 Qurratulain Aftab1†
Qurratulain Aftab1† Marc Mesnil2†Emmanuel Ojefua3Alisha Poole3Jenna Noordenbos3Pierre-Olivier Strale1Chris Sitko3Caitlin Le3Nikolay Stoynov4
Marc Mesnil2†Emmanuel Ojefua3Alisha Poole3Jenna Noordenbos3Pierre-Olivier Strale1Chris Sitko3Caitlin Le3Nikolay Stoynov4 Leonard J. Foster4Wun-Chey Sin2
Leonard J. Foster4Wun-Chey Sin2 Christian C. Naus2
Christian C. Naus2 Vincent C. Chen3*
Vincent C. Chen3*- 1Department of Cellular and Physiological Sciences, Life Sciences Institute, University of British Columbia, Vancouver, BC, Canada
- 2Signalisation et Transports Ioniques Membranaires (STIM), CNRS ERL 7003, University of Poitiers, Poitiers, France
- 3Department of Chemistry, Brandon University, Brandon, MB, Canada
- 4Department of Biochemistry and Molecular Biology, Centre for High-Throughput Biology, University of British Columbia, Vancouver, BC, Canada
Extracellular matrix (ECM) remodeling, degradation and glioma cell motility are critical aspects of glioblastoma multiforme (GBM). Despite being a rich source of potential biomarkers and targets for therapeutic advance, the dynamic changes occurring within the extracellular environment that are specific to GBM motility have yet to be fully resolved. The gap junction protein connexin43 (Cx43) increases glioma migration and invasion in a variety of in vitro and in vivo models. In this study, the upregulation of Cx43 in C6 glioma cells induced morphological changes and the secretion of proteins associated with cell motility. Demonstrating the selective engagement of ECM remodeling networks, secretome analysis revealed the near-binary increase of osteopontin and matrix metalloproteinase-3 (MMP3), with gelatinase and NFF-3 assays confirming the proteolytic activities. Informatic analysis of interactome and secretome downstream of Cx43 identifies networks of glioma motility that appear to be synergistically engaged. The data presented here implicate ECM remodeling and matrikine signals downstream of Cx43/MMP3/osteopontin and ARK1B10 inhibition as possible avenues to inhibit GBM.
Introduction
Gliomas are cancers of the central nervous system that arise from glial-like precursor or dedifferentiated glial cells (Parkin et al., 2001; Furnari et al., 2007). The most aggressive subtype, glioblastoma multiforme (GBM), is categorized as a World Health Organization grade IV astrocytoma. Despite radical treatment, patients with GBM have an extremely poor prognosis with a median survival of 12–15 months (Parkin et al., 2001; Louis et al., 2016). GBM tumors are comprised of a heterogeneous population of tumor and host cells, that are characterized by angiogenesis, proliferation and evasion of apoptosis. High-grade gliomas further demonstrate a high propensity to invade. Furthermore, hypoxic conditions within a growing mass initiates patterns of gene expression that promotes GBM tumors to seek new niches within the CNS to expand (Demuth and Berens, 2004). The close examination of actively moving glioma demonstrates a mechanism of mutual exclusivity between the pathways regulating invasion and proliferation. In this regard GBMs appear to have a switch-like ability to delay entry into the cell cycle to migrate; while highly proliferative tumors have a disproportionate number of cells that do not invade (Giese and Westphal, 1996; Giese et al., 1996; Giese, 2003). The identification of molecular signatures of cell proliferation vs. migration in glioma would be of significant interest.
As a natural barrier for migrating glioma cells, the regulation of the extracellular matrix (ECM) and the secretome has received much attention as avenues to inhibit GBM (Levental et al., 2009; Wu et al., 2010; Cox and Erler, 2011; Lu et al., 2012). To facilitate invasion, cells of GBM are known to regulate secretion and alter the ECM of host tissues (Nakada et al., 2007; Lu et al., 2012). Previous work by Yoon et al. (2014) examined the secretome of U373MG cells, correlated to hypoxic and normoxic conditions to identify stanniocalcins (STC1 and 2), and insulin-like growth factors binding proteins that correlate with glioma grade. To help discern the secretome signature of invasive GBM, Formolo et al. provided an in-depth analysis of U118, LN18, T98, and U87 cells to identify more than 1100 proteins, among which the non-proteolytic factor chitinase-3-like protein 1 (CH3L1) was found to be linked to the invasion of U87 cells (Formolo et al., 2011).
As invasion and tumor growth are critical aspects underlying glioma, there is intense interest in identifying the molecular pathways of GBM. Previous studies in glioma have shown that Cx43 suppresses proliferation (Zhu et al., 1991; Huang R.P. et al., 1998; Fu et al., 2004). However, a growing number of recent reports have implicated Cx43 with increases in migration and invasion (Kameritsch et al., 2012; Naus et al., 2016). Gap junction proteins have also been linked to the regulation of the cytoskeleton, and the migration of neural crest cells in developing mice (Huang G.Y. et al., 1998). Cx43 is postulated to modulate these pathways via calcium signal propagation (Frisch et al., 2003), protein–protein interactions (Theiss and Meller, 2002; Bates et al., 2007; Wall et al., 2007), the establishment of intercellular junctions/channels (Wei et al., 2005; Laird, 2009) and cell-to-cell adhesion (Elias et al., 2007). In this study, we chose the rat glioma cell line C6 which expresses very low levels of endogenous Cx43 and its clone C6-13 which express high levels of exogenous Cx43 (Zhu et al., 1991). Providing evidence of paracrine signals, the application of C6-13 conditioned media increased the mobility of parental C6 cells. Owing to differences in Cx43 expression and behaviors that are typical of aggressive glioma, we sought to compare the secretomes of C6/C6-13 cells by quantitative proteomics (nanoHPLC-MS/MS). The conditioned media of C6-13 cells was found to contain significant increases in the pro-migratory proteins osteopontin and matrix metalloproteinase-3 (MMP3). This level of expression was confirmed by Western blotting and high-resolution multiple reaction monitoring (HR-MRM). Viscosity and gelatinase activity assays, alongside our use of intramolecularly quenched fluorogenic MMP3 substrates (NFF-3) finds the ECM of C6-13 cells undergoes intense remodeling. The data presented here implicate ECM activities and matrikine signals downstream of Cx43/MMP3/osteopontin as possible avenues to inhibit GBM.
Materials and Methods
Cells and Cell Culture
The rat C6 line (American Type Culture Collection, ATCC) maintains many characteristics of GBM (Grobben et al., 2002) and naturally expresses low levels of Cx43 (Naus et al., 1991). The choice of these cells as a glioma model is motivated by their extensive use, and recognition of cancer stem cell behavior (Zheng et al., 2007), including their ability to generate GBMs in rats upon injection in the brain (Beljebbar et al., 2010). C6-13 is a C6 clone stably expressing Cx43 that demonstrates enhanced gap junctional intercellular communication, and restricted proliferation (Zhu et al., 1991). These cells have been in routine use by ourselves and others to monitor the cellular and biochemical aspects of gap junctions – including trafficking, turnover, and regulated growth (Charles et al., 1992; Naus et al., 1993; Stout et al., 2002; Fu et al., 2004; Chen et al., 2006, 2012). Glioma cells were maintained at 37°C, 95% air and 5% CO2 within a humidified incubator. Cells were cultivated in high glucose (4.0 g/L) Dulbecco’s Modified Eagles Medium (DMEM; Thermo Fisher Scientific) containing L-glutamine (4.0 mM), antibiotics (penicillin, 100 IU/mL; streptomycin, 100 μg/mL) with 10% fetal bovine serum (FBS; Gibco).
Migration (Wound Healing) Assays
Cells (C6/C6-13) were seeded at 2 × 105 cells in 12-well plates (Nunc, NucleonTM) and incubated (37°C, 95% air/5% CO2) for 3 days. Cultures at ∼80% confluence were scraped with a micropipette tip (P200, 1.1 mm OD), rinsed with phosphate buffered saline (PBS), and incubated with fresh media. Cell migration was accessed in serum-free (-FBS) or complete (+FBS) DMEM. For conditioned media transfer scrape-wound experiments, cells were taken through the same conditions, but on 6-well plates (Nunc, NucleonTM). From 90% confluent cultures (DMEM, 10% FBS, 10 cm plates), conditioned media were prepared by thoroughly washing cells twice with 10 mL of PBS, followed by the application of 12 mL of fresh, serum-free media (24 h, 37°C, 95% air/5% CO2). Conditioned medium (2 mL/well) was transferred to C6/C6-13 cells and immediately taken through scrape wound procedures. At 4, 8, and 24 h intervals, travel distances were monitored after methanol fixation (10 min) and crystal violet stain. Images were taken on an inverted microscope (Motic AE31) equipped with a digital camera and processing software (Motic Images Plus 2.0). Pictures of 3 random areas were imaged. Scrape areas (pixels) were measured using ImageJ (Schneider et al., 2012). Areas of migration (area = y ∗ x) were converted to migration distance in the y-direction. As the dimensions of the field of view and length of the scrape (x-direction) were known and held constant, the migration distances (y = area/x) and velocities (v = y/time) were readily calculated. Distance values were expressed as a mean value, ± standard error of the mean (SEM). Distances of glioma travel were compared by Student’s t-test, with a significant difference at ∗∗p ≤ 0.01 and ∗∗∗p ≤ 0.001. Statistical tests were performed using Microsoft Excel and presented in GraphPad Prism (San Diego, CA, United States) or DataGraph (Visual Data Tools Inc., United States).
Immunofluorescence
C6/C6-13 cells were seeded on coverslips (12 mm glass, Thermofisher Scientific, 2 × 104 cells). Confluent cultures were taken through scrape-wound procedures, rinsed with PBS, formalin fixed, blocked with BSA (2%) and permeabilized with 0.3% Triton X-100 (in 2% BSA). Cells were then exposed for 1 h to primary antibodies directed against actin (goat polyclonal, 1:200, Santa Cruz Biotechnology) or Ki-67 (rabbit polyclonal; 1:100; Santa Cruz Biotechnology). Coverslips were incubated with anti-rabbit or anti-goat secondary antibodies (1:500) linked to FITC or TRITC and washed with PBS. Coverslips were mounted with antifade medium with DAPI (Thermofisher/Life Technologies). Cells residing at scrape borders were imaged under fluorescence and DIC microscopy (Zeiss, Axioskop, Germany).
Conditioned Media Viscosity Measurement
Viscosity of C6/C6-13 conditioned media were measured using a capillary viscometer that constructed using a 18-gauge needle and a 1 mL syringe. Viscometer flow-through was collected under gravity using a standard beaker in a biocontainment hood. The laminar flow of 1 mL of media (24 h conditioning) was measured using a standard stopwatch. Between measurements, the viscometer was washed in 70% ethanol and dried.
Secretome Analysis
C6/C6-13 cells were seeded in two 15 cm dishes (Nunc) containing each 20 mL of complete DMEM. At 80% confluence cell cultures were rinsed twice with serum-free DMEM (10 mL) and maintained under serum-free conditions for 24 h. Possible differences in C6/C6-13 cell death due to FBS-free media (after 24 h) was determined by the trypan blue exclusion test (Strober, 2001). Here, dead/suspended cells in spent media were aspirated, collected, and combined with adherent cells that were released by trypsinization. Cells collected by centrifugation were rinsed twice with serum-free DMEM and incubated for 3 min in trypan blue stain (0.4%). After rinsing twice with fresh DMEM, cells were counted under light microscopy using a hemocytometer. No differences in cell survival between C6/C6-13 (∼98%) was observed (Supplementary Figure S1).
For secretome isolation, conditioned media was collected in 50 mL tubes, treated with protease inhibitor cocktail (1 tablet/10 mL; Roche Applied Science, Mannheim, Germany), centrifuged (5500 rpm; 10 min; 4°C) and decanted to remove insoluble debris. Proteins were precipitated with 25 mL of ethanol (4°C, 2 h) supplemented with 40 μL/mL sodium acetate (2.5 M; pH 5.0). Tubes were centrifuged (5500 rpm; 30 min; 4°C), decanted and isolated pellets rinsed with ice cold ethanol (to remove residual protease inhibitor). Pellets were suspended in 1 mL of trypsin digestion buffer (1% sodium deoxycholate and 50 mM NH4HCO3 at pH 8). Total protein quantity was determined by BCA (Pierce/Thermo Fisher Scientific, Rockford, IL, United States). Proteins (1 mg) were then reduced with DTT (1 μg/50 μg of proteins; 30 min at 37°C) and alkylated with iodoacetamide (IAA; 5 μg/50 μg of proteins, 20 min at 37°C). Protein digestions were conducted overnight (37°C) with sequencing-grade modified trypsin (1 μg/50 μg protein; Promega, Madison, WI, United States). Peptides were enriched by C18 solid-phase extraction (International Sorbent Technology Ltd., United Kingdom) and dried. The pellets were suspended in 15 μL of sodium acetate (0.5 M; pH 5.0) and peptides were isotopically modified with formaldehyde (dimethylation) at lysine and n-termini (Hsu et al., 2003; Boersema et al., 2008; Chen et al., 2012). In brief, 15 μL of “heavy” (CD2O) and “light” (CH2O) formaldehyde (200 mM) were used to label secretome peptides from C6-13 and C6 cells, respectively. Sodium cyanoborohydride (1.0 M) was added (1.5 μL/sample, 40 min in the dark; 20°C), with a second round of labeling performed at pH to 7.5 using NaOH (aqueous, 1.0 M). Reactions were quenched with 3.0 M NH4Cl (15 μL/sample; 10 min in the dark; 20°C). Samples were suspended in buffer (3% acetonitrile, 1% trifluoroacetic acid, 0.5% acetic acid) and processed with C18 stage-tips. Samples were collected in a 96-microwell plate, dried and suspended in 3.2 μL of sample buffer. Labeled peptides were separated using an Agilent 1100 nanoHPLC using an autosampler (4°C). Separations were conducted on a 150 mm × 75 μm ID column (Dr Maisch, 3 μm, ReproSil-Pur 120 C18-AC), mobile phase A – 0.5% aqueous acetic acid, mobile phase B – 80% acetonitrile in 0.5% aqueous acetic acid, gradient from 6% B to 80% B at 0.2 μL/min from 20 to 85 min). Peptides were analyzed by a high-resolution linear trapping quadrupole-orbital trapping mass spectrometer (LTQ-OrbitrapXL, Thermofisher, Germany) with a generation of 1 full scan MS (Orbitrap, m/z 300-2,000, R = 60000), followed by 5 data-dependent CID MS/MS scans in the LTQ (minimum signal 1000 counts, isolation 2.0 Da, normalized collision energy of 35.0, accepting charge states +2, +3, and +4). Active dynamic exclusion was set to 3 min after the acquisition of 3 spectra. Raw datafiles associated with this study have been uploaded to ProteomeXchange.org (accession PXD012175).
MS Data Analysis
Raw files were processed with MaxQuant software, version 1.5.5.1 (Cox and Mann, 2008) and Mascot (Matrix Science v.2.5) against the Rat Uniprot database (UP000002494, uploaded March 25, 2017). Trypsin enzyme specificity with peptides having a minimum length of 7 amino acids were considered. A maximum of one missed cleavage was allowed. Carbamidomethyl cysteine (fixed), alongside oxidation of methionine (variable) were enabled modifications for database searches. A 1% false discovery rate with the additional constraint of having been observed by both Mascot Server (v.2.6, Matrix Science) and Andromeda (MaxQuant) search engines. Relative quantitation of heavy and light peptide ratios reported by this study are based on extracted ion chromatograms (XICs) carrying the dimethyl modification. Protein identification required a minimum of two unique peptides having been observed within at least 2 biological replicates. Raw, MzXml, and mgf files, alongside and MaxQuant Protein outputs have been made available (PXD0121751). Statistical significance of differentially expressed proteins were interrogated using Perseus (v.1.60.7) (Tyanova and Cox, 2018). The Student t-test (two-tailed, unpaired, equal variance) was used to calculate significance (P ≤ 0.05). The threshold for significant change in expression was set to a FPR of 5%.
High-Resolution Multiple Reaction Monitoring (HR-MRM)
Label-free quantitation of C6/C6-13 secretome peptides were performed by HR-MRM using an Agilent 1200 nanoHPLC interfaced to a quadrupole time-of-flight mass spectrometer (QTOF, Agilent 6530). Peptides were subjected to fragmentation by collision induced dissociation (CID): osteopontin (VAEFGSSEEK, collision energy = 24.64 V), MMP3 (ESVDSAIER, 15.3 V) fibronectin (FTNIGPDTMR, CE 26.05 V). Peptides were separated using a flow rate of 300 nL/min on a 20 cm × 100 μm ID column packed with C18 (Dr Maisch, 3 μm, ReproSil-Pur) using a linear gradient of 5–35% B (99.9% acetonitrile, 0.1% formic acid) over a span of 78 min (HPLC buffer A: 99.9% water, 0.1% formic acid). Peptides were quantified using Skyline (v.4.1.0) (Pino et al., 2017). Full-scan HR-MRM spectra were verified by spectral matching (NIST Rat spectral libraries)2 and amino acid database search (<1%FDR, Mascot v2.6, Matrix Science). Data files (.MzML, .mgf, Skyline files) associated with this analysis have been submitted to proteomexchange.org (PXD012175).
SDS-PAGE and Western Blot Analysis
Western blot analyses were carried out on conditioned media of C6 or C6-13 cells (80% confluency) in 15 cm dishes and incubated for 24 h in serum-free DMEM. The concentration of secretome and cellular lysates proteins were determined by BCA (Pierce/Thermo Fisher Scientific, Rockford, IL, United States). Proteins from C6/C6-13 whole cell lysates or conditioned media (-FBS) were separated on 4–12% gradient mini-gels (150 V, 2 h). Gel separated proteins were transferred onto PVDF (Bio-Rad; 35 V, 1 h) and blocked (1 h) in a 5% (w/v) solution of non-fat milk powder in TBST (20 mM Tris-HCl, pH 7.4; 150 mM NaCl and 0.2% Tween-20). Membranes were incubated for 4 h with diluted rabbit polyclonal anti-MMP3 antibody (1:1000, 17873-1-AP, ProteinTech Group), or goat polyclonal anti-osteopontin (1:1000; Affinity BioReagents) antibodies in 5.0 mL of TBST containing 1% milk. Membranes were washed three times with TBST (10 min) and incubated with horseradish peroxidase-conjugated donkey anti-rabbit or anti-goat IgG (1:3000) in 1% milk TBST for 1 h, washed (TBST, 3 × 10 min). Blots were processed using standard film (ECL, GE Healthcare) or a C-DiGit Blot Scanner (ECL, Li-Cor). Thus, to ensure the validity of our loading procedures, we quantified total protein using a second SDS-PAGE gel. These Coomassie-stained gels demonstrated equivalent loads (Supplementary Figure S1) and transfer efficiency was monitored by staining membranes with ponceau S (data not shown). Transfer efficiency was measured using Ponceau S stain. Signals from ECL and Ponceau S (not shown) were quantified using Li-Cor Bioscience Image Studio Software (v.5.2).
MMP Zymography
Protocols used for zymography were adapted from Mendes et al. (2005). In brief, 2.5 × 105 cells were cultured in 6-well plates grown to 80% confluence. Conditioned media (24 h, 650 μL) proteins were concentrated using a 30 K MWCO (Pall Microsep) centrifugal spin filter and quantified by BCA. Concentrates were normalized (100 μg/lane) and diluted using SDS-PAGE loading buffer lacking DTT, without boiling. Electrophoresis was conducted at 4°C in 10% SDS-PAGE gels supplemented with 0.1% gelatin (Thermo Fisher Scientific). Post-separation, gels were rinsed in 18 Mohm water, followed by zymography wash buffer (incubation buffer containing 2.5% Triton-X100, 2 × 40 min) with rocking (5 min). In a sealed vessel, gels were completely submerged in incubation buffer (50 mM Tris-HCl, pH = 7.5, 5 mM CaCl2, 1 μM ZnCl2, 0.02% Brij-35, and 0.02% NaN3) for 24 h at 37°C. Gels were exposed to Coomassie G250 (0.5%, 1 h), distained in 18 Mohm water and 2% acetic acid fixative and imaged. Gelatinolytic activity was observed as a clear band against a blue background. Images were captured using a digital camera (α6000, Sony) and a standard light box. Zymography experiments were performed in triplicate.
Fluorogenic Assay for MMP3 Activity
Cells were plated at a concentration of 1.0 × 105 per well (6-well plates) and grown for 24 h in 2 mL of DMEM containing 10% FBS. DMEM (Sigma, D2902, 2 mL) lacking FBS was then added and allowed to condition the media for 24 h. Conditioned media was then collected and distributed into a 96-well assay plate preloaded with 15 μL of a stock solution containing NFF-3 (100 μM in DMSO). Conditioned or control media (300 μL) was added to each well (final concentration NFF-3 = 4.76 μM, MW = 1675.8 g/mole) using a standard 96 well plate. MMP3 activity was monitored at 37°C using a multi-well plate fluorimeter (SpectraMax M2, Molecular Devices). The proteolytic activity of C6/C6-13 conditioned media was measured relative to the background fluorescence produced by unconditioned DMEM. Wavelengths for excitation (325 nm) and fluorescence emission (393 nm) were measured every 10 min over 6 h.
Bioinformatics and Analysis of Functional Networks
Functional classification and cellular outcomes of expression were evaluated through bioinformatics using Ingenuity Pathways Analysis (IPA, Qiagen) (Kramer et al., 2014). Datasets re-examined here include our previous Cx43 protein–protein interactions originating from C6-13 (Chen et al., 2012). This dataset is available via BioGrid3 (Stark et al., 2006) and the original publication (Chen et al., 2012) and proteomeXchange.org (PXD012175). Synergistic functions common to Cx43 interactome and secretome were determined using statistically enriched cellular pathways in IPA. Here, statistical significance of C6/C6-13 datasets (secretome and Cx43 protein–protein interactions) was determined by comparison to networks obtained from similar-sized pool of random proteins. Databases used for this analysis integrated the BIND, DIP, MIPS, BIOGRID, INTACT, and COGNIA, alongside the curated IPA Knowledgebase (Kramer et al., 2014). In addition to network enrichments, we utilized Causal Analysis feature in IPA (a.k.a. Z-score Activation) to explain/predict the direction and impact Cx43 interactors and secretome protein expression have on increasing/decreasing cell migration and predicted cellular functions. While still depending on statistics, the IPA Causal Analysis approach leverages knowledge about genetic function and rather than solely relying on network association (Kramer et al., 2014). Data from our integrated analysis of the C6/C6-13 secretome and protein–protein interactions of Cx43 were presented using Microsoft Excel. Proteins found within the secretomes of C6/C6-13 were also evaluated using the Human Cancer Secretome Database (Feizi et al., 2015) and SignalP (Petersen et al., 2011; Nielsen, 2017).
Results
Cx43 Expression Increases Migration of C6 Glioma Cells
The expression of Cx43 is widely regarded as a potent inhibitor of glioma growth (Zhu et al., 1991; Naus et al., 1992; Sin et al., 2012). The C6 clone 13 line (C6-13) demonstrates a dramatic reduction of cell proliferation, in both in vitro (Zhu et al., 1991) and in vivo models of GBM (Naus et al., 1992). Glioma motility was monitored using the scrape wound healing assay (Figures 1A–F). At 24 h serum (Figure 1C) and serum-free (Figure 1E) conditions, scrape wound gaps were fully closed by C6-13, whereas C6 closure was only obtained after growth stimulation with FBS (Figures 1D,F). Despite having restricted cell proliferation (Zhu et al., 1991; Naus et al., 1992), wounds for C6-13 were consistently closed, an effect that was independent of FBS (Figure 1E compared to Figure 1F). In these images C6-13 are also distinguishable – polarized and elongated in the direction of migration, whereas C6 cells appeared smaller and compact. Due to the complete closure of C6-13 at 24 h, wound closures at 4 and 8 h were measured to determine the absolute distance and velocity (Figures 1G,H). This analysis confirmed the motility of C6-13 (21 μm/h) is significantly higher than C6 control (11 μm/h).
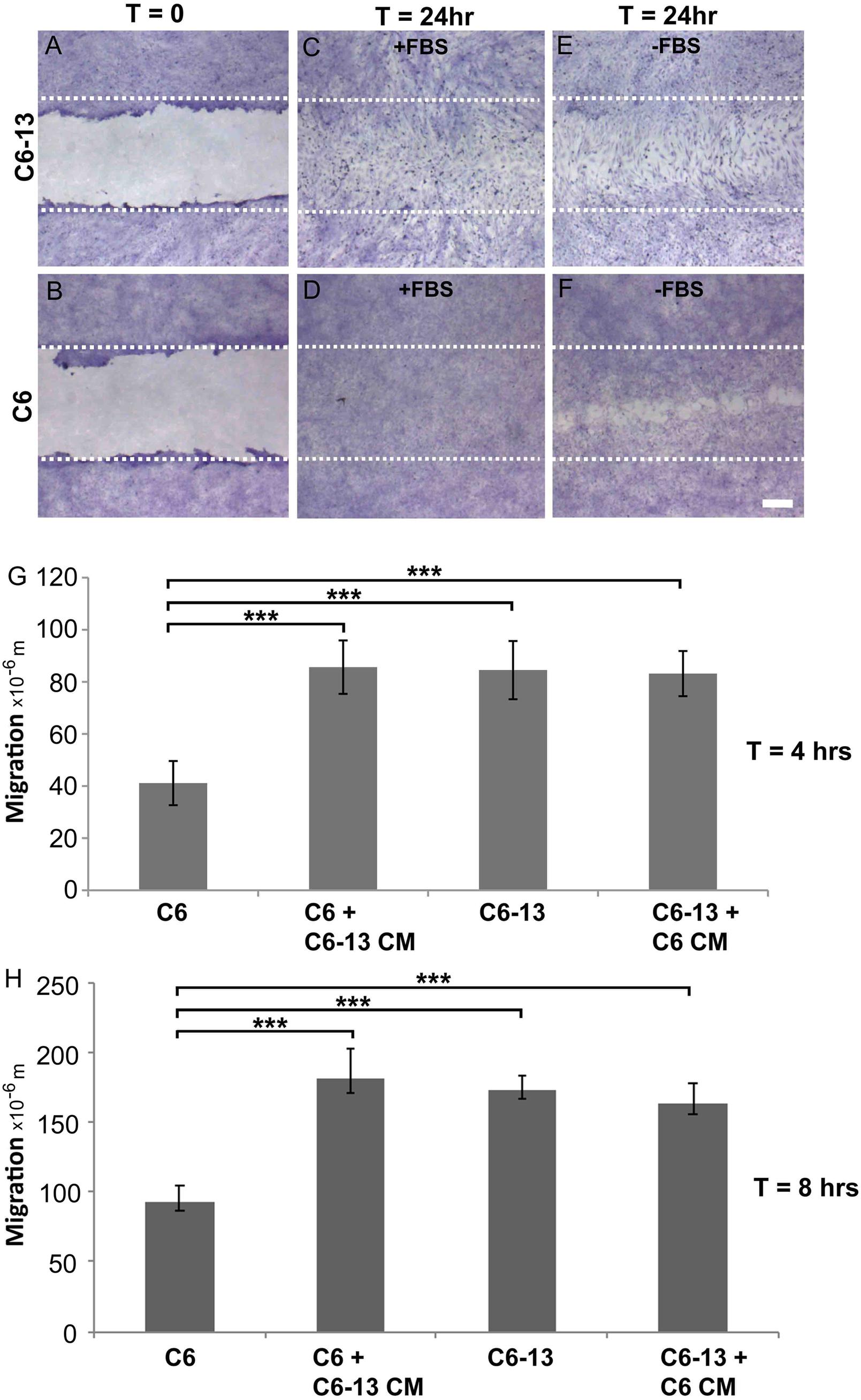
Figure 1. (A–D) Connexin expression increases migratory phenotype. (A,B) Confluent cultures of C6 and C6-13 cells underwent procedures for scrape-wound with representative images captured at T = 0 h. (C,D) At T = 24 h, in the presence of FBS, both C6 and C6-13 cells resulted in complete closures of the wound. Differences in cell density and morphology are noted. C6-13 cells are elongated, even within the unscraped regions that indicated their movement into the newly created space. Under the same conditions, this greatly contrasted C6 cells that appeared small and compact. (E,F) To differentiate effects of migration from cell proliferation, C6 and C6-13 cells were monitored in the absence of growth factors contained within FBS. Full recovery was only observed for C6-13, but not for C6. The white lines represent the limits of the original scrape. Cells were fixed with methanol and stained with crystal violet. Bar: 250 μm. (G,H) Cell migration monitored T = 4 and T = 8 h in the absence of FBS ± conditioned media (CM) from alternate cell type. Migration of C6 cells was less than C6-13 cells at both 4 and 8 h. C6 exposed to C6-13 conditioned media increase migration to levels similar to C6-13. C6-13 cell were not impacted by C6 conditioned media. Results are shown from three different sets of experiments, encompassing 6 scrapes and 18 measurements. Distance values were expressed as a mean value, with error bars representing the (±) standard error of the mean. Significance levels: ∗∗∗p ≤ 0.001.
Conditioned Media of C6-13 Contains Pro-Migratory Paracrine Signals
To determine the influence of extracellular factors produced by high motility gliomas, we next carried out conditioned media transfer experiments. While the conditioned media from C6 cells demonstrated little-to-no impact, the secretome of C6-13 cells was found to increase the motility of the parental line (Figures 1G,H). These findings suggest C6-13 factor that promote glioma migration.
Morphological Assessment Reveals Changes in the Actin Cytoskeleton
Cancer cells rely on their actin cytoskeleton to orchestrate movements through the ECM via the generation of protrusions in their plasma membranes (Machesky, 2008). Under subconfluent conditions, cells were examined by light microscopy. At low densities, C6 cells were elongated presenting fewer cellular contacts (Figure 2A). C6-13 cells on the other hand were larger and flat, with numerous plasma membrane ruffles that resembled lamellipodia and/or filopodia-like structures (Figure 2B). To examine the actin cytoskeleton during migration, cells were subjected to wound-healing prior to fixation and actin staining. Here, the stellate cytoskeletons of C6 cells (Figure 2C) greatly contrasted with the extended appearance and directional alignment of C6-13 (Figure 2D). Morphological features observed with C6-13 are consistent with aggressive glioma cells.
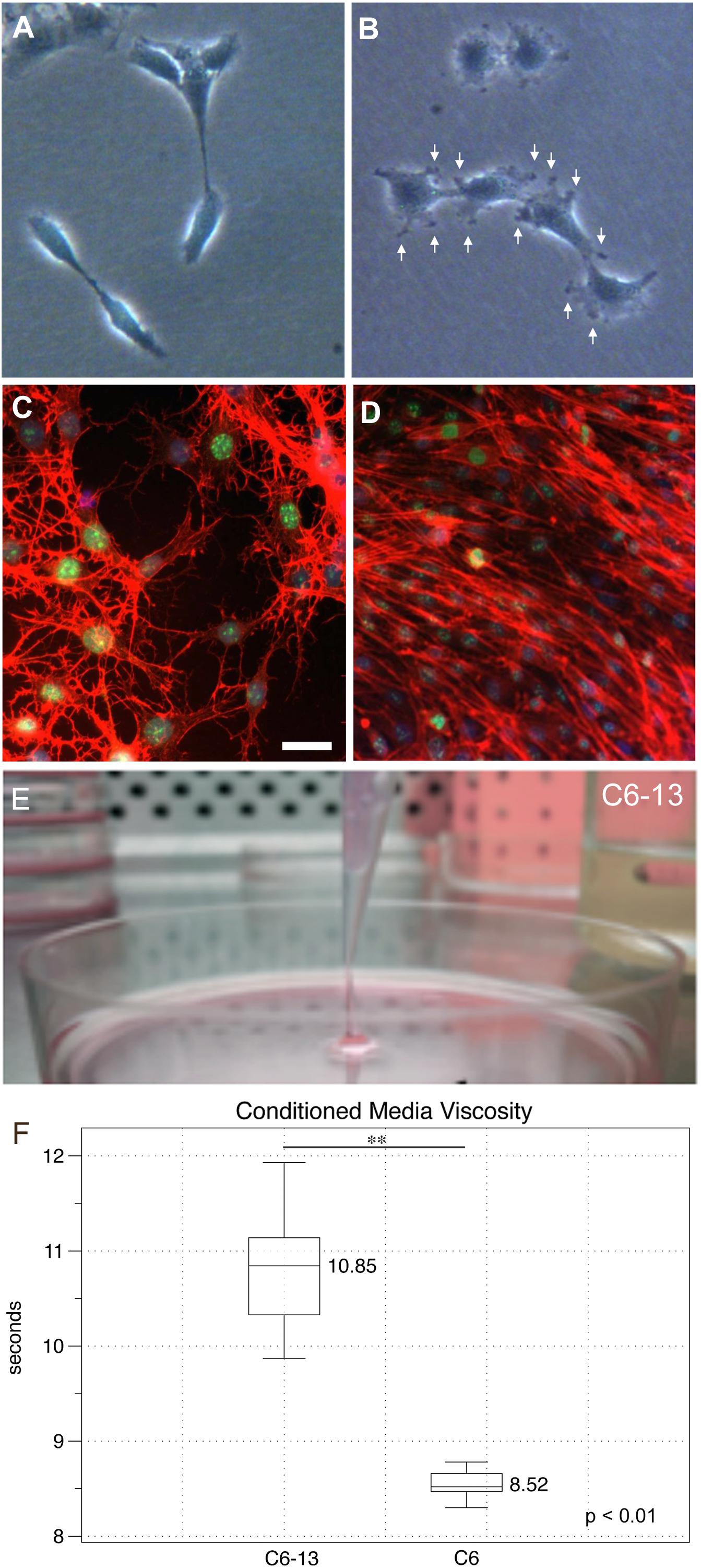
Figure 2. Morphology, actin organization and conditioned media of C6/C6-13. Subconfluent cultures of C6 (A) and C6-13 (B) cells. C6 present a spindle appearance, while C6-13 are flat and exhibit membrane ruffles and protrusions (arrows). (C,D) Actin (red) organization under scrape-wound conditions (-FBS, 15 h). (C) C6 actin stress fibres presented a stellate, random orientation at the wound gap. (D) C6-13 actin fibres were aligned between cells and parallel with the movement into the space. Nuclei were co-stained by DAPI (blue) and the cell proliferation marker Ki-67 (green). Bar: 20 μm. (E) C6-13 conditioned medium exhibited a higher viscosity that was readily observed during aspirations. (F) Conditioned media viscosity measurements. Box and whisker plot of timed laminar flow demonstrate increased viscosity of C6-13 conditioned media. Whiskers designate absolute max/min values (N = 8); significance using Student’s T (∗∗p ≤ 0.01).
Secretome Changes Associated With C6 Glioma Motility
During exchanges of spent media, we noted that C6-13 produced a viscous medium that resembled a loose gel. We were able to aspirate this gel-like media at distances >2 cm from the surface of the culture plate (Figure 2E). Timed viscosity measurement demonstrated significant increases in the mechanical resistance of C6-13 conditioned media (Figure 2F).
Based on these observations, we next sought to investigate the proteins differentially secreted by C6/C6-13. In four biological replicates, peptides were covalently modified by “light” or “heavy” dimethyl (Chen et al., 2012). The full list of proteins identified are provided (Supplementary Table S1). Significance (p ≤ 0.05) was accessed by Student’s T using Perseus (Tyanova and Cox, 2018). Volcano plot and table of differentially regulated proteins are shown in Figures 3A,F. This analysis revealed significant increases in 5 proteins: matrix metalloproteinase-3 (MMP3, >+8.5-fold), osteopontin (SPP1, >+7.4-fold), aldo-keto reductase family 1, member B10 (ARK1B10 >+6.4-fold), serpin family E member 2 (Serpine2, +2.5-fold) and phosphoglycerate kinase 1 (PGK1, +2.4-fold); and significant decreases in 12 proteins: elastin microfibril interface-located protein 1 (EMILIN1, < -15.5-fold), collagen type XII alpha 1 chain (COL12A1, < -11.3-fold), reelin (RELN, < -10.2-fold), dystroglycan 1 (DAG1, < -8.8-fold), chondroitin sulfate proteoglycan 4 (CSPG4, -6.0-fold), histone cluster 2 H4 family member A (Hist2h4, -5.9-fold), cadherin 2 (CDH2, -4.9-fold), transcobalamin-2 (TCN2, -3.8-fold), serpin family F member 1 (SERPINF1, -3.0-fold), N-acylsphingosine amidohydrolase 1 (ASAH1, -2.9-fold), tubulin beta chain (TUBB, -2.4-fold), and out at first homolog (OAF, -2.2-fold). Representative MS and MS/MS spectra are provided: reelin (RELN, GENVQFQWK, Ratio < -10, Figure 3B), MMP3 (VWEEVTPLTFSR, Ratio > +10, Figure 3C), and osteopontin (HSDAVATWLKPDPSQK, Ratio > +10, Figure 3D). SDS-PAGE/Western blot was employed to confirm expression. MMP3 and osteopontin were quantified as “heavy-only” peptides suggesting their sole expression with C6-13. Peptides form fibronectin from this dataset demonstrated a ratio of ∼1:1 suggesting their equivalent expression between C6 and C6-13 cells. Relative measurement of osteopontin by Western blot (Figure 3E) confirmed increases this protein in C6-13 whole cell lysates and conditioned media. A summary of secretome proteins significantly changed with Cx43 expression is provided in Figure 3F.
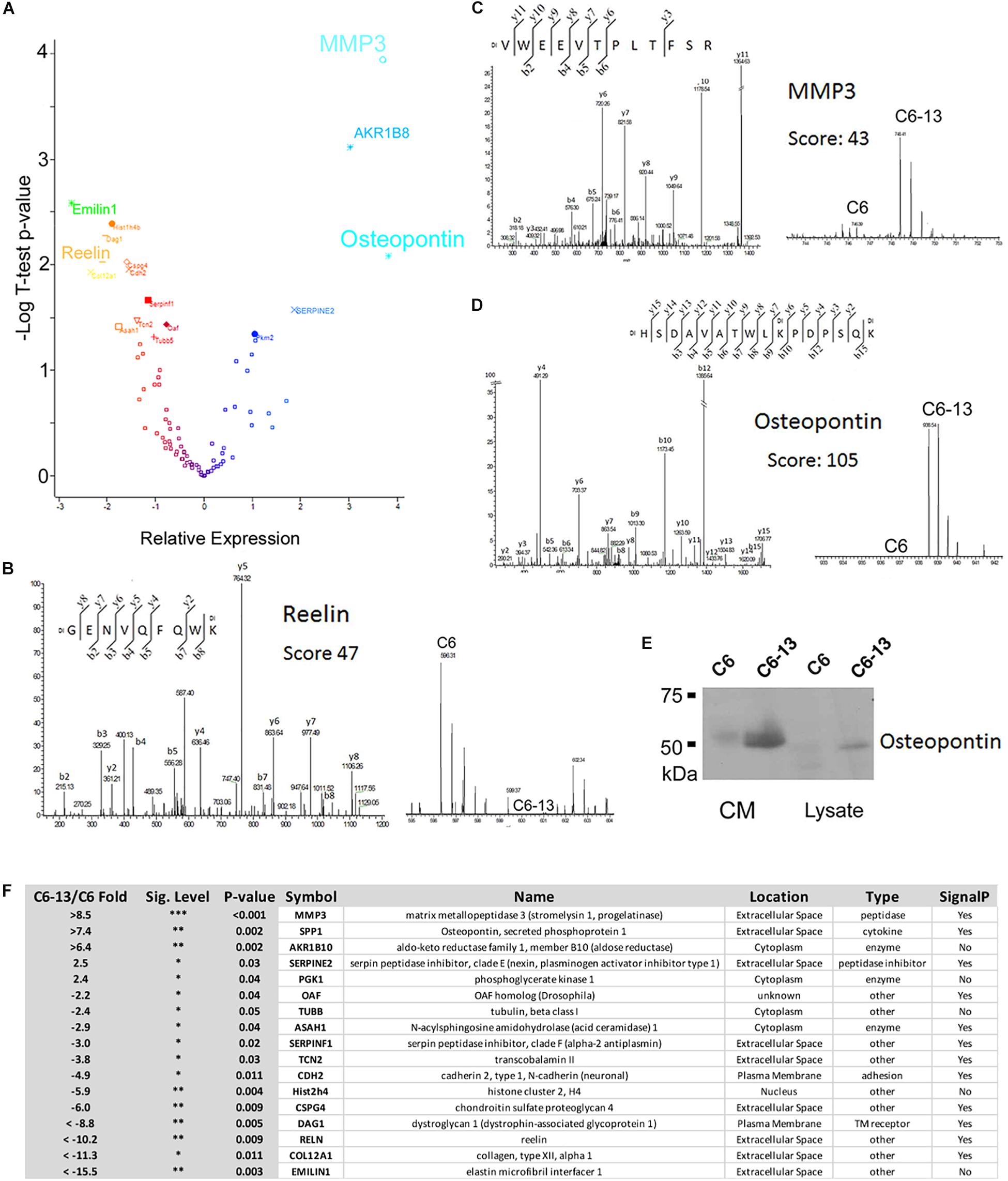
Figure 3. Secretome analysis identifies differentially expressed proteins. (A) Volcano plot of secretome proteins (T-test P-value, and log2 ratio) to identify differential expression. These proteins include reelin, MMP3 and osteopontin. Select MS/MS and MS spectra: (B) Reelin; (C) MMP3; and (D) Osteopontin showing identity and differential increases with C6/C6-13. (E) SDS-PAGE/Western blot of lysates and conditioned media (CM) supports increases in osteopontin in C6-13. (F) Table summarizing proteins found to be differentially expressed: fold-change ratio, P-value, anticipated cellular location, function/enzyme type and presence of a protein secretion signal.
Targeted Detection of Osteopontin and MMP3 by HR-MRM
Motivated by these findings we sought to validate the expression of osteopontin and MMP3 by HR-MRM (Shi et al., 2016). HR-MRM detections were based on LC retention time, parent ion mass-to-charge (m/z), CID collision energy (Q2) and full-scan high resolution MS/MS (TOF) spectra. Chromatographic traces from 3 biological replicates (Figure 4) demonstrate osteopontin and MMP3 in the media of C6-13, and the near-absence of these proteins in the media of C6 cells (Figures 4C,F). Chromatographic traces, sequences, and m/z (parent and selected daughter b/y-ions) are shown (Figures 4B,E,H). Peptides from fibronectin were used to normalize total signal area from these chromatographic profiles (Figure 4G).
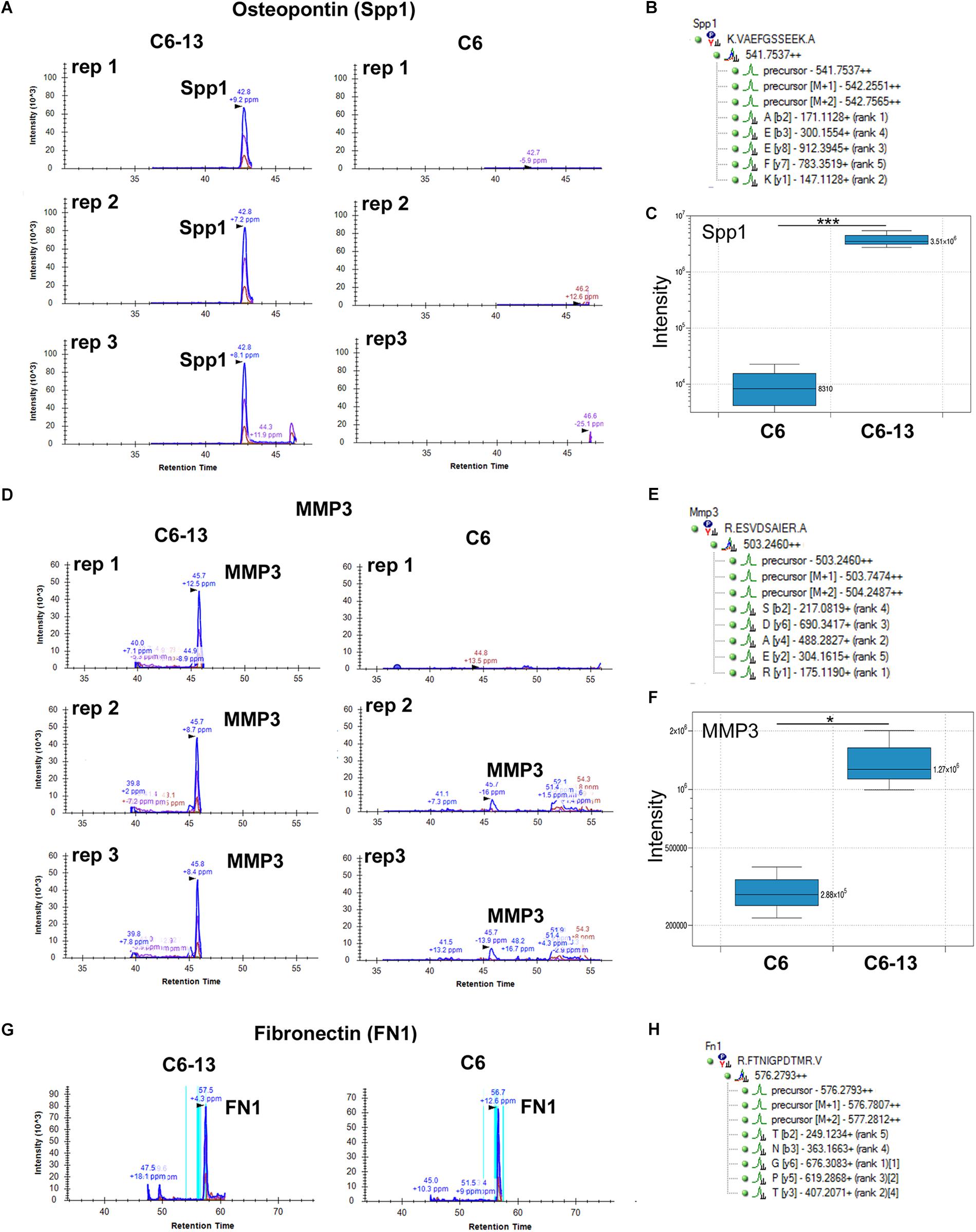
Figure 4. Binary increases of osteopontin and MMP3 in high motility C6-13. Targeted detection of (A) osteopontin/SPP1 and (D) MMP3 monitored by HR-MRM in biological triplicates of C6/C6-13 conditioned media. (B,E) Sequence, parent and fragment ions (m/z) used to monitor MMP3/osteopontin proteins. Significant increases of (C) osteopontin and (F) MMP3 are observed with C6-13 secretome (∗p ≤ 0.05, ∗∗∗p ≤ 0.001). (G,H) Fibronectin observed in C6/C6-13 was used to monitor sample load and signal normalization.
MMP3-Mediated Proteolytic Activity in Motile Glioma
MMP3 was robustly observed in the conditioned media of C6-13, but lacking with C6 cells (Figures 4D,F). To confirm these findings, we next utilized a polyclonal rabbit antibody (ProteinTech Group, 17873-1-AP) raised against the full-length sequence of MMP3 (minus the secretion signal peptide, A.A. 1-25). Consistent with our datum thus far, MMP3 was observed in the conditioned media of C6-13, but not C6 cells (Figure 5A). To ensure equal loading, BCA quantified protein were accessed using parallel run SDS-PAGE gels stained with Coomassie (Supplementary Figure S1). Using discovery and targeted proteomics (HR-MRM, QTOF), and SDS-PAGE/Western blot analysis, these findings demonstrate osteopontin and MMP3 are selectively expressed with C6-13.
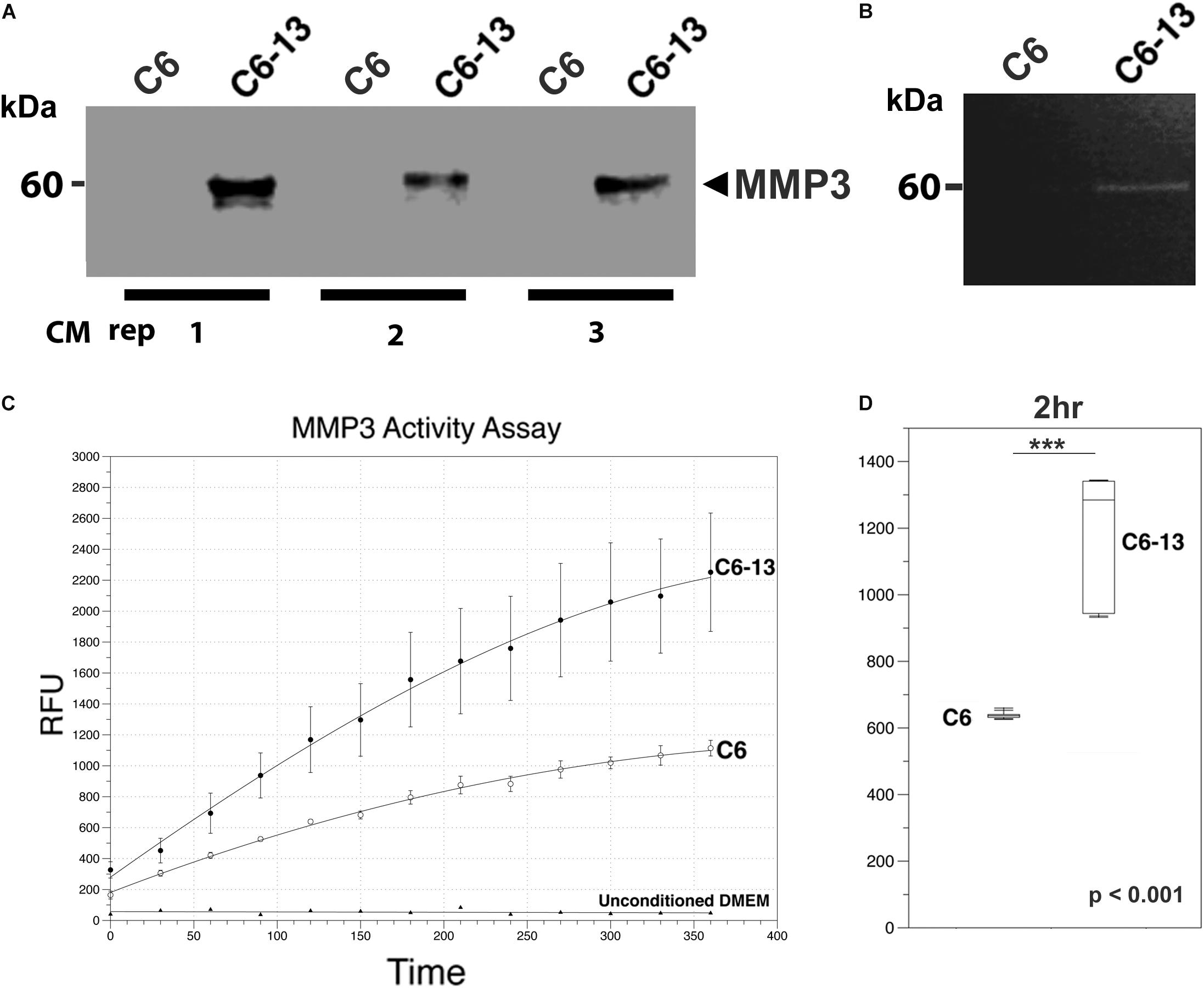
Figure 5. MMP3 is active in the ECM of high motility glioma. (A) SDS-PAGE/Western blot demonstrates the binary increases of MMP3 in C6-13 vs. C6 glioma conditioned media (CM) in 3 biological replicates. (B) Increases in gelatinase activity associated with MMP3 is observed in the conditioned media of C6-13, but not C6 cells. (C) Cleavage of the MMP3-specific fluorgenic substrate (NFF-3) demonstrates increased proteolytic activity in C6-13 CM. Limited cleavage of NFF-3 is observed with C6. Fresh, unconditioned media served as a negative control for each time point. (D) Representative, quantitative values from the 2 h time point demonstrates robust increases in MMP3 activity with C6-13.
Based on these findings, we next asked if the expression of MMP3 translated into proteinase activities within the conditioned medium of C6-13. To address this, we conducted a zymographic analysis and confirmed the gelatinolytic activity corresponding to the mass of MMP3 detected by Western blotting (representative figure shown, Figure 5B). To provide additional lines of evidence, we also monitored the protease using the fluorogenic substrate NFF-3 (excitation/emission = 325/393 nm) (Nagase et al., 1994). NFF-3 is a highly specific substrate having high selectively for MMP3 (kcat/Km = 218,000 s-1 M-1), and to a much lesser degree, MMP9 (kcat/Km = 10,000 s-1 M-1) (Nagase et al., 1994). Over the course of 6 h, the conditioned medium of C6-13 demonstrated significant, robust activity (Figures 5C,D). As some NFF-3 signal was observed with C6, suggests the presence of other MMPs (Zhang et al., 2003; Sahin et al., 2005). This analysis demonstrates increased rates of C6-13 migration coincides with dramatic increases in MMP3 and proteolytic activity within the ECM.
Biological Function, Localization, and Cellular Pathways
How the ECM is dynamically engaged in invasive glioma is poorly understood. Our experiments demonstrate that C6-13 cells increase viscosity and secrete promigratory factors that appear to engage pathways for glioma migration. We used an integrated proteomic and informatic strategy to examine the functional pathways and networks that are downstream of Cx43. Of the 91 total proteins identified by our secretome study (full list provided, Supplementary Table S1), 84 were annotated within the IPA Knowledge Base and considered further. SignalP (version 4.1, stringency 0.6)4 (Petersen et al., 2011; Nielsen, 2017) observed 45 of these proteins having signals consistent with their production and export via the protein secretion pathway (Supplementary Table S1). Further inspection of our data using the Human Cancer Secretome Database (HCSD)5 (Feizi et al., 2015) finds 98.8% (83/84) of C6/C6-13 proteins have been identified within the secretome of other cancers (Feizi et al., 2015); with 88% (74/84) observed with other GBM studies (Kashyap et al., 2010; Formolo et al., 2011; Gupta et al., 2013; Supplementary Table S1). Secretomes of both cell lines had strong functional links to Cellular Movement (P = 2.15 × 10-16), Cell Death and Survival (2.13 × 10-15), Cancer (1.87 × 10-12), and PTM (post-translational modification) (2.46 × 10-11) (Figures 6A,C). Examination for the Cellular Movement subcategory identifies proteins linked to the Migration, Invasion, and Scattering associated with the secretome of C6/C6-13 (Figure 6B).
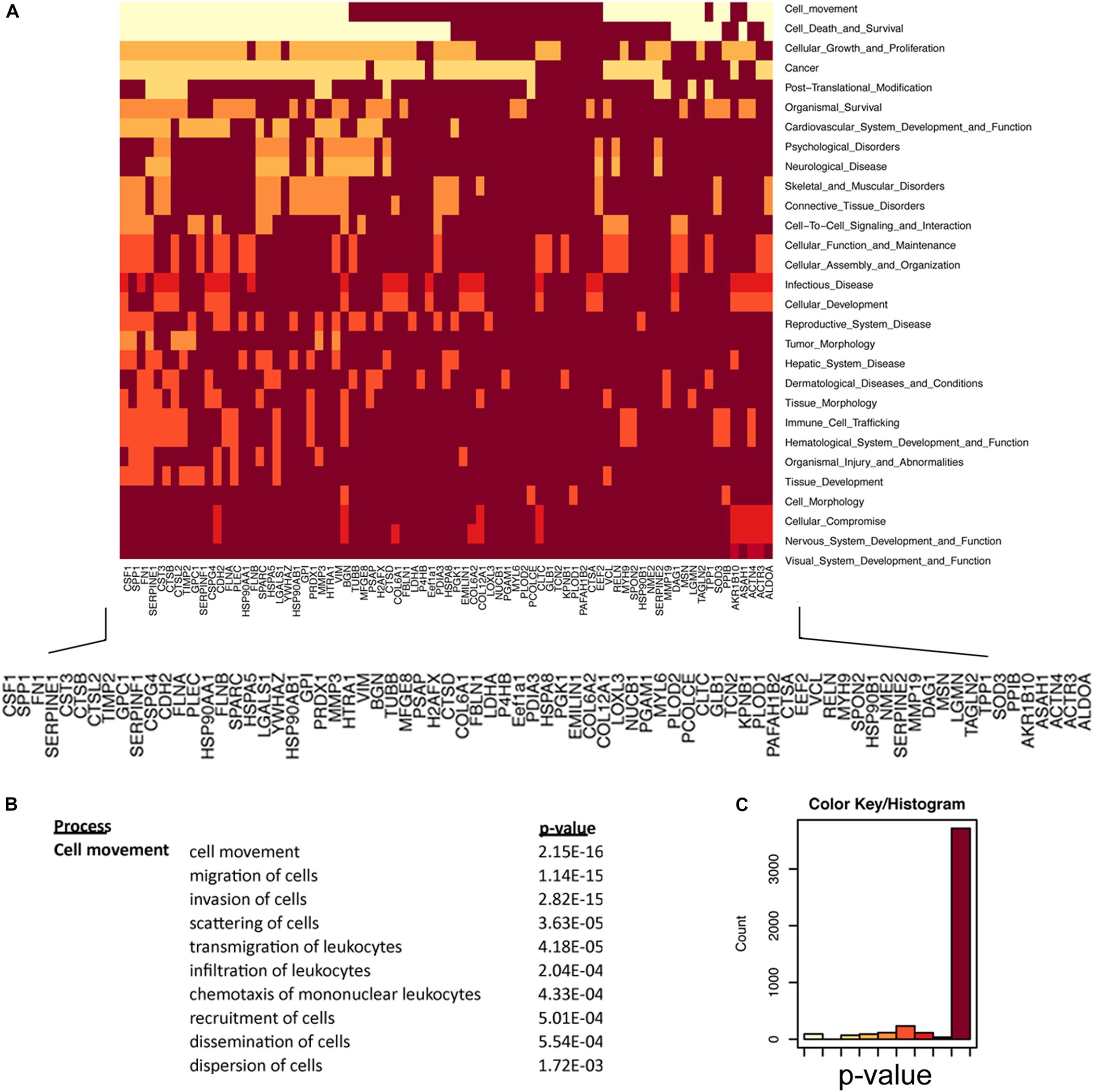
Figure 6. Regulators of cellular movement are significantly enriched in glioma secretome. (A) Heat map representation of secretome proteins and functional relationships. Lighter colors in this heat map group proteins that are statistically enriched for the listed cell function (y-axis). Protein network enrichment and statistical significance were determined by Ingenuity PathwaysTM. (B) Cell Movement (P = 2.15 × 10-16) was the most statistically significant network encoded by the secretome of C6/C6-13 cells. Migration subnetworks (and P-values) comprising the Cellular Movement category. (C) Color bin histogram representing the distribution of both significant (x-axis: left, light) and non-significant (right, dark) protein networks represented in Figure 6A.
Previous studies in glioma have demonstrated Cx43 has roles in the suppression of tumor growth (Zhu et al., 1991; Huang R.P. et al., 1998; Fu et al., 2004) and increases invasion (Kameritsch et al., 2012; Naus et al., 2016). Despite this growing body of evidence implicating connexins with GBM malignancy, the mechanism of how Cx43 impacts glioma is poorly understood. We have found C6-13 demonstrates behaviors typical of aggressive glioma – increased migration, changes in the actin cytoskeleton, increased expression of osteopontin and MMP3, and ECM remodeling via increased of MMP activity. The 17 differentially expressed secretome proteins were evaluated to determine links to phenotypic changes that were observed with Cx43. As outlined in Table 1, the top 6 functional annotations and disease processes differentiated by the Cx43 secretome (by rank) Metastasis (P = 5.51 × 10-7, Activation Z-Score +0.132), Aggregation of Cancer Cells (P = 3.36 × 10-6), Disruption of Cytoskeleton (P = 4.00 × 10-6), Metastasis of Tumor cell lines (P = 4.99 × 10-6, Activation Z-Score = +0.132), Breast/Colorectal Cancer (P = 5.57–5.97 × 10-6) and Advanced Malignant Tumor (P = 9.13 × 10-6, Activation Z-Score = +0.297).
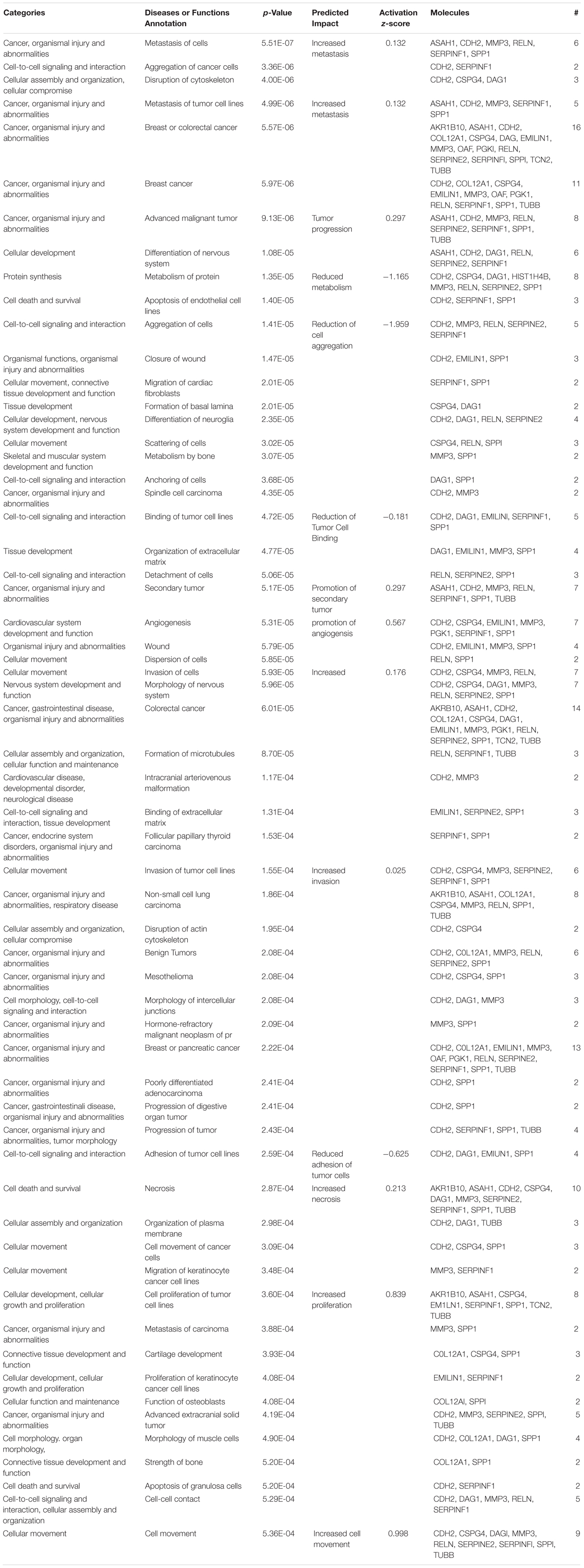
Table 1. Pathways analysis of the 17 differentially regulated secretome proteins found in C6-13. Where possible, the activation Z-score functions of the ingenuity pathways tool was used to predict the biological impact of C6-13 expression.
To determine Cx43 intracellular signals, we next conducted a retrospective analysis of immunoprecipitated Cx43–protein interactions obtained from C6-13 glioma cells (Chen et al., 2012). Figure 7 summarizes the functional annotations and disease processes for Cx43 (secretome, Figure 7A) and the connexin proteome (Cx43–protein interactions, Figure 7B). Top functions associated with Cx43 interaction in order of rank implicate networks for Cell Death and Survival (P = 2.99 × 10-11), Migration (5.15 × 10-9), Cellular Movement (6.79 × 10-9), Proliferation (3.16 × 10-10), and Apoptosis (3.06 × 10-9).
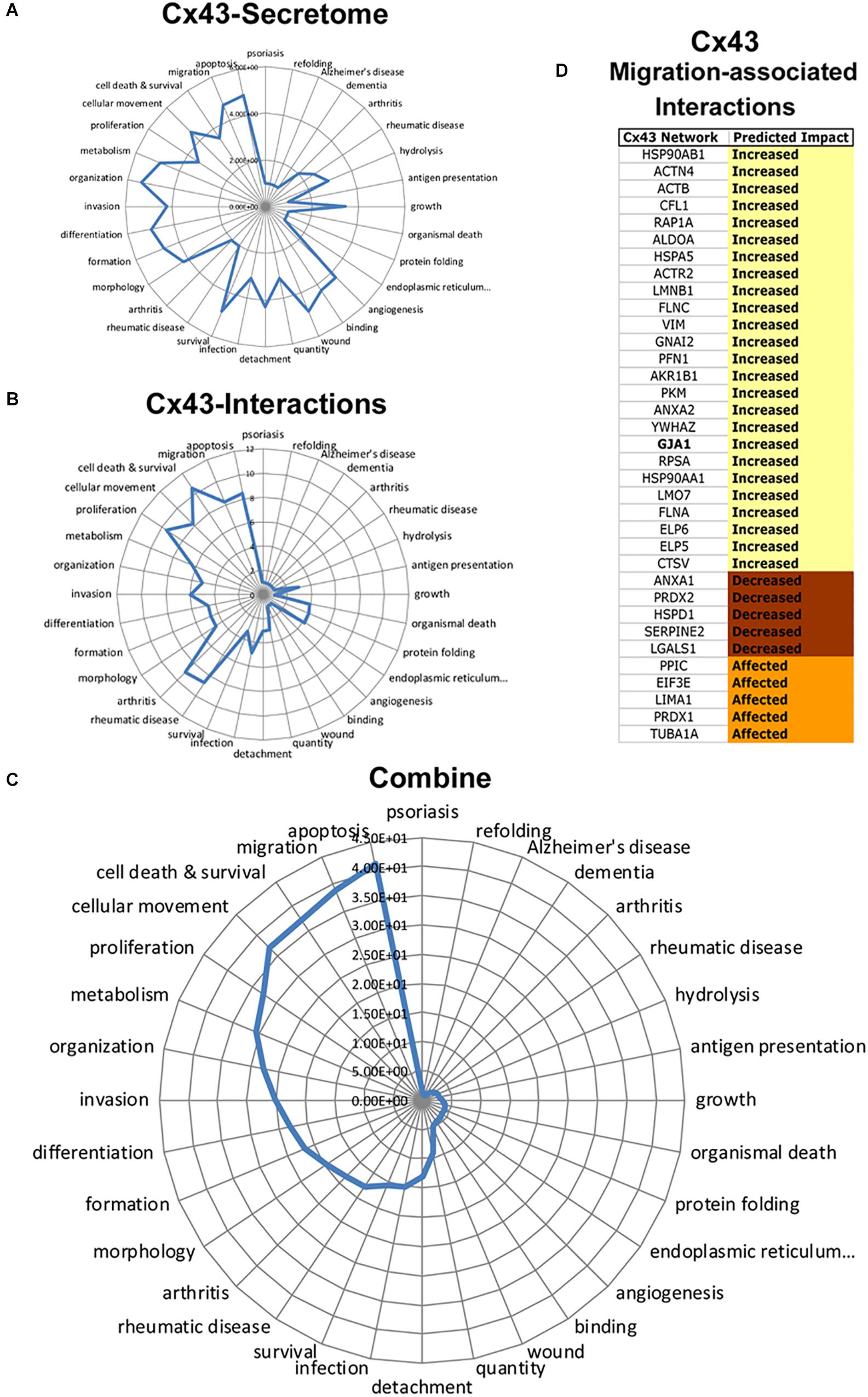
Figure 7. Integrated analysis of Cx43 secretome and Cx43 protein–protein interactions reveals synergistic networks. Radar plots of pathways associated with the Cx43 (A) secretome, and (B) interactome plotted as -log P-value. C) Analysis of Cx43 networks demonstrates the coherence of “Apoptosis,” “Migration,” “Cell Death and Survival,” “Cellular Movement,” and “Proliferation” between Cx43-protein interactions and secretome. (D) Downstream impact analysis (IPA) identifies interactions of Cx43 implicated with migration. Based on IPA informatics (Activation Z-Score) the majority of these interactions are expected to increase migration.
To determine synergistic impact of the Cx43 secretome and protein–protein interactions, we next utilized an approach to calculate overlapping P-values:
Synergistic networks identified by this approach find cellular pathways of: Apoptosis (Synergistic Score = 41.3), Migration (38.9), Cell Death and Survival (37.3), Cellular Movement (36.9), Proliferation (32.7), and Metabolism (30.8) (Figure 7C). Downstream Effect Analysis (Kramer et al., 2014) of interactions linked to migration demonstrated an overall abundance of factors that are expected to increase migration (25/35, 71.4%, Figure 7D), with the remaining predicted to decrease (5/35, 14.3%) or generally affecting migration (5/35, 14.3%). These findings predict the intersecting pathways of Cx43 conspire to increase migration in glioma by concentrating pro-migratory proteins to the cortical surface of the cell and the degradation and remodeling of the ECM.
Discussion
Cancer cell invasion requires the coordination of cellular programs regulating epithelial-mesenchymal transition, motility, and remodeling of the ECM. The most prominent feature underlying GBM malignancy is drug resistance and the ability of tumor cells to invade. A key goal of glioma research has been to seek a deeper understanding of the tumor microenvironment with the overall goal of developing approaches to limit infiltration (Giese and Westphal, 1996). C6 cells expressing connexin demonstrate lower cell proliferation that maintain a commonly held view of Cx43 being an inhibitor of GBM (Zhu et al., 1991). However, a number of recent reports have uncovered a “connexin paradox” in which Cx43 is a promoter of glioma malignancy (Sin et al., 2012; Naus et al., 2016). Amongst these studies, Cx43 has been found to increase migration and invasion, and resistance to the frontline chemotherapeutic agent Temozolomide (TMZ) (Gielen et al., 2013; Munoz et al., 2014; Murphy et al., 2016; Grek et al., 2018; Wang et al., 2018). Based on this body of knowledge, peptidomimetics that modulate the functions of Cx43 are currently being evaluated as adjuvants to address drug resistance in GBM (Grek et al., 2018). A better understanding of how Cx43 impacts GBM may provide avenues and novel approaches for therapeutic design.
The microenvironment influences tumor growth and behavior, including the progression and invasion of GBM. The microenvironment represents a highly dynamic niche by which glioma cells takeover the cellular programs that are normally reserved for development, tissue remodeling and repair (Quail and Joyce, 2017). As a complex meshwork, the tumor microenvironment is represented by stromal and tumor cell-derived cytokines, growth factors, enzymes and matrix proteins. Increases in glioma migration/invasion often coincide with aggressive cellular phenotypes, including alterations in cell morphology, cytoskeleton and adhesion (Lin et al., 2002). Increases in glioma migration are frequently accompanied by the secretion of regulatory matricellular proteins such as tenascin (Deryugina and Bourdon, 1996) and osteopontin/SPP1 (Jan et al., 2010). Cx43 has been shown to promote migration by wound healing (Bates et al., 2007), TranswellTM assay (Bates et al., 2007) and brain slices (Oliveira et al., 2005), and regulate expression of MMP-2, MMP-9 (Zhang et al., 2003), osteopontin (Naus et al., 2000), CCN1 (Naus et al., 2000), and CCN3/NOV (Sin et al., 2008). Despite the growing body of evidence linking the gap junction protein to the control of migration and the ECM, the mechanisms of Cx43-induced migration have yet to be resolved. The findings presented here provide several lines of evidence by which Cx43 may influence GBM. In particular, this study corroborates changes in glioma motility, morphology and cytoskeletal organization with expression of Cx43 (Bates et al., 2007; Crespin et al., 2010), and is also the first study to uncover secretome and ECM mechanisms downstream of Cx43 as a driver of malignancy. Providing insight, we noted C6-13 significantly changed the viscosity and thickness of conditioned media. Indeed, the tension of cell substrates has been shown to increase migration of a number of human and rat gliomas, including the C6 line (Hegedus et al., 2006; Ulrich et al., 2009). Changes in viscosity would also foreseeably limit free diffusion of soluble factors (enzymes, chemokines, cytokines and matrikines) resulting in the formation of concentration gradients that maybe used as chemotactic and/or repulsive signals (Manini et al., 2018). Such mechanisms occurring with C6-13 may be used to guide migration and direction of glioma invasion.
To better understand the paracrine signals in glioma, we sought to compare proteins within the conditioned media of C6/C6-13 using stable-isotope labeling/high resolution LC-MS. Global proteins identified were categorized into 7 groups: peptidases, peptidase inhibitors, cytokines, adhesion, and transmembrane receptors/other (Supplementary Table S1). Secretome proteins accessed by IPA identified the enrichment of cellular processes: (1) Cell Movement, (2) Proliferation, and (3) Survival. Dissemination of the cell movement category (P = 2.15 × 10-16) revealed networks having close linkages to migration (1.14 × 10-15) and invasion (2.82 × 10-15). While the migration/invasion pathways linked to the global secretome is intriguing, it is important to note the majority of proteins identified (97/114) were considered to be unchanged between high and low motility cells (Supplementary Table S1). Indeed, the majority (88%) of the proteins characterized have been identified within other GBM secretomes studies, presumably with gliomas of differing rates of mobility. Such congruence between secretomes suggest the ECMs of high-grade gliomas are “primed” – but remain quiescent in the absence of specific cell migration signals. In light of an accumulating body of evidence linking connexins to GBM malignancy, including our observation of Cx43 conditioned media increases glioma motility, it is reasonable to suggest the Cx43 and/or ECM proteins downstream of it may serve this role. In particular, we identify 17 (5 up- and 12 down-regulated proteins) differentiating the secretome of C6-13 (Figure 3F). Indeed, Z-Score Activation implicates the Cx43 as a driver of cell migration, tumor progression and malignancy (Table 1). Of the cellular functions reporting pathway activation direction, the Cx43 secretome is predicted to increase Metastasis, Tumor Progression, Secondary Tumor Formation, Angiogenesis, and Invasion; and decreased probability of Protein Metabolism, Aggregation, and Tumor Cell Binding. These findings predict the involvement of the Cx43 secretome in migration and malignancy of GBM.
Our study shows that high motility gliomas expressing Cx43 differ from their parental counterparts by the upregulation of MMP3, osteopontin, aldo-keto reductase family 1 member B10 (ARK1B10), and to a lesser degree SERPINE2 (2.5-fold, P = 0.03), and phosphoglycerate kinase 1 (2.4-fold, P = 0.04). As migratory behavior is a hallmark of GBM, we focused on components having links to this process. In particular, there is a strong correlation between patient outcome with the activity of proteases residing within the extracellular space (Friedl and Wolf, 2008; Ren et al., 2015; Sun et al., 2015; Gong et al., 2016; Jia et al., 2017). The activity of ECM proteases such as MMPs are widely considered to be major determinants of patient outcome (Lu et al., 2012). Signals making up the MMP protease-web include the activation/inactivation of growth factors, shedding of cell surface adhesion molecules, the release of ECM-bound cytokines and growth factors, and the activation of cryptic peptides (Lopez-Otin and Overall, 2002). Cryptic peptides signals (a.k.a. matrikines and matricryptins) are generated by the precision degradation of the ECM (e.g., collagen and laminin), and matricellular proteins, such as osteopontin and SPARC (Wells et al., 2015; Eckhard et al., 2016). A perfect example of matrikine signals regulating migration/invasion is the signaling nexus mediated by MMP3/osteopontin (Agnihotri et al., 2001). Osteopontin is a secreted, sialic acid-rich glyco/phosphoprotein (Denhardt and Noda, 1998) that plays a major role in cancer progression (Chong et al., 2012), and the in vivo invasion of glioma cells (Jan et al., 2010). MMP3-cleaved osteopontin acquires new functions, one of which is to regulate adhesive and migratory stimuli through cell surface integrins (Agnihotri et al., 2001; Kon et al., 2014). Matricryptic signals encoded by MMP3/osteopontin have been shown to expose RGD sequences that bind integrins and thus affect adhesion, migration, and survival (Ruoslahti, 1996). Suggesting Cx43 may engage similar pathways, quantitative proteomic datasets identified the dramatic, near binary (on/off) expression of MMP3 and osteopontin. These increases were substantiated by SDS-PAGE/Western blot and targeted HR-MRM analyses. Our findings are consistent with the observation of MMP3 at invasive fronts of GBM tumors, and the reduction of invasion potential after MMP3 loss (Jin et al., 2013). Based on this data we propose matrikine signals downstream of Cx43/MMP3/osteopontin as a possible avenue to inhibit GBM.
Synthesized as inactive zymogens, cross-talk between proteases are required for the removal of the auto-inhibitory domain of MMPs, resulting in the catalytic activation and amplification of degradative signals (Sevenich and Joyce, 2014). These activities are further potentiated by interactions with other proteins, including natural inhibitors of MMP (e.g., TIMPs) (Wang et al., 2000; Lu et al., 2004). In light of the multiple layers of MMP control, we sought to survey the activities of the enzyme within the ECM materials. This study is the first to implicate ECM-remodeling and the protease-web down-stream of MMP3 with Cx43 motility. Here, gelatin zymography and loss of MMP3-specific substrates confirmed this activity within the conditioned media of C6-13 cells. In keeping with this view, it is important to note a significant number of ECM components including collagen alpha 12 (COL12A1), reelin (RELN), elastin microfibril interface 1 (EMILIN), and chondroitin sulfate proteoglycan (CSPG4) (Figure 3F) were lost and/or down-regulated with Cx43. We speculate such losses, especially within the context of high MMP3 activity, may encode molecular events underlying movement in C6 glioma cells. Experiments to investigate MMP3 substrates are currently underway.
In addition to MMP3/osteopontin, our proteomic datum identifies the up-regulation of ARK1B10 within the Cx43 secretome. The AKRs are a super family of NADPH-dependent oxidoreductases that metabolize a wide variety of carbohydrates, steroids, prostaglandins, aldehydes, ketones, and xenobiotic compounds (Matsunaga et al., 2012). AKR1B10 is a 36 KDa reductase that shares a 71% identity and α/β-barrel topology with the better studied AKR1B1 (Gallego et al., 2007). The AKR1 subfamily of enzymes have a critical role in the detoxification of cytotoxic compounds by the reduction of aromatic and aliphatic aldehydes, carbonyl compounds, and drug ketones (Matsunaga et al., 2012). Studies employing drug resistant cell lines have provided some evidence implicating members of the ARK superfamily with the acquisition of therapeutic resistance in a variety of cancers (Matsunaga et al., 2012). In medullablastoma and colon cancers, AKR1B10 has been shown to be involved with the metabolic deactivation of the chemotherapeutic agents cyclophosphamide (Bacolod et al., 2008) and mitomycin-c (Matsunaga et al., 2011). Our identification of AKR1B10 within the Cx43 secretome provides clues into possible mechanisms of GBM malignancy related to motility/invasion and TMZ resistance (Gielen et al., 2013; Munoz et al., 2014; Murphy et al., 2016; Grek et al., 2018; Wang et al., 2018). The cytotoxic effects of TMZ are largely mediated by a key carbonyl that undergoes decarboxylation and ring opening to produce the methyldiazonium ion that in turn alkylates DNA leading to cell death (Saleem et al., 2003). Although the exact role of ARK1B10 in TMZ metabolism is currently unknown, the chemical conversion of this carbonyl group to an alcohol (or further reduced form), would inhibit decarboxylation and thus methyldiazonium formation. Furthermore, these findings of ARK1B10 in the Cx43 secretome also open the possibility of gliomas utilizing Cx43 to not only increase migration but additionally set up ECM defenses to inactivate therapeutic agents prior to their arrival at gliomas. Monitoring the metabolic conversion of TMZ in the secretome and media of cells expressing Cx43/ARK1B10, alongside the testing of ARK1/ARK1B10 inhibitors (Huang et al., 2016) as possible adjuvants, would seem to be logical next steps.
Given our observed changes in the glioma secretome, plasma membrane protrusions and the reorganization of the actin cytoskeleton, we sought to characterize networks associated with Cx43. We anticipate changes occurring with Cx43 expression would be accompanied by a significant rewiring of signaling networks regulating glioma. Examinations of Cx43/gap junction channel-dead mutants, truncations, and pharmacological gap junction channel blockers have isolated the effects of Cx43-induced migration to the long c-terminal tail (Bates et al., 2007; Crespin et al., 2010) - the principle site of of Cx43 protein-protein interaction (Giepmans, 2004). Here, a large variety of regulatory factors are known to co-localize with Cx43 at gap junctions (Giepmans, 2004; Laird, 2009). We therefore examined C6-13/Cx43 co-IPs (Chen et al., 2012). Cx43 protein–protein interactions demonstrated the statistical enrichment of pathways having known functions in cell migration. Migration-associated Cx43 interactome highlights components of the actin-based cytoskeleton (alpha-actinin-4, actin (ACTB, ACTR2), filamin, and vimentin) alongside regulators of cytoskeletal dynamics (cofilin and profilin, Figure 7D). These proteins belong to a pool of accessory proteins that are mechanistically responsible for the scaffolding, polymerization and depolymerization of actin filaments, including the crawling and locomotion of cancer cells (Yamaguchi and Condeelis, 2007; Pollard and Cooper, 2009). This overlap of proteins with Cx43 suggest the gap junction protein may influence migration by the trafficking and/or scaffolding of cytoskeletal proteins to the plasma membrane or sites of intercellular adhesion. This study identifies secretome/protein–protein interaction networks that further strengthen Cx43 as a driver of motility and therapeutic resistance in glioma.
Author Contributions
QA, MM, and VC designed the study and wrote the manuscript. NS, LF and VC conducted mass spectrometry. JN and VC conducted MS data analysis and informatics. P-OS and MM conducted microscopy and migration assays. EO, AP, CS and CL performed cell culture, zymography and NFF-3 assays. CN, LF, W-CS and VC provided reagents, feedback and conceptual advice. CN, W-CS, LF, and VC secured funding and research infrastructure. All authors discussed the results and commented on the manuscript at all stages.
Funding
VC was supported by the National Science and Engineering Research Council of Canada (NSERC, RGPIN-2018-04261), the Brandon University Research Committee (BURC), and the Brandon University Faculty of Science. CN, W-CS, and LF were supported by the Canadian Institute for Health Research. CN was supported by the Canada Research Chairs (CRC) Program. Infrastructure support from the Canadian Foundation for Innovation, and Computational Resources from the BC Proteomics Network are also acknowledged. MM was supported by the Institute of Health Research and Ligue Nationale Contre le Cancer (Comités Charente-Maritime, Deux-Sèvres, Morbihan, Vienne; France). QA was supported by a NSERC graduate fellowship. AP was supported by a CIHR Banting and Best Graduate Fellowship. EO was supported by a Manitoba Graduate Fellowship. JN and CL were supported by a Bruce and Jane Forest Memorial Scholarship.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.00143/full#supplementary-material
Abbreviations
Cx43, connexin43; ECM, extracellular matrix; GBM, glioblastoma multiforme; TMZ, Temozolomide.
Footnotes
- ^proteomeXchange.org
- ^https://chemdata.nist.gov
- ^thebiogrid.org
- ^http://www.cbs.dtu.dk/services/SignalP/
- ^www.cancersecretome.org
References
Agnihotri, R., Crawford, H. C., Haro, H., Matrisian, L. M., Havrda, M. C., and Liaw, L. (2001). Osteopontin, a novel substrate for matrix metalloproteinase-3 (stromelysin-1) and matrix metalloproteinase-7 (matrilysin). J. Biol. Chem. 276, 28261–28267. doi: 10.1074/jbc.M103608200
Bacolod, M. D., Lin, S. M., Johnson, S. P., Bullock, N. S., Colvin, M., Bigner, D. D., et al. (2008). The gene expression profiles of medulloblastoma cell lines resistant to preactivated cyclophosphamide. Curr. Cancer Drug Targets 8, 172–179. doi: 10.2174/156800908784293631
Bates, D. C., Sin, W. C., Aftab, Q., and Naus, C. C. (2007). Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia 55, 1554–1564. doi: 10.1002/glia.20569
Beljebbar, A., Dukic, S., Amharref, N., and Manfait, M. (2010). Ex vivo and in vivo diagnosis of C6 glioblastoma development by Raman spectroscopy coupled to a microprobe. Anal. Bioanal. Chem. 398, 477–487. doi: 10.1007/s00216-010-3910-6
Boersema, P. J., Aye, T. T., van Veen, T. A., Heck, A. J., and Mohammed, S. (2008). Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 8, 4624–4632. doi: 10.1002/pmic.200800297
Charles, A. C., Naus, C. C., Zhu, D., Kidder, G. M., Dirksen, E. R., and Sanderson, M. J. (1992). Intercellular calcium signaling via gap junctions in glioma cells. J. Cell Biol. 118, 195–201. doi: 10.1083/jcb.118.1.195
Chen, V. C., Kristensen, A. R., Foster, L. J., and Naus, C. C. (2012). Association of connexin43 with E3 ubiquitin ligase TRIM21 reveals a mechanism for gap junction phosphodegron control. J. Proteome Res. 11, 6134–6146. doi: 10.1021/pr300790h
Chen, V. C., Li, X., Perreault, H., and Nagy, J. I. (2006). Interaction of zonula occludens-1 (ZO-1) with alpha-actinin-4: application of functional proteomics for identification of PDZ domain-associated proteins. J. Proteome Res. 5, 2123–2134. doi: 10.1021/pr060216l
Chong, H. C., Tan, C. K., Huang, R. L., and Tan, N. S. (2012). Matricellular proteins: a sticky affair with cancers. J. Oncol. 2012:351089. doi: 10.1155/2012/351089
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi: 10.1038/nbt.1511
Cox, T. R., and Erler, J. T. (2011). Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis. Model. Mech. 4, 165–178. doi: 10.1242/dmm.004077
Crespin, S., Bechberger, J., Mesnil, M., Naus, C. C., and Sin, W. C. (2010). The carboxy-terminal tail of connexin43 gap junction protein is sufficient to mediate cytoskeleton changes in human glioma cells. J. Cell Biochem. 110, 589–597. doi: 10.1002/jcb.22554
Demuth, T., and Berens, M. E. (2004). Molecular mechanisms of glioma cell migration and invasion. J. Neurooncol, 70, 217–228. doi: 10.1007/s11060-004-2751-6
Denhardt, D. T., and Noda, M. (1998). Osteopontin expression and function: role in bone remodeling. J. Cell Biochem. Suppl. 30–31, 92–102. doi: 10.1002/(SICI)1097-4644(1998)72:30/31+<92::AID-JCB13>3.0.CO;2-A
Deryugina, E. I., and Bourdon, M. A. (1996). Tenascin mediates human glioma cell migration and modulates cell migration on fibronectin. J. Cell Sci. 109(Pt 3), 643–652.
Eckhard, U., Huesgen, P. F., Schilling, O., Bellac, C. L., Butler, G. S., Cox, J. H., et al. (2016). Active site specificity profiling of the matrix metalloproteinase family: proteomic identification of 4300 cleavage sites by nine MMPs explored with structural and synthetic peptide cleavage analyses. Matrix Biol. 49, 37–60. doi: 10.1016/j.matbio.2015.09.003
Elias, L. A., Wang, D. D., and Kriegstein, A. R. (2007). Gap junction adhesion is necessary for radial migration in the neocortex. Nature 448, 901–907. doi: 10.1038/nature06063
Feizi, A., Banaei-Esfahani, A., and Nielsen, J. (2015). HCSD: the human cancer secretome database. Database 2015:bav051. doi: 10.1093/database/bav051
Formolo, C. A., Williams, R., Gordish-Dressman, H., MacDonald, T. J., Lee, N. H., and Hathout, Y. (2011). Secretome signature of invasive glioblastoma multiforme. J. Proteome Res. 10, 3149–3159. doi: 10.1021/pr200210w
Friedl, P., and Wolf, K. (2008). Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res. 68, 7247–7249. doi: 10.1158/0008-5472.CAN-08-0784
Frisch, C., Theis, M., De Souza Silva, M. A., Dere, E., Sohl, G., Teubner, B., et al. (2003). Mice with astrocyte-directed inactivation of connexin43 exhibit increased exploratory behaviour, impaired motor capacities, and changes in brain acetylcholine levels. Eur. J. Neurosci. 18, 2313–2318. doi: 10.1046/j.1460-9568.2003.02971.x
Fu, C. T., Bechberger, J. F., Ozog, M. A., Perbal, B., and Naus, C. C. (2004). CCN3 (NOV) interacts with connexin43 in C6 glioma cells: possible mechanism of connexin-mediated growth suppression. J. Biol. Chem. 279:36943–36950. doi: 10.1074/jbc.M403952200
Furnari, F. B., Fenton, T., Bachoo, R. M., Mukasa, A., Stommel, J. M., Stegh, A., et al. (2007). Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 21, 2683–2710 doi: 10.1101/gad.1596707
Gallego, O., Ruiz, F. X., Ardevol, A., Dominguez, M., Alvarez, R., de Lera, A. R., et al. (2007). Structural basis for the high all-trans-retinaldehyde reductase activity of the tumor marker AKR1B10. Proc. Natl. Acad. Sci. U.S.A. 104, 20764–20769. doi: 10.1073/pnas.0705659105
Gielen, P. R., Aftab, Q., Ma, N., Chen, V. C., Hong, X., Lozinsky, S., et al. (2013). Connexin43 confers temozolomide resistance in human glioma cells by modulating the mitochondrial apoptosis pathway. Neuropharmacology 75, 539–548. doi: 10.1016/j.neuropharm.2013.05.002
Giepmans, B. N. (2004). Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 62, 233–245. doi: 10.1016/j.cardiores.2003.12.009
Giese, A. (2003). Glioma invasion–pattern of dissemination by mechanisms of invasion and surgical intervention, pattern of gene expression and its regulatory control by tumorsuppressor p53 and proto-oncogene ETS-1. Acta Neurochir. Suppl. 88, 153–162. doi: 10.1007/978-3-7091-6090-9_21
Giese, A., Loo, M. A., Tran, N., Haskett, D., Coons, S. W., and Berens, M. E. (1996). Dichotomy of astrocytoma migration and proliferation. Int. J. Cancer 67, 275–282. doi: 10.1002/(SICI)1097-0215(19960717)67:2<275::AID-IJC20>3.0.CO;2-9
Giese, A., and Westphal, M. (1996). Glioma invasion in the central nervous system. Neurosurgery 39, 235–250; discussion 50–52.
Gong, L., Wu, D., Zou, J., Chen, J., Chen, L., Chen, Y., et al. (2016). Prognostic impact of serum and tissue MMP-9 in non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget 7, 18458–18468. doi: 10.18632/oncotarget.7607
Grek, C. L., Sheng, Z., Naus, C. C., Sin, W. C., Gourdie, R. G., and Ghatnekar, G. G. (2018). Novel approach to temozolomide resistance in malignant glioma: connexin43-directed therapeutics. Curr. Opin. Pharmacol. 41, 79–88. doi: 10.1016/j.coph.2018.05.002
Grobben, B., De Deyn, P. P., and Slegers, H. (2002). Rat C6 glioma as experimental model system for the study of glioblastoma growth and invasion. Cell Tissue Res. 310, 257–270. doi: 10.1007/s00441-002-0651-7
Gupta, M. K., Polisetty, R. V., Ramamoorthy, K., Tiwary, S., Kaur, N., Uppin, M. S., et al. (2013). Secretome analysis of Glioblastoma cell line–HNGC-2. Mol. Biosyst. 9, 1390–1400. doi: 10.1039/c3mb25383j
Hegedus, B., Marga, F., Jakab, K., Sharpe-Timms, K. L., and Forgacs, G. (2006). The interplay of cell-cell and cell-matrix interactions in the invasive properties of brain tumors. Biophys. J. 91, 2708–2716. doi: 10.1529/biophysj.105.077834
Hsu, J. L., Huang, S. Y., Chow, N. H., and Chen, S. H. (2003). Stable-isotope dimethyl labeling for quantitative proteomics. Anal. Chem. 75, 6843–6852. doi: 10.1021/ac0348625
Huang, G. Y., Cooper, E. S., Waldo, K., Kirby, M. L., Gilula, N. B., and Lo, C. W. (1998). Gap junction-mediated cell-cell communication modulates mouse neural crest migration. J. Cell Biol. 143, 1725–1734
Huang, R. P., Fan, Y., Hossain, M. Z., Peng, A., Zeng, Z. L., and Boynton, A. L. (1998). Reversion of the neoplastic phenotype of human glioblastoma cells by connexin 43 (cx43). Cancer Res. 58, 5089–5096.
Huang, L., He, R., Luo, W., Zhu, Y. S., Li, J., Tan, T., et al. (2016). Aldo-keto reductase family 1 member B10 inhibitors: potential drugs for cancer treatment. Recent Pat. Anticancer Drug Discov. 11, 184–196. doi: 10.2174/1574892811888160304113346
Jan, H. J., Lee, C. C., Shih, Y. L., Hueng, D. Y., Ma, H. I., Lai, J. H., et al. (2010). Osteopontin regulates human glioma cell invasiveness and tumor growth in mice. Neuro Oncol. 12, 58–70. doi: 10.1093/neuonc/nop013
Jia, H., Zhang, Q., Liu, F., and Zhou, D. (2017). Prognostic value of MMP-2 for patients with ovarian epithelial carcinoma: a systematic review and meta-analysis. Arch. Gynecol. Obstet. 295, 689–696. doi: 10.1007/s00404-016-4257-9
Jin, X., Jin, X., Sohn, Y. W., Yin, J., Kim, S. H., Joshi, K., et al. (2013). Blockade of EGFR signaling promotes glioma stem-like cell invasiveness by abolishing ID3-mediated inhibition of p27(KIP1) and MMP3 expression. Cancer Lett. 328, 235–242. doi: 10.1016/j.canlet.2012.09.005
Kameritsch, P., Pogoda, K., and Pohl, U. (2012). Channel-independent influence of connexin 43 on cell migration. Biochim. Biophys, Acta 1818, 1993–2001. doi: 10.1016/j.bbamem.2011.11.016
Kashyap, M. K., Harsha, H. C., Renuse, S., Pawar, H., Sahasrabuddhe, N. A., Kim, M. S., et al. (2010). SILAC-based quantitative proteomic approach to identify potential biomarkers from the esophageal squamous cell carcinoma secretome. Cancer Biol. Ther. 10, 796–810. doi: 10.4161/cbt.10.8.12914
Kon, S., Nakayama, Y., Matsumoto, N., Ito, K., Kanayama, M., Kimura, C., et al. (2014). A novel cryptic binding motif, LRSKSRSFQVSDEQY, in the oxC-terminal fragment of MMP-3/7-cleaved osteopontin as a novel ligand for alpha9beta1 integrin is involved in the anti-type II collagen antibody-induced arthritis. PLoS One 9:e116210. doi: 10.1371/journal.pone.0116210
Kramer, A., Green, J., Pollard, J. Jr., and Tugendreich, S. (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. doi: 10.1093/bioinformatics/btt703
Laird, D. W. (2009). The gap junction proteome and its relationship to disease. Trends Cell Biol. 20, 92–101. doi: 10.1016/j.tcb.2009.11.001
Levental, K. R., Yu, H., Kass, L., Lakins, J. N., Egeblad, M., Erler, J. T., et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. doi: 10.1016/j.cell.2009.10.027.
Lin, J. H., Takano, T., Cotrina, M. L., Arcuino, G., Kang, J., Liu, S., et al. (2002). Connexin 43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J. Neurosci. 22, 4302–4311. doi: 10.1523/JNEUROSCI.22-11-04302.2002
Lopez-Otin, C., and Overall, C. M. (2002). Protease degradomics: a new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 3, 509–519. doi: 10.1038/nrm858
Louis, D. N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W. K., et al. (2016). The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 131, 803–820. doi: 10.1007/s00401-016-1545-1
Lu, K. V., Jong, K. A., Rajasekaran, A. K., Cloughesy, T. F., and Mischel, P. S. (2004). Upregulation of tissue inhibitor of metalloproteinases (TIMP)-2 promotes matrix metalloproteinase (MMP)-2 activation and cell invasion in a human glioblastoma cell line. Lab. Invest. 84, 8–20. doi: 10.1038/labinvest.3700003
Lu, P., Weaver, V. M., and Werb, Z. (2012). The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol. 196, 395–406. doi: 10.1083/jcb.201102147
Machesky, L. M. (2008). Lamellipodia and filopodia in metastasis and invasion. FEBS Lett. 582, 2102–2111. doi: 10.1016/j.febslet.2008.03.039
Manini, I., Caponnetto, F., Bartolini, A., Ius, T., Mariuzzi, L., Di Loreto, C., et al. (2018). Role of microenvironment in glioma invasion: what we learned from in vitro models. Int. J. Mol. Sci. 19:E147. doi: 10.3390/ijms19010147
Matsunaga, T., Wada, Y., Endo, S., Soda, M., El-Kabbani, O., and Hara, A. (2012). Aldo-keto reductase 1B10 and its role in proliferation capacity of drug-resistant cancers. Front. Pharmacol. 3:5. doi: 10.3389/fphar.2012.00005
Matsunaga, T., Yamane, Y., Iida, K., Endo, S., Banno, Y., El-Kabbani, O., et al. (2011). Involvement of the aldo-keto reductase, AKR1B10, in mitomycin-c resistance through reactive oxygen species-dependent mechanisms. Anticancer Drugs 22, 402–408. doi: 10.1097/CAD.0b013e3283448df0
Mendes, O., Kim, H. T., and Stoica, G. (2005). Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin. Exp. Metastasis. 22, 237–246. doi: 10.1007/s10585-005-8115-6
Munoz, J. L., Rodriguez-Cruz, V., Greco, S. J., Ramkissoon, S. H., Ligon, K. L., and Rameshwar, P. (2014). Temozolomide resistance in glioblastoma cells occurs partly through epidermal growth factor receptor-mediated induction of connexin 43. Cell Death Dis. 5:e1145. doi: 10.1038/cddis.2014.111
Murphy, S. F., Varghese, R. T., Lamouille, S., Guo, S., Pridham, K. J., Kanabur, P., et al. (2016). Connexin 43 inhibition sensitizes chemoresistant glioblastoma cells to temozolomide. Cancer Res. 76, 139–149. doi: 10.1158/0008-5472.CAN-15-1286
Nagase, H., Fields, C. G., and Fields, G. B. (1994). Design and characterization of a fluorogenic substrate selectively hydrolyzed by stromelysin 1 (matrix metalloproteinase-3). J. Biol. Chem. 269, 20952–20957.
Nakada, M., Nakada, S., Demuth, T., Tran, N. L., Hoelzinger, D. B., and Berens, M. E. (2007). Molecular targets of glioma invasion. Cell Mol. Life Sci. 64, 458–478. doi: 10.1007/s00018-007-6342-5
Naus, C. C., Aftab, Q., and Sin, W. C. (2016). Common mechanisms linking connexin43 to neural progenitor cell migration and glioma invasion. Semin. Cell Dev. Biol. 50, 59–66. doi: 10.1016/j.semcdb.2015.12.008
Naus, C. C., Bechberger, J. F., Caveney, S., and Wilson, J. X. (1991). Expression of gap junction genes in astrocytes and C6 glioma cells. Neurosci. Lett. 126, 33–36. doi: 10.1016/0304-3940(91)90364-Y
Naus, C. C., Bond, S. L., Bechberger, J. F., and Rushlow, W. (2000). Identification of genes differentially expressed in C6 glioma cells transfected with connexin43. Brain Res. Brain Res. Rev. 32, 259–266. doi: 10.1016/S0165-0173(99)00087-9
Naus, C. C., Elisevich, K., Zhu, D., Belliveau, D. J., and Del Maestro, R. F. (1992). In vivo growth of C6 glioma cells transfected with connexin43 cDNA. Cancer Res. 52, 4208–4213.
Naus, C. C., Hearn, S., Zhu, D., Nicholson, B. J., and Shivers, R. R. (1993). Ultrastructural analysis of gap junctions in C6 glioma cells transfected with connexin43 cDNA. Exp. Cell Res. 206, 72–84. doi: 10.1006/excr.1993.1122
Nielsen, H. (2017). Predicting secretory proteins with signalP. Methods Mol. Biol. 1611, 59–73. doi: 10.1007/978-1-4939-7015-5_6
Oliveira, R., Christov, C., Guillamo, J. S., de Bouard, S., Palfi, S., Venance, L., et al. (2005). Contribution of gap junctional communication between tumor cells and astroglia to the invasion of the brain parenchyma by human glioblastomas. BMC Cell. Biol. 6:7. doi: 10.1186/1471-2121-6-7
Parkin, D. M., Bray, F., Ferlay, J., and Pisani, P. (2001). Estimating the world cancer burden: Globocan 2000. Int. J. Cancer 94, 153–156. doi: 10.1002/ijc.1440.
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Pino, L. K., Searle, B. C., Bollinger, J. G., Nunn, B., MacLean, B., and MacCoss, M. J. (2017). The Skyline ecosystem: informatics for quantitative mass spectrometry proteomics. Mass Spectrom Rev. doi: 10.1002/mas.21540 [Epub ahead of print].
Polisetty, R. V., Gupta, M. K., Nair, S. C., Ramamoorthy, K., Tiwary, S., Shiras, A., et al. (2011). Glioblastoma cell secretome: analysis of three glioblastoma cell lines reveal 148 non-redundant proteins. J. Proteomics. 74, 1918–1925. doi: 10.1016/j.jprot.2011.05.002
Pollard, T. D., and Cooper, J. A. (2009). Actin, a central player in cell shape and movement. Science 326, 1208–1212. doi: 10.1126/science.1175862
Quail, D. F., and Joyce, J. A. (2017). The microenvironmental landscape of brain tumors. Cancer Cell 31, 326–341. doi: 10.1016/j.ccell.2017.02.009
Ren, F., Tang, R., Zhang, X., Madushi, W. M., Luo, D., Dang, Y., et al. (2015). Overexpression of MMP family members functions as prognostic biomarker for breast cancer patients: a systematic review and meta-analysis. PLoS One 10:e0135544. doi: 10.1371/journal.pone.0135544
Ruoslahti, E. (1996). RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 12, 697–715. doi: 10.1146/annurev.cellbio.12.1.697
Sahin, A., Velten, M., Pietsch, T., Knuefermann, P., Okuducu, A. F., Hahne, J. C., et al. (2005). Inactivation of Ets 1 transcription factor by a specific decoy strategy reduces rat C6 glioma cell proliferation and mmp-9 expression. Int. J. Mol. Med. 15, 771–776. doi: 10.3892/ijmm.15.5.771
Saleem, A., Brown, G. D., Brady, F., Aboagye, E. O., Osman, S., Luthra, S. K., et al. (2003). Metabolic activation of temozolomide measured in vivo using positron emission tomography. Cancer Res 63, 2409–2415.
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH image to imageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Sevenich, L., and Joyce, J. A. (2014). Pericellular proteolysis in cancer. Genes Dev. 28, 2331–2347. doi: 10.1101/gad.250647.114
Shi, T., Song, E., Nie, S., Rodland, K. D., Liu, T., and Qian, W. J., et al. (2016). Advances in targeted proteomics and applications to biomedical research. Proteomics 16, 2160–2182. doi: 10.1002/pmic.201500449
Sin, W. C., Bechberger, J. F., Rushlow, W. J., and Naus, C. C. (2008). Dose-dependent differential upregulation of CCN1/Cyr61 and CCN3/NOV by the gap junction protein Connexin43 in glioma cells. J. Cell Biochem. 103, 1772–1782. doi: 10.1002/jcb.21571
Sin, W. C., Crespin, S., and Mesnil, M. (2012). Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta 1818, 2058–2067. doi: 10.1016/j.bbamem.2011.10.022
Stark, C., Breitkreutz, B. J., Reguly, T., Boucher, L., Breitkreutz, A., and Tyers, M. (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539. doi: 10.1093/nar/gkj109
Stout, C. E., Costantin, J. L., Naus, C. C., and Charles, A. C. (2002). Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 277, 10482–10488. doi: 10.1074/jbc.M109902200
Strober, W. (2001). Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 21, A.3B.1–A.3B.2. doi: 10.1002/0471142735.ima03bs21
Sun, D. W., Zhang, Y. Y., Qi, Y., Zhou, X. T., and Lv, G. Y. (2015). Prognostic significance of MMP-7 expression in colorectal cancer: a meta-analysis. Cancer Epidemiol. 39, 135–142. doi: 10.1016/j.canep.2015.01.009
Theiss, C., and Meller, K. (2002). Microinjected anti-actin antibodies decrease gap junctional intercellular commmunication in cultured astrocytes. Exp. Cell Res. 281, 197–204. doi: 10.1006/excr.2002.5652
Tyanova, S., and Cox, J. (2018). Perseus: a bioinformatics platform for integrative analysis of proteomics data in cancer research. Methods Mol. Biol. 1711, 133–148. doi: 10.1007/978-1-4939-7493-1_7
Ulrich, T. A., de Juan Pardo, E. M., and Kumar, S. (2009). The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 69, 4167–4174. doi: 10.1158/0008-5472.CAN-08-4859
Wall, M. E., Otey, C., Qi, J., and Banes, A. J. (2007). Connexin 43 is localized with actin in tenocytes. Cell Motil. Cytoskeleton 64, 121–130. doi: 10.1002/cm.20170
Wang, L., Peng, Y., Peng, J., Shao, M., Ma, L., Zhu, Z., et al. (2018). Tramadol attenuates the sensitivity of glioblastoma to temozolomide through the suppression of Cx43mediated gap junction intercellular communication. Int. J. Oncol. 52, 295–304. doi: 10.3892/ijo.2017.4188
Wang, Z., Juttermann, R., Soloway, P. D. (2000). TIMP-2 is required for efficient activation of proMMP-2 in vivo. J. Biol. Chem. 275, 26411–26415. doi: 10.1074/jbc.M001270200
Wei, C. J., Francis, R., Xu, X., and Lo, C. W. (2005). Connexin43 associated with an oxN-cadherin-containing multiprotein complex is required for gap junction formation in NIH3T3 cells. J. Biol. Chem. 280, 19925–19936. doi: 10.1074/jbc.M412921200
Wells, J. M., Gaggar, A., and Blalock, J. E. (2015). MMP generated matrikines. Matrix Biol. 44–46, 122–129. doi: 10.1016/j.matbio.2015.01.016
Wu, C. C., Hsu, C. W., Chen, C. D., Yu, C. J., Chang, K. P., Tai, D. I., et al. (2010). Candidate serological biomarkers for cancer identified from the secretomes of 23 cancer cell lines and the human protein atlas. Mol. Cell Proteomics 9, 1100–1117. doi: 10.1074/mcp.M900398-MCP200
Yamaguchi, H., and Condeelis, J. (2007). Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 1773, 642–652. doi: 10.1016/j.bbamcr.2006.07.001
Yoon, J. H., Kim, J., Kim, K. L., Kim, D. H., Jung, S. J., Lee, H., et al. (2014). Proteomic analysis of hypoxia-induced U373MG glioma secretome reveals novel hypoxia-dependent migration factors. Proteomics 14, 1494–1502. doi: 10.1002/pmic.201300554
Zhang, W., Nwagwu, C., Le, D. M., Yong, V. W., Song, H., and Couldwell, W. T. (2003). Increased invasive capacity of connexin43-overexpressing malignant glioma cells. J. Neurosurg. 99, 1039–1046. doi: 10.3171/jns.2003.99.6.1039
Zheng, X., Shen, G., Yang, X., and Liu, W. (2007). Most C6 cells are cancer stem cells: evidence from clonal and population analyses. Cancer Res. 67, 3691–3697. doi: 10.1158/0008-5472.CAN-06-3912
Keywords: secretome, MMP3, osteopontin, ARK1B10, glioma, migration, connexin43, gap junction
Citation: Aftab Q, Mesnil M, Ojefua E, Poole A, Noordenbos J, Strale P-O, Sitko C, Le C, Stoynov N, Foster LJ, Sin W-C, Naus CC and Chen VC (2019) Cx43-Associated Secretome and Interactome Reveal Synergistic Mechanisms for Glioma Migration and MMP3 Activation. Front. Neurosci. 13:143. doi: 10.3389/fnins.2019.00143
Received: 27 September 2017; Accepted: 07 February 2019;
Published: 19 March 2019.
Edited by:
Valery I. Shestopalov, Bascom Palmer Eye Institute, University of Miami Health System, United StatesReviewed by:
Marco Vanoni, Università degli Studi di Milano Bicocca, ItalyAndrew K. Ottens, Virginia Commonwealth University, United States
Copyright © 2019 Aftab, Mesnil, Ojefua, Poole, Noordenbos, Strale, Sitko, Le, Stoynov, Foster, Sin, Naus and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vincent C. Chen, Y2hlbnZAYnJhbmRvbnUuY2E=
†Shared first authorship