 Mark W. Logue1,2,3,4
Mark W. Logue1,2,3,4 Daniel Lancour3John Farrell3Irina Simkina3M. Daniele Fallin5
Daniel Lancour3John Farrell3Irina Simkina3M. Daniele Fallin5 Kathryn L. Lunetta4
Kathryn L. Lunetta4 Lindsay A. Farrer3,4,6,7*
Lindsay A. Farrer3,4,6,7*- 1National Center for Posttraumatic Stress Disorder (PTSD), United States Department of Veterans Affairs, Boston Healthcare System, Boston, MA, United States
- 2Department of Psychiatry, Boston University School of Medicine, Boston University, Boston, MA, United States
- 3Biomedical Genetics, Department of Medicine, Boston University School of Medicine, Boston University, Boston, MA, United States
- 4Department of Biostatistics, Boston University School of Public Health, Boston University, Boston, MA, United States
- 5Department of Mental Health, Johns Hopkins Bloomberg School of Public Health, Johns Hopkins University, Baltimore, MD, United States
- 6Departments of Neurology and Ophthalmology, Boston University School of Medicine, Boston University, Boston, MA, United States
- 7Department of Epidemiology, Boston University School of Public Health, Boston, MA, United States
The genetic architecture of late-onset Alzheimer disease (AD) in African Americans (AAs) differs from that in persons of European ancestry. In addition to APOE, genome-wide association studies (GWASs) of AD in AA samples have implicated ABCA7, COBL, and SLC10A2 as AA-AD risk genes. Previously, we identified by whole exome sequencing a small number of AA AD cases and subsequent genotyping in a large AA sample of AD cases and controls association of AD risk with a pair of rare missense variants in AKAP9. In this study, we performed targeted deep sequencing (including both introns and exons) of approximately 100 genes previously linked to AD or AD-related traits in an AA cohort of 489 AD cases and 472 controls to find novel AD risk variants. We observed association with an 11 base-pair frame-shift loss-of-function (LOF) variant in ABCA7 (rs567222111) for which the evidence was bolstered when combined with data from a replication AA cohort of 484 cases and 484 controls (OR = 2.42, p = 0.022). We also found association of AD with a rare 9 bp deletion (rs371245265) located very close to the AKAP9 transcription start site (rs371245265, OR = 10.75, p = 0.0053). The most significant findings were obtained with a rare protective variant in F5 (OR = 0.053, p = 6.40 × 10-5), a gene that was previously associated with a brain MRI measure of hippocampal atrophy, and two common variants in KIAA0196 (OR = 1.51, p<8.6 × 10-5). Gene-based tests of aggregated rare variants yielded several nominally significant associations with KANSL1, CNN2, and TRIM35. Although no associations passed multiple test correction, our study adds to a body of literature demonstrating the utility of examining sequence data from multiple ethnic populations for discovery of new and impactful risk variants. Larger sample sizes will be needed to generate well-powered epidemiological investigations of rare variation, and functional studies are essential for establishing the pathogenicity of variants identified by sequencing.
Introduction
Studies of common genetic variants have identified many gene loci that influence risk of late-onset Alzheimer disease (AD) in persons of European ancestry (EA), most notably the APOE 𝜀2 and 𝜀4 alleles which confer strong protective and deleterious effects, respectively (Saunders et al., 1993; Corder et al., 1994), as well as more than 20 modest effect loci (odds ratios between 1.1 and 1.3) including BIN1, CR1, ABCA7, CLU, PICALM, and the MS4A gene region (Lambert et al., 2013). Extensions of these findings and the contributions of additional loci have emerged from investigations of non-EA cohorts, African Americans (AAs) in particular (Reitz et al., 2013a; Mez et al., 2017). The risk of AD is greater in AAs than EAs, however, paradoxically, 𝜀4 has a weaker effect in AAs than EAs (Farrer et al., 1997; Reitz et al., 2013a). These observations and greater genetic diversity among persons with African ancestry suggest that the genetic architecture for AD includes some variants and loci that differ from EAs. Several genome-wide association studies (GWAS) in AAs (Logue et al., 2011; Kamboh et al., 2012; Reitz et al., 2013a) confirmed the role of several genes identified initially in EAs, most notably APOE and ABCA7. The association peak in ABCA7 is ascribed to different SNPs in EAs (rs4147929) and AAs (rs115550680; Lambert et al., 2013; Reitz et al., 2013a). Gene resequencing studies have revealed multiple rare ABCA7 deletions causing missense loss-of-function (LOF) mutations in EAs (Cukier et al., 2016; N’Songo et al., 2017). Cukier et al. (2016) identified a 44 base pair (bp) frameshift deletion in ABCA7 (rs142076058) that is in linkage disequilibrium (LD) with rs115550680 and thus may be the functional variant underlying the observed association. A recent exome sequencing investigation in an AA cohort of 198 AD cases and 304 controls examined 20 putative AD risk genes implicated by GWAS in EAs, and found nominally (uncorrected) significant associations with two ABCA7 variants (rs3764647 and rs3752239) and with gene-based tests of coding variants in MS4A6A, PTK2B, and ZCWPW1 (N’Songo et al., 2017).
Novel AD loci have been identified in other studies of AA samples. Mez et al. (2017) identified GWAS significant associations with SNPs in COBL (rs112404845) and SLC10A2 (rs16961023) in a GWAS using an informed conditioning approach. A WES study of seven AA cases followed by genotyping using a staged design in AA cohorts containing 422 cases and 394 controls (stage 1) and 1,037 cases and 1,869 controls (stage 2) identified association with two rare AA-specific highly correlated variants in AKAP9, rs144662445 (OR = 2.75) and rs149979685 (OR = 3.61) (Logue et al., 2014).
These studies confirm the utility of examining African-descent samples to identify new AD risk variants in known AD genes as well as novel AD loci. In this study, we performed targeted sequencing in a discovery cohort containing approximately 1,000 AAs to identify new potentially causal variants in risk genes previously implicated in AD risk in AAs (ABCA7, AKAP9, COBL, MS4A6A, PTK2B, SLC10A2, and ZCWPW1) or in AD and related traits in other populations.
Materials and Methods
Samples
The targeted gene sequencing sample included AA subjects primarily from two cohorts: the Multi-Institutional Research on Alzheimer Genetic Epidemiology (MIRAGE, 113 AD cases, 131 controls) Study (Green et al., 2002) and the Genetic and Environmental Risk Factors for Alzheimer Disease Among African Americans (GenerAAtions, 222 AD cases, 190 controls) Study (Logue et al., 2011). MIRAGE is a family-based study of clinic-based AD cases and their first-degree relatives. The GenerAAtions study includes unrelated individuals ascertained through the Henry Ford Health System. In addition, we obtained DNA samples and phenotypic data from the National Cell Repository for Alzheimer Disease (NCRAD) that were aggregated from the Ibadan/Indianapolis (INDY) study (Hendrie et al., 1995; Sahota et al., 1997; Gureje et al., 2006), the African American Alzheimer’s Disease Genetics (AAG) study (Meier et al., 2012), the National Institute on Aging Alzheimer’s Disease Centers (ADC) (Jun et al., 2010), and the National Institute on Aging Late-Onset Alzheimer’s Disease (NIA-LOAD) Family Study (Lee et al., 2008). The Indianapolis/Ibadan study comprises elderly AA residents from Indianapolis (community dwelling and nursing home residents) and African-descent residents of Ibadan, Nigeria. The AAG study and ADC cohort include cases and controls ascertained at more than 30 sites across the United States. The NIA-LOAD Study includes families with multiple AD cases and unaffected members and an independent set of cognitively screened controls.
The discovery cohort included 489 cases and 472 controls from the MIRAGE and GenerAAtions studies supplemented with 154 cases and 151 controls from the AAG and Ibadan studies. The replication cohort consisted of additional samples from the AAG, ADC, Indy/Ibadan, and NIA-LOAD studies (484 AD cases, 484 controls). Characteristics of the discovery and replication cohorts are presented in Table 1. Further details about subject ascertainment and classification, including genetic screening for ancestry mismatches, were reported elsewhere (Reitz et al., 2013a). The diagnosis of AD in all cohorts was made according to established criteria (McKhann et al., 2011) and all controls were screened to be cognitively normal.
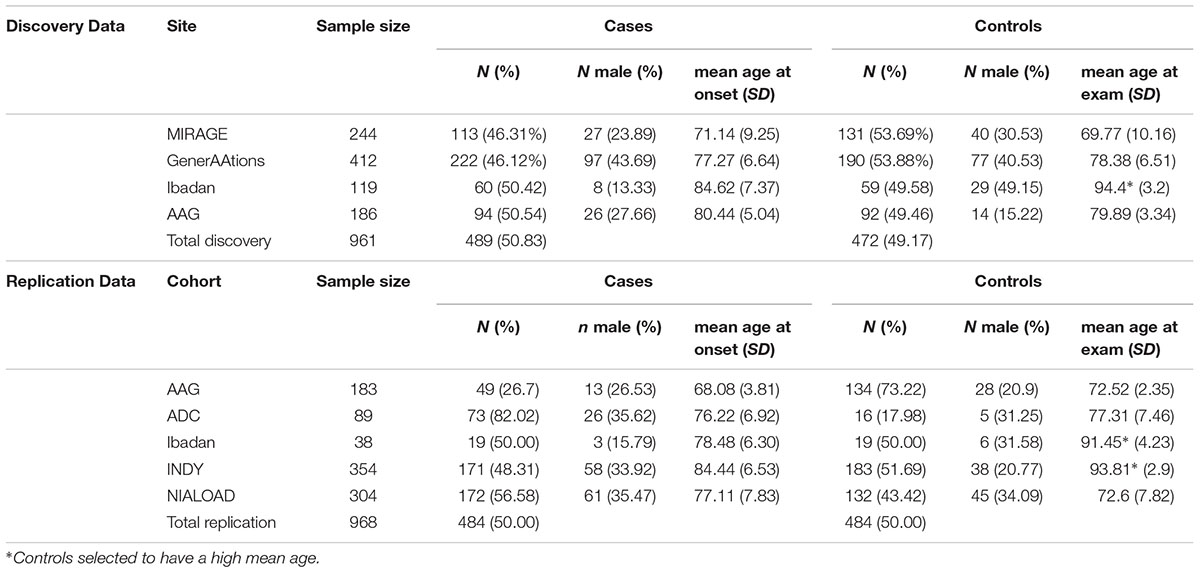
TABLE 1. Sample size and demographics for discovery and replication cohorts.
Sequencing Methods
The samples in the discovery cohort were sent to the McDonnell Genome Institute at Washington University1 for targeted sequencing. The targeted regions included genes previously associated with AD in AAs (ABCA7, AKAP9, COBL, MS4A6A, PTK2B, SLC10A2, and ZCWPW1) and approximately 100 other provisional and confirmed genes and regions that were identified by candidate gene and GWAS approaches in studies of AD and AD-related traits (Saunders et al., 1993; Farrer et al., 2000; Meng et al., 2006; Rogaeva et al., 2007; Vardarajan et al., 2012; Lambert et al., 2013; Reitz et al., 2013b; Jun et al., 2014; Logue et al., 2014; Wetzel-Smith et al., 2014; Jun et al., 2016; Chung et al., 2017; Mez et al., 2017) (Supplementary Table S1). Nimblegen probes (Roche Nimblegen, Madison, WI, United States) were generated to cover all non-repetitive exonic, intronic, and intergenic sequence and 5,000 bp upstream and 1,000 bp downstream of gene boundaries including all isoforms totaling approximately nine Mb of genomic sequence. Only exons were targeted for SLC10A2 and COBL because these associations (Mez et al., 2017) were not known at the time the capture design was proposed and the limited amount of genomic sequence that could be added to the capture at this stage. The capture design included 10,906 capture targets and had 92.7% estimated coverage of 122 targeted regions, with gaps due to repetitive sequence.
Samples were assessed for volume and concentration by the Genome Center using either a Qubit or a VarioSkan assay prior to sequencing. All but eight had >250 ng starting material. The sequencing was done in two waves. The first wave included 667 samples from the MIRAGE and GenerAAtions cohorts. Libraries were captured in sets of 66 and 67 samples per pool and each pool was run in two lanes of an Illumina Hiseq2500 1T platform. The remaining discovery cohort samples were sequenced in the second wave using the same capture probes in pools of 90 samples each, and each sample was run on 2 lanes of an Illumina HiSeq4000 platform. Valid sequence data were available for a discovery cohort including 489 cases and 472 controls. The median number of reads per sample was 14,322,643 (range 6,175,120–25,567,585). The median number of reads was greater for the samples run on the HiSeq4000 platform (median reads/sample for batch 1 = 12,809,719, median reads/sample for batch 2 = 16,804,253). In batch 1, the number of reads/sample for the MIRAGE Study samples (median = 12,331,728) was significantly less than for the GenerAAtions samples (median = 17,217,602, P = 3.51 × 10-5). The number of reads per sample for the second batch of sequencing did not vary by cohort (p = 0.17). Importantly, the number of reads per sample was not associated with AD status in either batch or in the combined discovery sample (all p > 0.3). Across samples, the median percentage of bases with more than 10 reads was 94.60 and the mean coverage depth was 155.7.
Sanger Sequencing
Genotyping for the ABCA7 deletion polymorphism rs567222111 was performed in the replication sample by GENEWIZ (GENEWIZ LLC, South Plainfield NJ, United States2) using bi-directional Sanger sequencing. Sequencing was repeated for samples that did not yield a reliable genotype call in the first run. Validity of the Sanger sequencing assay was demonstrated by verifying genotype calls for 10 samples which had been identified as having the deletion by targeted-sequencing.
Data Processing and Quality Control
The 126 bp paired-end reads were aligned to the GRCh37 +Decoy reference with BWA MEM version 0.7.10-r789. Variant genotypes were jointly called within the targeted regions using the GATK 3.7 pipeline. The “best practices” pipeline included steps for duplicate removal, local realignment near indels, base quality score recalibration, and variant quality score recalibration. GATK yielded calls for 230,595 variants. Annotation of the variants was performed with SnpEff and SnpSift version 4.3i (Cingolani et al., 2012). According to SnpEff, these variants mapped to 151 protein-coding genes. Variants that were not assigned a “PASS” rating by GATK (n = 11,808) were excluded from association analyses. We also excluded variants in the HLA region (n = 24,297) due to difficulties in mapping the repetitive sequence and variants in the APOE region (n = 197) due to difficulties discerning associations in this region that are independent of APOE (Jun et al., 2017). However, we did use sequence calls to derive APOE isoform genotypes for QC purposes (see description below). Another 5,147 variants occurring only in subjects with missing phenotype information were excluded. After these filtering steps, 189,145 variants remained. From this point forward, the pipeline differed for single variant association tests and the gene based tests. For the single variant test, variants observed only once (n = 66,278) were excluded. Genotypes with quality scores <30 were set to missing and variants with a missing rate of >20% were excluded (n = 18,526). After these filtering steps, there remained 104,341 variants for analyses. For the gene based tests, we included singleton variants but excluded variants with a mapping quality of less than 30 (n = 3,748 of 189,145). We excluded variants with minor allele frequency (MAF) in the discovery cohort >5% (32,935). One hundred seventy-three of these variants labeled as “High Impact” according to SNPeff (includes LOF variants and deletions) and 1,079 missense SNPs predicted to be possibly or probably damaging according to Polyphen2 (Adzhubei et al., 2010) were included in the gene-based analyses.
As a quality control check, we compared genotypes for APOE and two rare AKAP9 missense variants (rs144662445 and rs149979685) in MIRAGE cohort subjects that were generated previously by direct genotyping to those derived from targeted sequencing. The two methods agreed for 236 of 237 APOE genotype calls. Among 190 subjects with overlapping genotype and sequencing data for the AKAP9 variants, rare variant calls in three individuals (each with both variants) were concordant.
Statistical Analysis
We applied a hypothesis-driven four-stage design which prioritized variants most likely to have high impact on transcript structure or function in order to minimize the penalty associated with performing more than 100,000 tests. Specifically, variants were selected for analysis if they were (1) predicted to result in loss of function according to the SNPeff annotation, which includes nonsense (stop site) and splice site variants, out of frame deletions/insertions, and large exon-removing deletions (MacArthur et al., 2012), (2) predicted to be a missense variant according to SNPeff, and (3) within 50 base pairs (bp) of transcription start sites (position determined via the Eukaryotic Promotor Database3). We then examined (4) all variants (intronic and exonic) regardless of potential impact. To avoid model instability that can occur with logistic or GEE or mixed models when applied to rare variants, association of AD with individual variants was evaluated using a X2 case:control allele test without continuity correction as implemented in PLINK v1.9 (Chang et al., 2015). For particular variants of interest identified in the allele test, we additionally checked for bias due to relatedness within the MIRAGE cohort as well as potential effects due to population substructure by computing a WALD test using a logistic mixed model in the R GMMAT package (Chen et al., 2016) including as covariates the first three principle components (PCs) for ancestry. The GMMAT package incorporates information from the relationship matrix which we computed from the genetic data in PLINK v1.9 based on 4,569 common (MAF>5%) variants from the sequence data remaining after trimming for LD (plink filter –indep-pairwise 5 20.04). PCs were also computed based on common LD-trimmed SNPs using PLINK. Gene based tests were performed for the 151 protein coding genes (as identified by the SNPeff annotation) using the variable threshold burden test (Price et al., 2010) and the collapsing burden test methods (Li and Leal, 2008) implemented in EPACTS4 which incorporates information about related subjects in the sample. The correlation matrix for related subjects for the gene based test was estimated from the sequence data. LD estimates for 1000 genomes data were obtained using LDlink5. LD estimates for the sequencing results from the AD cohort were estimated using PLINK v1.9 with the –rsq dprime option.
This study, involving use of repository data and biospecimens, was approved by the Boston University Institutional Review Board.
Results
Genes Previously Associated With AD in AAs
LOF Variants
In the seven genes previously associated with AD in AAs, nominally significant associations were observed for a rare LOF variant in MS4A6A observed only in controls (rs140130948, p = 0.013) and an 11 bp ABCA7 deletion (rs567222111, OR = 3.57, p = 0.038) which had an estimated allele frequency (AF) of 1.1% in cases and 0.32% in controls (Table 2). This association remained significant in the mixed model adjusting for relatedness within the sample and including PCs for ancestry (OR = 3.65, p = 0.049). This deletion had a stronger impact on AD risk than the more common 44 bp ABCA7 deletion (rs142076058) which was previously reported to be associated with AD in an AA cohort (OR = 1.81) (Cukier et al., 2016) but not in our sample (OR = 1.27, p = 0.16). The evidence for association with rs567222111 in the replication sample was not significant, but had the same effect direction (OR for the deletion = 1.84, p = 0.22), and the significance in the combined discovery and replication samples was greater than in the discovery sample alone (OR = 2.42, p = 0.022). No LOF variants were observed in AKAP9, COBL, PTK2B, SLC10A2, or ZCWPW1.
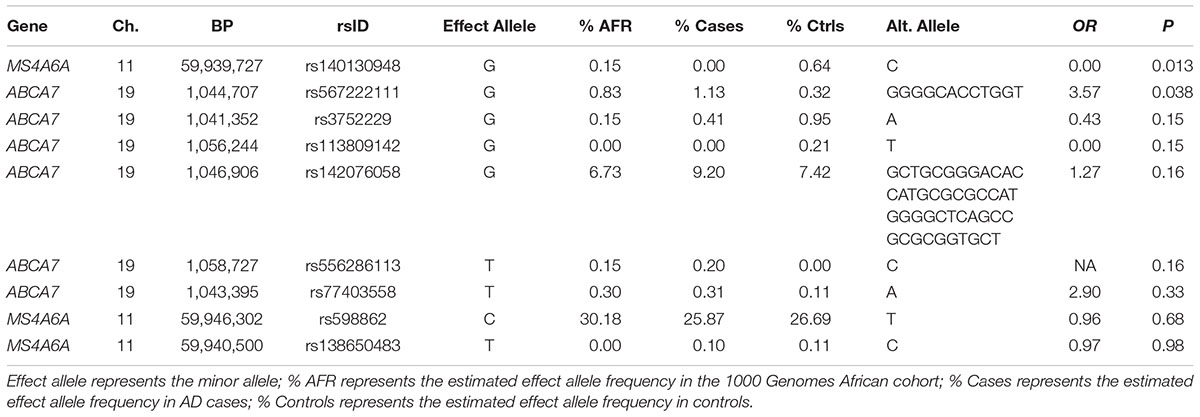
TABLE 2. Association between AD and loss of function (LOF) variants observed in African American AD genes.
Missense Variants
Association tests were nominally significant for 14 of 172 missense variants tested in the seven genes previously associated with AD in AAs including a common SNP in ABCA7 (rs5985184, p = 0.0043) and the rare missense variants in AKAP9, rs149979685 (OR = 10.73, p = 0.0046) and rs144662445 (OR = 6.35, p = 0.0054), previously identified in a sample that overlaps substantially with the discovery cohort in this study (Logue et al., 2014) (Table 3). Our analysis also confirmed the previously reported association for one of the common ABCA7 missense SNPs noted in N’Songo et al., 2017 (rs3764647, OR = 1.29 for minor allele, p = 0.017), but not the rare coding variant (rs3752239, OR = 0.39, p = 0.24). Consistent with prior results (Logue et al., 2014), the association with the rare AKAP9 variants was significant in a mixed model which adjusted for relatedness within the sample with ancestry PCs as covariates (for rs149979685 OR = 10.53, p = 0.025 and for rs144662445 OR = 6.25, p = 0.016).
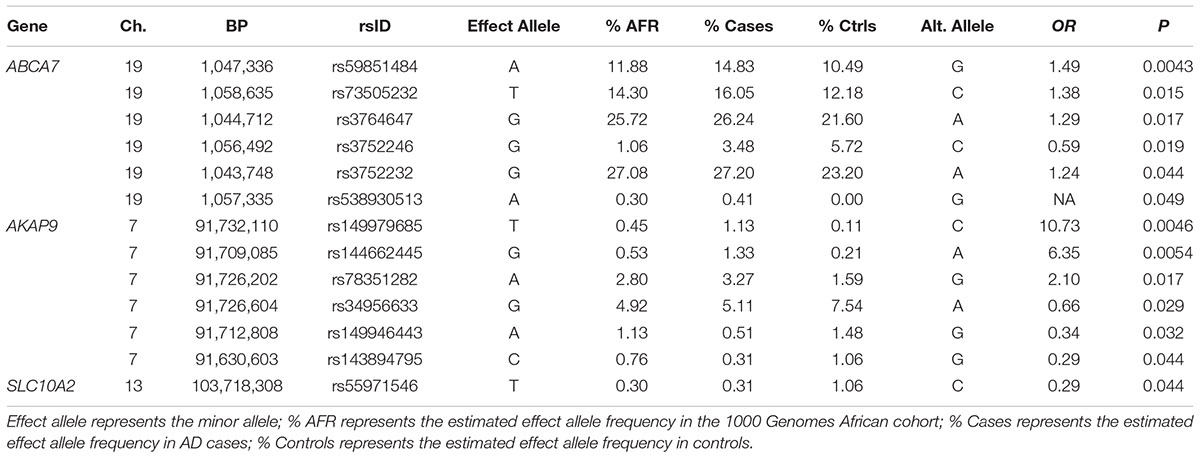
TABLE 3. Nominally significant missense variants in the 7 AA-AD genes.
Regulatory Variants
We also examined potentially regulatory variants in the AD genes implicated in AAs. Association was tested with variants in regulatory regions for the two primary AKAP9 isoforms. One variant identified near the TSS of the shorter isoform was not associated with AD (p = 0.66). Significant association was identified with a rare nine bp deletion (rs371245265) located near the TSS for the longer AKAP9 isoform (OR for the deletion = 6.37, p = 0.0053). Prompted by the similarity of allele frequencies between this deletion and the previously identified coding AD risk variants (rs144662445 and rs149979685), we checked the 1000 genomes phase 3 African population data and confirmed high LD between rs371245265 and both rs144662445 (r2 = 0.86) and rs149979685 (r2 = 1). Consistent with this information, all 17 discovery sample subjects with the rs371245265 deletion were also carriers of the rs144662445 minor allele, and 14 of these subjects were also carriers of the rs149979685 minor allele. As noted for rs149979685, the association with rs371245265 remained significant in a model adjusting for relatedness and ancestry (OR = 6.30, p = 0.016). Nominally significant associations were also observed with three common potentially regulatory SNPs in ZCWPW1. The most significant of these three was rs10693652, a 2 bp deletion which was more common in controls than cases (OR for the deletion = 0.75, p = 0.0042). The sole ABCA7 variant and 13 PTK2B variants located in TSSs were not associated with AD. Regulatory variants in COBL and SLC10A2 could not be evaluated because the custom capture design for these loci included exons only.
Other Variants
Examination of the full complement of variation in these genes (n = 4,325) including 342 variants in ABCA7, 1,445 in AKAP9, 167 in COBL, 204 in MS4A6A, 1,874 in PTK2B, 37 variants in SLC10A2, and 256 variants in ZCWPW1 revealed many nominally significant associations (Table 4). The most significant association was observed with a rare SNP in PTK2B (rs115828696, MAF = 0.0020 in AD cases and 0.18 in controls) which was protective (OR for the minor allele A = 0.11, p = 0.00041). A strong protective effect was also identified with a common SNP in ZCWPW1 (OR = 0.67, p = 0.0013). Genotypes were not available for several of the previously implicated AA-specific risk SNPs including ABCA7 rs115550680 (Reitz et al., 2013a,b) which is located in a repetitive region and was not captured by the design. The COBL rs112404845 and SLC10A2 rs16961023 variants (Mez et al., 2017) are outside of the coding regions and, thus, were not assessed.
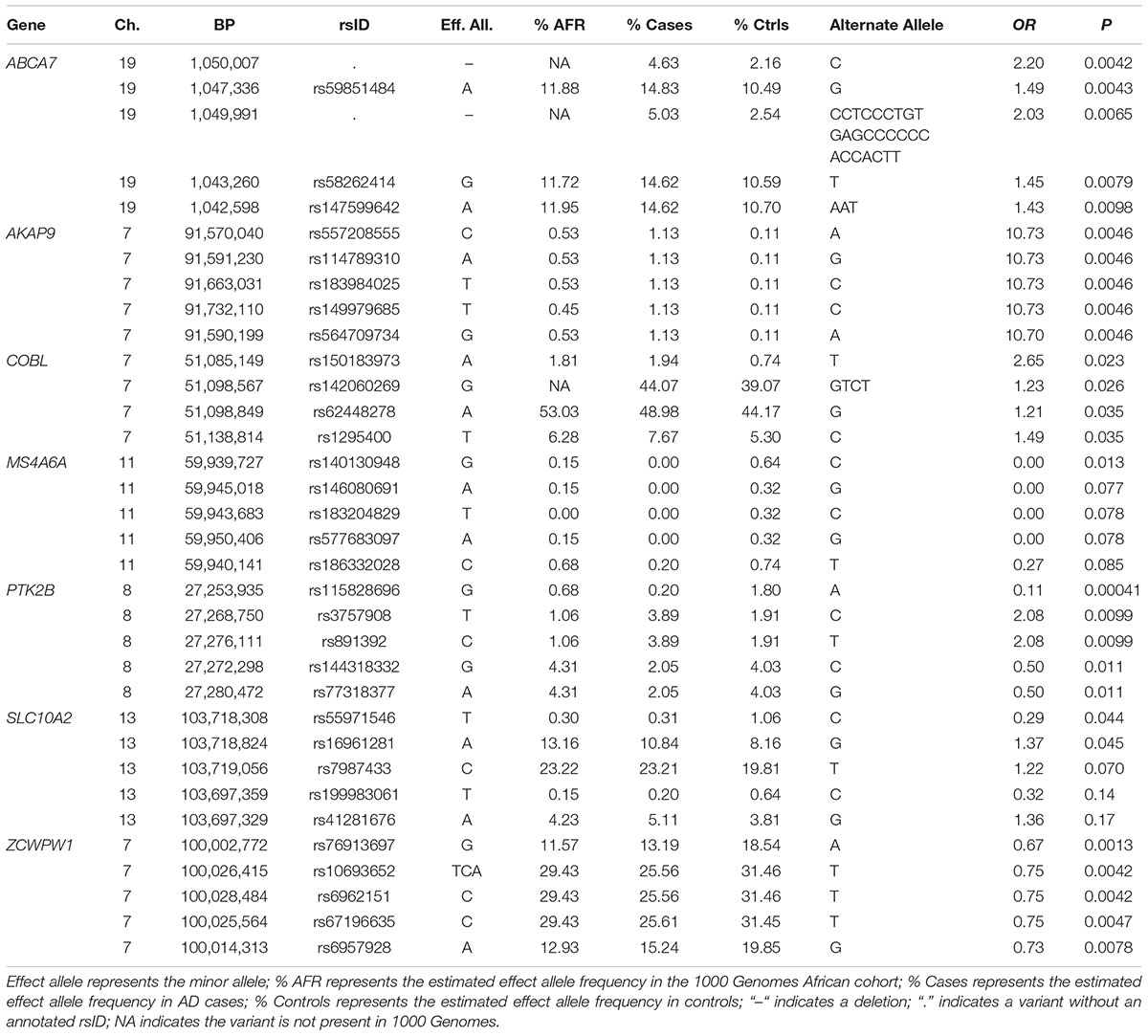
TABLE 4. Top-ranked association results in previously established AA AD-risk genes.
Genes Previously Associated With AD in Other Ancestry Groups
Of the 104,341 variants observed in all targeted regions that were tested for association with AD, 29 were annotated as LOF variants. Only the previously noted MS4A6A and ABCA7 variants (rs140130948 and rs567222111) were nominally significant (Table 5). The most significant association findings among 1,067 missense variants were obtained with five common highly correlated variants in PILRB that showed a protective effect (0.0010 < p < 0.0017; estimated OR for minor alleles varied from 0.65 to 0.67; Table 6). Restricting the analysis to potentially regulatory variants, a protective common indel near the TSS of ZCWPW1 (rs10693652, OR for the minor allele = 0.75, p = 0.0042) and the rare risk indel near the TSS of AKAP9 (rs536714523) noted above were the most significant of the 223 variants tested (Table 7). Finally, examination of the entire set of 104,341 variants identified in the targeted sequencing experiments yielded significant associations with multiple loci (Table 8), most notably a rare protective variant in F5 (rs2027885, OR for minor allele A = 0.053, p = 6.40 × 10-5), a gene that was previously associated with a MRI measure of hippocampal atrophy (Melville et al., 2012), and two common variants in KIAA0196 (p < 8.6 × 10-5; Table 8). Out of the 151 protein-coding genes, nominally significant gene-based associations were found with six genes using the CMC test and with three genes using the VT test (Table 9). The most significant of these results was KANSL1 (p = 0.013). None of the seven previously established AD risk genes in AAs were significant (p > 0.05).
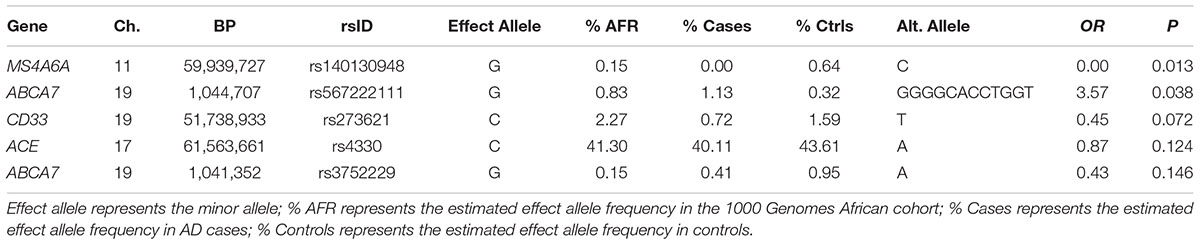
TABLE 5. Top loss of function variants from all sequenced genes (out of 29 LOF variants examined).
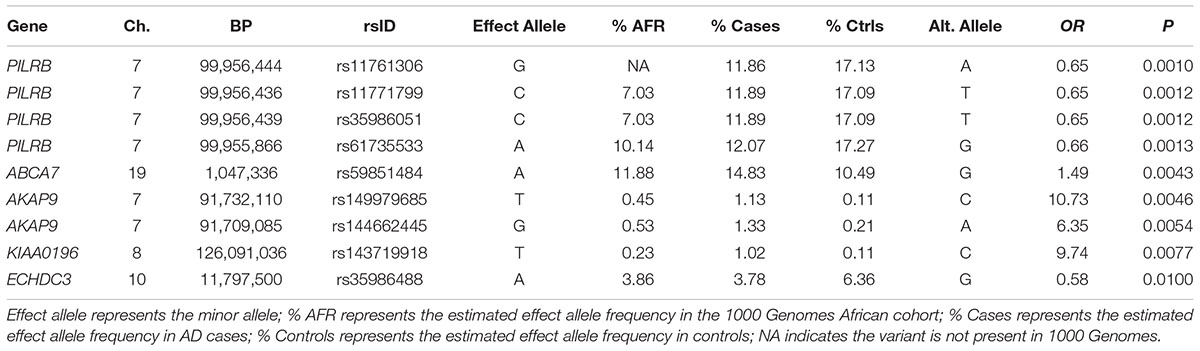
TABLE 6. Top missense variants from all sequenced genes (out of 1,067 missense variants examined).
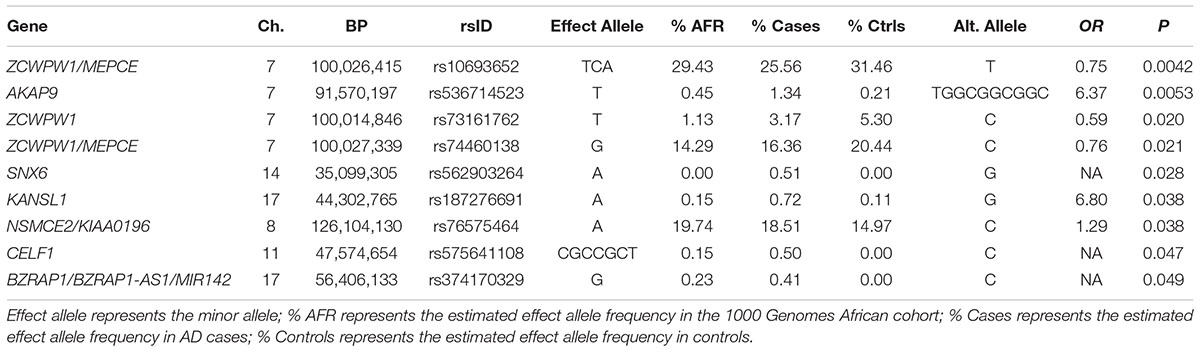
TABLE 7. Top potentially regulatory variants from all sequenced genes (out of 223).
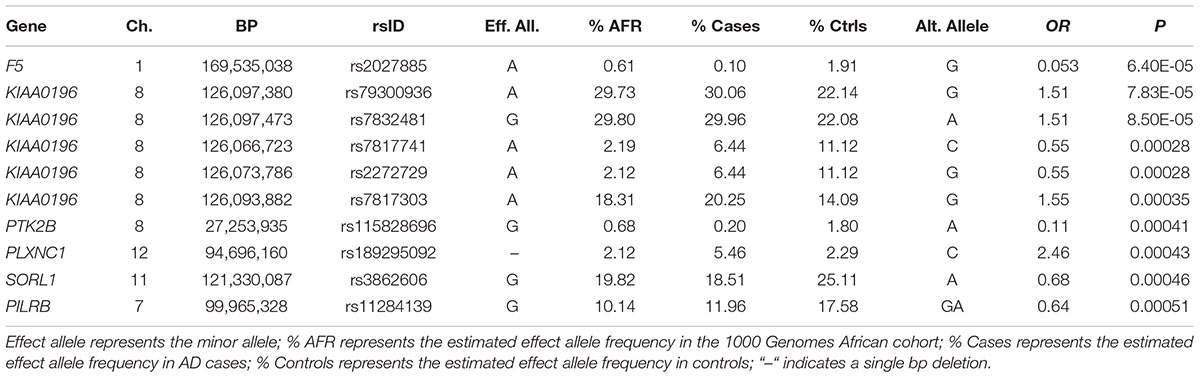
TABLE 8. Top variants from all genes (out of 104,341 variants examined).
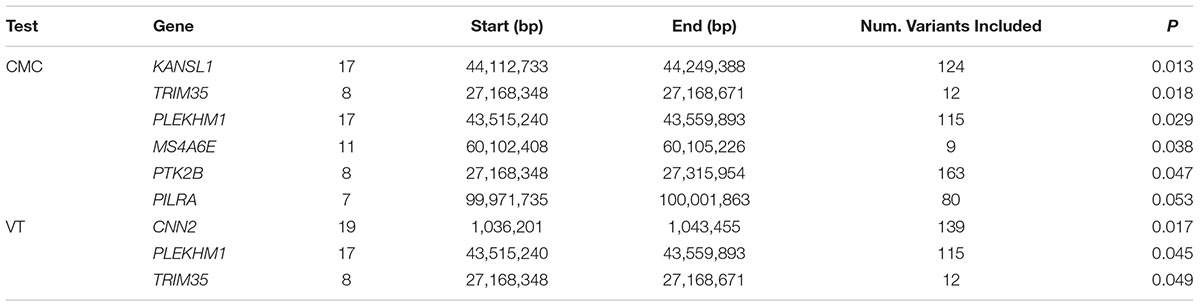
TABLE 9. Nominally significant gene-based burden tests of association with AD.
Discussion
We performed targeted gene sequencing in an AA cohort containing 489 AA AD cases and 472 cognitively normal controls and found evidence of association with several novel variants in genes that were previously implicated with AD risk in AAs including a deletion causing LOF of ABCA7 (rs567222111). We subsequently genotyped this deletion in an independent cohort containing 484 AD cases and 484 controls, and the association with this large effect variant (OR = 2.42) became more significant in the combined sample. Another notable novel association was identified with a rare 9 bp. deletion (rs371245265) located near the TSS of AKAP9. We also confirmed previously reported associations with missense variants in ABCA7 (rs3764647) and AKAP9 (rs149979685 and rs144662445). Gene-based tests of aggregated rare variants yielded several associations, most significantly with KANSL1, CNN2, and TRIM35.
The association with the AKAP9 regulatory region variant rs371245265 calls into question whether the previously identified AKAP9 missense variants (rs149979685 and rs144662445) are causally related to AD because all of these variants are in high LD. Previous analysis of the background haplotype harboring rs149979685 and rs144662445 and spanning an 800 kb region including five genes showed that no other coding variants could explain the association with these AKAP9 missense variants (Logue et al., 2014). However, it remains possible that the rs371245265 variant has a regulatory effect on AKAP9 expression, and this variant alone or in conjunction with the missense variants, could underlie the observed association with AD risk. Because these three rare variants most often co-occur, it is unlikely that the potentially causal effects of these variants will be disentangled by epidemiological studies. Recently, we observed significantly higher phosphorylation and greater post-translational modifications of Tau protein in lymphoblastoid cells from subjects having at least one of the missense variants, a finding that was independent of the disease status of the cell donors (Ikezu et al., 2018). However, since these subjects also have the potentially regulatory variant, experimental studies will be necessary to determine whether this variant does indeed have a regulatory effect and in particular which of the three variants account for the observed effect on Tau phosphorylation.
Our observed novel association with a rare 11 bp loss of function frameshift deletion (rs567222111, Leu396fs) in a gene encoding one of the ATP-binding cassette transporter proteins (ABCA7) adds to a growing list of AD-associated LOF mutations in this gene (Farrer, 2015). The most remarkable of these is a 7 bp deletion, causing a frameshift mutation (Glu709fs) that was detected in 11 out of 772 unrelated patients but not in 757 controls from the Flanders region of northern Belgium (Cuyvers et al., 2015). Association of this mutation with AD has also been observed in several other European ancestry populations (Steinberg et al., 2015). Cukier et al reported association of AD and a relatively common 44 bp LOF deletion in ABCA7 (rs142076058, Ser587fs, OR = 2.13) in an AA cohort that is non-overlapping with our study sample (Cukier et al., 2016). This deletion was observed in the current study, but had a smaller effect on AD risk (OR = 1.27, p = 0.16). Of note, the frameshift mutation identified in our study occurs earlier in the amino acid sequence (position 396) than the Belgian (position 709) or other AA (position 587) frameshift mutations and thus may yield a more seriously impaired protein than these other mutations, but this will have to be confirmed experimentally.
Surprisingly, expanding the analyses from the relatively small set of genes that were implicated in previous studies of AAs to the larger set of AD genes that were established in other populations yielded relatively few significant results, the most significant of which is a rare protective variant (rs2027885) in the gene encoding the blood clotting protein Factor 5 (F5, OR = 0.053, p = 6.40 × 10-5). A GWAS of a brain MRI measure of hippocampal atrophy in a MIRAGE Study sample composed primarily of AD and control subjects of European ancestry and a smaller group of AAs (many of which are included in this study) found genome-wide significant association with several common SNPs spanning portions of F5 and its immediate neighbor, SELP, that was supported by evidence in both populations (Melville et al., 2012). Although there is scant genetic evidence linking F5 to AD, it has been shown that factor V activating protein in Russell’s viper venom destabilizes amyloid-β aggregates as revealed from a thioflavin T assay (Bhattacharjee and Bhattacharyya, 2013).
Our findings contrast those of another recent exome sequencing study of AD in a smaller sample of AAs (198 AD cases and 304 controls) which focused exclusively on 20 loci reaching genome-wide significance in a very large GWAS of European ancestry cohorts (N’Songo et al., 2017). The previous study found nominally significant associations with two variants in ABCA7 (rs3764647 and rs3752239) and in gene-based tests of coding variants in MS4A6A, PTK2B, and ZCWPW1. We observed association with rs3764647 (p = 0.017), but did not replicate the association with rs3752239 or the gene-based associations. On the other hand, gene-based tests of aggregated rare variants in KANSL1, TRIM35, MS4A6E, and PILRA were nominally significant in our study. Differences in findings may be due in part to the use of exome sequencing by N’Songo et al. (2017) versus sequencing of complete gene regions in our study which allowed detection of association with potentially functional variants in regulatory regions and introns that influence transcription and splicing, as well as with structural variants that span non-coding regions.
Our findings should be interpreted cautiously. None of our findings remain significant after correcting for the total number of tests performed in the study. Our sample size was not large enough to detect associations with rare variants exerting modest effects with experiment-wide significance. Also, our primary analyses of individual variants did not account for the correlated structure of our dataset which included many related individuals. Our study highlights the difficulty of obtaining statistically significant results with rare variants, especially those with frequencies less than 1%. It is essential to replicate our findings in independent AA samples, and sufficiently large samples will become available eventually through the efforts of large consortia including the Alzheimer’s Disease Genetics Consortium and Alzheimer’s Disease Sequencing Project. In addition, experimental studies are needed to establish functionally relevant roles of these genes and variants in AD pathogenesis.
With these concerns in mind, the goal of this study was to identify variants with supporting genetic evidence and predicted functional impact for examination in relevant biological systems. Given the previously identified relationship between loss of function mutations in ABCA7 and AD (Cuyvers et al., 2015; Farrer, 2015; Steinberg et al., 2015; Cukier et al., 2016) and genetic and biological evidence for a role of rare AKAP9 variants in AD (Logue et al., 2014; Ikezu et al., 2018), the novel ABCA7 coding region deletion (rs567222111) and the potentially regulatory AKAP9 deletion (rs371245265) are the most compelling findings for future studies.
Data Availability
The unprocessed sequence data generated for this study can be found in the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site (https://www.niagads.org/).
Author Contributions
MWL, DL, LAF, and KLL contributed to the study design. JF cleaned and processed the sequence data. IS extracted and performed quality control on the DNA specimens used for sequencing and genotyping. MWL and DL performed analyses of the data and prepared the results for presentation. MWL and LAF drafted the manuscript. All authors contributed to the editing and revision of the manuscript.
Funding
This study was funded in part by grants from the National Institute on Aging (NIA, R01-AG048927, P30-AG13846, U01-AG032984, UF1-AG046198, and RF1-AG057519). Some biological samples used in this study were obtained from the National Cell Repository for Alzheimer’s Disease (NCRAD) at Indiana University funded by NIA grant U24-AG021886.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2018.00592/full#supplementary-material
TABLE S1 | Targeted regions including provisional and confirmed loci associated with AD and related phenotypes.
Footnotes
- ^http://genome.wustl.edu
- ^http://www.genewiz.com
- ^http://epd.vital-it.ch/index.php
- ^http://genome.sph.umich.edu/wiki/EPACTS
- ^https://analysistools.nci.nih.gov/LDlink/
References
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Bhattacharjee, P., and Bhattacharyya, D. (2013). Factor V activator from Daboia russelli russelli venom destabilizes beta-amyloid aggregate, the hallmark of Alzheimer disease. J. Biol. Chem. 288, 30559–30570. doi: 10.1074/jbc.M113.511410
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi: 10.1186/s13742-015-0047-8
Chen, H., Wang, C., Conomos, M. P., Stilp, A. M., Li, Z., Sofer, T., et al. (2016). Control for population structure and relatedness for binary traits in genetic association studies via logistic mixed models. Am. J. Hum. Genet. 98, 653–666. doi: 10.1016/j.ajhg.2016.02.012
Chung, J., Wang, X., Maruyama, T., Ma, Y., Zhang, X., Mez, J., et al. (2017). Genome-wide association study of Alzheimer’s disease endophenotypes at prediagnosis stages. Alzheimers Dement 14, 623–633. doi: 10.1016/j.jalz.2017.11.006
Cingolani, P., Platts, A., Wang le, L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Corder, E. H., Saunders, A. M., Risch, N. J., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 7, 180–184. doi: 10.1038/ng0694-180
Cukier, H. N., Kunkle, B. W., Vardarajan, B. N., Rolati, S., Hamilton-Nelson, K. L., Kohli, M. A., et al. (2016). ABCA7 frameshift deletion associated with Alzheimer disease in African Americans. Neurol. Genet. 2:e79. doi: 10.1212/NXG.0000000000000079
Cuyvers, E., De Roeck, A., Van den Bossche, T., Van Cauwenberghe, C., Bettens, K., Vermeulen, S., et al. (2015). Mutations in ABCA7 in a Belgian cohort of Alzheimer’s disease patients: a targeted resequencing study. Lancet Neurol. 14, 814–822. doi: 10.1016/S1474-4422(15)00133-7
Farrer, L. A. (2015). Expanding the genomic roadmap of Alzheimer’s disease. Lancet Neurol. 14, 783–785. doi: 10.1016/S1474-4422(15)00146-5
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease meta analysis consortium. JAMA 278, 1349– 1356.
Farrer, L. A., Sherbatich, T., Keryanov, S. A., Korovaitseva, G. I., Rogaeva, E. A., Petruk, S., et al. (2000). Association between angiotensin-converting enzyme and Alzheimer disease. Arch. Neurol. 57, 210–214.
Green, R. C., Cupples, L. A., Go, R., Benke, K. S., Edeki, T., Griffith, P. A., et al. (2002). Risk of dementia among white and African American relatives of patients with Alzheimer disease. JAMA 287, 329–336.
Gureje, O., Ogunniyi, A., Baiyewu, O., Price, B., Unverzagt, F. W., Evans, R. M., et al. (2006). APOE epsilon4 is not associated with Alzheimer’s disease in elderly Nigerians. Ann. Neurol. 59, 182–185. doi: 10.1002/ana.20694
Hendrie, H. C., Hall, K. S., Hui, S., Unverzagt, F. W., Yu, C. E., Lahiri, D. K., et al. (1995). Apolipoprotein E genotypes and Alzheimer’s disease in a community study of elderly African Americans. Ann. Neurol. 37, 118–120. doi: 10.1002/ana.410370123
Ikezu, T., Chen, C., DeLeo, A. M., Zeldich, E., Fallin, M. D., Kanaan, N. M., et al. (2018). Tau phosphorylation is impacted by rare akap9 mutations associated with Alzheimer Disease in African Americans. Neuroimmune Pharmacol. 13, 254–264. doi: 10.1007/s11481-018-9781-x
Jun, G., Asai, H., Zeldich, E., Drapeau, E., Chen, C., Chung, J., et al. (2014). PLXNA4 is associated with Alzheimer disease and modulates tau phosphorylation. Ann. Neurol. 76, 379–392. doi: 10.1002/ana.24219
Jun, G., Ibrahim-Verbaas, C. A., Vronskaya, M., Lambert, J. C., Chung, J., Naj, A. C., et al. (2016). A novel Alzheimer disease locus located near the gene encoding tau protein. Mol. Psychiatry 21, 108–117. doi: 10.1038/mp.2015.23
Jun, G., Naj, A. C., Beecham, G. W., Wang, L. S., Buros, J., Gallins, P. J., et al. (2010). Meta-analysis confirms CR1, CLU, and PICALM as Alzheimer Disease risk loci and reveals interactions With APOE genotypes. Arch. Neurol. 67, 1473–1484. doi: 10.1001/archneurol.2010.201
Jun, G. R., Chung, J., Mez, J., Barber, R., Beecham, G. W., Bennett, D. A., et al. (2017). Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement 13, 727–738. doi: 10.1016/j.jalz.2016.12.012
Kamboh, M. I., Demirci, F. Y., Wang, X., Minster, R. L., Carrasquillo, M. M., Pankratz, V. S., et al. (2012). Genome-wide association study of Alzheimer’s disease. Transl. Psychiatry 2:e117. doi: 10.1038/tp.2012.45
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Lee, J. H., Cheng, R., Graff-Radford, N., Foroud, T., and Mayeux, R. (2008). Analyses of the National Institute on aging late-onset Alzheimer’s disease family study: implication of additional loci. Arch. Neurol. 65, 1518–1526. doi: 10.1001/archneur.65.11.1518
Li, B., and Leal, S. M. (2008). Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 83, 311–321. doi: 10.1016/j.ajhg.2008.06.024
Logue, M. W., Schu, M., Vardarajan, B. N., Buros, J., Green, R. C., Go, R. C., et al. (2011). A comprehensive genetic association study of Alzheimer disease in African Americans. Arch. Neurol. 68, 1569–1579. doi: 10.1001/archneurol.2011.646
Logue, M. W., Schu, M., Vardarajan, B. N., Farrell, J., Bennett, D. A., Buxbaum, J. D., et al. (2014). Two rare AKAP9 variants are associated with Alzheimer’s disease in African Americans. Alzheimers Dement 10, 609.e11–618.e11. doi: 10.1016/j.jalz.2014.06.010
MacArthur, D. G., Balasubramanian, S., Frankish, A., Huang, N., Morris, J., Walter, K., et al. (2012). A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828. doi: 10.1126/science.1215040
McKhann, G. M., Knopman, D. S., Chertkow, H., Hyman, B. T., Jack, C. R. Jr., Kawas, C. H., et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269. doi: 10.1016/j.jalz.2011.03.005
Meier, I. B., Manly, J. J., Provenzano, F. A., Louie, K. S., Wasserman, B. T., Griffith, E. Y., et al. (2012). White matter predictors of cognitive functioning in older adults. J. Int. Neuropsychol. Soc. 18, 414–427. doi: 10.1017/S1355617712000227
Melville, S. A., Buros, J., Parrado, A. R., Vardarajan, B., Logue, M. W., Shen, L., et al. (2012). Multiple loci influencing hippocampal degeneration identified by genome scan. Ann. Neurol. 72, 65–75. doi: 10.1002/ana.23644
Meng, Y., Baldwin, C. T., Bowirrat, A., Waraska, K., Inzelberg, R., Friedland, R. P., et al. (2006). Association of polymorphisms in the angiotensin-converting enzyme gene and Alzheimer disease in an Israeli-Arab community. Am. J. Hum. Genet. 78, 871–877. doi: 10.1086/503687
Mez, J., Chung, J., Jun, G., Kriegel, J., Bourlas, A. P., Sherva, R., et al. (2017). Two novel loci, COBL and SLC10A2, for Alzheimer’s disease in African Americans. Alzheimers Dement 13, 119–129. doi: 10.1016/j.jalz.2016.09.002
N’Songo, A., Carrasquillo, M. M., Wang, X., Burgess, J. D., Nguyen, T., Asmann, Y. W., et al. (2017). African American exome sequencing identifies potential risk variants at Alzheimer disease loci. Neurol. Genet. 3:e141. doi: 10.1212/NXG.0000000000000141
Price, A. L., Kryukov, G. V., de Bakker, P. I., Purcell, S. M., Staples, J., Wei, L. J., et al. (2010). Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 86, 832–838. doi: 10.1016/j.ajhg.2010.04.005
Reitz, C., Jun, G., Naj, A., Rajbhandary, R., Vardarajan, B. N., Wang, L. S., et al. (2013a). Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 309, 1483–1492. doi: 10.1001/jama.2013.2973
Reitz, C., Tosto, G., Vardarajan, B., Rogaeva, E., Ghani, M., Rogers, R. S., et al. (2013b). Independent and epistatic effects of variants in VPS10-d receptors on Alzheimer disease risk and processing of the amyloid precursor protein (APP). Transl. Psychiatry 3:e256. doi: 10.1038/tp.2013.13
Rogaeva, E., Meng, Y., Lee, J. H., Gu, Y.-J., Zou, F., Kawarai, T., et al. (2007). The sortilin-related receptor SORL1 is functionally and genetically associated with Alzheimer’s disease. Nat. Genet. 39, 168–177.
Sahota, A., Yang, M., Gao, S., Hui, S. L., Baiyewu, O., Gureje, O., et al. (1997). Apolipoprotein E-associated risk for Alzheimer’s disease in the African-American population is genotype dependent. Ann. Neurol. 42, 659–661. doi: 10.1002/ana.410420418
Saunders, A. M., Strittmatter, W. J., Schmechel, D., George-Hyslop, P. H., Pericak-Vance, M. A., Joo, S. H., et al. (1993). Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472.
Steinberg, S., Stefansson, H., Jonsson, T., Johannsdottir, H., Ingason, A., Helgason, H., et al. (2015). Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat. Genet. 47, 445–447. doi: 10.1038/ng.3246
Vardarajan, B. N., Bruesegem, S. Y., Harbour, M. E., Inzelberg, R., Friedland, R., St George-Hyslop, P., et al. (2012). Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol. Aging 33, 2231.e15–2231.e30. doi: 10.1016/j.neurobiolaging.2012.04.020
Keywords: Alzheimer disease, sequencing, AKAP9, ABCA7, rare variant, African Americans
Citation: Logue MW, Lancour D, Farrell J, Simkina I, Fallin MD, Lunetta KL and Farrer LA (2018) Targeted Sequencing of Alzheimer Disease Genes in African Americans Implicates Novel Risk Variants. Front. Neurosci. 12:592. doi: 10.3389/fnins.2018.00592
Received: 05 March 2018; Accepted: 07 August 2018;
Published: 27 August 2018.
Edited by:
Celeste Karch, Washington University in St. Louis, United StatesReviewed by:
Sheng Chih Jin, Yale University, United StatesManav Kapoor, Icahn School of Medicine at Mount Sinai, United States
Copyright © 2018 Logue, Lancour, Farrell, Simkina, Fallin, Lunetta and Farrer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lindsay A. Farrer, ZmFycmVyQGJ1LmVkdQ==