 Antonio Benítez-Burraco
Antonio Benítez-Burraco Ryo Kimura
Ryo Kimura- 1Department of Spanish, Linguistics, and Theory of Literature (Linguistics), Faculty of Philology, University of Seville, Seville, Spain
- 2Department of Anatomy and Developmental Biology, Graduate School of Medicine, Kyoto University, Kyoto, Japan
Williams syndrome (WS) is a clinical condition, involving cognitive deficits and an uneven language profile, which has been the object of intense inquiry over the last decades. Although WS results from the hemideletion of around two dozen genes in chromosome 7, no gene has yet been probed to account for, or contribute significantly to, the language problems exhibited by the affected people. In this paper we have relied on gene expression profiles in the peripheral blood of WS patients obtained by microarray analysis and show that several robust candidates for language disorders and/or for language evolution in the species, all of them located outside the hemideleted region, are up- or downregulated in the blood of subjects with WS. Most of these genes play a role in the development and function of brain areas involved in language processing, which exhibit structural and functional anomalies in people with this condition. Overall, these genes emerge as robust candidates for language dysfunction in WS.
Introduction
Williams syndrome (WS) is a clinical condition resulting from a hemizygous deletion of 1.5 to 1.8 Mb on 7q11.23, which encompasses nearly 30 genes (Korenberg et al., 2000; Pober et al., 2010). The affected people exhibit a distinctive behavioral and cognitive profile, with enhanced sociability, mental retardation, impaired spatial cognition, and spared musical abilities (Reilly et al., 1990; Udwin and Yule, 1991; Bellugi et al., 1999; Galaburda et al., 2002; Levitin et al., 2005). Language abilities are significantly preserved in people with WS compared to other neurodevelopmental disorders, to the extent that this syndrome has often been used to support the view that language can be teased apart from other aspects of cognition. Nonetheless, recent, fine-grained analyses of WS language have concluded that WS language is delayed or impaired across different levels compared to the neurotypical population (Karmiloff-Smith and Mills, 2006; Brock, 2007; Mervis and Becerra, 2007; Martens et al., 2008 for good reviews). Specifically, children with WS experience problems with irregular word forms and complex syntax; likewise, they have problems with word definitions, although they usually excel on expressive vocabulary (including semantic organization and fluency) (Volterra et al., 1996; Mervis et al., 1999; Purser et al., 2011; Van Den Heuvel et al., 2016; see Mervis and Becerra, 2007 for discussion). However, as with other aspects of the cognitive profile of this condition, no robust gene-to-phenotype associations have been established in the language domain. To date, the most promising candidates for language dysfunction in WS are GTF2I, BAZ1B, and LIMK1. In particular, GTF2I, which encodes a regulator of transcription, has been repeatedly related to the behavioral and cognitive disabilities that are typically found in this condition and that have an impact on language function (Morris et al., 2003; Tassabehji et al., 2005; Sakurai et al., 2011; Hoeft et al., 2014). Its adjacent paralog, GTF2IRD1, has been related to altered vocalizations among other features (Howard et al., 2012). Interestingly too, BAZ1B haploinsufficiency explains almost 50% of transcriptional dysregulation in WS neurons, with BAZ1B target genes being enriched in functions related to neurogenesis and neuron differentiation (Lalli et al., 2016). Regarding LIMK1, it regulates synaptic plasticity and long-term memory (Todorovski et al., 2015), and its hemideletion has been hypothesized to account for the observed deficits in spatial cognition in combination with other genes (Gray et al., 2006; Smith et al., 2009). Still, these potential links with aspects of language (dys)function seem quite vague, particularly if one considers our remarkable understanding of the genetic underpinnings of human language, language disorders, and language evolution (see Scharff and White, 2004; Li and Bartlett, 2012; Benítez-Burraco, 2013; Graham et al., 2015; Fisher, 2017; Murphy and Benítez-Burraco, 2017, 2018 for reviews). Examining how robust candidate genes for language disorders and language evolution behave in people with WS should help refine our view of the molecular causes of the language deficits attested in this condition. One general reason supporting this approach is the deep link that exists between evolution and (abnormal) development, in the spirit of evo-devo theories. One specific reason supporting this approach is that although in WS the number of hemideleted genes is small, changes in the dosage of hundreds, or even thousands, of other genes can be expected, with a potential impact on language abilities, in the spirit of omnigenic theories of complex diseases (Boyle et al., 2017; Peedicayil and Grayson, 2018). Recently Kimura et al. (2018) confirmed that the dysregulation of several co-expression modules involving dozens of genes outside of the 7q11.23 region seemingly accounts for the complex phenotypes observed in WS patients. Importantly, they found BCL11A, a gene associated with speech disorders, among the hub genes in the top WS-related modules.
In this paper we have conducted a more focused research on the potential dysregulation of genes related to language outside the WS region as a possible explanation of the distinctive language profile of the affected people. Similarly to Kimura et al. (2018), we have relied on gene expression profiles in peripheral blood of WS patients obtained by microarray analysis. We have found that significant differences exist in the blood of subjects with WS compared to neurotypical controls in the expression levels of robust candidates for language development, language evolution, and language impairment.
Methods
The list of core candidates for language (abnormal) development and language evolution (Supplementary Table S1) encompasses two subsets of genes. The first subset consists of strong candidates for language disorders, in particular, developmental dyslexia (DD) and specific language disorder (SLI), as listed by Paracchini et al. (2016), Pettigrew et al. (2016) and Chen et al. (2017). The second subset consists of strong candidates for language evolution, as compiled by Boeckx and Benítez-Burraco (2014a,b) and Benítez-Burraco and Boeckx (2015). These are genes involved in the globularization of the human skull/brain and the cognitive changes accounting for our species-specific ability to learn and use languages (aka our language- readiness). Overall, the genes comprising this second subset fulfill several criteria. First, they have changed (and/or interact with genes that have changed) after our split from Neanderthals/Denisovans, including changes in their coding regions and/or their epigenetic profile. Second, they play some known role in brain development, regionalization, wiring, and/or function. Third, they are candidates for language dysfunction in broad cognitive disorders, particularly, autism spectrum disorder (ASD) and schizophrenia (SZ) (see Benítez-Burraco and Murphy, 2016; Murphy and Benítez-Burraco, 2016, 2017 for details about their role in language processing).
The gene expression profiling data of peripheral blood were obtained from our previous study (Kimura et al., 2018), available at the Gene Expression Omnibus (GSE89594). Briefly, total RNA from 32 WS patients and 30 healthy controls were analyzed using an Agilent Human GE v2 8×60K Microarray (Agilent Technologies). After the normalization step, differentially expressed genes (DEG) were calculated using the Limma R package (Smyth, 2005). The Benjamini-Hochberg method was used to evaluate the false discovery rate (FDR) (Benjamini and Hochberg, 1995). DEG were defined as FDR < 0.05 and the |fold change (FC)| > 1.2. Gene list enrichment analysis was performed using Fisher’s exact test. All the expressed genes were used as the background gene list.
Results
We found that candidates for language (abnormal) development and language evolution are significantly dysregulated in the blood of subjects with WS (p = 1.1e-7 by Fisher’s exact test). Figure 1 shows the genes that are significantly up- or down-regulated compared to controls (FDR < 0.05, |FC| > 1.2).
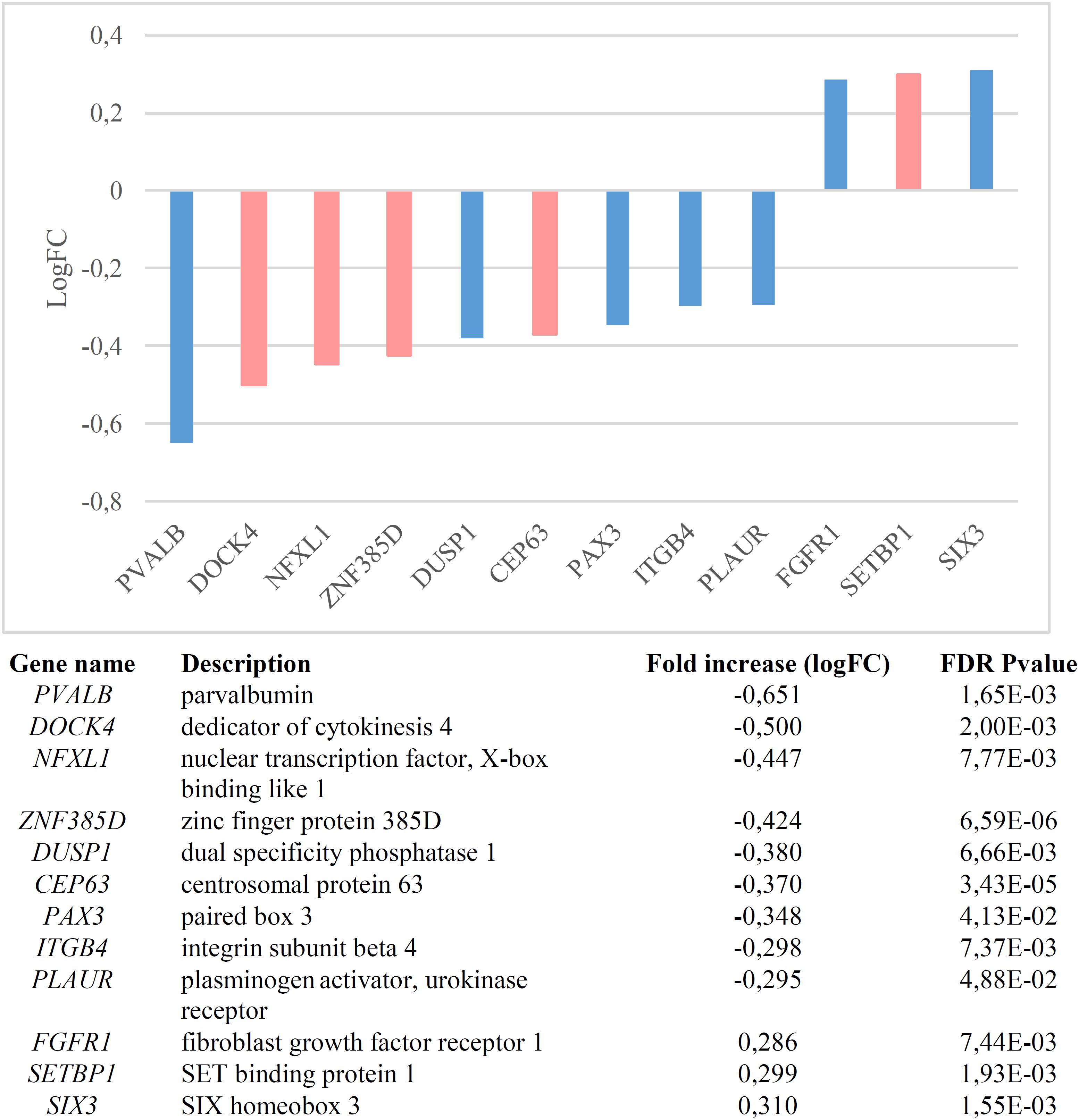
Figure 1. Genes significantly dysregulated in the blood of subjects with WS (FDR < 0.05, |FC| > 1.2). Candidate genes for language disorders (SLI, DD) are displayed in light red, whereas candidates for language evolution are colored in light blue.
In order to check the specificity of this set of genes in relation to language we conducted a functional enrichment analysis with Enrichr (amp.pharm.mssm.edu/Enrichr; Chen et al., 2013; Kuleshov et al., 2016), which showed that they are significantly related to biological processes, molecular functions, and abnormal phenotypes of interest for language (Table 1). Finally, these genes were predicted to be preferentially expressed in body parts important for language processing or for language development, particularly, the cerebellum and the thalamus (Table 1 and Supplementary Table S2). We now provide a detailed discussion of our results.
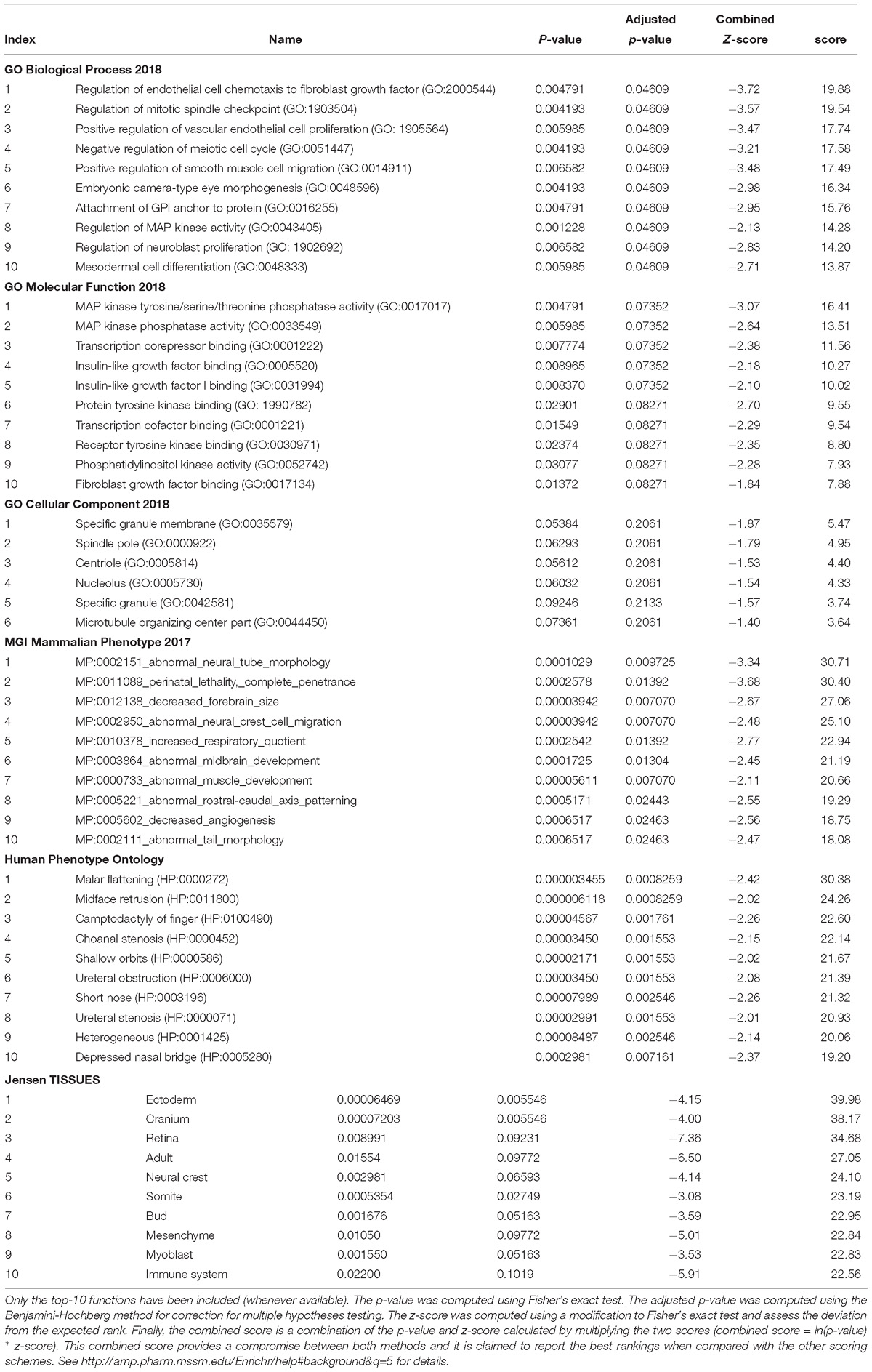
Table 1. Functional enrichment analysis according to Enrichr of the set of genes significantly dysregulated in the blood of subjects with WS.
Discussion
Functional Characterization of Individual Genes
Nearly one third of the language-related genes found downregulated in the blood of subjects with WS are candidates for DD (DOCK4, ZNF385D, and CEP63) and/or for SLI (DOCK4, NFXL1). As other members of the Dock family, DOCK4 regulates cytoskeleton assembly and cell adhesion and migration (Gadea and Blangy, 2014). Specifically, DOCK4 has been shown to be involved in neuronal migration and neurite differentiation (Ueda et al., 2008; Xiao et al., 2013), via interaction with the actin-binding protein cortactin (Ueda et al., 2013). Knockdown of Dock4 in mice abolishes commissural axon attraction by Shh (Makihara et al., 2018). The gene has been related to neuronal migration and neurite outgrowth anomalies linked to DD (Shao et al., 2016), although it is also associated with ASD (Pagnamenta et al., 2010) and SZ (Alkelai et al., 2012). GWAs have associated markers in ZNF385D to the co-occurrence of reading disability and language impairment (Eicher et al., 2013), but also to negative symptoms in SZ (Xu et al., 2013). CEP63 is required for normal spindle assembly, being involved in maintaining centriole number and establishing the order of events in centriole formation (Brown et al., 2013). Besides its association with DD (Einarsdottir et al., 2015), the gene is also a candidate for primary microcephaly (Marjanović et al., 2015), a feature that is commonly found in subjects with WS (Jernigan and Bellugi, 1990; Schmitt et al., 2001; Thompson et al., 2005; Jackowski et al., 2009). Finally, variants of NFXL1, which is predicted to encode a transcription factor, confer a risk for SLI (Villanueva et al., 2015). The gene is highly expressed in the cerebellum (Nudel, 2016).
Regarding the candidates for language evolution that we have found downregulated in the blood of subjects with WS, DUSP1 is involved in vocal learning in songbirds (Horita et al., 2010, 2012). PVALB encodes a calcium-binding protein that is structurally and functionally similar to calmodulin and that is involved in hippocampal plasticity, learning and memory (Donato et al., 2013). Interestingly enough, the inactivation of Pvalb-expressing interneurons in the auditory cortex alters response to sound, strengthening forward suppression and altering its frequency dependence (Phillips et al., 2017). Inhibition of PVALB-expressing GABAergic interneurons results in complex behavioral changes related to the behavioral phenotype of people with SZ (Brown et al., 2015). Importantly, some of the key changes that contributed to the emergence of our language-readiness involved GABAergic signaling (discussed in detail in Boeckx and Benítez-Burraco, 2014b), which are vital for oscillatory processes underlying language processing (Bae et al., 2010; see Murphy and Benítez-Burraco, 2018 for details). Reduction in PVALB expression in interneurons has also been found in mouse models of ASD (Filice et al., 2016), specifically, in the Cntnap2-/- model (Lauber et al., 2018). CNTNAP2 is a direct target of FOXP2, the renowned “language gene” (Vernes et al., 2008; Adam et al., 2017), and regulates language development in non-pathological populations too (Whitehouse et al., 2011; Whalley et al., 2011, Kos et al., 2012). Also mice lacking PLAUR have significantly fewer neocortical parvalbumin-containing GABAergic interneurons, with this reduction correlating with impaired social interactions (Bruneau and Szepetowski, 2011). PLAUR is a target of FOXP2 too (Roll et al., 2010), but also an effector of SRPX2, another of FOXP2’s targets (Royer-Zemmour et al., 2008) and a candidate for speech dyspraxia (Roll et al., 2006). Concerning PAX3, this gene is expressed in the neural crest and is a candidate for Waardenburg syndrome, a clinical condition entailing sensorineural hearing loss and developmental delay (Tassabehji et al., 1992; Chen et al., 2010). Finally, ITGB4 encodes the integrin beta 4 subunit, a receptor for the laminins, including FLNA (Travis et al., 2004), an actin-binding protein needed for cytoskeleton remodeling and neuronal migration (Fox et al., 1998) FLNA binds CMIP (Fox et al., 1998), a candidate for SLI (Newbury et al., 2009). Interestingly enough, ITGB4 is one of the proteins bearing fixed changes in humans compared to extinct hominins (Pääbo, 2014; Supplementary Table S1).
Lastly, among the genes found to be upregulated in the blood on WS subjects, we found the SLI candidate SETBP1, as well as FGFR1 and SIX3. SETBP1 is also a candidate for Schinzel-Giedion syndrome, a clinical condition entailing occasional epilepsy and severe developmental delay (Ko et al., 2013; Miyake et al., 2015). Mutations on this gene have been associated as well to behavioral and social deficits (Coe et al., 2014). The Integrative Nuclear FGFR1 Signaling (INFS) has been hypothesized to be one of the neurodevelopmental pathways on which multiple SZ candidates converge, regulating numerous neurotransmitter systems and neural circuits (Stachowiak et al., 2013). Finally, SIX3 contributes to regulate the relative size of the telencephalon versus the thalamus (Lavado et al., 2008; Sylvester et al., 2010). Interestingly, Six3 regulates Shh (Jeong et al., 2008), one robust candidate for microcephaly that has been positively selected in the human lineage (Dorus et al., 2004), but it also interacts with several genes relevant for our language-ready brain (Benítez-Burraco and Boeckx, 2015).
Functional Characterization of the Set of Dysregulated Genes
The results of our functional enrichment analyses (Table 1) show that the language-related genes that are dysregulated in the blood of people with WS mainly contribute to the cytoskeleton activity, being significantly involved in cell proliferation and migration, including neuroblast proliferation. Regarding their molecular function, they typically participate in protein modification, particularly via (tyrosine) kinase phosphatase and (tyrosine) kinase binding activities, but also in gene regulation, via transcription cofactor binding. Interestingly, these genes are significantly associated to aberrant processes impacting on brain development, like abnormal neural tube morphology and neural crest cell migration, as well as decreased forebrain size and abnormal midbrain development. Likewise, they are associated to clinical symptoms mostly impacting on craniofacial morphology, like malar flattening, midface retrusion, shallow orbits, or depressed nasal bridge. Finally, these genes are predicted to be preferentially expressed in the ectoderm, the cranium, the retina, and the neural crest. According to the Human Brain Transcriptome Database1 all these genes are expressed in the brain, particularly in the thalamus and the cerebellum (Supplementary Table S2). The thalamus functions as a sort of relay center to connect many brain areas involved in language processing (Wahl et al., 2008; Murdoch, 2010; David et al., 2011) and changes in the thalamus have been claimed to contribute to the evolutionary emergence of our language-ready brain (see Boeckx and Benítez-Burraco, 2014b for details). Similarly, the cerebellum plays a key role in language processing and is impaired in language-related pathologies (Vias and Dick, 2017; Mariën and Borgatti, 2018). People with WS exhibit cerebellar volume alterations that are seemingly associated with their cognitive, affective and motor distinctive features (Osório et al., 2014). In the same vein, the thalamus exhibits structural and functional differences with the neurotypical population, including disproportionately reduced volumes and decreased gray matter (Chiang et al., 2007; Campbell et al., 2009), as well as enhanced thalamic activity (Mobbs et al., 2007; Bódizs et al., 2012).
Conclusion
To conclude, it is true that deciphering the exact molecular causes of language dysfunction in WS is still pending, particularly, because at present none of the genes hemideleted in this condition has been demonstrated to play a central role in language processing. Nonetheless, in this paper we have shown that the genes that are dysregulated in subjects with WS are significantly enriched in core candidates for language disorders and language evolution. These genes emerge as robust candidates for language dysfunction in WS. Future research should try to delve into the expression patterns of these genes in the brain of people with WS, as well as into their role in neurotypical brain development. Likewise, altering these genes in animal models of WS should help gaining a better understanding of their biological role and ultimately, of their contribution to language dysfunction in WS.
Data Availability
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE89594.
Author Contributions
AB-B conceived and wrote the manuscript. RK conducted the expression studies and analyzed the data. Both authors contributed to manuscript revision, and read and approved the submitted version.
Funding
This research was funded by the Spanish Ministry of Economy and Competitiveness [Grant FFI2016-78034-C2-2-P (AEI/FEDER, UE) to AB-B].
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.00258/full#supplementary-material
Footnotes
References
Adam, I., Mendoza, E., Kobalz, U., Wohlgemuth, S., and Scharff, C. (2017). CNTNAP2 is a direct FoxP2 target in vitro and in vivo in zebra finches: complex regulation by age and activity. Genes Brain Behav. 16, 635–642. doi: 10.1111/gbb.12390
Alkelai, A., Lupoli, S., Greenbaum, L., Kohn, Y., Kanyas-Sarner, K., Ben-Asher, E., et al. (2012). DOCK4 and CEACAM21 as novel schizophrenia candidate genes in the Jewish population. Int. J. Neuropsychopharmacol. 15, 459–469. doi: 10.1017/S1461145711000903
Bae, M. H., Bissonette, G. B., Mars, W. M., Michalopoulos, G. K., Achim, C. L., Depireux, D. A., et al. (2010). Hepatocyte growth factor (HGF) modulates GABAergic inhibition and seizure susceptibility. Exp. Neurol. 221, 129–135. doi: 10.1016/j.expneurol.2009.10.011
Bellugi, U., Lichtenberger, L., Mills, D., Galaburda, A., and Korenberg, J. R. (1999). Bridging cognition, the brain and molecular genetics: evidence from Williams syndrome. Trends Neurosci. 5, 197–207. doi: 10.1016/S0166-2236(99)01397-1
Benítez-Burraco, A. (2013). “Genetics of language: roots of specific language deficits,” in The Cambridge Handbook of Biolinguistics, eds C. Boeckx and K. K. Grohmann (Cambridge: Cambridge University Press), 375–412.
Benítez-Burraco, A., and Boeckx, C. (2015). Possible functional links among brain- and skull-related genes selected in modern humans. Front. Psychol. 6:794. doi: 10.3389/fpsyg.2015.00794
Benítez-Burraco, A., and Murphy, E. (2016). The oscillopathic nature of language deficits in autism: from genes to language evolution. Front. Hum. Neurosci. 10:120. doi: 10.3389/fnhum.2016.00120
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bódizs, R., Gombos, F., and Kovács, I. (2012). Sleep EEG fingerprints reveal accelerated thalamocortical oscillatory dynamics in Williams syndrome. Res. Dev. Disabil. 33, 153–164. doi: 10.1016/j.ridd.2011.09.004
Boeckx, C., and Benítez-Burraco, A. (2014a). Globularity and language-readiness: generating new predictions by expanding the set of genes of interest. Front. Psychol. 5:1324. doi: 10.3389/fpsyg.2014.01324
Boeckx, C., and Benítez-Burraco, A. (2014b). The shape of the human language-ready brain. Front. Psychol. 5:282. doi: 10.3389/fpsyg.2014.00282
Boyle, E. A., Li, Y. I., and Pritchard, J. K. (2017). An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186. doi: 10.1016/j.cell.2017.05.038
Brock, J. (2007). Language abilities in Williams syndrome: a critical review. Dev. Psychopathol. 19, 97–127. doi: 10.1017/S095457940707006X
Brown, J. A., Ramikie, T. S., Schmidt, M. J., Báldi, R., Garbett, K., Everheart, M. G., et al. (2015). Inhibition of parvalbumin-expressing interneurons results in complex behavioral changes. Mol. Psychiatry 20, 1499–1507. doi: 10.1038/mp.2014.192
Brown, N. J., Marjanović, M., Lüders, J., Stracker, T. H., and Costanzo, V. (2013). Cep63 and cep152 cooperate to ensure centriole duplication. PLoS One 8:e69986. doi: 10.1371/journal.pone.0069986
Bruneau, N., and Szepetowski, P. (2011). The role of the urokinase receptor in epilepsy, in disorders of language, cognition, communication and behavior, and in the central nervous system. Curr. Pharm. Des. 17, 1914–1923. doi: 10.2174/138161211796718198
Campbell, L. E., Daly, E., Toal, F., Stevens, A., Azuma, R., Karmiloff-Smith, A., et al. (2009). Brain structural differences associated with the behavioural phenotype in children with Williams syndrome. Brain Res. 1258, 96–107. doi: 10.1016/j.brainres.2008.11.101
Chen, E. Y., Tan, C. M., Kou, Y., Duan, Q., Wang, Z., Meirelles, G. V., et al. (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14:128. doi: 10.1186/1471-2105-14-128
Chen, H., Jiang, L., Xie, Z., Mei, L., He, C., Hu, Z., et al. (2010). Novel mutations of PAX3, MITF, and SOX10 genes in Chinese patients with type I or type II Waardenburg syndrome. Biochem. Biophys. Res. Commun. 397, 70–74. doi: 10.1016/j.bbrc.2010.05.066
Chen, X. S., Reader, R. H., Hoischen, A., Veltman, J. A., Simpson, N. H., Francks, C., et al. (2017). Next-generation sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci. Rep. 7:46105. doi: 10.1038/srep46105
Chiang, M. C., Reiss, A. L., Lee, A. D., Bellugi, U., Galaburda, A. M., Korenberg, J. R., et al. (2007). 3D pattern of brain abnormalities in Williams syndrome visualized using tensor-based morphometry. Neuroimage 36, 1096–1109. doi: 10.1016/j.neuroimage.2007.04.024
Coe, B. P., Witherspoon, K., Rosenfeld, J. A., van Bon, B. W., Vulto-van Silfhout, A. T., Bosco, P., et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 46, 1063–1071. doi: 10.1038/ng.3092
David, O., Maess, B., Eckstein, K., and Friederici, A. D. (2011). Dynamic causal modeling of subcortical connectivity of language. J. Neurosci. 31, 2712–2717. doi: 10.1523/JNEUROSCI.3433-10.2011
Donato, F., Rompani, S. B., and Caroni, P. (2013). Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature 504, 272–276. doi: 10.1038/nature12866
Dorus, S., Vallender, E. J., Evans, P. D., Anderson, J. R., Gilbert, S. L., Mahowald, M., et al. (2004). Accelerated evolution of nervous system genes in the origin of Homo sapiens. Cell 119, 1027–1040. doi: 10.1016/j.cell.2004.11.040
Eicher, J. D., Powers, N. R., Miller, L. L., Akshoomoff, N., Amaral, D. G., Bloss, C. S., et al. (2013). Genome-wide association study of shared components of reading disability and language impairment. Genes Brain Behav. 12, 792–801. doi: 10.1111/gbb.12085
Einarsdottir, E., Svensson, I., Darki, F., Peyrard-Janvid, M., Lindvall, J. M., Ameur, A., et al. (2015). Mutation in CEP63 co-segregating with developmental dyslexia in a Swedish family. Hum. Genet. 134, 1239–1248. doi: 10.1007/s00439-015-1602-1
Filice, F., Vörckel, K. J., Sungur, A. Ö., Wöhr, M., and Schwaller, B. (2016). Reduction in parvalbumin expression not loss of the parvalbumin-expressing GABA interneuron subpopulation in genetic parvalbumin and shank mouse models of autism. Mol. Brain 9:10. doi: 10.1186/s13041-016-0192-8
Fisher, S. E. (2017). Evolution of language: lessons from the genome. Psychon. Bull. Rev. 24, 34–40. doi: 10.3758/s13423-016-1112-8
Fox, J. W., Lamperti, E. D., Eksioglu, Y. Z., Hong, S. E., Feng, Y., Graham, D. A., et al. (1998). Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325. doi: 10.1016/S0896-6273(00)80651-0
Gadea, G., and Blangy, A. (2014). Dock-family exchange factors in cell migration and disease. Eur. J. Cell Biol. 93, 466–477. doi: 10.1016/j.ejcb.2014.06.003
Galaburda, A. M., Holinger, D. P., Bellugi, U., and Sherman, G. F. (2002). Williams syndromeneuronal size and neuronal-packing density in primary visual cortex. Arch. Neurol. 59, 1461–1467. doi: 10.1001/archneur.59.9.1461
Graham, S. A., Deriziotis, P., and Fisher, S. E. (2015). Insights into the genetic foundations of human communication. Neuropsychol. Rev. 25, 3–26. doi: 10.1007/s11065-014-9277-2
Gray, V., Karmiloff-Smith, A., Funnell, E., and Tassabehji, M. (2006). In-depth analysis of spatial cognition in Williams syndrome: a critical assessment of the role of the LIMK1 gene. Neuropsychologia 44, 679–685. doi: 10.1016/j.neuropsychologia.2005.08.007
Hoeft, F., Dai, L., Haas, B. W., Sheau, K., Mimura, M., Mills, D., et al. (2014). Mapping genetically controlled neural circuits of social behavior and visuo-motor integration by a preliminary examination of atypical deletions with Williams syndrome. PLoS One 9:e104088. doi: 10.1371/journal.pone.0104088
Horita, H., Kobayashi, M., Liu, W. C., Oka, K., Jarvis, E. D., and Wada, K. (2012). Specialized motor-driven Dusp1 expression in the song systems of multiple lineages of vocal learning birds. PLoS One 7:e42173. doi: 10.1371/journal.pone.0042173
Horita, H., Wada, K., Rivas, M. V., Hara, E., and Jarvis, E. D. (2010). The dusp1 immediate early gene is regulated by natural stimuli predominantly in sensory input neurons. J. Comp. Neurol. 518, 2873–2901. doi: 10.1002/cne.22370
Howard, M. L., Palmer, S. J., Taylor, K. M., Arthurson, G. J., Spitzer, M. W., Du, X., et al. (2012). Mutation of Gtf2ird1 from the Williams-Beuren syndrome critical region results in facial dysplasia, motor dysfunction, and altered vocalisations. Neurobiol. Dis. 45, 913–922. doi: 10.1016/j.nbd.2011.12.010
Jackowski, A. P., Rando, K., Maria de Araújo, C., Del Cole, C. G., Silva, I., and Tavares de Lacerda, A. L. (2009). Brain abnormalities in Williams syndrome: a review of structural and functional magnetic resonance imaging findings. Eur. J. Paediatr. Neurol. 13, 305–316. doi: 10.1016/j.ejpn.2008.07.002
Jeong, Y., Leskow, F. C., El-Jaick, K., Roessler, E., Muenke, M., Yocum, A., et al. (2008). Regulation of a remote Shh forebrain enhancer by the Six3 homeoprotein. Nat. Genet. 40, 1348–1353. doi: 10.1038/ng.230
Jernigan, T. L., and Bellugi, U. (1990). Anomalous brain morphology on magnetic resonance images in Williams syndrome and Down syndrome. Arch. Neurol. 47, 529–533. doi: 10.1001/archneur.1990.00530050049011
Karmiloff-Smith, A., and Mills, D. L. (2006). “Williams Syndrome,” in Encyclopedia of Language and Linguistics, ed. K. Brown (Oxford: Elsevier) 585–589. doi: 10.1016/B0-08-044854-2/04181-X
Kimura, R., Swarup, V., Tomiwa, K., Gandal, M. J., Parikshak, N. N., Funabiki, Y., et al. (2018). Integrative network analysis reveals biological pathways associated with Williams syndrome. J. Child Psychol. Psychiatry doi: 10.1111/jcpp.12999 [Epub ahead of print].
Ko, J. M., Lim, B. C., Kim, K. J., Hwang, Y. S., Ryu, H. W., Lee, J. H., et al. (2013). Distinct neurological features in a patient with Schinzel-Giedion syndrome caused by a recurrent SETBP1 mutation. Childs Nerv. Syst 29, 525–529. doi: 10.1007/s00381-013-2047-2
Korenberg, J. R., Chen, X. N., Hirota, H., Lai, Z., Bellugi, U., Burian, D., et al. (2000). Genome structure and cognitive map of Williams syndrome. J. Cogn. Neurosci. 12(Suppl. 1), 89–107. doi: 10.1162/089892900562002
Kos, M., van den Brink, D., Snijders, T. M., Rijpkema, M., Franke, B., Fernandez, G., et al. (2012). CNTNAP2 and language processing in healthy individuals as measured with ERPs. PLoS One 7:e46995. doi: 10.1371/journal.pone.0046995
Kuleshov, M. V., Jones, M. R., Rouillard, A. D., Fernandez, N. F., Duan, Q., Wang, Z., et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. doi: 10.1093/nar/gkw377
Lalli, M. A., Jang, J., Park, J. H., Wang, Y., Guzman, E., Zhou, H., et al. (2016). Haploinsufficiency of BAZ1B contributes to Williams syndrome through transcriptional dysregulation of neurodevelopmental pathways. Hum. Mol. Genet. 25, 1294–1306. doi: 10.1093/hmg/ddw010
Lauber, E., Filice, F., and Schwaller, B. (2018). Dysregulation of parvalbumin expression in the Cntnap2-/- mouse model of Autism spectrum disorder. Front. Mol. Neurosci. 11:262. doi: 10.3389/fnmol.2018.00262
Lavado, A., Lagutin, O. V., and Oliver, G. (2008). Six3 inactivation causes progressive caudalization and aberrant patterning of the mammalian diencephalon. Development 135, 441–450. doi: 10.1242/dev.010082
Levitin, D. J., Cole, K., Lincoln, A., and Bellugi, U. (2005). Aversion, awareness, and attraction: investigating claims of hyperacusis in the Williamssyndrome phenotype. J. Child Psychol. Psychiatry 46, 514–523. doi: 10.1111/j.1469-7610.2004.00376.x
Li, N., and Bartlett, C. W. (2012). Defining the genetic architecture of human developmental language impairment. Life Sci. 90, 469–475. doi: 10.1016/j.lfs.2012.01.016
Makihara, S., Morin, S., Ferent, J., Côté, J. F., Yam, P. T., and Charron, F. (2018). Polarized dock activity drives Shh-mediated axon guidance. Dev. Cell 46:410-425.e7. doi: 10.1016/j.devcel.2018.07.007
Mariën, P., and Borgatti, R. (2018). Language and the cerebellum. Handb. Clin. Neurol. 154, 181–202. doi: 10.1016/B978-0-444-63956-1.00011-4
Marjanović, M., Sánchez-Huertas, C., Terré, B., Gómez, R., Scheel, J. F., Pacheco, S., et al. (2015). CEP63 deficiency promotes p53-dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nat. Commun. 6:7676. doi: 10.1038/ncomms8676
Martens, M. A., Wilson, S. J., and Reutens, D. C. (2008). Research Review: Williams syndrome: acritical review of the cognitive, behavioral, and neuroanatomical phenotype. J. Child Psychol. Psychiatry 49, 576–608. doi: 10.1111/j.1469-7610.2008.01887.x
Mervis, C. B., and Becerra, A. M. (2007). Language and communicative development in Williams syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 13, 3–15.
Mervis, C. B., Morris, C. A., Bertrand, J., and Robinson, E. F. (1999). “Williams syndrome: findings from an integrated program of research,” in Neurodevelopmental Disorders, ed. H. Tager-Flusberg (Cambridge, MA: The MIT Press), 65–110.
Miyake, F., Kuroda, Y., Naruto, T., Ohashi, I., Takano, K., and Kurosawa, K. (2015). West syndrome in a patient with Schinzel-Giedion syndrome. J. Child Neurol. 30, 932–936. doi: 10.1177/0883073814541468
Mobbs, D., Eckert, M. A., Menon, V., Mills, D., Korenberg, J., Galaburda, A. M., et al. (2007). Reduced parietal and visual cortical activation during global processing in Williams syndrome. Dev. Med. Child Neurol. 49, 433–438. doi: 10.1111/j.1469-8749.2007.00433.x
Morris, C. A., Mervis, C. B., Hobart, H. H., Gregg, R. G., Bertrand, J., Ensing, G. J., et al. (2003). GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am. J. Med. Genet. 123A, 45–59. doi: 10.1002/ajmg.a.20496
Murdoch, B. (2010). The cerebellum and language: historical perspective and review. Cortex 46, 858–868. doi: 10.1016/j.cortex.2009.07.018
Murphy, E., and Benítez-Burraco, A. (2016). Bridging the gap between genes and language deficits in schizophrenia: an oscillopathic approach. Front. Hum. Neurosci. 10:422. doi: 10.3389/fnhum.2016.00422
Murphy, E., and Benítez-Burraco, A. (2017). Language deficits in schizophrenia and autism as related oscillatory connectomopathies: an evolutionary account. Neurosci. Biobehav. Rev. 83, 742–764. doi: 10.1016/j.neubiorev.2016.07.029
Murphy, E., and Benítez-Burraco, A. (2018). Toward the language oscillogenome. Front. Psychol. 9:1999. doi: 10.3389/fpsyg.2018.01999
Newbury, D. F., Winchester, L., Addis, L., Paracchini, S., Buckingham, L. L., Clark, A., et al. (2009). CMIP and ATP2C2 modulate phonological short-term memory in language impairment. Am. J. Hum. Genet. 85, 264–72. doi: 10.1016/j.ajhg.2009.07.004
Nudel, R. (2016). An investigation of NFXL1, a gene implicated in a study of specific language impairment. J. Neurodev. Disord. 8:13. doi: 10.1186/s11689-016-9146-9
Osório, A., Soares, J. M., Prieto, M. F., Vasconcelos, C., Fernandes, C., Sousa, S., et al. (2014). Cerebral and cerebellar MRI volumes in Williams syndrome. Res. Dev. Disabil. 35, 922–928. doi: 10.1016/j.ridd.2013.12.014
Pääbo, S. (2014). The human condition-a molecular approach. Cell 157, 216–226. doi: 10.1016/j.cell.2013.12.036
Pagnamenta, A. T., Bacchelli, E., de Jonge, M. V., Mirza, G., Scerri, T. S., Minopoli, F., et al. (2010). Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol. Psychiatry 68, 320–328. doi: 10.1016/j.biopsych.2010.02.002
Paracchini, S., Diaz, R., and Stein, J. (2016). “Advances in dyslexia genetics—new insights into the role of brain asymmetries,” in Advances in Genetics 96, eds T. Friedmann, J. C. Dunlap, and S. F. Goodwin (London: Academic Press),53–97.
Peedicayil, J., and Grayson, D. R. (2018). An epigenetic basis for an omnigenic model of psychiatric disorders. J. Theor. Biol. 443, 52–55. doi: 10.1016/j.jtbi.2018.01.027
Pettigrew, K. A., Frinton, E., Nudel, R., Chan, M. T., Thompson, P., Hayiou-Thomas, M. E., et al. (2016). Further evidence for a parent-of-origin effect at the NOP9 locus on language-related phenotypes. J. Neurodev. Disord. 8:24. doi: 10.1186/s11689-016-9157-6
Phillips, E. A. K., Schreiner, C. E., and Hasenstaub, A. R. (2017). Cortical interneurons differentially regulate the effects of acoustic context. Cell Rep. 20, 771–778. doi: 10.1016/j.celrep.2017.07.001
Pober, B. R., Wang, E., Caprio, S., Petersen, K. F., Brandt, C., Stanley, T., et al. (2010). High prevalence of diabetes and pre-diabetes in adults with Williams syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 154C, 291–298. doi: 10.1002/ajmg.c.30261
Purser, H. R., Thomas, M. S., Snoxall, S., Mareschal, D., and Karmiloff-Smith, A. (2011). Definitions versus categorization: assessing the development of lexico-semantic knowledge in Williams syndrome. Int. J. Lang. Commun. Disord. 46, 361–373. doi: 10.3109/13682822.2010.497531
Reilly, J., Klima, E. S., and Bellugi, U. (1990). Once more with feeling: affect and language in atypical populations. Dev. Psychopathol. 2, 367–391. doi: 10.1017/S0954579400005782
Roll, P., Rudolf, G., Pereira, S., Royer, B., Scheffer, I. E., Massacrier, A., et al. (2006). SRPX2 mutations in disorders of language cortex and cognition. Hum. Mol. Genet. 15, 1195–1207. doi: 10.1093/hmg/ddl035
Roll, P., Vernes, S. C., Bruneau, N., Cillario, J., Ponsole-Lenfant, M., Massacrier, A., et al. (2010). Molecular networks implicated in speech-related disorders: FOXP2 regulates the SRPX2/uPAR complex. Hum. Mol. Genet. 19, 4848–4860. doi: 10.1093/hmg/ddq415
Royer-Zemmour, B., Ponsole-Lenfant, M., Gara, H., Roll, P., Lévêque, C., Massacrier, A., et al. (2008). Epileptic and developmental disorders of the speech cortex: ligand/receptor interaction of wild-type and mutant SRPX2 with the plasminogen activator receptor uPAR. Hum. Mol. Genet. 17, 3617–3630. doi: 10.1093/hmg/ddn256
Sakurai, T., Dorr, N. P., Takahashi, N., McInnes, L. A., Elder, G. A., and Buxbaum, J. D. (2011). Haploinsufficiency of Gtf2i, a gene deleted in Williams Syndrome, leads to increases in social interactions. Autism Res. 4, 28–39. doi: 10.1002/aur.169
Scharff, C., and White, S. A. (2004). Genetic components of vocal learning. Ann. N. Y. Acad. Sci. 1016, 325–347. doi: 10.1196/annals.1298.032
Schmitt, J. E., Eliez, S., Bellugi, U., and Reiss, A. L. (2001). Analysis of cerebral shape in Williams syndrome. Arch. Neurol. 58, 283–287. doi: 10.1001/archneur.58.2.283
Shao, S., Kong, R., Zou, L., Zhong, R., Lou, J., Zhou, J., et al. (2016). The roles of genes in the neuronal migration and neurite outgrowth network in developmental dyslexia: single- and multiple-risk genetic variants. Mol. Neurobiol. 53, 3967–3975. doi: 10.1007/s12035-015-9334-8
Smith, A. D., Gilchrist, I. D., Hood, B., Tassabehji, M., and Karmiloff-Smith, A. (2009). Inefficient search of large-scale space in Williams syndrome: further insights on the role of LIMK1 deletion in deficits of spatial cognition. Perception 38, 694–701. doi: 10.1068/p6050
Smyth, G. K. (2005). “Limma: linear models for microarray data,” in Bioinformatics and Computational Biology Solutions Using R and Bioconductor, eds R. Gentleman, V. Carey, S. Dudoit, R. Irizarry, and W. Huber (New York: Springer), 397–420. doi: 10.1007/0-387-29362-0_23
Stachowiak, M. K., Kucinski, A., Curl, R., Syposs, C., Yang, Y., Narla, S., et al. (2013). Schizophrenia: a neurodevelopmental disorder-integrative genomic hypothesis and therapeutic implications from a transgenic mouse model. Schizophr. Res. 143, 367–376. doi: 10.1016/j.schres.2012.11.004
Sylvester, J. B., Rich, C. A., Loh, Y. H., van Staaden, M. J., Fraser, G. J., and Streelman, J. T. (2010). Brain diversity evolves via differences in patterning. Proc. Natl. Acad. Sci. U.S.A. 107, 9718–9723. doi: 10.1073/pnas.1000395107
Tassabehji, M., Hammond, P., Karmiloff-Smith, A., Thompson, P., Thorgeirsson, S. S., Durkin, M. E., et al. (2005). GTF2IRD1 in craniofacial development of humans and mice. Science 310, 1184–1187. doi: 10.1126/science.1116142
Tassabehji, M., Read, A. P., Newton, V. E., Harris, R., Balling, R., Gruss, P., et al. (1992). Waardenburg’s syndrome patients have mutations in the human homologue of the Pax-3 paired box gene. Nature 355, 635–636. doi: 10.1038/355635a0
Thompson, P. M., Lee, A. D., Dutton, R. A., Geaga, J. A., Hayashi, K. M., Eckert, M. A., et al. (2005). Abnormal cortical complexity and thickness profiles mapped in Williams syndrome. J. Neurosci. 25, 4146–4158. doi: 10.1523/JNEUROSCI.0165-05.2005
Todorovski, Z., Asrar, S., Liu, J., Saw, N. M., Joshi, K., Cortez, M. A., et al. (2015). LIMK1 regulates long-term memory and synaptic plasticity via the transcriptional factor CREB. Mol. Cell. Biol. 35, 1316–1328. doi: 10.1128/MCB.01263-14
Travis, M. A., van der Flier, A., Kammerer, R. A., Mould, A. P., Sonnenberg, A., and Humphries, M. J. (2004). Interaction of filamin A with the integrin beta 7 cytoplasmic domain: role of alternative splicing and phosphorylation. FEBS Lett. 569, 185–190. doi: 10.1016/j.febslet.2004.04.099
Udwin, O., and Yule, W. (1991). A cognitive and behavioural phenotype in Williams syndrome. J. Clin. Exp. Neuropsychol. 13, 232–244. doi: 10.1080/01688639108401040
Ueda, S., Fujimoto, S., Hiramoto, K., Negishi, M., and Katoh, H. (2008). Dock4 regulates dendritic development in hippocampal neurons. J. Neurosci. Res. 86, 3052–3061. doi: 10.1002/jnr.21763
Ueda, S., Negishi, M., and Katoh, H. (2013). Rac GEF Dock4 interacts with cortactin to regulate dendritic spine formation. Mol. Biol. Cell 24, 1602–1613. doi: 10.1091/mbc.E12-11-0782
Van Den Heuvel, E., Manders, E., Swillen, A., and Zink, I. (2016). Developmental trajectories of structural and pragmatic language skills in school-aged children with Williams syndrome. J. Intellect. Disabil. Res. 60, 903–919. doi: 10.1111/jir.12329
Vernes, S. C., Newbury, D. F., Abrahams, B. S., Winchester, L., Nicod, J., Groszer, M., et al. (2008). A functional genetic link between distinct developmental language disorders. N. Engl. J. Med. 359:2337. doi: 10.1056/NEJMoa0802828
Vias, C., and Dick, A. S. (2017). Cerebellar contributions to language in typical and atypical development: a review. Dev. Neuropsychol. 42, 404–421. doi: 10.1080/87565641.2017.1334783
Villanueva, P., Nudel, R., Hoischen, A., Fernández, M. A., Simpson, N. H., Gilissen, C., et al. (2015). Exome sequencing in an admixed isolated population indicates NFXL1 variants confer a risk for specific language impairment. PLoS Genet. 11:e1004925. doi: 10.1371/journal.pgen.1004925
Volterra, V., Capirci, O., Pezzini, G., Sabbadini, L., and Vicari, S. (1996). Linguistic abilities in Italian children with Williams syndrome. Cortex 32, 663–677. doi: 10.1016/S0010-9452(96)80037-2
Wahl, M., Marzinzik, F., Friederici, A. D., Hahne, A., Kupsch, A., Schneider, G. H., et al. (2008). The human thalamus processes syntactic and semantic language violations. Neuron 59, 695–707. doi: 10.1016/j.neuron.2008.07.011
Whalley, H. C., O’Connell, G., Sussmann, J. E., Peel, A., Stanfield, A. C., Hayiou-Thomas, M. E., et al. (2011). Genetic variation in CNTNAP2 alters brain function during linguistic processing in healthy individuals. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B, 941–948. doi: 10.1002/ajmg.b.31241
Whitehouse, A. J., Bishop, D. V., Ang, Q. W., Pennell, C. E., and Fisher, S. E. (2011). CNTNAP2 variants affect early language development in the general population. Genes Brain Behav. 10, 451–456. doi: 10.1111/j.1601-183X.2011.00684.x
Xiao, Y., Peng, Y., Wan, J., Tang, G., Chen, Y., Tang, J., et al. (2013). The atypical guanine nucleotide exchange factor Dock4 regulates neurite differentiation through modulation of Rac1 GTPase and actin dynamics. J. Biol. Chem. 288, 20034–20045. doi: 10.1074/jbc.M113.458612
Keywords: Williams syndrome, blood transcriptional profile, language disorders, language evolution, candidate genes
Citation: Benítez-Burraco A and Kimura R (2019) Robust Candidates for Language Development and Evolution Are Significantly Dysregulated in the Blood of People With Williams Syndrome. Front. Neurosci. 13:258. doi: 10.3389/fnins.2019.00258
Received: 10 January 2019; Accepted: 05 March 2019;
Published: 26 March 2019.
Edited by:
Robin Clark, University of Pennsylvania, United StatesReviewed by:
Alfredo Ardila, Florida International University, United StatesMarco Smolla, University of Pennsylvania, United States
Copyright © 2019 Benítez-Burraco and Kimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Benítez-Burraco, YWJlbml0ZXo4QHVzLmVz