 Dongze Zhang
Dongze Zhang Robert L. Muelleman
Robert L. Muelleman Yu-Long Li
Yu-Long Li- Department of Emergency Medicine, University of Nebraska Medical Center, Omaha, NE, USA
Chronic heart failure (CHF) affects approximately 5.7 million people in the United States. Increasing evidence from both clinical and experimental studies indicates that the sensitivity of arterial baroreflex is blunted in the CHF state, which is a predictive risk factor for sudden cardiac death. Normally, the arterial baroreflex regulates blood pressure and heart rate through sensing mechanical alteration of arterial vascular walls by baroreceptor terminals in the aortic arch and carotid sinus. There are aortic baroreceptor neurons in the nodose ganglion (NG), which serve as the main afferent component of the arterial baroreflex. Functional changes of baroreceptor neurons are involved in the arterial baroreflex dysfunction in CHF. In the CHF state, circulating angiotensin II (Ang II) and local Ang II concentration in the NG are elevated, and AT1R mRNA and protein are overexpressed in the NG. Additionally, Ang II-superoxide-NFκB signaling pathway regulates the neuronal excitability of aortic baroreceptors through influencing the expression and activation of Nav channels in aortic baroreceptors, and subsequently causes the impairment of the arterial baroreflex in CHF. These new findings provide a basis for potential pharmacological interventions for the improvement of the arterial baroreflex sensitivity in the CHF state. This review summarizes the mechanisms responsible for the arterial baroreflex dysfunction in CHF.
Introduction
Chronic heart failure (CHF), the most common type of heart diseases, affects approximately 5.7 million people in the United States. During the past several decades, about 870,000 new cases were diagnosed and more than 58,309 individuals died from CHF in the United States each year (Mozaffarian et al., 2015). CHF is characterized by autonomic dysfunction, including withdraw of the parasympathetic tone and overactivation of the sympathetic tone (Creager et al., 1986; Saul et al., 1988; Porter et al., 1990), which is closely related to mortality in patients with CHF (Gronda et al., 2014). Impairment of the baroreflex sensitivity is directly associated with this autonomic dysfunction (Creager, 1992). Many studies have demonstrated that the arterial baroreflex sensitivity is attenuated in both clinical and experimental CHF (White, 1981; Floras, 1993; Frenneaux, 2004; Pinna et al., 2005; Ruttanaumpawan et al., 2008), which is a predictive risk factor for sudden cardiac death (Kleiger et al., 1987) and is associated with mortality of CHF (Nolan et al., 1998; Cygankiewicz et al., 2008; Boogers et al., 2011; Hauptman et al., 2012). Although the precise mechanisms responsible for blunted arterial baroreflex in the CHF state are not fully understood, activation of the arterial baroreflex (as a long-term therapeutic approach) has been shown to have numerous beneficial effects on CHF treatments in both clinical and experimental studies (Zucker et al., 2007; Sabbah, 2012; Doumas et al., 2014; Gronda et al., 2014; Halbach et al., 2014; Iliescu et al., 2014; Liao et al., 2014; Madershahian et al., 2014; Schmidli et al., 2014).
In general, the arterial baroreflex is a homeostatic mechanism by which arterial blood pressure and heart rate are changed through sensing the alteration of tension in arterial vascular walls. The arterial baroreflex arc is composed mainly of an afferent component, a central neural component, and autonomic neuroeffector components. Arterial baroreceptors are the main afferent component of arterial baroreflex arc, which are sensors located in the arterial blood vessels including aortic baroreceptor neurons in the NG and carotid baroreceptor neurons in the petrosal ganglion. They sense the mechanical alteration of arterial vascular walls through the baroreceptor terminals and increase the excitation of these afferent baroreceptors. This excitatory signal is integrated in the baroreceptors by local endogenous substances, and then the sensory information is communicated to the dorsal medial nucleus tractus solitary (NTS). The NTS in the brain recognizes the information from the baroreceptors and finally evokes the parasympathoexcitatory and sympathoinhibitory responses (Czachurski et al., 1982; Spyer et al., 1997; Benarroch, 2008). Each component of the arterial baroreflex arc might be involved in the impairment of the baroreflex function in CHF. It has been demonstrated that central autonomic pathways and neuroeffectors contribute to baroreflex dysfunction in CHF (Zucker et al., 1995; Zucker and Liu, 2000; Pan et al., 2007; Schultz, 2009; Wang et al., 2014). However, the impairment of arterial baroreceptors also is a major obstacle to influence the arterial baroreflex function in CHF (Dibner-Dunlap and Thames, 1989; Wang et al., 1990, 1991a,b; Rondon et al., 2006). Indeed, recent studies have shown that the functional impairment of aortic baroreceptor neurons located in the NG is involved in the baroreflex dysfunction in CHF (Tu et al., 2010; Zhang et al., 2014). In this brief review, therefore, we will focus mainly on our work to discuss the contribution of aortic baroreceptors in blunted arterial baroreflex in CHF, especially the possible cellular and molecular mechanisms responsible for the alteration of aortic baroreceptors.
Voltage-gated Sodium (Nav) Channel Remodeling in Aortic Baroreceptors Contributes to Blunted Baroreflex Sensitivity in CHF
Based on the fact that CHF is a chronic, progressive cardiovascular disease, it is possible that morphological changes and/or functional alterations in aortic baroreceptor neurons might be involved in the impaired baroreflex sensitivity in CHF. Morphological data from our published study have shown that both total neuron number and ratio of A-type/C-type neurons in the NG have no significant difference between CHF and sham rats (Tu et al., 2010). Using a dog model of CHF, Wang, et al. also measured the density of A-type and C-type nerve fibers in the carotid sinus and found that there is no change in A-/C-fiber ratio and total fiber density in CHF dogs, compared to sham dogs (Wang et al., 1996). These results from the morphological measurement support the view that cellular and molecular changes rather than morphological alterations in aortic baroreceptors could be involved in the arterial baroreflex dysfunction in the CHF state.
It is well-established that voltage-gated ion channels (e.g., sodium, calcium, and potassium voltage-gated channels) exist in the transmembrane of excitable cells, and these types of ion channels can affect cell excitability including neuronal excitation. Using path-clamp and molecular biological techniques, all major subtypes of sodium, calcium, and potassium voltage-gated channels have been functionally characterized in NG neurons including aortic baroreceptor neurons. These major subtypes of voltage-gated ion channels include: (1) tetrodotoxin (TTX)-sensitive and TTX-resistant Nav channels; (2) N-type, L-type, T-type, and R-type calcium voltage-gated channels; (3) 4-aminopyridine-sensitive, tetraethylammonium-sensitive, and calcium-activated potassium voltage-gated channels (Li et al., 1998; Lancaster et al., 2002; Schild and Kunze, 2012; Tatalovic et al., 2012; Xu et al., 2015).
Nav channels are responsible for initiation and propagation of the action potential in neurons including primary viscerosensory neurons (Yu and Catterall, 2003; Ritter et al., 2009). Thus, far, nine α-subunits (Nav1.1–1.9) of Nav channels have been functionally characterized. Each Nav channel subunit has particular tissue localization, consistent with a distinct role for each Nav channel subunit in mammalian physiology. In general, Nav channels in primary afferent neurons are separated into TTX-sensitive Nav channels and TTX-resistant Nav channels. TTX-sensitive Nav channels are characterized by low activation threshold, fast activating and inactivating Nav channels, which include Nav 1.1, Nav 1.2, Nav 1.3, Nav 1.4, Nav 1.6, and Nav 1.7 channels. TTX resistant Nav channels are characterized by high activation threshold, slow activating, and inactivating Nav channels, which include Nav 1.5, Nav 1.8, and Nav 1.9 channels (Waxman et al., 1999; Yu and Catterall, 2003; Catterall et al., 2005). Based on the sensitivity of neurons to TTX, aortic baroreceptor neurons in the NG could be separated into A-type and C-type neurons (Schild and Kunze, 1997). TTX totally blocks Nav currents in A-type aortic baroreceptor neurons, whereas TTX partially inhibits Nav currents in C-type aortic baroreceptor neurons. TTX-blocked Nav currents are defined as TTX-sensitive Nav currents and remained Nav currents are defined as TTX-resistant Nav currents. Although Nav 1.7, Nav 1.8, and Nav 1.9 channels are abundantly expressed in the NG (Waxman et al., 1999; Baker and Wood, 2001; Cummins et al., 2007; Kwong et al., 2008; Tu et al., 2010), their distribution in the NG is different. TTX-sensitive Nav 1.7 channels are located in the cell transmembrane of both A-type and C-type nodose neurons, while TTX-resistant Nav 1.8 and Nav 1.9 channels are only expressed in the cell transmembrane of C-type nodose neurons (Tu et al., 2010).
Although the correlation of voltage-gated Nav channels with initiation of the action potential and propagation of the neuronal discharge in primary sensory neurons has been discussed in many studies (Schild et al., 1994; Yoshida, 1994; Schild and Kunze, 1997; Matsutomi et al., 2006; Patrick Harty and Waxman, 2007), there is little known about the role of Nav channels in determining the activity of baroreceptor neurons and arterial baroreflex, especially in the CHF state. One study from Shen et al. has shown that intravenous administration of Nav channel enhancer restores the blunted baroreflex sensitivity in conscious dogs with CHF (Shen et al., 2005). Therefore, the alteration of Nav channels is closely associated with blunted baroreflex sensitivity in the CHF state. Real-time RT-PCR, western blot, and immunofluorescent staining data in our study have demonstrated that the expression (mRNA and protein) of Nav 1.7, Nav 1.8, and Nav 1.9 channels is decreased in the NG from CHF rats (Tu et al., 2010). Additionally, patch-clamp data have also shown that densities of both TTX-sensitive and TTX-resistant Nav currents recorded in isolated aortic baroreceptor neurons are reduced in CHF rats. Furthermore, the suppression of cell excitability is observed in aortic baroreceptor neurons from CHF rats. When a Nav channel activator, rATX II was administered in aortic baroreceptor neurons from CHF rats, it significantly restored CHF-decreased Nav current density and cell excitability of aortic baroreceptor neurons (Tu et al., 2010). Therefore, the involvement of reduced Nav channels in the aortic baroreceptor dysfunction in the CHF state is further evidenced by our study mentioned above (Tu et al., 2010).
To evaluate the role of aortic baroreceptors in the arterial baroreflex in the CHF state, we employed two methods. One method is to examine the changes in blood pressure and heart rate when the aortic depressor nerve is electrically stimulated. There are three advantages in this method for measurement of the baroreflex function. Firstly, rat aortic depressor nerves do not contain functional chemoreceptor afferent fibers for the generation of arterial chemoreflex, which means that there are only baroreceptor afferent fibers in the rat aortic depressor nerve to convey the electrical signal to the central nervous system (Sapru et al., 1981; Fan et al., 1996; Kobayashi et al., 1999). Secondly, a directly electrical stimulation of the rat aortic depressor nerve can bypass aortic depressor nerve terminals and aortic arterial vascular walls to induce the arterial baroreflex. Thirdly, by varying the frequency of electrical stimulus, reflex responses to activating A-type and C-type afferent fibers can be differentiated. However, a disadvantage of this technique is that a physiological substrate for the aortic baroreceptor activation is not represented. Another method is to measure reflex changes in heart rate and cardiac sympathetic nerve activity in response to changes in arterial blood pressure. The advantage for this method is that a physiological stimulation (changes in arterial blood pressure) is used to activate the arterial baroreflex. A major limitation in this approach is that possible alterations in the mechanotransduction process at the barosensory nerve terminal and the arterial vascular elasticity may also play a role in the arterial baroreflex function. Our previous in vivo studies have demonstrated that the arterial baroreflex is significantly depressed in CHF rats whatever electrical stimulation of the aortic depressor nerve or change in the arterial blood pressure is used to induce the arterial baroreflex (Tu et al., 2010; Zhang et al., 2014). Additionally, baroreceptor nerve stimulation-induced baroreflex sensitivity was markedly improved in CHF rats when the NG was treated by rATX II (a Nav channel activator) (Tu et al., 2010). However, the local treatment of rATX II did not normalize the Nav current density and neuronal excitability of aortic baroreceptors, and arterial baroreflex sensitivity in CHF rats toward the level seen in sham rats, suggesting that other mechanisms might be involved in this process. In physiological and pathophysiological conditions, acute changes of the ion channel kinetics and chronic alterations of the ion channel expression are two major factors to modulate the ion channel function. Based on the inability of rATX to improve the expression of Nav channels, we consider that low level of Nav channel expression in the NG from CHF rats might explain the above results. So far we cannot identify the contribution of each Nav channel subunit to the cell excitability of aortic baroreceptor neurons and baroreflex sensitivity, because no specific Nav channel activators are available for Nav 1.7, Nav 1.8, and Nav 1.9 channels. These experimental results indicate that the remodeling of Nav channels including the lower expression of Nav channels and the decrease of Nav currents could reduce the neuronal excitability of aortic baroreceptors and induce resultant impairment of the arterial baroreflex sensitivity in the CHF state.
Currently, there is no information available about the changes of voltage gated-calcium channels and potassium channels in aortic baroreceptor neurons in the CHF state. Therefore, we cannot rule out the involvement of these ion channels in the alteration of aortic baroreceptors and the arterial baroreflex dysfunction in CHF.
Mitochondria-derived Superoxide Overproduction Mediates the Decreased Nav Currents and Cell Excitability in Baroreceptor Neurons in CHF
The mitochondrial electron transport chain contains several mitochondrial complex enzymes, which constitutes the main source of superoxide in physiological and pathophysiological conditions (McCord, 1993; Cadenas and Davies, 2000; Turrens, 2003; Balaban et al., 2005; Adam-Vizi and Chinopoulos, 2006; Murphy, 2009). Under physiological conditions, the mitochondrial electron transport chain transfers electrons to molecular oxygen for ATP production. Only a tiny leakage of electrons (1–2%) from the mitochondrial electron transport chain produces a low level of superoxide (McCord, 1993; Cadenas and Davies, 2000). The low level of superoxide is essential for normal cellular metabolism (Fattman et al., 2003). However, in pathophysiological conditions, the inhibition of mitochondrial complex enzymes (mitochondrial oxidative system) and/or the reduction of manganese superoxide dismutase (MnSOD, mitochondrial antioxidative system) can elevate the mitochondria-derived superoxide level (Robinson, 1998; Cadenas and Davies, 2000; Wallace, 2001; Murphy, 2009). Previous studies demonstrated that superoxide overproduction was primarily a consequence of the reduction in cellular mitochondrial complex I activity in patients with inherited mitochondrial complex I deficiency (Pitkanen and Robinson, 1996; Verkaart et al., 2007).
Our previous study has shown that CHF significantly reduces the protein expression and activity of mitochondrial complex enzymes (complex I, II, and III) and MnSOD in the NG including aortic baroreceptors (Tu et al., 2012). At the same time, the mitochondrial superoxide production in the NG from CHF rats was also increased. These results demonstrate the association of mitochondrial complex enzyme and MnSOD dysfunctions with elevation of the mitochondria-derived superoxide in NG neurons from CHF rats. To analyze the correlation between elevation of the mitochondrial-derived superoxide and reduced Nav channel activation and cell excitability in baroreceptor neurons from CHF rats, adenoviral MnSOD (Ad.MnSOD) gene was transfected into the NG in our study (Tu et al., 2012). Our data demonstrated that transfection of the Ad.MnSOD gene into the NG restored the protein expression of MnSOD, reduced the mitochondria-derived superoxide, and reversed the expression and current density of Nav channels and the cell excitability in aortic baroreceptor neurons from CHF rats. These data strongly suggest that elevation of the mitochondria-derived superoxide contributes to the reduced Nav currents and the suppression of neuronal excitability in CHF aortic baroreceptor neurons. Although transfection of the Ad.MnSOD gene completely restored expression of the MnSOD protein, it did not normalize the mitochondrial-derived superoxide, and the protein expression and activation of Nav channels in CHF aortic baroreceptor neurons toward the level seen in sham neurons. This inconsistency might be explained by following possibilities. Firstly, as mentioned above, both oxidative and antioxidative systems in the mitochondria could affect the mitochondria-derived superoxide. Recovering the ability of scavenging mitochondrial superoxide through Ad.MnSOD gene-induced overexpression of the MnSOD protein is insufficient to scavenge mitochondrial oxidative system-derived superoxide overproduction, because the function of mitochondrial complex enzymes is not improved in our study. Secondly, the cytosolic superoxide production system (such as NADPH oxidase components) also exists in the NG (Li and Zheng, 2011). Cytosolic superoxide and other endogenous factors might also mediate CHF-reduced Nav channel activity in aortic baroreceptor neurons. Additionally, transfection of the Ad.MnSOD gene into the NG also significantly restored CHF-blunted arterial baroreflex function, measured by responses of blood pressure and heart rate to electrical stimulation of the aortic depressor nerve, and reflex changes of heart rate and cardiac sympathetic nerve activity in response to changes of arterial blood pressure (Zhang et al., 2014). These data clearly indicate that the mitochondria-derived superoxide overproduction in aortic baroreceptors contributes to the impairment of the arterial baroreflex sensitivity in CHF.
Overall, elevation of the endogenous mitochondria-derived superoxide is involved in the reduced Nav current density and cell excitability in CHF aortic baroreceptor neurons through acutely decreasing the activation of Nav channels and chronically reducing the protein expression of Nav channels. Subsequently, the mitochondrial superoxide overproduction is further associated with the impairment of the arterial baroreflex function in the CHF state. Thus, far, there have been very few studies explored how superoxide modulates electrophysiological properties and expression of ion channels, especially no report focusing on the mitochondria-derived superoxide. Usually an inside-out or outside-out single-channel patch-clamp recording is used to measure the direct regulatory effect of superoxide on the single-channel open probability. However, loss of the mitochondria in a single-channel recording prevents us from measuring the direct effect of the mitochondria-derived superoxide on Nav channels. Therefore, exploring the mechanisms underlying the acute modulation of the mitochondrial superoxide in Nav channels will require the development of advanced techniques. As regards the mechanism(s) responsible for modulation of the mitochondrial superoxide in expression of Nav channels, we discuss the details below.
Regulatory Effect of Nuclear Factor κB (NFκB) p65 on Nav Channel Expression and Cell Excitability in Aortic Baroreceptor Neurons in CHF
NFκB, a transcription factor, can regulate the expression of a number of genes involved in pathophysiological states, such as inflammatory disease and heart failure (Frantz et al., 2003; Valen, 2004; Israël, 2010; Van der Heiden et al., 2010). NFκB consists of five structurally related proteins, namely RelA (p65), RelB, c-Rel, p50, and p52. The p65/p50 heterodimer is the most abundant and widely expressed form of NFκB (Hoffmann and Baltimore, 2006). In the resting state, NFκB presents a silent form in the cytosol through tight binding to the specific inhibitor of κBα (IκBα) (Hoffmann and Baltimore, 2006; Israël, 2010). In response to multiple stimuli in pathophysiological conditions, IκB molecules are phosphorylated on Ser32 and Ser36 residues by activation of IKKβ kinases (Kabe et al., 2005; Häcker and Karin, 2006). The serine-phosphorylated IκB is ubiquitinated and degraded (Karin and Ben-Neriah, 2000; Kabe et al., 2005). As a consequence, NFκB binds to specific sites on DNA, and induces transcription of numerous target genes after NFκB is activated and translocated from the cytoplasm to the nucleus (Israël, 2010).
Although many studies have discovered the role of NFκB in target gene transcription, very few studies focus on the involvement of NFκB in regulating ion channel gene transcription. Shang et al. found that NFκB could directly bind to the SCN5A promoter, which was involved in angiotensin II/hydrogen peroxide-induced down-transcription of Nav1.5 channels (Shang et al., 2008). Therefore, NFκB may be involved in mitochondrial superoxide-lowered activation and expression of Nav channels in baroreceptor neurons in the CHF state. Our recent study has shown that the IKK–IκB–NFκB signaling pathway exists in rat NG (Zhang et al., 2014). We also found that CHF increased the phosphorylated IKK, degraded the IκBα, and enhanced the phosphorylated NFκB p65 in the NG. More importantly, our study further confirmed that CHF enhanced the ability of NFκB p65 binding to the Nav 1.7 promoter in the NG. These results provide the molecular evidence that the activation of NFκB p65 is associated with the change of Nav 1.7 channels in nodose neurons from CHF rats. Additionally, to further clarify whether the activation of NFκB p65 is involved in CHF-decreased Nav channels and neuronal excitability in baroreceptor neurons, NFκB p65 shRNA gene was in vivo transfected into CHF nodose neurons in our study. Transfection of NFκB p65 shRNA into the NG not only normalized the phosphorylated NFκB p65 protein, but also significantly increased the protein expression and current density of Nav channels in CHF rats (Zhang et al., 2014), which indicates that NFκB p65 shRNA gene upregulates the protein expression of Nav channels in the NG from CHF rats through inhibiting the phosphorylated NFκB p65 protein. Although current studies do not unveil the detail molecular mechanisms of how NFκB p65 shRNA increases the protein expression of Nav channels in the NG from CHF rats, hyperactivation of NFκB is considered to downregulate the protein expression and current density of Nav channels in nodose neurons from CHF rats, which is inconsistent with the common conception that NFκB binding with target gene activates the gene transcription (Ghosh et al., 1998; Valen et al., 2001; Frantz et al., 2003; McKenna and Wright, 2015). Furthermore, we also observed that transfection of NFκB p65 shRNA into the NG significantly improved the cell excitability of aortic baroreceptor neurons and resultant arterial baroreflex sensitivity in CHF rats (Zhang et al., 2014). These data suggest that activation of the NFκB signaling is involved in CHF-induced downregulation of the Nav 1.7 channel, suppression of the aortic baroreceptor neuronal excitability, and impairment of the arterial baroreflex function.
As mentioned above, transfection of Ad.MnSOD gene into nodose neurons reduces CHF-induced elevation of the mitochondrial superoxide, reverses CHF-decreased activation of the Nav channel and neuronal excitability in aortic baroreceptors (Tu et al., 2012), and improves CHF-impaired arterial baroreflex sensitivity (Zhang et al., 2014). Additionally, transfection of Ad.MnSOD gene also inhibits CHF-induced augmentation of the phosphorylated NFκB p65 in the NG tissue (Zhang et al., 2014). However, transfection of NFκB p65 shRNA does not affect the superoxide level in the NG from CHF rats (Zhang et al., 2014). From these data, we can deduce that inhibition of NFκB p65 improves the aortic baroreceptor function and arterial baroreflex sensitivity even if a high level of superoxide is preserved in the NG from CHF rats. Based on these results, we consider that superoxide overproduction-induced impairment of the aortic baroreceptor neuron and abnormality of the arterial baroreflex function in CHF rats is attributed to activation of the NFκB p65 in the NG. However, the exact mechanisms by which superoxide induces activation of the NFκB p65 in nodose neurons from CHF rats are yet unclear. In human endothelial cells, protein kinase C is involved in superoxide-induced NFκB activation (Ogata et al., 2000). It has also been reported that superoxide mediates interleukin-1β–induced IκBα degradation and consequent NFκB activation in bovine articular chondrocytes (Mendes et al., 2003). the IKK pathway also links superoxide with NFκB activation (Kabe et al., 2005). In our study, CHF increased the phosphorylated IKKβ, decreased the total IκBα, and enhanced the phosphorylated NFκB p65 in nodose neurons (Zhang et al., 2014). Therefore, it is possible that superoxide regulates activation of the NFκB p65 in nodose neurons from CHF rats through multiple signal-transduction pathways.
In our study, we only measured the modulatory role of superoxide-NFκB signaling in Nav 1.7 channels in rat nodose neurons, because Nav 1.7 channels are expressed in all nodose neurons (A-type and C-type nodose neurons) as a predominant Nav channel α-subunit, but Nav 1.8 and Nav 1.9 channels are located only in C-type nodose neurons (Tu et al., 2010). Thus, far there is no report regarding NFκB binding sites on rat Nav 1.8 and Nav 1.9 channel promoters. However, we realize that future studies addressing the influence of superoxide-NFκB signaling on Nav 1.8 and Nav 1.9 channels in the CHF state are absolutely needed because Nav 1.8 and Nav 1.9 channels also have an important role in the baroreceptor function (Tu et al., 2010).
Angiotensin II Signaling Pathway and Reduced Activation of the Nav Channels in the Aortic Baroreceptor Neurons in CHF
Angiotensin (Ang) II has been recognized as a physiologically active peptide in multiple tissues including the NG (Allen et al., 1988a; Touyz, 2005). The physiological effects of Ang II including the maintenance of fluid homeostasis and blood pressure have been reported (Peach, 1977; Harris and Navar, 1985; Navar et al., 1996). Normally, Ang II receptors located on the cell membranes mediate these physiological actions of Ang II (Mehta and Griendling, 2007). It has been well-documented that circulating and tissue Ang II levels are increased in CHF patients and animal models of CHF (Liu et al., 2000; Roig et al., 2000; Cardin et al., 2003; van de Wal et al., 2006). Allen, et al. found that Ang II receptor binding sites exist in somata of nodose neurons and transport to terminals of nodose neurons (Allen et al., 1988a,b). Electrophysiological study also revealed that exogenous Ang II has the direct neuronal effect on nodose neurons through AT1R (Widdop et al., 1992). Moreover, some studies have demonstrated that Ang II down-regulates the arterial baroreflex function (Lee and Lumbers, 1981; Guo and Abboud, 1984; Garner et al., 1987).
Our recent study has also confirmed the involvement of Ang II in CHF-induced arterial baroreflex abnormality (Zhang et al., 2015). In our study, overexpression of the AT1R mRNA and protein and elevation of the local Ang II concentration in the NG from CHF rats were observed (Zhang et al., 2015). Additionally, local microinjection of losartan into the NG significantly improved CHF-attenuated arterial baroreflex sensitivity, whereas this drug did not change the arterial baroreflex sensitivity in sham rats. Furthermore, local application of exogenous Ang II in the NG from sham rats mimicked CHF to depress the arterial baroreflex function (Zhang et al., 2015). These results suggest that elevation of endogenous Ang II with AT1R overexpression in the NG contributes to the aortic baroreceptor dysfunction and subsequent down-regulation of the arterial baroreflex sensitivity in CHF rats, although a physiological level of endogenous Ang II does not affect the baroreceptor function and arterial baroreflex sensitivity. However, the local microinjection of losartan into the NG did not fully normalize the arterial baroreflex sensitivity in CHF rats toward the level seen in sham rats. We understand that the mechanism(s) responsible for CHF-blunted arterial baroreflex sensitivity are very complicated. The influence of endogenous Ang II on other cardiovascular reflex afferents [i.e., muscle reflex afferents, cardiac sympathetic afferents, and chemoreflex afferents (Khan and Sinoway, 2000; Li et al., 2006; Wang et al., 2007, 2008; Michelini et al., 2015)] and the interaction of these cardiovascular reflex afferents with baroreceptor afferents at the level of the NTS might also be potential factors to contribute to the arterial baroreflex dysfunction in the CHF state. Additionally, the effect of Ang II on central regions might be also accounted for the impairment of the arterial baroreflex function in CHF. Llewellyn et al. have reported that a high level of Ang II is detected in the plasma of CHF rats (Llewellyn et al., 2014). In particular, some studies have found that CHF induces overexpression of the AT1R in several central regions including rostral ventrolateral medulla, nucleus tractus solitarius, paraventricular nucleus, and subfornical organ, etc (Liu et al., 2006; Wang et al., 2008; Zheng et al., 2009; Zucker et al., 2009; Llewellyn et al., 2014). It has also been shown that Ang II also plays an important role in regulation of the cardiovascular system through these central regions (Casto and Phillips, 1984, 1986; Zhu et al., 2004; Liu et al., 2006; Wang et al., 2008; Zheng et al., 2009; Zucker et al., 2009; Llewellyn et al., 2014).
As mentioned above, reduced expression and activation of Nav channels are involved in attenuation of the baroreceptor neuronal excitability and resultant impairment of the arterial baroreflex sensitivity in CHF rats. Therefore, it is possible that the Nav channel is a potential target associated with the regulatory effect of Ang II on the aortic baroreceptor function. In isolated aortic baroreceptor neurons of sham rats, application of exogenous Ang II acutely inhibits Nav currents, and pretreatment of losartan totally abolishes the inhibitory effect of Ang II on Nav currents (Zhang et al., 2015), which supports the view that the acute inhibitory effect of Ang II on Nav currents mediates Ang II-attenuated arterial baroreflex function. However, losartan alone did not change Nav currents in isolated aortic baroreceptor neurons of CHF rats although this drug markedly improved the arterial baroreflex function in anesthetized CHF rats (Zhang et al., 2015). This discrepancy is explained by the fact that isolated aortic baroreceptor neurons of CHF rats loss the in vivo environment in which circulating (plasma) Ang II and paracrine release of Ang II from local tissue (the NG) are elevated in CHF rats (Llewellyn et al., 2014; Zhang et al., 2015). Based on these results, we consider that CHF-elevated endogenous Ang II with overexpression of AT1R inhibits activation of Nav channels in aortic baroreceptor neurons and further contributes to attenuated arterial baroreflex sensitivity in CHF rats.
Previous study has shown that Ang II binds with AT1 receptors to cause superoxide production mainly through activation of NADPH oxidase (Touyz and Berry, 2002). However, there are no previous reports of how Ang II mediates CHF-induced hypoactivation of Nav channels in aortic baroreceptors. In most cells including neurons, the mitochondria serve as the main source of superoxide production (Turrens, 2003). Ang II significantly elevated the mitochondria-derived superoxide in neurons (Yin et al., 2010; Case et al., 2013), leading to a series of downstream effects including modulation of the ion channel activation and neuronal firing rate (Zhu et al., 1999; Sun et al., 2005; Zimmerman et al., 2005; Yin et al., 2010). In particular, the mitochondria-derived superoxide overproduction mediates decreased Nav currents and neuronal excitability in aortic baroreceptors from CHF rats (Tu et al., 2012). Based on these studies, it is reasonable to conclude that Ang II-induced inactivation of the Nav channel might be linked to the mitochondrial superoxide overproduction in aortic baroreceptor neurons in CHF.
Conclusion
This review summarizes the mechanisms responsible for attenuated baroreceptor function and impaired arterial baroreflex in the CHF state. The information presented in this review suggests that Ang II-superoxide-NFκB signaling pathway down-regulates the neuronal excitability of aortic baroreceptors through influencing the expression and activation of Nav channels on the cell transmembrane and subsequently causes the impairment of the arterial baroreflex in the CHF state (Figure 1). These new findings also reveal potential pharmacological targets for improving the arterial baroreflex function in the CHF state.
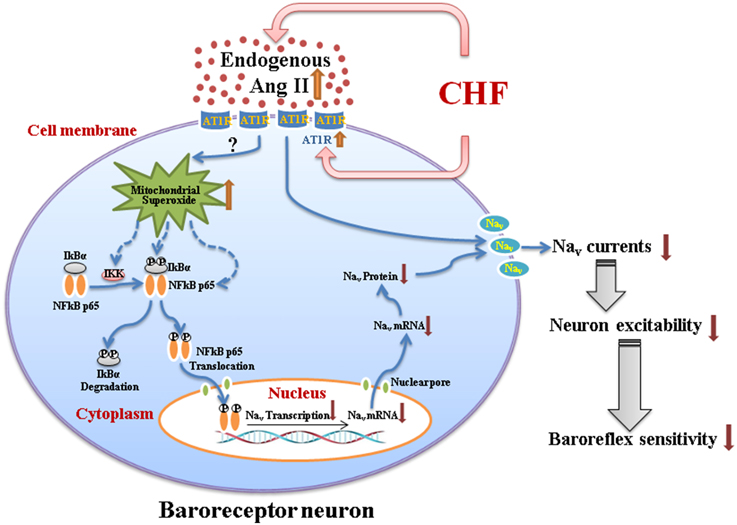
Figure 1. A schematic diagram illustrating the mechanisms responsible for impairment of the arterial baroreceptor function in chronic heart failure. CHF-elevated endogenous Ang II (plasma Ang II and tissue Ang II in the nodose ganglion) with overexpression of AT1R in baroreceptor neurons might induce the mitochondrial superoxide overproduction and the latter upregulates expression of the phosphorylated IKK, phosphorylated IκBα, and phosphorylated NFκB p65. The phosphorylated NFκB p65 translocates to the nucleus, and down-regulates the mRNA and protein expression of Nav channels. Additionally, endogenous Ang II with AT1R also directly inhibits the activation of Nav channels. As a consequence, reduced expression, and activation of Nav channels in baroreceptor neurons contribute to the impairment of aortic baroreceptors and the arterial baroreflex dysfunction in the CHF state. Briefly, Ang II-superoxide-NFκB signaling pathway down-regulates the aortic baroreceptor function through influencing the expression and activation of Nav channels in CHF. CHF, chronic heart failure; Ang II, Angiotensin II; AT1R, Angiotensin II type 1 receptor; Nav, voltage-gated sodium channel.
Funding
This work was supported by the National Institutes of Health's National Heart, Lung, and Blood Institute grant R01 HL-098503 (to YL).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adam-Vizi, V., and Chinopoulos, C. (2006). Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 27, 639–645. doi: 10.1016/j.tips.2006.10.005
Allen, A. M., Lewis, S. J., Verberne, A. J., and Mendelsohn, F. A. (1988a). Angiotensin receptors and the vagal system. Clin. Exp. Hypertens. A 10, 1239–1249.
Allen, A. M., McKinley, M. J., Oldfield, B. J., Dampney, R. A., and Mendelsohn, F. A. (1988b). Angiotensin II receptor binding and the baroreflex pathway. Clin. Exp. Hypertens. A 10(Suppl. 1), 63–78. doi: 10.3109/10641968809075964
Baker, M. D., and Wood, J. N. (2001). Involvement of Na+ channels in pain pathways. Trends Pharmacol. Sci. 22, 27–31. doi: 10.1016/S0165-6147(00)01585-6
Balaban, R. S., Nemoto, S., and Finkel, T. (2005). Mitochondria, oxidants, and aging. Cell 120, 483–495. doi: 10.1016/j.cell.2005.02.001
Benarroch, E. E. (2008). The arterial baroreflex: functional organization and involvement in neurologic disease. Neurology 71, 1733–1738. doi: 10.1212/01.wnl.0000335246.93495.92
Boogers, M. J., Veltman, C. E., and Bax, J. J. (2011). Cardiac autonomic nervous system in heart failure: imaging technique and clinical implications. Curr. Cardiol. Rev. 7, 35–42. doi: 10.2174/157340311795677725
Cadenas, E., and Davies, K. J. (2000). Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 29, 222–230. doi: 10.1016/S0891-5849(00)00317-8
Cardin, S., Li, D., Thorin-Trescases, N., Leung, T. K., Thorin, E., and Nattel, S. (2003). Evolution of the atrial fibrillation substrate in experimental congestive heart failure: angiotensin-dependent and -independent pathways. Cardiovasc. Res. 60, 315–325. doi: 10.1016/j.cardiores.2003.08.014
Case, A. J., Li, S., Basu, U., Tian, J., and Zimmerman, M. C. (2013). Mitochondrial-localized NADPH oxidase 4 is a source of superoxide in angiotensin II-stimulated neurons. Am. J. Physiol. Heart Circ. Physiol. 305, H19–H28. doi: 10.1152/ajpheart.00974.2012
Casto, R., and Phillips, M. I. (1984). Cardiovascular actions of microinjections of angiotensin II in the brain stem of rats. Am. J. Physiol. 246, R811–R816.
Casto, R., and Phillips, M. I. (1986). Angiotensin II attenuates baroreflexes at nucleus tractus solitarius of rats. Am. J. Physiol. 250, R193–R198.
Catterall, W. A., Goldin, A. L., and Waxman, S. G. (2005). International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol. Rev. 57, 397–409. doi: 10.1124/pr.57.4.4
Creager, M. A. (1992). Baroreceptor reflex function in congestive heart failure. Am. J. Cardiol. 69, 10G–15G. doi: 10.1016/0002-9149(92)91250-8
Creager, M. A., Faxon, D. P., Cutler, S. S., Kohlmann, O., Ryan, T. J., and Gavras, H. (1986). Contribution of vasopressin to vasoconstriction in patients with congestive heart failure: comparison with the renin-angiotensin system and the sympathetic nervous system. J. Am. Coll. Cardiol. 7, 758–765. doi: 10.1016/S0735-1097(86)80333-3
Cummins, T. R., Sheets, P. L., and Waxman, S. G. (2007). The roles of sodium channels in nociception: implications for mechanisms of pain. Pain 131, 243–257. doi: 10.1016/j.pain.2007.07.026
Cygankiewicz, I., Zareba, W., Vazquez, R., Vallverdu, M., Gonzalez-Juanatey, J. R., Valdes, M., et al. (2008). Heart rate turbulence predicts all-cause mortality and sudden death in congestive heart failure patients. Heart Rhythm 5, 1095–1102. doi: 10.1016/j.hrthm.2008.04.017
Czachurski, J., Lackner, K. J., Ockert, D., and Seller, H. (1982). Localization of neurones with baroreceptor input in the medial solitary nucleus by means of intracellular application of horseradish peroxidase in the cat. Neurosci. Lett. 28, 133–137. doi: 10.1016/0304-3940(82)90141-0
Dibner-Dunlap, M. E., and Thames, M. D. (1989). Baroreflex control of renal sympathetic nerve activity is preserved in heart failure despite reduced arterial baroreceptor sensitivity. Circ. Res. 65, 1526–1535. doi: 10.1161/01.RES.65.6.1526
Doumas, M., Faselis, C., Kokkinos, P., Anyfanti, P., Tsioufis, C., and Papademetriou, V. (2014). Carotid baroreceptor stimulation: a promising approach for the management of resistant hypertension and heart failure. Curr. Vasc. Pharmacol. 12, 30–37. doi: 10.2174/15701611113119990138
Fan, W., Reynolds, P. J., and Andresen, M. C. (1996). Baroreflex frequency-response characteristics to aortic depressor and carotid sinus nerve stimulation in rats. Am. J. Physiol. 271, H2218–H2227.
Fattman, C. L., Schaefer, L. M., and Oury, T. D. (2003). Extracellular superoxide dismutase in biology and medicine. Free Radic. Biol. Med. 35, 236–256. doi: 10.1016/S0891-5849(03)00275-2
Floras, J. S. (1993). Clinical aspects of sympathetic activation and parasympathetic withdrawal in heart failure. J. Am. Coll. Cardiol. 22, 72A–84A. doi: 10.1016/0735-1097(93)90466-e
Frantz, S., Fraccarollo, D., Wagner, H., Behr, T. M., Jung, P., Angermann, C. E., et al. (2003). Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc. Res. 57, 749–756. doi: 10.1016/S0008-6363(02)00723-X
Frenneaux, M. P. (2004). Autonomic changes in patients with heart failure and in post-myocardial infarction patients. Heart 90, 1248–1255. doi: 10.1136/hrt.2003.026146
Garner, M. G., Phippard, A. F., Fletcher, P. J., Maclean, J. M., Duggin, G. G., Horvath, J. S., et al. (1987). Effect of angiotensin II on baroreceptor reflex control of heart rate in conscious baboons. Hypertension 10, 628–634. doi: 10.1161/01.HYP.10.6.628
Ghosh, S., May, M. J., and Kopp, E. B. (1998). NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16, 225–260. doi: 10.1146/annurev.immunol.16.1.225
Gronda, E., Seravalle, G., Brambilla, G., Costantino, G., Casini, A., Alsheraei, A., et al. (2014). Chronic baroreflex activation effects on sympathetic nerve traffic, baroreflex function, and cardiac haemodynamics in heart failure: a proof-of-concept study. Eur. J. Heart Fail. 16, 977–983. doi: 10.1002/ejhf.138
Guo, G. B., and Abboud, F. M. (1984). Angiotensin II attenuates baroreflex control of heart rate and sympathetic activity. Am. J. Physiol. 246, H80–H89.
Häcker, H., and Karin, M. (2006). Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006:re13. doi: 10.1126/stke.3572006re13
Halbach, M., Hickethier, T., Madershahian, N., and Müller-Ehmsen, J. (2014). Baroreflex activation therapy: a new treatment option for heart failure with reduced ejection fraction. Expert. Rev. Cardiovasc. Ther. 12, 1465–1469. doi: 10.1586/14779072.2014.979790
Harris, P. J., and Navar, L. G. (1985). Tubular transport responses to angiotensin. Am. J. Physiol. 248, F621–F630.
Hauptman, P. J., Schwartz, P. J., Gold, M. R., Borggrefe, M., van veldhuisen, D. J., Starling, R. C., et al. (2012). Rationale and study design of the increase of vagal tone in heart failure study: INOVATE-HF. Am. Heart J. 163, 954–962. doi: 10.1016/j.ahj.2012.03.021
Hoffmann, A., and Baltimore, D. (2006). Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 210, 171–186. doi: 10.1111/j.0105-2896.2006.00375.x
Iliescu, R., Tudorancea, I., and Lohmeier, T. E. (2014). Baroreflex activation: from mechanisms to therapy for cardiovascular disease. Curr. Hypertens. Rep. 16:453. doi: 10.1007/s11906-014-0453-9
Israël, A. (2010). The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2:a000158. doi: 10.1101/cshperspect.a000158
Kabe, Y., Ando, K., Hirao, S., Yoshida, M., and Handa, H. (2005). Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid. Redox. Signal. 7, 395–403. doi: 10.1089/ars.2005.7.395
Karin, M., and Ben-Neriah, Y. (2000). Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 18, 621–663. doi: 10.1146/annurev.immunol.18.1.621
Khan, M. H., and Sinoway, L. I. (2000). Muscle reflex control of sympathetic nerve activity in heart failure: the role of exercise conditioning. Heart Fail. Rev. 5, 87–100. doi: 10.1023/A:1009802308872
Kleiger, R. E., Miller, J. P., Bigger, J. T. Jr., and Moss, A. J. (1987). Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am. J. Cardiol. 59, 256–262. doi: 10.1016/0002-9149(87)90795-8
Kobayashi, M., Cheng, Z. B., Tanaka, K., and Nosaka, S. (1999). Is the aortic depressor nerve involved in arterial chemoreflexes in rats? J. Auton. Nerv. Syst. 78, 38–48. doi: 10.1016/S0165-1838(99)00054-5
Kwong, K., Carr, M. J., Gibbard, A., Savage, T. J., Singh, K., Jing, J., et al. (2008). Voltage-gated sodium channels in nociceptive versus non-nociceptive nodose vagal sensory neurons innervating guinea pig lungs. J. Physiol. 586, 1321–1336. doi: 10.1113/jphysiol.2007.146365
Lancaster, E., Oh, E. J., Gover, T., and Weinreich, D. (2002). Calcium and calcium-activated currents in vagotomized rat primary vagal afferent neurons. J. Physiol. 540, 543–556. doi: 10.1113/jphysiol.2001.013121
Lee, W. B., and Lumbers, E. R. (1981). Angiotensin and the cardiac baroreflex response to phenylephrine. Clin. Exp. Pharmacol. Physiol. 8, 109–117. doi: 10.1111/j.1440-1681.1981.tb00141.x
Li, Y. L., Xia, X. H., Zheng, H., Gao, L., Li, Y. F., Liu, D., et al. (2006). Angiotensin II enhances carotid body chemoreflex control of sympathetic outflow in chronic heart failure rabbits. Cardiovasc. Res. 71, 129–138. doi: 10.1016/j.cardiores.2006.03.017
Li, Y. L., and Zheng, H. (2011). Angiotensin II-NADPH oxidase-derived superoxide mediates diabetes-attenuated cell excitability of aortic baroreceptor neurons. Am. J. Physiol. Cell Physiol. 301, C1368–C1377. doi: 10.1152/ajpcell.00214.2011
Li, Z., Chapleau, M. W., Bates, J. N., Bielefeldt, K., Lee, H. C., and Abboud, F. M. (1998). Nitric oxide as an autocrine regulator of sodium currents in baroreceptor neurons. Neuron 20, 1039–1049. doi: 10.1016/S0896-6273(00)80484-5
Liao, K., Yu, L., He, B., Huang, B., Yang, K., Saren, G., et al. (2014). Carotid baroreceptor stimulation prevents arrhythmias induced by acute myocardial infarction through autonomic modulation. J. Cardiovasc. Pharmacol. 64, 431–437. doi: 10.1097/FJC.0000000000000135
Liu, D., Gao, L., Roy, S. K., Cornish, K. G., and Zucker, I. H. (2006). Neuronal angiotensin II type 1 receptor upregulation in heart failure: activation of activator protein 1 and Jun N-terminal kinase. Circ. Res. 99, 1004–1011. doi: 10.1161/01.RES.0000247066.19878.93
Liu, J. L., Irvine, S., Reid, I. A., Patel, K. P., and Zucker, I. H. (2000). Chronic exercise reduces sympathetic nerve activity in rabbits with pacing-induced heart failure: a role for angiotensin II. Circulation 102, 1854–1862. doi: 10.1161/01.CIR.102.15.1854
Llewellyn, T. L., Sharma, N. M., Zheng, H., and Patel, K. P. (2014). Effects of exercise training on SFO-mediated sympathoexcitation during chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 306, H121–H131. doi: 10.1152/ajpheart.00534.2013
Madershahian, N., Scherner, M., Müller-Ehmsen, J., Halbach, M., Hickethier, T., Velden, R., et al. (2014). Baroreflex activation therapy in patients with pre-existing implantable cardioverter-defibrillator: compatible, complementary therapies. Europace 16, 861–865. doi: 10.1093/europace/eut403
Matsutomi, T., Nakamoto, C., Zheng, T., Kakimura, J., and Ogata, N. (2006). Multiple types of Na(+) currents mediate action potential electrogenesis in small neurons of mouse dorsal root ganglia. Pflugers Arch. 453, 83–96. doi: 10.1007/s00424-006-0104-3
McCord, J. M. (1993). Human disease, free radicals, and the oxidant/antioxidant balance. Clin. Biochem. 26, 351–357. doi: 10.1016/0009-9120(93)90111-I
McKenna, S., and Wright, C. J. (2015). Inhibiting IkappaBbeta-NFkappaB signaling attenuates the expression of select pro-inflammatory genes. J. Cell Sci. 128, 2143–2155. doi: 10.1242/jcs.168351
Mehta, P. K., and Griendling, K. K. (2007). Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 292, C82–C97. doi: 10.1152/ajpcell.00287.2006
Mendes, A. F., Caramona, M. M., Carvalho, A. P., and Lopes, M. C. (2003). Differential roles of hydrogen peroxide and superoxide in mediating IL-1-induced NF-kappa B activation and iNOS expression in bovine articular chondrocytes. J. Cell Biochem. 88, 783–793. doi: 10.1002/jcb.10428
Michelini, L. C., O'Leary, D. S., Raven, P. B., and Nóbrega, A. C. (2015). Neural control of circulation and exercise: a translational approach disclosing interactions between central command, arterial baroreflex, and muscle metaboreflex. Am. J. Physiol. Heart Circ. Physiol. 309, H381–H392. doi: 10.1152/ajpheart.00077.2015
Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., Cushman, M., et al. (2015). Heart disease and stroke statistics-2015 update: a report from the american heart association. Circulation 131, e29–e322. doi: 10.1161/CIR.0000000000000157
Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/BJ20081386
Navar, L. G., Inscho, E. W., Majid, S. A., Imig, J. D., Harrison-Bernard, L. M., and Mitchell, K. D. (1996). Paracrine regulation of the renal microcirculation. Physiol. Rev. 76, 425–536.
Nolan, J., Batin, P. D., Andrews, R., Lindsay, S. J., Brooksby, P., Mullen, M., et al. (1998). Prospective study of heart rate variability and mortality in chronic heart failure: results of the United Kingdom heart failure evaluation and assessment of risk trial (UK-heart). Circulation 98, 1510–1516. doi: 10.1161/01.CIR.98.15.1510
Ogata, N., Yamamoto, H., Kugiyama, K., Yasue, H., and Miyamoto, E. (2000). Involvement of protein kinase C in superoxide anion-induced activation of nuclear factor-kappa B in human endothelial cells. Cardiovasc. Res. 45, 513–521. doi: 10.1016/S0008-6363(99)00364-8
Pan, Y. X., Gao, L., Wang, W. Z., Zheng, H., Liu, D., Patel, K. P., et al. (2007). Exercise training prevents arterial baroreflex dysfunction in rats treated with central angiotensin II. Hypertension 49, 519–527. doi: 10.1161/01.HYP.0000256955.74461.93
Patrick Harty, T., and Waxman, S. G. (2007). Inactivation properties of sodium channel Nav1.8 maintain action potential amplitude in small DRG neurons in the context of depolarization. Mol. Pain 3:12. doi: 10.1186/1744-8069-3-12
Peach, M. J. (1977). Renin-angiotensin system: biochemistry and mechanisms of action. Physiol. Rev. 57, 313–370.
Pinna, G. D., Maestri, R., Capomolla, S., Febo, O., Robbi, E., Cobelli, F., et al. (2005). Applicability and clinical relevance of the transfer function method in the assessment of baroreflex sensitivity in heart failure patients. J. Am. Coll. Cardiol. 46, 1314–1321. doi: 10.1016/j.jacc.2005.06.062
Pitkanen, S., and Robinson, B. H. (1996). Mitochondrial complex I deficiency leads to increased production of superoxide radicals and induction of superoxide dismutase. J. Clin. Invest. 98, 345–351. doi: 10.1172/JCI118798
Porter, T. R., Eckberg, D. L., Fritsch, J. M., Rea, R. F., Beightol, L. A., Schmedtje, J. F. Jr., et al. (1990). Autonomic pathophysiology in heart failure patients. Sympathetic-cholinergic interrelations. J. Clin. Invest. 85, 1362–1371. doi: 10.1172/JCI114580
Ritter, A. M., Martin, W. J., and Thorneloe, K. S. (2009). The voltage-gated sodium channel Nav1.9 is required for inflammation-based urinary bladder dysfunction. Neurosci. Lett. 452, 28–32. doi: 10.1016/j.neulet.2008.12.051
Robinson, B. H. (1998). The role of manganese superoxide dismutase in health and disease. J. Inherit. Metab. Dis. 21, 598–603. doi: 10.1023/A:1005427323835
Roig, E., Perez-Villa, F., Morales, M., Jimenez, W., Orus, J., Heras, M., et al. (2000). Clinical implications of increased plasma angiotensin II despite ACE inhibitor therapy in patients with congestive heart failure. Eur. Heart J. 21, 53–57. doi: 10.1053/euhj.1999.1740
Rondon, E., Brasileiro-Santos, M. S., Moreira, E. D., Rondon, M. U., Mattos, K. C., Coelho, M. A., et al. (2006). Exercise training improves aortic depressor nerve sensitivity in rats with ischemia-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 291, H2801–H2806. doi: 10.1152/ajpheart.01352.2005
Ruttanaumpawan, P., Gilman, M. P., Usui, K., Floras, J. S., and Bradley, T. D. (2008). Sustained effect of continuous positive airway pressure on baroreflex sensitivity in congestive heart failure patients with obstructive sleep apnea. J. Hypertens. 26, 1163–1168. doi: 10.1097/HJH.0b013e3282fb81ed
Sabbah, H. N. (2012). Baroreflex activation for the treatment of heart failure. Curr. Cardiol. Rep. 14, 326–333. doi: 10.1007/s11886-012-0265-y
Sapru, H. N., Gonzalez, E., and Krieger, A. J. (1981). Aortic nerve stimulation in the rat: cardiovascular and respiratory responses. Brain Res. Bull. 6, 393–398. doi: 10.1016/S0361-9230(81)80009-3
Saul, J. P., Arai, Y., Berger, R. D., Lilly, L. S., Colucci, W. S., and Cohen, R. J. (1988). Assessment of autonomic regulation in chronic congestive heart failure by heart rate spectral analysis. Am. J. Cardiol. 61, 1292–1299. doi: 10.1016/0002-9149(88)91172-1
Schild, J. H., Clark, J. W., Hay, M., Mendelowitz, D., Andresen, M. C., and Kunze, D. L. (1994). A- and C-type rat nodose sensory neurons: model interpretations of dynamic discharge characteristics. J. Neurophysiol. 71, 2338–2358.
Schild, J. H., and Kunze, D. L. (1997). Experimental and modeling study of Na+ current heterogeneity in rat nodose neurons and its impact on neuronal discharge. J. Neurophysiol. 78, 3198–3209.
Schild, J. H., and Kunze, D. L. (2012). Differential distribution of voltage-gated channels in myelinated and unmyelinated baroreceptor afferents. Auton. Neurosci. 172, 4–12. doi: 10.1016/j.autneu.2012.10.014
Schmidli, J., von Allmen, R. S., and Mohaupt, M. G. (2014). Electrical carotid baroreceptor stimulation. Wien. Med. Wochenschr. 164, 508–514. doi: 10.1007/s10354-014-0329-2
Schultz, H. D. (2009). Nitric oxide regulation of autonomic function in heart failure. Curr. Heart Fail. Rep. 6, 71–80. doi: 10.1007/s11897-009-0012-x
Shang, L. L., Sanyal, S., Pfahnl, A. E., Jiao, Z., Allen, J., Liu, H., et al. (2008). NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am. J. Physiol. Cell Physiol. 294, C372–C379. doi: 10.1152/ajpcell.00186.2007
Shen, W., Gill, R. M., Zhang, J. P., Jones, B. D., Corbly, A. K., and Steinberg, M. I. (2005). Sodium channel enhancer restores baroreflex sensitivity in conscious dogs with heart failure. Am. J. Physiol. Heart Circ. Physiol. 288, H1508–H1514. doi: 10.1152/ajpheart.00337.2004
Spyer, K. M., Lambert, J. H., and Thomas, T. (1997). Central nervous system control of cardiovascular function: neural mechanisms and novel modulators. Clin. Exp. Pharmacol. Physiol. 24, 743–747. doi: 10.1111/j.1440-1681.1997.tb02125.x
Sun, C., Sellers, K. W., Sumners, C., and Raizada, M. K. (2005). NAD(P)H oxidase inhibition attenuates neuronal chronotropic actions of angiotensin II. Circ. Res. 96, 659–666. doi: 10.1161/01.RES.0000161257.02571.4b
Tatalovic, M., Glazebrook, P. A., and Kunze, D. L. (2012). Expression of the P/Q (Cav2.1) calcium channel in nodose sensory neurons and arterial baroreceptors. Neurosci. Lett. 520, 38–42. doi: 10.1016/j.neulet.2012.05.026
Touyz, R. M. (2005). Reactive oxygen species as mediators of calcium signaling by angiotensin II: implications in vascular physiology and pathophysiology. Antioxid. Redox. Signal. 7, 1302–1314. doi: 10.1089/ars.2005.7.1302
Touyz, R. M., and Berry, C. (2002). Recent advances in angiotensin II signaling. Braz. J. Med. Biol. Res. 35, 1001–1015. doi: 10.1590/S0100-879X2002000900001
Tu, H., Liu, J., Zhu, Z., Zhang, L. B., Pipinos, I. I., and Li, Y. L. (2012). Mitochondria-derived superoxide and voltage-gated sodium channels in baroreceptor neurons from chronic heart failure rats. J. Neurophysiol. 107, 591–602. doi: 10.1152/jn.00754.2011
Tu, H., Zhang, L., Tran, T. P., Muelleman, R. L., and Li, Y. L. (2010). Reduced expression and activation of voltage-gated sodium channels contributes to blunted baroreflex sensitivity in heart failure rats. J. Neurosci. Res. 88, 3337–3349. doi: 10.1002/jnr.22483
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. doi: 10.1113/jphysiol.2003.049478
Valen, G. (2004). Signal transduction through nuclear factor kappa B in ischemia-reperfusion and heart failure. Basic Res. Cardiol. 99, 1–7. doi: 10.1007/s00395-003-0442-7
Valen, G., Yan, Z. Q., and Hansson, G. K. (2001). Nuclear factor kappa-B and the heart. J. Am. Coll. Cardiol. 38, 307–314. doi: 10.1016/S0735-1097(01)01377-8
Van der Heiden, K., Cuhlmann, S., Luong le, A., Zakkar, M., and Evans, P. C. (2010). Role of nuclear factor kappaB in cardiovascular health and disease. Clin. Sci. 118, 593–605. doi: 10.1042/CS20090557
van de Wal, R. M., Plokker, H. W., Lok, D. J., Boomsma, F., van der Horst, F. A., van Veldhuisen, D. J., et al. (2006). Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. Int. J. Cardiol. 106, 367–372. doi: 10.1016/j.ijcard.2005.02.016
Verkaart, S., Koopman, W. J., van Emst-de Vries, S. E., Nijtmans, L. G., van den Heuvel, L. W., Smeitink, J. A., et al. (2007). Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim. Biophys. Acta 1772, 373–381. doi: 10.1016/j.bbadis.2006.12.009
Wallace, D. C. (2001). A mitochondrial paradigm for degenerative diseases and ageing. Novartis. Found. Symp. 235, 247–263. doi: 10.1002/0470868694.ch20
Wang, W., Chen, J. S., and Zucker, I. H. (1990). Carotid sinus baroreceptor sensitivity in experimental heart failure. Circulation 81, 1959–1966. doi: 10.1161/01.CIR.81.6.1959
Wang, W., Chen, J. S., and Zucker, I. H. (1991a). Carotid sinus baroreceptor reflex in dogs with experimental heart failure. Circ. Res. 68, 1294–1301. doi: 10.1161/01.RES.68.5.1294
Wang, W., Chen, J. S., and Zucker, I. H. (1991b). Postexcitatory depression of baroreceptors in dogs with experimental heart failure. Am. J. Physiol. 260, H1160–H1165.
Wang, W., Han, H. Y., and Zucker, I. H. (1996). Depressed baroreflex in heart failure is not due to structural change in carotid sinus nerve fibers. J. Auton. Nerv. Syst. 57, 101–108. doi: 10.1016/0165-1838(95)00107-7
Wang, W. Z., Gao, L., Pan, Y. X., Zucker, I. H., and Wang, W. (2007). AT1 receptors in the nucleus tractus solitarii mediate the interaction between the baroreflex and the cardiac sympathetic afferent reflex in anesthetized rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R1137–R1145. doi: 10.1152/ajpregu.00590.2006
Wang, W. Z., Gao, L., Wang, H. J., Zucker, I. H., and Wang, W. (2008). Interaction between cardiac sympathetic afferent reflex and chemoreflex is mediated by the NTS AT1 receptors in heart failure. Am. J. Physiol. Heart Circ. Physiol. 295, H1216–H1226. doi: 10.1152/ajpheart.00557.2008
Wang, Y., Seto, S. W., and Golledge, J. (2014). Angiotensin, I. I., sympathetic nerve activity and chronic heart failure. Heart Fail. Rev. 19, 187–198. doi: 10.1007/s10741-012-9368-1
Waxman, S. G., Dib-Hajj, S., Cummins, T. R., and Black, J. A. (1999). Sodium channels and pain. Proc. Natl. Acad. Sci. U.S.A. 96, 7635–7639. doi: 10.1073/pnas.96.14.7635
White, C. W. (1981). Abnormalities in baroreflex control of heart rate in canine heart failure. Am. J. Physiol. 240, H793–H799.
Widdop, R. E., Krstew, E., and Jarrott, B. (1992). Electrophysiological responses of angiotensin peptides on the rat isolated nodose ganglion. Clin. Exp. Hypertens. A 14, 597–613. doi: 10.3109/10641969209036210
Xu, W. X., Ban, T., Wang, L. Q., Zhao, M., Yin, L., Li, G., et al. (2015). KCa1.1 beta4-subunits are not responsible for iberiotoxin-resistance in baroreceptor neurons in adult male rats. Int. J. Cardiol. 178, 184–187. doi: 10.1016/j.ijcard.2014.10.128
Yin, J. X., Yang, R. F., Li, S., Renshaw, A. O., Li, Y. L., Schultz, H. D., et al. (2010). Mitochondria-produced superoxide mediates angiotensin II-induced inhibition of neuronal potassium current. Am. J. Physiol. Cell Physiol. 298, C857–C865. doi: 10.1152/ajpcell.00313.2009
Yoshida, S. (1994). Tetrodotoxin-resistant sodium channels. Cell. Mol. Neurobiol. 14, 227–244. doi: 10.1007/BF02088322
Yu, F. H., and Catterall, W. A. (2003). Overview of the voltage-gated sodium channel family. Genome Biol. 4:207. doi: 10.1186/gb-2003-4-3-207
Zhang, D., Liu, J., Tu, H., Muelleman, R. L., Cornish, K. G., and Li, Y. L. (2014). In vivo transfection of manganese superoxide dismutase gene or nuclear factor kappaB shRNA in nodose ganglia improves aortic baroreceptor function in heart failure rats. Hypertension 63, 88–95. doi: 10.1161/HYPERTENSIONAHA.113.02057
Zhang, D., Liu, J., Zheng, H., Tu, H., Muelleman, R. L., and Li, Y. L. (2015). Effect of angiotension II on voltage-gated sodium currents in aortic baroreceptor neurons and arterial baroreflex sensitivity in heart failure rats. J. Hypertens. 33, 1401–1410. doi: 10.1097/HJH.0000000000000563
Zheng, H., Li, Y. F., Wang, W., and Patel, K. P. (2009). Enhanced angiotensin-mediated excitation of renal sympathetic nerve activity within the paraventricular nucleus of anesthetized rats with heart failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1364–R1374. doi: 10.1152/ajpregu.00149.2009
Zhu, G. Q., Gao, L., Li, Y., Patel, K. P., Zucker, I. H., and Wang, W. (2004). AT1 receptor mRNA antisense normalizes enhanced cardiac sympathetic afferent reflex in rats with chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 287, H1828–H1835. doi: 10.1152/ajpheart.01245.2003
Zhu, M., Gelband, C. H., Posner, P., and Sumners, C. (1999). Angiotensin II decreases neuronal delayed rectifier potassium current: role of calcium/calmodulin-dependent protein kinase II. J. Neurophysiol. 82, 1560–1568.
Zimmerman, M. C., Sharma, R. V., and Davisson, R. L. (2005). Superoxide mediates angiotensin II-induced influx of extracellular calcium in neural cells. Hypertension 45, 717–723. doi: 10.1161/01.HYP.0000153463.22621.5e
Zucker, I. H., Hackley, J. F., Cornish, K. G., Hiser, B. A., Anderson, N. R., Kieval, R., et al. (2007). Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension 50, 904–910. doi: 10.1161/HYPERTENSIONAHA.107.095216
Zucker, I. H., and Liu, J. L. (2000). Angiotensin II–nitric oxide interactions in the control of sympathetic outflow in heart failure. Heart Fail. Rev. 5, 27–43. doi: 10.1023/A:1009894007055
Zucker, I. H., Schultz, H. D., Patel, K. P., Wang, W., and Gao, L. (2009). Regulation of central angiotensin type 1 receptors and sympathetic outflow in heart failure. Am. J. Physiol. Heart Circ. Physiol. 297, H1557–H1566. doi: 10.1152/ajpheart.00073.2009
Keywords: angiotensin II, baroreflex, baroreceptor, heart failure, nodose ganglion, nuclear factor κB, sodium channel, superoxide
Citation: Zhang D, Muelleman RL and Li Y-L (2015) Angiotensin II-superoxide-NFκB signaling and aortic baroreceptor dysfunction in chronic heart failure. Front. Neurosci. 9:382. doi: 10.3389/fnins.2015.00382
Received: 31 August 2015; Accepted: 02 October 2015;
Published: 16 October 2015.
Edited by:
J. Kevin Shoemaker, The University of Western Ontario, CanadaReviewed by:
Noah J. Marcus, Des Moines University, USARenata Maria Lataro, University of São Paulo, Brazil
Copyright © 2015 Zhang, Muelleman and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yu-Long Li, eXVsb25nbGlAdW5tYy5lZHU=