 Marcela Cárdenas-Tueme1
Marcela Cárdenas-Tueme1 Larisa Montalvo-Martínez2Roger Maldonado-Ruiz2
Larisa Montalvo-Martínez2Roger Maldonado-Ruiz2 Alberto Camacho-Morales2,3Diana Reséndez-Pérez1*
Alberto Camacho-Morales2,3Diana Reséndez-Pérez1*- 1Departamento de Biología Celular y Genética, Facultad de Ciencias Biológicas, Universidad Autónoma de Nuevo León, San Nicolás de los Garza, Mexico
- 2Departamento de Bioquímica, Facultad de Medicina, Universidad Autónoma de Nuevo León, San Nicolás de los Garza, Mexico
- 3Centro de Investigación y Desarrollo en Ciencias de la Salud, Unidad de Neurometabolismo, Universidad Autónoma de Nuevo León, San Nicolás de los Garza, Mexico
Maternal overnutrition modulates body weight, development of metabolic failure and, potentially, neurodegenerative susceptibility in the offspring. Overnutrition sets a chronic pro-inflammatory profile that integrates peripheral and central immune activation nodes, damaging neuronal physiology and survival. Innate immune cells exposed to hypercaloric diets might experience trained immunity. Here, we address the role of maternal overnutrition as a trigger for central and peripheral immune training and its contribution to neurodegeneration and the molecular nodes implicated in the Nod-like receptor protein 3 (NLRP3) inflammasome pathway leading to immune training. We propose that maternal overnutrition leads to peripheral or central immune training that favor neurodegenerative susceptibility in the offspring.
Introduction
According to the World Health Organization, nearly 39% of adults were overweight in 2016, and 13% were obese. Epidemiological data from human catastrophes such as the Dutch famine (1944), the siege of Leningrad (1942–1944), the great Chinese famine (1958–1961), and the Överkalix study (1890-present) propose that changes in nutrient intake such as fasting or overnutrition during pregnancy and lactation are associated with metabolic and behavioral disorders in the offspring (Vaiserman, 2017). Maternal nutritional programing can influence offspring body weight, adiposity, and development of metabolic syndrome in diverse ways (Friedman, 2018; Patro Golab et al., 2018). For instance, nutritional programing adversely impacts both maternal and offspring health, increasing susceptibility to show metabolic abnormalities later in life such as obesity, dyslipidemia, type 2 diabetes (T2D) mellitus and hypertension, as well as behavioral disorders related to schizophrenia, autism, and compulsive eating (Shapiro et al., 2017; Friedman, 2018; Patro Golab et al., 2018; Derks et al., 2019).
Recent experimental evidence shows that exposure to hypercaloric diets programs the immune nodes that modulate metabolism and neuronal survival during embryogenesis. For instance, the immune response is involved in the balance and maintenance of an adequate metabolic state; however, conditions of altered metabolic demand promote a pro-inflammatory state (Christ et al., 2018). On this context, overnutrition might trigger an epigenetic process called “trained immunity” in innate immune cells, such as microglia in the central nervous system (CNS) and macrophages or natural killer cells in the periphery, which results in enhanced immune responses following infections or immune stimulation. The epigenetic mechanisms that mediate trained immunity are better understood in macrophages and monocytes, and a few have been described in microglia. Wendeln et al. (2018) demonstrated that microglia, just like peripheral macrophages, can be trained to confer long-lasting memory. In microglia, “trained immunity” is dependent on chromatin remodeling and activation of inflammatory signaling pathways (NF-κB, JNK, and ERK1/2). Also, by knocking down Hdac1/2 or Tak1 in long-lived CX3CR1 + cells (most of them microglia) cytokine levels were reduced, suggesting that epigenetic reprograming of inflammatory responses is key to accomplish immune training (Wendeln et al., 2018).
In this review, we address the role of maternal nutritional programing on central and peripheral immune training and crosstalk during positive metabolic scenarios, as well as their potential relevance in neurodegenerative susceptibility in the offspring.
Central and Peripheral Immune System Communication
The innate immune system is the first line of defense against pathogens and tissue damage; it includes physical barriers such as the skin, and specific cell types that modulate the inflammatory response such as macrophages, microglia, and complement proteins. The inflammatory response consists of an innate cellular system and humoral responses that occur during injury to restore homeostasis (Ciaccia, 2010). In this scenario, microglia, play the role of the innate immune system in the brain, infiltrating during early embryogenesis, coordinating neurogenesis, synaptic pruning, connectome establishment, and acting as major antigen-presenting cells (Colonna and Butovsky, 2017; Maldonado-Ruiz et al., 2017). In healthy brains, microglia remain in a ramified stated, and when activated, they enlarge their cell body, change to a phagocytic state, release pro-inflammatory cytokines, and increase antigen presentation and ROS production, leading to neuroinflammation (Colonna and Butovsky, 2017). As shown in Figure 1, under an altered physiological scenario, peripheral immune cells infiltrate the brain, become pro-inflammatory and secrete cytokines. This causes an exacerbated immune response by M1-activated microglia which leads to neuroinflammation, favoring protein aggregation and brain damage consistent with neurodegenerative pathologies (Colonna and Butovsky, 2017).
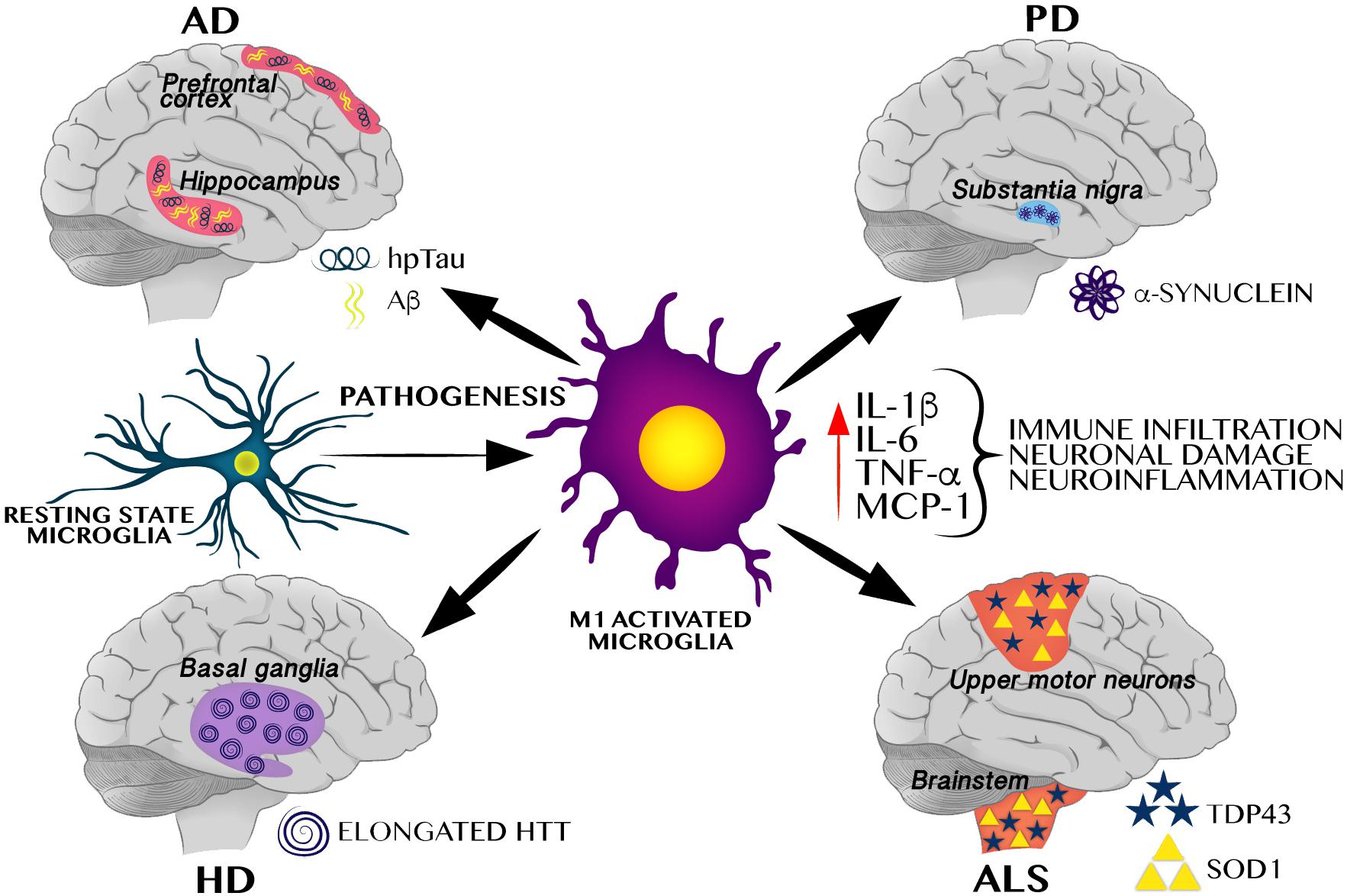
Figure 1. M1 Microglial activation induces a pro-inflammatory cytokines favoring neuroinflammation and protein aggregation-related neurodegenerative pathologies. M1 activated microglia induces a pro-inflammatory profile, including increase of IL-1β, TNF-α, MCP-1, and IL-6, all of these cytokines have been identified in different neurodegenerative pathologies including AD, PD, HD, and ALS. These neurodegenerative pathologies are characterized by selective protein aggregation, including amyloid beta (Aβ) and hyperphosphorylated Tau (hpTau) in AD, α-synuclein in PD, elongated huntingtin (HTT) in HD, and TAR DNA binding protein 43 (TDP43) and superoxide dismutase 1 (SOD1) in ALS.
In the past years, the concept of the CNS as an immune-privileged site has changed, and interactions between the peripheral and central immune system have been demonstrated. Furthermore, these interactions are necessary to maintain homeostasis in the CNS, which was once thought to be impossible. For instance, the myeloid cells such as the macrophages and dendritic cells (DCs) in the meninges (dura, arachnoid, and pia mater), choroid plexus, and perivascular spaces in the CNS parenchyma can mount a robust protective and restorative response when necessary.
In addition, there is a strong relationship between inflammation and metabolic disorders in the immune response against infection. It is known that the immune response is involved in the balance and maintenance of an adequate metabolic state; however, it can have adverse effects under conditions of altered metabolic demand (Agrawal, 2014). In this way, positive energy balance during overnutrition promotes an inflammatory state in the CNS. For instance, saturated fatty acids from the diet are considered powerful candidates to trigger immune response in the peripheral system and in the CNS. However, how central immunity communicates with the peripheral immune response during overnutrition remains to be clarified (Maldonado-Ruiz et al., 2017). Based on this complex and delicate regulation, we next address how much the cells of the immune system can be influenced by selective diets.
Maternal Programing by Exposure to High-Fat-High-Sugar Diets Activates Immunity and Regulates Neurodegenerative Susceptibility
Experimental data in animal models and humans have contributed to understand the effect of caloric overnutrition during pregnancy on immunity and neurodegenerative susceptibility later in life. The nervous system develops through a sophisticated and precise process from embryonic stages through puberty and adulthood. Proliferation, migration and differentiation of major brain cell types followed by rapid synaptogenesis integrate and establish a selective and efficient brain connectome. The immune system shares a time-dependent development and maturation with the nervous system during embryogenesis and post-natal periods (Estes and McAllister, 2016), which renders these systems extremely responsive to environmental factors, potentially modulating central and peripheral cross-talk and neurodegenerative susceptibility.
Initial experimental evidence in animal models and humans showing defects in glucose homeostasis such as T2D, have reported to contribute to neurodegeneration. For instance, rats fed a high-fat/glucose die develop insulin resistance and exhibit impaired spatial learning ability, reduced hippocampal dendritic spine density, and reduced long-term potentiation in the CA1 region due to glucotoxicity or altered insulin signaling (Stranahan et al., 2008). Moreover, Alzheimer’s disease (AD) transgenic mice models fed with a high-fat diet showed T2D-like peripheral insulin resistance but also more severe cognitive deficits compared to normoglycemic AD mice (Procaccini et al., 2016), demonstrating the possible interplay between defects in glucose metabolism and AD development. Of note, patients with T2D show two-fold higher risk to develop AD than normal subjects (Ott et al., 1999).
Experimental and clinical data support the hypothesis that inflammation itself might be a risk factor for neurodegeneration during AD (Ott et al., 1999; Bertram et al., 2007; Takahashi et al., 2007; Tartaglione et al., 2016). For instance, Ling Z. et al. (2004) and Ling Z. D. et al. (2004) exposed rats to LPS prenatally and then administered with a subtoxic dose of the dopaminergic (DA) neurotoxin rotenone (1.25 mg/kg per day for 14 days) when 16 months old. Of note, prenatal LPS and postnatal rotenone exposure exacerbate DA cell loss compared with the effects of single LPS or rotenone exposure. Also, initial genomic analysis of AD patients identified several risk genes that encode proteins involved in neural repair and remyelination and in the modulation of microglial responses, including phagocytosis (Bertram et al., 2007; Takahashi et al., 2007).
Obesity or maternal programing by exposure to high-fat-high-sugar diets in animal models and humans is in fact associated with overactivation of the immune system, triggering a process of chronic inflammation named “meta-inflammation” or “metabolic inflammation,” which ultimately links central and peripheral immune activation (O’Brien et al., 2017). For instance, maternal overnutrition of high-fat-high-sugar diets during pregnancy in murine models programs metabolic and hormonal settings that modulate neuronal development, axonal pruning, synaptic plasticity and connectome establishment during embryogenesis (Bilbo and Tsang, 2010; Morin et al., 2017; Mucellini et al., 2019). Also, mice born from obese dams have shown significant metabolic alterations including higher body fat, altered serum leptin levels, pancreatic islet hypertrophy, and lower expression of metabolism-related genes (Bilbo and Tsang, 2010; Gomes et al., 2018; Mucellini et al., 2019). In addition, human obesity is associated with the low-grade peripheral immune activation characteristic of an acute phase reaction, including C-reactive protein and serum amyloid A (SAA), an increase of cytokines, such as tumor necrosis factor (TNF-α), IL-1β, and IL-6 in serum, and an increase in white cell counts (Esser et al., 2014). In fact, systemic immune activation in psoriasis, a skin disorder, efficiently favors neuroinflammation (O’Brien et al., 2017). There are also other evidences in humans verifying that obesity is closely associated with neurodegenerative susceptibility including Alzheimer’s, Parkinson’s, and Huntington’s diseases and nervous system sclerosis (Procaccini et al., 2016), and an increase in the Body Mass Index in healthy mothers is negatively associated with white matter development in their offspring (Ou et al., 2015; Procaccini et al., 2016; Widen et al., 2018). These results suggest that positive energy balance such as during obesity or maternal overnutrition leads to neurodegenerative susceptibility, which potentially involves an immune activation pathway.
Initial reports of maternal programing by overnutrition using murine models identified sex-specific differences in fetal size and gene expression signatures in fetal-brains, showing that male offspring are the most affected. Importantly, the offspring displays a pro-inflammatory gene signature as the top regulated gene pathway (Edlow et al., 2016a). Later on, these results were confirmed in humans in a pilot study that analyzed the amniotic fluid from obese and lean pregnant women on the second trimester; in this study, 205 genes were found differentially regulated, where the lipid regulator, Apolipoprotein D, was the most upregulated gene (9-fold). Also, genes involved in apoptotic cell death were significantly downregulated particularly within pathways involving the cerebral cortex, such as the Serine/threonine kinase 24 ATPase (STK24). These biomarkers also correlated with the activation of the transcriptional regulators estrogen receptor, FOS, and STAT3, suggesting a pro-estrogenic, pro-inflammatory milieu (Edlow et al., 2014). In addition, this molecular signature was then confirmed in a prospective case-control study were 701 differentially regulated genes were identified, integrating a neurodegeneration gene signature, including Occludin (OCLN), Kinesin Family Member 14 (KIF14), and Nuclear receptor subfamily 2, group E, member 1 (NR2E1) (Edlow et al., 2016b). Upregulation of OCLN and KIF14 genes have been previously associated to AD and vascular dementia in post-mortem human brain tissue (Romanitan et al., 2007), and in negative regulation of neuron apoptosis and regulation of myelination (Fujikura et al., 2013), respectively. Also, NR2E1 is implicated in neurogenesis and neuronal differentiation modulating aggressive behavior and fear response (Young et al., 2002). Notably, all of these gene alterations also correlated with a pro-inflammatory signature of upregulated genes: chemokine (C-C motif) receptor 6 (CCR6), O-linked N-acetylglucosamine (GlcNAc) transferase (OGT), chemokine (C-C motif) receptor 2 (CCR2), caspase 4, apoptosis-related cysteine peptidase (CASP4), toll-like receptor 1 (TLR1), nucleoporin 107 kDa (NUP107), decapping enzyme, scavenger (D), among others (Edlow et al., 2016b). Together, maternal nutritional programing by overnutrition activates the central and peripheral immune systems that intimately communicate with each other to modulate neuroinflammation, thus increasing neurodegenerative susceptibility in offspring.
We will now discuss the experimental data addressing the potential role of maternal nutritional programing on the Nod-like receptor protein 3 (NLRP3) inflammasome pathway activation and its effects on neurodegeneration.
Potential Role of the Nod-Like Receptor Protein 3 Inflammasome Pathway on Neurodegenerative Susceptibility by Nutrient Over Supply
The NLRP3- inflammasome pathway is linked to a danger-associated molecular pattern released from damaged or dying neurons that bind and activate the Toll-like receptor (TLR) –dependent myeloid differentiation primary response protein MyD88 (MYD88)–nuclear factor-κB (NF-κB) pathway. At first, the TLR-MYD88-NF-κB pathway positively produces pro-IL-1β and NLRP3 synthesis, activating a positive feedback loop. Then, negative stimuli, including changes in potassium efflux or reactive oxygen species, trigger the inflammasome assembly and processing of pro-IL-1β into IL-1β by caspase 1 activation (Heneka et al., 2018). Finally, the NF-κB transcription factor also regulates a variety of different processes, including stress response and a pro-inflammatory profile activation.
Initial reports by Christ et al. (2018) identified that murine models exposed to high-fat-high-sugar diets set an epigenetic program that primes B lymphocytes into an exacerbated pro-inflammatory phenotype. These, become much more responsive under physiologic stimuli which depend on the NLRP3-inflammasome pathway (Christ et al., 2018). Selective lipid species, such as palmitate and stearate, as well as, carbohydrates have been identified to activate the NLRP3-inflammasome pathway (Wen et al., 2011; Ann et al., 2018). For instance, saturated lipids from diet intake such as palmitic and stearic acids promote IL-1β release from bone marrow-derived macrophages of rodents and humans, respectively (Wen et al., 2011; L’homme et al., 2013), an effect replicated in murine macrophages (Ann et al., 2018). Of note, immune activation by palmitic and stearic acids precisely depends on the NLRP3-inflammasome pathway (Wen et al., 2011; L’homme et al., 2013). Conversely, unsaturated fatty acids, including oleate and linoleate, prevent IL-1β release and are unable to activate the NLRP3-inflammasome pathway in human monocytes/macrophages (L’homme et al., 2013; Sui et al., 2016). Also, a high-fructose diet in mice positively activates the NLRP3-inflammasome pathway and IL-1β release in human macrophage and liver cell lines, which correlates with neutrophil infiltration (Mastrocola et al., 2016; Nigro et al., 2017; Choe and Kim, 2017). Finally, the role of metabolic species regulating the NLRP3-inflammasome and neurodegeneration was recently evidenced by showing that the 25-hydroxycholesterol also activates the NLRP3-inflammasome pathway, promoting progressive neurodegeneration in X-linked adrenoleukodystrophy, a nervous disease with cerebral inflammatory demyelination (Jang et al., 2016). Moreover, the NLRP3 inflammasome pathway has been identified to contribute to PD (Fan et al., 2017; Gordon et al., 2018; Lee et al., 2018), AD and ALS (Heneka et al., 2013; Johann et al., 2015), HD (Glinsky, 2008), as well as to behavioral alterations in mice at later stages, such as anhedonia (Zhu et al., 2017), anxiety (Lei et al., 2017) and depression-like behavior (Pan et al., 2014; Su et al., 2017).
Altogether, the evidence suggests that overnutrition during pregnancy might promote microglia activation, which correlates with peripheral pro-inflammatory profiles and brain abnormalities in the offspring that are related with neurodegenerative susceptibility. We next discuss the role of diet-induced central and peripheral immune training on neurodegeneration.
Does Central and Peripheral Innate Immune Training by NLRP3 Inflammasome Contributes to Neurodegeneration?
Trained immunity is a selective innate immune memory that induces an enhanced response to an inflammatory stimulus in innate immune cells (for instance, microglia), with a much stronger effect than that observed in basal conditions. Trained immunity depends of signal imprinting on transcription factors and epigenetic reprograming (Figure 2A). At this state, microglia in the CNS, or macrophages, leukocytes, natural killer cells, etc., in the peripheral system undergo apparent changes in their morphology, elevated secretion of cytokines and other inflammatory mediators, as well as substantial epigenetic program settings (Netea et al., 2016). Recent reports in different models and also in humans have confirmed robust innate immune training of myeloid cells following a pro-inflammatory challenge (Arts et al., 2018; Bekkering et al., 2018; Christ et al., 2018; Kaufmann et al., 2018; Mitroulis et al., 2018). For instance, exposure to a brief high-fat diet for 3 days promoted the activation of the NLRP3 inflammasome pathway, leading to central immune training, which potentially associates to deficits in long-term memory formation in mice (Sobesky et al., 2016). Also, peripherally applied inflammatory stimuli induce acute immune training in microglia, which correlates with differential epigenetic reprograming that modulates the microglial Tak1 or Hdac1/2 genes. Notably, microglia immune training persists for at least 6 months and increases neuronal death in an animal model of AD and ischemia (Wendeln et al., 2018). These results support the importance of central immune training as a positive regulator of neurodegenerative susceptibility that, during nutritional programing, depends on the NLRP3 inflammasome pathway, as we mentioned before (Christ et al., 2018).
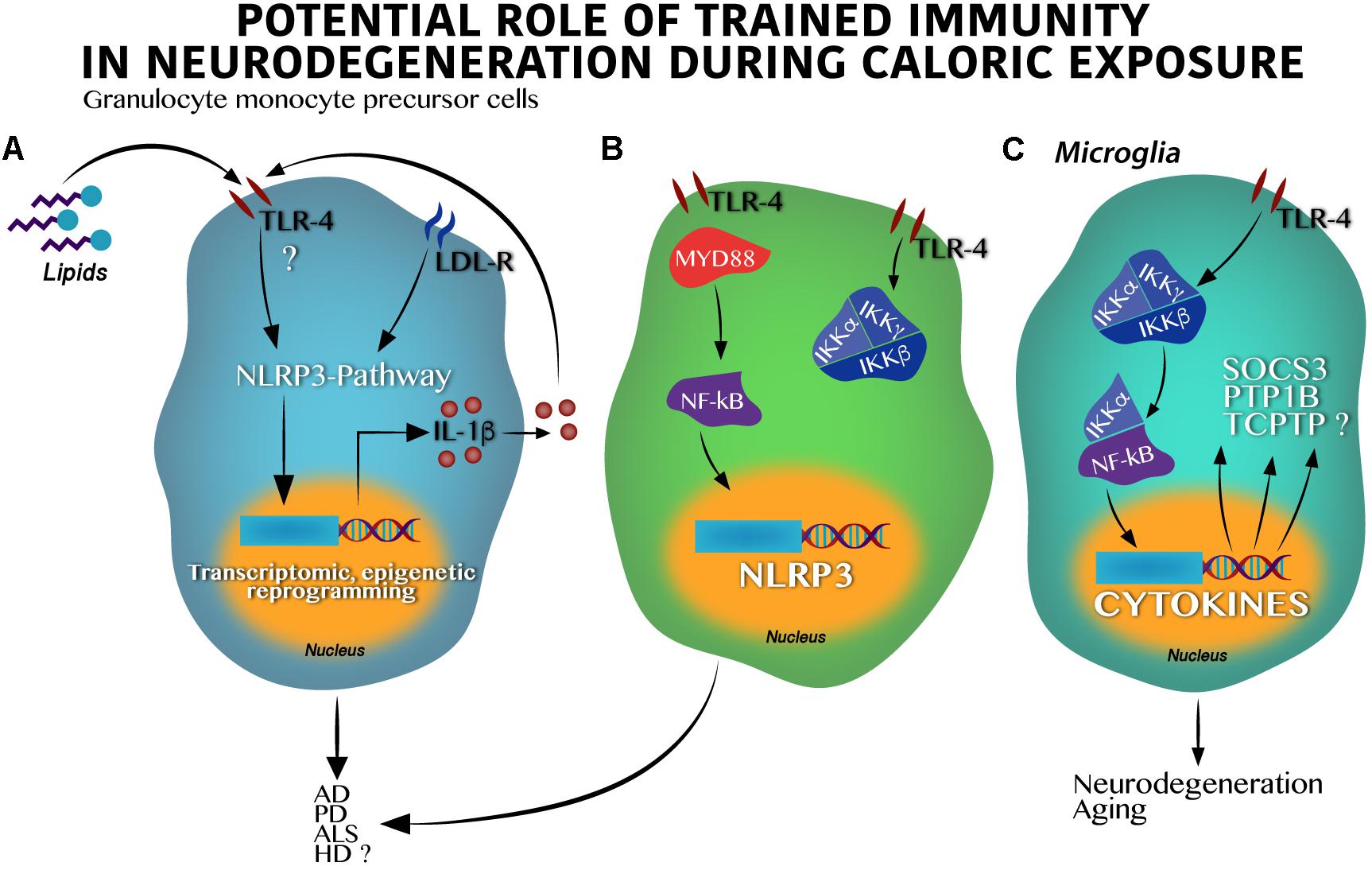
Figure 2. Toll-like receptor 4 activation by hypercaloric diet regulates inflammasome activation in innate cells leading to neurodegeneration. (A) In granulocyte monocyte precursor cells lipids activate Toll-like receptor 4 (TLR-4) leading to the inflammasome-pathway (NLRP3) activation, epigenetic, and transcriptomic changes in nucleus and IL-1β synthesis and release. IL-1β itself might stimulate an autocrine loop activating the TLR-4. (B) Classic activation of the IKK kinase complex (IKKα and IKKβ) by lipids in granulocyte monocyte precursor cells over supply leads to the NF-κB transcription factor activation and translocation into the nucleus positively regulating the NLRP3 synthesis. Dual activation of the (A) and (B) pathways result in neuronal death found in AD, PD, HD, and potentially in ALS. (C) Activation of the TLR-4 in microglia stimulates the IKK kinase complex following by the NF-κB activation and translocation into the nucleus leading to cytokines, suppressor of cytokine signaling 3 (SOCS3), protein-tyrosine phosphatase 1B (PTP1B) and probably, T-cell protein tyrosine phosphatase (TCPTP) synthesis, which altogether promote neurodegeneration and aging.
Major advances demonstrating the role of the NLRP3 inflammasome pathway on neurodegenerative susceptibility have been identified in animal models. In fact, it is believed that the NLRP3 inflammasome pathway goes beyond IL-1β synthesis, activating the function of the NF-κB factor and modulating neurodegenerative susceptibility. At first, experimental studies using murine models propose that NF-κB seems to have a role in the development of synaptic plasticity and neuronal survival (Monje et al., 2003), however, over activation of the IKKβ/NF-kB binomial might set the scenario toward a new pro-inflammatory state involving TNF-α through the paracrine actions modulating neuronal survival (Figure 2B). For instance, microglia IKKβ/NF-κB ablation in the hypothalamus reverts cognitive decline in rodents (Zhang et al., 2013), whereas IKKβ/NF-κB activation under long-time exposure to a hypercaloric diet depletes and impairs neuronal differentiation of adult hypothalamic neural stem cells (Li et al., 2012).
In the context of overnutrition, the IKKβ/NF-kB pathway (Figure 2C) seems to activate metabolic-related neurodegenerative markers including the suppressor of cytokine signaling 3 (SOCS3), the tyrosine-protein phosphatase non-receptor type 1 (PTP1B), and potentially, the T-cell protein tyrosine phosphatase (TCPTP) (Milanski et al., 2009). SOCS3 is implicated in neuronal damage during aging (Yadav et al., 2005; DiNapoli et al., 2010), and is also activated during Wallerian degeneration in injured peripheral nerves (Girolami et al., 2010; McNamee et al., 2010). The PTP1B marker leads to neuronal damage in the developing brain (Liu et al., 2019) and is actively implicated in apoptotic neuronal death in a high-glucose in vitro model (Arroba et al., 2016). Of note, PTP1B inhibition significantly inactivates GSK-3β-suppressing, amyloid β (Aβ)-induced tau phosphorylation and ameliorates spatial learning and memory in an animal model of AD (Kanno et al., 2016). On the other hand, the TCPTP marker was initially identified as a contributor to leptin resistance in the hypothalamus during obesity in rodents (Loh et al., 2011) and as a modulator of the homeostasis of body glucose (Dodd et al., 2015, 2018). Recently, TCPTP was reported to modulate cytokine release by the expression of connexin 30 (CX30) and connexin 43 (CX43) from astrocytes in a reciprocal interplay with PTP1B following LPS administration (Debarba et al., 2018). In fact, connexin43 or pannexin1 in astrocytes has been demonstrated to promote activation of the inflammasome in glial cells (Yi et al., 2016) and, its blockade with boldine, inhibited hemichannel activity in astrocytes and microglia without affecting gap junctional communication in culture and acute hippocampal slices. Also, when tested in animal models, AD mice with long-term oral administration of boldine prevented the increase in glial hemichannel activity, astrocytic Ca2+ signal, ATP and glutamate release which in turn, alleviated hippocampal neuronal suffering (Yi et al., 2017). These evidences allow us to propose that the IKKβ/NF-kB pathway activation during overnutrition integrates the PTP1B – TCPTP cross-talk which might be potentially implicated in neuronal survival. Together, given that overnutrition is associated to immune training by regulating the NF-κB activation linked to the NLRP3 inflammasome pathway (Christ et al., 2018), we propose that maternal nutritional programing by exposure to hypercaloric diets during pregnancy might set microglia immune training by activation of the IKKβ/NF-kB pathway, amplifying its effects on the SOCS3, the PTP1B, and the TCPTP (Figure 2C). Altogether, these proteins might increase susceptibility to neurodegenerative diseases in the offspring at later stages.
Potential molecular mechanisms that regulate peripheral immune training linked to NLRP3 inflammasome and neurodegeneration are being elucidated. Qiao et al. (2018) identified that hepatic NLRP3 inflammasome inhibition by in vivo siRNA administration decreases the TNF-α, IL-β, IL-12, and IL-18 serum levels. Of note, inhibition of the hepatic NLRP3 inflammasome partially prevents dopaminergic neuronal death in the substantia nigra of a murine Parkinson model by decreasing the pro-inflammatory profile in plasma (Qiao et al., 2018). It seems that immune activation, at least for viral infections, depends on CXCR6, a chemokine receptor of hepatic NK cells involved in the persistence of cellular memory (Netea et al., 2011). This suggests a potential direct link between peripheral immune training in the liver and neuroinflammation that leads to neurodegeneration in the CNS (Figure 3A).
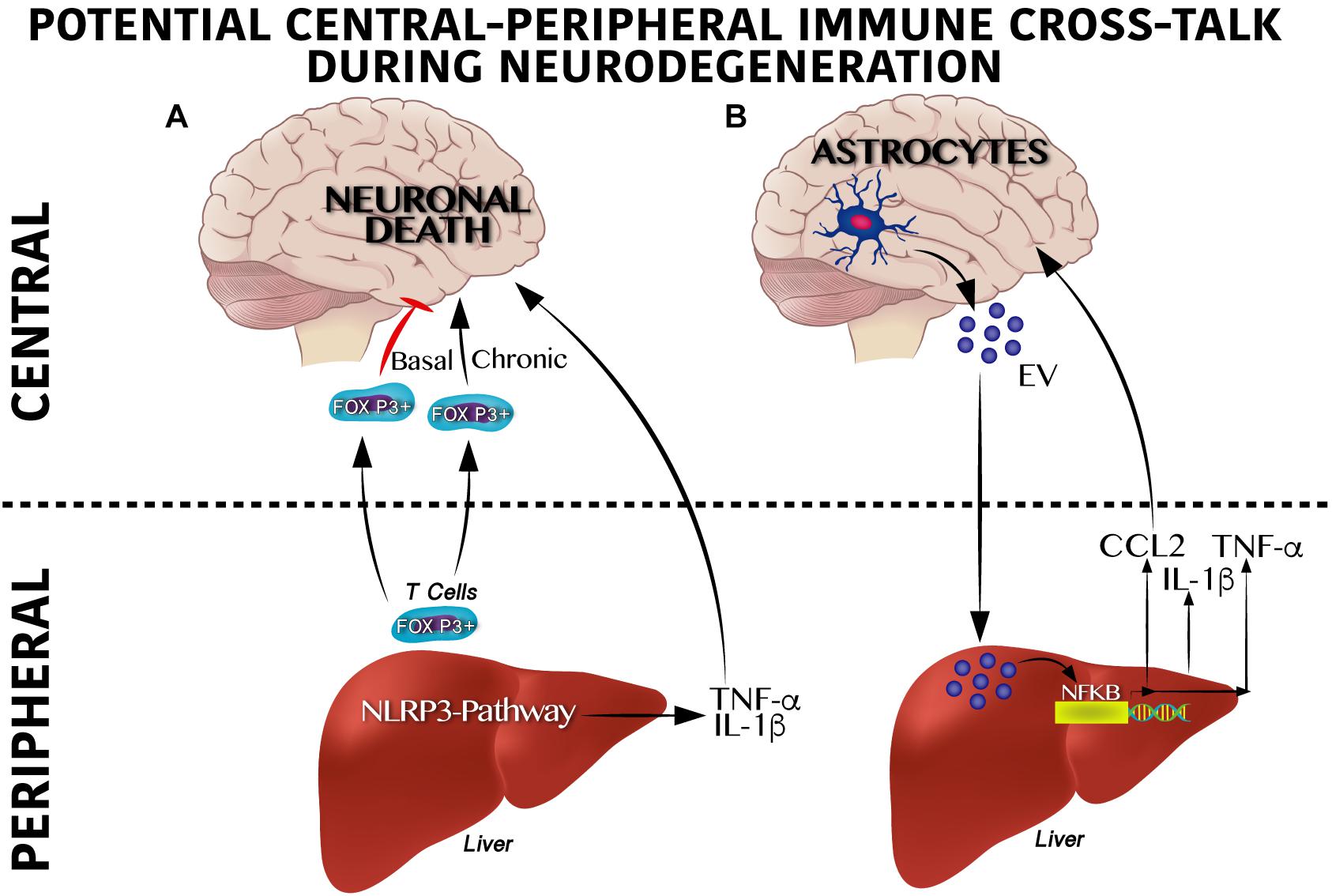
Figure 3. Cross-talk between peripheral and central immune system on neurodegenerative susceptibility. (A) Peripheral immune system regulates neurodegeneration. Under a physiological scenario, Foxp3 + T cells are released from the liver to circulation reaching the central nervous system (CNS) positively modulating neuronal survival, whereas soon after a chronic activation Foxp3 + T cells become overactive promoting neuronal death. Likewise, activation of the inflammasome pathway in the liver produces IL-1β and TNF-α which exacerbate neurodegeneration. (B) Central immune system regulates neurodegeneration. Astrocytes secrete extracellular vesicles (EV) to the circulation reaching the liver and positively modulating the NF-κB transcriptional activation program, which includes IL-1β, TNF-α and CCL2 synthesis and release. This pro-inflammatory profile has been reported to contribute centrally to neuronal damage.
Moreover, initial central immune training and its cross-talk with peripheral immunity also seems to be involved in neurodegeneration. For instance, recently, a substantial accumulation of peripheral FOXP3 + regulatory T cells in the mouse brain following ischemic stroke (Ito et al., 2019) was described. Of note, central FOXP3 + regulatory T cells accumulation inhibits neurotoxic astrogliosis by releasing amphiregulin, a low-affinity ligand of the epidermal growth factor receptor (Ito et al., 2019). Central FOXP3 + regulatory T cell accumulation appears to have a binary role on neuronal survival (Figure 3A). Transient depletion of FOXP3 + regulatory T cells decrease amyloid-β plaque accumulation, reverts the neuroinflammatory response and improves cognitive performance in the 5XFAD AD mouse model, therefore is beneficial under acute central inflammation (Baruch et al., 2015). Conversely, peripheral immune training in the liver might also be regulated by central immune inflammation (Figure 3B). Initial reports show that extracellular vesicles released from astrocytes and microglia during central immune training might be implicated in the peripheral-central immune cross-talk (Wang et al., 2017; Yang et al., 2018). For instance, in murine models, intracerebral injection of IL-1β promotes the release of extracellular vesicles from astrocytes, leading to leukocyte migration into the brain and regulation of the liver cytokines profile by inhibition of the peroxisome proliferator-activated receptor α (Dickens et al., 2017). It is important to mention that extracellular vesicles released by central immune activation depend on glutamine modulation by glutaminase enzyme activation (Dickens et al., 2017; Wang et al., 2017; Wu et al., 2018). In fact, substantial programing of metabolic related pathways coordinates innate immunity. For instance, M1 pro-inflammatory state in macrophages depends largely on glycolytic metabolism, impairment of oxidative phosphorylation and disruption of the Krebs cycle; glycolysis activation during immune training was recently reported to be dependent on the mevalonate pathway (Bekkering et al., 2018). Conversely, macrophages M2 phenotype, which are involved in tissue repair and homeostasis, are dependent on the Krebs cycle (Netea et al., 2011). These results suggest that a shift from oxidative phosphorylation toward glycolysis seems to be a potential metabolic node to regulated immune training. In line with this, the Akt/mTOR/HIF-1α–dependent pathway and the brain neutral sphingomyelin hydrolase 2 have been reported to be essential for trained immunity (Cheng et al., 2014; Saeed et al., 2014; Dickens et al., 2017).
A pro-inflammatory profile has been demonstrated to contribute to four major neurodegenerative pathologies and either suppressing or ameliorating this inflammatory process can have a good outcome for these diseases. According to this, we next discuss four of the major neurodegenerative pathologies in which inflammation has been proven to play a key role.
Central and Peripheral Inflammatory Nodes in Neurodegenerative Diseases
Active Immunity in Alzheimer Disease (AD)
Alzheimer disease is the most common type of senile dementia. Worldwide, approximately 50 million people are living with dementia and this number will triple in the next 40 years (Venigalla et al., 2015). AD is characterized by Aβ aggregation and intracellular bundles of hyperphosphorylated Tau protein (Colonna and Butovsky, 2017), which contribute to cognitive decline (Heneka et al., 2015).
High levels of TNF-α have also been identified in murine models of AD that resemble the classical behavioral abnormalities of the disease in humans (Gabbita et al., 2012; Kinney et al., 2018). Blocking the TNF-α pathway decreases central immune infiltration and prevents AD neurochemical and behavioral phenotypes (Tweedie et al., 2012; Prasad Gabbita et al., 2015), suggesting that central–peripheral cross-talk may be implicated in the disease. Notably, experimental studies using transgenic AD models have demonstrated that non-steroidal anti-inflammatory drugs (NSAIDs) can reduce AD pathology (Kinney et al., 2018). In addition, not only vertebrates seem to have dysregulation of the immune system during AD: In a D. melanogaster AD-like model, there is an upregulation of all five antimicrobial peptides (AMPs) related to inflammatory genes, suggesting the activation of innate immunity in AD-like flies. To counteract this neuroinflammation, they fed the flies with Gardenia jasminoides extracts (iridoid glycosides and aglycones, such as gardenoside, crocin, geniposide, and genipin) whose components can rescue cognitive deficits in a memory impairment mouse model (Nam and Lee, 2013) and show neuroprotective effect in cell culture (Sun et al., 2014). This experiment demonstrated that even though these extracts did not alter the Aβ concentration, they regulated the expression of immune-related genes (Drosomycin, Diptericine, Attacin, Cecropin, Mtk) in the brain of the flies and ameliorated memory deficits (Wei-Wei et al., 2017). The amelioration of memory loss and the counteraction of inflammation occurs through the modulation of the activity of acetylcholine esterase and the regulation the genes in apoptosis for neuroprotection, which have been recently accounted for in glycosides and aglycones present in Gardenia jasminoides extracts (Sun et al., 2014). Moreover, the robust effect of central-peripheral immune activation on AD-like neuropathology is also simulated using prenatal immune activation by the bacterial endotoxin lipopolysaccharide (LPS) or polyinosinic:polycytidylic acid (PolyI:C) (Chen et al., 2011; Krstic et al., 2012; Giovanoli et al., 2015, 2016) these findings suggest that prenatal immune activation may be an important environmental risk factor that can cause hippocampus-related cognitive and synaptic deficits in the absence of chronic inflammation across aging.
Initial reports in humans have identified a proinflammatory profile of cytokines such as TNF-α, IL-6, and IL-1β in the prefrontal cortex, hippocampus and serum of AD subjects (Cacabelos et al., 1994; Mrak and Griffin, 2005; Calsolaro and Edison, 2016). In fact, the risk for progression from mild cognitive impairment to AD and cognitive decline is higher in subjects with elevated IL-6 and TNF-α levels in the cerebral spinal fluid (Heneka et al., 2015). In the 1980s, when neuroinflammation was described in AD and immune-related cells where described to be close to Aβ plaques, several large epidemiological and observational studies were published concluding that long-term use of NSAIDs showed protective qualities against developing AD. However, recent studies have identified variable outcomes and no convincing evidence regarding the positive effect of NSAIDs in AD (Kinney et al., 2018). Despite these experimental evidences, it is still unknown whether elevated levels of IL-1β, IL-6 and TNF-α lead directly to neurodegeneration in humans (Figure 1).
Active Immunity in Parkinson’s Disease (PD)
Parkinson’s disease is a the second most common neurodegenerative condition, affecting about 1.6% of people over 60 years old; it acts by promoting dopaminergic neuronal death in the substantia nigra pars compacta (Bassani et al., 2015). PD shows intraneuronal aggregates (called Lewy bodies) formed by α-synuclein that contribute to dopaminergic neuronal death in the basal ganglia, leading to the classical parkinsonian motor symptoms (De Virgilio et al., 2016). Neurodegeneration in PD is associated with the activation of microglia (Heneka et al., 2015), increased TNF-α, IL-1β, and IL-6 levels in the midbrain, serum and cerebral spinal fluid (Hofmann et al., 2009; Montgomery and Bowers, 2012) and activation of astrocytes (Wang et al., 2015).
Potential systemic triggers of immune activation in PD have identified that maternal immune activation in murine models reproduces the neurochemical and neuropathological symptoms of the disease (Wang et al., 2009; Vuillermot et al., 2012). Also, TNF-α and IL-6 levels correlate with dopaminergic neuronal death in the PD brain – it seems that both cytokines show a dual role on neurodegeneration and neurophysiology (Figure 1). Another in vivo example in which neuroinflammation is involved in neurodegeneration is the D. melanogaster Rotenone PD-like model. Lakkappa et al. (2018) used a new compound, PTUPB [(4-(5-phenyl-3-3-3-(4-trifluoromethyl-phenyl)-ureido-propyl-pyrazol-1-yl)-benzenesulfonamide)], that inhibited neuroinflammation and in turn prevented the loss of dopaminergic neurons, slightly ameliorating the PD-like pathology.
Similar to the animal models, post-mortem PD patients show infiltration of T-lymphocytes into the substantia nigra (Montgomery and Bowers, 2012). The immune hypothesis as a tentative cause of PD is still active given that, similar to AD genetic risk factors, PD involves the LRRK2, NR4A2, PARK7, and CD74 genes, which are capable of regulating immune function and microglial activation (Pandey et al., 2009). In summary, the involvement of immune responses in PD is not fully understood, but they influence the inflammation response and seem to affect disease progression.
Active Immunity in Huntington’s Disease (HD)
Huntington’s disease is an autosomal-dominant neurodegenerative disease caused by a CAG trinucleotide repeat expansion that gives place to a polyglutamine region in the huntingtin protein (HTT). HD patients show a positive pro-inflammatory profile (Figure 1) correlated with the disease’s progression, including increases of IL-1β, IL-6, and TNF-α in the striatum, cerebral spinal fluid and plasma that were also confirmed in mouse models (Rocha et al., 2016). Central immune activation in HD subjects was confirmed using positron emission tomography (PET) and in pre-symptomatic HD-gene carriers (Rocha et al., 2016). Even though pharmacologic immune modulation using the XPro1595 TNF-α inhibitor has shown neuroprotection against the cytokine-induced neurotoxicity in primary R6/2 neurons and human neurons derived from iPSCs of HD patients (Hsiao et al., 2014), the information available is contradictory based on one report describing that neuroinflammation seems a consequence rather than a cause of neurodegeneration in late HD (Vinther-Jensen et al., 2016).
Active Immunity in Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis is the most prevalent kind of motor neuron disease in adults, affecting 4–6 per 100,000 people, and is considered a fatal neurodegenerative disease (Filippi and Agosta, 2016). ALS is characterized by the degeneration of motor spinal neurons, brain stem and primary motor cortex mainly associated to a neuroinflammatory response by central (microglial) and peripheral (T-cells) immune activation and infiltration into the affected brain regions (Evans et al., 2013; Filippi and Agosta, 2016). Central immune activation also occurs in patients with either sporadic or familiar ALS, as well as in transgenic models of the disease (Geloso et al., 2017). Immune activation has also been reported in a model of ALS in D. melanogaster, in which the flies were supplemented with potent anti-inflammatory natural extracts of Withania somnifera (Wse) and Mucuna pruriens (Mpe), which significantly rescued climbing impairment (De Rose et al., 2017). Also, ALS patients and mouse models of the disease have elevated levels of TNFα, IL-6, and IL-1β in blood and cerebrospinal fluid. In addition to TNF-α upregulation in ALS, TNFR1, and TNFR2 transcripts have also been found to be overexpressed in ALS patients compared with non-neurological controls (Han et al., 2015; Tortarolo et al., 2017). Similar to Alzheimer, Parkinson and Huntington, modulation of inflammatory cytokine–dependent pathways prevent neuronal death in ALS models. As we commented previously, the contribution of TNFα or IL-6 (Figure 1) as etiological causes of ALS pathophysiology remains controversial because of their physiological functions or dual role on the central and peripheral immune activation or as a neurotrophic factors (Tortarolo et al., 2017).
Altogether, these results propose a very new and not-yet-understood pathway that integrates metabolism, central and peripheral immune training and cross-talk which might actively modulate neurodegeneration.
Conclusion
Maternal nutritional programing and immune training in microglia or peripheral innate cells are complex and require a profound integration of glycolytic and oxidative metabolism as well as the reprograming of molecular signatures that modulate susceptibility to neuronal damage at earlier stages of development in the offspring. Peripheral or central innate immune training during maternal nutritional programing might integrate a significant node that regulates neurodegenerative susceptibility in the offspring. Potential integration of the NLRP3 inflammasome and the IKKβ/NF-kB pathways in microglia at early stages, and peripheral or central immune cross-talk at later stages, including the FOXP3 + regulatory T cells or the extracellular vesicles, might actively regulate neuroinflammation and neuronal death/survival. This information allows us to propose that maternal nutritional programing leads to peripheral or central immune training, favoring neurodegenerative susceptibility.
Author Contributions
MC-T, LM-M, RM-R, AC-M, and DR-P: conceptualization and writing – review and editing. AC-M and DR-P: supervision and visualization.
Funding
This work was funded by the National Council of Science and Technology in Mexico (CONACYT), 650620 CONACYT for MC-T, 582196 CONACYT for LM-M, and 573686 CONACYT for RM-R.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Alejandra Arreola-Triana for her support on editing this manuscript and Arlette Yaressi Vinaja-Romero for her figure artworks.
References
Agrawal, N. K. (2014). Targeting inflammation in diabetes: newer therapeutic options. World J. Diabetes 5, 697–710. doi: 10.4239/wjd.v5.i5.697
Ann, S. J., Kim, K. K., Cheon, E. J., Noh, H. M., Hwang, I., Yu, J. W., et al. (2018). Palmitate and minimally-modified low-density lipoprotein cooperatively promote inflammatory responses in macrophages. PLoS One 13:e0193649. doi: 10.1371/journal.pone.0193649
Arroba, A. I., Mazzeo, A., Cazzoni, D., Beltramo, E., Hernández, C., Porta, M., et al. (2016). Somatostatin protects photoreceptor cells against high glucose–induced apoptosis. Mol. Vis. 22, 1522–1531.
Arts, R. J. W., Moorlag, S. J. C. F. M., Novakovic, B., Li, Y., Wang, S. Y., Oosting, M., et al. (2018). BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe 23, 89–100. doi: 10.1016/j.chom.2017.12.010
Baruch, K., Rosenzweig, N., Kertser, A., Deczkowska, A., Sharif, A. M., Spinrad, A., et al. (2015). Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 6:7967. doi: 10.1038/ncomms8967
Bassani, T. B., Vital, M. A., and Rauh, L. K. (2015). Neuroinflammation in the pathophysiology of Parkinson’s disease and therapeutic evidence of anti-inflammatory drugs. Arq. Neuropsiquiatr. 73, 616–623. doi: 10.1590/0004-282X20150057
Bekkering, S., Arts, R. J. W., Novakovic, B., Kourtzelis, I., van der Heijden, C. D. C. C., Li, Y., et al. (2018). Metabolic induction of trained immunity through the mevalonate pathway. Cell 172, 135.e9–146.e9. doi: 10.1016/j.cell.2017.11.025
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat. Genet. 39, 17–23. doi: 10.1038/ng1934
Bilbo, S. D., and Tsang, V. (2010). Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 24, 2104–2115. doi: 10.1096/fj.09-144014
Cacabelos, R., Alvarez, X. A., Fernandez-Novoa, L., Franco, A., Mangues, R., Pellicer, A., et al. (1994). Brain interleukin-1 beta in Alzheimer’s disease and vascular dementia. Methods Find. Exp. Clin. Pharmacol. 16, 141–151.
Calsolaro, V., and Edison, P. (2016). Neuroinflammation in Alzheimer’ s disease: current evidence and future directions. Alzheimer’s Dement. 12, 719–732. doi: 10.1016/j.jalz.2016.02.010
Chen, G. H., Wang, H., Yang, Q. G., Tao, F., Wang, C., and Xu, D. X. (2011). Acceleration of age-related learning and memory decline in middle-aged CD-1 mice due to maternal exposure to lipopolysaccharide during late pregnancy. Behav. Brain Res. 218, 267–279. doi: 10.1016/j.bbr.2010.11.001
Cheng, S. C., Quintin, J., Cramer, R. A., Shepardson, K. M., Saeed, S., Kumar, V., et al. (2014). MTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345:1250684. doi: 10.1126/science.1250684
Choe, J. Y., and Kim, S. K. (2017). Quercetin and ascorbic acid suppress fructose-induced NLRP3 inflammasome activation by blocking intracellular shuttling of TXNIP in human macrophage cell lines. Inflammation 40, 980–994. doi: 10.1007/s10753-017-0542-4
Christ, A., Günther, P., Lauterbach, M. A. R., Duewell, P., Biswas, D., Pelka, K., et al. (2018). Western diet Triggers NLRP3-dependent innate immune reprogramming. Cell 172, 162.e14–175.e14. doi: 10.1016/j.cell.2017.12.013
Ciaccia, L. (2010). in Fundamentals of Inflammation, eds D. W. C. N. Serhan, and P. A. Ward (New York, NY: Cambridge University Press), doi: 10.1097/00000433-198206000-00020
Colonna, M., and Butovsky, O. (2017). Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 35, 441–468. doi: 10.1146/annurev-immunol-051116-052358
De Rose, F., Marotta, R., Talani, G., Catelani, T., Solari, P., Poddighe, S., et al. (2017). Differential effects of phytotherapic preparations in the hSOD1 Drosophila melanogaster model of ALS. Sci. Rep. 7:41059. doi: 10.1038/srep41059
De Virgilio, A., Greco, A., Fabbrini, G., Inghilleri, M., Rizzo, M. I., Gallo, A., et al. (2016). Parkinson’s disease: autoimmunity and neuroinflammation. Autoimmun. Rev. 15, 1005–1011. doi: 10.1016/j.autrev.2016.07.022
Debarba, L. K., Vechiato, F. M. V., Veida-Silva, H., Borges, B. C., Jamur, M. C., Antunes-Rodrigues, J., et al. (2018). The role of TCPTP on leptin effects on astrocyte morphology. Mol. Cell. Endocrinol. 482, 62–69. doi: 10.1016/j.mce.2018.12.010
Derks, I. P. M., Hivert, M.-F., Rifas-Shiman, S. L., Gingras, V., Young, J. G., Jansen, P. W., et al. (2019). Associations of prenatal exposure to impaired glucose tolerance with eating in the absence of hunger in early adolescence. Int. J. Obes. 43, 1903–1913. doi: 10.1038/s41366-018-0296-296
Dickens, A. M., Tovar-Y-Romo, L. B., Yoo, S. W., Trout, A. L., Bae, M., Kanmogne, M., et al. (2017). Astrocyte-shed extracellular vesicles regulate the peripheral leukocyte response to inflammatory brain lesions. Sci. Signal. 10:eaai7696. doi: 10.1126/scisignal.aai7696
DiNapoli, V. A., Benkovic, S. A., Li, X., Kelly, K. A., Miller, D. B., Rosen, C. L., et al. (2010). Age exaggerates proinflammatory cytokine signaling and truncates signal transducers and activators of transcription 3 signaling following ischemic stroke in the rat. Neuroscience 170, 633–644. doi: 10.1016/j.neuroscience.2010.07.011
Dodd, G. T., Decherf, S., Loh, K., Simonds, S. E., Wiede, F., Balland, E., et al. (2015). Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell 160, 88–104. doi: 10.1016/j.cell.2014.12.022
Dodd, G. T., Lee-Young, R. S., Brüning, J. C., and Tiganis, T. (2018). TCPTP regulates insulin signaling in AgRP neurons to coordinate glucose metabolism with feeding. Diabetes 67, 1246–1257. doi: 10.2337/db17-1485
Edlow, A. G., Guedj, F., Pennings, J. L. A., Sverdlov, D., Neri, C., and Bianchi, D. W. (2016a). Males are from Mars, and females are from Venus: sex-specific fetal brain gene expression signatures in a mouse model of maternal diet-induced obesity. Am. J. Obstet. Gynecol. 214, 620.e1–620.e4. doi: 10.1016/j.ajog.2016.02.054
Edlow, A. G., Hui, L., Wick, H. C., Fried, I., and Bianchi, D. W. (2016b). Assessing the fetal effects of maternal obesity via transcriptomic analysis of cord blood: a prospective case-control study. BJOG 123, 180–189. doi: 10.1111/1471-0528.13795
Edlow, A. G., Vora, N. L., Hui, L., Wick, H. C., Cowan, J. M., and Bianchi, D. W. (2014). Maternal obesity affects fetal neurodevelopmental and metabolic gene expression: a pilot study. PLoS One 9:e88661. doi: 10.1371/journal.pone.0088661
Esser, N., Legrand-Poels, S., Piette, J., Scheen, A. J., and Paquot, N. (2014). Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 105, 141–150. doi: 10.1016/j.diabres.2014.04.006
Estes, M. L., and McAllister, A. K. (2016). Maternal immune activation: implications for neuropsychiatric disorders. Science 353, 772–777. doi: 10.1126/science.aag3194
Evans, M. C., Couch, Y., Sibson, N., and Turner, M. R. (2013). Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 53, 34–41. doi: 10.1016/j.mcn.2012.10.008
Fan, Z., Liang, Z., Yang, H., Pan, Y., Zheng, Y., and Wang, X. (2017). Tenuigenin protects dopaminergic neurons from inflammation via suppressing NLRP3 inflammasome activation in microglia. J. Neuroinflammation 14:256. doi: 10.1186/s12974-017-1036-x
Filippi, M., and Agosta, F. (2016). Does neuroinflammation sustain neurodegeneration in ALS? Neurology 87, 2508–2509. doi: 10.1212/WNL.0000000000003441
Friedman, J. E. (2018). Developmental programming of obesity and diabetes in mouse, monkey, and man in 2018: Where are we headed? Diabetes 67, 2137–2151. doi: 10.2337/dbi17-0011
Fujikura, K., Setsu, T., Tanigaki, K., Abe, T., Kiyonari, H., Terashima, T., et al. (2013). Kif14 mutation causes severe brain malformation and hypomyelination. PLoS One 8:e53490. doi: 10.1371/journal.pone.0053490
Gabbita, S. P., Srivastava, M. K., Eslami, P., Johnson, M. F., Kobritz, N. K., Tweedie, D., et al. (2012). Early intervention with a small molecule inhibitor for tumor nefosis factor-α prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J. Neuroinflammation 9:99.
Geloso, M. C., Corvino, V., Marchese, E., Serrano, A., Michetti, F., and D’Ambrosi, N. (2017). The dual role of microglia in ALS: mechanisms and therapeutic approaches. Front. Aging Neurosci. 9:242. doi: 10.3389/fnagi.2017.00242
Giovanoli, S., Notter, T., Richetto, J., Labouesse, M. A., Vuillermot, S., Riva, M. A., et al. (2015). Late prenatal immune activation causes hippocampal deficits in the absence of persistent inflammation across aging. J. Neuroinflammation 12:221. doi: 10.1186/s12974-015-0437-y
Giovanoli, S., Weber-Stadlbauer, U., Schedlowski, M., Meyer, U., and Engler, H. (2016). Prenatal immune activation causes hippocampal synaptic deficits in the absence of overt microglia anomalies. Brain. Behav. Immun. 55, 25–38. doi: 10.1016/j.bbi.2015.09.015
Girolami, E. I., Bouhy, D., Haber, M., Johnson, H., and David, S. (2010). Differential expression and potential role of SOCS1 and SOCS3 in Wallerian degeneration in injured peripheral nerve. Exp. Neurol. 233, 173–182. doi: 10.1016/j.expneurol.2009.06.018
Glinsky, G. V. (2008). SNP-guided microRNA maps (MirMaps) of 16 common human disorders identify a clinically accessible therapy reversing transcriptional aberrations of nuclear import and inflammasome pathways. Cell Cycle 7, 3564–3576. doi: 10.4161/cc.7.22.7073
Gomes, R. M., Bueno, F. G., Schamber, C. R., de Mello, J. C. P., de Oliveira, J. C., Francisco, F. A., et al. (2018). Maternal diet-induced obesity during suckling period programs offspring obese phenotype and hypothalamic leptin/insulin resistance. J. Nutr. Biochem. 61, 24–32. doi: 10.1016/j.jnutbio.2018.07.006
Gordon, R., Albornoz, E. A., Christie, D. C., Langley, M. R., Kumar, V., Mantovani, S., et al. (2018). Inflammasome inhibition prevents -synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 10:eaah4066. doi: 10.1126/scitranslmed.aah4066
Han, L., Zhang, D., Tao, T., Sun, X., Liu, X., Zhu, G., et al. (2015). The role of N-Glycan modification of TNFR1 in inflammatory microglia activation. Glycoconj. J. 32, 685–693. doi: 10.1007/s10719-015-9619-9611
Heneka, M. T., Carson, M. J., Khoury, J., El Gary, E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5.Neuroinflammation
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Heneka, M. T., McManus, R. M., and Latz, E. (2018). Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 19, 610–621. doi: 10.1038/s41583-018-0055-7
Hofmann, K. W., Schuh, A. F. S., Saute, J., Townsend, R., Fricke, D., Leke, R., et al. (2009). Interleukin-6 serum levels in patients with Parkinson’s disease. Neurochem. Res. 34, 1401–1404. doi: 10.1007/s11064-009-9921-z
Hsiao, H.-Y., Chiu, F.-L., Chen, C.-M., Wu, Y.-R., Chen, H.-M., Chen, Y.-C., et al. (2014). Inhibition of soluble tumor necrosis factor is therapeutic in Huntington’s disease. Hum. Mol. Genet. 23, 4328–4344. doi: 10.1093/hmg/ddu151
Ito, M., Komai, K., Mise-Omata, S., Iizuka-Koga, M., Noguchi, Y., Kondo, T., et al. (2019). Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565, 246–250. doi: 10.1038/s41586-018-0824-5
Jang, J., Park, S., Jin Hur, H., Cho, H. J., Hwang, I., Pyo Kang, Y., et al. (2016). 25-hydroxycholesterol contributes to cerebral inflammation of X-linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nat. Commun. 7:13129. doi: 10.1038/ncomms13129
Johann, S., Heitzer, M., Kanagaratnam, M., Goswami, A., Rizo, T., Weis, J., et al. (2015). NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63, 2260–2273. doi: 10.1002/glia.22891
Kanno, T., Tsuchiya, A., Tanaka, A., and Nishizaki, T. (2016). Combination of PKCε activation and PTP1B inhibition effectively suppresses Aβ-induced GSK-3β activation and tau phosphorylation. Mol. Neurobiol. 53, 4787–4797. doi: 10.1007/s12035-015-9405-x
Kaufmann, E., Sanz, J., Dunn, J. L., Khan, N., Mendonça, L. E., Pacis, A., et al. (2018). BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell 172, 176.e19–190.e19. doi: 10.1016/j.cell.2017.12.031
Kinney, J. W., Bemiller, S. M., Murtishaw, A. S., Leisgang, A. M., Salazar, A. M., and Lamb, B. T. (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 4, 575–590. doi: 10.1016/j.trci.2018.06.014
Krstic, D., Madhusudan, A., Doehner, J., Vogel, P., Notter, T., Imhof, C., et al. (2012). Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J. Neuroinflammation 9:151. doi: 10.1186/1742-2094-9-151
Lakkappa, N., Krishnamurthy, P. T., Yamjala, K., Hwang, S. H., Hammock, B. D., and Babu, B. (2018). Evaluation of antiparkinson activity of PTUPB by measuring dopamine and its metabolites in Drosophila melanogaster: LC–MS/MS method development. J. Pharm. Biomed. Anal. 149, 457–464. doi: 10.1016/j.jpba.2017.11.043
Lee, E., Hwang, I., Park, S., Hong, S., Hwang, B., Cho, Y., et al. (2018). MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 26, 213–228. doi: 10.1038/s41418-018-0124-5
Lei, Y., Chen, C. J., Yan, X. X., Li, Z., and Deng, X. H. (2017). Early-life lipopolysaccharide exposure potentiates forebrain expression of NLRP3 inflammasome proteins and anxiety-like behavior in adolescent rats. Brain Res. 1671, 43–54. doi: 10.1016/j.brainres.2017.06.014
L’homme, L., Esser, N., Riva, L., Scheen, A., Paquot, N., Piette, J., et al. (2013). Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J. Lipid Res. 54, 2998–3008. doi: 10.1194/jlr.M037861
Li, J., Tang, Y., and Cai, D. (2012). IKKβ/NF-κB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat. Cell Biol. 14, 999–1012. doi: 10.1038/ncb2562
Ling, Z., Chang, Q. A., Tong, C. W., Leurgans, S. E., Lipton, J. W., and Carvey, P. M. (2004). Rotenone potentiates dopamine neuron loss in animals exposed to lipopolysaccharide prenatally. Exp. Neurol. 190, 373–383. doi: 10.1016/j.expneurol.2004.08.006
Ling, Z. D., Chang, Q., Lipton, J. W., Tong, C. W., Landers, T. M., and Carvey, P. M. (2004). Combined toxicity of prenatal bacterial endotoxin exposure and postnatal 6-hydroxydopamine in the adult rat midbrain. Neuroscience 124, 619–628. doi: 10.1016/j.neuroscience.2003.12.017
Liu, B., Ou, G., Chen, Y., and Zhang, J. (2019). Inhibition of protein tyrosine phosphatase 1B protects against sevoflurane-induced neurotoxicity mediated by ER stress in developing brain. Brain Res. Bull. 146, 28–39. doi: 10.1016/j.brainresbull.2018.12.006
Loh, K., Fukushima, A., Zhang, X., Galic, S., Briggs, D., Enriori, P. J., et al. (2011). Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 14, 684–699. doi: 10.1016/j.cmet.2011.09.011
Maldonado-Ruiz, R., Fuentes-Mera, L., and Camacho, A. (2017). Central modulation of neuroinflammation by neuropeptides and energy-sensing hormones during obesity. Biomed Res. Int. 2017:7949582. doi: 10.1155/2017/7949582
Mastrocola, R., Collino, M., Penna, C., Nigro, D., Chiazza, F., Fracasso, V., et al. (2016). Maladaptive modulations of nlrp3 inflammasome and cardioprotective pathways are involved in diet-induced exacerbation of myocardial ischemia/reperfusion injury in mice. Oxid. Med. Cell. Longev. 2016:3480637. doi: 10.1155/2016/3480637
McNamee, E. N., Ryan, K. M., Griffin, ÉW., González-Reyes, R. E., Ryan, K. J., Harkin, A., et al. (2010). Noradrenaline acting at central β-adrenoceptors induces interleukin-10 and suppressor of cytokine signaling-3 expression in rat brain: implications for neurodegeneration. Brain. Behav. Immun. 24, 660–671. doi: 10.1016/j.bbi.2010.02.005
Milanski, M., Degasperi, G., Coope, A., Morari, J., Denis, R., Cintra, D. E., et al. (2009). Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J. Neurosci. 29, 359–370. doi: 10.1523/JNEUROSCI.2760-08.2009
Mitroulis, I., Ruppova, K., Wang, B., Chen, L. S., Grzybek, M., Grinenko, T., et al. (2018). Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell 172, 147.e12–161.e12. doi: 10.1016/j.cell.2017.11.034
Monje, M. L., Toda, H., and Palmer, T. D. (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765. doi: 10.1126/science.1088417
Montgomery, S. L., and Bowers, W. J. (2012). Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J. Neuroimmune Pharmacol. 7, 42–59. doi: 10.1007/s11481-011-9287-2
Morin, J.-P., Rodríguez-Durán, L. F., Guzmán-Ramos, K., Perez-Cruz, C., Ferreira, G., Diaz-Cintra, S., et al. (2017). Palatable hyper-caloric foods impact on neuronal plasticity. Front. Behav. Neurosci. 11:19. doi: 10.3389/fnbeh.2017.00019
Mrak, R. E., and Griffin, W. S. T. (2005). Potential inflammatory biomarkers in Alzheimer’s disease. J. Alzheimers. Dis. 8, 369–375. doi: 10.3233/jad-2005-8406
Mucellini, A. B., Laureano, D. P., Silveira, P. P., and Sanvitto, G. L. (2019). Maternal and post-natal obesity alters long-term memory and hippocampal molecular signaling of male rat. Brain Res. 1708, 138–145. doi: 10.1016/j.brainres.2018.12.021
Nam, Y., and Lee, D. (2013). Ameliorating effect of Zhizi (Fructus Gardeniae) extract and its glycosides on scopolamine-induced memory impairment. J. Tradit. Chinese Med. 33, 223–227. doi: 10.1016/s0254-6272(13)60129-6
Netea, M. G., Joosten, L. A. B., Latz, E., Mills, K. H. G., Natoli, G., Stunnenberg, H. G., et al. (2016). Trained immunity: a program of innate immune memory in health and disease. Science 352:aaf1098. doi: 10.1126/science.aaf1098
Netea, M. G., Quintin, J., and Van Der Meer, J. W. M. (2011). Trained immunity: a memory for innate host defense. Cell Host Microbe 9, 355–361. doi: 10.1016/j.chom.2011.04.006
Nigro, D., Menotti, F., Cento, A. S., Serpe, L., Chiazza, F., Dal Bello, F., et al. (2017). Chronic administration of saturated fats and fructose differently affect SREBP activity resulting in different modulation of Nrf2 and Nlrp3 inflammasome pathways in mice liver. J. Nutr. Biochem. 42, 160–171. doi: 10.1016/j.jnutbio.2017.01.010
O’Brien, P. D., Hinder, L. M., Callaghan, B. C., and Feldman, E. L. (2017). Neurological consequences of obesity. Lancet Neurol. 16, 465–477.
Ott, A., Stolk, R. P., Van Harskamp, F., Pols, H. A. P., Hofman, A., and Breteler, M. M. B. (1999). Diabetes mellitus and the risk of dementia: the rotterdam study. Neurology 53, 1937–1942. doi: 10.1212/wnl.53.9.1937
Ou, X., Thakali, K. M., Shankar, K., Andres, A., and Badger, T. M. (2015). Maternal adiposity negatively influences infant brain white matter development. Obesity (Silver Spring) 23, 1047–1054. doi: 10.1002/oby.21055
Pan, Y., Chen, X. Y., Zhang, Q. Y., and Kong, L. D. (2014). Microglial NLRP3 inflammasome activation mediates IL-1β-related inflammation in prefrontal cortex of depressive rats. Brain. Behav. Immun. 41, 90–100. doi: 10.1016/j.bbi.2014.04.007
Pandey, A. K., Agarwal, P., Kaur, K., and Datta, M. (2009). MicroRNAs in diabetes: tiny players in big disease. Cell. Physiol. Biochem. 23, 221–232. doi: 10.1159/000218169
Patro Golab, B., Santos, S., Voerman, E., Lawlor, D. A., Jaddoe, V. W., Gaillard, R., et al. (2018). Influence of maternal obesity on the association between common pregnancy complications and risk of childhood obesity: an individual participant data meta-analysis. Lancet Child Adolesc. Heal. 2, 812–821. doi: 10.1016/S2352-4642(18)30273-6
Prasad Gabbita, S., Johnson, M. F., Kobritz, N., Eslami, P., Poteshkina, A., Varadarajan, S., et al. (2015). Oral TNFα modulation alters neutrophil infiltration, improves cognition and diminishes tau and amyloid pathology in the 3xtgad mouse model. PLoS One 10:e0137305. doi: 10.1371/journal.pone.0137305
Procaccini, C., Santopaolo, M., Faicchia, D., Colamatteo, A., Formisano, L., De Candia, P., et al. (2016). Role of metabolism in neurodegenerative disorders. Metabolism 65, 1376–1390. doi: 10.1016/j.metabol.2016.05.018
Qiao, C., Zhang, Q., Jiang, Q., Zhang, T., Chen, M., Fan, Y., et al. (2018). Inhibition of the hepatic Nlrp3 protects dopaminergic neurons via attenuating systemic inflammation in a MPTP/p mouse model of Parkinson’s disease. J. Neuroinflammation 15:193. doi: 10.1186/s12974-018-1236-z
Rocha, N. P., Ribeiro, F. M., Furr-Stimming, E., and Teixeira, A. L. (2016). Neuroimmunology of huntington’s disease: revisiting evidence from human studies. Mediators Inflamm. 2016:8653132. doi: 10.1155/2016/8653132
Romanitan, M. O., Popescu, B. O., Winblad, B., Bajenaru, O. A., and Bogdanovic, N. (2007). Occludin is overexpressed in Alzheimer’s disease and vascular dementia. J. Cell. Mol. Med. 11, 569–579. doi: 10.1111/j.1582-4934.2007.00047.x
Saeed, S., Quintin, J., Kerstens, H. H. D., Rao, N. A., Aghajanirefah, A., Matarese, F., et al. (2014). Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345:1251086. doi: 10.1126/science.1251086
Shapiro, A. L. B., Sauder, K. A., Tregellas, J. R., Legget, K. T., Gravitz, S. L., Ringham, B. M., et al. (2017). Exposure to maternal diabetes in utero and offspring eating behavior: the EPOCH study. Appetite 116, 610–615. doi: 10.1016/j.appet.2017.05.005
Sobesky, J. L., DAngelo, H. M., Weber, M. D., Anderson, N. D., Frank, M. G., Watkins, L. R., et al. (2016). Glucocorticoids mediate short-term high-fat diet induction of neuroinflammatory priming, the NLRP3 inflammasome, and the danger signal HMGB1. eNeuro 3, e0113–e0116. doi: 10.1523/ENEURO.0113-16.2016
Stranahan, A. M., Norma, E. D., Lee, K., Cutler, R. G., Telljohann, R., Egan, J. M., et al. (2008). Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 23, 1–7. doi: 10.1038/jid.2014.371
Su, W. J., Zhang, Y., Chen, Y., Gong, H., Lian, Y. J., Peng, W., et al. (2017). NLRP3 gene knockout blocks NF-κB and MAPK signaling pathway in CUMS-induced depression mouse model. Behav. Brain Res. 322(Pt A), 1–8. doi: 10.1016/j.bbr.2017.01.018
Sui, Y. H., Luo, W. J., Xu, Q. Y., and Hua, J. (2016). Dietary saturated fatty acid and polyunsaturated fatty acid oppositely affect hepatic NOD-like receptor protein 3 inflammasome through regulating nuclear factor-kappa B activation. World J. Gastroenterol. 22, 2533–2544. doi: 10.3748/wjg.v22.i8.2533
Sun, P., Ding, H., Liang, M., Li, X., Mo, W., Wang, X., et al. (2014). Neuroprotective effects of geniposide in SH-SY5Y cells and primary hippocampal neurons exposed to A β 42. Biomed Res. Int. 2014:284314. doi: 10.1155/2014/284314
Takahashi, K., Prinz, M., Stagi, M., Chechneva, O., and Neumann, H. (2007). TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 4:e124. doi: 10.1371/journal.pmed.0040124
Tartaglione, A. M., Venerosi, A., and Calamandrei, G. (2016). “Early-life toxic insults and onset of sporadic neurodegenerative diseases—An overview of experimental studies. Curr. Top. Behav. Neurosci. 29, 231–264. doi: 10.1007/7854_2015_416
Tortarolo, M., Lo Coco, D., Veglianese, P., Vallarola, A., Giordana, M. T., Marcon, G., et al. (2017). Amyotrophic lateral sclerosis, a multisystem pathology: insights into the role of TNF α. Mediators Inflamm. 2017:2985051. doi: 10.1155/2017/2985051
Tweedie, D., Ferguson, R. A., Fishman, K., Frankola, K. A., Van Praag, H., Holloway, H. W., et al. (2012). Tumor necrosis factor-α synthesis inhibitor 3,6’-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflammation 9, 575.
Vaiserman, A. M. (2017). Early-life nutritional programming of type 2 diabetes: experimental and quasi-experimental evidence. Nutrients 9:e236. doi: 10.3390/nu9030236
Venigalla, M., Gyengesi, E., Sharman, M. J., and Münch, G. (2015). Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 95, 63–74. doi: 10.1016/j.neuint.2015.10.011
Vinther-Jensen, T., Börnsen, L., Budtz-Jørgensen, E., Ammitzbøll, C., Larsen, I. U., Hjermind, L. E., et al. (2016). Selected CSF biomarkers indicate no evidence of early neuroinflammation in Huntington disease. Neurol. Neuroimmunol. Neuroinflamm. 3:e287. doi: 10.1212/NXI.0000000000000287
Vuillermot, S., Joodmardi, E., Perlmann, T., Ove Ogren, S., Feldon, J., and Meyer, U. (2012). Prenatal immune activation interacts with genetic Nurr1 deficiency in the development of attentional impairments. J. Neurosci. 32, 436–451. doi: 10.1523/JNEUROSCI.4831-11.2012
Wang, K., Ye, L., Lu, H., Chen, H., Zhang, Y., Huang, Y., et al. (2017). TNF-α promotes extracellular vesicle release in mouse astrocytes through glutaminase. J. Neuroinflammation 14:87.
Wang, Q., Liu, Y., and Zhou, J. (2015). Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 4, 1–9.
Wang, S., Yan, J. Y., Lo, Y. K., Carvey, P. M., and Ling, Z. (2009). Dopaminergic and serotoninergic deficiencies in young adult rats prenatally exposed to the bacterial lipopolysaccharide. Brain Res. 1265, 196–204. doi: 10.1016/j.brainres.2009.02.022
Wei-Wei, M., Ye, T., Yan-Ying, W., and I-Feng, P. (2017). Effects of Gardenia jasminoides extracts on cognition and innate immune response in an adult Drosophila model of Alzheimer’s disease. Chin. J. Nat. Med. 15, 899–904. doi: 10.1016/S1875-5364(18)30005-0
Wen, H., Gris, D., Lei, Y., Jha, S., Zhang, L., Huang, M. T. H., et al. (2011). Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415. doi: 10.1038/ni.2022
Wendeln, A., Degenhardt, K., Kaurani, L., Gertig, M., Ulas, T., Jain, G., et al. (2018). Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338. doi: 10.1038/s41586-018-0023-4
Widen, E. M., Kahn, L. G., Cirillo, P., Cohn, B., Kezios, K. L., and Factor-Litvak, P. (2018). Prepregnancy overweight and obesity are associated with impaired child neurodevelopment. Matern. Child Nutr. 14:e12481. doi: 10.1111/mcn.12481
Wu, B., Liu, J., Zhao, R., Li, Y., Peer, J., Braun, A. L., et al. (2018). Glutaminase 1 regulates the release of extracellular vesicles during neuroinflammation through key metabolic intermediate alpha-ketoglutarate. J. Neuroinflammation 15:79. doi: 10.1186/s12974-018-1120-x
Yadav, A., Kalita, A., Dhillon, S., and Banerjee, K. (2005). JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J. Biol. Chem. 280, 31830–31840. doi: 10.1074/jbc.M501316200
Yang, Y., Boza-Serrano, A., Dunning, C. J. R., Clausen, B. H., Lambertsen, K. L., and Deierborg, T. (2018). Inflammation leads to distinct populations of extracellular vesicles from microglia. J. Neuroinflammation 15:168. doi: 10.1186/s12974-018-1204-7
Yi, C., Ezan, P., Fernández, P., Schmitt, J., Sáez, J. C., Giaume, C., et al. (2017). Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 65, 1607–1625. doi: 10.1002/glia.23182
Yi, C., Mei, X., Ezan, P., Mato, S., Matias, I., Giaume, C., et al. (2016). Astroglial connexin43 contributes to neuronal suffering in a mouse model of Alzheimer’s disease. Cell Death Differ. 23, 1691–1701. doi: 10.1038/cdd.2016.63
Young, K. A., Berry, M. L., Mahaffey, C. L., Saionz, J. R., Hawes, N. L., Chang, B., et al. (2002). Fierce: a new mouse deletion of Nr2e1; violent behaviour and ocular abnormalities are background-dependent. Behav. Brain Res. 132, 145–158. doi: 10.1016/s0166-4328(01)00413-2
Zhang, G., Li, J., Purkayastha, S., Tang, Y., Zhang, H., Yin, Y., et al. (2013). Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 497, 211–216. doi: 10.1038/nature12143
Keywords: maternal programing, neurodegeneration, inflammation, immune training, cytokines
Citation: Cárdenas-Tueme M, Montalvo-Martínez L, Maldonado-Ruiz R, Camacho-Morales A and Reséndez-Pérez D (2020) Neurodegenerative Susceptibility During Maternal Nutritional Programing: Are Central and Peripheral Innate Immune Training Relevant? Front. Neurosci. 14:13. doi: 10.3389/fnins.2020.00013
Received: 18 November 2019; Accepted: 08 January 2020;
Published: 04 February 2020.
Edited by:
Kathleen C. Page, Bucknell University, United StatesReviewed by:
Juan C. Saez, Pontifical Catholic University of Chile, ChileNafisa M. Jadavji, Midwestern University, United States
Copyright © 2020 Cárdenas-Tueme, Montalvo-Martínez, Maldonado-Ruiz, Camacho-Morales and Reséndez-Pérez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Diana Reséndez-Pérez, ZGlhbmEucmVzZW5kZXpwckB1YW5sLmVkdS5teA==; ZGlhcmVzZW5kZXpAZ21haWwuY29t