 Gautam Wali1,2*
Gautam Wali1,2* Erandhi Liyanage1
Erandhi Liyanage1 Nicholas F. Blair1,2Ratneswary Sutharsan3,4
Nicholas F. Blair1,2Ratneswary Sutharsan3,4 Jin-Sung Park1,5
Jin-Sung Park1,5 Alan Mackay-Sim2,3†Carolyn M. Sue1,2*†
Alan Mackay-Sim2,3†Carolyn M. Sue1,2*†- 1Department of Neurogenetics, Kolling Institute of Medical Research, The University of Sydney, Sydney, NSW, Australia
- 2Sydney Medical School, The University of Sydney, Sydney, NSW, Australia
- 3Griffith Institute for Drug Discovery, Griffith University, Nathan, QLD, Australia
- 4Queensland Brain Institute, The University of Queensland, Brisbane, QLD, Australia
- 5Department of Experimental Animal Research, Biomedical Research Institute, Seoul National University Hospital, Seoul, South Korea
Hereditary spastic paraplegia (HSP) is a group of inherited disorders characterized by progressive spasticity and paralysis of the lower limbs. Autosomal dominant mutations in SPAST gene account for ∼40% of adult-onset patients. We have previously shown that SPAST patient cells have reduced organelle transport and are therefore more sensitive to oxidative stress. To test whether these effects are present in neuronal cells, we first generated 11 induced pluripotent stem (iPS) cell lines from fibroblasts of three healthy controls and three HSP patients with different SPAST mutations. These cells were differentiated into FOXG1-positive forebrain neurons and then evaluated for multiple aspects of axonal transport and fragmentation. Patient neurons exhibited reduced levels of SPAST encoded spastin, as well as a range of axonal deficits, including reduced levels of stabilized microtubules, lower peroxisome transport speed as a consequence of reduced microtubule-dependent transport, reduced number of peroxisomes, and higher density of axon swellings. Patient axons fragmented significantly more than controls following hydrogen peroxide exposure, suggesting for the first time that the SPAST patient axons are more sensitive than controls to the deleterious effects of oxidative stress. Treatment of patient neurons with tubulin-binding drugs epothilone D and noscapine rescued axon peroxisome transport and protected them against axon fragmentation induced by oxidative stress, showing that SPAST patient axons are vulnerable to oxidative stress-induced degeneration as a consequence of reduced axonal transport.
Introduction
Hereditary spastic paraplegia (HSP) is an inherited neurological disorder that predominantly affects the corticospinal tracts, leading to lower limb weakness, spasticity, and paralysis (Salinas et al., 2008). Mutations in the SPAST gene are the most common cause of HSP, accounting for over 40% of autosomal dominant cases (Vandebona et al., 2012; Ruano et al., 2014). Spastin, the protein encoded by SPAST, is a microtubule severing protein that helps control microtubule stability and the cellular processes dependent on these structures, including organelle transport (Errico et al., 2002; McDermott et al., 2003). Histopathological studies suggest that deficits in organelle transport might be responsible for degeneration of the corticospinal tract axons, a common pathological feature of SPAST HSP cases (McDermott et al., 2000; Wharton et al., 2003; Orlacchio et al., 2004; Salinas et al., 2008; Duning et al., 2010), although this hypothesis has not been tested.
Multiple studies have consistently indicated that neurons from SPAST patients, post mortem and in vitro, have deficits in axon transport. For example, post-mortem spinal cord analyses of two patients with missense and frameshift SPAST mutations showed distinct axonal swellings containing abnormal neurofilament and accumulated mitochondria (Kasher et al., 2009). In three SPAST HSP cases, mitochondria were distributed more densely around the periphery of the neuronal soma of lower and upper motor neurons, in contrast to being uniformly distributed throughout the neuronal soma in healthy controls (McDermott et al., 2003). Similarly, induced pluripotent stem (iPS) cell-induced neurons from a patient with a mutation in intron 4 of the SPAST gene (c.683-1G > T) had abnormal axon swellings containing mitochondria and stable microtubules (i.e., acetylated α-tubulin) (Denton et al., 2014). Patient axons displayed reduced mitochondria motility, with fewer moving in retrograde compared to control axons (Denton et al., 2014). In a second study, glutamatergic neurons differentiated from iPS cells from two patients with the same non-sense mutation in the SPAST gene (c.1684C > T) (p.R562X), also had fewer axon mitochondria moving in retrograde compared to controls (Havlicek et al., 2014). Restoring axonal transport in SPAST HSP patients is therefore a promising therapeutic target.
The unique architecture of neurons, with axons extending up to 1 m in length, makes them more dependent on microtubules for organelle transport than other cell types. Unlike smaller cells and neuronal cell bodies that depend on both actin and microtubule cytoskeletons for organelle transport, organelle transport within axons is primarily dependent on microtubules (Barlan and Gelfand, 2017). Therefore, it would be expected that any condition that reduces microtubules would have a larger impact on neurons than other cell types. We have previously shown that olfactory neurosphere (ONS)-derived cells from SPAST patients have reduced levels of stabilized microtubules, leading to reduced organelle transport and thereby making them more sensitive to oxidative stress (Wali et al., 2016). In the present study, to test if these deficits are present in neurons and measure the impact the deficits have on neuronal function, we first generated six iPS cell lines from three SPAST patients and five iPS cell lines from three healthy controls. These were differentiated into forebrain neurons to act as a model for HSP and will here on be referred to as patient and control neurons. We then characterized several aspects of the axon transport and showed that patient neurons had less stabilized microtubules, reduced microtubule-dependent peroxisome transport, reduced number of peroxisomes, and a higher density of axon swellings. We found that patient axons exhibited increased axon fragmentation when compared to healthy control neurons. Finally, we show that two tubulin-binding drugs, epothilone D and noscapine, can rescue microtubule-dependent peroxisome axon transport in patient axons and protect them against the effects of oxidative stress. This represents a significant step toward the development of effective treatments for SPAST-related HSP.
Materials and Methods
Detailed methods are in Supplementary Material.
Participants
Hereditary spastic paraplegia patients in this study were reviewed and examined by a movement disorders specialist (CS). All patients exhibited similar clinical symptoms, typical of adult onset HSP, and were subsequently identified with SPAST mutations. Details of their clinical phenotypes are reported elsewhere (Vandebona et al., 2012) and the mutation identified in each patient is listed in Supplementary Table S1. Our study involving human participants was reviewed and approved by Human Research Ethics Committee affiliated to the Northern Sydney Local Health District, New South Wales government, Australia. The ethics committee reference number: RESP/15/314. The patients/participants provided their written informed consent to participate in this study.
Generation and Characterization of Induced Pluripotent Stem Cell Lines
Skin fibroblasts were cultured and reprogrammed to iPS cells using the Sendai virus approach. iPS cell pluripotency was assessed by quantifying the expression of SSEA-4 and Sox2 by flow cytometry and immunostaining. TaqMan® hPSC Scorecard (Thermofisher, A15872) assay was used to predict tri-lineage differentiation potential. Differentiation ability of iPS cells to the three germ layers was assessed by spontaneous differentiation of iPS cells to embryoid bodies (Abraham et al., 2010) and the expression of β III-tubulin, GATA4, and brachyury.
Neuronal Differentiation of Induced Pluripotent Stem Cell Lines
All iPS cell lines were differentiated to FOXG1-positive forebrain neurons following a previously published protocol (Chambers et al., 2009; Maroof et al., 2013). Confirmation of neuronal differentiation was quantified in the cultures by immunofluorescence labeling with neuronal marker: β III-tubulin; synaptic marker, SYN1; and forebrain neuron marker, FOXG1.
Phenotyping of Differentiated Patient Derived Neurons
(A) Spastin protein expression was assessed in all patient and control differentiated neuronal cultures using western blot analysis. (B) Axon swellings were quantified in images of axons immune-labeled with β III-tubulin. (C) Peroxisome imaging and analysis in living cells were quantified in patient and control cell lines using automated imaging and analysis as described previously (Wali et al., 2016). To test the effect of tubulin-binding drugs on peroxisome trafficking speeds, we quantified peroxisome speed in untreated control axons, untreated patient axons, and in patient axons treated with the tubulin-binding drugs, 2 nM epothilone D or 10 μM noscapine for 24 h prior to live cell imaging (Fan et al., 2014). (D) The effect of oxidative stress on axon fragmentation was quantified in axons exposed to H2O2. To induce oxidative stress, we used 20 μM H2O2, that causes axon fragmentation but not cell death (Bilsland et al., 2010; Fang et al., 2012). Patient and control axons were treated with H2O2 for 3, 6, and 12 h with media change every hour, after which axon degeneration was quantified by image analysis by measuring degeneration index (DI) as described previously (Sasaki et al., 2009). To determine whether the tubulin-binding drugs could rescue axon degeneration induced by oxidative stress, patient cells were treated with 2 nM epothilone D or 10 μM noscapine for 24 h prior to H2O2 treatment and DI was measured. Axon degeneration was measured as described previously (Sasaki et al., 2009). Briefly, bright-field images were binarized so that pixel intensity of the axon regions was black and all other regions white. The total number of black pixels was defined as the “total axon area.” The degenerated axons were detected using the ImageJ particle analyzer module and quantified the area of small fragments (size = 20–10,000 pixels). DI was calculated as the ratio of fragmented axon area over total axon area. This assay generates a score of 0 for completely intact axons and 1 for completely fragmented axons.
Statistical Analysis
Data are expressed as mean ± SEM. SPSS Statistics Version 24 (IBM), Prism (GraphPad), and Stata IC Version 12.1 software (STATA Corp., College Station, TX, United States) were used to analyze data and plot graphs. In all graphs, the individual clones from the same cell lines have the same shape and are shown as filled or patterned.
Results
Generation of iPS Cell Lines From Patient and Control-Derived Skin Fibroblasts
Fibroblasts were reprogrammed to iPS cells using the Sendai virus system (Figure 1A). We established five control and six patient iPS cell lines (multiple clones, details in Supplementary Table S1). All iPS cell lines expressed pluripotency markers SSEA4 and SOX2 (Figures 1B–E). Scorecard gene expression assay demonstrated that all iPS cell lines had gene expression associated with pluripotency and predicted differentiation capacity into neuroectoderm lineage (Supplementary Figure S1). Differentiation ability of iPS lines was confirmed by inducing embryoid body differentiation. iPS cell lines differentiated into cells positive for all germ layer markers, β III-tubulin, brachyury, and GATA-binding protein 4 (Figures 1F,G). No difference was seen in the pluripotency or differentiation ability between patient and control iPS cell lines (Figures 1E,G). No iPS cell lines had any chromosomal abnormality (Supplementary Figure S2A). The presence of SPAST mutations was confirmed after reprogramming by sequencing DNA from all patient iPS cell lines (Supplementary Figure S2B).
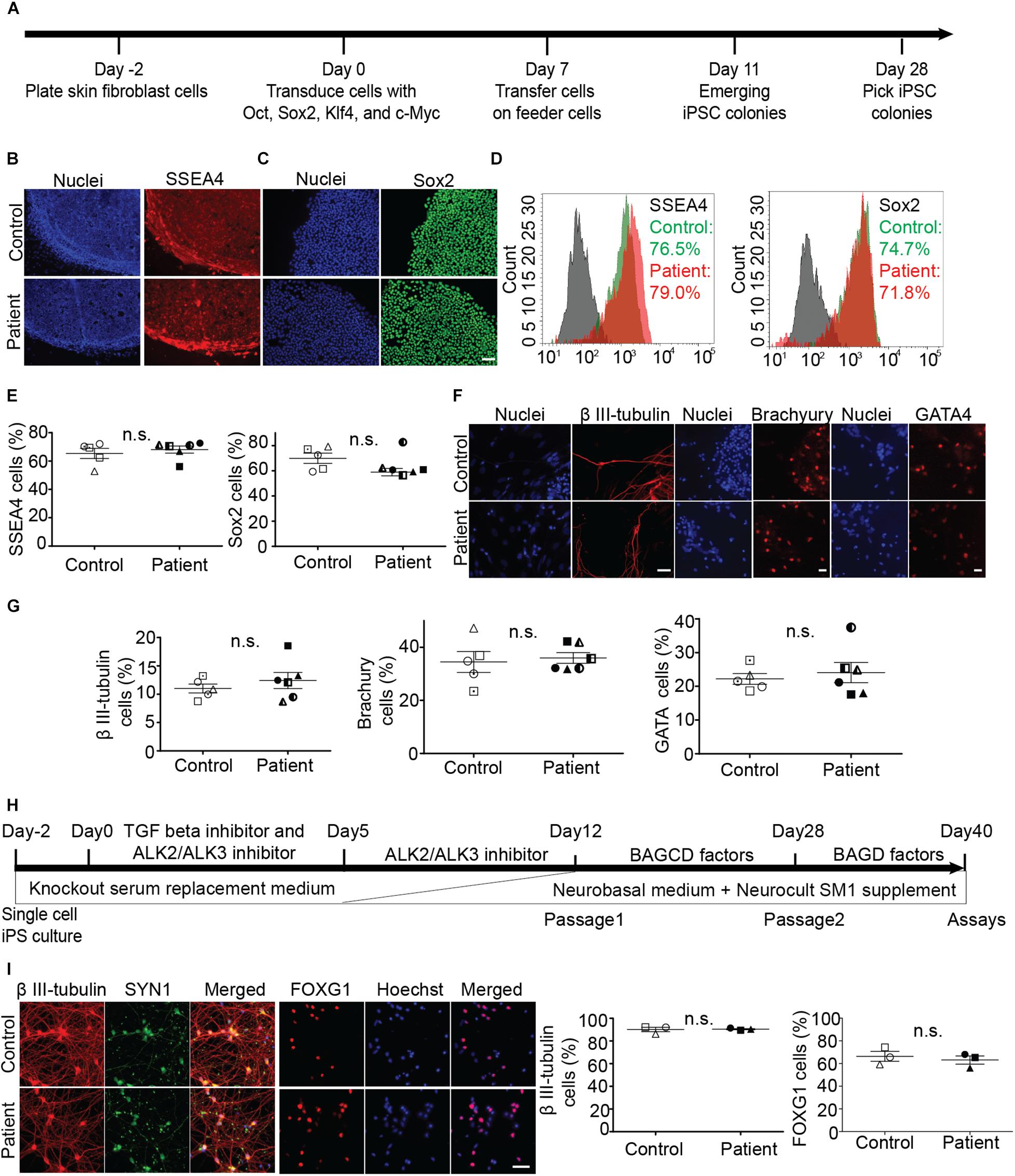
Figure 1. Generation and characterization of iPS cell lines and differentiation into neurons. (A) Timeline of reprogramming skin fibroblasts to iPS cells. (B–E) Expression of pluripotency markers SSEA-4 and Sox2 in patient and control iPS cells and group means (+/− SEM). (D) Flowcytometry based analysis of control and patient cells. (F,G) Embryoid body cells expressing three germ layer markers: β III-tubulin (ectoderm), brachyury (mesoderm), GATA4 (endoderm). (H) Timeline of iPS cell differentiation to forebrain neurons. Brain-derived neurotrophic factor (B), ascorbic acid (A), glial cell-derived neurotrophic factor (G), DAPT (D), and dibutyryl cAMP (C). (I) Expression of neuronal differentiation markers and quantification of neurons immuno-labeled for β III-tubulin and FOXG1 expression. Scale bar: 50 microns.
Differentiation of iPS Cell Lines Into Forebrain Neurons
Induced pluripotent stem cells were differentiated to forebrain neurons using a dual SMAD inhibition protocol (Figure 1H), previously described (Chambers et al., 2009). By day 40 of differentiation (Figure 1I), patient and control iPS cell lines generated neurons that were positive for β-tubulin type III, a neuronal marker; FOX G1, a forebrain neuron marker; and SYN1, a neuronal synaptic marker (Figure 1I). There was no difference in the differentiation ability between patient and control cell lines (Figure 1I).
Spastin Was Reduced in Patient Neurons
Compared to control neurons, patient neurons had reduced expression of spastin (Figure 2A). Western blot analysis showed that spastin expression in patient neurons was about 50% of control neurons (Figure 2B; control cells: 100 ± 8.59%, patient cells: 45.1 ± 2.89%, t = 6.54, df = 9, p = 0.0001).
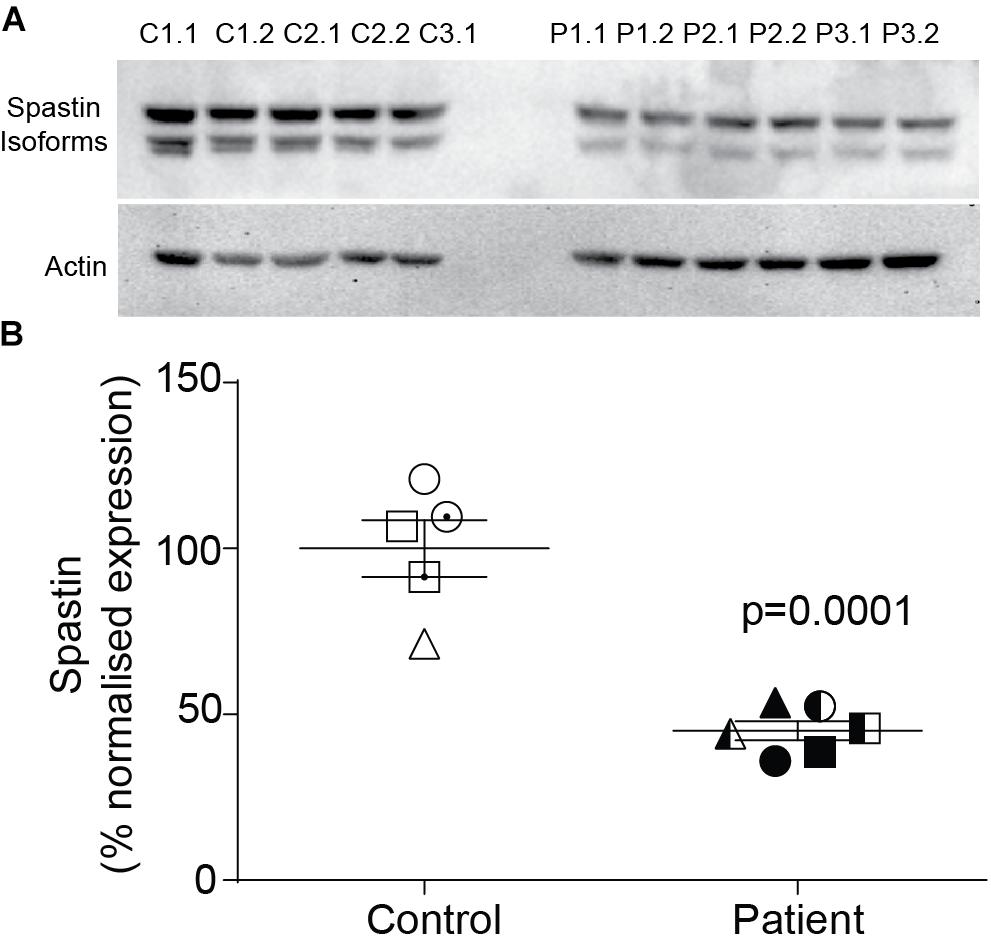
Figure 2. Spastin expression is reduced in neurons of SPAST patients. (A) Western blot analysis of spastin expression in patient and control neurons showing both isoforms. (B) Expression of spastin isoforms relative to actin.
Reduced Acetylated α-Tubulin Expression and Increased Axon Swellings in Patient Axons
Patient and control axons were identified as long processes immunostained for the neuron marker, β III-tubulin (Figures 3A,B). Levels of stabilized microtubules were quantified by measuring the fluorescence intensity of acetylated α-tubulin protein (Figures 3A‘,B‘). Compared to control axons, patient axons had reduced expression of acetylated α-tubulin (Figure 3C; control cells: 100 ± 8.76%, patient cells: 51.63 ± 5.06%, t = 4.991, df = 9, p = 0.0004).
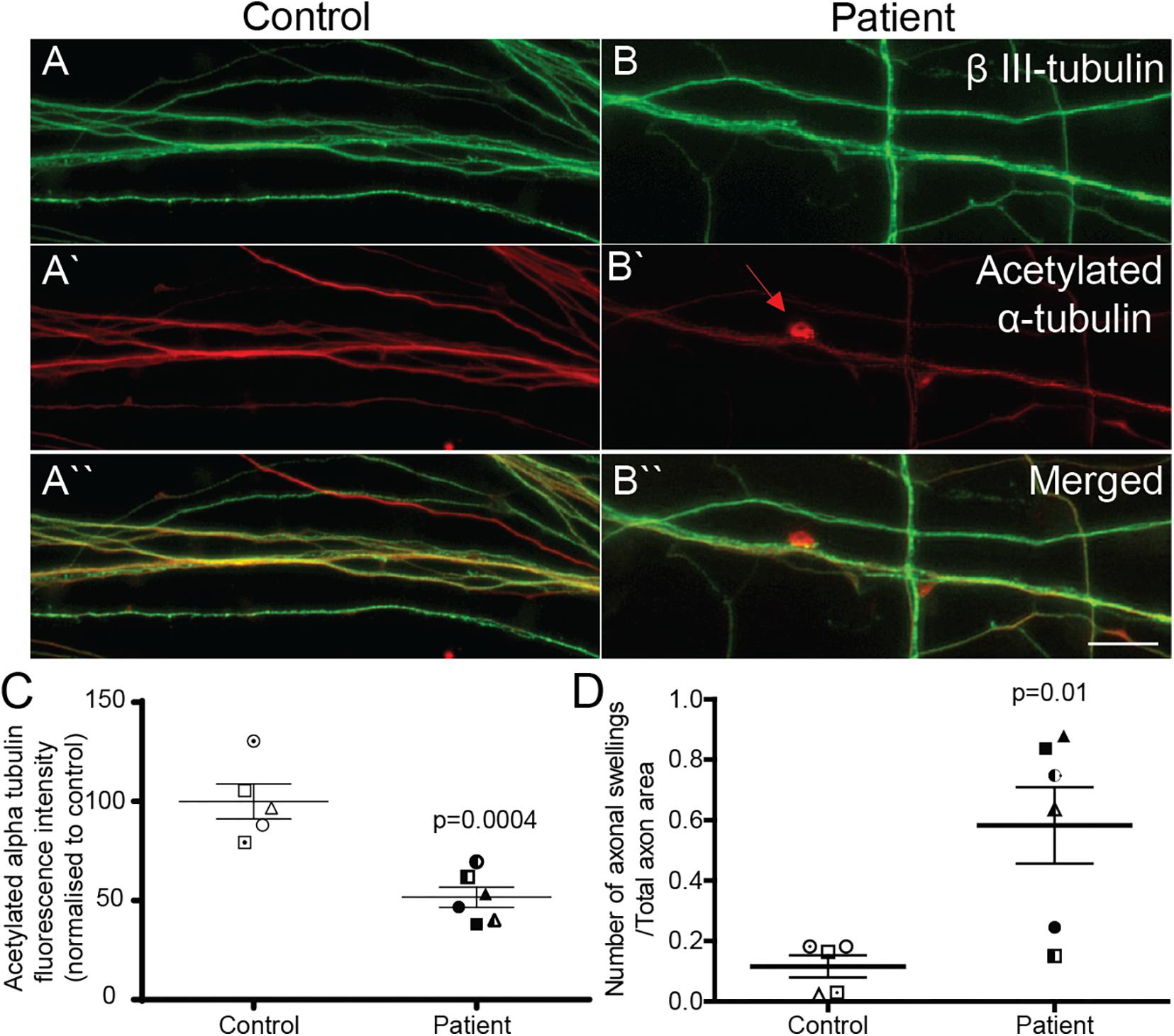
Figure 3. Acetylated α-tubulin expression is reduced and axon swellings are increased in SPAST patient neurons. Control (A,A‘,A“) and patient (B,B‘,B“) axons co-immunostained for β III-tubulin, neuronal marker, and acetylated α-tubulin. Scale bar = 100 μm. Arrows, axon swelling. (C) Quantification of fluorescence intensity of acetylated tubulin in control and patient axons. (D) Quantification of axon swelling density in control and patient axons.
Axon swellings in control axons were rarely observed (Figure 3A‘), but were often observed in patient axons (Figure 3B‘). Axon swelling density is the ratio of the number of axonal swellings to total axon area. The density of axon swellings was significantly greater in patient axons than in controls (Figure 3D; control axon swelling density: 0.1163 ± 0.037, patient axon swelling density: 0.5828 ± 0.1267, t = 3.240, df = 9, p = 0.01).
Reduced Number of Peroxisomes in Patient Axons
To test if patient axons have reduced number of organelles whose transport is dependent on microtubules, we quantified the number of peroxisomes in control and patient axons. Compared to control axons, patient axons had 28% fewer peroxisomes (Supplementary Figure S3; peroxisomes per control axon: 79.60 ± 8.524, peroxisomes per patient axon: 57.11 ± 6.907, t = 2.074, df = 9, p < 0.05).
Reduced Microtubule-Dependent Peroxisome Axon Transport
Peroxisome transport is essential to maintain axon integrity (Bottelbergs et al., 2010) as they regulate oxidative state via detoxification of hydrogen peroxide by catalase (Antonenkov et al., 2010). To test if peroxisome transport along patient axons is impaired, we performed live cell imaging and tracked their movement. Peroxisome movement along the axons was quantified using time-lapse imaging of GFP-labeled peroxisomes (Figures 4A–C). Peroxisomes exhibit local Brownian movements (indicated by peroxisome P1) and saltatory movements along the axon (indicated by peroxisome P2). The time-lapse movies were analyzed using automated image analysis software, IMARIS. Figure 4D shows peroxisomes identified as “spots” (red dots) and their paths of movement as identified by the analysis software. The speeds of peroxisome transport were calculated for about 1500 peroxisomes from six patient- and five control iPS cell lines differentiated neurons (∼130 peroxisomes per cell line).
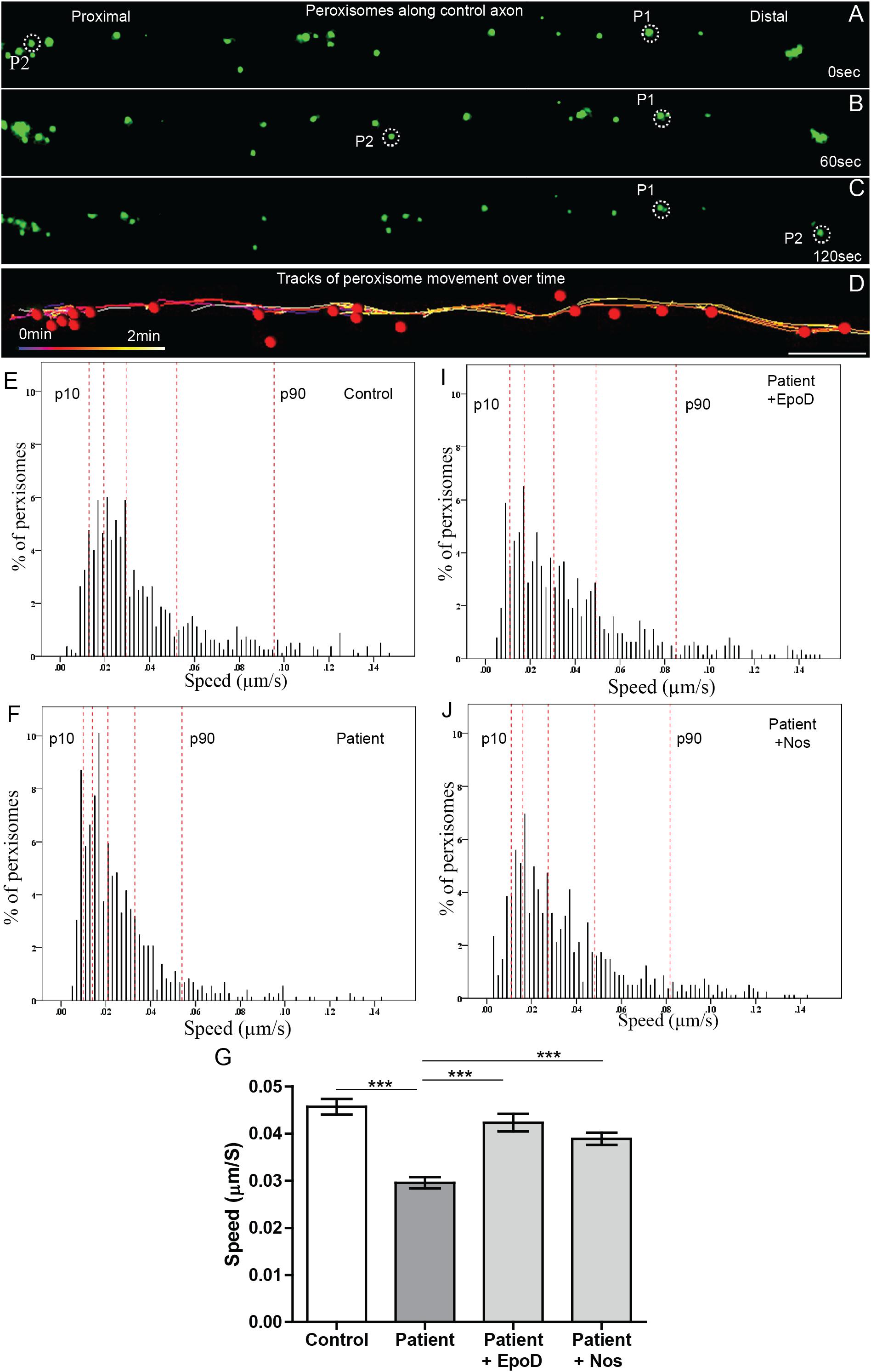
Figure 4. Peroxisome transport is reduced in SPAST patient axons and rescued by tubulin-binding drugs. (A,B,C) Time-lapse images of peroxisomes (green) in control axon at 0, 60, and 120 s (times shown on right). (D) Peroxisomes in A (red dots) and their paths of movement (colored lines) identified by the automated analysis software. Scale bar = 10 μm for all images. Frequency distributions of peroxisome speeds of: (E) untreated control group, (F) untreated patient group, (I) epothilone D-treated patient group, and (J) noscapine-treated patient group. The red dotted lines indicate the 10th, 25th, 50th, 75th, and 90th percentiles of the frequency distributions. (G) Means of peroxisome speed distributions. ***Different from the untreated patient group p < 0.0001.
Peroxisomes exhibited typical Brownian and saltatory movements as described in olfactory neural stem cells and other mammalian cells (Figures 4A–C and Supplementary Videos S1, S2) (Huybrechts et al., 2009; Wali et al., 2016). The frequency distribution of peroxisome speeds from patient and control axons had similar positively skewed non-normal distributions (skewness > 1) (Figures 4E,F), indicating that most peroxisomes traveled at slow speeds with few peroxisomes traveling at fast speeds. Fast peroxisome transport (with speeds above 90th percentile) is microtubule dependent (Wiemer et al., 1997; Wali et al., 2016). The average speed of peroxisome transport in patient axons was 35% less than in control axons (control peroxisome speed: 0.046 ± 0.001 μm/s; patient peroxisome speed: 0.028 ± 0.001 μm/s). Most of this difference appeared to be due to the reduced number of fast moving peroxisomes with concomitant increase in slow moving peroxisomes in patient cells, seen in the shift to the left of the frequency distribution of peroxisome speeds and the changes in the speeds represented by the percentiles (Figures 4E,F). Quantile regression analysis of the frequency distributions showed a significant reduction (p < 0.001) at all percentiles in mean peroxisome speeds in the patient group compared to control group (Table 1A) based on analysis of speeds of 715 peroxisomes from five control cell lines and 671 peroxisomes from six untreated patient cell lines. The biggest difference is seen in the fastest peroxisomes where the 90th percentile of the patient peroxisome speeds is approximately equivalent to the 75th percentile of the control peroxisome speeds (Figures 4E,F).
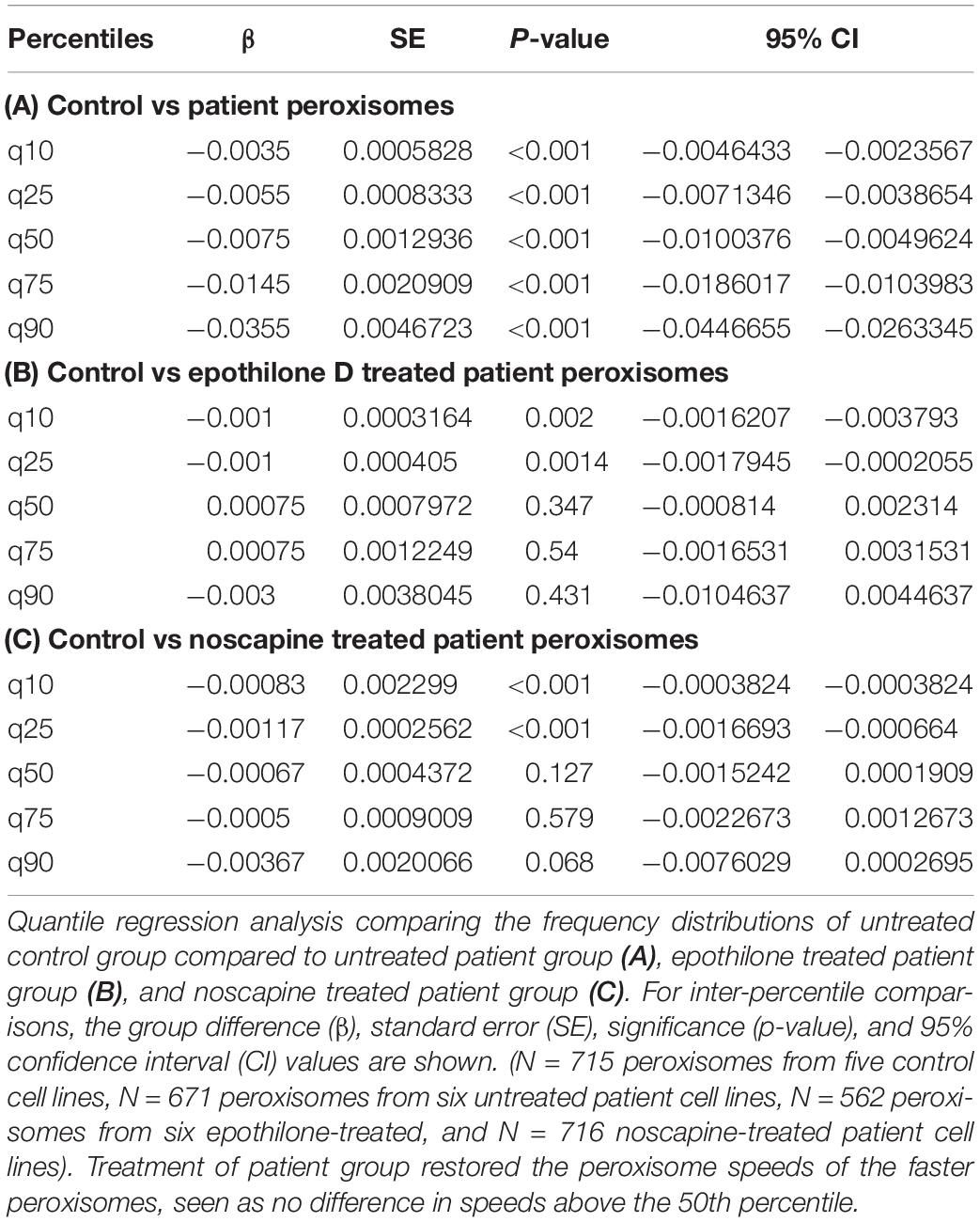
Table 1. Quantile regression of frequency distributions of peroxisome speeds.
Tubulin-Binding Drugs Rescued Peroxisome Transport in Patient Axons
To test if tubulin-binding drugs can rescue the reduced peroxisome axon transport in patient axons, patient axons were treated with tubulin-binding drugs epothilone D and noscapine and their peroxisome transport was compared to peroxisome transport of control axons. Both drug treatments significantly increased the average peroxisome speeds in patient axons back to control axon levels (p < 0.001) (Figures 4E–G,I,J). Drug treatments shifted frequency distributions of peroxisome speeds to the right, increasing the numbers of fast-moving peroxisomes after drug treatment to values similar to controls (Figure 4E). Quantile regression analysis of the frequency distributions showed a rightward shift in the 50th percentile and above, indicated by the lack of difference in these percentiles after drug treatment compared the controls (Tables 1B,C), based on analysis of speeds of 562 peroxisomes from six epothilone-treated and 716 peroxisomes from six noscapine-treated patient cell lines.
Patient Axons Were More Susceptible to Fragmentation Induced by Hydrogen Peroxide
It has been previously established that damage to the microtubule network of cells, particularly neurons, sensitizes the cell to further damage by oxidation (Fukui, 2016). To examine whether the microtubule defects observed here in SPAST patient neurons recapitulates this effect, we exposed the neurons to oxidative stress induced with hydrogen peroxide. Axon fragmentation was subsequently quantitated using an image-based assay, as previously described (Sasaki et al., 2009). Healthy axons are continuous, whereas degenerating axons are fragmented or beaded, and in this assay axon fragmentation is quantified and reported as the DI, which ranges from 0 (intact axons) to 1 (completely beaded/fragmented axons) (Sasaki et al., 2009). At baseline, the untreated control and untreated patient axons were intact (Figures 5A,B). After exposure to hydrogen peroxide, patient and control axons fragmented with fragmentation increasing with time (Figures 5C–H,I). A repeated measures analysis of variance showed a significant main effect of disease status (p = 0.0003) and a significant main effect for hydrogen peroxide treatment (p < 0.0001). Dunnett’s post hoc multiple comparison indicated a significant increase in axon degeneration by patient neurons over controls after exposure to hydrogen peroxide for 3 h (p < 0.01) and 6 h (p < 0.05).
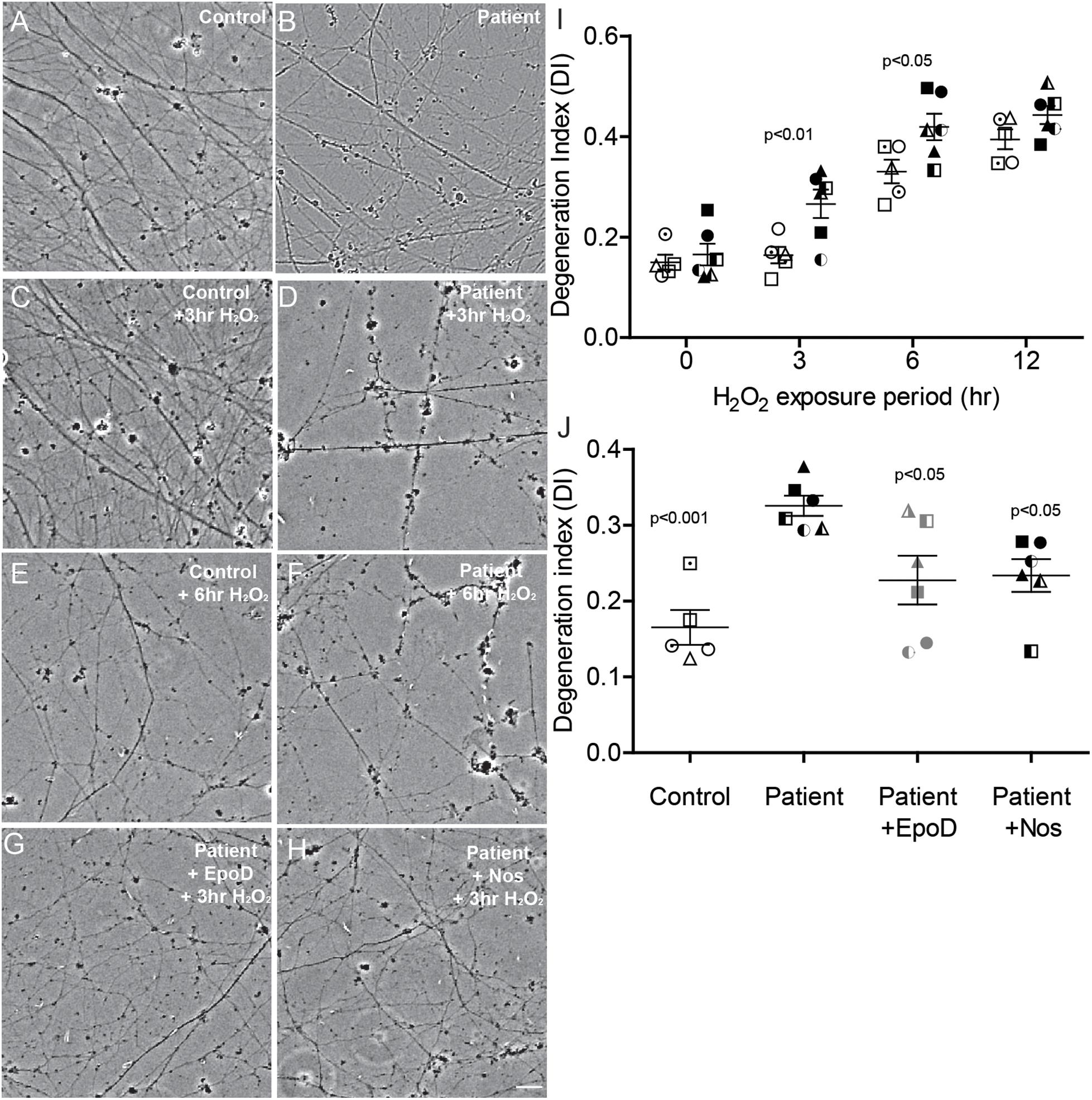
Figure 5. Axon degeneration under oxidative stress increased in SPAST patients and is rescued by tubulin-binding drugs. (A,B) Untreated control and patient axons. Control and patient axons exposed to 20 μM H2O2 for 3 h (C,D) and 6 h (E,F). Patient axons treated with epothilone D (G) or noscapine (H) for 24 h before exposing to 20 μM H2O2 for 3 h. Scale bar = 25 μm. I: Degeneration index (DI) at different periods of treatment H2O2 exposure. Patient DIs (filled symbols) were significantly different from control DIs (open symbols) at 3 and 6 h. (J) DIs of untreated controls (open symbols) and untreated patients (filled symbols) and patient cultures treated with epothilone D or noscapine for 24 h prior to 3 h exposure to 20 μM H2O2. Statistics shown for differences between untreated patient axons and other groups.
Tubulin-Binding Drugs Reduced Hydrogen Peroxide-Induced Axon Fragmentation in Patient Axons
Patient cultures treated with epothilone D or noscapine for 24 h before exposure to hydrogen peroxide for 3 h had axon fragmentation similar to untreated controls (Figures 5A,G,H). DI was significantly greater in untreated patient axons than in untreated control axons (DI: 0.326 ± 0.013 vs 0.165 ± 0.023, respectively; Figure 5J). Treatment with epothilone D and noscapine decreased DI in patient axons toward untreated control axon levels (Figure 5J). ANOVA indicated a significant effect of treatment (p < 0.01). Dunnett’s post hoc multiple comparison tests indicate that untreated patient axon degeneration was significantly greater than control axon degeneration (p < 0.001). DI of patient axons was significantly reduced by treatment with epothilone D (p < 0.05) and noscapine (p < 0.05).
Discussion
We show here that iPS cell-induced neurons from a variety of SPAST HSP patients carrying different mutations, including frameshift, missense, and exonic deletion mutations, share common axon pathologies of reduced stabilized microtubules, increased density of axon swellings, reduced microtubule-dependent peroxisome transport, and reduced number of peroxisomes. Patient axons did not show signs of degeneration, evaluated by quantifying axon fragmentation under basal conditions. However, patient axons fragmented significantly more than controls following hydrogen peroxide exposure, suggesting for the first time that the SPAST patient axons are more sensitive than controls to the deleterious effects of oxidative stress.
Our finding of reduced axonal transport in iPS-neurons derived from SPAST patients concurs with findings of studies involving post-mortem spinal cord analysis (Kasher et al., 2009) and iPS-neurons (Denton et al., 2014; Havlicek et al., 2014), showing that we were able to recapitulate HSP pathology in our neurons. Patient axons have less spastin, encoded by SPAST. This effect is commonly seen in patient-derived iPS-neurons (Denton et al., 2014; Havlicek et al., 2014) and ONS cells (Abrahamsen et al., 2013) and presumed to be a haploinsufficiency effect (Abrahamsen et al., 2013). Since spastin is involved in maintaining microtubule dynamics, reduced spastin is likely to affect cellular processes dependent on microtubules including organelle transport. Consistent with this, patient axons have reduced mean speed of peroxisome transport compared to controls. The reduced mean speed of peroxisome transport was a consequence of reduced number of fast microtubule-dependent peroxisome movements, as treatment of patient axons with tubulin-binding drugs, increased the number of fast microtubule-dependent peroxisome movements and subsequently increased the mean peroxisome transport speeds in patient axons to levels comparable with control axons, thus rescuing the axonal transport deficit. Also using patient ONS cells, we have previously shown that the same concentration of tubulin-binding drugs epothiloneD (2 nM) and noscapine (10 μM) used here increase mean speed of peroxisome transport by increasing the levels of stabilized microtubules (Fan et al., 2014). It is possible that both the tubulin-binding drugs may have independent mechanisms by which they increase peroxisome transport speed and increasing levels of stabilized microtubules is one possibility. For example, organelle transport speed can be increased by reintegration of microtubule lattice with drug treatment in addition to increased stabilized microtubules (Kan et al., 2011).
It has been previously established that damage to the microtubule network of cells, particularly neurons, sensitizes the cell to further damage by oxidation (Fukui, 2016). Is this the case with SPAST patient neurons? Oxidative stress arises from a significant increase in concentrations of reactive oxygen species and/or a decrease in the detoxification mechanisms. Coincidently, both cellular organelles, peroxisomes and mitochondria, whose transport is shown to be reduced in SPAST HSP, here and elsewhere (Abrahamsen et al., 2013; Denton et al., 2014; Havlicek et al., 2014), are involved in maintaining reactive oxygen species levels in cells. It is likely that slow or reduced transport of peroxisomes/mitochondria could limit the availability of these organelles, particularly at the axon distal ends, making them unavailable to manage increased reactive oxygen species when necessary. Consistent with this, exposure to hydrogen peroxide induced axon fragmentation in both patient and control axons; however, patient axons displayed significantly increased fragmentation. There was no difference between the patient and control axons at baseline, showing that oxidative stress can have deleterious effects on SPAST patient axons. Treatment of patient axons with the same concentrations of epothilone D and noscapine drugs that rescued the axonal transport deficit also protected the patient axons from oxidative stress-induced increased fragmentation showing for the first time that SPAST patient axons are vulnerable to oxidative stress-induced degeneration as a consequence of reduced axonal transport.
We show here that reduced axon transport and oxidative stress-induced axonal fragmentation are common axon pathologies in iPS-neurons derived from a variety of SPAST patients carrying different mutations. This understanding is relevant when testing drug candidates to rescue axon pathologies. A common drug treatment may be effective across HSP patients carrying different SPAST mutations, as seen here with both epothilone D and noscapine. We believe this is an extremely promising strategy to find a candidate drug that can rescue SPAST related axon pathologies in vitro.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
Our study involving human participants was reviewed and approved by the Human Research Ethics Committee affiliated to the Northern Sydney Local Health District, New South Wales government, Australia. The ethics committee reference number: RESP/15/314. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
GW, AM-S, and CS conceived and designed the experiments and wrote the main manuscript text. GW generated and charecterized all the iPS cell lines, differentiated iPS cells to forebrain neurons, and performed all assays described in the manuscript. EL provided assistance with the western blot assay. NB provided advice and assistance with iPS generation and charecterization. J-SP performed the DNA sequencing experiments. RS assisted with live cell peroxisome transport assay data. GW, AM-S, and RS performed the statistical analysis. AM-S, CS, and GW provided funding for the research. All authors contributed to and edited the manuscript.
Funding
This work was supported by grants to AM-S, CS, and GW from the Hereditary Spastic Paraplegia Research Foundation Inc. and the Spastic Paraplegia Foundation Inc. CS was supported by an NHMRC Practitioner Fellowship (App1136800).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We express our thanks to the patients who consented to contribute to this study. We thank Dr. Daniel R. Whiten for critically reading the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2020.00401/full#supplementary-material
References
Abraham, S., Sheridan, S. D., Laurent, L. C., Albert, K., Stubban, C., Ulitsky, I., et al. (2010). Propagation of human embryonic and induced pluripotent stem cells in an indirect co-culture system. Biochem. Biophys. Res. Commun. 393, 211–216. doi: 10.1016/j.bbrc.2010.01.101
Abrahamsen, G., Fan, Y., Matigian, N., Wali, G., Bellette, B., Sutharsan, R., et al. (2013). A patient-derived stem cell model of hereditary spastic paraplegia with SPAST mutations. Dis. Model. Mech. 6, 489–502. doi: 10.1242/dmm.010884
Antonenkov, V., Grunau, S., Ohlmeier, S., and Hiltunen, J. (2010). Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 13, 525–537. doi: 10.1089/ars.2009.2996
Barlan, K., and Gelfand, V. I. (2017). Microtubule-based transport and the distribution, tethering, and organization of organelles. Cold Spring Harb. Perspect. Biol. 9:a025817. doi: 10.1101/cshperspect.a025817
Bilsland, L. G., Sahai, E., Kelly, G., Golding, M., Greensmith, L., and Schiavo, G. (2010). Deficits in axonal transport precede ALS symptoms in vivo. Proc. Natl. Acad. Sci. U.S.A. 107, 20523–20528. doi: 10.1073/pnas.1006869107
Bottelbergs, A., Verheijden, S., Hulshagen, L., Gutmann, D. H., Goebbels, S., Nave, K. A., et al. (2010). Axonal integrity in the absence of functional peroxisomes from projection neurons and astrocytes. Glia 58, 1532–1543. doi: 10.1002/glia.21027
Chambers, S. M., Fasano, C. A., Papapetrou, E. P., Tomishima, M., Sadelain, M., and Studer, L. (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. doi: 10.1038/nbt.1529
Denton, K. R., Lei, L., Grenier, J., Rodionov, V., Blackstone, C., and Li, X.-J. (2014). Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 32, 414–423. doi: 10.1002/stem.1569
Duning, T., Warnecke, T., Schirmacher, A., Schiffbauer, H., Lohmann, H., Mohammadi, S., et al. (2010). Specific pattern of early white-matter changes in pure hereditary spastic paraplegia. Mov. Disdord. 25, 1986–1992. doi: 10.1002/mds.23211
Errico, A., Ballabio, A., and Rugarli, E. (2002). Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum. Mol. Genet. 11, 153–163. doi: 10.1093/hmg/11.2.153
Fan, Y., Wali, G., Sutharsan, R., Bellette, B., Crane, D. I., Sue, C. M., et al. (2014). Low dose tubulin-binding drugs rescue peroxisome trafficking deficit in patient-derived stem cells in hereditary spastic paraplegia. Biol. Open 3, 494–502. doi: 10.1242/bio.20147641
Fang, C., Bourdette, D., and Banker, G. (2012). Oxidative stress inhibits axonal transport: implications for neurodegenerative diseases. Mol. Neurodegen. 7:29. doi: 10.1186/1750-1326-7-29
Fukui, K. (2016). Reactive oxygen species induce neurite degeneration before induction of cell death. J. Clin. Biochem. Nutr. 59, 155–159. doi: 10.3164/jcbn.16-34
Havlicek, S., Kohl, Z., Mishra, H., Prots, I., Eberhardt, E., Denguir, N., et al. (2014). Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 23, 2527–2541. doi: 10.1093/hmg/ddt644
Huybrechts, S. J., Van Veldhoven, P. P., Brees, C., Mannaerts, G. P., Los, G. V., and Fransen, M. (2009). Peroxisome dynamics in cultured mammalian cells. Traffic 10, 1722–1733. doi: 10.1111/j.1600-0854.2009.00970.x
Kan, R., Yurttas, P., Kim, B., Jin, M., Wo, L., Lee, B., et al. (2011). Regulation of mouse oocyte microtubule and organelle dynamics by PADI6 and the cytoplasmic lattices. Dev. Biol. 350, 311–322. doi: 10.1016/j.ydbio.2010.11.033
Kasher, P., De Vos, K., Wharton, S., Manser, C., Bennett, E. J., Bingley, M., et al. (2009). Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem. 110, 34–44. doi: 10.1111/j.1471-4159.2009.06104.x
Maroof, A. M., Keros, S., Tyson, J. A., Ying, S. W., Ganat, Y. M., Merkle, F. T., et al. (2013). Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12, 559–572. doi: 10.1016/j.stem.2013.04.008
McDermott, C., Grierson, A., Wood, J., Bingley, M., Wharton, S. B., Bushby, K. M., et al. (2003). Hereditary spastic paraparesis: disrupted intracellular transport associated with spastin mutation. Ann. Neurol. 54, 748–759. doi: 10.1002/ana.10757
McDermott, C., White, K., Bushby, K., and Shaw, P. (2000). Hereditary spastic paraparesis: a review of new developments. J. Neurol. Neurosurg. Psychiatry 69, 150–160. doi: 10.1136/jnnp.69.2.150
Orlacchio, A., Kawarai, T., Totaro, A., Errico, A., St George-Hyslop, P. H., Rugarli, E. I., et al. (2004). Hereditary spastic paraplegia: clinical genetic study of 15 families. Arch. Neurol. 61, 849–855.
Ruano, L., Melo, C., Silva, M. C., and Coutinho, P. (2014). The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42, 174–183. doi: 10.1159/000358801
Salinas, S., Proukakis, C., Crosby, A., and Warner, T. (2008). Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 7, 1127–1138. doi: 10.1016/S1474-4422(08)70258-8
Sasaki, Y., Vohra, B. P. S., Lund, F. E., and Milbrandt, J. (2009). Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J. Neurosci. 29, 5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009
Vandebona, H., Kerr, N., Liang, C., and Sue, C. (2012). SPAST mutations in Australian patients with hereditary spastic paraplegia. Int. Med. J. 42, 1342–1347. doi: 10.1111/j.1445-5994.2012.02941.x
Wali, G., Sutharsan, R., Fan, Y., Stewart, R., Velasquez, J. T., Sue, M. C., et al. (2016). Mechanism of impaired microtubule-dependent peroxisome trafficking and oxidative stress in SPAST-mutated cells from patients with hereditary spastic paraplegia. Sci. Rep. 6:27004. doi: 10.1038/srep27004
Wharton, S. B., McDermott, C. J., Grierson, A. J., Wood, J. D., Gelsthorpe, C., Ince, P. G., et al. (2003). The cellular and molecular pathology of the motor system in hereditary spastic paraparesis due to mutation of the spastin gene. J. Neuropathol. Exp. Neurol. 62, 1166–1177. doi: 10.1093/jnen/62.11.1166
Keywords: hereditary spastic paraplegia, SPAST, axon transport, peroxisomes, axon degeneration, epothilone D, noscapine
Citation: Wali G, Liyanage E, Blair NF, Sutharsan R, Park J-S, Mackay-Sim A and Sue CM (2020) Oxidative Stress-Induced Axon Fragmentation Is a Consequence of Reduced Axonal Transport in Hereditary Spastic Paraplegia SPAST Patient Neurons. Front. Neurosci. 14:401. doi: 10.3389/fnins.2020.00401
Received: 14 November 2019; Accepted: 01 April 2020;
Published: 07 May 2020.
Edited by:
Vincenzo La Bella, University of Palermo, ItalyReviewed by:
Giorgio Casari, Vita-Salute San Raffaele University, ItalyAntonio Orlacchio, Santa Lucia Foundation (IRCCS), Italy
Copyright © 2020 Wali, Liyanage, Blair, Sutharsan, Park, Mackay-Sim and Sue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gautam Wali, Z2F1dGFtLndhbGlAc3lkbmV5LmVkdS5hdQ==; Carolyn M. Sue, Y2Fyb2x5bi5zdWVAc3lkbmV5LmVkdS5hdQ==
†These authors share senior authorship