 Fotis Tsetsos1
Fotis Tsetsos1 Shanmukha S. Padmanabhuni1
Shanmukha S. Padmanabhuni1 John Alexander1
John Alexander1 Iordanis Karagiannidis1
Iordanis Karagiannidis1 Margaritis Tsifintaris1
Margaritis Tsifintaris1 Apostolia Topaloudi1
Apostolia Topaloudi1 Dimitrios Mantzaris1
Dimitrios Mantzaris1 Marianthi Georgitsi1,2
Marianthi Georgitsi1,2 Petros Drineas3
Petros Drineas3 Peristera Paschou1*
Peristera Paschou1*- 1Department of Molecular Biology and Genetics, Democritus University of Thrace, Alexandroupolis, Greece
- 2Laboratory of General Biology, Department of Medicine, Aristotle University of Thessaloniki, Thessaloniki, Greece
- 3Computer Science Department, Rensselaer Polytechnic Institute, Troy, NY, USA
Gilles de la Tourette Sydrome (TS) is a childhood onset neurodevelopmental disorder, characterized phenotypically by the presence of multiple motor and vocal tics. It is often accompanied by multiple psychiatric comorbidities, with Attention Deficit/Hyperactivity Disorder (ADHD) among the most common. The extensive co-occurrence of the two disorders suggests a shared genetic background. A major step toward the elucidation of the genetic architecture of TS was undertaken by the first TS Genome-wide Association Study (GWAS) reporting 552 SNPs that were moderately associated with TS (p < 1E-3). Similarly, initial ADHD GWAS attempts and meta-analysis were not able to produce genome-wide significant findings, but have provided insight to the genetic basis of the disorder. Here, we examine the common genetic background of the two neuropsychiatric phenotypes, by meta-analyzing the 552 top hits in the TS GWAS with the results of ADHD first GWASs. We identify 19 significant SNPs, with the top four implicated genes being TBC1D7, GUCY1A3, RAP1GDS1, and CHST11. TBCD17 harbors the top scoring SNP, rs1866863 (p:3.23E-07), located in a regulatory region downstream of the gene, and the third best-scoring SNP, rs2458304 (p:2.54E-06), located within an intron of the gene. Both variants were in linkage disequilibrium with eQTL rs499818, indicating a role in the expression levels of the gene. TBC1D7 is the third subunit of the TSC1/TSC2 complex, an inhibitor of the mTOR signaling pathway, with a central role in cell growth and autophagy. The top genes implicated by our study indicate a complex and intricate interplay between them, warranting further investigation into a possibly shared etiological mechanism for TS and ADHD.
1. Introduction
Gilles de la Tourette Syndrome (TS) is a childhood onset neuropsychiatric disorder characterized by a multitude of motor and vocal tics that last longer than a year. Its international prevalence has been estimated to be approximately 1% (Robertson et al., 2009). A recent systematic review and meta-analysis on the population prevalence of TS, refined its prevalence estimate in children to 0.3–1% (Scharf et al., 2015). It presents a significant gender bias, with 73% of its patients being male, and the male patients being more likely to develop comorbid disorders (Robertson et al., 2015). TS is often associated with other neuropsychiatric disorders, including Attention Deficit/Hyperactivity Disorder (ADHD), Obsessive Compulsive Disorder (OCD), depression and anxiety (Robertson, 2006).
The first genome-wide association study (GWAS) on TS was undertaken by the Tourette Syndrome Association International Consortium for Genetics (TSAICG) (Scharf et al., 2013). In their primary analysis, no SNPs achieved an association p-value of genome-wide significance, however this study provided the basis for subsequent studies, as the top signals that attained a p < 10−3 were found to be significantly enriched for functional variants. The Gilles de la Tourette Syndrome GWAS Replication Initiative (GGRI) undertook a replication (Paschou et al., 2014) of the first GWAS study, by selecting the top LD-independent SNPs and additional SNPs singificantly enriched in eQTL or mQTLs for genotyping in 609 European TS patients and 610 ancestry-matched controls, recruited from different European countries and Canada. This replication study enriched the significance of the selected SNPs and provided more evidence toward the genetic aetiology of TS.
On the other hand, initial GWAS attempts on ADHD also did not yield genome-wide significant results (Neale et al., 2008; Mick et al., 2010; Neale et al., 2010a; Lesch et al., 2008). To that end, a meta-analysis was conducted by Neale et al. (2010b), aggregating the results of the previous GWAS projects and meta-analyzing them. This meta-analysis could not produce any significant results either, but, similar to the TS GWAS, it set the groundwork for the elucidation of the genetic background of ADHD.
The relationship of TS with ADHD is well established (Karagiannidis et al., 2016). Individuals with ADHD commonly present tics, and in individuals with TS and tics, ADHD is a significant commorbidity. ADHD occurs in a significant proportion of TS patients, ranging from 21 to 90% in studied cohorts (Robertson, 2006). This phenotypic association is a major indication of a common genetic background between the two disorders. Furthermore, a recent study investigating the genetic correlation among neuropsychiatric and neurological disease based on GWAS results for each disorder, also recovered a genetic correlation between TS and ADHD (Anttila et al., 2016).
This is the first study to attempt to identify a shared genetic component between TS and ADHD. We used summary statistics from the latest large-scale genomic efforts to unravel the genetic background of TS and ADHD and derived the combined effects of shared polymorphisms between the two datasets, highlighting genes and pathways that may play a role in the shared etiology between the two disorders.
2. Materials and Methods
2.1. Data Sources
For our study we focused on the combination of the known effects of SNPs on the phenotypes of TS and ADHD.
Scharf et al in their study (Scharf et al., 2013) performed a GWAS and meta-analysis on a total of 1285 cases and 4964 ancestry-matched controls of European ancestry, genotyped on 484,295 SNPs. The dataset was analyzed in three split cohorts and was subsequently meta-analyzed. The study reported 552 SNPs associated with TS that acquired a p < 10−3.
We acquired the ADHD meta-GWAS whole-genome summary statistics from the study conducted by the ADHD subgroup of the Psychiatric GWAS consortium (Neale et al., 2010b; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). The total sample size consisted of 896 cases, 2455 controls and 2064 trios genotyped and then imputed to 1,230,536 SNPs.
We used the publically available top SNPs with a p < 10−3 associated with TS (Scharf et al., 2013) and meta-analyzed them with the results of the ADHD meta-analysis (Neale et al., 2010b; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013). We identified 489 SNPs that were overlapping between the two sources to proceed with the meta-analysis.
2.2. Meta-Analytical Procedure
We combined the effects of the SNPs in each phenotype, following a meta-analytic approach, assuming a fixed-effects model, using the Z-Scores as the effect and the number of cases in each study as the weight. The heterogeneity of each analyzed SNP was assessed using Cochran's Q-test and I2 statistic. The analysis was performed using the METAL (Willer et al., 2010) software. The significance threshold was set using the Bonferroni correction for multiple testing.
2.3. Annotation and Functional Significance
We proceeded to analyze the significant SNPs using the ENSEMBL Variant Effect Predictor (McLaren et al., 2010) to annotate and explore the possible functional characteristics of the associated variants. The genomic positions of the variants were converted to the GRCh38 assembly coordinates. For the investigation of the allelic frequencies, and the linkage disequilibrium (LD) patterns we used data from the 1000Genomes project (1000 Genomes Project Consortium et al., 2015) and the LDlink software (Machiela and Chanock, 2014). To investigate the association of the variants and their respective genes with tissue expression levels we used the GTEx portal (The GTEx Consortium, 2013) and the Expression Atlas database (Petryszak et al., 2014). The annotation and exploration of the genomic structure of the identified loci was further assisted by the use of LdOOKUP, developed by Shaun Purcell (https://purces04.u.hpc.mssm.edu/ldookup/ldookup.cgi).
We have uploaded all codes necessary to confirm our conclusions and they can be found at https://github.com/ftsetsos/tsadhdmeta2016.
3. Results
The meta-analysis produced 19 significant SNPs, out of the total 489 tested. The significance threshold was set using the Bonferroni correction for multiple testing, setting the significance level at a p-value of 0.0001022.
Of these 19 SNPs, five attained the lowest p-values, coupled with no evidence of confounding heterogeneity (I2 = 0). The tested SNPs that present the most significant heterogeneity (Het p < 0.05) were the ones that had achieved a p < 0.05 in the original ADHD meta-analysis. The annotation showed that the majority of the significant variants are located in introns, two are in regulatory regions, while three are intergenic.
The first and third top hits (rs1866863, p:3.23E-07 and rs2458304, p:2.54E-06) reside on a LD-block of 54.19 kb on the 6p24.1 region. They show significant linkage disequilibrium between them (D′: 0.909, R2: 0.725). The former is a variant located in the regulatory region downstream of the TBC1D7 gene, and the latter is an intron variant in the same gene. Both are in LD with the rs499818 eQTL (R2: 0.59 and 0.48 respectively), suggesting an interplay with the expression levels of the gene.
Chromosome 4 hosts the second (rs2705462, p:1.44E-06), the fourth (rs17561798, p:9.89E-06), and two lower-ranked variants(rs477897, p:8.65E-05 and rs2285084, p:1.00E-04), each residing in four distinct, LD-independent loci. The most significant variant, rs2705462, is located in the intergenic region upstream of the GUCY1A3 gene in the 4q32.1 region on a LD-block of 46.63 kb. The next variant, rs17561798, resides in the 4q23 region and is an intron variant inside the RAP1GDS1 gene. The variant rs477897 is located within an intron of ADD1 in the region 4p16.3 captures an area of 125.59 kb, implicating the genes H3BP2, ADD1, MFSD10, NOP14. The intron variant rs2285084 is located in the gene ANXA10. The gene is included in the locus 4q32.3 in a high LD region of 330.00 kb that contains also the genes ANXA10, DDX60, DDX60L.
The fifth most significant SNP, rs1650137 (p:1.76E-05), is located on 12q23.3 in an intron of the gene CHST11. This region is inside a LD-block that extends for 39.68 kb. The variant rs2246417 came up as the sixth most significant (p:1.95E-05), residing in the locus 21q22.3 in a LD-block of 16.29 kb, within an intron of the LINC00316 gene.
The variant rs11716445 (p: 8.01E-05) resides in the 3p21.31 region. This region is characterized by a very large area with an extended high-LD block of 1941.64 kb and it contains 70 genes, with the first genes being PLXNB1, CCDC51, TMA7, ATRIP, TREX1, and ending with HYAL2, TUSC2, RASSF1, ZMYND10, NPRL2. The variant itself is located in the intron of the RHOA gene. It is one of the lower-ranked variants that achieved minimal confounding by heterogeneity.
The locus 7p21.3 hosts the intergenic variants rs13244651 (p:4.11E-05) and rs17531553 (p:7.08E-05) that are part of a LD-block sized at 103.93 kb, albeit with no known genes close to them, and no strong suggestive results for any direct functional implication.
Two genomic regions on chromosome 9 are implicated by our results. A region of 58.16 kb in the 9p24.2 locus contains the variants rs1007021 (p: 4.38E-05) and rs1007022 (p:7.68E-05) in the introns of KCNV2, showing strong LD between them (D′: 1.000 R2:0.803). The region 9q31.1 contains the intergenic variant rs7858600 (p:5.30E-05) inside a region of 27.35 kb.
In the locus 10q21.1, the intergenic variant rs1896373 (p:7.46E-05) captures a region of 47.58 kb, in strong LD with the rs1919459 eQTL (R2: 0.97) that is associated with the regulation of DKK1. The variant rs4789936 (p:8.92E-05) is located in the 17q25.3 locsus, in a LD-region 34.19 kb, and is an intron variant of the gene TIMP2 while on it is non-coding exon variant in the gene CEP295NL. In the locus 16q12.1, the variant rs7203818 (p:1.01E-04) resides in a LD-block of 21.12 kb within an intron of ZNF423.
On chromosome 13, the LD-associated variants rs7336083 and rs9319159 (D′: 0.974 R2: 0.897) represent an LD-block of 292.34 kb and reside in the introns of the LINC00351 gene. The result is mostly driven by the p-value attained in the TS meta-analysis and there is evidence of significant confounding caused by heterogeneity.
We summarize the results of the meta-analysis on Table 1. In Table 2, we provide the annotation we generated for each significant variant, and in Table 3 we provide the LD regions associated with the variants. The full results of the meta-analysis on the 489 tested SNPs are described in more detail in the Supplementary Material.
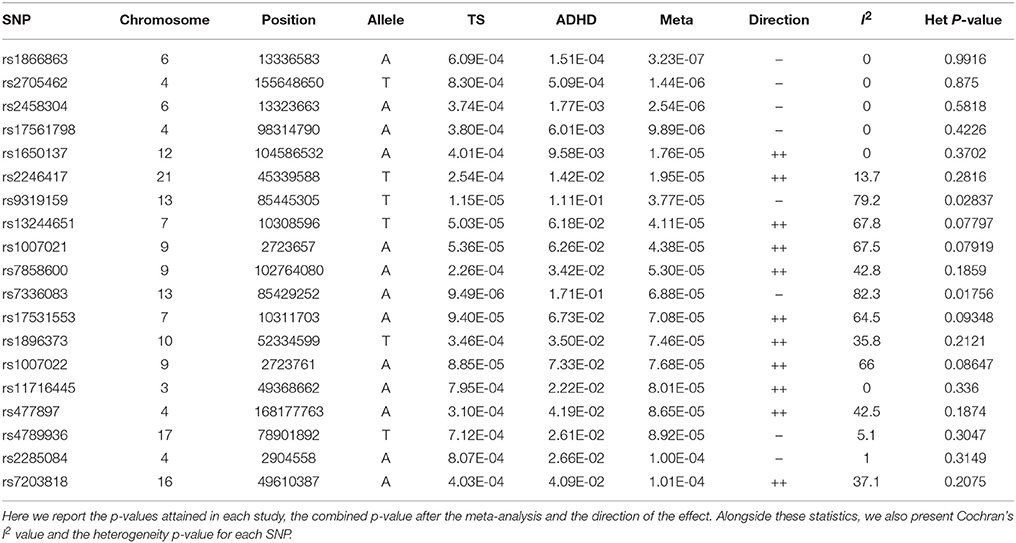
Table 1. Significant results of the meta-analysis.
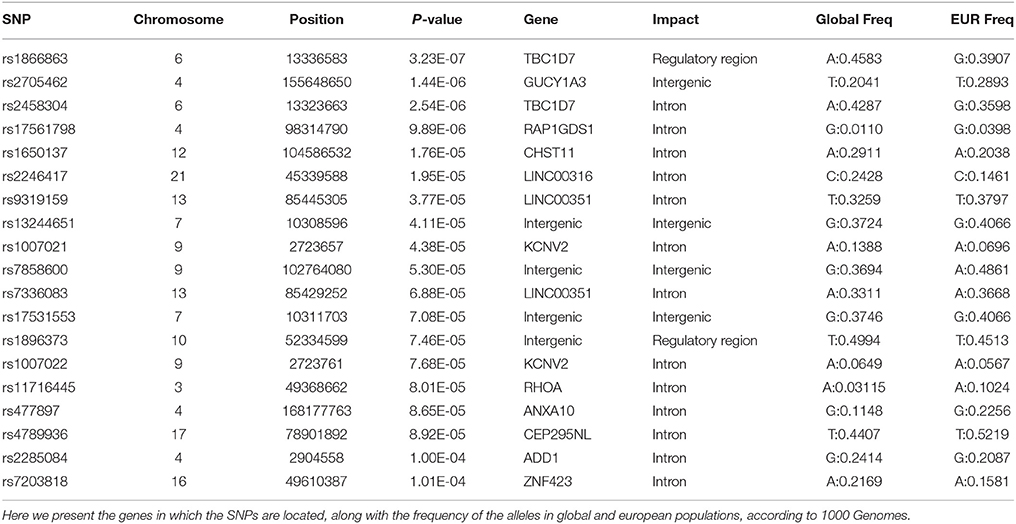
Table 2. Functional annotation of the significant SNPs of the meta-analysis.
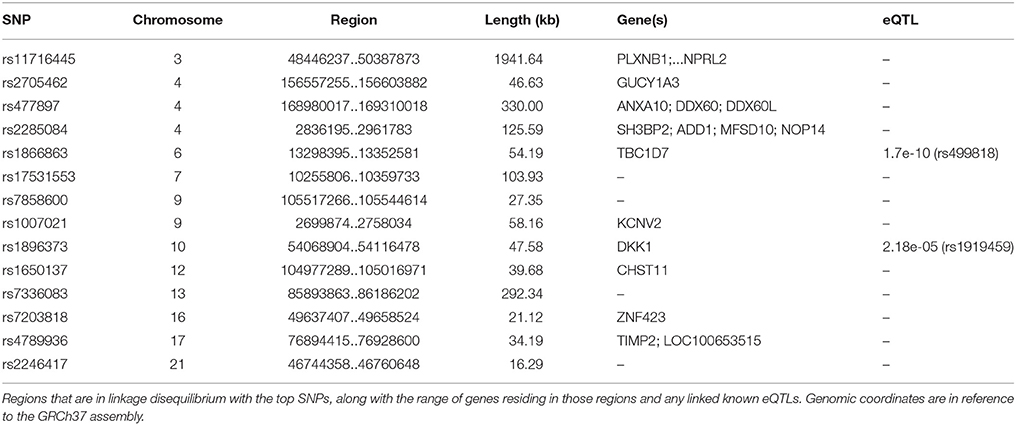
Table 3. LD-regions that are captured by the top SNPs in our study.
4. Discussion
This is the first study to identify shared genetic factors underlying TS and ADHD, two closely related and often co-occurring neuropsychiatric disorders (Karagiannidis et al., 2016). We meta-analyzed 489 of the top hit SNPs in the first TS GWAS, that had also been tested in ADHD published GWASs and meta-analysis. Our own meta-analysis highlights genes that may play a role in the shared etiology between TS and ADHD. 19 SNPs attained in the meta-analysis a p-value lower than the significance threshold, as denoted by the Bonferroni correction approach for multiple testing. All significant SNPs had the same direction of effect, which is indicative of a shared mechanism of disease development. A minority of those had not presented any association with ADHD in the original ADHD meta-analysis, with the resulting combined p-value being driven mostly by the p-value acquired from the TS study.
The five most significant SNPs had achieved moderate association p-values in the original ADHD study, and thus attained high p-values with no heterogeneity-based confounding in our meta-analysis, becoming strong candidates for the shared genomic background of the disorders.
TBC1D7 (TBC1 Domain Family, Member 7) is a prominent gene in our results, with two variants achieving the top and the third best p-values in our study. The top scoring SNP is located in a regulatory region downstream of the gene, while the third top is located within an intron of the gene. The associated variants have demonstrated linkage disequilibrium with a known eQTL for the expression of the gene, further substantiating their implication into the regulation of the expression profile of the gene. Expression profiling in Expression Atlas and GTEx show significant overexpression in the brain, the heart, the testis and in blood cells. The product of the gene is the third subunit of the TSC1-TSC2 complex with a Rheb-GAP activity, and is ubiquitously present in the complex (Dibble et al., 2012). An eQTL for TBC1D7 has been significantly associated with migraine and migraine without aura in a study of 23,285 individuals with migraine and 95,425 population-matched controls (Anttila et al., 2013). Interestingly, Anttila et al (Anttila et al., 2016) also picked up a genetic correlation between TS and migraine.
The presence of TBC1D7 in the TSC1/2 complex creates a suggestive functional link between the proteins. The role of the TSC1/2 complex is indicative of TBC1D7's role in the brain and neuropsychiatric disease, as an important component of the active complex. The TSC1 (Tuberous Sclerosis 1) and TSC2 (Tuberous Sclerosis 2) genes have an important role in the aetiology of Tuberous Sclerosis Complex (TSC). TSC is a neurodevelopmental disorder that typically presents with tumours of the brain, skin, heart, lungs, and kidneys, but also neurological disorders such as epilepsy, cognitive disability and autism. The TSC1/2 complex acts as an inhibitor of the mechanistic target of rapamycin (mTOR) signaling pathway which plays a central role in cell growth, proliferation, autophagy and thus also neurodevelopment (Henske et al., 2016). The TSC pathway regulates neuronal structure and function, and is sensitive to gene-dosage effects, showing degrees of haploinsufficiency (Tavazoie et al., 2005). TSC1 has also been implicated in bipolar disorder, without attaining genome-wide significance (Scott et al., 2009). Furthermore, TSC1 has been shown to have a neuroprotective role in hippocampal regions of the brain, protecting against ischemic events (Papadakis et al., 2013).
RAP1GDS1 (RAP1, GTP-GDP dissociation stimulator 1) is a GDP/GTP exchange protein with GTPase activity (Riess et al., 1993). It is located on chromosome 4 and is the third top locus to be implicated in the shared genetic background, with the associated variant residing in the intron of the gene. It is significantly overexpressed in brain and nervous tissues. RAP1GDS1 has been shown to interact with RHO (Ras homolog gene family, member A), that has also been implicated in this study, in a cascade involving interactions with multiple signaling proteins (Vikis et al., 2002; Berg et al., 2010; Hamel et al., 2011). CHST11 (carbohydrate chondroitin 4 sulfotransferase 11) is involved in the sulfation of chondroitin (Klppel, 2010), which is a key element of the brain matrix (Kwok et al., 2012). It is expressed in areas of the brain, including the hippocampus and the caudate nucleus. GUCY1A3 (Guanylate cyclase soluble subunit alpha-3) functions as the main receptor for nitric oxide, and has been implicated in Moyamoya disease, a disease causing constriction in arteries and brain ischemic events (Wallace et al., 2016).
5. Conclusion
We investigate, for the first time, the common genetic background between TS and ADHD on a genomewide scale and provide evidence that specific genes may underlie both disorders. The implicated variants lie on genes that appear to have a complex interplay between them. The main theme of the results is the Ras signaling cascade in the brain, with TBC1D7 and RAP1GDS1 being key elements of the brain signaling pathways. Interestingly, an additional theme emerging from the data, is related to brain ischemic response, with GUCY1A3 and the TSC1/2 complex (which includes TBC1D7) as implicated as factors. Intriguingly, one of our top hits, TBC1D7, implicates the mTOR signaling pathway and autophagy processes (Dibble et al., 2012). Furthermore, our analysis also points to CHST11, which has been shown to regulate the brain extracellular matrix, by affecting the chondroitin sulfation levels. Therefore, further investigation in the role of the respective genes in the shared genetic aetiology of TS and ADHD is warranted. Our results provide an intriguing insight into the shared mechanism of common neuropsychiatric disorders.
Author Contributions
FT, SP, JA, IK, MT, AT, DM, MG, PD performed data analysis and interpretation, and participated in manuscript writing. PP designed and supervised the study, performed data analysis and interpretation, and participated in manuscript writing. All authors read and approved the final version to be published and agreed to be accountable for all aspects of the work.
Funding
This study was supported by the TS-EUROTRAIN and the EMTICS projects, funded by the European Union under the Seventh Framework People Programme (TS-EUROTRAIN, FP7-PEOPLE-2012-ITN, Grant Agr.No.316978 EMTICS, FP7-HEALTH, Grant Agr.No. 278367).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a current collaboration with one of the authors PP and states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
We would like to thank the Psychiatric Genetics Consortium, the Tourette Syndrome Association International Consortium for Genetics and the TS/OCD working group of the Psychiatric Genetics Consortium.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2016.00340
References
1000 Genomes Project Consortium, Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
Anttila, V., Bulik-Sullivan, B., Finucane, H. K., Bras, J., Duncan, L., Escott-Price, V., et al. (2016). Analysis of shared heritability in common disorders of the brain. bioRxiv. doi: 10.1002/ajmg.b.32323. [Epub ahead of print].
Anttila, V., Winsvold, B. S., Gormley, P., Kurth, T., Bettella, F., McMahon, G., et al. (2013). Genome-wide meta-analysis identifies new susceptibility loci for migraine. Nat. Genet. 45, 912–917. doi: 10.1038/ng.2676
Berg, T. J., Gastonguay, A. J., Lorimer, E. L., Kuhnmuench, J. R., Li, R., Fields, A. P., et al. (2010). Splice variants of SmgGDS control small gtpase prenylation and membrane localization. J. Biol. Chem. 285, 35255–35266. doi: 10.1074/jbc.M110.129916
Cross-Disorder Group of the Psychiatric Genomics Consortium (2013). Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379. doi: 10.1016/S0140-6736(12)62129-1
Dibble, C. C., Elis, W., Menon, S., Qin, W., Klekota, J., Asara, J. M., et al. (2012). TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 47, 535–546. doi: 10.1016/j.molcel.2012.06.009
Hamel, B., Monaghan-Benson, E., Rojas, R. J., Temple, B. R. S., Marston, D. J., Burridge, K., et al. (2011). SmgGDS is a guanine nucleotide exchange factor that specifically activates RhoA and RhoC. J. Biol. Chem. 286, 12141–12148. doi: 10.1074/jbc.M110.191122
Henske, E. P., Jóźwiak, S., Kingswood, J. C., Sampson, J. R., and Thiele, E. A. (2016). Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2:16035. doi: 10.1038/nrdp.2016.35
Karagiannidis, I., Tsetsos, F., Padmanabhuni, S. S., Alexander, J., Georgitsi, M., and Paschou, P. (2016). The genetics of Gilles de la Tourette syndrome: a common aetiological basis with comorbid disorders? Curr. Behav. Neurosci. Rep. 1–14. doi: 10.1007/s40473-016-0088-z
Klppel, M. (2010). The roles of chondroitin-4-sulfotransferase-1 in development and disease. Prog. Mol. Biol. Transl. Sci. 93, 113–132. doi: 10.1016/S0140-6736(12)62129-1
Kwok, J. C. F., Warren, P., and Fawcett, J. W. (2012). Chondroitin sulfate: a key molecule in the brain matrix. Int. J. Biochem. Cell Biol. 44, 582–586. doi: 10.1016/j.biocel.2012.01.004
Lesch, K. P., Timmesfeld, N., Renner, T. J., Halperin, R., Röser, C., Nguyen, T. T., et al. (2008). Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J. Neural Transm. 115, 1573–1585. doi: 10.1007/s00702-008-0119-3
Machiela, M. J., and Chanock, S. J. (2014). LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557. doi: 10.1093/bioinformatics/btv402
McLaren, W., Pritchard, B., Rios, D., Chen, Y., Flicek, P., and Cunningham, F. (2010). Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26, 2069–2070. doi: 10.1093/bioinformatics/btq330
Mick, E., Todorov, A., Smalley, S., Hu, X., Loo, S., Todd, R. D., et al. (2010). Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 49, 898–905.e3. doi: 10.1016/j.jaac.2010.02.014
Neale, B. M., Lasky-Su, J., Anney, R., Franke, B., Zhou, K., Maller, J. B., et al. (2008). Genome-wide association scan of attention deficit hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1337–1344. doi: 10.1002/ajmg.b.30866
Neale, B. M., Medland, S., Ripke, S., Anney, R. J. L., Asherson, P., Buitelaar, J., et al. (2010a). Case-control genome-wide association study of attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 49, 906–920. doi: 10.1016/j.jaac.2010.06.007
Neale, B. M., Medland, S. E., Ripke, S., Asherson, P., Franke, B., Lesch, K.-P., et al. (2010b). Meta-analysis of genome-wide association studies of attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 49, 884–897. doi: 10.1016/j.jaac.2010.06.008
Papadakis, M., Hadley, G., Xilouri, M., Hoyte, L. C., Nagel, S., McMenamin, M. M., et al. (2013). Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat. Med. 19, 351–357. doi: 10.1038/nm.3097
Paschou, P., Yu, D., Gerber, G., Evans, P., Tsetsos, F., Davis, L. K., et al. (2014). Genetic association signal near NTN4 in Tourette syndrome. Ann. Neurol. 76, 310–315. doi: 10.1002/ana.24215
Petryszak, R., Burdett, T., Fiorelli, B., Fonseca, N. A., Gonzalez-Porta, M., Hastings, E., et al. (2014). Expression Atlas update - A database of gene and transcript expression from microarray- and sequencing-based functional genomics experiments. Nucleic Acids Res. 42, D926–D932. doi: 10.1093/nar/gkt1270
Riess, O., Epplen, C., Siedlaczck, I., and Epplen, J. T. (1993). Chromosomal assignment of the human smg GDP dissociation stimulator gene to human chromosome 4q21-q25. Hum. Genet. 92, 629–630. doi: 10.1007/BF00420952
Robertson, M. M. (2006). Attention deficit hyperactivity disorder, tics and Tourette's syndrome: the relationship and treatment implications. A commentary. Eur. Child Adolesc. Psychiatry 15, 1–11. doi: 10.1007/s00787-006-0505-z
Robertson, M. M., Cavanna, A. E., and Eapen, V. (2015). Gilles de la Tourette syndrome and disruptive behavior disorders: prevalence, associations, and explanation of the relationships. J. Neuropsychiatry Clin. Neurosci. 27, 33–41. doi: 10.1176/appi.neuropsych.13050112
Robertson, M. M., Eapen, V., and Cavanna, A. E. (2009). The international prevalence, epidemiology, and clinical phenomenology of Tourette syndrome: a cross-cultural perspective. J. Psychosom. Res. 67, 475–483. doi: 10.1016/j.jpsychores.2009.07.010
Scharf, J. M., Miller, L. L., Gauvin, C. A., Alabiso, J., Mathews, C. A., and Ben-Shlomo, Y. (2015). Population prevalence of Tourette syndrome: a systematic review and meta-analysis. Mov. Disord. 30, 221–228. doi: 10.1002/mds.26089
Scharf, J. M., Yu, D., Mathews, C. A., Neale, B. M., Stewart, S. E., Fagerness, J. A., et al. (2013). Genome-wide association study of Tourette's syndrome. Mol. Psychiatry 18, 721–728. doi: 10.1038/mp.2012.69
Scott, L. J., Muglia, P., Kong, X. Q., Guan, W., Flickinger, M., Upmanyu, R., et al. (2009). Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc. Natl. Acad. Sci. U.S.A. 106, 7501–7506. doi: 10.1073/pnas.0813386106
Tavazoie, S. F., Alvarez, V. A., Ridenour, D. A., Kwiatkowski, D. J., and Sabatini, B. L. (2005). Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734. doi: 10.1038/nn1566
The GTEx Consortium (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585. doi: 10.1038/ng.2653
Vikis, H. G., Stewart, S., and Guan, K.-L. (2002). SmgGDS displays differential binding and exchange activity towards different Ras isoforms. Oncogene 21, 2425–2432. doi: 10.1038/sj.onc.1205306
Wallace, S., Guo, D.-C., Regalado, E., Mellor-Crummey, L., Bamshad, M., Nickerson, D. A., et al. (2016). Disrupted nitric oxide signaling due to GUCY1A3 mutations increases risk for moyamoya disease, achalasia and hypertension. Clin. Genet. doi: 10.1111/cge.12739. [Epub ahead of print].
Keywords: Tourette Syndrome, ADHD, meta-analysis, cross-disorder, TBC1D7, GUCY1A3, RAP1GDS1, CHST11
Citation: Tsetsos F, Padmanabhuni SS, Alexander J, Karagiannidis I, Tsifintaris M, Topaloudi A, Mantzaris D, Georgitsi M, Drineas P and Paschou P (2016) Meta-Analysis of Tourette Syndrome and Attention Deficit Hyperactivity Disorder Provides Support for a Shared Genetic Basis. Front. Neurosci. 10:340. doi: 10.3389/fnins.2016.00340
Received: 31 March 2016; Accepted: 06 July 2016;
Published: 22 July 2016.
Edited by:
Kirsten R. Müller-Vahl, Hannover Medical School, GermanyReviewed by:
Thomas V. Fernandez, Yale School of Medicine, USASuhash Chakraborty, Hindustan Aeronautics Limited Hospital, India
Copyright © 2016 Tsetsos, Padmanabhuni, Alexander, Karagiannidis, Tsifintaris, Topaloudi, Mantzaris, Georgitsi, Drineas and Paschou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peristera Paschou, cHBhc2Nob3VAbWJnLmR1dGguZ3I=