 Ya-Nan Song1Jie-Qiong Li1Chen-Chen Tan1Hui-Fu Wang1
Ya-Nan Song1Jie-Qiong Li1Chen-Chen Tan1Hui-Fu Wang1 Meng-Shan Tan1Xi-Peng Cao2
Meng-Shan Tan1Xi-Peng Cao2 Jin-Tai Yu1,2
Jin-Tai Yu1,2 Lan Tan1*the Alzheimer’s Disease Neuroimaging Initiative†
Lan Tan1*the Alzheimer’s Disease Neuroimaging Initiative†- 1Department of Neurology, Qingdao Municipal Hospital, Qingdao University, Qingdao, China
- 2Clinical Research Center, Qingdao Municipal Hospital, Qingdao University, Qingdao, China
A coding missense mutation (rs3747742) in triggering receptor expressed on myeloid cell-like 2 (TREML2) has been recently proposed as an important protective factor against Alzheimer’s disease (AD). However, the link between TREML2 and AD pathology remains unclear. Therefore, we explored the association of TREML2 rs3747742 with cognitive function, neuroimaging biomarkers and cerebrospinal fluid (CSF) biomarkers related to AD, including CSF total-tau (T-tau), phosphor-tau (P-tau), and amyloid-β (Aβ1-42). As for cognitive function, related cognitive scores of Clinical Dementia Rating Sum of Boxes (CDRSB), Alzheimer’s Disease Assessment Scale-cognitive section 11 (ADAS-cog 11), Mini-Mental State Examination (MMSE), and Rey Auditory-Verbal Learning Test (RAVLT) were extracted. We used a multiple linear regression model to examine the association of TREML2 rs3747742 with the baseline variables. Furthermore, we also calculated the change rate of above variables influenced by TREML2 rs3747742 via applying a mixed-effects model over a 4-year follow-up. In this analysis, a total of 1,306 individuals from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database were included. Finally, we observed that only in AD patients, but not in normal controls or mild cognitive impairment (MCI) individuals, TREML2 rs3747742 exhibited a strong association with CSF total-tau levels at baseline (β = -22.1210, p = 0.0166) and 4-year follow-up (β = -0.3961, p = 0.0115). Furthermore, no associations were found with CSF Aβ1-42 levels, P-tau levels, neuroimaging biomarkers and cognitive function neither for baseline variables nor for longitudinal data. Thus, this study indicated that TREML2 mediated the risk of AD through influencing AD-related neurodegeneration (abnormal T-tau levels) but not P-tau levels and Aβ pathology.
Introduction
Characterized by intracellular tau neurofibrillary tangles and extracellular amyloid-β (Aβ) plaques, Alzheimer’s disease (AD) is the most prevalent age-dependent dementia, accompanied by deteriorating cognitive function (Hardy and Selkoe, 2002; Jiang et al., 2012). AD has been suggested as a multifactorial disorder, and the role of genetics in AD pathology has been accepted widely (Querfurth and LaFerla, 2010; Jiang et al., 2012; Karch et al., 2014). Moreover, many biomarkers such as cerebrospinal fluid (CSF) Aβ1-42 and tau proteins have emerged as intermediate phenotype approaches in genetic analyses of risk for us to explore the association between genetic variation and process of AD. In the past few years, many genetic variants associated with late-onset AD, such as APOE, BIN1, PICALM, PLD3, and NME8, have been identified to affect the process of AD pathology via modifying CSF Aβ1-42 and tau levels (Schjeide et al., 2011; Liu et al., 2014; Wang et al., 2015; Liu et al., 2016; Wang et al., 2016).
A missense mutation (rs3747742-C) in TREML2 exhibited the highest linkage disequilibrium (LD) with rs9381040, which is an inter-genic SNP between TREM2 and TREML2 showing association with AD risk in the International Genomics of Alzheimer’s Project (Lambert et al., 2013; Benitez et al., 2014). In the previous studies, TREML2 rs3747742 was identified as a protective factor against AD in Caucasians (Benitez et al., 2014), which was also verified by Jiang et al. in Han Chinese population (Jiang et al., 2017). However, little is known about the mechanism by which this genetic mutation modifies the risk of AD. This study is the first one designed to analyze the role of TREML2 rs3747742 in the pathogenesis of AD by examining the relation of rs3747742 with CSF proteins, neuroimaging biomarkers and cognitive function in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database.
Materials and Methods
ADNI Database and Subjects
Subjects included in our study were obtained from the ADNI database1, which was launched in 2003 as a longitudinal study and recruited participants from almost 63 sites across the United States and Canada (Mueller et al., 2005). Detailed clinical information on the ADNI cohort has been reported previously (Petersen et al., 2010). Here, we restricted our ADNI cohort to general participants (N = 1,306), cognitively normal (CN) (N = 374), mild cognitive impairment (MCI) (N = 705), and AD subjects (N = 227) who underwent CSF protein examinations, neuroimaging biomarker measurements and cognitive function tests. Informed consent according to the Declaration of Helsinki was signed by all participants or their authorized representatives.
Genetic Data
The Illumina Human 610-Quad Bead Chip (including 620,901 SNP and CNV markers) and Illumina Human Omini Express Bead Chip (including 730,525 SNP and CNV markers) were applied for genotyping of ADNI subjects (Saykin et al., 2010). Then these genotype data was made available for ADNI website via sample verification and quality control bioinformatics. Here, we obtained the genotype data of TREML2 (rs3747742) (N = 1,306) from the ADNI database. The data for our study were extracted from the ADNI database.
CSF Biomarker Data
Data for CSF biomarkers, including CSF Aβ1-42, total tau (T-tau), and phosphor-tau (P-tau), was obtained from the ADNI database. The acquisition and measurement of CSF data have been described previously (Olsson et al., 2005). These CSF proteins were examined using the xMAP Luminex platform with Innogenetics/Fujirebio AlzBio3 immunoassay kits.
Neuroimaging Data
The neuroimaging data, including MRI volumes of brain structures, and FDG-PET of cerebral metabolic rate for glucose (CMRgl), was from the ADNI database. The methods for acquisition and processing of cerebral image can be found in prior publications (Desikan et al., 2006). Here, many regions of interest (ROI) analysis, such as brain ventricles, hippocampus, and entorhinal cortex were conducted to calculate their associations with TREML2 genotypes.
Neuropsychological Test
To test the influence of TREML2 rs3747742 on cognitive function, we extracted related cognitive scores of CDRSB, ADAS-cog 11, MMSE, and RAVLT in this study. Finally, the analyses of these baseline cognitive scores and their longitudinal changed scores over 4 years were conducted.
Statistical Methods
We used Kruskal-Wallis rank sum test to examine the differences in clinical and demographic characteristics of participants included in our analysis. We performed a multiple linear regression model and a mixed-effects model to explore the associations of TREML2 genotypes with baseline and longitudinal variables, respectively. All above analyses were adjusted for age, gender, education, and APOE𝜀4 status. Both outcome variables were normalized to z scores to facilitate the comparison between modalities. Statistical significance was considered to have been achieved when p < 0.05. Data analyses were performed using R version 3.4.1 statistical software.
Results
Demographic Analysis
The demographic characteristics and clinical data of the participants are summarized in Table 1. A total of 1,306 subjects (74.07±7.16 years, 572 women) were recruited. Specifically, 374 CN individuals (74.94 ± 5.53 years, 186 women), 705 MCI patients (73.18 ± 7.59 years, 284 women), and 227 AD patients (75.40 ± 7.72 years, 102 women) were included in this study. As expected, the three groups (CN, MCI, and AD subjects) showed significant differences in MMSE scores and CSF protein levels.
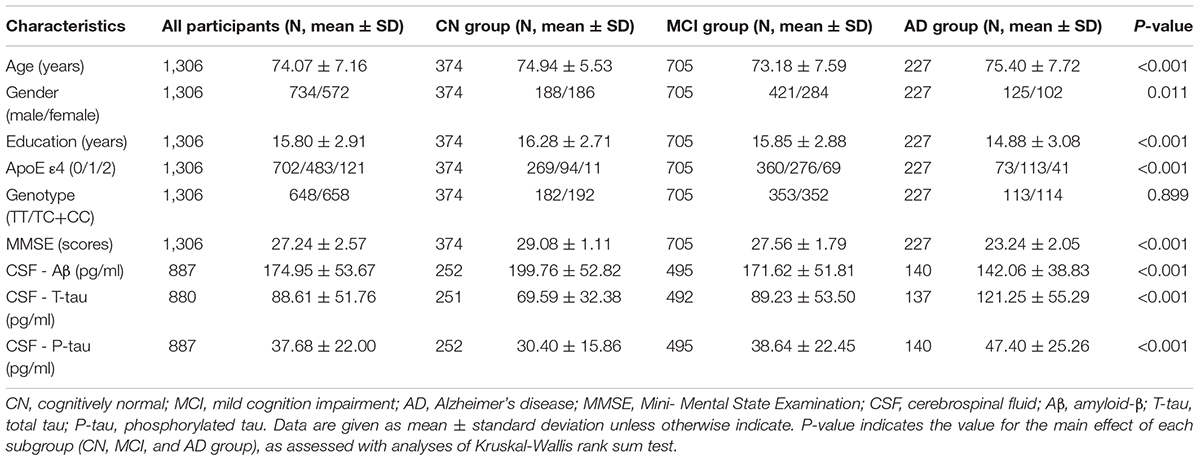
Table 1. The characteristics of included subjects at baseline.
TREML2 rs3747742 and Baseline Variants
In our analysis, we examined the association of a missense mutation (rs3747742) in TREML2 with CSF proteins, neuroimaging biomarkers and cognitive function among the total participants as well as three clinically diagnosed groups at baseline. Finally, the results indicated that in total participants and AD patients rs3747742 was associated with the level of CSF T-tau, but not with CSF Aβ1-42 level, P-tau level, neuroimaging biomarkers and cognitive function (Table 2 and Supplementary Table 1). In total participants, subjects with CC and TC genotypes had lower T-tau levels (β = -7.7764, p = 0.0143) when adjusted for age, gender, education, and APOE𝜀4 status. In addition, AD patients with C allele (CC, TC) had lower T-tau levels than those with TT genotype (β = -22.1210, p = 0.0166) when adjusted for age, gender, education, and APOE𝜀4 status (Figure 1A).
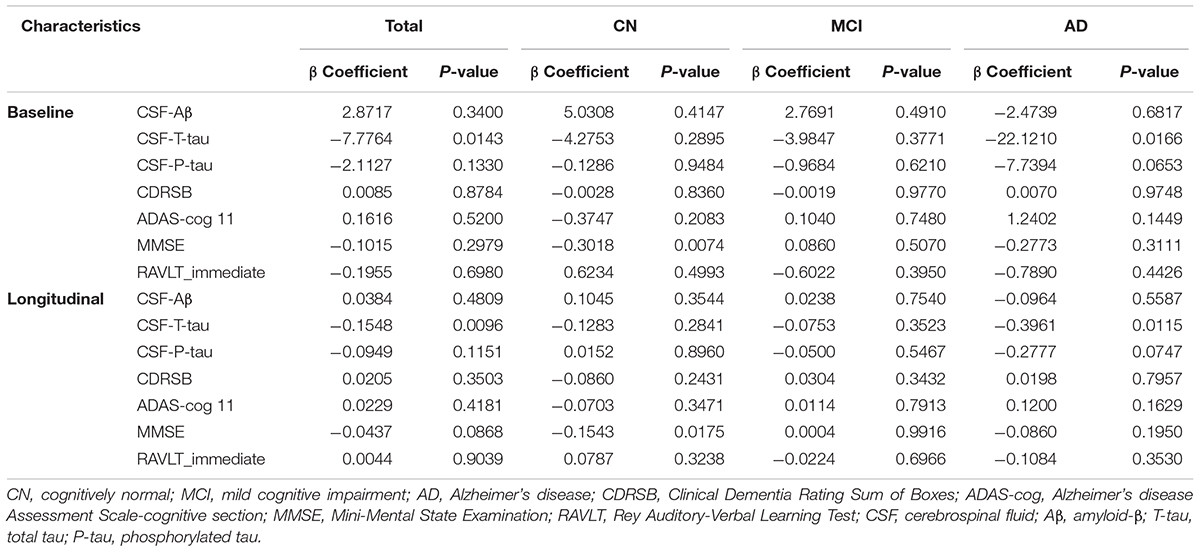
Table 2. The correlations of rs3747742 with CSF proteins and cognitive function.
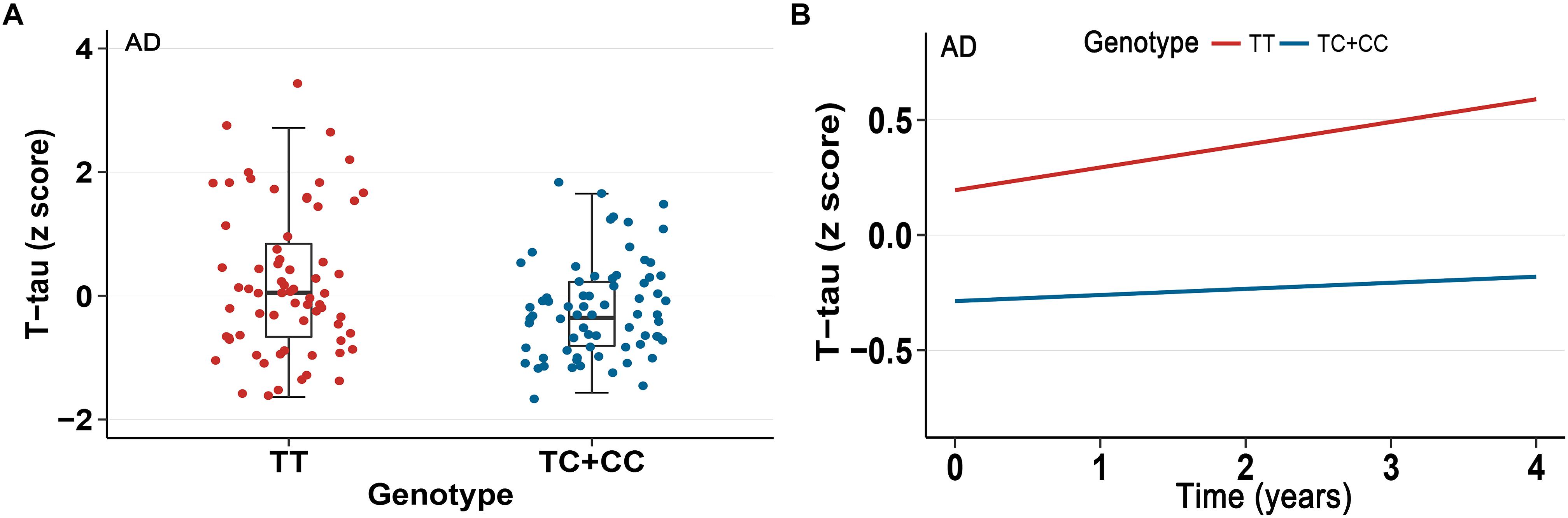
Figure 1. Association between rs3747742 and cerebrospinal fluid (CSF) T-tau levels in AD group. (A) Data from multiple linear regression analysis adjusted for age, gender, education, and ApoE𝜀4 status indicated the correlation of rs3747742 with CSF T-tau levels at baseline. (B) Data from mixed-effects model adjusted for age, gender, education, and ApoE𝜀4 status indicated the correlation of rs3747742 with longitudinal concentration changes of CSF T-tau during a 4-year follow-up.
TREML2 rs3747742 and Longitudinal Changes
Furthermore, we also tested the correlations of TREML2 rs3747742 with longitudinal changes of CSF protein levels, neuroimaging biomarkers and cognition over a 4-year follow-up. We found that total participants (β = -0.1548, p = 0.0096) (Table 2) and AD patients (β = -0.3961, p = 0.0115) with C allele (CC, TC) showed a slower rate of change in CSF T-tau levels than TT allele carriers after controlling for age, gender, education and APOE𝜀4 status (Figure 1B), but no associations were found with CSF Aβ1-42 levels, CSF P-tau levels, neuroimaging biomarkers and cognitive function (Table 2 and Supplementary Table 1).
Discussion
This study investigates the association of TREML2 rs3747742 with CSF protein levels, neuroimaging biomarkers and cognition in total participants as well as CN, MCI and AD subjects. Our main finding was that the coding missense mutation of TREML2 (rs3747742-C) was closely related to decreased baseline CSF T-tau concentrations and slower rate of longitudinal changes in CSF T-tau levels during the 4 years follow-up in AD patients. Located at TREML2, rs3747742 shows the highest LD (r2= 0.73, D′ = 0.86) with the GWAS SNP rs9381040 (Benitez et al., 2014), which was confirmed to be the top-significant SNP (p = 6.3 × 10-7) around TREML2 (Lambert et al., 2013). It was previously reported that both of the above two minor alleles of rs9381040 (β = -0.02, p = 4.11 × 10-4) and rs3747742 (β = -0.02, p = 1.4 × 10-4) were strongly associated with CSF P-tau levels and lower risk of AD (p = 1.21 × 10-5, CI = 0.88–0.95; p = 8.66 × 10-5, CI = 0.89–0.96) (Benitez et al., 2014). In the present analysis, we demonstrated rs3747742 was not associated with CSF Aβ1-42 level, which was consistent with the result of Benitez et al. (2014). Furthermore, we also detected that rs3747742 was not correlated to CSF P-tau levels but was associated with CSF T-tau levels. This discrepancy may be due to the differences in the grouping of subjects in the two studies. Specifically, the previous analysis was performed in two groups, including the AD group and the control group, but in this study we divided participants into three different clinical subgroups (AD, MCI, and CN groups) (Benitez et al., 2014). Another reason may be the too small sample size of AD subjects in this study, which caused bias in the results of the study. Therefore, more large-scale studies are warranted to confirm this conclusion.
Previous studies have demonstrated that as the functional coding mutation in TREML2 gene, rs3747742 can encode related proteins and exhibit association with the risk of AD (Cruchaga et al., 2013). TREM2 and TREML2 are structurally similar proteins encoded by the same gene cluster on chromosome 6, which have the opposite effect on the risk of AD. A recent study showed that TREM2 and TREML2 could strictly regulate microglial proliferation, whose dysfunctions may contribute to AD pathogenesis via impairing brain innate immunity (Zheng et al., 2016). TREML2 is a single-pass type I transmembrane protein containing an extracellular Ig-like type V domain and a potential cytoplasmic +xxPxxP SH3-binding motif, which can mediate signal transduction through its cytoplasmic tail (King et al., 2006). Stimulated by inflammatory factors, TREML2 up-regulates the expression of neutrophils and macrophages associated with immune responses, a process that activates immune-related cells to respond to inflammatory stimuli, thereby amplifying the inflammatory response (Klesney-Tait et al., 2006). As for the correlation between neuroinflammation and tau protein, numerous studies shown that proinflammatory cytokines (including IL-1, IL-6, and TNF-α) produced by reactive microglia can induce the pathological modification of tau protein (Li et al., 2003; Quintanilla et al., 2004; Gorlovoy et al., 2009), resulting in neurodegeneration. In addition, it was previously reported that neurodegeneration can influence inflammatory response (Zilka et al., 2009; Stozicka et al., 2010). Also, neuroinflammation mediated by microglia and astrocytes can cause neuronal damage and even death by affecting intracellular mitochondrial function (Wilkins and Swerdlow, 2016). In 2018, the ATN classification system for the diagnostic criteria for AD clearly indicated that CSF T-tau is one of the biomarkers for neuronal damage or neurodegeneration [marked as (N)] (Jack et al., 2016). Furthermore, it also has been speculated that endogenous intracellular tau can be released outside the cells after neurodegeneration (Gomez-Ramos et al., 2006). Soluble extracellular tau can promote neurotoxicity (Gomez-Ramos et al., 2008, 2009) and the release of proinflammatory cytokines, such as IL-1, IL-6, and TNF-α (Kovac et al., 2011). Thus, interactions between tau protein, inflammatory cytokines, and neurodegeneration can lead to the generation of AD pathology. TREML2 will expand the immune-related neuroinflammatory phase to exacerbate this pathological process. Besides, we observed that TREML2 rs3747742 exhibited a strong association with CSF total tau levels at baseline and 4-year follow up only in AD patients, but not in CN or MCI individuals. We thought the distinct outcomes among three groups about the association between rs3747742 and CSF total tau levels may be due to the disease status. Evidence has shown that disease status may affect the association between genetic variation and gene expression (Rhinn et al., 2013). Therefore, disease status may also affect the association between rs3747742 and CSF total tau levels at baseline and 4-year follow-up. However, the specific mechanism by which TREML2 rs3747742 affects the expression and function of related proteins with various disease status needs further study and interpretation.
In summary, our findings showed that TREML2 genetic mutation (rs3747742-C) was associated with CSF T-tau levels in AD patients, suggesting this mutation plays an important role in AD-related neurodegeneration. Neurodegeneration is a common pathway in various neurodegenerative diseases. However, as for other neurodegenerative disease, such as Parkinson’s disease (PD), one study discovered that 24 PD susceptibility variants which identified in GWAS previously were not associated with TREML2 (Nalls et al., 2014; Chan et al., 2016). Thus, more independent researches with large sample size and diverse ethnicity are required to confirm the role of TREML2 in AD pathology.
Author Contributions
J-TY, LT, and Y-NS designed the study, ADNI conducted the subject recruitment and data collection. Y-NS, J-QL, and H-FW analyzed the data. Y-NS, J-TY, C-CT, and M-ST interpreted the findings of study. Y-NS, J-TY, J-QL, and X-PC wrote the manuscript.
Funding
This work was supported by grants from the National Key R&D Program of China (2016YFC1305803), the Shandong Provincial Outstanding Medical Academic Professional Program, Taishan Scholars Program of Shandong Province (ts201511109, tsqn20161078, and tsqn20161079), Qingdao Key Health Discipline Development Fund, Qingdao Outstanding Health Professional Development Fund, and Shandong Provincial Collaborative Innovation Center for Neurodegenerative Disorders, Research Award Fund for Outstanding Young and Middle-aged Scientists of Shandong Province (BS2015SW006), Projects of medical and health technology development program in Shandong Province (2015WSA02054). Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.00455/full#supplementary-material
Footnotes
References
Benitez, B. A., Jin, S. C., Guerreiro, R., Graham, R., Lord, J., Harold, D., et al. (2014). Missense variant in TREML2 protects against Alzheimer’s disease. Neurobiol. Aging 35, 1510.e19–1526. doi: 10.1016/j.neurobiolaging.2013.12.010
Chan, G., White, C. C., Winn, P. A., Cimpean, M., Replogle, J. M., Glick, L. R., et al. (2016). Trans-pQTL study identifies immune crosstalk between Parkinson and Alzheimer loci. Neurol. Genet. 2:e90. doi: 10.1212/nxg.0000000000000090
Cruchaga, C., Kauwe, J. S., Harari, O., Jin, S. C., Cai, Y., Karch, C. M., et al. (2013). GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 78, 256–268. doi: 10.1016/j.neuron.2013.02.026
Desikan, R. S., Segonne, F., Fischl, B., Quinn, B. T., Dickerson, B. C., Blacker, D., et al. (2006). An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 31, 968–980. doi: 10.1016/j.neuroimage.2006.01.021
Gomez-Ramos, A., Diaz-Hernandez, M., Cuadros, R., Hernandez, F., and Avila, J. (2006). Extracellular tau is toxic to neuronal cells. FEBS Lett. 580, 4842–4850. doi: 10.1016/j.febslet.2006.07.078
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Diaz-Hernandez, J. I., Miras-Portugal, M. T., and Avila, J. (2009). Characteristics and consequences of muscarinic receptor activation by tau protein. Eur. Neuropsychopharmacol. 19, 708–717. doi: 10.1016/j.euroneuro.2009.04.006
Gomez-Ramos, A., Diaz-Hernandez, M., Rubio, A., Miras-Portugal, M. T., and Avila, J. (2008). Extracellular tau promotes intracellular calcium increase through M1 and M3 muscarinic receptors in neuronal cells. Mol. Cell Neurosci. 37, 673–681. doi: 10.1016/j.mcn.2007.12.010
Gorlovoy, P., Larionov, S., Pham, T. T., and Neumann, H. (2009). Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J. 23, 2502–2513. doi: 10.1096/fj.08-123877
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Jack, C. R. Jr., Bennett, D. A., Blennow, K., Carrillo, M. C., Feldman, H. H., Frisoni, G. B., et al. (2016). A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547. doi: 10.1212/wnl.0000000000002923
Jiang, T., Wan, Y., Zhou, J. S., Tan, M. S., Huang, Q., Zhu, X. C., et al. (2017). A missense variant in TREML2 reduces risk of Alzheimer’s disease in a han chinese population. Mol. Neurobiol. 54, 977–982. doi: 10.1007/s12035-016-9706-8
Jiang, T., Yu, J. T., and Tan, L. (2012). Novel disease-modifying therapies for Alzheimer’s disease. J. Alzheimers Dis. 31, 475–492. doi: 10.3233/jad-2012-120640
Karch, C. M., Cruchaga, C., and Goate, A. M. (2014). Alzheimer’s disease genetics: from the bench to the clinic. Neuron 83, 11–26. doi: 10.1016/j.neuron.2014.05.041
King, R. G., Herrin, B. R., and Justement, L. B. (2006). Trem-like transcript 2 is expressed on cells of the myeloid/granuloid and B lymphoid lineage and is up-regulated in response to inflammation. J. Immunol. 176, 6012–6021. doi: 10.4049/jimmunol.176.10.6012
Klesney-Tait, J., Turnbull, I. R., and Colonna, M. (2006). The TREM receptor family and signal integration. Nat. Immunol. 7, 1266–1273. doi: 10.1038/ni1411
Kovac, A., Zilka, N., Kazmerova, Z., Cente, M., Zilkova, M., and Novak, M. (2011). Misfolded truncated protein tau induces innate immune response via MAPK pathway. J. Immunol. 187, 2732–2739. doi: 10.4049/jimmunol.1100216
Lambert, J. C., Ibrahim-Verbaas, C. A., Harold, D., Naj, A. C., Sims, R., Bellenguez, C., et al. (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 45, 1452–1458. doi: 10.1038/ng.2802
Li, Y., Liu, L., Barger, S. W., and Griffin, W. S. (2003). Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 23, 1605–1611. doi: 10.1523/jneurosci.23-05-01605.2003
Liu, Y., Tan, L., Wang, H. F., Liu, Y., Hao, X. K., Tan, C. C., et al. (2016). Multiple effect of APOE genotype on clinical and neuroimaging biomarkers across Alzheimer’s disease spectrum. Mol. Neurobiol. 53, 4539–4547. doi: 10.1007/s12035-015-9388-7
Liu, Y., Yu, J. T., Wang, H. F., Hao, X. K., Yang, Y. F., Jiang, T., et al. (2014). Association between NME8 locus polymorphism and cognitive decline, cerebrospinal fluid and neuroimaging biomarkers in Alzheimer’s disease. PLoS One 9:e114777. doi: 10.1371/journal.pone.0114777
Mueller, S. G., Weiner, M. W., Thal, L. J., Petersen, R. C., Jack, C. R., Jagust, W., et al. (2005). Ways toward an early diagnosis in Alzheimer’s disease: the Alzheimer’s Disease neuroimaging initiative (ADNI). Alzheimers Dement. 1, 55–66. doi: 10.1016/j.jalz.2005.06.003
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 46, 989–993. doi: 10.1038/ng.3043
Olsson, A., Vanderstichele, H., Andreasen, N., De Meyer, G., Wallin, A., Holmberg, B., et al. (2005). Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin. Chem. 51, 336–345. doi: 10.1373/clinchem.2004.039347
Petersen, R. C., Aisen, P. S., Beckett, L. A., Donohue, M. C., Gamst, A. C., Harvey, D. J., et al. (2010). Alzheimer’s disease neuroimaging initiative (ADNI): clinical characterization. Neurology 74, 201–209. doi: 10.1212/WNL.0b013e3181cb3e25
Querfurth, H. W., and LaFerla, F. M. (2010). Alzheimer’s disease. N. Engl. J. Med. 362, 329–344. doi: 10.1056/NEJMra0909142
Quintanilla, R. A., Orellana, D. I., Gonzalez-Billault, C., and Maccioni, R. B. (2004). Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 295, 245–257. doi: 10.1016/j.yexcr.2004.01.002
Rhinn, H., Fujita, R., Qiang, L., Cheng, R., Lee, J. H., and Abeliovich, A. (2013). Integrative genomics identifies APOE epsilon4 effectors in Alzheimer’s disease. Nature 500, 45–50. doi: 10.1038/nature12415
Saykin, A. J., Shen, L., Foroud, T. M., Potkin, S. G., Swaminathan, S., Kim, S., et al. (2010). Alzheimer’s Disease neuroimaging initiative biomarkers as quantitative phenotypes: genetics core aims, progress, and plans. Alzheimers Dement. 6, 265–273. doi: 10.1016/j.jalz.2010.03.013
Schjeide, B. M., Schnack, C., Lambert, J. C., Lill, C. M., Kirchheiner, J., Tumani, H., et al. (2011). The role of clusterin, complement receptor 1, and phosphatidylinositol binding clathrin assembly protein in Alzheimer disease risk and cerebrospinal fluid biomarker levels. Arch. Gen. Psychiatry 68, 207–213. doi: 10.1001/archgenpsychiatry.2010.196
Stozicka, Z., Zilka, N., Novak, P., Kovacech, B., Bugos, O., and Novak, M. (2010). Genetic background modifies neurodegeneration and neuroinflammation driven by misfolded human tau protein in rat model of tauopathy: implication for immunomodulatory approach to Alzheimer’s disease. J. Neuroinflammation 7:64. doi: 10.1186/1742-2094-7-64
Wang, C., Tan, L., Wang, H. F., Yu, W. J., Liu, Y., Jiang, T., et al. (2015). Common variants in PLD3 and correlation to amyloid-related phenotypes in Alzheimer’s disease. J. Alzheimers Dis. 46, 491–495. doi: 10.3233/jad-150110
Wang, H. F., Wan, Y., Hao, X. K., Cao, L., Zhu, X. C., Jiang, T., et al. (2016). Bridging integrator 1 (BIN1) genotypes mediate Alzheimer’s disease risk by altering neuronal degeneration. J. Alzheimers Dis. 52, 179–190. doi: 10.3233/jad-150972
Wilkins, H. M., and Swerdlow, R. H. (2016). Relationships between mitochondria and neuroinflammation: implications for Alzheimer’s disease. Curr. Top. Med. Chem. 16, 849–857. doi: 10.2174/1568026615666150827095102
Zheng, H., Liu, C. C., Atagi, Y., Chen, X. F., Jia, L., Yang, L., et al. (2016). Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging 42, 132–141. doi: 10.1016/j.neurobiolaging.2016.03.004
Keywords: TREML2, neurodegeneration, Alzheimer’s disease, CSF biomarkers, Alzheimer’s disease neuroimaging initiative
Citation: Song Y-N, Li J-Q, Tan C-C, Wang H-F, Tan M-S, Cao X-P, Yu J-T, Tan L and the Alzheimer’s Disease Neuroimaging Initiative (2019) TREML2 Mutation Mediate Alzheimer’s Disease Risk by Altering Neuronal Degeneration. Front. Neurosci. 13:455. doi: 10.3389/fnins.2019.00455
Received: 04 September 2018; Accepted: 23 April 2019;
Published: 15 May 2019.
Edited by:
Yan Huang, Harvard Medical School, United StatesReviewed by:
Guiyou Liu, Tianjin Institute of Industrial Biotechnology (CAS), ChinaRossen Donev, MicroPharm Ltd., United Kingdom
Copyright © 2019 Song, Li, Tan, Wang, Tan, Cao, Yu, Tan and the Alzheimer’s Disease Neuroimaging Initiative. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Tan, ZHIudGFubGFuQDE2My5jb20=
†Data used in preparation for this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.