 Daniel J. Shepherd
Daniel J. Shepherd Shih-Yen Tsai2
Shih-Yen Tsai2- 1Neuroscience Institute, Loyola University Chicago Health Sciences Division, Maywood, IL, USA
- 2Research Service, Edward Hines Jr. VA Hospital, Hines, IL, USA
- 3Department of Mathematics and Statistics, Loyola University Chicago, Chicago, IL, USA
- 4Department of Molecular Pharmacology and Therapeutics, Loyola University Chicago Health Sciences Division, Maywood, IL, USA
Ischemic stroke is a leading cause of adult disability, including cognitive impairment. Our laboratory has previously shown that treatment with function-blocking antibodies against the neurite growth inhibitory protein Nogo-A promotes functional recovery after stroke in adult and aged rats, including enhancing spatial memory performance, for which the hippocampus is critically important. Since spatial memory has been linked to hippocampal neurogenesis, we investigated whether anti-Nogo-A treatment increases hippocampal neurogenesis after stroke. Adult rats were subject to permanent middle cerebral artery occlusion followed 1 week later by 2 weeks of antibody treatment. Cellular proliferation in the dentate gyrus was quantified at the end of treatment, and the number of newborn neurons was determined at 8 weeks post-stroke. Treatment with both anti-Nogo-A and control antibodies stimulated the accumulation of new microglia/macrophages in the dentate granule cell layer, but neither treatment increased cellular proliferation or the number of newborn neurons above stroke-only levels. These results suggest that anti-Nogo-A immunotherapy does not increase post-stroke hippocampal neurogenesis.
Introduction
Cognitive impairment is a recognized sequela of ischemic stroke (Gottesman and Hillis, 2010). Our laboratory has previously shown that treatment with function-blocking antibodies against the neurite growth-inhibitory protein Nogo-A (anti-Nogo-A immunotherapy) improves spatial memory performance after stroke in aged rats (Gillani et al., 2010), but a cellular mechanism of efficacy has not yet been identified. We and others have previously demonstrated that anti-Nogo-A immunotherapy stimulates dendritic and axonal remodeling and increases dendritic spine density in the contralesional sensorimotor cortex after stroke (Papadopoulos et al., 2002, 2006; Wiessner et al., 2003; Seymour et al., 2005; Tsai et al., 2007, 2011; Lindau et al., 2014). These neuroplastic changes may underlie the sensorimotor recovery seen in anti-Nogo-A treated animals (Papadopoulos et al., 2002, 2006; Wiessner et al., 2003; Seymour et al., 2005; Tsai et al., 2007, 2011; Lindau et al., 2014; reviewed by Kumar and Moon, 2013), as silencing of newly sprouted axonal connections ablates the sensorimotor recovery promoted by anti-Nogo-A treatment (Wahl et al., 2014). However, no changes in dendritic complexity or spine density were found in anti-Nogo-A-treated animals in pyramidal neurons of CA1 or CA3 or in dentate granule cells, despite spatial memory improvement, suggesting an alternate mechanism of efficacy (Gillani et al., 2010). We and other groups have likewise reported that anti-Nogo-A treatment enhances recovery from hemispatial neglect after aspiration lesion of the medial agranular cortex (Brenneman et al., 2008) and recovery of cognitive function after traumatic brain injury (Lenzlinger et al., 2005; Marklund et al., 2007), positioning Nogo-A as a promising therapeutic target for improving cognition after brain injury.
Nogo-A is a transmembrane protein with two main inhibitory domains (Nogo-66 and Nogo-A-Δ20), and acts primarily by activating two different cell surface receptors. Nogo-66 binds to the Nogo receptor NgR1, leading to activation of the small GTPase RhoA and subsequent activation of Rho-associated protein kinase (ROCK). Nogo-A-Δ20 activates the previously characterized sphingosine-1-phosphate receptor S1PR2, which also activates RhoA/ROCK and may also influence gene expression. Both receptors have been found to play a role in mediating structural and synaptic plasticity (Schwab and Strittmatter, 2014).
Several studies have linked hippocampal neurogenesis and spatial memory performance on the Morris water maze (reviewed by Garthe and Kempermann, 2013), and interventions that increase neurogenesis have also been shown to improve Morris water maze performance after brain injury, including stroke (Wurm et al., 2007; Meng et al., 2014). Whether Nogo-A plays a direct role in adult hippocampal neurogenesis is unknown. However, a previous study reported that mice deficient for the Nogo receptor NgR1 exhibit increased hippocampal neurogenesis and reduced cognitive impairment after traumatic brain injury (Tong et al., 2013). Furthermore, at the molecular level, the key Nogo-A signaling mediators RhoA and ROCK play a suppressive role in hippocampal neurogenesis (Keung et al., 2011; Christie et al., 2013; reviewed by Vadodaria and Jessberger, 2013). Nogo-A signaling has also been shown to inhibit nerve growth factor-mediated CREB phosphorylation in vitro (Joset et al., 2010), whereas CREB phosphorylation is important for the maturation and survival of newborn dentate granule cells, including after stroke (Zhu et al., 2004; Jagasia et al., 2009). These studies raise the question of whether antibody-mediated Nogo-A neutralization could lead to alterations in neurogenesis, which may in turn contribute to cognitive recovery after stroke.
The goal of this study was to determine whether Nogo-A neutralization enhanced post-stroke hippocampal neurogenesis. Our results showed that while infusion of both anti-Nogo-A and control antibodies led to the accumulation of new microglia/macrophages in the hippocampus, Nogo-A neutralization did not affect the number of newborn neurons in the dentate gyrus after stroke. Therefore, enhanced neurogenesis is unlikely to contribute to the improvement in spatial memory that we previously reported after stroke and anti-Nogo-A immunotherapy.
Materials and Methods
Animal Subjects
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Hines Veterans Affairs Hospital. A total of 42 adult male Long-Evans black hooded rats (Harlan, Indianapolis, IN), 12 weeks of age at study initiation, were used. See Table 1 for an overview of experimental design. Animals were housed in pairs on a 12 h light-dark cycle with ad lib food and water.
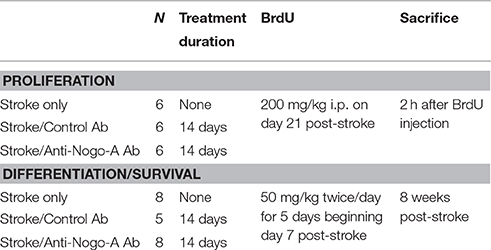
Table 1. Overview of experimental groups.
Middle Cerebral Artery Occlusion
Rats were anesthetized with 2% isoflurane in oxygen. Distal middle cerebral artery occlusion was performed as described previously (Chen et al., 1986; Papadopoulos et al., 2002). An incision through the scalp and temporalis muscle was made, followed by a craniotomy to expose the middle cerebral artery (MCA). The left MCA was then ligated with 10-0 suture and bisected. After making a midline ventral neck incision, the left common carotid artery (CCA) was permanently ligated with 4-0 suture, and the right CCA was occluded for 1 h using an aneurysm clip. Body temperature was maintained at 37°C throughout the procedure by a thermoregulator and heating pad. After incisions were closed, animals were allowed to recover in their home cage. Sham surgery animals were anesthetized for an equivalent duration and given neck and scalp incisions.
Anti-Nogo-A Treatment Antibody Production and Purification
The hybridoma cell line for the mouse monoclonal anti-Nogo-A antibody 11C7 was provided by Prof. Martin Schwab (Brain Research Institute, University of Zurich). The cells were grown in Hybridoma-SFM (Gibco, Waltham, MA) using the CELLLine multi-chamber cell cultivation system (BD Biosciences, San Jose, CA) according to manufacturer's protocol. The 11C7 antibody was purified from antibody-containing medium by Protein-G column chromatography (Pierce, Waltham, MA). Coomassie blue staining of purified antibody separated on denaturing polyacrylamide gels routinely showed only two bands corresponding to heavy and light chains. For infusion, purified 11C7 was diluted to 2.5 mg/mL in sterile phosphate-buffered saline.
Intracerebroventricular Antibody Treatment
One week following stroke (a delay in treatment that still improves functional recovery, Seymour et al., 2005; Gillani et al., 2010), rats were anesthetized with isoflurane and implanted with a subcutaneous osmotic minipump (Alzet model 2ML2; Durect Corporation, Cupertino, CA) connected to a cannula leading to the ipsilesional lateral cerebral ventricle, as previously done. Either anti-Nogo-A mouse IgG1 (antibody 11C7) or a control antibody raised against a non-mammalian peptide (anti-cyclosporine A; mouse IgG1, a generous gift from Novartis International AG; Craveiro et al., 2013), both 2.5 μg/μL, were infused at a rate of 5 μL/h (as previously done, Markus et al., 2005; Gillani et al., 2010) for 14 days. At the end of the treatment period, pumps were removed under isoflurane anesthesia.
5-Bromo-2′-Deoxyuridine (BrdU) Injections
BrdU (Sigma-Aldrich Co., St. Louis, MO) was dissolved at 20 mg/mL in sterile saline plus 0.007 N NaOH and sterilized by passing through a 0.22 μm syringe filter. Rats were injected intraperitoneally according to one of two injection schedules. To measure cellular proliferation, rats were injected with a single dose of 200 mg/kg body weight BrdU (a saturating dose, even in animals with increased hippocampal neurogenesis; Eadie et al., 2005) and killed 2 h after injection. For long-term phenotype analysis of proliferating cells, rats were injected with 50 mg/kg BrdU twice per day for 5 consecutive days, beginning 7 days after stroke.
Tissue Collection and Preparation
Animals were euthanized by overdose with Euthasol (390 mg/kg i.p., Virbac, Fort Worth, TX) and transcardially perfused with cold heparinized saline followed by 4% paraformaldehyde (PFA). Brains were extracted and post-fixed overnight in 4% PFA, cryoprotected in 30% sucrose until sinking, and embedded in OCT on dry ice. 40 μm sections were cut using a Leica CM1850 cryostat and stored in cryoprotectant solution at −20°C until use.
Histology
For BrdU immunostaining, tissue sections were mounted on plus-charged slides, dried at room temperature overnight, and then immersed in 99–100°C 10 mM sodium citrate pH 6 for 15 min (Tang et al., 2007). Slides were then placed in sodium phosphate buffer (PB) and sections carefully removed from the slides using a razor blade, allowing subsequent staining steps to be performed free-floating. Sections were then incubated in primary antibodies diluted in PB pH 7.4 plus 0.2% Tween 20 overnight at 4°C with gentle agitation. After extensive washing in PB/0.2% Tween-20, tissue was incubated in secondary antibody (conjugated to either biotin or fluorophores) diluted in PB/0.2% Tween-20 for 2 h at room temperature (see Table 2 for a list of antibodies and dilutions used in this study). S1PR2 immunostaining was detected by avidin-biotin peroxidase complex (VectaStain Elite ABC kit; Vector Laboratories, Burlingame, CA) followed by AlexaFluor 568 tyramide signal amplification (Thermo Fisher T20949) per manufacturer's instructions. For fluorescence microscopy, nuclei were counterstained with DAPI. For chromogenic detection, sections incubated in biotinylated secondary antibody were then incubated in avidin-biotin complex (Vector Laboratories) for 1 h and reacted in nickel-enhanced 3,3′diaminobenzidine (DAB, Sigma-Aldrich Co.). Fluorescent immunostained tissue was mounted on gelatin-subbed slides and coverslipped with Fluoromount G mounting media (Southern Biotech, Birmingham, AL). DAB tissue was mounted on gelatin-subbed slides, dehydrated in graded ethanols, cleared in xylene, and coverslipped with Permount (Fisher Scientific, Waltham, MA).
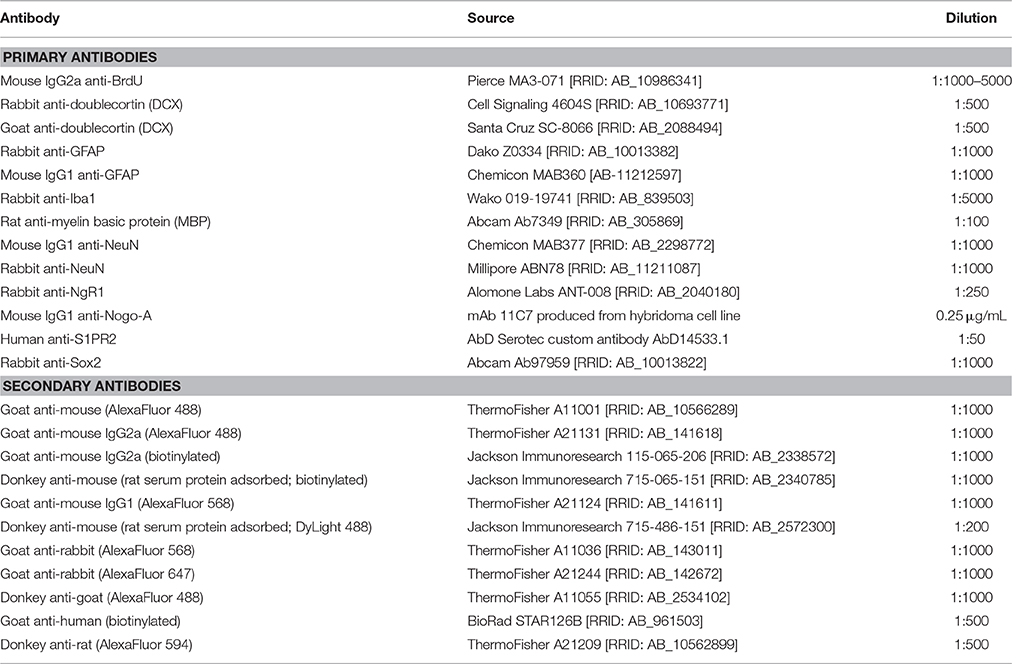
Table 2. Antibodies used for immunofluorescence and immunohistochemistry.
Quantification
All quantification was performed by an investigator blind to experimental group.
Cellular Proliferation
Six 40 μm sections per subject (n = 6 per group) encompassing the dorsal DG (every 12th section beginning at the rostral appearance of the dentate granule cell layer, between −2 and −4.8 mm with respect to bregma; Paxinos and Watson, 1998) were immunostained for BrdU and examined using bright-field microscopy on a Leica DM4000B microscope with a 40x/0.75 NA objective. Exhaustive cell counts were performed by manually counting all BrdU+ nuclei in the subgranular zone (SGZ) and basal layers of the granule cell layer (GCL) (within approximately 3 nuclei from the interface between the dentate granule cell layer and polymorphic layer) of the dorsal hippocampus bilaterally. Cell counts were multiplied by 12 to estimate the total number of proliferating cells.
BrdU+ Cell Counts at 8 Weeks Post-stroke
Six 40 μm sections (every 12th section beginning at the rostral appearance of the dentate granule cell layer; stroke/control antibody: n = 5; stroke only and stroke/anti-Nogo-A antibody: n = 8) were stained for BrdU, lightly counterstained with toluidine blue to identify the GCL, mounted, and coverslipped. BrdU+ nuclei within the dorsal DG GCL of each section were exhaustively counted using a 40x/0.75 NA objective and multiplied by 12 to estimate the total number of BrdU+ cells. Counts were then normalized to GCL volume using Cavalieri's principle (see below).
Measurement of GCL Volume
The toluidine blue-stained tissue sections used for measuring total BrdU+ cells at 8 weeks post-stroke (6 sections total per subject) were imaged using MBF StereoInvestigator software. The Cavalieri Estimator probe was applied to measure GCL area and estimate the total volume of the GCL within the dorsal DG encompassed by the six sections.
Quantification of Newborn Cell Phenotypes
A total of three 40 μm sections per subject (every 24th section beginning at the rostral appearance of the GCL [bregma −2 mm] and proceeding caudally) were stained for BrdU plus NeuN, Iba1, or Sox2, counterstained with DAPI, and examined on a Leica SPE confocal microscope using a 63x/1.3 NA oil immersion objective.
Due to the dense cellularity of the GCL and poor penetration of the NeuN antibody that confounded co-expression analysis in the middle of the tissue section, analysis of BrdU/NeuN co-labeling was restricted to near the outer surfaces of the tissue where NeuN expression was unambiguous. Approximately 50 cells per dentate gyrus per side were examined in each subject. Workflow was as follows: BrdU-positive cells were identified by first scanning the tissue with the appropriate excitation laser until positive nuclei within the GCL were identified. Then a single optical section was acquired with 1 Airy unit pinhole size, and channels merged to identify (1) total BrdU+ cells, and (2) the number of BrdU+ cells that were positive for either NeuN, Iba1, or Sox2. When co-labeling was not clear from a single optical section, z-stacks were acquired to disambiguate the labels.
Estimates of the total numbers of new neurons were calculated by multiplying the total number of BrdU+ cells by the proportion of BrdU+ cells expressing each marker.
Treatment Antibody Distribution and Fluorescence Intensity
Infused treatment antibody was detected using either a chromogen (DAB) or a fluorescent secondary antibody. For chomogenic detection, sections were incubated in a biotinylated anti-mouse IgG secondary antibody (rat serum protein adsorbed) overnight at 4°C (1:1000 in sodium phosphate buffer plus 0.3% Triton X100), followed by incubation in avidin-biotin peroxidase complex (Vector) and reaction in nickel-enhanced DAB. For visualization of the reaction product, the staining intensities of scanned tissue sections were then remapped in ImageJ (Schindelin et al., 2012) using the “Fire” look up table.
For fluorescence intensity analysis, three tissue sections through the dorsal DG (a 1 in 24 series) from an untreated, 7-day treated (3 subjects each from control antibody and anti-Nogo-A groups) and 8 weeks post-stroke (3 subjects each from control antibody and anti-Nogo-A groups) were washed in sodium phosphate buffer (PB) and incubated in DyLight-488-conjugated donkey anti-mouse (rat serum protein adsorbed) secondary antibody (Jackson Immunoresearch, West Grove, PA; 1:200 in PB/0.3% Triton X100) for 90 min at room temperature. Sections were washed in PB and then mounted on gelatin-subbed slides and coverslipped in Fluoromount G. Z stacks through the entire thickness of each tissue section were acquired using a 10x objective on a Leica SPE confocal microscope at equivalent parts of the DG in each tissue section. All image acquisition settings were kept constant. Image stacks were imported into ImageJ and compressed to maximum intensity Z projections. The mean gray value of the tissue was then measured in each section using ImageJ and averaged to yield a single intensity value for each hippocampus per subject. The tissue from the untreated (stroke-only) subject was used to determine background fluorescence, which is a combination of tissue autofluorescence and any potential non-specific binding of the fluorescent anti-mouse secondary antibody.
Lesion Analysis
For each subject, a 1 in 24 tissue section series throughout each brain (excluding olfactory bulbs and cerebellum) was mounted on gelatin-subbed slides and stained with toluidine blue. Slides were then scanned at high resolution using a flatbed scanner and imported into Adobe Photoshop CS3, where the number of pixels in the intact and lesioned hemispheres was measured. To compute a lesion size as a percentage of the intact hemisphere, the total number of pixels in the lesioned hemisphere was subtracted from the total number of pixels in the intact hemisphere, and divided by the total intact hemisphere pixel number. This method therefore calculates the size of the missing, lesioned tissue.
Statistics
Statistical analysis was performed using Minitab 17 and SAS 9.4 software. When appropriate, data were analyzed using one-way ANOVA, equal-variance t-tests or paired t-tests; when assumptions were not supported, unequal-variance ANOVA or t-tests or non-parametric tests (Wilcoxon rank-sum or Kruskal-Wallis) were used. Details regarding significance testing for each experiment can be found in Supplementary Table 1.
Results
Nogo-A Is Expressed by Immature Neurons in the Normal Adult Dentate Gyrus (DG)
To determine whether neural precursor cells in the subgranular zone and granule cell layer (GCL) of the DG may be potential direct cellular targets of anti-Nogo-A immunotherapy, we performed double-label immunofluorescent staining using the Nogo-A-specific antibody 11C7 and antibodies to cell type-specific markers (Figures 1, 2). Strong Nogo-A expression was found in immature (doublecortin [DCX]-positive) neurons in various stages of development. Both radially-oriented (more mature) cells with more complex arborizations (Figure 2A) and tangentially-oriented (transitioning, less mature progenitors) (Figure 2B; Kempermann et al., 2004) were positive for Nogo-A. Nogo-A expression was especially enriched in the apical dendrites of radially-oriented DCX+ cells.
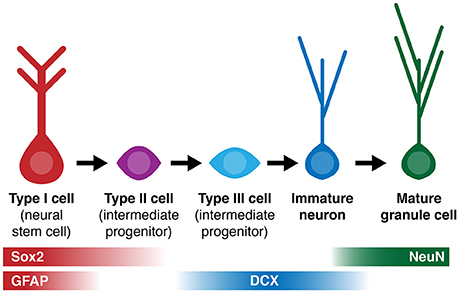
Figure 1. Simplified diagram of cell lineage progression and stage-specific expression of markers (Sox2, GFAP, DCX, NeuN) referenced in this study.
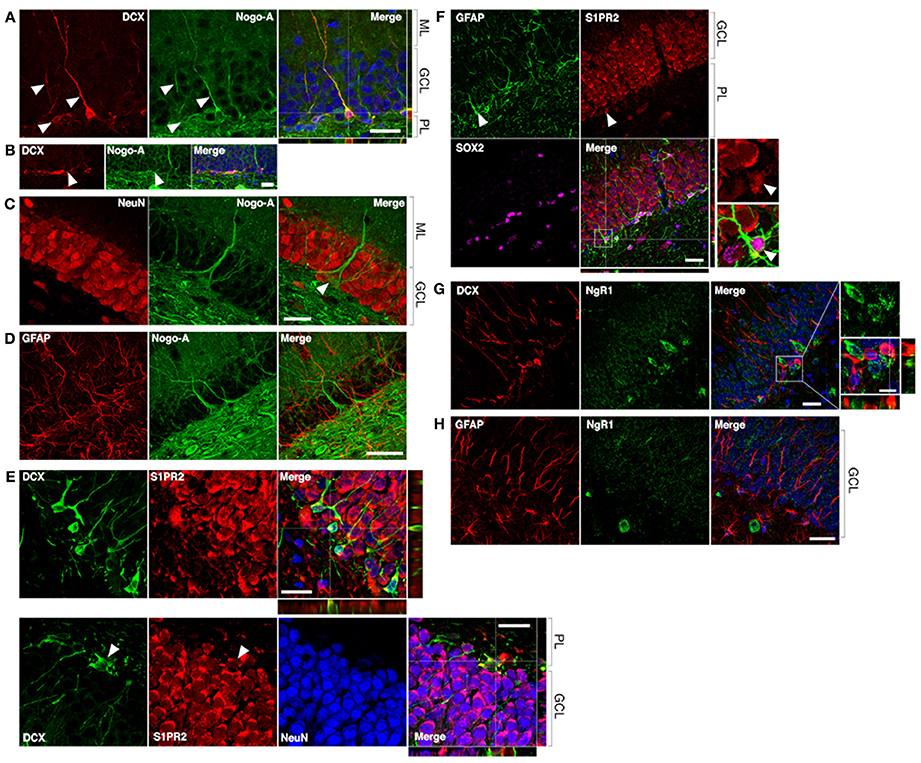
Figure 2. Nogo-A is expressed by immature neurons in the adult dentate gyrus. (A) Nogo-A is expressed in the processes and somata of immature neurons (arrowheads), which are also positive for doublecortin (DCX). Scale bar: 25 μm. (B) Horizontally-oriented DCX+/Nogo-A+ neuroblast. Scale bar: 25 μm. (C) Nogo-A expression is not appreciable in NeuN+ mature granule cells, the majority of NeuN+ cells in the GCL. However, putative basket cells (arrow) label strongly for Nogo-A. Scale bar: 25 μm. (D) Nogo-A immunoreactivity is not detectable in GFAP+ stem cells or astrocytes. Scale bar: 50 μm. (E) S1PR2 is broadly expressed in the GCL, including DCX+ immature neurons and NeuN+ mature neurons. Scale bar: 20 μm. (F) S1PR2 expression by GFAP+/Sox2+ neural stem cells in the SGZ. Scale bar: 25 μm. (G) Lack of NgR1 expression by DCX+ immature neurons. Scale bars: 25 μm; 10 μm (inset). (H) Lack of NgR1 expression by GFAP+ neural stem cells in the SGZ. Scale bar: 25 μm. ML, molecular layer; GCL, granule cell layer; PL, polymorphic layer.
In contrast, Nogo-A expression by mature dentate granule cells within the GCL was not appreciable by immunofluorescence (Figrue 2C), consistent with a previous report (Huber et al., 2002), suggesting transient expression of Nogo-A during the development of adult-born dentate granule cells. Strong Nogo-A expression was observed in large, pyramidal NeuN+ cells at the GCL/polymorphic layer interface (putative basket cells) (Figure 2C, arrowhead), while Nogo-A was not detectable in GFAP+ putative stem cells or astrocytes of the subgranular zone (Figure 2D).
A recently identified receptor for the Nogo-A Δ20 domain, sphingosine-1-phosphate receptor 2 (Kempf et al., 2014), was found to be widely expressed in the DG GCL (as reported by Akahoshi et al., 2011), including in the cell bodies of DCX+ cells (Figure 2E, top and bottom panels). S1PR2 staining typically did not occupy the entirety of the DCX+ cell bodies, possibly suggesting targeting to distinct subcellular domains. Mature NeuN+ granule cell bodies were likewise positive for S1PR2 (Figure 2E, bottom panel; arrowheads: additional DCX/S1PR2 co-expression). GFAP+/Sox2+ cells located in the subgranular zone (putative stem cells) appeared to be weakly S1PR2 positive relative to the stronger S1PR2 expression seen in mature dentate granule cells (Figure 2F).
Lastly, we examined the expression of the Nogo-66 receptor NgR1 in hippocampal neural precursor cells. Throughout the dentate GCL and SGZ, NgR1 expression was observed primarily in a punctate pattern, with more distinctly labeled cell bodies less frequently seen. We found no clear evidence of NgR1 expression by either DCX+ immature neurons or GFAP+ astrocytes or stem cells in the SGZ (Figures 2G,H).
Lesion Size Is Not Affected by Antibody Treatment
As the size of the stroke lesion may affect neurogenesis, we measured lesion sizes at both 21 and 56 days post-stroke. Stroke lesions in all experimental groups were unilateral and similar in location, encompassing the dorsolateral cortex and extending from primary motor cortex rostrally through auditory and visual cortices caudally (Figure 3A). Little to no infarction of the underlying white matter or subcortical structures was evident, consistent with previous observations using this model (Gillani et al., 2010). At all-time points, the hippocampus was grossly intact upon brain cryosectioning, but occasionally appeared distorted on the side ipsilateral to the stroke lesion, possibly due to distention of the cerebral ventricles. Lesion sizes were not different among the three treatment groups at either time point assessed (Figure 3B).
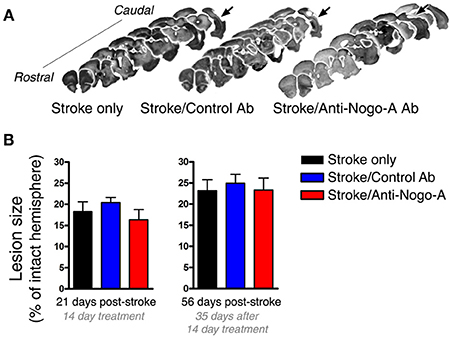
Figure 3. Lesion size is not affected by antibody treatment. (A) Representative images of lesion (arrows) location and size from each experimental group at 8 weeks post-stroke. (B) Lesion size did not differ among groups at either 21 or 56 days post-stroke.
Infused Treatment Antibody Penetrates the Hippocampus
Treatment antibodies penetrated into the hippocampal parenchyma as assessed by immunostaining for mouse IgG after 3 days of treatment (Papadopoulos et al., 2002; Weinmann et al., 2006; Tsai et al., 2007; Figures 4A,B). Treatment antibody was detected in the hippocampus after 3, 7, and 14 days of treatment. Five weeks after pump removal (7 weeks after treatment initiation), both control and anti-Nogo-A antibodies appeared to have been substantially cleared, and were no longer detectable by immunofluorescence above background levels in the DG (Figure 4C).
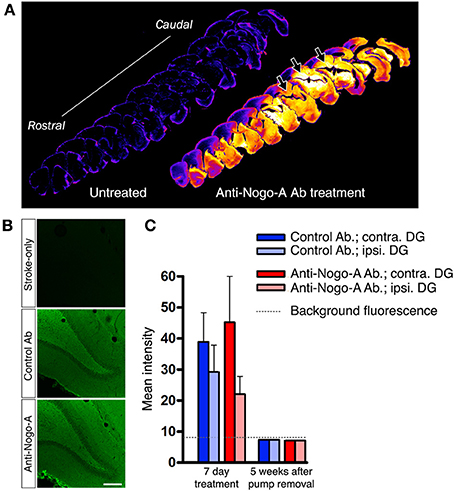
Figure 4. Infused treatment antibody penetrates hippocampus. (A) Low magnification images of anti-mouse IgG immunostaining in a stroke-only, untreated subject (left; negative control) and an anti-Nogo-A-treated subject after 3 days of treatment (right). Staining intensities have been remapped, where brighter/hotter colors represent increased signal intensity. Abundant treatment antibody can be seen in the hippocampus (arrows). (B) Immunofluorescence staining for mouse IgG after 3 days of antibody infusion shows diffuse, uniform penetration of anti-Nogo-A and control antibodies in the dentate gyrus (DG), whereas only background fluorescence is evident in untreated MCAO rats. Scale bar: 200 μm. (C) Quantification of mean fluorescence intensity shows expected elevated signal intensity after 7 days of treatment, whereas at 5 weeks after treatment cessation, the signal is not detectable above background fluorescence by direct immunofluorescence. Contra., contralesional; Ipsi., ipsilesional; DG, Dentate gyrus.
Anti-Nogo-A Treatment Does Not Alter Cellular Proliferation in the Subgranular Zone (SGZ)
Cellular proliferation was measured after 14 days of treatment (i.e., at 21 days post-stroke) by injecting rats with a single dose of BrdU and euthanizing 2 h later (Figures 5A,B). The number of proliferating cells in anti-Nogo-A-treated subjects was not significantly different vs. stroke-only or control antibody-treated controls in either the ipsilesional or contralesional SGZ (Figure 5C; Table 3).
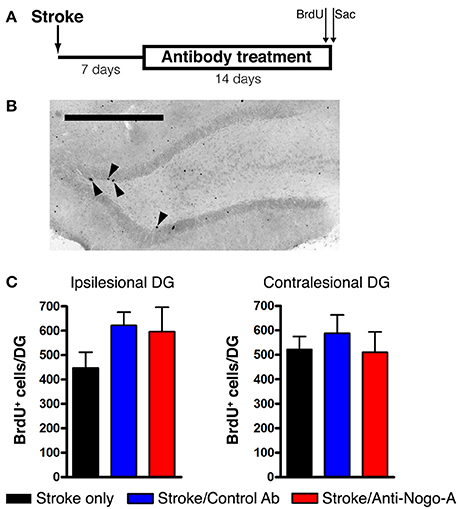
Figure 5. Cellular proliferation in the subgranular zone and basal granule cell layer is not altered by anti-Nogo-A treatment after stroke. (A) Overview of BrdU injection strategy to measure cellular proliferation. (B) Representative image of BrdU+ nuclei (arrowheads) in the ipsilesional DG of a stroke-only subject at 21 days post-stroke. Scale bar: 500 μm. (C) Total numbers of BrdU+ nuclei in the SGZ and basal GCL of the ipsilesional (left) and contralesional (right) DG. Error bars indicate SEM.

Table 3. Cellular proliferation in the subgranular zone after stroke and anti-Nogo-A immunotherapy.
Both Control Antibody and Anti-Nogo-A Antibody Treatment Stimulate the Accumulation of New Microglia/Macrophages, but Not New Neurons, in the Dentate Granule Cell Layer (GCL)
To analyze the phenotypes of newborn cells in the GCL, rats were administered multiple injections of BrdU beginning 7 days after stroke and euthanized for analysis 7 weeks thereafter (Figure 6A). Stroke itself led to a significant increase in the total number of BrdU-positive cells (i.e., cells that had proliferated between days 7 and 11 post-stroke and survived approximately 6–7 weeks thereafter) in the ipsilesional vs. the contralesional GCL (Figures 6B,C; Supplementary Table 2). In both control antibody and anti-Nogo-A treatment groups, more BrdU+ cells were found in the GCL compared to the stroke-only group, but were also more generally distributed throughout the DG (Figures 6B,C). As in the stroke-only group, both antibody-treated groups showed higher numbers of BrdU+ cells in the ipsilesional vs. contralesional GCL (Figure 6C). However, the proportion of BrdU+ cells co-labeled for NeuN (i.e., new neurons) in the GCL was lower in antibody-treated groups (Figure 6F; Supplementary Table 3), such that the total numbers of newborn neurons was not statistically different among groups (Figure 6G; Table 4).
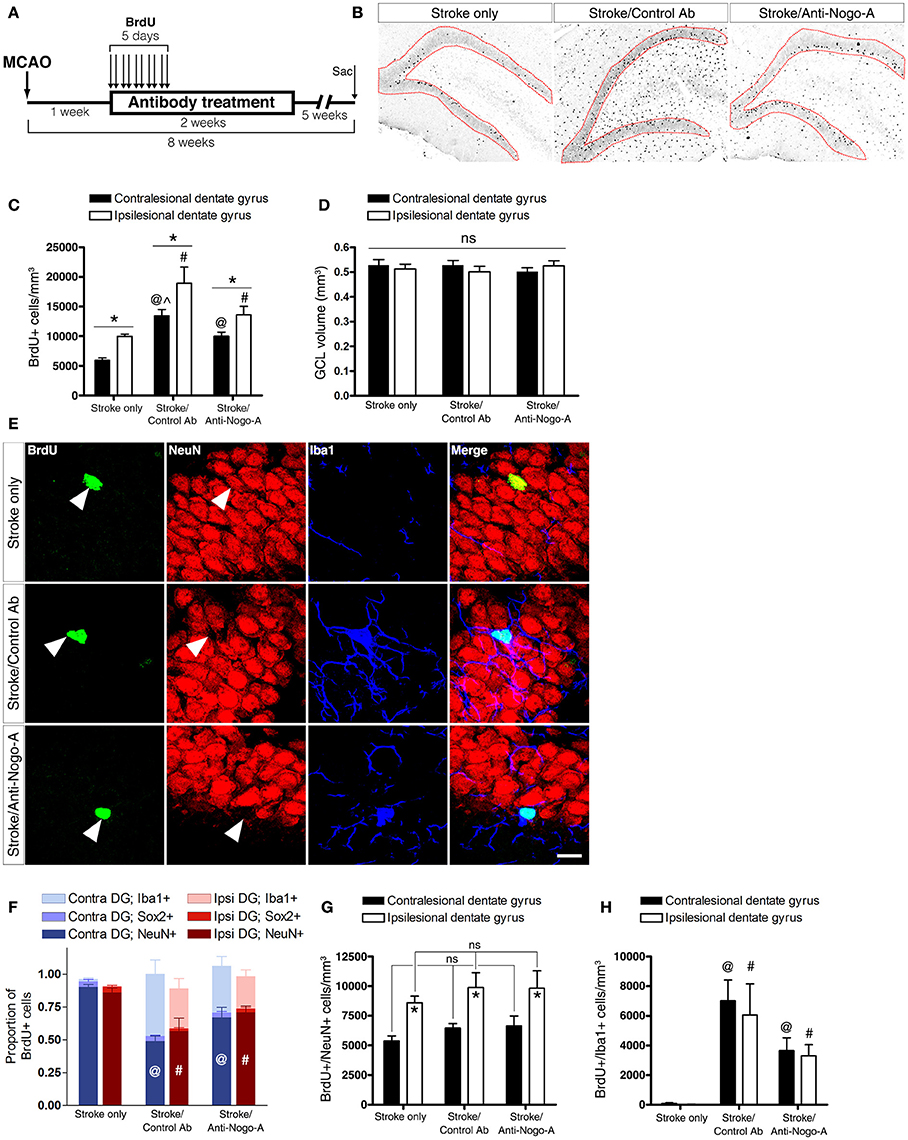
Figure 6. Both anti-Nogo-A and control antibody treatment induce long-lasting accumulation of new microglia/macrophages without altering neurogenesis. (A) Overview of BrdU injection strategy to measure the differentiation and survival of proliferating cells. (B) Representative images of BrdU immunoreactivity in the ipsilesional dentate gyrus at 8 weeks post-stroke. Newborn cells are evident throughout the DG in both control and anti-Nogo-A antibody groups. The granule cell layer, where cells were counted, is outlined in red. (C) Total BrdU+ nuclei in the contralesional (black bars) and ipsilesional (white bars) GCLs. *p < 0.05, ipsilesional vs. contralesional DG (within treatment group); @p < 0.05, vs. stroke-only contralesional DG; #p < 0.05, vs. stroke-only ipsilesional DG; ∧p < 0.05, vs. stroke/anti-Nogo-A contralesional GCL. (D) The volume of the GCL in which BrdU+ nuclei were counted (in panel “C”) was not significantly different among groups. (E) Representative image of newborn neurons (BrdU+/NeuN+) and microglia/macrophages (BrdU+/Iba1+) in the GCL. Scale bar: 10 μm. (F) Proportions of newborn cells of each phenotype (neuron [NeuN+], microglia/macrophage [Iba1+], neural stem/progenitor cell or astrocyte [Sox2+]). @p < 0.05, vs. stroke-only contralesional DG NeuN+ proportion; #p < 0.05, vs. stroke-only ipsilesional DG NeuN+ proportion. (G) Total number of new neurons in the GCL. *p < 0.05, ipsilesional vs. contralesional DG (within treatment group). (H) Total number of new Iba1+ microglia/macrophages in the GCL. @p < 0.05, vs. stroke-only contralesional DG; #p < 0.05, vs. stroke-only ipsilesional DG. All error bars indicate SEM.
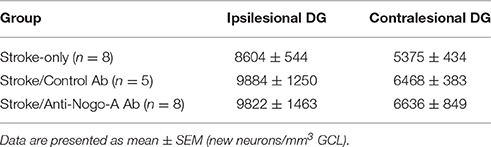
Table 4. Total numbers of newborn neurons in the dentate granule cell layer (GCL) at 8 weeks post-stroke.
The total volume of the GCL in the area of cell counting in each subject was estimated using Cavalieri's principle. No significant differences were found either among groups or within groups between the ipsilesional and contralesional GCLs when compared at 8 weeks post-stroke (Figure 6D).
Nearly all BrdU+/NeuN– cells in both control antibody and anti-Nogo-A treatment groups were positive for Iba1 (Figures 6E,H; Supplementary Table 4), identifying these cells as microglia/macrophages. Resident microglia were found in the DG of untreated rats, as previously reported (reviewed by Gemma and Bachstetter, 2013), but only rarely had incorporated BrdU.
At 8 weeks post-stroke, only a small percentage (approximately 2–5%) of BrdU+ cells in each group was positive for Sox2, which labels neural stem cells, early intermediate progenitors, and mature astrocytes in the adult rat brain (Komitova and Eriksson, 2004; Yu et al., 2014).
Discussion
Anti-Nogo-A immunotherapy improves spatial memory after stroke in aged rats, but a cellular mechanism of efficacy has not been identified (Gillani et al., 2010). This study was conducted to determine whether Nogo-A neutralization enhances post-stroke neurogenesis in the dentate gyrus.
We first performed multiple-label immunofluorecent staining to determine whether Nogo-A is expressed by neural precursor cells in the adult DG, thereby identifying possible direct treatment targets. Nogo-A was found to be expressed by doublecortin (DCX)-positive immature neurons, but not stem cells or mature dentate granule cells. To our knowledge, this is the first report of Nogo-A expression in immature neurons of the adult dentate gyrus. This transient expression suggests a stage-specific role of Nogo-A expression in adult hippocampal neuronal development, similar to what has been reported in the adult subventricular zone (Rolando et al., 2012) and during embryonic and early post-natal development (Huber et al., 2002; Aloy et al., 2007; Mingorance-Le Meur et al., 2007; Mathis et al., 2010; Schwab, 2010). Notably, per many of these reports, Nogo-A is expressed by migratory neurons. While we do not directly address the normal physiological role of cell surface and/or intracellular Nogo-A in DG neurogenesis here, we may infer from these previous studies that Nogo-A could play a role in migration of neuronal precursors in the adult DG (Deng et al., 2010; Sun et al., 2015) or in the morphogenesis of new DG neurons (Petrinovic et al., 2013; Kurowska et al., 2014).
Expression of the recently identified receptor for the Nogo-A Δ20 domain, S1PR2, was broadly observed in the dentate granule cell layer, including by immature (DCX+) and mature dentate granule cells. Qualitatively, staining appeared to be stronger in the mature granule cells. Therefore, it is possible that upregulation of S1PR2 begins at the immature neuron stage and persists throughout maturation. Furthermore, we found evidence of S1PR2 expression in GFAP+/Sox2+ cells in the SGZ (putative neural stem cells).
In contrast, NgR1 expression was not clearly seen in neural precursors in the SGZ/DG. We observed punctate NgR1 labeling throughout the GCL, possibly indicative of synaptic localization, as NgR1 has been localized both pre- and post-synaptically (Lee et al., 2008). Cell bodies labeled distinctly for NgR1 were seen infrequently in the GCL, but these cells did not co-express either DCX or GFAP, suggesting against NgR1 expression by immature neurons or neural stem cells.
Examining treatment antibody distribution, we showed that intracerebroventricularly infused antibody entered the hippocampal parenchyma, but was undetectable by immunofluorescence 5 weeks after cessation of treatment. Therefore, direct exposure of target tissue to infused antibody is transient. As we did not analyze antibody distribution at earlier time points after treatment cessation, we cannot conclude that complete antibody clearance requires the full 5 weeks. However, our findings are in line with a previous report noting a reduction in anti-Nogo-A antibody in the brain parenchyma just 1 week after the end of treatment (Marklund et al., 2007). These results raise the possibility that rapid clearance of the antibody from the brain may limit the full potential of anti-Nogo-A antibodies to promote functional recovery, and that a longer treatment duration may be further clinically beneficial.
Despite Nogo-A expression by DCX+ immature neurons (as seen in fixed tissue sections; Figure 2A), we were unable to discern by immunofluorescent histology whether the infused anti-Nogo-A treatment antibody had bound to this cell type in vivo. Therefore, it is unclear whether Nogo-A is expressed at the surface of immature neurons in the DG. While Nogo-A is expressed both intracellularly and at the surface of several cell types, including oligodendrocytes and dorsal root ganglion neurons (Caroni and Schwab, 1988; Dodd et al., 2005), Nogo-A was found to be intracellular in a human dopaminergic neuron line (Kurowska et al., 2014). Therefore, subcellular localization of Nogo-A may be cell type-specific. Regardless, intracellular localization of Nogo-A in DCX+ immature neurons would not preclude an indirect effect of anti-Nogo-A treatment antibody on their function. For example, antibody neutralization of surface Nogo-A could block Nogo-A signaling to immature neurons from neighboring cells or myelin.
After inducing a large cortical stroke followed 1 week later by 2 weeks of antibody treatment, we measured the number of proliferating cells in the SGZ. Anti-Nogo-A treatment did not significantly alter the number of proliferating cells in either the ipsilesional or contralesional subgranular zone (SGZ) and basal granule cell layer (GCL) of the dorsal DG. Furthermore, we found no evidence of earlier, transient effects after either 3 or 7 days of treatment (data not shown), suggesting that neural precursor proliferation is unaffected by surface Nogo-A neutralization.
We then investigated the types of cells that were produced after stroke and survived long-term. We found that the proportion of long-lived newborn cells that were positive for NeuN (i.e., new neurons) was approximately 86–90% in the stroke-only group, similar to findings in a previous report (Kluska et al., 2005). Given the increase in total BrdU+ cells in the ipsilesional GCL, this indicates a significant increase in the number of new neurons in the ipsilesional vs. contralesional DG. This result is consistent with reports of increased hippocampal neurogenesis in numerous animal models of stroke, including transient global ischemia (Liu et al., 1998; Kee et al., 2001), transient middle cerebral artery occlusion (Jin et al., 2001; Zhu et al., 2003, 2004), photothrombotic cortical stroke (Kluska et al., 2005), and distal middle cerebral artery occlusion (Matsumori et al., 2006). In contrast, only a small number of BrdU+ cells at 8 weeks post-stroke were Sox2-positive, indicating relatively scant production of new, long-lived neural stem cells and astrocytes.
Both control antibody- and anti-Nogo-A-treated groups exhibited robust accumulation of new Iba1-positive microglia/macrophages in the GCL. In contrast, newborn microglia/macrophages were found very rarely in the GCL of stroke-only subjects. Several potential mechanisms behind the observed accumulation of new microglia/macrophages may be considered. First, the absence of differences in lesion size between treated and untreated groups argues against a direct effect of the lesion itself. Cannulae for antibody delivery are implanted in the lateral cerebral ventricle, and may in rare cases puncture the hippocampal fimbria. However, the fact that BrdU+ cells were generally elevated bilaterally and more uniformly distributed, rather than clustered around a cannula track, makes it unlikely that the observed response was a reaction to mechanical injury. On the other hand, infusion of mouse antibody into the rat CNS could potentially induce a microglial/macrophage response through either recognition of the antibody as a foreign protein, or binding and activation of microglia/macrophage-expressed Fc receptors. Antibody immunogenicity in human patients should be reduced by the use of human antibodies (Nelson et al., 2010), which are currently in use in anti-Nogo-A clinical trials for spinal cord injury and have so far shown an encouraging safety profile (Zörner and Schwab, 2010). While to our knowledge direct demonstration of rat FcR-mouse IgG binding has not been demonstrated, cross-species FcR binding has been reported between more phylogenetically distant species (Lubeck et al., 1985), and FcR cross linking has been shown to stimulate macrophage proliferation (Luo et al., 2010).
The mechanism responsible for improved spatial memory after stroke and anti-Nogo-A treatment is not yet fully understood. While our previous work did not find an effect of anti-Nogo-A treatment on dendritic complexity in CA1, CA3, or DG GCL neurons, a subsequent study noted dendritic alterations in these subfields after acute treatment of hippocampal slice cultures with anti-Nogo-A antibody (Zagrebelsky et al., 2010). These changes were evident after just 4 days of antibody treatment, a much shorter time course than in our previous study, in which histological analysis was performed 10 weeks after the end of treatment. Therefore, it is possible that in vivo anti-Nogo-A antibody treatment after stroke leads to rapid changes in dendritic growth that may be pruned back over time (Andres et al., 2011).
Intriguingly, several studies have shown that Nogo-A and its receptors NgR1 and S1PR2 can regulate cognitive function and synaptic plasticity. Transgenic Nogo-A knockdown rats exhibit subtle spatial memory deficits in certain tasks (Petrasek et al., 2014a,b), while mice overexpressing NgR1 show impaired spatial memory performance in the Morris water maze (Karlsson et al., 2016), suggesting that the proper balance of Nogo-A signaling, including during development, is necessary for optimal cognitive function. These effects may also depend on whether Nogo-A signaling perturbation is chronic (as in the case of Nogo-A- or NgR1-transgenic animals), or acute (after neutralizing antibody or blocking peptide treatment). For example, CA3-CA1 long-term potentiation (LTP) was unaffected by null mutation of NgR1 (in the absence of FGF2) (Lee et al., 2008), whereas acute application of an NgR1 blocking antibody enhanced LTP (Delekate et al., 2011). However, both Nogo-A knockdown rats (Tews et al., 2013) and acute hippocampal slices treated with anti-Nogo-A antibodies (Delekate et al., 2011; Kellner et al., 2016) exhibited enhanced CA3-CA1 LTP, suggesting different roles of the ligand (Nogo-A) and receptor (NgR1) in the proper development and function of hippocampal circuitry. Given these findings, it is possible that Nogo-A neutralization improves spatial memory after stroke through a mechanism involving enhanced synaptic plasticity.
Lastly, other properties related to newborn neuron function that we did not examine, including connectivity, synaptogenesis, or morphogenesis, rather than the total number of newborn neurons, may be altered by Nogo-A neutralization. The Nogo receptor NgR1 negatively regulates synaptogenesis and dendritic complexity during hippocampal development (Wills et al., 2012), raising the possibility of a similar role in adult hippocampal neurogenesis. Future studies examining these changes in adult-born neurons after anti-Nogo-A treatment may be enlightening.
In conclusion, our results suggest that anti-Nogo-A immunotherapy does not significantly alter hippocampal neurogenesis after focal cortical stroke in adult rats. We cannot rule out that treatment may induce differences in neurogenesis specifically in aged rats, which were used in our previous study showing efficacy of anti-Nogo-A treatment in improving spatial memory after stroke. However, the present results suggest that different mechanisms outside of enhanced neurogenesis are more likely to underlie this recovery. These results add to our understanding of the scope and limitations of anti-Nogo-A immunotherapy, which are vitally important as anti-Nogo-A antibodies continue to be used in human clinical trials.
Author Contributions
Designed experiments: DS, GK. Performed experiments: DS, ST, RF. Analyzed and interpreted data: DS, ST, TO, GK. Wrote the manuscript: DS, ST, TO, RF, GK.
Funding
This work was supported by grant 5I01RX000828 from the US Department of Veterans Affairs Rehabilitation Research and Development service to GK, and American Heart Association 15PRE24470136 to DS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Novartis for providing the anti-cyclosporine A control antibody used in this study, and Prof. Martin Schwab for providing the 11C7 hybridoma cell line and for helpful comments regarding this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fnins.2016.00467
References
Akahoshi, N., Ishizaki, Y., Yasuda, H., Murashima, Y. L., Shinba, T., Goto, K., et al. (2011). Frequent spontaneous seizures followed by spatial working memory/anxiety deficits in mice lacking sphingosine 1-phosphate receptor 2. Epilepsy Behav. 22, 659–665. doi: 10.1016/j.yebeh.2011.09.002
Aloy, E. M., Weinmann, O., Pot, C., Kasper, H., Dodd, D. A., Rülicke, T., et al. (2007). Synaptic destabilization by neuronal Nogo-A. Brain Cell Bio 35, 137–157. doi: 10.1007/s11068-007-9014-3
Andres, R. H., Horie, N., Slikker, W., Keren-Gill, H., Zhan, K., Sun, G., et al. (2011). Human neural stem cells enhance structural plasticity and axonal transport in the ischaemic brain. Brain 134, 1777–1789. doi: 10.1093/brain/awr094
Brenneman, M. M., Wagner, S. J., Cheatwood, J. L., Heldt, S. A., Corwin, J. V., Reep, R. L., et al. (2008). Nogo-A inhibition induces recovery from neglect in rats. Behav. Brain Res. 187, 262–272. doi: 10.1016/j.bbr.2007.09.018
Caroni, P., and Schwab, M. E. (1988). Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron 1, 85–96. doi: 10.1016/0896-6273(88)90212-7
Chen, S. T., Hsu, C. Y., Hogan, E. L., Maricq, H., and Balentine, J. D. (1986). A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke 17, 738–743. doi: 10.1161/01.STR.17.4.738
Christie, K. J., Turbic, A., and Turnley, A. M. (2013). Adult hippocampal neurogenesis, Rho kinase inhibition and enhancement of neuronal survival. Neuroscience 247, 75–83. doi: 10.1016/j.neuroscience.2013.05.019
Craveiro, L. M., Weinmann, O., Roschitzki, B., Gonzenbach, R. R., Zörner, B., Montani, L., et al. (2013). Infusion of anti-Nogo-A antibodies in adult rats increases growth and synapse related proteins in the absence of behavioral alterations. 250C, 52–68. doi: 10.1016/j.expneurol.2013.09.015
Delekate, A., Zagrebelsky, M., Kramer, S., Schwab, M. E., and Korte, M. (2011). NogoA restricts synaptic plasticity in the adult hippocampus on a fast time scale. Proc. Natl. Acad. Sci. U.S.A. 108, 2569–2574. doi: 10.1073/pnas.1013322108
Deng, W., Aimone, J. B., and Gage, F. H. (2010). New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat. Rev. Neurosci. 11, 339–350. doi: 10.1038/nrn2822
Dodd, D. A., Niederoest, B., Bloechlinger, S., Dupuis, L., Loeffler, J.-P., and Schwab, M. E. (2005). Nogo-A, -B, and -C are found on the cell surface and interact together in many different cell types. J. Biol. Chem. 280, 12494–12502. doi: 10.1074/jbc.M411827200
Eadie, B. D., Redila, V. A., and Christie, B. R. (2005). Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dendritic complexity, and spine density. J. Comp. Neurol. 486, 39–47. doi: 10.1002/cne.20493
Garthe, A., and Kempermann, G. (2013). An old test for new neurons: refining the Morris water maze to study the functional relevance of adult hippocampal neurogenesis. Front. Neurosci. 7:63. doi: 10.3389/fnins.2013.00063
Gemma, C., and Bachstetter, A. D. (2013). The role of microglia in adult hippocampal neurogenesis. Front. Cell. Neurosci. 7:229. doi: 10.3389/fncel.2013.00229
Gillani, R. L., Tsai, S.-Y., Wallace, D. G., O'Brien, T. E., Arhebamen, E., Tole, M., et al. (2010). Cognitive recovery in the aged rat after stroke and anti-Nogo-A immunotherapy. Behav. Brain Res. 208, 415–424. doi: 10.1016/j.bbr.2009.12.015
Gottesman, R. F., and Hillis, A. E. (2010). Predictors and assessment of cognitive dysfunction resulting from ischaemic stroke. Lancet Neurol. 9, 895–905. doi: 10.1016/S1474-4422(10)70164-2
Huber, A. B., Weinmann, O., Brösamle, C., Oertle, T., and Schwab, M. E. (2002). Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J. Neurosci. 22, 3553–3567.
Jagasia, R., Steib, K., Englberger, E., Herold, S., Faus-Kessler, T., Saxe, M., et al. (2009). GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J. Neurosci. 29, 7966–7977. doi: 10.1523/JNEUROSCI.1054-09.2009
Jin, K., Minami, M., Lan, J. Q., Mao, X. O., Batteur, S., Simon, R. P., et al. (2001). Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. U.S.A. 98, 4710–4715. doi: 10.1073/pnas.081011098
Joset, A., Dodd, D. A., Halegoua, S., and Schwab, M. E. (2010). Pincher-generated Nogo-A endosomes mediate growth cone collapse and retrograde signaling. J. Cell Biol. 188, 271–285. doi: 10.1083/jcb.200906089
Karlsson, T. E., Smedfors, G., Brodin, A. T. S., Åberg, E., Mattsson, A., Högbeck, I., et al. (2016). NgR1: a tunable sensor regulating memory formation, synaptic, and dendritic plasticity. Cereb. Cortex 26, 1804–1817. doi: 10.1093/cercor/bhw007
Kee, N. J., Preston, E., and Wojtowicz, J. M. (2001). Enhanced neurogenesis after transient global ischemia in the dentate gyrus of the rat. Exp. Brain Res. 136, 313–320. doi: 10.1007/s002210000591
Kellner, Y., Fricke, S., Kramer, S., Iobbi, C., Wierenga, C. J., Schwab, M. E., et al. (2016). Nogo-A controls structural plasticity at dendritic spines by rapidly modulating actin dynamics. Hippocampus 26, 816–831. doi: 10.1002/hipo.22565
Kempermann, G., Jessberger, S., Steiner, B., and Kronenberg, G. (2004). Milestones of neuronal development in the adult hippocampus. Trends Neurosci. 27, 447–452. doi: 10.1016/j.tins.2004.05.013
Kempf, A., Tews, B., Arzt, M. E., Weinmann, O., Obermair, F. J., Pernet, V., et al. (2014). The sphingolipid receptor S1PR2 is a receptor for Nogo-A repressing synaptic plasticity. PLoS Biol. 12:e1001763. doi: 10.1371/journal.pbio.1001763
Keung, A. J., de Juan-Pardo, E. M., Schaffer, D. V., and Kumar, S. (2011). Rho GTPases mediate the mechanosensitive lineage commitment of neural stem cells. Stem Cells 29, 1886–1897. doi: 10.1002/stem.746
Kluska, M. M., Witte, O. W., Bolz, J., and Redecker, C. (2005). Neurogenesis in the adult dentate gyrus after cortical infarcts: effects of infarct location, N-methyl-D-aspartate receptor blockade and anti-inflammatory treatment. Neuroscience 135, 723–735. doi: 10.1016/j.neuroscience.2005.06.082
Komitova, M., and Eriksson, P. S. (2004). Sox-2 is expressed by neural progenitors and astroglia in the adult rat brain. Neurosci. Lett. 369, 24–27. doi: 10.1016/j.neulet.2004.07.035
Kumar, P., and Moon, L. D. F. (2013). Therapeutics targeting Nogo-A hold promise for stroke restoration. CNS Neurol. Disord. Drug Targets 12, 200–208. doi: 10.2174/1871527311312020006
Kurowska, Z., Brundin, P., Schwab, M. E., and Li, J. Y. (2014). Intracellular Nogo-A facilitates initiation of neurite formation in mouse midbrain neurons in vitro. Neuroscience 256, 456–466. doi: 10.1016/j.neuroscience.2013.10.029
Lee, H., Raiker, S. J., Venkatesh, K., Geary, R., Robak, L. A., Zhang, Y., et al. (2008). Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J. Neurosci. 28, 2753–2765. doi: 10.1523/JNEUROSCI.5586-07.2008
Lenzlinger, P. M., Shimizu, S., Marklund, N., Thompson, H. J., Schwab, M. E., Saatman, K. E., et al. (2005). Delayed inhibition of Nogo-A does not alter injury-induced axonal sprouting but enhances recovery of cognitive function following experimental traumatic brain injury in rats. Neuroscience 134, 1047–1056. doi: 10.1016/j.neuroscience.2005.04.048
Lindau, N. T., Bänninger, B. J., Gullo, M., Good, N. A., Bachmann, L. C., Starkey, M. L., et al. (2014). Rewiring of the corticospinal tract in the adult rat after unilateral stroke and anti-Nogo-A therapy. Brain 137, 739–756. doi: 10.1093/brain/awt336
Liu, J., Solway, K., Messing, R. O., and Sharp, F. R. (1998). Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J. Neurosci. 18, 7768–7778.
Lubeck, M. D., Steplewski, Z., Baglia, F., Klein, M. H., Dorrington, K. J., and Koprowski, H. (1985). The interaction of murine IgG subclass proteins with human monocyte Fc receptors. J. Immunol. 135, 1299–1304.
Luo, Y., Pollard, J. W., and Casadevall, A. (2010). Fcgamma receptor cross-linking stimulates cell proliferation of macrophages via the ERK pathway. J. Biol. Chem. 285, 4232–4242. doi: 10.1074/jbc.M109.037168
Marklund, N., Bareyre, F. M., Royo, N. C., Thompson, H. J., Mir, A. K., Grady, M. S., et al. (2007). Cognitive outcome following brain injury and treatment with an inhibitor of Nogo-A in association with an attenuated downregulation of hippocampal growth-associated protein-43 expression. J. Neurosurg. 107, 844–853. doi: 10.3171/JNS-07/10/0844
Markus, T. M., Tsai, S.-Y., Bollnow, M. R., Farrer, R. G., O'Brien, T. E., Kindler-Baumann, D. R., et al. (2005). Recovery and brain reorganization after stroke in adult and aged rats. Ann. Neurol. 58, 950–953. doi: 10.1002/ana.20676
Mathis, C., Schröter, A., Thallmair, M., and Schwab, M. E. (2010). Nogo-a regulates neural precursor migration in the embryonic mouse cortex. Cereb. Cortex 20, 2380–2390. doi: 10.1093/cercor/bhp307
Matsumori, Y., Hong, S. M., Fan, Y., Kayama, T., Hsu, C. Y., Weinstein, P. R., et al. (2006). Enriched environment and spatial learning enhance hippocampal neurogenesis and salvages ischemic penumbra after focal cerebral ischemia. Neurobiol. Dis. 22, 187–198. doi: 10.1016/j.nbd.2005.10.015
Meng, Y., Chopp, M., Zhang, Y., Liu, Z., An, A., Mahmood, A., et al. (2014). Subacute intranasal administration of tissue plasminogen activator promotes neuroplasticity and improves functional recovery following traumatic brain injury in rats. PLoS ONE 9:e106238. doi: 10.1371/journal.pone.0106238
Mingorance-Le Meur, A., Zheng, B., Soriano, E., and del Río, J. A. (2007). Involvement of the myelin-associated inhibitor Nogo-A in early cortical development and neuronal maturation. Cereb. Cortex 17, 2375–2386. doi: 10.1093/cercor/bhl146
Nelson, A. L., Dhimolea, E., and Reichert, J. M. (2010). Development trends for human monoclonal antibody therapeutics. Nat. Rev. Drug Discov. 9, 767–774. doi: 10.1038/nrd3229
Papadopoulos, C. M., Tsai, S.-Y., Alsbiei, T., O'Brien, T. E., Schwab, M. E., and Kartje, G. L. (2002). Functional recovery and neuroanatomical plasticity following middle cerebral artery occlusion and IN-1 antibody treatment in the adult rat. Ann. Neurol. 51, 433–441. doi: 10.1002/ana.10144
Papadopoulos, C. M., Tsai, S.-Y., Cheatwood, J. L., Bollnow, M. R., Kolb, B. E., Schwab, M. E., et al. (2006). Dendritic plasticity in the adult rat following middle cerebral artery occlusion and Nogo-a neutralization. Cereb. Cortex 16, 529–536. doi: 10.1093/cercor/bhi132
Paxinos, G., and Watson, C. (1998). The Rat Brain in Stereotaxic Coordinates, 4th Edn. San Diego, CA: Academic Press.
Petrasek, T., Prokopova, I., Bahnik, S., Schönig, K., Berger, S., Vales, K., et al. (2014a). Nogo-A downregulation impairs place avoidance in the Carousel maze but not spatial memory in the Morris water maze. Neurobiol. Learn. Mem. 107, 42–49. doi: 10.1016/j.nlm.2013.10.015
Petrasek, T., Prokopova, I., Sladek, M., Weissova, K., Vojtechova, I., Bahnik, S., et al. (2014b). Nogo-A-deficient transgenic rats show deficits in higher cognitive functions, decreased anxiety, and altered circadian activity patterns. Front. Behav. Neurosci. 8:90. doi: 10.3389/fnbeh.2014.00090
Petrinovic, M. M., Hourez, R., Aloy, E. M., Dewarrat, G., Gall, D., Weinmann, O., et al. (2013). Neuronal Nogo-A negatively regulates dendritic morphology and synaptic transmission in the cerebellum. Proc. Natl. Acad. Sci. U.S.A. 110, 1083–1088. doi: 10.1073/pnas.1214255110
Rolando, C., Parolisi, R., Boda, E., Schwab, M. E., Rossi, F., and Buffo, A. (2012). Distinct roles of Nogo-a and Nogo receptor 1 in the homeostatic regulation of adult neural stem cell function and neuroblast migration. J. Neurosci. 32, 17788–17799. doi: 10.1523/JNEUROSCI.3142-12.2012
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schwab, M. E. (2010). Functions of Nogo proteins and their receptors in the nervous system. Nat. Rev. Neurosci. 11, 799–811. doi: 10.1038/nrn2936
Schwab, M. E., and Strittmatter, S. M. (2014). Nogo limits neural plasticity and recovery from injury. Curr. Opin. Neurobiol. 27, 53–60. doi: 10.1016/j.conb.2014.02.011
Seymour, A. B., Andrews, E. M., Tsai, S.-Y., Markus, T. M., Bollnow, M. R., Brenneman, M. M., et al. (2005). Delayed treatment with monoclonal antibody IN-1 1 week after stroke results in recovery of function and corticorubral plasticity in adult rats. J. Cereb. Blood Flow Metab. 25, 1366–1375. doi: 10.1038/sj.jcbfm.9600134
Sun, G. J., Zhou, Y., Stadel, R. P., Moss, J., Yong, J. H. A., Ito, S., et al. (2015). Tangential migration of neuronal precursors of glutamatergic neurons in the adult mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 112, 9484–9489. doi: 10.1073/pnas.1508545112
Tang, X., Falls, D. L., Li, X., Lane, T., and Luskin, M. B. (2007). Antigen-retrieval procedure for bromodeoxyuridine immunolabeling with concurrent labeling of nuclear DNA and antigens damaged by HCl pretreatment. J. Neurosci. 27, 5837–5844. doi: 10.1523/JNEUROSCI.5048-06.2007
Tews, B., Schönig, K., Arzt, M. E., Clementi, S., Rioult-Pedotti, M.-S., Zemmar, A., et al. (2013). Synthetic microRNA-mediated downregulation of Nogo-A in transgenic rats reveals its role as regulator of synaptic plasticity and cognitive function. Proc. Natl. Acad. Sci. U.S.A. 110, 6583–6588. doi: 10.1073/pnas.1217665110
Tong, J., Liu, W., Wang, X., Han, X., Hyrien, O., Samadani, U., et al. (2013). Inhibition of Nogo-66 receptor 1 enhances recovery of cognitive function after traumatic brain injury in mice. J. Neurotrauma 30, 247–258. doi: 10.1089/neu.2012.2493
Tsai, S.-Y., Markus, T. M., Andrews, E. M., Cheatwood, J. L., Emerick, A. J., Mir, A. K., et al. (2007). Intrathecal treatment with anti-Nogo-A antibody improves functional recovery in adult rats after stroke. Exp. Brain Res. 182, 261–266. doi: 10.1007/s00221-007-1067-0
Tsai, S.-Y., Papadopoulos, C. M., Schwab, M. E., and Kartje, G. L. (2011). Delayed anti-nogo-a therapy improves function after chronic stroke in adult rats. Stroke 42, 186–190. doi: 10.1161/STROKEAHA.110.590083
Vadodaria, K. C., and Jessberger, S. (2013). Maturation and integration of adult born hippocampal neurons: signal convergence onto small Rho GTPases. Front. Synaptic Neurosci. 5:4. doi: 10.3389/fnsyn.2013.00004
Wahl, A. S., Omlor, W., Rubio, J. C., Chen, J. L., Zheng, H., Schröter, A., et al. (2014). Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science 344, 1250–1255. doi: 10.1126/science.1253050
Weinmann, O., Schnell, L., Ghosh, A., Montani, L., Wiessner, C., Wannier, T., et al. (2006). Intrathecally infused antibodies against Nogo-A penetrate the CNS and downregulate the endogenous neurite growth inhibitor Nogo-A. Mol. Cell. Neurosci. 32, 161–173. doi: 10.1016/j.mcn.2006.03.007
Wiessner, C., Bareyre, F. M., Allegrini, P. R., Mir, A. K., Frentzel, S., Zurini, M., et al. (2003). Anti-Nogo-A antibody infusion 24 hours after experimental stroke improved behavioral outcome and corticospinal plasticity in normotensive and spontaneously hypertensive rats. J. Cereb. Blood Flow Metab. 23, 154–165. doi: 10.1097/01.WCB.0000040400.30600.AF
Wills, Z. P., Mandel-Brehm, C., Mardinly, A. R., McCord, A. E., Giger, R. J., and Greenberg, M. E. (2012). The nogo receptor family restricts synapse number in the developing hippocampus. Neuron 73, 466–481. doi: 10.1016/j.neuron.2011.11.029
Wurm, F., Keiner, S., Kunze, A., Witte, O. W., and Redecker, C. (2007). Effects of skilled forelimb training on hippocampal neurogenesis and spatial learning after focal cortical infarcts in the adult rat brain. Stroke 38, 2833–2840. doi: 10.1161/STROKEAHA.107.485524
Yu, D. X., Marchetto, M. C., and Gage, F. H. (2014). How to make a hippocampal dentate gyrus granule neuron. Development 63, 199–209. doi: 10.1242/dev.096776
Zagrebelsky, M., Schweigreiter, R., Bandtlow, C. E., Schwab, M. E., and Korte, M. (2010). Nogo-A stabilizes the architecture of hippocampal neurons. J. Neurosci. 30, 13220–13234. doi: 10.1523/JNEUROSCI.1044-10.2010
Zhu, D.-Y., Lau, L., Liu, S. H., Wei, J. S., and Lu, Y. M. (2004). Activation of cAMP-response-element-binding protein (CREB) after focal cerebral ischemia stimulates neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. U.S.A. 101, 9453–9457. doi: 10.1073/pnas.0401063101
Zhu, D.-Y., Liu, S. H., Sun, H. S., and Lu, Y. M. (2003). Expression of inducible nitric oxide synthase after focal cerebral ischemia stimulates neurogenesis in the adult rodent dentate gyrus. J. Neurosci. 23, 223–229.
Keywords: stroke, neurogenesis, Nogo-A, immunotherapy, myelin-associated inhibitor
Citation: Shepherd DJ, Tsai S-Y, O'Brien TE, Farrer RG and Kartje GL (2016) Anti-Nogo-A Immunotherapy Does Not Alter Hippocampal Neurogenesis after Stroke in Adult Rats. Front. Neurosci. 10:467. doi: 10.3389/fnins.2016.00467
Received: 03 June 2016; Accepted: 28 September 2016;
Published: 18 October 2016.
Edited by:
Carlos P. Fitzsimons, University of Amsterdam, NetherlandsReviewed by:
Muriel Koehl, French Institute of Health and Medical Research, FranceJuan Manuel Encinas, Achucarro Basque Center for Neuroscience and Ikerbasque, Spain
Copyright © 2016 Shepherd, Tsai, O'Brien, Farrer and Kartje. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel J. Shepherd, ZGpzaGVwaGVyZEBsdWMuZWR1