 Irene Meester
Irene Meester Gerardo Francisco Rivera-Silva
Gerardo Francisco Rivera-Silva Francisco González-Salazar
Francisco González-Salazar- 1Laboratory of Tissue Engineering and Regenerative Medicine, Basic Sciences Department, University of Monterrey, San Pedro Garza García, Mexico
- 2Laboratory of Cellular Physiology, Northeast Center of Research, Mexican Institute of Social Security, Monterrey, Mexico
The fibromyalgia syndrome (FMS) is characterized by chronic widespread pain, sleep disturbances, fatigue, and cognitive alterations. A limited efficacy of targeted treatment and a high FMS prevalence (2–5% of the adult population) sums up to high morbidity. Although, altered nociception has been explained with the central sensitization hypothesis, which may occur after neuropathy, its molecular mechanism is not understood. The marked female predominance among FMS patients is often attributed to a psychosocial predisposition of the female gender, but here we will focus on sex differences in neurobiological processes, specifically those of the immune system, as various immunological biomarkers are altered in FMS. The activation of innate immune sensors is compatible with a neuropathy or virus-induced autoimmune diseases. Considering sex differences in the immune system and the clustering of FMS with autoimmune diseases, we hypothesize that the female predominance in FMS is due to a neuropathy-induced autoimmune pathophysiology. We invite the scientific community to verify the autoimmune hypothesis for FMS.
Introduction
As long as the pathophysiology of the FMS is not elucidated, the diagnosis (Wolfe et al., 2016; Arnold et al., 2019) and the treatment (Macfarlane et al., 2017) will remain inadequate. Many consider FMS to be psychosomatic (Lami et al., 2018) and there are still physicians who do not recognize the disorder. Although the name indicates a fibromuscular affection and the syndrome is classified as a rheumatic disorder, FMS is treated as a neurological problem, in accordance with the currently most accepted hypothesis: central sensitization (Staud et al., 2009). The history of FMS not only reveals the confusion (Inanici and Yunus, 2004) but also the importance of (1) inflammation, (2) a neuropathic type of pain, (3) referred pain after irritation or damage of the paraspinal ligaments, (4) increased substance P levels in cerebrospinal fluid (CSF), and (5) an etiology of trauma and/or infection accompanied by mental stress, which are all consistent with neuroinflammation. Female predominance and clustering with autoimmune diseases were recognized in the historical review (Inanici and Yunus, 2004), but suggestions of autoimmunity markers were omitted (Jacobsen et al., 1990; Klein et al., 1992), despite being actual at the time of the review. Still, it seems that autoimmune susceptibility accompanies FMS. We propose that FMS is a neuropathy-induced autoimmune disease directed to nervous tissue. As autoimmunity is sex biased (Beeson, 1994), the autoimmune hypothesis may explain the female prevalence observed in FMS.
The focus of the paper is to present the biological data from which this hypothesis emerges, followed by how it may explain central sensitization and the sleep alteration that characterize FMS. Next, we describe the mechanisms of immunological self-tolerance and how it can be breached, as well as the well-known sex differences in the immune system, which explains why women are more susceptible to develop certain autoimmunity disorders. We reflect on the complexities of proving the hypothesis and offer suggestions to verify the hypothesis.
Fibromyalgia: Introducing the Autoimmune Hypothesis
Fibromyalgia syndrome is characterized by unexplained chronic (>3 months) widespread pain accompanied by moderate to severe sleep problems and/or fatigue (Arnold et al., 2019). Fatigue upon awakening has been associated with altered sleep wave patterns, especially a lack of slow-wave sleep (Roizenblatt et al., 2001). A myriad of additional symptoms tends to accompany the disease, amongst them cognitive difficulties, depression, irritable bowel, irritable bladder, restless legs, dry mouth and eyes, and altered sense perception (Arnold et al., 2019). Primary FMS is not accompanied by another chronic pain disorder, whereas secondary FMS develops as a co-morbidity of another dominant chronic disease, commonly an autoimmune disease (Häuser et al., 2015). FMS prevalence among the adult population ranges from 0.8–5% worldwide, depending on the geographical area, case definition, and assessment method (Johnston et al., 2013; Jackson et al., 2015). FMS occurs in the pediatric population, generally beginning with the onset of puberty (Gedalia et al., 2000), but the highest prevalence is among middle-aged women. The female-to-male ratio ranges from 1:1 to 30:1, but a worldwide average is about 3:1 in both the pediatric (Gedalia et al., 2000) and adult populations (Queiroz, 2013).
Although FMS is classified as a musculoskeletal disease, the currently most accepted hypothesis of pathogenesis, central sensitization (Staud et al., 2009), is neurobiology-based and supported by empirical and impartial evidence (Maestu et al., 2013; Sluka and Clauw, 2016). As the etiology and pathophysiology of FMS remain elusive, FMS treatment is directed to symptom management, which includes inhibition of an overreacting CNS (Macfarlane et al., 2017). In general, 30% of the patients report a 30% improvement because of treatment (Häuser et al., 2015). This modest efficacy suggests that the pharmacological treatment does not target the cause.
We hypothesize that FMS is a neuropathy-induced autoimmunity directed against nervous tissue. The autoimmune hypothesis provides a mechanistic explanation for the central sensitization hypothesis and thus the two hypotheses are compatible. Considering sex differences in the immune system, the autoimmune hypothesis may explain female predominance among FMS patients.
The autoimmune hypothesis emerged from the following observations. First, the epidemiological profile of FMS is similar to the one of autoimmunity as both peak among middle-aged women (Beeson, 1994). Second, FMS co-occurs with a cluster of autoimmune diseases e.g., sicca syndrome, SLE, rheumatoid arthritis, irritable bowel syndrome, thyroiditis, interstitial cystitis/painful bladder syndrome, and restless legs syndrome co-occur. Autoantibodies ‘specific’ for aforementioned autoimmune diseases tend to be shared rather than unique. When they are detected in FMS, a corresponding autoimmunity is diagnosed and FMS is redefined as secondary FMS (Hervier et al., 2009). Still, secondary FMS reveals autoimmune susceptibility (Buskila and Sarzi-Puttini, 2008; Giacomelli et al., 2013; Haliloglu et al., 2017). Specific antibodies for FMS have been reported (Supplementary Table 1), but they are neither consolidated nor generally accepted (Werle et al., 2001; Giacomelli et al., 2013). Third, there is overlap in the clinical profile of FMS and certain autoimmune diseases, with respect to complex genetic and environmental risk factors. The latter include infections (Smatti et al., 2019) and stress due to traumatic experiences (Sharif et al., 2018). Stress and certain personality characteristics associate positively with autoimmune diseases, FMS, and other chronic diseases in retrospective studies with selected controls (Martin et al., 1996; Lami et al., 2018). But whereas stress and personality are considered precipitating factors or consequences in autoimmune diseases (Mitsonis et al., 2009; Hassett and Clauw, 2010), they are interpreted as the cause in FMS (Lami et al., 2018) despite a lack of convincing evidence demonstrating a causal relation (Häuser and Henningsen, 2014). Similarly, there is no convincing evidence that a certain personality causes pain (Naylor et al., 2017). Fourth, although routinely screened inflammatory biomarkers in FMS samples (such as C-reactive protein levels and erythrocyte sedimentation velocity) tend to be within the normal clinical range, these and other immune markers are significantly different from the ones of healthy controls in a research setting (Kadetoff et al., 2012; Xiao et al., 2013; Pernambuco et al., 2015; Mendieta et al., 2016; Bäckryd et al., 2017; Ciregia et al., 2018). Although data on inflammatory biomarkers are not consisted among studies, they correlate weakly with clinical variables (Ernberg et al., 2018), or can be explained by comorbid conditions, the general impression is that chronic inflammation occurs in FMS (Ernberg et al., 2018). Similarly, routine leukocyte counts are within the normal range, but specific lymphocyte subgroups, not screened in clinical routine studies, are altered in FMS patients. Compared to age-matched healthy controls, female FMS patients had higher proportions of CD57+ natural killer cells (NK) (17.1% vs. 11.3%) and CD5+ B cells (6.46% vs. 2.5%) (Russell et al., 1999) but lower CD56+ NK (Landis et al., 2004). Case-control observational whole-genome expression studies among women revealed altered expression of immune pathways and markers of tissue destruction (Lukkahatai et al., 2015; Jones et al., 2016). These expression studies did not confirm gene polymorphisms that had been identified in genome association studies (Park and Lee, 2017). The gene association studies often had a selection bias and did not clarify why the genetic susceptibility would only lead to a disorder later in life. In this respect, the HLA alleles (Branco et al., 1996; Yunus et al., 1999) form an exception, as their impact depends on an interaction between genetic and environmental factors. HLA genes have an essential role in the immune system. Thus, the emerging picture is that a combination of genetic predisposition, a precipitating event (infections, trauma, autoimmune diseases or other reasons of necrosis) (Jiao et al., 2015), and immune dysregulation due to psychological stress (Takahashi et al., 2018) may convert autotolerance or pre-existing occult autoimmunity into overt autoimmunity, but the autoreactive component remains elusive (Figure 1). Importantly, though the precipitation event may be transitory, autoimmunity is a response of the adaptive immune system and is chronic. The sex bias in the immune system may explain the female preponderance of FMS.
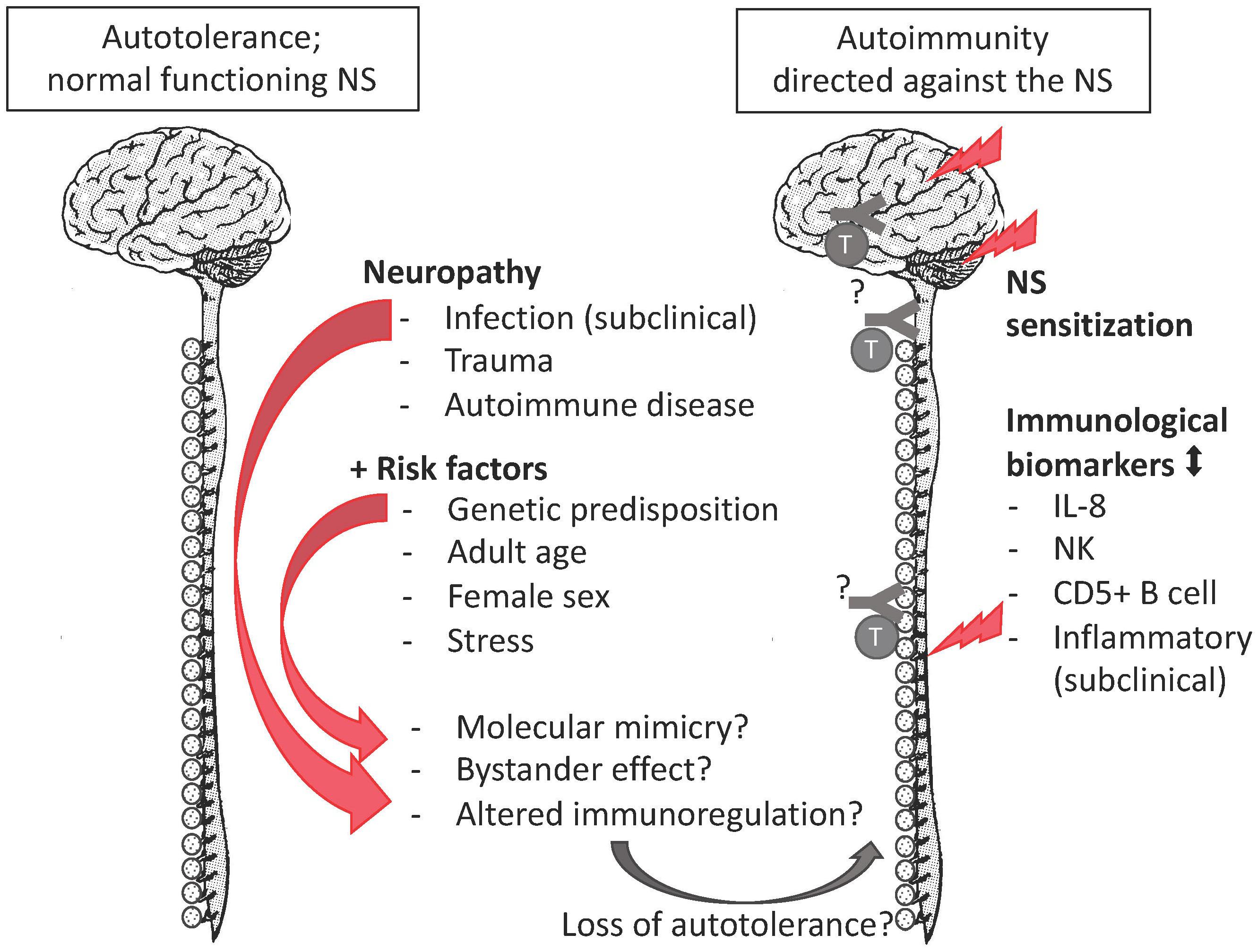
Figure 1. The autoimmune hypothesis for FMS. FMS complies with all mentioned risk factors of autoimmune disease, as well as with research biomarkers of an altered immune response. The missing pieces (indicated by “?”) are the evidence of autoantibodies or autoreactive lymphocytes against nervous tissue.
Why Does Central Sensitization Occur?
Pain perception not only depends on the pain stimulus, but also on the emotional and psychosocial state at a certain moment (Rhudy et al., 2010; Finnern et al., 2018). Both human and animal studies reveal greater pain sensitivity among females than males for most pain modalities (Rhudy et al., 2010; Kisler et al., 2016; Melchior et al., 2016; Aufiero et al., 2017; Kosek et al., 2018). The gender/sex bias in pain perception in various animal species denotes the importance of biological processes and thus sex differences therein. Unfortunately, pain studies aimed at other aspects than a sex or gender bias seldom report outcome variables according to sex or gender and only mention the proportion of males or females among study participants (Supplementary Table 1). As a consequence, although the sex-neutral neurophysiology of the pain pathway is well-described in several reviews (Basbaum et al., 2009; Zeilhofer et al., 2012; Peirs et al., 2015; Kendroud and Hanna, 2019) (Figure 2), little is known about sex differences in pain processing, except for modulation by sex-related hormones (Taleghany et al., 1999; Vincent and Tracey, 2008; Artero-Morales et al., 2018). These biological aspects are at least as important as the psychological aspect (Foo et al., 2017). Though sex differences in functional pain processing in itself are interesting, for FMS the focus is on pathological pain processing, which has been explained with the central sensitization hypothesis.
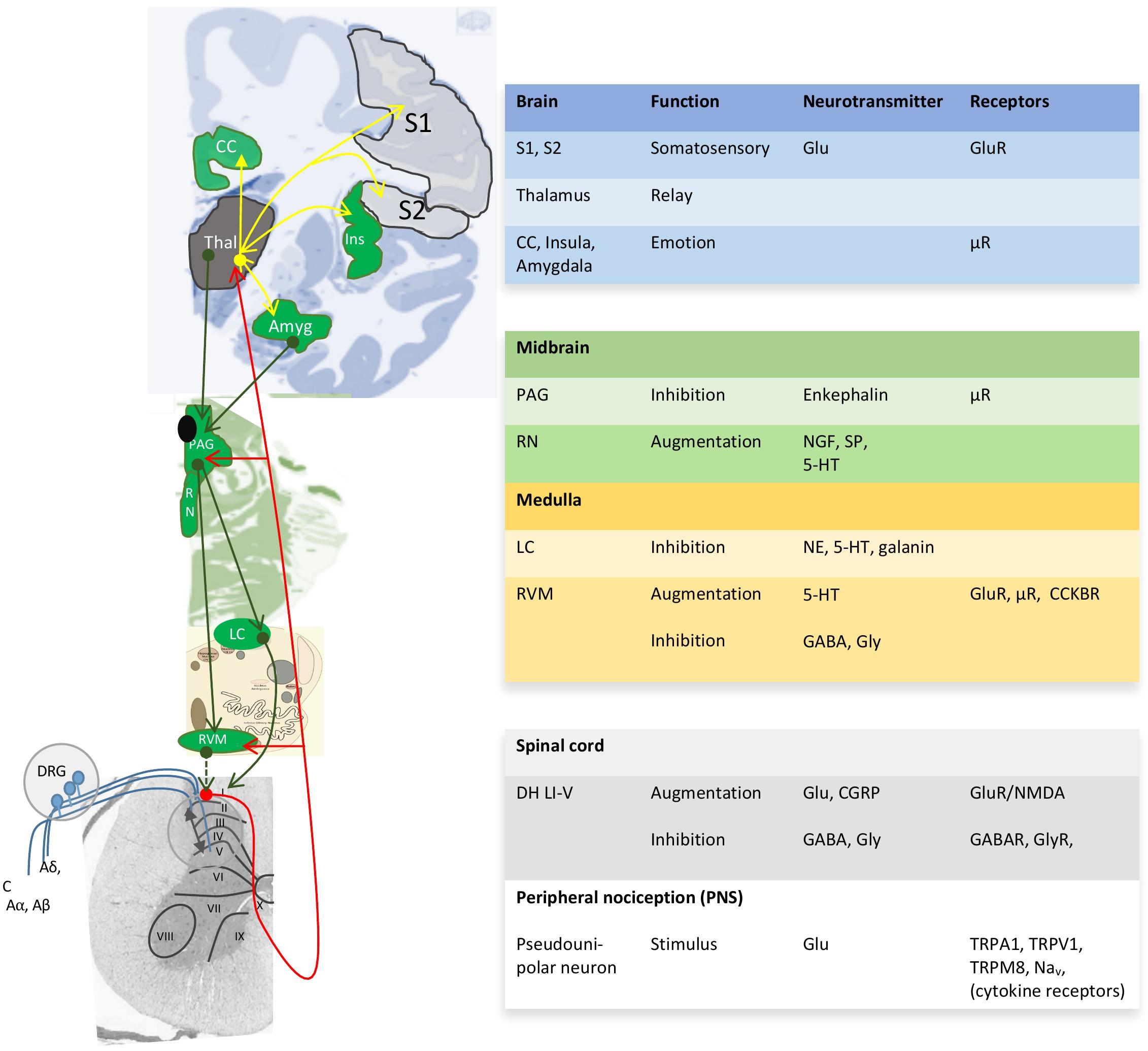
Figure 2. Neuroanatomy and chemistry of the central modulation of pain. Blue projections, incoming signals from 1st order neurons; red projections, ascending projections from 2nd order neurons toward thalamus (Thal) and cortical areas; yellow projections, projections for 3rd order neurons to cortical areas for awareness; green projections, descending projections that modulate the pain pathway. I-X, Reddit layers within the gray matter of the spinal cord; ↔, Integration of modulatory ascending and descending information in the dorsal horn Reddit laminae I-V (DH LI-V). 5-HT, serotonin; Aα, Aβ, Aδ y C, incoming nerves with decreasing levels of myelination; Amyg, amygdala; CC, cingulate cortex; CCKBR, cholecystokinin B receptor; CGRP, calcitonin gene-related peptide; DRG, dorsal root ganglia; Glu, glutamate; Ins, insula; LC, locus ceruleus; Nav, voltage-gated sodium channels; NE, norepinephrine; NGF, nerve growth factor; PAG, periaqueductal gray; RN, raphe nucleus; RVM, rostroventral medulla; S1, S, somatosensory areas 1 and 2; SP, substance P; TRP, transient receptor potential sensitive to nociceptive stimulus; μR, μ-opioid receptor with high affinity for enkephalins and beta-endorphin. Image based on (Tracey and Mantyh, 2007; Allen Human Brain Atlas, 2010; Ossipov et al., 2010).
Central sensitization was originally described as an increased electrophysiological activity in the dorsal horn in both a polyarthritic (Menétrey and Besson, 1982) and a post-injury male rat model (Woolf, 1983). Importantly, in both models a peripheral tissue injury triggered off alterations in dorsal horn neurons so that they augment pain signaling to normal input, even from low-threshold Aβ mechanoreceptors (Woolf, 1991). Besides, in animal models, sex differences in pain processing, i.e., at a biological level, could only be detected after neuropathy (Sorge et al., 2015) and involved the immune system.
Central sensitization has become the most accepted hypothesis to explain FMS mainly because peripheral sensitization due to autoimmunity is discarded because FMS does not comply with the following criteria of inflammation: (a) the presence of blood inflammatory biomarkers according to common clinical criteria, (b) responsiveness to non-steroid anti-inflammatory cyclooxygenase inhibitors. The former is debatable once research findings on immunological biomarkers (as mentioned in the previous section) are considered. The latter only indicates that FMS is not cyclooxygenase dependent, but discredits neither the involvement of other inflammatory and immune response pathways, nor does it nullify the possibility of neurogenic pain. Failure to comply with inflammatory criteria have not prevented the recognition of Graves’ disease (Baruah and Bhattacharya, 2012) and MS as autoimmune diseases (Luzzio and Dangond, 2018). Furthermore, the need for inflammatory biomarkers does not apply to secondary FMS because the accompanying autoimmune disease will generate not only inflammatory biomarkers but also a continuous nociceptive input. In secondary FMS, the CNS changes appear to improve when nociceptive input is removed (Sluka and Clauw, 2016). The point is that the nociceptive input may also be present in primary FMS, but we are ignorant of it. FMS pain is said to be idiopathic, but the burning, nagging, excruciating pain that is characteristic of FMS (Inanici and Yunus, 2004) is consistent with neuropathy. Histopathological studies have been performed on muscular and connective tissues (Inanici and Yunus, 2004), but not on dorsal root ganglia and central nervous tissue along the pain pathway. History teaches that myasthenia gravis was considered an idiopathic paralysis. Only after having established the involvement of the immune system were pathological immune infiltrates observed in muscle tissue (Hughes, 2005). There are indications that neuropathy is present in FMS patients (Üçeyler et al., 2013; Ramírez et al., 2015; Krumina et al., 2019). FMS pain may reflect a neuropathy of a currently unknown etiology, which is compatible with the central sensitization hypothesis. Latremoliere and Woolf (2009) reviewed neuroplasticity at the molecular and cellular level to explain central sensitization in pain hypersensitivity. They underscore the fundamental contribution of an inflammatory or neuropathic event to initiate the central sensitization.
Rat spinal cord slices exposed to pro-inflammatory cytokines display patch-clamp recordings that are congruent with the central sensitization hypothesis (Kawasaki et al., 2008), suggesting that the immune system plays a role; but, as is the case too often, proper controls were missing. A mouse study revealed the essential role of the immune sensor TLR8 in the maintenance of neuropathic pain. After nerve injury, TLR8 levels increased in the small IB4+ neurons in the dorsal root ganglia and in the ipsilateral dorsal horn. Subsequent intrathecal or intradermal injection of TLR8 agonists (VTX-2337 and miR-21) induced mechanical allodynia, and increased excitability of neurons in the dorsal root ganglia, accompanied by the expression of inflammatory mediators like interleukin 1 beta, interleukin 6, and tumor necrosis factor alpha. These effects were absent or reduced in TLR8 knock-out mice (Zhang et al., 2018). Aforementioned study did not report on sex differences, but in humans, TLR8 is an X-linked gene that may escape X-inactivation leading to a dosage difference between men and women (Umiker et al., 2014). TLR8 is especially important because of its pro-autoimmunity potential (Peng et al., 2005). A recent study with BALB/c and C57BL/6 mice suggests that circulating immunoglobulin G (IgG)-type immune complexes may directly mediate hyperalgesia at the level of dorsal root ganglia (Bersellini Farinotti et al., 2019) where macrophages and neurons have receptors for IgG1 and IgG2b (Bersellini Farinotti et al., 2019). If this mechanism were to be confirmed in humans, it would make women more vulnerable, because women are more inclined to humoral adaptive immune responses than men (see Section Sex Differences in the Immune System). Another mouse study revealed sex differences in pain processing. Intrathecal stimulation of the immune sensor of danger TLR4 induced mechanical allodynia in male, but not in female mice. At a cellular level, microglia in the spinal cord proliferated in both sexes after peripheral nerve injury, but only male microglia upregulate the immune sensor of danger, P2RX4 (Mapplebeck et al., 2016). P2RX4 detects nucleotides, mainly ATP, released after CNS stress or injury (Di Virgilio and Sarti, 2018). Activation of P2RX4 receptors leads to release of pro-inflammatory interleukin 1β and brain-derived neurotropic factor, which promote pain hypersensitivity. The inhibition of microglia in the spinal cord reversed allodynia only in male rodents. The female pain process requires more investigation (Sorge et al., 2015). As for humans, sex-biased pain because of knee osteoarthritis could be explained by differences in immune signaling molecules, interleukin 8 and monocyte-chemoattractant protein-1 (Kosek et al., 2018). Interleukin 8 is one of the immunological biomarkers most consistently associated with FMS (Kosek et al., 2015). Thus, sex differences in pathological pain processing mainly involves immune sensors and immune cells. And as we will see in Section “Sex Differences in the Immune System,” the immune system presents sex differences.
Besides, the sex bias of central sensitization remains to be elucidated. We hypothesize that autoimmunity directed to the CNS, either toxic or stimulatory, explains not only central sensitization, but also the female predominance of FMS and the lack of peripheral inflammatory biomarkers.
Why Is Slow-Wave Sleep Missing?
The neurobiology of sleep and the regulation of the daily sleep-wake cycle have been reviewed with a clinical perspective by España and Scammell (2011). Gender and sex differences in sleep health have been recognized but major gaps continue to exist is areas of sleep regulation, the epidemiology of sleep problems, diagnosis and treatment (Mallampalli and Carter, 2014). Two sexually dimorphic areas, the preoptic area and suprachiasmatic nucleus (Hofman and Swaab, 1989; Hofman et al., 1996) have been associated with sleep problems (España and Scammell, 2011). However, it seems that the sexual dimorphic nucleus of the preoptic area is dedicated to sexual and parental behavior (Rosenblatt et al., 1996) rather than to sleep regulation. More recently, another brain area, the anterior cingulate cortex, has been involved in both primary insomnia (Yan et al., 2018) and FMS (Jensen et al., 2009), but neither study analyzed sex differences. Still, sleep differs between men and women and may contribute to a sex-biased risk for sleep disorders (Mallampalli and Carter, 2014) and, consequently, for FMS.
Polysomnography studies have revealed a variety of sleep disturbances in FMS patients (Choy, 2015). Especially, the phase of deep sleep is reduced and otherwise affected. Instead of the synchronized and therefore high-amplitude slow waves characteristic of the δ rhythm, desynchronized low-amplitude high-frequency waves characteristic of the α rhythm interfere, generating a pattern known as α-δ sleep (Choy, 2015). Deep sleep is considered to be important for memory consolidation and restoration processes. Abnormal deep sleep and other sleep disturbances may contribute to the development of chronic pain (Finan et al., 2013) and FMS (Mork and Nilsen, 2012).
Taking together the difficulty to maintain slow-wave sleep and the peak symptomology upon awakening, it seems as if the regulation of the different sleep phases may be involved. The well-known somnogenic adenosine seems to have a special role in the regulation of the slow-wave sleep phase as revealed by studies with male C57BL/6 mice (Oishi et al., 2017). Extracellular adenosine accumulation activates the adenosine A1 receptor which inhibit arousal and induces slow-wave sleep. Another adenosine receptor, A2A receptor, can induce slow-wave sleep, but can be overruled by a motivation stimulus, like hunger or stress (Lazarus, et al. 2019). These receptors are found in the nucleus accumbens. Thus, the nucleus accumbens, already known for being part of the reward circuit, has a role in the control of the slow-wave sleep phase via adenosine receptors. A variety of enzymes and adenosine and nucleotide transporters in both neurons and astroglia are important for extracellular adenosine levels in the micro-environment of the nucleus accumbens. Interestingly, the A2A receptor also regulates naive T cell development in the thymus and its maintenance in the periphery (Cekic et al., 2013). In summary, adenosine, associated metabolites and involved enzymes and transporters may be important in slow-wave sleep and FMS, but further research is necessary.
Autoimmune Diseases: a Confused Immune System
Activation and Tolerance in the Immune System
The definition of autoimmune disease is relatively straightforward: a “disease that results when the immune system [..] mistakenly attacks the body’s own tissues” (Wein, 2013). In practice it is more complicated as non-symptomatic healthy persons tend to have autoantibodies (Tan et al., 1997), which are eliminated before doing harm (Nagele et al., 2013). For a disease to be classified as autoimmune there must be detectable autoantibodies or autoreactive T-cells in amounts sufficiently higher than non-symptomatic controls and they must explain the symptoms or present a strong epidemiological association with the symptoms. These requirements are fulfilled for all recognized autoimmune diseases, most often because of autoantibodies whether or not in combination with immune infiltrates (Dornmair et al., 2003). In autoimmune diseases that cluster with FMS, such as rheumatoid arthritis, Sjögren’s syndrome, and SLE, specific and diagnostic autoantibodies have been identified. Autoantibodies against cryptic nuclear, cytoplasmic and proteolipid protein antigens are shared in various autoimmune diseases (Suurmond and Diamond, 2015; Fayyaz et al., 2016).
Instead of being due to one specific cause, autoimmune diseases develop when risk factors accumulate. Central to autoimmune diseases is the loss of tolerance to autoantigens. Tolerance of the immune system is the non-activation of the immune response. Upon contact with a substance, particle or pathogen our defense system must decide to attack or to be tolerant. Essential for this decision is recognition, which is different for the three levels of protection of our defense system.
The first line of defense is a biophysicochemical barrier that does not need activation nor recognition. The second level of protection is provided by the innate immune system, a fast-reacting system without memory. Memory is not required as similar cell types or humoral factors share the same receptors for dangers and will attack the same patterns of danger. This in contrast with the third level of protection, provided by T lymphocytes and B lymphocytes. Lymphocytes have unique receptors, so that few will react upon a pathogenic invasion. T lymphocytes are activated by antigen presenting cells of the innate immune system, that provide instructions according to the danger pattern that were encountered. When selected and activated, lymphocytes generate a clone for future memory and effector cells that are either cytotoxic (Tc) or differentiate in a variety of mediators (helper T cells, Th) that potentiate different components of the innate and adaptive immune systems, while effector B lymphocytes (B) become plasma cells (plasma) that produce antibodies. If a tolerance response is erroneously converted into an active immune response, the memory of the adaptive immune response will respond with immune hypersensitivity upon future challenges.
Activation and Tolerance in the Innate Immune System
The tolerance mechanism of the innate immune system is passive; i.e., the innate immune system is only activated upon the detection of a limited number of non-self molecular patterns associated with pathogens (PAMPs) or endogenous damage (DAMPs). Hereto, innate immune cells share a limited set of PAMP recognition receptors. Depending on the pattern recognized, an appropriate action is initiated (Hoebe et al., 2004). Everything that is not a PAMP or DAMP is automatically tolerated by the innate immune system. In general, loss of auto-tolerance is not due to breaches of tolerance of the innate immune system.
Activation and Tolerance in the Adaptive Immune System
Both recognition/activation and tolerance are more complex in the adaptive immune system (Schwartz, 2012). The adaptive immune system is able to recognize a huge number of structures (known as antigenic determinants or epitopes) because of an enormous variety of specific receptors, T-cell receptors (TCRs) and B-cell receptors, that differ among lymphocytes. As a consequence, when a noxious antigen invades the body, only few specific lymphocytes will react. Clonal proliferation of a triggered lymphocyte generates a specific ‘army’ composed of effector lymphocytes (to eliminate current danger) and memory lymphocytes to provide a faster and stronger immune response for a future challenge with the same antigen.
As the specificity of the TCR and B-cell receptors is generated at random, these receptors may recognize harmless xenoantigens and autoantigens (Klein et al., 2014). To avoid allergies and autoimmune diseases, the adaptive immune system has three control mechanisms: (a) central tolerance, (b) peripheral tolerance, and (c) major histocompatibility complex-restricted activation of T cells, i.e., a T cell can only be activated when its cognate antigen is presented by a major histocompatibility complex molecule.
HLA Presents Protein-Associated Epitopes to the Adaptive Immune System
In humans, the major histocompatibility complex is known as HLA. The HLA genes of class I (A, B, and C) and II (DP, DQ, and DR) are the most polymorphic genes of the human genome and provide a molecular identity to an individual. All nucleated cells of the human body express HLA-I, although leukocytes express them in larger amounts, while HLA-II is expressed by specialized antigen-presenting cells. HLA-I presents antigens to CD8 + cytotoxic T cells, while HLA-II presents antigens to CD4+ helper T cells (Th) (Hoebe et al., 2004). As antigen presentation to T cells is HLA-restricted, only body-own cells will be protected. The polymorphism of the HLA molecules has an impact on the quality of the immune response. Certain HLA-antigen complexes facilitate the immune response of an individual and others not. Thus, the individuals of a population differ in their protective capacity and their susceptibility to autoimmune diseases. The antigen presentation process is designed for large proteins, preferentially in a particulate presentation. Therefore, electrolytes, sugars, lipids (e.g., sex hormones), nucleic acids, peptides (e.g., neuropeptides), and other small molecules (e.g., neurotransmitters) are passively tolerated. On the other hand, tolerance to protein autoantigens is an active, energy-consuming, and highly controlled process with central and peripheral mechanisms.
Mechanisms of Central Tolerance
Central tolerance is established in the primary lymphatic organs, most importantly, in the thymus, where developing T cells or thymocytes reorganize their genome at random to express a unique TCR, either xenoreactive or autoreactive. The thymic epithelial cells function as ‘teachers’ of the thymocytes. Hereto, the thymic epithelial cells express “AIRE),” a transcription factor that facilitates the expression of organ-specific autoantigens in the thymus. The autoantigens are presented in combination with HLA to the thymocytes. To survive, thymocytes should recognize HLA (positive selection) but not recognize autoantigens (negative selection). Simplified, autoreactive thymocytes have two fates (Klein et al., 2014): (1) they die by apoptosis, in case of a strong and long antigen-TCR interaction, and (2) they become regulatory T cells (nTreg), when the antigen-TCR interaction is of intermediate strength and length (Azzam et al., 2001; Ohkura et al., 2013). Thymocytes that do not react with autoantigens or only shortly and with low-affinity are liberated as naïve Th or cytotoxic T cells to protect against danger in the periphery.
As not all autoantigens are expressed in the thymus in sufficient amounts, autoreactive naive T cells may circulate in the periphery, a phenomenon known as ignorance.
Mechanisms of Peripheral Tolerance
Peripheral tolerance complements central tolerance by any of the following mechanisms: (1) clonal deletion of autoreactive T cells by apoptosis, (2) the peripheral induction of Treg (iTreg) under the influence of transforming growth factor beta, (3) anergy, i.e., a reversible inactive state of the T cell when the antigen presenting cells do not provide a costimulatory signal (Mueller, 2010), and (4) ignorance, i.e., the amount of auto-antigen is insufficient to induce either tolerance or an immune response. The choice for any of these tolerance options depends on the abundance, strength and duration of the TCR-antigen interactions, but it is conditioned by the absence of a danger signal. As the T cell has no information about the type of danger recognized by its at-random-generated TCR, this information is provided by the antigen presenting cell. Depending on the type of PRR activated the antigen presenting cell provides a costimulatory signal and instructions about the desired type of response (Th1, Th2, or Th17 response) via cytokines (Kaiko et al., 2008). Apart from these major tolerance processes, immune responses are fine-tuned by many stimulatory, inhibitory, and modulatory molecules (e.g., CD5) (Sigal, 2012) and cells, e.g., NKT cells (Dasgupta and Kumar, 2016) within the immune system, as well as neuroendocrine peptides and hormones beyond the immune system (Carniglia et al., 2017). In summary, the adaptive system mainly attacks large protein (>10 kDa) antigens and tolerance to large self-proteins is a complex and highly regulated process.
Loss of Tolerance and Development of Autoimmune Diseases
Tolerance may be breached for any of the following reasons or a combination of them. First of all, molecular mimicry between an auto-antigen and a pathogenic antigen may confuse the immune system. A well-known example is the Guillain-Barré syndrome due to autoantibodies directed to peripheral nerves because of cross-reaction between nerve autoantigens and certain pathogen antigens, especially Campylobacter jejuni, Epstein-Barr virus, cytomegalovirus (Jacobs et al., 1998).
Secondly, bystander autoantigens that are presented in combination with danger signals due to concurrent infections or physical trauma may overcome tolerance, which may play a role in the development of autoimmune diseases (Gestermann et al., 2018). The bystander mechanism may also explain how a ‘founder’ or primary autoimmune disease may lead to secondary autoimmune diseases. Cell lysis due to inflammation of the primary autoimmune disease liberates multiple cryptic autoantigens (mitochondrial antigens, phospholipid antigens, ribonucleoproteins, and other cytoplasmic autoantigens) that provide DAMPs. And the cycle repeats itself: novel autoantigens in combination with danger signals may lead to multiple autoreactive clones of lymphocytes. Cryptic autoantigens liberated during necrosis are shared by different autoimmune diseases (Suurmond and Diamond, 2015). Although the etiology is not proven, latent viral infections associate with MS (Virtanen et al., 2014). A reactivating latent viral infection may cause not only minor damage to nervous tissue but also a breach in tolerance either by molecular mimicry or the bystander effect. Multiple cryptic autoantigens are liberated due to repeated tissue damage, so that polyautoimmunity develops. In MS, oligoclonals are directed against a mixture of autoantigens of cellular debris (Brändle et al., 2016). Initially, MS can be more or less controlled by interferon treatment (Multiple Sclerosis Therapy Consensus Group, 2008), which has antiviral activity, until the polyautoimmune disease has become autosustainable and the relapsing-remitting MS patient turns into a secondary-progressive MS patient. In MS, just as in FMS, inflammatory biomarkers of the blood tend to be within the normal range (Luzzio and Dangond, 2018) as the inflammatory process occurs localized, i.e., behind the blood-brain barrier. Aforementioned process probably plays a role in many autoimmune diseases but have been recognized for only a few. The clinical importance is high because antimicrobial treatment may resolve the initial infection, once the autoimmune disease has developed it may be too late (Mukherjee et al., 2018). Alternatively, it explains why steroid anti-inflammatory treatment worsens (Clark et al., 1985) rather than improves the symptoms.
Thirdly, there is a genetic predisposition for most autoimmune diseases, especially in the HLA genes, probably as a consequence of affinity issues between the HLA molecule and the presented antigen (Bodis et al., 2018). HLA-DR2, DR3, DR4, DQ6, and DQ8 are associated with SLE, sicca syndrome, and rheumatoid arthritis (Mangalam et al., 2013). Furthermore, gene polymorphisms in complement factors may diminish autoantibody clearance (Macedo and Isaac, 2016).
Fourthly, CD5 expression fine-tunes the adaptive immune response in a complex way (Sigal, 2012). CD5 on T or B lymphocytes may either facilitate or inhibit an adaptive immune response depending on avidity issues (Domingues et al., 2016). In general, CD5+ B cells have been related with increased susceptibility for autoimmune disease, while the opposite applies to T cells (Tarakhovsky et al., 1995; Pers et al., 1999; Hawiger et al., 2004).
Fifthly, stress dysregulates the immune system (Sharif et al., 2018). Acute stress enhances catecholamines and circulating leukocytes, which facilitates a stronger immune response. Chronic stress, on the other hand, is immunosuppressive. The stress hormone cortisol diminishes leukocyte numbers and suppresses leukocyte function (Dhabhar, 2008). The stress hormones epinephrine and cortisol induce a rapid leukocyte redistribution. Although the exact mechanisms of this phenomenon remains to be elucidated, clinical and epidemiological data convincingly and consistency reveal an association between chronically stressed people and vulnerability to and resurgence of infections and autoimmune diseases (Reiche et al., 2004).
Sixthly, the immune response is age dependent (Elisia et al., 2017) and age has is a risk factor for loss of central tolerance. In over 60% of autoimmune cases, the onset of symptoms was in the fourth and fifth decade of life, with a median onset at 37.5 years of age (Euesden et al., 2017). A detection bias may play a role in the age effect. Autoimmune disease may not be apparent at the onset, because the progression is slow and their biochemical, physiological or visual detection not obvious. For example, Sjögren syndrome, an autoimmune disease affecting the glands, becomes apparent in older individuals, because destruction of the glands is slow and symptoms of dryness are only experienced when most of the glands are destroyed (Hsu and Mountz, 2003). Importantly, the most essential organ for central tolerance, the thymus, undergoes profound age-associated atrophy (Lynch et al., 2009). Thymic decline is clearly associated with the presence of sex steroids (Heng et al., 2005).
And finally, being female is a risk factor for many autoimmune diseases. A sex bias in the immune system is well-documented and will be described in more detail in the next section.
The unfortunate co-occurrence of aforementioned risk factors elicits an autoimmune disease. It is important to highlight that the initial trigger may be transitory, but the induced autoimmune response is chronic due to the memory of the adaptive immune response. Aforementioned risk factors all apply to FMS (see Section Fibromyalgia: Introducing the Autoimmune Hypothesis and Figure 1).
Sex Differences in the Immune System
The burden of infectious diseases and the incidence of cancer, allergies, and autoimmune disease differs between men and women. Though this phenomenon can be partially explained by a differential exposure of men and women to environmental risk factors, sex differences in the immune system are evident and well-known. The expression of many immune system-associated proteins and activation of immune cells differs between the sexes as have been reviewed elsewhere (Klein and Flanagan, 2016; Rainville et al., 2018). Well-known is a Th1 bias in male and a Th2 bias in female (Klein and Flanagan, 2016). The latter facilitates the natural passive transfer of immunity from mother to fetus, as immunoglobulins G are able to pass the placenta to protect the fetus. Furthermore, as of adulthood, the number of innate and adaptive leukocytes is higher in females than in age-matched males (Urquhart et al., 2008), except for Treg and innate lymphoid cells, including NK, which are more abundant in males (Klein and Flanagan, 2016). NK have functions beyond cytotoxicity including an important role in the regulation of immune homeostasis and inflammation (Vitale et al., 2005). During aging, the diverse NK population changes gradually; the proportion of immunoregulatory CD56hi NK diminishes in favor of highly differentiated cytotoxic CD57+ NK. This redistribution may explain functional changes in NK cells with healthy aging (Gayoso et al., 2011). The dysregulation of NK and NKT cells is associated with allergies and autoimmune diseases (Nielsen et al., 2013; Tahrali et al., 2018). Here, we highlight that the cellular immune response tends to predominate in men and the humoral immune response in women. Furthermore, adult men tend to have more immunoregulatory cells than women.
Differential Effect of Sex Hormones on Leukocyte Behavior
Sex hormones differentially influence the behavior of leukocytes. There is a differential distribution of sex hormones receptors among leukocytes. For example, CD4+ T cells have high levels of the estrogen receptor alpha but low estrogen receptor β levels, whereas the opposite applies for Treg. CD8+ Tc have low expression levels for both types of estrogen receptors, whereas NK express both receptors highly. In mice, high estrogen levels signal via estrogen receptor α and induce antiviral type 1 interferon and NK cells, as well as Treg. Signaling via estrogen receptor β has opposing effect and results in diminished Treg activation. Furthermore, the transmembrane G protein-coupled estrogen receptor, which induces rapid signaling is highly expressed by certain leukocytes (CD4+ Th, Treg, B cells, and macrophages) (Koenig et al., 2017). Estrogen diminishes neutrophil infiltrates and protects against the harmful effects of the innate response (Shimizu et al., 2008; Ritzel et al., 2013). Progesterone and testosterone promote monocyte recruitment and an androgen receptor antagonist reduced monocyte recruitment (Toyoda et al., 2012; Sutti and Tacke, 2018). On the other hand, testosterone treatment reduced immunoglobulin M and immunoglobulin G production and as such diminishes peripheral humoral adaptive immunity (Kanda et al., 1996). The effects of sex hormones are complex because of opposing effects depending on concentration (Hughes, 2012), the variety in metabolic forms that modulate the immune response, and relative concentrations of various sex hormones. Our intention is not to unravel the intricate interactions between the sex hormones and immune system, but to highlight its existence. The area requires more investigation, but it seems that testosterone procures central tolerance. Taken together, testosterone and progesterone downregulate peripheral (humoral) adaptive immunity, but facilitate peripheral innate immunity (Hughes and Clark, 2007; Lai et al., 2012), whereas the opposite applies for estrogen as it is associated with peripheral innate immune suppression, stronger humoral responses, and weaker central tolerance (Kovats, 2015).
Sex Differences in Immune Tolerance
As mentioned above, the thymus has an essential role in central tolerance. Thymic involution is different between the sexes, especially after the onset of puberty, with a more prominent decline in males than in females, so that the adult female thymus contains more thymocytes and has a higher thymic output than the male thymus (Gui et al., 2012). However, the T cells liberated from the female thymus may have been less well ‘educated’ as the female thymic epitheliocytes express less AIRE and fewer autoantigens (mRNA and protein) than the male ones (Dragin et al., 2016). Low expression levels of AIRE associate with autoimmune disease (Sato et al., 2002; Liu et al., 2014).
The sex hormones, estrogen and dihydrotestosterone (the main active metabolite of testosterone), regulate AIRE expression in opposite directions. At physiological doses, estrogen diminishes AIRE expression, whereas dihydrotestosterone increases AIRE expression (Dragin et al., 2016). Dihydrotestosterone treatment in an experimental animal model of MS, upregulated AIRE and tissue-specific antigen expression in the thymus, improved negative selection of autoreactive T cells, and diminished the severity of autoimmune disease (Zhu et al., 2016). A cross-sectional population-based study revealed that a higher estradiol/testosterone ratio associated significantly with autoimmune thyroid disease among Chinese men (Chen et al., 2017). Interestingly, a clinical phase I/II pilot study among 12 female FMS patients revealed that a 28-day treatment with testosterone gel significantly decreased pain, stiffness, and fatigue (White et al., 2015). Altogether, the efficacy of the thymus depends strongly on age and sex hormones. Male sex hormones seem to compensate for thymic involution with an increased AIRE expression, whereas female sex hormones contribute to diminished tolerance and increased vulnerability to autoimmune diseases.
Sex Chromosomes and Immune-Associated Genes
The major genetic difference between men and women are the sex chromosomes. Men are XY, whereas women are XX. To enable the pairing of the X and Y chromosomes during male meiosis, small pseudo-autosomal regions are present at the extremes of both the X and Y chromosome. In the pseudo-autosomal regions, the X and Y chromosomes encode the same genes (Mangs and Morris, 2007). For non-pseudoautosomal region genes, males will express the genes of the unique X chromosome, whereas female cells perform at random X inactivation as a dosage compensation mechanism. X inactivation is clonally maintained and generates a functional mosaic organism for X chromosome-encoded genes (Rubtsov et al., 2010). Importantly, X-chromosome inactivation is not an all-or-non-phenomenon; about 10–15% of the X chromosome-encoded genes escape X-inactivation in humans, and a mouse model of accelerated aging revealed increased reactivation with age (Berletch et al., 2011).
Although it has been stated that the X chromosome encodes a disproportionally large number of immune-associated genes (Bianchi et al., 2012), so far no scientifically sound supporting evidence has been published. Still, the increased susceptibility for immune hypersensitivities of men with Klinefelter’s syndrome (XXY) or women with Turner syndrome (X-) reveals the importance of dosage of X-linked immune system-associated genes (Mortensen et al., 2009; Sawalha et al., 2009). An elegant study that used the four genome model (XXSry+, XYSry+, XX, XYSry–) in two gonadectomized mouse models for autoimmune disease demonstrated a dosage effect of the X chromosome in SJL mice. Interestingly, the dosage effect was not observed in C57BL/6 mice (Smith-Bouvier et al., 2008). The genetic background of C57BL/6 mice contrast with the one of SJL mice in terms of susceptibility for murine cytomegalovirus and autoimmune disease. In contrast to SJL, C57BL/6 has a bias toward a Th1 response and high NK activity (Sellers et al., 2012; Song and Hwang, 2017). This cellular immune response protects against viral infections.
Two X-linked genes are especially associated with humoral autoimmune disease to cryptic antoantigens. TLR7 and TLR8 (both in band Xp22.2) encode endosomal immune sensors that sense microbial and endogenous RNA (Umiker et al., 2014). TLR7 expression displays a dosage disequilibrium in biallelic B lymphocytes of women and men with Klinefelter’s syndrome. These biallelic B lymphocytes switch more easily to IgG (Souyris et al., 2018), which is consistent with SLE symptom severity in TLR7 + as compared to TLR7-deficient C57BL/6 mice (Desnues et al., 2014). In a 564Igi mouse model of SLE, a dosage difference in TLR8 determined the sex bias in anti-RNA IgG antibodies, which were higher in female than male mice (Umiker et al., 2014). 564Igi mice are especially susceptible to autoimmunity because of diminished somatic hypermutation (McDonald et al., 2017). The release of miR-21 due to neuropathy stimulates TLR8 signaling in the dorsal root ganglia, which leads to hyperexcitability and pain (Zhang et al., 2018). Importantly, TLR8 activation can reverse tolerant Treg into aggressive forms (Peng et al., 2005). Thus, X-linked RNA immune sensors may be activated in neuropathy and favor autoimmunity rather than immune tolerance.
Autoimmunity to the Nervous System. What Triggers It Off?
The CNS used to be considered an immunoprivileged organ, and therefore little susceptible to autoimmune issues. But this viewpoint has changed over the last two decades upon the detection of autoantibodies against a variety of nervous system autoantigens. As expected, most autoantibodies are directed against large, complex protein autoantigens (Table 1) rather than against small non-protein molecules. Currently, among patients with mental illnesses, the serum prevalence of autoantibodies against nervous tissue antigens is 11–17%, which may be an underestimation (Graus and Santamaría, 2017).

Table 1. Autoantibodies against nervous tissue autoantigens.
After neuropathy (due to infection or a lesion), intracellular macromolecules that previously were encrypted autoantigens may be exposed and targeted by autoantibodies (Totsch and Sorge, 2017). The latter probably occurs in MS, where oligoclonals target ubiquitous intracellular proteins (Brändle et al., 2016). Oligoclonal bands are interpreted as immunoglobulins that are produced intrathecally, i.e., inside the CNS (Luzzio and Dangond, 2018). Oligoclonal bands occur in 95% of MS patients (Halbgebauer et al., 2016). Other targets of MS autoantibodies are myelin-associated autoantigens (Link et al., 1990) and viral antigens (Virtanen et al., 2014). Although no specific virus is considered to be the causative agent of MS, viruses may be direct or indirect risk factors. The latter via molecular mimicry and/or bystander activation (Virtanen and Jacobsen, 2012) as described in Section “Loss of Tolerance and Development of Autoimmune Diseases.” Various psychiatric diseases are considered to be caused by either an autoimmune process or an infection (Singh and Trevick, 2016; Dubey et al., 2018). We propose that in many cases both occur; the infection would be the initiation event and autoimmunity a consequence. Still they may occur simultaneously, especially when involving opportunistic pathogens. A variety of herpes viruses are opportunistic, pandemic, and neurotropic. Depending on the geographic location, 40–100% of the adult population is infected. A primary infection establishes a lifelong latent infection, which reactivates intermittently without obvious disease symptoms, except for immunocompromised persons. Stress may be a trigger for ‘asymptomatic’ reactivation. Among herpes viruses, cytomegalovirus seems especially apt to alter the immune response into autoimmunity (Andersen and Andersen, 1978; Varani and Landini, 2011; Halenius and Hengel, 2014), while Epstein-Barr virus-transformed lymphocytes tend to produce autoantibodies (Garzelli et al., 1984). In case FMS etiology involves neuropathy by reactivating latent pathogens, the unresponsiveness to corticosteroid treatment is understood. Considering the heterogeneity of FMS patients, other infections or neuropathic events should not be ruled out as possible triggers of autoimmunity.
The Missing Piece: Evidence of Autoimmune Components Specific for FMS
Autoantibodies against intracellular antigen, nervous and muscle tissue have been reported in FMS patients (Supplementary Table 1), but their role in FMS pathogenesis is controversial. We suggest to screen nervous tissue involved in the pain pathway, both CNS (including the pituitary and pineal glands) and peripheral nervous tissue (dorsal root ganglia) with patient samples (blood and CSF), to complete the missing evidence (Figure 1). A variety of conceptual and technical issues may complicate the detection of autoantibodies. The lack of tissue lesions should not be interpreted as the absence of autoimmunity, as autoantibodies may be stimulatory as in Graves’ disease (Yeung and Habra, 2018). Screening of autoantibodies should not be limited to blood, as autoantibodies or oligoclonals may be limited to CSF when neuropathic symptoms predominate (Luzzio and Dangond). Furthermore, pleocytosis of leukocytes in CSF should be evaluated. Autoantibody screening on certain tissues may yield false negatives when the autoantibodies are directed against other tissues than the ones that are screened. Screening on animal tissues may yield false negatives when the human autoantigens are sufficiently different from the animal forms. Screening on fixed tissues may yield false negatives because the appropriate antigen retrieval method was not applied. Also, the autoimmune response may be cellular rather than humoral. A conceptual or interpretation issue are prodromal autoantibodies; tissue destruction mediated by prodromal autoantibodies remains asymptomatic until the overcapacity of the targeted organ has been lost (Arbuckle et al., 2003; Hayashi et al., 2008; Haller-Kikkatalo et al., 2017). During the prodromal period, autoantibodies run the risk to be interpreted as false positives. Longitudinal follow-up studies of patients with prodromal autoantibodies would be interesting. And finally, because of the heterogeneity among FMS patients, a certain etiology or pathogenesis may be limited to a subgroup of FMS patients (Jacobsen et al., 1990; Purnamawati et al., 2018). The worst scenario would be that the detected pathogenesis is discarded because it does not apply to a sufficiently high proportion of FMS patients. To avoid this situation, stratification or clustering of FMS patients is recommendable. Despite the aforementioned, the challenge is not impossible; the detection of anti-IgLON5 is exemplary (Sabater et al., 2014). We recommend a similar screening technique to verify whether an autoimmune process is involved in the pathogenesis of FMS.
Conclusion
The clinical profile of FMS displays a strong overlap with certain autoimmune diseases. In fibromyalgia, physical or mental stress may constitute a precipitating factor or a consequence rather than a cause, similar to the situation in autoimmune diseases. Stress may debilitate the immune system and allow for reactivation of a latent (viral) infection, which may cause neuroinflammation or neuropathy and facilitate autoimmune phenomena. However, different from most autoimmune diseases, common clinical serum markers of inflammation are within the normal range in FMS. Still, altered immunological biomarkers, especially CD57 and IL-8 levels, are compatible with a viral infection or autoimmune mechanism. Sex differences in the immune system would explain a sex bias in FMS prevalence. If convincing evidence for an autoimmune process were detected for FMS, diagnostic tests and effective therapies could be developed. Blood and CSF should be screened for autoantibodies and/or autoreactive lymphocytes. Screening for autoantibodies directed to peripheral nervous tissues and CNS should include dorsal root ganglia, the spinal cord, pituitary gland and pineal gland, should be screened as possible targets for autoantibodies and autoreactive lymphocytes.
Author Contributions
All authors complied with the ICMJE criteria for authorship of contribution in conception or data acquisition, drafting or revising the manuscript, approval of the final document, and agreement with all aspects of the document. The contributions of GR-S were mainly in the neuroscience and pain part, the ones of FG-S in the immune system section, and IM in the FMS and autoimmune aspects, as well as the coordination of the publication aspects.
Funding
This work was supported by the University of Monterrey (UDEM); grant number UIN17536.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. David Hafner for reviewing the document and the University of Monterrey (UDEM) for grant UIN17536.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2019.01414/full#supplementary-material
Abbreviations
AIRE, autoimmune regulator; CNS, central nervous system; CSF, cerebrospinal fluid; FMS, fibromyalgia syndrome; HLA, human leukocyte antigen; MS, multiple sclerosis; NK, natural killer cell; SLE, systemic lupus erythematosus; TCR, T cell receptor; TLR, Toll-like receptor; Th, helper T lymphocyte; Treg, suppressor T lymphocyte.
References
Allen Human Brain Atlas (2010). © 2010 Allen Institute for Brain Science. [Internet]. Seattle, WA: Allen Institute.
Andersen, P., and Andersen, H. K. (1978). Immunoglobulin levels and specific viral antibodies in relation to smooth-muscle antibodies in cytomegalovirus infection. J. Clin. Lab. Immunol. 1, 233–236.
Arbuckle, M. R., McClain, M. T., Rubertone, M. V., Scofield, R. H., Dennis, G. J., James, J. A., et al. (2003). Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 349, 1526–1533. doi: 10.1056/NEJMoa021933
Arnold, L. M., Bennett, R. M., Crofford, L. J., Dean, L. E., Clauw, D. J., Goldenberg, D. L., et al. (2019). AAPT diagnostic criteria for fibromyalgia. J. Pain 20, 611–628. doi: 10.1016/j.jpain.2018.10.008
Artero-Morales, M., González-Rodríguez, S., and Ferrer-Montiel, A. (2018). TRP channels as potential targets for sex-related differences in migraine pain. Front. Mol. Biosci. 5:73. doi: 10.3389/fmolb.2018.00073
Aufiero, M., Stankewicz, H., Quazi, S., Jacoby, J., and Stoltzfus, J. (2017). Pain perception in latino vs. caucasian and male vs. female patients: is there really a difference? West. J. Emerg. Med. 18, 737–742. doi: 10.5811/westjem.2017.1.32723
Azzam, H. S., DeJarnette, J. B., Huang, K., Emmons, R., Park, C. S., Sommers, C. L., et al. (2001). Fine tuning of TCR signaling by CD5. J. Immunol. 166, 5464–5472. doi: 10.4909/jimmunol.166.9.5464
Bäckryd, E., Tanum, L., Lind, A. L., Larsson, A., and Gordh, T. (2017). Evidence of both systemic inflammation and neuroinflammation in fibromyalgia patients, as assessed by a multiplex protein panel applied to the cerebrospinal fluid and to plasma. J. Pain Res. 10, 515–525. doi: 10.2147/JPR.S128508
Baruah, M. P., and Bhattacharya, B. (2012). Significant role of serum CRP in differentiating inflammatory from non-inflammatory causes of thyrotoxicosis. Indian J. Endocrinol. Metab. 16, 976–981. doi: 10.4103/2230-8210.103002
Basbaum, A. I., Bautista, D. M., Scherrer, G., and Julius, D. (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. doi: 10.1016/j.cell.2009.09.028
Beeson, P. B. (1994). Age and sex associations of 40 autoimmune diseases. Am. J. Med. 96, 457–462. doi: 10.1016/0002-9343(94)90173-2
Berletch, J. B., Yang, F., Xu, J., Carrel, L., and Disteche, C. M. (2011). Genes that escape from X inactivation. Hum. Genet. 130, 237–245. doi: 10.1007/s00439-011-1011-z
Bersellini Farinotti, A., Wigerblad, G., Nascimento, D., Bas, D. B., Morado Urbina, C., Nandakumar, K. S., et al. (2019). Cartilage-binding antibodies induce pain through immune complex-mediated activation of neurons. J. Exp. Med. 216, 1904–1924. doi: 10.1084/jem.20181657
Bianchi, I., Lleo, A., Gershwin, M. E., and Invernizzi, P. (2012). The X chromosome and immune associated genes. J. Autoimmun. 38, J187–J192. doi: 10.1016/j.jaut.2011.11.012
Bodis, G., Toth, V., and Schwarting, A. (2018). Role of human leukocyte antigens (HLA) in autoimmune disease. Rheumatol. Ther. 5, 5–20. doi: 10.1007/s40744-018-0100-z
Branco, J. C., Tavares, V., Abreu, I., Correia, M. M., and Macahdoa Cetano, J. A. (1996). HLA studies in fibromyalgia. J. Musculoskelet. Pain 4, 21–27. doi: 10.1300/J094v04n03_03
Brändle, S. M., Obermeier, B., Senel, M., Bruder, J., Mentele, R., Khademi, M., et al. (2016). Distinct oligoclonal band antibodies in multiple sclerosis recognize ubiquitous self-proteins. Proc. Natl. Acad. Sci. U.S.A. 113, 7864–7869. doi: 10.1073/pnas.1522730113
Buskila, D., and Sarzi-Puttini, P. (2008). Fibromyalgia and autoimmune diseases: the pain behind autoimmunity. Isr. Med. Assoc. J. 10, 77–78.
Carniglia, L., Ramírez, D., Durand, D., Saba, J., Turati, J., Caruso, C., et al. (2017). Neuropeptides and microglial activation in inflammation, pain, and neurodegenerative diseases. Mediators Inflamm. 2017:5048616. doi: 10.1155/2017/5048616
Caturegli, P., Newschaffer, C., Olivi, A., Pomper, M. G., Burger, P. C., and Rose, N. R. (2005). Autoimmune hypophysitis. Endocr. Rev. 26, 599–614. doi: 10.1210/er.2004-0011
Cekic, C., Sag, D., Day, Y. J., and Linden, J. (2013). Extracellular adenosine regulates naive T cell development and peripheral maintenance. J. Exp. Med. 210, 2693–2706. doi: 10.1084/jem.20130249
Chen, Y., Chen, Y., Xia, F., Wang, N., Chen, C., Nie, X., et al. (2017). A higher ratio of estradiol to testosterone is associated with autoimmune thyroid disease in males. Thyroid 27, 960–966. doi: 10.1089/thy.2016.0661
Choy, E. H. (2015). The role of sleep in pain and fibromyalgia. Nat. Rev. Rheumatol. 11, 513–520. doi: 10.1038/nrrheum.2015.56
Ciregia, F., Giacomelli, C., Giusti, L., Boldrini, C., Piga, I., Pepe, P., et al. (2018). Putative salivary biomarkers useful to differentiate patients with fibromyalgia. J. Proteom. 190, 44–54. doi: 10.1016/j.jprot.2018.04.012
Clark, S., Tindall, E., and Bennett, R. M. (1985). A double blind crossover trial of prednisone versus placebo in the treatment of fibrositis. J. Rheumatol. 12, 980–983.
Dasgupta, S., and Kumar, V. (2016). Type II NKT cells: a distinct CD1d-restricted immune regulatory NKT cell subset. Immunogenetics 68, 665–676. doi: 10.1007/s00251-016-0930-1
Desnues, B., Macedo, A. B., Rousset-Queval, A., Bonnardel, J., Henri, S., Demaria, O., et al. (2014). TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc. Natl. Acad. Sci. U.S.A. 111, 1497–1502. doi: 10.1073/pnas.1314121111
Dhabhar, F. S. (2008). Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection versus immunopathology. Allergy Asthma Clin. Immunol. 4, 2–11. doi: 10.1186/1710-1492-4-1-2
Di Virgilio, F., and Sarti, A. C. (2018). Microglia P2X4 receptors as pharmacological targets for demyelinating diseases. EMBO Mol. Med. 10:e9369. doi: 10.15252/emmm.201809369
Domingues, R. G., Lago-Baldaia, I., Pereira-Castro, I., Fachini, J. M., Oliveira, L., Drpic, D., et al. (2016). CD5 expression is regulated during human T-cell activation by alternative polyadenylation, PTBP1, and miR-204. Eur. J. Immunol. 46, 1490–1503. doi: 10.1002/eji.201545663
Dornmair, K., Goebels, N., Weltzien, H. U., Wekerle, H., and Hohlfeld, R. (2003). T-cell-mediated autoimmunity: novel techniques to characterize autoreactive T-cell receptors. Am. J. Pathol. 163, 1215–1226. doi: 10.1016/S0002-9440(10)63481-5
Dragin, N., Bismuth, J., Cizeron-Clairac, G., Biferi, M. G., Berthault, C., Serraf, A., et al. (2016). Estrogen-mediated downregulation of AIRE influences sexual dimorphism in autoimmune diseases. J. Clin. Invest. 126, 1525–1537. doi: 10.1172/JCI81894
Dubey, D., Pittock, S. J., Kelly, C. R., McKeon, A., Lopez-Chiriboga, A. S., Lennon, V., et al. (2018). Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann. Neurol. 83, 166–177. doi: 10.1002/ana.25131
Elisia, I., Lam, V., Hofs, E., Li, M. Y., Hay, M., Cho, B., et al. (2017). Effect of age on chronic inflammation and responsiveness to bacterial and viral challenges. PLoS One 12:e0188881. doi: 10.1371/journal.pone.0188881
Ernberg, M., Christidis, N., Ghafouri, B., Bileviciute-Ljungar, I., Löfgren, M., Bjersing, J., et al. (2018). Plasma cytokine levels in fibromyalgia and their response to 15 weeks of progressive resistance exercise or relaxation therapy. Mediators Inflamm. 2018:3985154. doi: 10.1155/2018/3985154
España, R. A., and Scammell, T. E. (2011). Sleep neurobiology from a clinical perspective. Sleep 34, 845–858. doi: 10.5665/SLEEP.1112
Euesden, J., Danese, A., Lewis, C. M., and Maughan, B. (2017). A bidirectional relationship between depression and the autoimmune disorders - New perspectives from the National Child Development Study. PLoS One 12:e0173015. doi: 10.1371/journal.pone.0173015
Fayyaz, A., Kurien, B. T., and Scofield, R. H. (2016). Autoantibodies in Sjögren’s syndrome. Rheum. Dis. Clin. North Am. 42, 419–434. doi: 10.1016/j.rdc.2016.03.002
Finan, P. H., Goodin, B. R., and Smith, M. T. (2013). The association of sleep and pain: an update and a path forward. J. Pain 14, 1539–1552. doi: 10.1016/j.jpain.2013.08.007
Finnern, M. M., Kleinböhl, D., Flor, H., Benrath, J., and Hölzl, R. (2018). Deconstructing chronicity of musculoskeletal pain: intensity-duration relations, minimal dimensions and clusters of chronicity. Scand. J. Pain 18, 363–377. doi: 10.1515/sjpain-2018-0021
Foo, Y. Z., Nakagawa, S., Rhodes, G., and Simmons, L. W. (2017). The effects of sex hormones on immune function: a meta-analysis. Biol. Rev. Camb. Philos. Soc. 92, 551–571. doi: 10.1111/brv.12243
Fukata, M., Yokoi, N., and Fukata, Y. (2018). Neurobiology of autoimmune encephalitis. Curr. Opin. Neurobiol. 48, 1–8. doi: 10.1016/j.conb.2017.07.012
Garzelli, C., Taub, F. E., Scharff, J. E., Prabhakar, B. S., Ginsberger-Fellner, F., and Notkins, A. L. (1984). Epstein-Barr virus-transformed lymphocytes produce monoclonal autoantibodies that react with antigens in multiple organs. J. Virol. 52, 722–725.
Gayoso, I., Sanchez-Correa, B., Campos, C., Alonso, C., Pera, A., Casado, J. G., et al. (2011). Immunosenescence of human natural killer cells. J. Innate Immun. 3, 337–343. doi: 10.1159/000328005
Gedalia, A., Garcia, C. O., Molina, J. F., Bradford, N. J., and Espinoza, L. R. (2000). Fibromyalgia syndrome: experience in a pediatric rheumatology clinic. Clin. Exp. Rheumatol. 18, 415–419.
Gestermann, N., Di Domizio, J., Lande, R., Demaria, O., Frasca, L., Feldmeyer, L., et al. (2018). Netting neutrophils activate autoreactive B cells in lupus. J. Immunol. 200, 3364–3371. doi: 10.4049/jimmunol.1700778
Giacomelli, C., Talarico, R., Bombardieri, S., and Bazzichi, L. (2013). The interaction between autoimmune diseases and fibromyalgia: risk, disease course and management. Expert Rev. Clin. Immunol. 9, 1069–1076. doi: 10.1586/1744666X.2013.849440
Graus, F., and Santamaría, J. (2017). Understanding anti-IgLON5 disease. Neurol. Neuroimmunol. Neuroinflamm. 4:e393. doi: 10.1212/NXI.0000000000000393
Gui, J., Mustachio, L. M., Su, D. M., and Craig, R. W. (2012). Thymus size and age-related thymic involution: early programming, sexual dimorphism, progenitors and stroma. Aging Dis. 3, 280–290.
Halbgebauer, S., Huss, A., Buttmann, M., Steinacker, P., Oeckl, P., Brecht, I., et al. (2016). Detection of intrathecal immunoglobulin G synthesis by capillary isoelectric focusing immunoassay in oligoclonal band negative multiple sclerosis. J. Neurol. 263, 954–960. doi: 10.1007/s00415-016-8094-3
Halenius, A., and Hengel, H. (2014). Human cytomegalovirus and autoimmune disease. Biomed. Res. Int. 472978. doi: 10.1155/2014/472978
Haliloglu, S., Ekinci, B., Uzkeser, H., Sevimli, H., Carlioglu, A., and Macit, P. M. (2017). Fibromyalgia in patients with thyroid autoimmunity: prevalence and relationship with disease activity. Clin. Rheumatol. 36, 1617–1621. doi: 10.1007/s10067-017-3556-2
Haller-Kikkatalo, K., Alnek, K., Metspalu, A., Mihailov, E., Metsküla, K., Kisand, K., et al. (2017). Demographic associations for autoantibodies in disease-free individuals of a European population. Sci. Rep. 7:44846. doi: 10.1038/srep44846
Hassett, A. L., and Clauw, D. J. (2010). The role of stress in rheumatic diseases. Arthrit. Res. Ther. 12:123. doi: 10.1186/ar3024
Häuser, W., Ablin, J., Fitzcharles, M. A., Littlejohn, G., Luciano, J. V., Usui, C., et al. (2015). Fibromyalgia. Nat. Rev. Dis. Primers 1:15022. doi: 10.1038/nrdp.2015.22
Häuser, W., and Henningsen, P. (2014). Fibromyalgia syndrome: a somatoform disorder. Eur. J. Pain 18, 1051–1059. doi: 10.1102/j.1532-2149.2014.00453.x
Hawiger, D., Masilamani, R. F., Bettelli, E., Kuchroo, V. K., and Nussenzweig, M. C. (2004). Immunological unresponsiveness characterized by increased expression of CD5 on peripheral T cells induced by dendritic cells in vivo. Immunity 20, 695–705. doi: 10.1016/j.immuni.2004.05.002
Hayashi, N., Koshiba, M., Nishimura, K., Sugiyama, D., Nakamura, T., Morinobu, S., et al. (2008). Prevalence of disease-specific antinuclear antibodies in general population: estimates from annual physical examinations of residents of a small town over a 5-year period. Mod. Rheumatol. 18, 153–160. doi: 10.1007/s10165-008-0028-1
Heng, T. S., Goldberg, G. L., Gray, D. H., Sutherland, J. S., Chidgey, A. P., and Boyd, R. L. (2005). Effects of castration on thymocyte development in two different models of thymic involution. J. Immunol. 175, 2982–2993. doi: 10.4049/jimmunol.175.5.2982
Hervier, B., Rimbert, M., Colonna, F., Hamidou, M. A., and Audrain, M. (2009). Clinical significance of anti-Ro/SSA-52 kDa antibodies: a retrospective monocentric study. Rheumatology 48, 964–967. doi: 10.1093/rheumatology/kep145
Hiyama, T. Y., Matsuda, S., Fujikawa, A., Matsumoto, M., Watanabe, E., Kajiwara, H., et al. (2010). Autoimmunity to the sodium-level sensor in the brain causes essential hypernatremia. Neuron 66, 508–522. doi: 10.1016/j.neuron.2010.04.017
Hoebe, K., Janssen, E., and Beutler, B. (2004). The interface between innate and adaptive immunity. Nat. Immunol. 5, 971–974. doi: 10.1038/ni1004-971
Hofman, M. A., and Swaab, D. F. (1989). The sexually dimorphic nucleus of the preoptic area in the human brain: a comparative morphometric study. J. Anat. 164, 55–72.
Hofman, M. A., Zhou, J. N., and Swaab, D. F. (1996). Suprachiasmatic nucleus of the human brain: an immunocytochemical and morphometric analysis. Anat. Rec. 244, 552–562. doi: 10.1002/(SICI)1097-0185(199604)244:4<552::AID-AR13<3.0.CO;2-O
Hsu, H. C., and Mountz, J. D. (2003). Origin of late-onset autoimmune disease. Immunol. Allergy Clin. North Am. 23, 65–82. doi: 10.1016/s0889-8561(02)00074-7
Hughes, G. C. (2012). Progesterone and autoimmune disease. Autoimmun. Rev. 11, A502–A514. doi: 10.1016/j.autrev.2011.12.003
Hughes, G. C., and Clark, E. A. (2007). Regulation of dendritic cells by female sex steroids: relevance to immunity and autoimmunity. Autoimmunity 40, 470–481. doi: 10.1080/08916930701464764
Hughes, T. (2005). The early history of myasthenia gravis. Neuromuscul. Disord. 15, 878–886. doi: 10.1016/j.nmd.2005.08.007
Inanici, F., and Yunus, M. B. (2004). History of fibromyalgia: past to present. Curr. Pain Headache Rep. 8, 369–378. doi: 10.1007/s11916-996-0010-6
Jackson, T., Thomas, S., Stabile, V., Han, X., Shotwell, M., and McQueen, K. (2015). Prevalence of chronic pain in low-income and middle-income countries: a systematic review and meta-analysis. Lancet 385:S10. doi: 10.1016/S0140-6736(15)60805-4
Jacobs, B. C., Rothbarth, P. H., van der Meché, F. G., Herbrink, P., Schmitz, P. I., de Klerk, M. A., et al. (1998). The spectrum of antecedent infections in Guillain-Barré syndrome: a case-control study. Neurology 51, 1110–1115. doi: 10.1212/wnl.51.4.1110
Jacobsen, S., Hoyer-Madsen, M., Danneskiold-Samsoe, B., and Wiik, A. (1990). Screeningn for autoantibodies in patients with primary fibromyalgia syndrome and a matched control group. APMIS 98, 655–658. doi: 10.1111/j.1699-0463.1990.tb04984.x
Jarius, S., and Wildemann, B. (2013). Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol. 23, 661–683. doi: 10.1111/bpa.12084
Jensen, K. B., Kosek, E., Petzke, F., Carville, S., Fransson, P., Marcus, H., et al. (2009). Evidence of dysfunctional pain inhibition in fibromyalgia reflected in rACC during provoked pain. Pain 144, 95–100. doi: 10.1016/j.pain.2009.03.018
Jiao, J., Vincent, A., Cha, S. S., Luedtke, C. A., Kim, C. H., and Oh, T. H. (2015). Physical trauma and infection as precipitating factors in patients with fibromyalgia. Am. J. Phys. Med. Rehabil. 94, 1075–1082. doi: 10.1097/PHM.0000000000000300
Johnston, S., Brenu, E. W., Staines, D., and Marshall-Gradisnik, S. (2013). The prevalence of chronic fatigue syndrome/myalgic encephalomyelitis: a meta-analysis. Clin. Epidemiol. 5, 105–110. doi: 10.2147/CLEP.S39876
Jones, K. D., Gelbart, T., Whisenant, T. C., Waalen, J., Mondale, T. S., Iklé, D. N., et al. (2016). Genome-wide expression profiling in the peripheral blood of patients with fibromyalgia. Clin. Exp. Rheumatol. 34, S89–S98.
Kadetoff, D., Lampa, J., Westman, M., Andersson, M., and Kosek, E. (2012). Evidence of central inflammation in fibromyalgia-increased cerebrospinal fluid interleukin-8 levels. J. Neuroimmunol. 242, 33–38. doi: 10.1016/j.jneuroim.2011.10.013
Kaiko, G. E., Horvat, J. C., Beagley, K. W., and Hansbro, P. M. (2008). Immunological decision-making: how does the immune system decide to mount a helper T-cell response? Immunology 123, 326–338. doi: 10.1111/j.1365-2567.2007.02719.x
Kanda, N., Tsuchida, T., and Tamaki, K. (1996). Testosterone inhibits immunoglobulin production by human peripheral blood mononuclear cells. Clin. Exp. Immunol. 106, 410–415. doi: 10.1046/j.1365-2249.1996.d01-842.x
Kawasaki, Y., Zhang, L., Cheng, J. K., and Ru-Rong, J. (2008). Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 28, 5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008
Kendroud, S., and Hanna, W. (2019). Physiology, Nociceptive Pathways. Treasure Island, FL: StatPearls Publishing.
Kisler, L. B., Granovsky, Y., Sinai, A., Sprecher, E., Shamay-Tsoory, S., and Weissman-Fogel, I. (2016). Sex dimorphism in a mediatory role of the posterior midcingulate cortex in the association between anxiety and pain sensitivity. Exp. Brain Res. 234, 3119–3131. doi: 10.1007/s00221-016-4710-9
Klein, L., Kyewski, B., Allen, P. M., and Hogquist, K. A. (2014). Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol. 14, 377–391. doi: 10.1038/nri3667
Klein, R., Bänsch, M., and Berg, P. A. (1992). Clinical relevance of antibodies against serotonin and gangliosides in patients with primary fibromyalgia syndrome. Psychoneuroendocrinology 17, 593–598. doi: 10.1016/0306-4530(92)90017-2
Klein, S. L., and Flanagan, K. L. (2016). Sex differences in immune responses. Nat. Rev. Immunol. 16, 626–638. doi: 10.1038/nri.2016.90
Koenig, A., Buskiewicz, I., and Huber, S. A. (2017). Age-associated changes in estrogen receptor ratios correlate with increased female susceptibility to coxsackievirus B3-induced myocarditis. Front. Immunol. 8:1585. doi: 10.3389/fimmu.2017.01585
Koga, M., Yuki, N., Takahashi, M., Saito, K., and Hirata, K. (1998). Close association of IgA anti-ganglioside antibodies with antecedent Campylobacter jejuni infection in Guillain-Barré and Fisher’s syndromes. J. Neuroimmunol. 81, 138–143. doi: 10.1016/s0165-5728(97)00168-9
Kosek, E., Altawil, R., Kadetoff, D., Finn, A., Westman, M., Le Maître, E., et al. (2015). Evidence of different mediators of central inflammation in dysfunctional and inflammatory pain–interleukin-8 in fibromyalgia and interleukin-1 β in rheumatoid arthritis. J. Neuroimmunol. 280, 49–55. doi: 10.1016/j.jneuroim.2015.02.002
Kosek, E., Finn, A., Ultenius, C., Hugo, A., Svensson, C., and Ahmed, A. S. (2018). Differences in neuroimmune signalling between male and female patients suffering from knee osteoarthritis. J Neuroimmunol. 321, 49–60. doi: 10.1016/j.jneuroim.2018.05.009
Kovats, S. (2015). Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol. 294, 63–69. doi: 10.1016/j.cellimm.2015.01.018
Krumina, A., Chapenko, S., Kenina, V., Mihailova, M., Logina, I., Rasa, S., et al. (2019). The role of HHV-6 and HHV-7 infections in the development of fibromyalgia. J Neurovirol. 25, 194–207. doi: 10.1007/s13365-018-0703-8
Kruse, J. L., Lapid, M. I, Lennon, V. A., Klein, C. J., Toole, O. O., Pittock, S. J., et al. (2015). Psychiatric autoimmunity: N-Methyl-D-aspartate receptor IgG and beyond. Psychosomatics 56, 227–241. doi: 10.1016/j.psym.2015.01.003
Lai, J. J., Lai, K. P., Zeng, W., Chuang, K. H., Altuwaijri, S., and Chang, C. (2012). Androgen receptor influences on body defense system via modulation of innate and adaptive immune systems: lessons from conditional AR knockout mice. Am. J. Pathol. 181, 1504–1512. doi: 10.1016/j.ajpath.2012.07.008
Lami, M. J., Martínez, M. P., Miró, E., Sánchez, A. I., and Guzmán, M. A. (2018). Catastrophizing, acceptance, and coping as mediators between pain and emotional distress and disability in fibromyalgia. J. Clin. Psychol. Med. Settings 25, 80–92. doi: 10.1007/s10880-018-9543-1
Landis, C. A., Lentz, M. J., Tsuji, J., Buchwald, D., and Shaver, J. L. (2004). Pain, psychological variables, sleep quality, and natural killer cell activity in midlife women with and without fibromyalgia. Brain Behav. Immun. 18, 304–313. doi: 10.1016/j.bbi.2003.11.001
Latremoliere, A., and Woolf, C. J. (2009). Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10, 895–926. doi: 10.1016/j.jpain.2009.06.01
Lazarus, M., Oishi, Y., Bjorness, T. E., and Greene, R. W. (2019). Gating and the need for sleep: dissociable effects of adenosine A1 and A2 receptors. Front. Neurosci. 13:740. doi: 10.3389/fnins.2019.00740
Levite, M., and Ganor, Y. (2008). Autoantibodies to glutamate receptors can damage the brain in epilepsy, systemic lupus erythematosus and encephalitis. Expert Rev. Neurother. 8, 1141–1160. doi: 10.1586/14737175.8.7.1141
Link, H., Baig, S., Olsson, O., Jiang, Y. P., Höjeberg, B., and Olsson, T. (1990). Persistent anti-myelin basic protein IgG antibody response in multiple sclerosis cerebrospinal fluid. J. Neuroimmunol. 28, 237–248. doi: 10.1016/0165-5728(90)90017-h
Liu, Y., Zhang, H., Zhang, P., Meng, F., Chen, Y., Wang, Y., et al. (2014). Autoimmune regulator expression in thymomas with or without autoimmune disease. Immunol. Lett. 161, 50–56. doi: 10.1016/j.imlet.2014.04.008
Lukkahatai, N., Walitt, B., Espina, A., Wang, D., and Saligan, L. N. (2015). Comparing genomic profiles of women with and without fibromyalgia. Biol. Res. Nurse 17, 373–383. doi: 10.1177/1099800415589785
Lynch, H. E., Goldberg, G. L., Chidgey, A., Van den Brink, M. R., Boyd, R., and Sempowski, G. D. (2009). Thymic involution and immune reconstitution. Trends Immunol. 30, 366–373. doi: 10.1016/j.it.2009.04.003
Macedo, A. C., and Isaac, L. (2016). Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front. Immunol. 7:55. doi: 10.3389/fimmu.2016.00055
Macfarlane, G. J., Kronisch, C., Dean, L. E., Atzeni, F., Häuser, W., Fluß, E., et al. (2017). EULAR revised recommendations for the management of fibromyalgia. Ann. Rheum. Dis. 76, 318–328. doi: 10.1136/annrheumdis-2016-209724
Maestu, C., Cortes, A., Vazquez, J. M., del Rio, D., Gomez-Arguelles, J. M., del Pozo, F., et al. (2013). Increased brain responses during subjectively-matched mechanical pain stimulation in fibromyalgia patients as evidenced by MEG. Clin. Neurophysiol. 124, 752–760. doi: 10.1016/j.clinph.2012.09.015
Mallampalli, M. P., and Carter, C. L. (2014). Exploring sex and gender differences in sleep health: a Society for Women’s health research report. J. Womens Health 23, 552–562. doi: 10.1089/jwh.2014.4816
Mangalam, A. K., Taneja, V., and David, C. S. (2013). HLA class II molecules influence susceptibility vs protection in inflammatory diseases by determining the cytokine profile. J. Immunol. 190, 513–519. doi: 10.4049/jimmunol.1201891
Mangs, A. H., and Morris, B. J. (2007). The human pseudoautosomal region (PAR): origin, function and future. Curr. Genomics 8, 129–136. doi: 10.2174/138920207780368141
Mapplebeck, J. C., Beggs, C., and Salter, M. W. (2016). Sex differences in pain: a tale of two immune cells. Pain 157, S2–S6. doi: 10.1097/j.pain.0000000000000389
Martin, M. Y., Bradley, L. A., Alexander, R. W., Alarcó, G. S., Triana-Alexander, M., Aaron, L. A., et al. (1996). Coping strategies predict disability in patients with primary fibromyalgia. Pain 68, 45–53. doi: 10.1016/s0304-3959(96)03179-x
McDonald, G., Medina, C. O., Pilichowska, M., Kearny, J. F., Shinkura, R., Selsing, E., et al. (2017). Accelerated systemic autoimmunity in the absence of somatic hypermutation in 564Igi: a mouse model of systemic lupus with knocked-in heavy and light chain genes. Front. Immunol. 8:1094. doi: 10.3389/fimmu.2017.010194
Melchior, M., Poisbeau, P., Gaumond, I., and Marchand, S. (2016). Insights into the mechanisms and the emergence of sex-differences in pain. Neuroscience 338, 63–80. doi: 10.1016/j.neuroscience.2016.05.007
Mendieta, D., De la Cruz-Aguilera, D. L., Barrera-Villalpando, M. I, Becerril-Villanueva, E., Arreola, R., Hernández-Ferreira, E., et al. (2016). IL-8 and IL-6 primarily mediate the inflammatory response in fibromyalgia patients. J. Neuroimmunol. 290, 22–25. doi: 10.1016/j.jneuroim.2015.11.011
Menétrey, D., and Besson, J. M. (1982). Electrophysiological characteristics of dorsal horn cells in rats with cutaneous inflammation resulting from chronic arthritis. Pain 13, 343–364. doi: 10.1016/0304-3959(82)90003-3
Mitsonis, C. I., Potagas, C., Zervas, I., and Sfagos, K. (2009). The effects of stressful life events on the course of multiple sclerosis: a review. Int. J. Neurosci. 119, 315–335. doi: 10.1080/00207450802480192
Mork, P. J., and Nilsen, T. I. (2012). Sleep problems and risk of fibromyalgia: longitudinal data on an adult female population in Norway. Arthrit. Rheum. 64, 281–284. doi: 10.1002/art.33346
Mortensen, K. H., Cleemann, L., Hjerrild, B. E., Nexo, E., Locht, H., Jeppesen, E. M., et al. (2009). Increased prevalence of autoimmunity in Turner syndrome–influence of age. Clin. Exp. Immunol. 156, 205–210. doi: 10.1111/j.1365-2249.2009.03895.x
Mueller, D. L. (2010). Mechanisms maintaining peripheral tolerance. Nat. Immunol. 11, 21–27. doi: 10.1038/ni.1817
Mukherjee, A., Jantsch, V., Khan, R., Hartung, W., Fischer, R., Jantsch, J., et al. (2018). Rheumatoid arthritis-associated autoimmunity due to. Front. Immunol. 9:2352. doi: 10.3389/fimmu.2018.02352
Multiple Sclerosis Therapy Consensus Group (2008). Basic and escalating immunomodulatory treatments in multiple sclerosis: current therapeutic recommendations. J. Neurol. 255, 1449–1463. doi: 10.1007/s0045-008-0061-1
Nagele, E. P., Han, M., Acharya, N. K., DeMarshall, C., Kosciuk, M. C., and Nagele, R. G. (2013). Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS One 8:e60726. doi: 10.1371/journal.pone.0060726
Naylor, B., Boag, S., and Gustin, S. M. (2017). New evidence for a pain personality? A critical review of the last 120 years of pain and personality. Scand. J. Pain 17, 58–67. doi: 10.1016/j.sjpain.2017.07.011
Nielsen, C. M., White, M. J., Goodier, M. R., and Riley, E. M. (2013). Functional significance of CD57 expression on human NK cells and relevance to disease. Front. Immunol. 4:422. doi: 10.3389/fimmu.2013.00422
Noridomi, K., Watanabe, G., Hansen, M. N., Han, G. W., and Chen, L. (2017). Structural insights into the molecular mechanisms of myasthenia gravis and their therapeutic implications. eLife 6:e23043. doi: 10.7554/eLife.23043
Ohkura, N., Kitagawa, Y., and Sakaguchi, S. (2013). Development and maintenance of regulatory T cells. Immunity 38, 414–423. doi: 10.1016/j.immuni.2013.03.002
Oishi, Y., Xu, Q., Wang, L., Zhang, B. J., Takahashi, K., Takata, Y., et al. (2017). Slow-wave sleep is controlled by a subset of nucleus accumbens core neurons in mice. Nat. Commun. 8:374. doi: 10.1038/s41467-017-00781-4
Ossipov, M. H., Dussor, G. O., and Porreca, F. (2010). Central modulation of pain. J. Clin. Invest. 120, 3779–3787. doi: 10.1172/JCI43766
Pang, Z., Kushiyama, A., Sun, J., Kikuchi, T., Yamazaki, H., Iwamoto, Y., et al. (2017). Glial fibrillary acidic protein (GFAP) is a novel biomarker for the prediction of autoimmune diabetes. FASEB J. 31, 4053–4063. doi: 10.1096/fj.201700110R
Park, D. J., and Lee, S. S. (2017). New insights into the genetics of fibromyalgia. Korean J. Intern. Med. 32, 984–995. doi: 10.3904/kjim.2016207
Peirs, C., Williams, S. P., Zhao, X., Walsh, C. E., Gedeon, J. Y., Cagle, N. E., et al. (2015). Dorsal horn circuits for persistent mechanical pain. Neuron 87, 797–812. doi: 10.1016/j.neuron.2015.07.029
Peng, G., Guo, Z., Kiniwa, Y., Voo, K. S., Peng, W., Fu, T., et al. (2005). Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science 309, 1380–1384. doi: 10.1126/science.1113401
Pernambuco, A. P., Schetino, L. P., Viana, R. S., Carvalho, L. S., and d’Ávila Reis, D. (2015). The involvement of melatonin in the clinical status of patients with fibromyalgia syndrome. Clin. Exp. Rheumatol. 33, S14–S19.
Pers, J. O., Jamin, C., Predine-Hug, F., Lydyard, P., and Youinou, P. (1999). The role of CD5-expressing B cells in health and disease (review). Int. J. Mol. Med. 3, 239–245.
Purnamawati, K., Ong, J. A., Deshpande, S., Tan, W. K., Masurkar, N., Low, J. K., et al. (2018). The importance of sex stratification in autoimmune disease biomarker research: a systematic review. Front. Immunol. 9:1208. doi: 10.3389/fimmu.2018.01208
Queiroz, L. P. (2013). Worlwide epidemiology of fibromyalgia. Curr. Pain Headache Rep. 17:356. doi: 10.1007/s11916-013-0356-6
Raddassi, K., Kent, S. C., Yang, J., Bourcier, K., Bradshaw, E. M., Seyfert-Margolis, V., et al. (2011). Increased frequencies of myelin oligodendrocyte glycoprotein/MHC class II-binding CD4 cells in patients with multiple sclerosis. J. Immunol. 187, 1039–1046. doi: 10.4049/jimmunol.1001543
Rainville, J. R., Tsyglakova, M., and Hodes, G. E. (2018). Deciphering sex differences in the immune system and depression. Front. Neuroendocrinol. 50, 67–90. doi: 10.1016/j.yfrne.2017.12.004
Ramírez, M., Martínez-Martínez, L. A., Hernández-Quintela, E., Velazco-Casapía, J., Vargas, A., and Martínez-Lavín, M. (2015). Small fiber neuropathy in women with fibromyalgia. An in vivo assessment using corneal confocal bio-microscopy. Semin. Arthrit. Rheum. 45, 214–219. doi: 10.1016/j.semarthrit.2015.03.003
Reiche, E. M., Nunes, S. O., and Morimoto, H. K. (2004). Stress, depression, the immune system, and cancer. Lancet Oncol. 5, 617–625. doi: 10.1016/S1470-2045(04)01597-9
Rhudy, J. L., Bartley, E. J., Williams, A. E., McCabe, K. M., Chandler, M. C., Russell, J. L., et al. (2010). Are there sex differences in affective modulation of spinal nociception and pain? J. Pain 11, 1429–1441. doi: 10.1016/j.jpain.2010.04.003
Ritzel, R. M., Capozzi, L. A., and McCullough, L. D. (2013). Sex, stroke, and inflammation: the potential for estrogen-mediated immunoprotection in stroke. Horm. Behav. 63, 238–253. doi: 10.1016/j.yhbeh.2012.04.007
Roizenblatt, S., Moldofsky, H., Beneditto-Silva, A. A., and Tufik, S. (2001). Alpha sleep characteristics in fibromyalgia. Arthrit. Rheumatol. 44, 222–230. doi: 10.1002/1529-0131(200101)44:1
Rosenblatt, J. S., Hazelwood, S., and Poole, J. (1996). Maternal behavior in rats: effects of medial preoptic area lesions and presence of maternal aggression. Horm. Behav. 30, 201–215. doi: 10.1006/hbeh.1996.0025
Rubtsov, A. V., Rubtsova, K., Kappler, J. W., and Marrack, P. (2010). Genetic and hormonal factors in female-biased autoimmunity. Autoimmun. Rev. 9, 494–498. doi: 10.1016/j.autrev.2010.02.008
Russell, I. J., Vipraio, G. A., Michalek, J. E., Craig, F. E., Kang, Y. K., and Richards, A. B. (1999). Lymphocyte markers and natural killer cell activity in fibromyalgia syndrome: effects of low-dose, sublingual use of human interferon-alpha. J. Interferon Cytokine Res. 19, 969–978. doi: 10.1089/107999099313523
Sabater, L., Gaig, C., Gelpi, E., Bataller, L., Lewerenz, J., Torres-Vega, E., et al. (2014). A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 13, 575–586. doi: 10.1016/S1474-4422(14)70051-1
Sato, K., Nakajima, K., Imamura, H., Deguchi, T., Horinouchi, S., Yamazaki, K., et al. (2002). A novel missense mutation of AIRE gene in a patient with autoimmune polyendocrinopathy, candidiasis and ectodermal dystrophy (APECED), accompanied with progressive muscular atrophy: case report and review of the literature in Japan. Endocr. J. 49, 625–633. doi: 10.1507/endocrj.49.625
Sawalha, A. H., Harley, J. B., and Scofield, R. H. (2009). Autoimmunity and Klinefelter’s syndrome: when men have two X chromosomes. J. Autoimmun. 33, 31–34. doi: 10.1016/j.jaut.2009.03.006
Schwartz, R. H. (2012). Historical overview of immunological tolerance. Cold Spring Harb. Perspect. Biol. 4:a006908. doi: 10.1101/cshperspect.aa006908
Sellers, R. S., Clifford, C. B., Treuting, P. M., and Brayton, C. (2012). Immunological variation between inbred laboratory mouse strains: points to consider in phenotyping genetically immunomodified mice. Vet. Pathol. 49, 32–43. doi: 10.1177/0300985811429314
Sharif, K., Watad, A., Coplan, L., Lichtbroun, B., Krosser, A., Bragazzi, N. L., et al. (2018). The role of stress in the mosaic of autoimmunity: an overlooked association. Autoimmun. Rev. 17, 967–983. doi: 10.1016/j.autrev.2018.04.005
Shimizu, T., Suzuki, T., Yu, H. P., Yokoyama, Y., Choudhry, M. A., Bland, K. I., et al. (2008). The role of estrogen receptor subtypes on hepatic neutrophil accumulation following trauma-hemorrhage: direct modulation of CINC-1 production by Kupffer cells. Cytokine 43, 88–92. doi: 10.1016/j.cyto.2008.04.001
Sigal, L. H. (2012). Basic science for the clinician 54: CD5. J. Clin. Rheumatol. 18, 83–88. doi: 10.1097/RHU.0b013e318247bc64
Singh, A., and Trevick, S. (2016). The epidemiology of global epilepsy. Neurol. Clin. 34, 837–847. doi: 10.1016/j.ncl.2016.06.015
Sluka, K. A., and Clauw, D. J. (2016). Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience 338, 114–129. doi: 10.1016/j.neuroscience.2016.06.006
Smatti, M. K., Cyprian, F. S., Nasrallah, G. K., Al Thani, A. A., Almishal, R. O., and Yassine, H. M. (2019). Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses 11:E762. doi: 10.3390/v11080762
Smith-Bouvier, D. L., Divekar, A. A., Sasidhar, S. D., Tiwari-Woodruff, S. K., King, J. K., Arnold, A. P., et al. (2008). A role for sex chromosome complement in the female bias in autoimmune disease. J. Exp. Med. 205, 1099–1108. doi: 10.1084/jrem.20080950
Song, H. K., and Hwang, D. Y. (2017). Use of C57BL/6N mice on the variety of immunological researches. Lab. Anim. Res. 33, 119–123. doi: 10.5625/lar.2017.33.2.119
Sorge, R. E., Mapplebeck, J. C. S., Rosen, S., Beggs, S., Taves, S., Alexander, J. K., et al. (2015). Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 18, 1081–1083. doi: 10.1038/nn.4053
Souyris, M., Cenac, C., Azar, P., Daviaud, D., Canivet, A., Grunenwald, S., et al. (2018). Sci. Immunology 3:eaa8855. doi: 10.1126/sciimmunol.aap8855
Staud, R., Nagel, S., Robinson, M. E., and Price, D. D. (2009). Enhanced central pain processing of fibromyalgia patients is maintained by muscle afferent input: a randomized, double-blind, placebo-controlled study. Pain 145, 96–104. doi: 10.1016/j.pain.2009.05.020
Sutti, S., and Tacke, F. (2018). Liver inflammation and regeneration in drug-induced liver injury: sex matters! Clin. Sci. 132, 609–613. doi: 10.1042/CS20171313
Suurmond, J., and Diamond, B. (2015). Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J. Clin. Invest. 125, 194–202. doi: 10.1127/JCI8084
Tahrali, I., Kucuksezer, U. C., Altintas, A., Uygunoglu, U., Akdeniz, N., Aktas-Cetin, E., et al. (2018). Dysfunction of CD3-CD16+CD56dim and CD3–CD16–CD56bright NK cell subsets in RR-MS patients. Clin. Immunol. 193, 88–97. doi: 10.1016/j.clim.2018.02.005
Takahashi, A., Flanigan, M. E., McEwen, B. S., and Russo, S. J. (2018). Aggression, social stress, and the immune system in humans and animal models. Front. Behav. Neurosci. 12:56. doi: 10.3389/fnbeh.2018.00056
Taleghany, N., Sarajari, S., DonCarlos, L. L., Gollapudi, L., and Oblinger, M. M. (1999). Differential expression of estrogen receptor alpha and beta in rat dorsal root ganglion neurons. J. Neurosci. Res. 57, 603–615. doi: 10.1002/(sici)1097-4547(19990901)57:5<603::aid-jnr3>3.0.co;2-r
Tan, E. M., Feltkamp, T. E., Smolen, J. S., Butcher, B., Dawkins, R., Fritzler, M. J., et al. (1997). Range of antinuclear antibodies in “healthy” individuals. Arthrit. Rheum. 40, 1601–1611. doi: 10.1002/1529-0131(199709)40:9
Tarakhovsky, A., Kanner, S. B., Hombach, J., Ledbetter, J. A., Müller, W., Killeen, N., et al. (1995). A role for CD5 in TCR-mediated signal transduction and thymocyte selection. Science 269, 535–537. doi: 10.1126/science.7542801
Totsch, S. K., and Sorge, R. E. (2017). Immune system involvement in specific pain conditions. Mol. Pain 13:1744806917724559. doi: 10.1177/1744806917724559
Toyoda, Y., Endo, S., Tsuneyama, K., Miyashita, T., Yano, A., Fukami, T., et al. (2012). Mechanism of exacerbative effect of progesterone on drug-induced liver injury. Toxicol. Sci. 126, 16–27. doi: 10.1093/toxsci/kfr326
Tracey, I., and Mantyh, P. W. (2007). The cerebral signature for pain perception and its modulation. Neuron 55, 3773–3791. doi: 10.1016/j.neuron.2007.07.012
Üçeyler, N., Zeller, D., Kahn, A. K., Kewenig, S., Kittel-Schneider, S., Schmid, A., et al. (2013). Small fibre pathology in patients with fibromyalgia syndrome. Brain 136, 1857–1867. doi: 10.1093/brain/awt053
Umiker, B. R., Andersson, S., Fernandez, L., Korgaokar, P., Larbi, A., Pilichowska, M., et al. (2014). Dosage of X-linked Toll-like receptor 8 determines gender differences in the development of systemic lupus erythematosus. Eur. J Immunol. 44, 1503–1516. doi: 10.1002/eji.201344283
Urquhart, N. E., Capildeo, K. D., Sargeant, L. A., Wharfe, G., Hisada, M., and Hanchard, B. (2008). White blood cell counts in healthy Jamaican adults. West. Indian Med. J. 57, 147–151.
Varani, S., and Landini, M. P. (2011). Cytomegalovirus-induced immunopathology and its clinical consequences. Herpesviridae 2:6. doi: 10.1186/2042-4280-2-6
Vincent, K., and Tracey, I. (2008). Hormones and their interaction with the pain experience. Rev. Pain 2, 20–24. doi: 10.1177/204946370800200206
Virtanen, J. O., and Jacobsen, S. (2012). Viruses and multiple sclerosis. CNS Neurol. Disord. Drug Targets 11, 528–544.
Virtanen, J. O., Wohler, J., Fenton, K., Reich, D. S., and Jacobson, S. (2014). Oligoclonal bands in multiple sclerosis reactive against two herpesviruses and association with magnetic resonance imaging findings. Mult. Scler. 20, 27–34. doi: 10.1177/1352458513490545
Vitale, M., Della Chiesa, M., Carlomagno, S., Pende, D., Aricò, M., Moretta, L., et al. (2005). NK-dependent DC maturation is mediated by TNFalpha and IFNgamma released upon engagement of the NKp30 triggering receptor. Blood 106, 566–571. doi: 10.1182/blood-2004-10-4035
Werle, E., Fischer, H. P., Müller, A., Fiehn, W., and Eich, W. (2001). Antibodies against serotonin have no diagnostic relevance in patients with fibromyalgia syndrome. J. Rheumatol. 28, 595–600.
White, H. D., Brown, L. A., Gyurik, R. J., Manganiello, P. D., Robinson, T. D., Hallock, L. S., et al. (2015). Treatment of pain in fibromyalgia patients with testosterone gel: pharmacokinetics and clinical response. Int. Immunopharmacol. 27, 249–256. doi: 10.1016/j.intimp.2015.05.016
Wolfe, F., Clauw, D. J., Fitzcharles, M. A., Goldenberg, D. L., Häuser, W., Katz, R. L., et al. (2016). 2016 Revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin. Arthrit. Rheum. 46, 319–329. doi: 10.1016/j.semarthrit.2016.08.012
Woolf, C. J. (1983). Evidence for a central component of post-injury pain hypersensitivity. Nature 306, 686–688. doi: 10.1038/306686a0
Xiao, Y., Haynes, W. L., Michalek, J. E., and Russell, I. J. (2013). Elevated serum high-sensitivity C-reactive protein levels in fibromyalgia syndrome patients correlate with body mass index, interleukin-6, interleukin-8, erythrocyte sedimentation rate. Rheumatol. Int. 33, 1259–1264. doi: 10.1007/s00296-012-2538-6
Yan, C. Q., Liu, C. Z., Wang, X., Huo, J. W., Zhou, P., Zhang, J., et al. (2018). Abnormal funtional connectivity of anterior cingulate cortex in patients with primary insomnia: a resting-state functional magnetic resonance imaging study. Front. Aging Neurosci. 10:167. doi: 10.3389/fnagi.2018.00167
Yunus, M. B., Khan, M. A., Rawlings, K. K., Green, J. R., Oson, J. M., and Shah, S. (1999). Genetic linkage analysis of multicase families with fibromyalgia syndrome. J. Rheumatol. 26, 408–412.
Zeilhofer, H. U., Wildner, H., and Yévenes, G. E. (2012). Fast synaptic inhibition in spinal sensory processing and pain control. Physiol. Rev. 92, 193–235. doi: 10.1152/physrev.00043.2010
Zhang, Z.-J., Guo, J.-S., Li, S.-S., Wu, W.-B., Cao, D.-L., Jiang, B.-C., et al. (2018). TLR and its endogeneous ligand miR-21 contribute to neuropathic pain in murine DRG. J. Exp. Med. 215, 3019–3037. doi: 10.1084/jrem.20180800
Keywords: autoimmune disease, central nervous system sensitization, fibromyalgia, pathophysiology, sex differences, widespread chronic pain
Citation: Meester I, Rivera-Silva GF and González-Salazar F (2020) Immune System Sex Differences May Bridge the Gap Between Sex and Gender in Fibromyalgia. Front. Neurosci. 13:1414. doi: 10.3389/fnins.2019.01414
Received: 29 November 2018; Accepted: 16 December 2019;
Published: 17 January 2020.
Edited by:
Annie Duchesne, University of Northern British Columbia, CanadaReviewed by:
Gillian Einstein, University of Toronto, CanadaIlke Coskun Benlidayi, Cukurova University, Turkey
Larry Culpepper, Boston University, United States
Reinhild Klein, University of Tübingen, Germany
Copyright © 2020 Meester, Rivera-Silva and González-Salazar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irene Meester, bWVlc3RlcmlyZW5lQGhvdG1haWwuY29t; ZWxpc2FiZXRoZC5tZWVzdGVyQHVkZW0uZWR1; Gerardo Francisco Rivera-Silva, Z2VyYXJkby5yaXZlcmFAdWRlbS5lZHU=