3806(96)00173-3
Pasternak, J. F., Predey, T. A., and Mikhael, M. A. (1991). Neonatal asphyxia: vulnerability of basal ganglia, thalamus and brain stem. Pediatr. Neurol. 7, 147–149. doi: 10.1016/0887-8994(91)90014-C
Pastuzko, A. (1994). Metabolic responses of the dopaminergic system during hypoxia in newborn brain. Biochem. Med. Metab. Biol. 51, 1–15. doi: 10.1006/bmmb.1994.1001
Peters-Golden, M., Canetti, C., Mancuso, P., and Coffey, M. J. (2005). Leucotrienes: under appreciated mediators of innate immunoresponse. J. Immunol. 174, 589–594.
Pittelli, M., Cavone, L., Lapucci, A., Oteri, C., Felici, R., Niccolai, E., et al. (2014). Nicotinamide phosphoribosyltransferase (NAMPT) activity is essential for survival of resting lymphocytes. Immunol. Cell Biol. 94, 191–199. doi: 10.1038/icb.2013.85
Roelfsema, V., Bennet, L., George, S., Wu, D., Guan, J., Veerman, M., et al. (2004). The window of opportunity for cerebral hypothermia and white matter injury after cerebral ischemia in near-term fetal sheep. J. Cereb. Blood Flow Metab. 24, 877–886. doi: 10.1097/01.WCB.0000123904.17746.92
Rojas-Mancilla, E., Neira-Pena, T., Kraus, C., Rivera, B., Perez, R., Alvarez, A., et al. (2013). “Perinatal asphyxia decreases neuronal branching and synaptogenesis in rat hippocampus. Implication of astrocyte reactivity and HIF-1α,” in XXVIth International Symposium on Cerebral Flow, Metabolism and Function and XIth International Conference on Quantification of Brain Function with PET (Shanghai).
Romijn, H. J., Hofman, M. A., and Gramsbergen, A. (1991). At what age is the developing cerebral cortex of the rat comparable to that of the full-term newborn human baby? Early Hum. Dev. 26, 61–67. doi: 10.1016/0378-3782(91)90044-4
Russo, I., Barlati, S., and Bosetti, F. (2011). Effects of neuroinflammation on the regenerative capacity of brain stem cells. J. Neurochem. 116, 947–956. doi: 10.1111/j.1471-4159.2010.07168.x
Sakakibara, Y., Mitha, A. P., Ogilvy, C. S., and Maynard, K. I. (2000). Post-treatment with nicotinamide (vitamin, B. (3)) reduces the infarct volume following permanent focal cerebral ischemia in female Sprague-Dawley and Wistar rats. Neurosci. Lett. 281, 111–114. doi: 10.1016/S0304-3940(00)00854-5
Saldeen, J., and Welsh, N. (1998). Nicotinamide-induced apoptosis in insulin producing cells is associated with cleavage of poly(ADP-ribose)polymerase. Mol. Cell. Endocrinol. 139, 99–107. doi: 10.1016/S0303-7207(98)00068-9
Sawaya, M. R., Prasad, R., Wilson, S. H., Kraut, J., and Pelletier, H. (1997). Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry 36, 11205–11215. doi: 10.1021/bi9703812
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Segalowitz, S. J., and Davies, P. L. (2004). Charting the maturation of the frontal lobe: an electrophysiological strategy. Brain Cogn. 55, 116–133. doi: 10.1016/S0278-2626(03)00283-5
Seidl, R., Stoeckler-Ipsiroglu, S., Rolinski, B., Kohlhauser, C., Herkner, K. R., Lubec, B., et al. (2000). Energy metabolism in graded perinatal asphyxia of the rat. Life Sci. 67, 421–435. doi: 10.1016/S0024-3205(00)00630-5
Seiger, Å., and Olson, L. (1973). Late prenatal ontogeny of central monoamine neurons in the rat: fluorescence histochemical observations. Z. Anat. Entwicklungsgesch. 140, 281–318. doi: 10.1007/BF00525058
Shetty, P. K., Galeffi, F., and Turner, D. A. (2013). Nicotinamide pre-treatment ameliorates NAD(H) hyperoxidation and improves neuronal function after severe hypoxia. Neurobiol. Dis. 31, 469–478. doi: 10.1016/j.nbd.2013.10.025
Simola, N., Bustamante, D., Pinna, A., Pontis, S., Morales, P., Morelli, M., et al. (2008). Acute perinatal asphyxia impairs non-spatial memory and alters motor coordination in adult male rats. Exp. Brain Res. 185, 595–601. doi: 10.1007/s00221-007-1186-7
Simsek, D., Furda, A., Gao, Y., Artus, J., Brunet, E., Hadjantonakis, A. K., et al. (2011). Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 471, 245–248. doi: 10.1038/nature09794
Skaper, S. D. (2003). Poly(ADP-ribosyl)ation enzyme-1 as a target for neuroprotection in acute central nervous system injury. Curr. Drug Targets CNS Neurol. Disord. 2, 279–291. doi: 10.2174/1568007033482733
Solberg, R., Andresen, J. H., ERscrig, R., Vento, M., and Saugstad, O. D. (2007). Resuscitation of hypoxic newborn piglets with oxygen induces a dose-dependent increase in markers of oxidation. Pediatr. Res. 62, 559–563. doi: 10.1203/PDR.0b013e318156e8aa
Sowell, E. R., Thompson, P. M., Holmes, C. J., Jernigan, T. L., and Toga, A. W. (1999). In vivo evidence for post-adolescent brain maturation in frontal and striatal regions. Nat. Neurosci. 2, 859–861. doi: 10.1038/13154
Strackx, E., Van den Hove, D. L., Prickaerts, J., Zimmermann, L., Steinbusch, H. W., Blanco, C. E., et al. (2010). Fetal asphyctic preconditioning protects against perinatal asphyxia-induced behavioral consequences in adulthood. Behav. Brain Res. 208, 343–351. doi: 10.1016/j.bbr.2009.11.040
Sung, P., Bailly, V., Weber, C., Thompson, L. H., Prakash, L., and Prakash, S. (1993). Human xeroderma pigmentosum group D gene encodes a DNA helicase. Nature 365, 852–855. doi: 10.1038/365852a0
Towfighi, J., Mauger, D., Vannucci, R. C., and Vannucci, S. J. (1997). Influence of age on the cerebral lesions in an immature rat model of cerebral hypoxia-ischemia: a light microscopic study. Dev. Brain Res. 100, 149–160. doi: 10.1016/S0165-3806(97)00036-9
Turunc Bayrakdar, E., Uyanikgil, Y., Kanit, L., Koylu, E., and Yalcin, A. (2014). Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Aβ (1-42)-induced rat model of Alzheimer's disease. Free Radic. Res. 48, 146–158. doi: 10.3109/10715762.2013.857018
Ullrich, O., Diestel, A., Evüpoglu, I. Y., and Nitsch, R. (2001). Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat. Cell Biol. 3, 1035–1042. doi: 10.1038/ncb1201-1035
Ungethüm, U., Chen, Y., Gross, J., Bjelke, B., Bolme, P., Eneroth, P., et al. (1996). Effects of perinatal asphyxia on the mesostriatal/mesolimbic dopamine system of neonatal and 4-week-old male rats. Exp. Brain Res. 112, 403–410. doi: 10.1007/BF00227946
Van de Berg, W. D., Kwaijtaal, M., de Louw, A. J., Lissone, N. P., Schmitz, C., Faull, R. L., et al. (2003). Impact of perinatal asphyxia on the GABAergic and locomotor system. Neuroscience 117, 83–96. doi: 10.1016/S0306-4522(02)00787-X
Van den Bergh, B. R., Mennes, M., Stevens, V., van der Meere, J., Borger, N., Stiers, P., et al. (2006). ADHD deficit as measured in adolescent boys with a continuous performance task is related to antenatal maternal anxiety. Pediatr. Res. 59, 78–82. doi: 10.1203/01.pdr.0000191143.75673.52
Van den Bergh, B. R., Mulder, E. J., Mennes, M., and Glover, V. (2005). Antenatal maternal anxiety and stress and the neurobehavioural development of the fetus and child: links and possible mechanisms. Neurosci. Biobehav. Rev. 29, 237–258. doi: 10.1016/j.neubiorev.2004.10.007
Van den Broek, M. P. H., Groenendaal, F., Egberts, A. C. G., and Rademaker, C. M. A. (2010). Effects of hypothermia on pharmacokinetics and pharmacodynamics. Clin. Pharmacokinet. 49, 277–294. doi: 10.2165/11319360-000000000-00000
Van Erp, T. G. M., Saleh, P. A., Rosso, P. A., Huttunen, M., Lönnqvist, J., Pirkola, T., et al. (2002). Contributions of genetic risk and fetal hypoxia to hippocampal volume in patients with schizophrenia or schizoaffective disorder, their unaffected siblings and healthy unrelated volunteers. Am. J. Psychiatry 159, 1514–1520. doi: 10.1176/appi.ajp.159.9.1514
Vannuci, S., and Hagberg, H. (2004). Hypoxia-ischemia in the immature brain. J. Exp. Biol. 207, 3149–3154. doi: 10.1242/jeb.01064
Venerosi, A., Cutuli, D., Chiarotti, F., and Calamandrei, G. (2006). C-section birth per se or followed by global asphyxia altered emotional behaviour in neonate and adult rats. Behav. Brain Res. 168, 56–63. doi: 10.1016/j.bbr.2005.10.010
Venerosi, A., Valanzano, A., Cirulli, F., Alleva, E., and Calamandrei, G. (2004). Acute global anoxia during C-section birth affects dopamine-mediated behavioural responses and reactivity to stress. Behav. Brain Res. 154, 155–164. doi: 10.1016/j.bbr.2004.02.008
Virag, L., and Szabo, C. (2002). The Therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev. 54, 375–429. doi: 10.1124/pr.54.3.375
Voorn, P., Kalsbeck, A., Jorritsma-Byham, B., and Groenewegen, H. J. (1988). The pre- and postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience 25, 857–887. doi: 10.1016/0306-4522(88)90041-3
Wan, F. J., Lin, H. C., Kang, B. H., Tseng, C. J., and Tung, C. S. (1999). D-amphetamine-induced depletion of energy and dopamine in the rat striatum is attenuated by nicotinamide pretreatment. Brain Res. Bull. 50, 167–171. doi: 10.1016/S0361-9230(99)00185-9
Wilson, S. H. (1998). Mammalian base excision repair and DNA polymerase β. Mutation Res. 407, 203–215. doi: 10.1016/S0921-8777(98)00002-0
Wyss, M. T., Jolivet, R., Buck, A., Magistretti, P. J., and Weber, B. (2011). In vivo evidence for lactate as a neuronal energy source. J. Neurosci. 31, 7477–7485. doi: 10.1523/JNEUROSCI.0415-11.2011
Yan, Q., Briehl, M., Crowley, C. L., Payne, C. M., Bernstein, H., and Bernstein, C. (1999). The NAD+ precursors, nicotinic acid and nicotinamide upregulate glyceraldehyde-3-phosphate dehydrogenase and glucose-6-phosphate dehydrogenase mRNA in Jurkat cells. Biochem. Biophys. Res. Commun. 255, 133–136. doi: 10.1006/bbrc.1999.0154
Yu, S. W., Poitras, M. F., Coombs, C., Bowers, W. J., Federoff, H. J., Poirier, G. C., et al. (2002). Mediation of poly(ADP-ribose) polymerase-1 dependent cell death by apoptosis-inducing factor. Science 297, 250–263. doi: 10.1126/science.1072221
Zhang, J., Pieper, A., and Snyder, S. H. (1995). Poly(ADP-ribose) synthase activation: an early indicator of neurotoxic DNA damage. J. Neurochem. 65, 1411–1414. doi: 10.1046/j.1471-4159.1995.65031411.x
Keywords: obstetric complications, neonatal hypoxic ischemic encephalopathy, sentinel proteins, poly(ADP-ribose) polymerase, plasticity, behavior, cognition, development
Citation: Herrera-Marschitz M, Neira-Pena T, Rojas-Mancilla E, Espina-Marchant P, Esmar D, Perez R, Muñoz V, Gutierrez-Hernandez M, Rivera B, Simola N, Bustamante D, Morales P and Gebicke-Haerter PJ (2014) Perinatal asphyxia: CNS development and deficits with delayed onset. Front. Neurosci. 8:47. doi: 10.3389/fnins.2014.00047
Received: 12 November 2013; Paper pending published: 11 December 2013;
Accepted: 22 February 2014; Published online: 26 March 2014.
Edited by:
Hari S. Sharma, Uppsala University, SwedenCopyright © 2014 Herrera-Marschitz, Neira-Pena, Rojas-Mancilla, Espina-Marchant, Esmar, Perez, Muñoz, Gutierrez-Hernandez, Rivera, Simola, Bustamante, Morales and Gebicke-Haerter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Herrera-Marschitz, Programme of Molecular and Clinical Pharmacology, ICBM, Medical Faculty, University of Chile, Santiago 7, PO Box 70.000, Chile e-mail:bWgtbWFyc2NoaXR6QG1lZC51Y2hpbGUuY2w=