 Emily F. Ozdowski
Emily F. Ozdowski Helena Abbott
Helena Abbott- Department of Biology, Duke University, Durham, NC, United States
Neurodegenerative mechanisms due to mutations in spastin currently center on neuronal defects, primarily in microtubule and endomembrane regulation. Spastin loss in Drosophila larvae compromises neuronal microtubule distribution, alters synaptic bouton morphology, and weakens synaptic transmission at glutamatergic neuromuscular junction (NMJ) synapses. Pak3, a p21-activated kinase that promotes actin polymerization and filopodial projections, is required for these spastin mutant defects; animals lacking both genes have normal NMJs. Here we show that Pak3 is expressed in central and peripheral glial populations, and reduction of Pak3 specifically in subperineurial glial cells is sufficient to suppress the phenotypes associated with spastin loss. Subperineurial glia in the periphery ensheathe motor neuron axons and have been shown to extend actin-based projections that regulate synaptic terminals during normal NMJ development. We find that these subperineurial glial projections are Pak3-dependent and nearly twice as frequent in spastin mutants, while in Pak3, spastin double mutants, neither glial projections nor synaptic defects are observed. Spastin deficiency thus increases Pak3-dependent subperineurial glia activity, which is in turn required for neuronal defects. Our results demonstrate a central role for Pak3-mediated, altered glial behavior in the neuronal defects due to spastin loss, and suggest that a similar reactive glia-mediated mechanism may underlie human AD-HSP pathogenesis.
Introduction
The identification of human SPAST as the most common gene mutated in Autosomal-Dominant Hereditary Spastic Paraplegia two decades ago was a major advance, enabling multiple model systems to be leveraged toward understanding the requirement for Spastin in nervous system function (Hazan et al., 1999). Spastin likely serves to coordinately regulate the microtubule cytoskeleton and membrane components (reviewed in Lumb et al., 2012), and although clear consensus is still in progress, several working hypotheses have emerged for its neuronal roles. Potential processes include neural stem cell proliferation (Jeong et al., 2019), remodeling the endoplasmic reticulum and other endomembrane components (e.g., Park et al., 2010; Allison et al., 2019), lipid droplet function (Papadopoulos et al., 2015; Chang et al., 2019), axon regeneration (Stone et al., 2012), axon outgrowth (Wood et al., 2006), axon transport (Tarrade et al., 2006; Kasher et al., 2009), and synaptic morphology and transmission (Sherwood et al., 2004; Trotta et al., 2004).
Drosophila spastin was identified in a forward screen for genes involved in nervous system development (Sherwood et al., 2004). Just as SPAST mutations impair the longest axons of the central nervous system in humans, diminishing patient mobility, homozygous deletion of Drosophila spastin (the spastin5.75 allele) impairs mobility in adult flies, with the distal-most limbs also appearing the weakest. These adult flies are rarely viable, and although homozygous null larvae appear healthy, electrophysiological analysis of the larval neuromuscular junction (NMJ), a well-established model for vertebrate glutamatergic central synapses (Collins and DiAntonio, 2007), reveals reduced synaptic transmission due to defects at the presynaptic terminal (Sherwood et al., 2004; Ozdowski et al., 2011). This functional weakening is accompanied by distinctive changes in presynaptic arbor morphology. In contrast to wild type NMJs, spastin5.75 larvae have more highly branched axon terminal arbors, with smaller and more numerous synaptic boutons that often form bunched, rather than linear, arrays. Subcellularly, the normally continuous microtubule cytoskeleton appears sparse or undetectable in mutant terminal boutons, suggesting that Spastin’s microtubule severing activity is required for the generation and/or penetration of microtubules into distal sites. Consistent with this model, recent in vitro studies have shown that Spastin promotes stabilization and subsequent growth of severed microtubules, in addition to its well-established severing activity (Vemu et al., 2018; Kuo et al., 2019). These subcellular, morphological, and functional phenotypes in spastin mutants are all significantly rescued by low-level, neuron-specific expression of fly – or human – wild type Spastin, demonstrating that Spastin function is well-conserved between flies and humans, and is required within neurons (Sherwood et al., 2004; Du et al., 2010).
To understand the molecular and cellular mechanisms underlying the consequences of spastin loss, we executed a forward genetic screen for modifiers of spastin mutant phenotypes and identified Pak3, a member of the p21-activated serine/threonine kinase family (Ozdowski et al., 2011). Pak proteins are conserved from amoebae to humans, and typically activated downstream of Rac and Cdc42 small GTPase signaling, leading to alterations in the actin cytoskeleton and phosphorylation of a wide range of other protein targets (reviewed in Kumar et al., 2017; Semenova and Chernoff, 2017). The Paks are particularly well-studied because of their demonstrated roles inducing cell motility, invasion, and metastasis in several cancers, as well as contributing to cardiac and neurological disorders. Humans have six PAK genes (PAK 1–6), segregated into two structurally distinct groups. The group I PAKs, characterized by highly conserved autoinhibitory (AID) and kinase domains, form inactive dimers that depend upon Rac or Cdc42 binding for relief of autoinhibition and subsequent kinase activity. The group II PAKs lack an AID and do not require RhoGTPase binding for kinase activity. Of the proteins encoded by the three Drosophila Pak genes, Pak, mushroom bodies tiny (mbt), and Pak3, Pak is orthologous to the group I vertebrate Paks, mbt falls clearly into the group II subfamily, and Pak3, while less similar than Pak, is also considered a group I Pak due to its clear AID and ∼60% amino acid identity in the kinase domain (Mentzel and Raabe, 2005). Both overexpression and knockdown of Drosophila Pak3 in various cell lines alters actin distribution and cell motility (Mentzel and Raabe, 2005; Asano et al., 2009; Ribeiro et al., 2014). In vivo, Pak3 is important in dorsal closure (Bahri et al., 2010), epidermal wound healing (Baek et al., 2012), podosome invasion during myoblast fusion (Duan et al., 2012), and border cell migration during oogenesis (Felix et al., 2015), all processes that require rearrangements of the actin cytoskeleton and changes in cell motility downstream of Rac signaling.
At the larval NMJ, ubiquitous, genetic loss of Pak3 does not significantly affect synapse form or function; however, Pak3 reduction in a spastin deficient background dramatically rescues the defects that characterize spastin mutants at this stage (Ozdowski et al., 2011). NMJ synapses in Pak3, spastin double mutants exhibit near-wild type morphology, with large and linearly-arrayed synaptic boutons. Subcellularly, continuity of the microtubule cytoskeleton within boutons is restored, and functionally, synaptic strength is indistinguishable from wild type levels. Absence of Pak3 thus renders these neurons resistant to spastin loss.
Given this striking rescue, we sought to understand how the presence of Pak3 confers spastin mutant phenotypes. We examined the Pak3 expression pattern and used tissue-specific knockdown to determine the cells in which it acts when spastin is lost. Surprisingly, Pak3 is not required in the motor neurons where Spastin functions, but rather, in the peripheral glia that ensheathe them. These glia provide essential support to the motor neurons, including extending filopodial projections that regulate the neuronal bouton arbor during development (Fuentes-Medel et al., 2009; Brink et al., 2012). We find that Pak3 promotes glial projections at the NMJ. Furthermore, in spastin mutants these glial projections are more numerous, while reduction of glial Pak3 suppresses both the projections and spastin mutant bouton morphology. Together, these data support a model in which spastin loss leads to increased Pak3-mediated glial activity, changing glial behavior such that they become toxic, rather than supportive, to synaptic terminals. Increasing evidence of a central role for reactive glial behavior in the progression of several major neurodegenerative diseases (Liddelow and Barres, 2017) makes these results particularly intriguing, as it suggests that pathogenesis in AD-HSP may be similarly mediated by reactive, toxic changes in a normally supportive subpopulation of glia.
Materials and Methods
Drosophila Stocks and Sources
Stocks and crosses were maintained on standard molasses-based food (Archon Scientific), except for experiments using the flower (fwe) loss of function alleles, fweDB56 and fweDB25 (gift of H. Bellen), which were raised on yeasted grape juice plates to facilitate recovery of the desired genotypes. All larvae and adults assayed were from crosses kept at 25°C. Unless otherwise specified, controls were w1118 flies, the common genetic background for transgenic strains; their NMJs are indistinguishable from wild type flies so are also referred to here as “wild type.” Driver lines were repo-GAL4 (gift of M. Freeman), gliotactin (gli)-GAL4 (gift of V. Auld), e22c-Gal4 spaghetti-squash (sqh)-GAL4 (gift of D. Kiehart), breathless (btl)-GAL4 (gift of M. Metzstein), and elavC155-GAL4, Mef2-GAL4, Mhc-GAL4, and nSyb-GAL4 (all from the Bloomington Drosophila Stock Center, BDSC). Pak3 alleles were PBac{RB}Pak3e00329, P{XP}Pak3d02472 (abbreviated Pak3d in Figures 3, 4; BDSC), GAL4 insertion line P{GawB}Pak3NP4472 (Kyoto Drosophila Genomics and Genetic Resources Center), and RNAi lines Pak3NIG.14895R–2 (National Institute of Genetics, NIG-FLY), Pak3GL00287 (P{TRiP.GL00287}attB, BDSC), and Pak3v39844 (P{GD8481}v39844, Vienna Drosophila Resource Center). Generation of the deletion allele Df(3R)Pak3 (abbreviated Pak3Df in Figures 3, 4), was previously described (Ozdowski et al., 2011). UAS-Pak3::GFP constructs (gift of B. Baum) were injected into Drosophila embryos and two lines established: one insertion on chromosome II, UAS-Pak350::GFP, and one on chromosome III, UAS-Pak332::GFP. Tissue-specific expression of UAS-Pak350::GFP rescues Pak3 mutant adult lethality, consistent with the transgene producing functional Pak3 protein (E.F.O., unpublished results). UAS-Pak332::GFP was used for Pak3 overexpression in glia. Tissue-specific GFP was expressed using UAS-mCD8::GFP or UAS-nls::GFP flies (BDSC).
Immunofluorescence
Wandering third-instar larvae were dissected in room temperature Phosphate Buffered Saline (PBS, Invitrogen) or HL3 Ringer’s medium without Ca2+ for < 20 min., or when quantifying glial projections, in HL3 Ringer’s containing 1.5 mM Ca2+ for <10 min. prior to fixation. Filets were then fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 30 min., washed in PBS with 0.2% Triton X-100 (PBST), and blocked in PBST with 5% normal goat serum, 0.01% bovine serum albumin, and 0.01% sodium azide for up to 2 h at room temperature. Filets were incubated overnight at 4°C in primary antibody diluted in block. Primary antibodies used were mouse polyclonal αPak3 (gift of S. Bahri, 1:250), rabbit αHRP (Jackson, 1:100), mouse αDLG (4F3 supernatant, Developmental Studies Hybridoma Bank [DSHB], 1:100), rat αElav (7E8A10, DSHB, 1:100), mouse αSlit (C555.6D, DSHB, 1:50), and mouse (mAb 3E6) or rabbit αGFP (Invitrogen, 1:300). Filets were then washed in PBST and incubated with the species-appropriate Alexa Fluor 488 or 568 secondary antibodies (Invitrogen) diluted 1:300 in block, for 2 h at room temperature or overnight at 4°C. After further washing, filets were mounted in Vectashield (Vector Laboratories) or ProLong Antifade (ThermoFisher).
Synaptic Bouton and Glial Quantification and Imaging
Slide-mounted filets were scored with the experimenter blinded to larval genotypes, using an inverted fluorescence compound microscope (Zeiss Axiophot with a 63X, 1.2NA oil immersion objective or Zeiss Axio Imager with a 100X, 1.4NA oil immersion objective). Representative confocal images were obtained using a Zeiss 880 Airyscan confocal microscope and 63X, 1.2NA oil immersion or a 20X, 0.8NA dry objective. Images were false-colored magenta in Adobe Photoshop to aid visualization by color blind readers. Abdominal muscle 4 was identified for each hemisegment and type 1b synaptic bouton numbers were recorded. Type 1b bouton identity was confirmed by double staining with anti-DLG whenever possible (in all larvae for which anti-GFP immunostaining was not needed); otherwise, they were distinguished from type 1s boutons based on size and branch morphology. Terminal boutons, a measurement of synaptic arbor branching, were defined as any synaptic bouton with only one connection. This included all termini along the arbor and at the distal tips, regardless of size. Subperineurial glial projections were visualized using anti-GFP to visualize gliotactin-GAL4-driven expression of UAS-mCD8::GFP. Glial morphology at each main axon branch was characterized as (1) blunt, ending at synaptic boutons, (2) broad lamellipodia extending over synaptic boutons, (3) long, thin filopodia (gliopods) extending over synaptic boutons, away from the axon, or to neighboring axons, or (4) rounded structures (gliobulbs) that resemble boutons. Numbers of gliopods and gliobulbs were combined as a measure of glial projections. Glial and bouton counts from individual hemisegments were averaged for each larva and all values plotted as bee-swarm superplots using GraphPad Prism (Lord et al., 2020). Statistical significance of larval means was determined by two-tailed Student’s t-test in Microsoft Excel (Figure 2 and Supplementary Figure S1) or one-way ANOVA with Sidak’s post hoc test for multiple comparisons in GraphPad Prism (Figures 3, 4). P-values are denoted as ns for p > 0.05, ∗ for 0.01 < p ≤ 0.05, ∗∗ for 0.001 < p ≤ 0.01, and ∗∗∗ for p ≤ 0.001.
Results
Pak3 Is Expressed in Larval Glia
To begin to identify the specific cells in which Pak3 loss prevents spastin mutant defects, we used a polyclonal antibody directed against the Drosophila Pak3 protein (kind gift of Sami Bahri; Bahri et al., 2010) to determine its endogenous expression pattern in larvae. Anti-Pak3 staining in wild type controls was strongly detected in the ventral nerve cord (VNC, analogous to the spinal cord of vertebrates) in a dashed pattern characteristic of midline glial cells (Figure 1A, arrow). Immunofluorescence was also seen in a thin layer on the surface of the larval brain, and along axonal projections emanating from the brain and ventral nerve cord (Figure 1A, arrowheads). The strength of the antibody signal correlated with predicted protein expression levels consistent with RNA disruption in different Pak3 alleles, supporting the specificity of the antibody for Pak3 (compare Figures 1A–C).
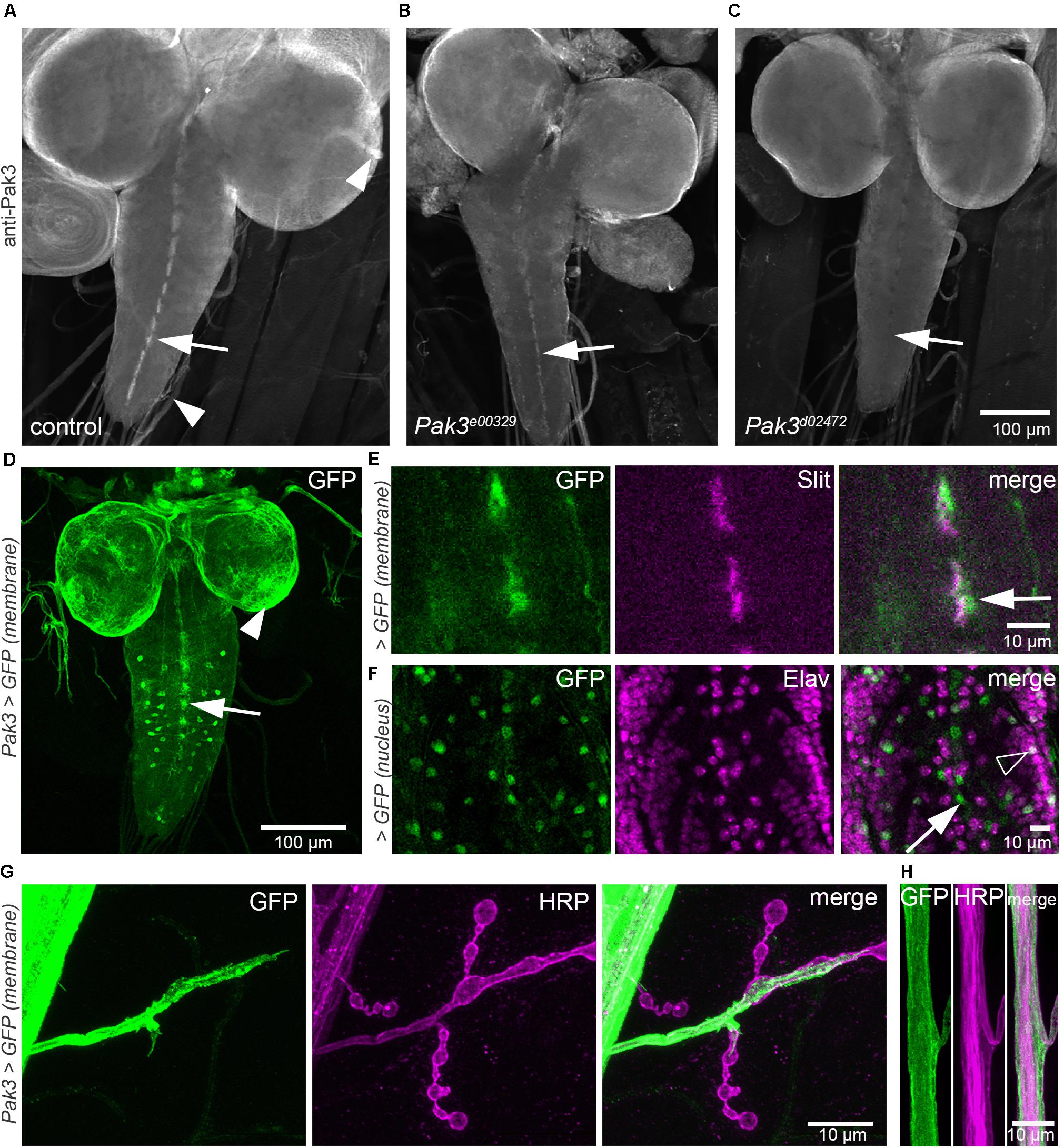
Figure 1. Pak3 is expressed in larval glia. (A–C) An anti-Pak3 polyclonal antibody reveals immunostaining consistent with glial cell expression: in larval ventral nerve cord (VNC) midline cells (A, arrow), along axonal projections emanating from the brain and VNC (A, arrowheads), and on the surface of the CNS. Expression is reduced in Pak3e00329 mutants (B) and almost undetectable in Pak3d02472 mutants (C), consistent with the severity of RNA disruption in these alleles and thus supporting antibody specificity. (D) A second method of observing Pak3 expression using the GAL4-UAS reporter system, Pak3-GAL4 NP4472 > UAS-mcD8::GFP, shows similar membrane GFP expression patterns (midline cells, arrow; surface cells, arrowhead), with additional cells visible in the VNC. (E) Midline membrane GFP expression (green) overlaps with anti-Slit (magenta; arrow), a marker for midline glia from embryonic through larval stages (Oliva et al., 2016). (F) Expression of nuclear-localized GFP (green) in animals with Pak3-GAL4 NP4472 > UAS-nls::GFP confirms that the midline cells are anti-Elav-negative, indicating non-neuronal identity (arrow), but other GFP-positive cells in the VNC are neuronal (open arrowhead). (G,H) Along motor axons and at neuromuscular junction (NMJ) synapses, strong membrane GFP expression (green) extends beyond regions staining with the neuronal marker HRP (magenta), appearing to wrap the axons as well as project over proximal synaptic boutons, consistent with Pak3 expression in peripheral glia.
Pak3 expression was also assessed using a Pak3 promoter and GFP reporter system. The Pak3 promoter-GAL4 transgenic line Pak3NP4472, in which the GAL4 gene is inserted 160 bp upstream of the Pak3 translational start site, was crossed to UAS-membrane GFP flies (UAS-mCD8::GFP; Lee and Luo, 1999). Larval progeny displayed strong GFP expression in cells that, similar to those revealed by the anti-Pak3 antibody, were arrayed in a dashed pattern characteristic of VNC midline glia (Figure 1D, arrow). Consistent with this identity, GFP-expressing cells at the midline colocalized with immunostaining for Slit, a protein secreted by midline glia (Figure 1E, arrow). They also failed to colocalize with anti-Elav, confirming their non-neuronal identity (Figure 1F, arrow). Also consistent with the anti-Pak3 antibody results, robust GFP signal was observed in glia covering the brain surface (Figure 1D, arrowhead) and ensheathing the motor axons between the ventral nerve cord and the periphery (Figures 1G,H). Pak3NP4472-driven GFP expression also revealed several other GFP-positive tissues that were not observed using the Pak3 polyclonal antibody, likely due to signal amplification conferred by both the GAL4-UAS system and immunostaining. These included a subset of Elav-positive neurons in the VNC (Figure 1F, open arrowhead), sensory neuron glia, and several tissues outside of the nervous system, particularly the trachea, oenocytes and salivary glands (data not shown). GFP expression was not detected in the larval body wall muscles, although Pak3 plays a significant role in myoblast fusion during embryonic stages (Duan et al., 2012). The expression pattern of the Pak3 promoter-GAL4 line thus corroborated the Pak3 antibody staining results, revealing surface, midline, and peripheral glia as major sites of Pak3 expression at this stage, but also suggested Pak3 functions in other cell types including neurons and trachea.
Glial Knockdown of Pak3 Rescues the spastin Mutant Phenotype
Given that loss of Pak3 in the whole animal rescues the synaptic phenotype of spastin mutants, we next narrowed Pak3 knockdown to specific tissues and looked for the same rescuing effect. Using tissue-specific GAL4 expression to drive RNAi targeting Pak3 in a spastin null background (spastin5.75; Sherwood et al., 2004), we assayed type Ib glutamatergic synaptic boutons on muscle 4 of each larval hemisegment, as the neuronal arbors on this muscle are relatively simple, easily observed, and representative of the NMJ arbors across body wall muscles. Bouton and branching frequencies, measured primarily by terminal bouton number, were quantified for each genotype (Figure 2); increases in both at the larval NMJ are spastin loss of function hallmarks. spastin5.75 mutants had typically small and clustered boutons, averaging over twice as many terminal boutons as compared to w1118 controls (Figures 2A,B,J; p = 4.6 × 10–4). Separate experiments in which the total number of boutons was scored instead of just the terminals yielded equivalent results (Supplementary Figure S1, w1118 vs. spas; p = 1.7 × 10–4). To identify an effective Pak3 RNAi line, we used e22c-GAL4, spaghetti squash-GAL4 (e22c,sqh-GAL4; Franke et al., 2005) to drive ubiquitous expression of candidate RNAi transgenes in the spastin5.75 background, with the expectation that effective Pak3 knockdown should recapitulate the suppression of spastin mutant phenotypes observed with genetic loss of Pak3. Of the publicly available lines we tested, Pak3NIG.14895R–2 (henceforth referred to as “Pak3RNAi” or “Pak3[RNAi]”) was the most effective. In the absence of a GAL4 driver it did not significantly affect the spastin mutation (Figures 2C,J; p = 2.3 × 10–4 compared to controls; Supplementary Figure 1, Pak3RNAi vs. Pak3RNAi, spas; p = 1.3 × 10–3). When ubiquitously expressed, however, Pak3RNAi in the spastin5.75 null background resulted in larger and more linearly arrayed (less branched) boutons resembling wild type rather than spastin5.75 mutants (compare Figure 2D to the wild type control in Figure 2A versus spastin5.75 in Figures 2B,C). Terminal bouton number in spastin5.75 mutants also decreased to wild type levels, although due to difficulties obtaining healthy e22c,sqh; spas paired controls (i.e., animals with the e22c,sqh-GAL4 driver and spastin mutation but no Pak3RNAi), this change did not reach statistical significance (Figure 2J; p = 0.13 for e22c,sqh; spas vs. e22c,sqh > Pak3RNAi, spas). However, comparison of e22c,sqh > Pak3RNAi, spas larvae to e22c,sqh > Pak3RNAi controls (ubiquitously expressing Pak3RNAi in a wild type, rather than spastin mutant background) showed no difference in bouton number, providing further support that ubiquitous expression of this Pak3RNAi transgene potently suppresses the effects of spastin mutation (Supplementary Figure S1, p = 0.67).
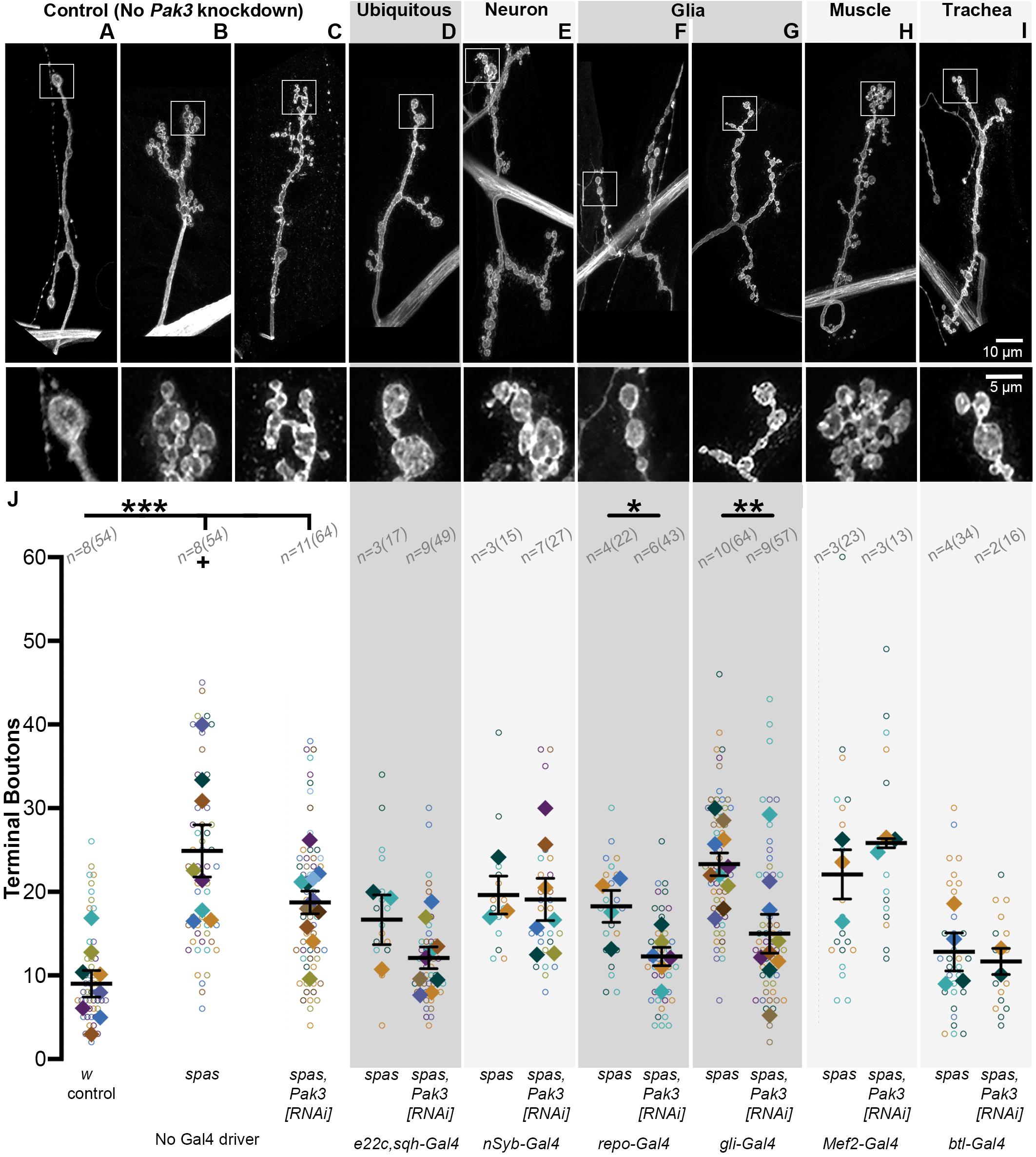
Figure 2. Glial, and specifically subperineurial glial, knockdown of Pak3 rescues spastin synaptic defects. (A–I) Anti-HRP staining shows neuronal morphology at the NMJ. The area within the box is magnified below. (A–C) are genetic background controls in which Pak3RNAi is not expressed via GAL4 activation (A wild type; B,C: spastin5.75 mutant); (D–I) are representative arbors from spastin5.75 mutants in which Pak3RNAi is expressed in the specified tissues. (A) Wild type synaptic boutons are arrayed linearly with few branches, while (B) spastin5.75 loss of function mutants have highly branched arbors with many small, bunched boutons. (C) In the absence of a GAL4 driver, the Pak3RNAi transgene has little effect on spastin5.75 morphology; terminal boutons remain significantly increased compared to wild type. (D) Ubiquitous expression of Pak3RNAi in the spastin5.75 background yields terminal boutons more similar in size, arrangement, and number to wild type. (E) Pak3RNAi expression in neurons does not rescue the spastin mutant phenotype, but (F,G) Pak3 knockdown in all except midline glia with repo-GAL4 (F), or in subperineurial glia with gliotactin-GAL4 (G), significantly rescues spastin5.75. (H,I) Knockdown of Pak3 in muscle (H) or trachea (I) show no difference in morphology from their spastin5.75 controls. (J) Synaptic morphologies are quantified by measuring the number of terminal boutons at each muscle 4 in a larva. In this and subsequent graphs, each open circle represents the quantity scored at an individual muscle; solid diamonds are the average of these counts for each larva, grouped by color; bars represent larval mean ± SEM; n(N) = number of larvae (number of muscles scored). “+” denotes two data points (y = 65 and 77) that fall outside of the y-axis range. The number of terminal boutons is statistically reduced compared to its driver-specific spastin5.75 control only when Pak3RNAi is expressed in glia or subperineurial glia. When compared to the spastin5.75 control, Pak3RNAi alleviates the spastin mutant phenotype if expressed ubiquitously, in all glia, or in subperineurial glia (p = 0.02). Statistical significance is calculated by Student’s t-test; asterisks signify p ≤ 0.001 (***), 0.001 < p ≤ 0.01 (**), or 0.01 < p ≤ 0.05 (*).
We then compared the consequence of Pak3 reduction using the pan-neuronal driver nSyb-GAL4 (Jenett et al., 2012), the glial driver repo-GAL4 (Sepp et al., 2001), the muscle drivers Mef2-GAL4 and Mhc-GAL4, and the tracheal driver btl-GAL4 (Shiga et al., 1996), as all of these cell types have been implicated in regulating Drosophila NMJ synapses and show evidence of Pak3 expression as discussed above. nSyb-GAL4-, Mef2-GAL4-, and Mhc-GAL4-mediated Pak3RNAi expression all failed to alleviate spastin5.75, excluding neurons (p = 0.90) and muscles (p = 0.28 and 0.79 for Mef2 and Mhc, respectively), as sites of Pak3’s deleterious effects (Figures 2E,H,J; Mhc-GAL4 data not shown, n = 3 and 4 larvae for control and experimental groups). Spastin mutant synapses were rescued only with glial-specific reduction of Pak3 using repo-GAL4, which drives expression in all glia except for those at the midline (Figures 2F,J; p = 1.9 × 10–3 for repo-GAL4, spas vs. repo-GAL4 > Pak3RNAi, spas). Repo-GAL4-mediated Pak3 knockdown restored synapse morphology of spastin5.75 mutants to wild type (Figure 2F), comparable to ubiquitous (e22c,sqh-GAL4-driven) knockdown (Figure 2D) or genetic loss of Pak3 (Ozdowski et al., 2011). Parallel experiments showed no significant difference in bouton number between glial Pak3 knockdown in wild type versus spastin mutant backgrounds, further supporting a requirement for glial Pak3 in the spastin mutant phenotype (Supplementary Figure S1, p = 0.07). Simultaneous expression of two independently generated Pak3 RNAi transgenes (Pak3GL0028 and Pak3v39844) with repo-GAL4 also suppressed the spastin5.75 phenotype (Supplementary Figure S2, Pak3GL0028,Pak3v39844, spas controls vs. repo-GAL4 > Pak3GL0028,Pak3v39844, spas; p = 1.3 × 10–3), and had no effect when expressed using the neuron-specific elav-GAL4 driver (Supplementary Figure S2, Pak3GL0028,Pak3v39844, UAS-Dcr2, spas controls vs. elavC155-GAL4 > Pak3GL0028,Pak3v39844, UAS-Dcr2, spas; p = 0.32), supporting that these results are specific to Pak3 reduction and not due to RNAi off-target interactions. We were unable to draw any conclusions regarding a role for trachea using this tissue-specific knockdown approach since the btl-GAL4 transgene alone with spastin5.75 had near-wild type morphology (Figures 2I,J). Regardless, together with the Pak3 expression pattern, these data substantiate glia as the major cell type in which Pak3 acts to cause neuronal defects when spastin is lost.
Pak3 Acts in the Subperineurial Glia
The Drosophila nervous system includes multiple glial cell types, each with distinct functions that bear striking molecular and behavioral parallels to vertebrate glial subtypes (reviewed in Freeman, 2015; Yildirim et al., 2019). Motor axons projecting from the larval VNC to the NMJ are ensheathed by three layers of glia. The outermost, perineurial glial (PG) layer, and middle, subperineurial glia (SPG) layer, effectively shield the axons from their surroundings, just as they do in the CNS where, as the “surface glia” (at least some of which express Pak3, see Figure 1), they form the blood-brain-barrier. The innermost layer, the wrapping glia, directly contact individual axons and mediate their homeostasis. All three layers ensheathe the motor axons along their lengths, typically ending at or near the first synaptic bouton at the NMJ. Beyond this point, the perineurial and subperineurial, but not wrapping, glial layers extend dynamic, actin-based processes that interact with synaptic arbors at the NMJ (Fuentes-Medel et al., 2009; Brink et al., 2012). We found that expression of Pak3RNAi using the subperineurial-specific glia driver gliotactin (gli)-GAL4 (Sepp et al., 2000) rescued the spastin mutant phenotype as effectively as repo-GAL4 or e22c,sqh-GAL4, reducing terminal bouton number by 35% and restoring wild type morphology (Figures 2G,J; p = 5.7 × 10–3 for gli-GAL4, spas vs. gli-GAL4 > Pak3RNAi, spas). Subperineurial glia can therefore account for the majority of spastin5.75 rescue conferred by Pak3 loss, supporting the idea that neuronal damage due to spastin loss requires Pak3 in the SPG.
SPG Projections Depend on Pak3 and Are Enhanced in spastin Mutants
How might Pak3 in subperineurial glia damage neuronal synapses? One possibility is that deleterious effects are mediated by the SPG projections that interact with the synaptic arbor. Given that Pak3 promotes actin polymerization and that these glial projections are actin-rich (Brink et al., 2012), we predicted that neurotoxic Pak3 in spastin mutants would be reflected in enhanced numbers of these glial projections. We used gli-GAL4 driven-expression of UAS-mCD8::GFP to visualize SPG membranes (Figures 3A–D, green) and anti-HRP immunostaining to label neurons (magenta) in (Figure 3A) wild type, (Figure 3B) Pak3Df/Pak3d02472 mutant, (Figure 3C) spastin5.75 mutant, or (Figure 3D) the Pak3Df/Pak3d02472, spastin5.75 double mutant backgrounds. These animals were dissected in physiological levels of Ca2+ since glial projections are highly Ca2+-sensitive (Brink et al., 2012). Gliopods (long, thin projections) and gliobulbs (rounded, resembling a bouton) were scored separately, but no genotype-dependent changes were observed between the two morphologies so they were combined. In the control background (“gli > GFP control” in Figure 3A), we observed on average 10 terminal boutons (Figure 3F) and a single SPG projection (Figure 3G) per muscle 4. Glial projections in Pak3Df/Pak3d02472 loss of function animals were rarer, averaging only one projection for every 5 muscles scored (Figures 3B,G; p = 0.017 compared to controls). This demonstrated that Pak3 is generally required for the formation of SPG projections (Figure 3G), but that these projections are not required for normal synaptic bouton arbors, which were no different from controls (Figure 3F, p = 0.97). In contrast, spastin5.75 mutants had twice as many projections compared to controls (Figures 3C,G; p = 0.011), consistent with increased Pak3-mediated glial activity when spastin is lost, and correlating with the increase in average terminal bouton numbers (Figures 3C,F; p = 8.0 × 10–3). Animals deficient for both spastin and Pak3 had near-wild type bouton numbers as expected (Figures 3D,F; p = 0.87), and similar to the Pak3 mutant alone, glial projections were virtually absent (Figures 3D,G; p > 0.99). Thus, SPG projections depend on Pak3, are enhanced in spastin mutants, which have aberrant synaptic arbors, and are sparse in Pak3, spastin double mutants, which have normal synaptic arbors.
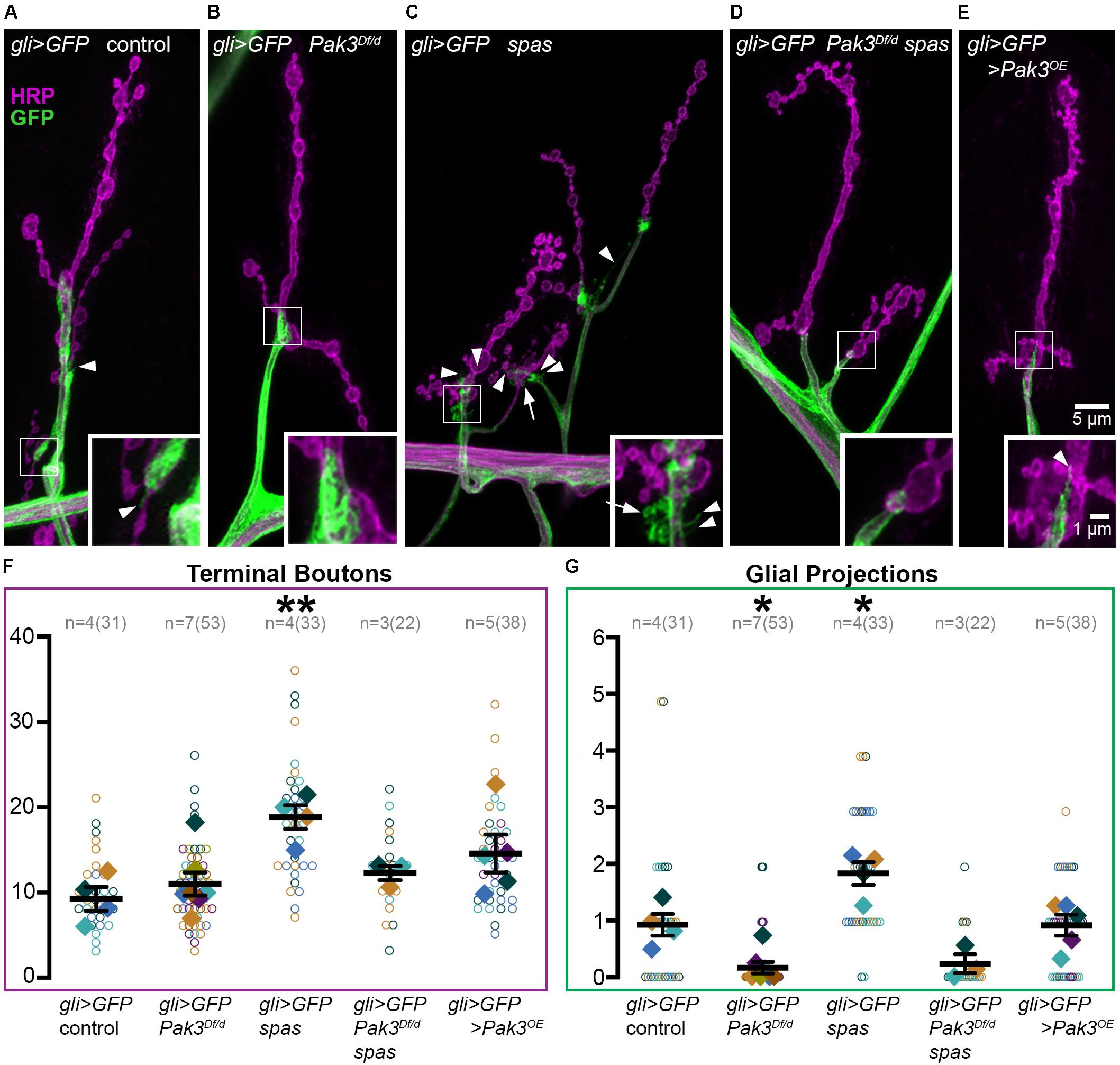
Figure 3. Subperineurial glia projections depend on Pak3 and are doubled in spastin mutants. (A–E) Anti-HRP shows neuronal morphology in magenta, anti-GFP shows glial morphology in green. (A) gli-GAL4, UAS-mCD8::GFP (gli > GFP) controls have linearly arrayed synaptic boutons and average one glial projection (arrowhead) per muscle 4. (B) Pak3 loss of function mutants, gli > GFP; Pak3Df/Pak3d02472 (abbreviated “Pak3Df/d”) have similarly arrayed bouton arbors but very few glial projections. (C) spastin5.75 loss of function, gli > GFP; spas, causes extra glial projections in addition to extra terminal boutons. These projections include thin glial filopodia (arrowheads) and round glial projections (arrows). (D) Consistent with these extra glial projections requiring Pak3 function, gli > GFP; Pak3Df, spas/Pak3d02472, spas double mutants display few glial projections, and have wild type bouton arbors. (E) Overexpression of Pak3 in subperineurial glia does not affect glial projections or terminal bouton numbers compared to controls, although small, clustered terminal boutons are observed. (F,G) Graphs of individual (open circles) and averaged (solid diamonds) values per larva for terminal boutons (F) and glial projections (G) are shown for each of the genotypes in (A–E). Significant differences relative to gli > GFP controls are calculated by one-way ANOVA followed by Sidak’s multiple comparisons test. P-values are denoted as * for 0.01 < p ≤ 0.05, and ** for 0.001 < p ≤ 0.01.
Given that Pak3 in SPG is necessary for spastin mutant synapses, we asked whether overexpression of Pak3 in wild type SPG is sufficient to elicit synaptic defects characteristic of spastin loss. Pak3 was overexpressed in GFP-labeled SPG using gli-GAL4 to drive expression of UAS-Pak3 and UAS-mCD8::GFP in an otherwise wild type background. UAS-Pak3 induces actin-rich projections in the synaptic bouton arbor when overexpressed in neurons (Ozdowski et al., 2011) and rescues adult lethality in Pak3 loss of function animals (E.F.O. unpublished results). Pak3 overexpression in SPG induced small boutons like those seen in spastin mutants (Figure 3E), but did not significantly increase terminal bouton number (Figure 3F, p = 0.21 compared to controls) or alter glial projections (Figures 3E,G; p > 0.99). Closer examination of the relationship between terminal bouton and glial projection numbers at individual muscles confirmed an absence of synapse-specific correlation between neuronal boutons and glial projections in wild type as well as spastin loss of function animals (Pearson’s correlation coefficient rwildtype = −0.06 and rspastin = −0.2), despite their strong association on average. While this could suggest that SPG projections are simply an epiphenomenon, their intimate association with the NMJ and role in sculpting synaptic arbors (Fuentes-Medel et al., 2009) argue for their functional relevance in this context.
Pak3-Mediated Toxicity Shows Specificity to spastin Loss
The striking efficacy of Pak3 removal in suppressing defects due to spastin loss suggested that Pak3-mediated changes in glial behavior might be a common mechanism underlying neuronal dysfunction. To test this possibility, we investigated genetic mutants in flower (fwe), which like spastin mutants, exhibit small, highly branched, supernumerary boutons (Figure 4; Yao et al., 2009, 2017). Mutants bearing these morphological hallmarks are thought to be functionally related, most likely in aspects of endosomal cycling (Dickman et al., 2006), and we reasoned that functionally related proteins are more likely to have conserved regulatory mechanisms. We compared the NMJs of trans-heterozygous loss of function mutants in fwe (fweDB56/fweDB25) with and without Pak3 loss of function, and found no rescue of terminal bouton number or overall morphology of fwe mutants with Pak3 loss (compare Figures 4C,D; p = 0.96). Loss of Pak3 in SPG thus does not act as a non-specific suppressor of synaptic defects; rather, its rescuing role is specific, at least in part, to the sequelae of spastin loss.
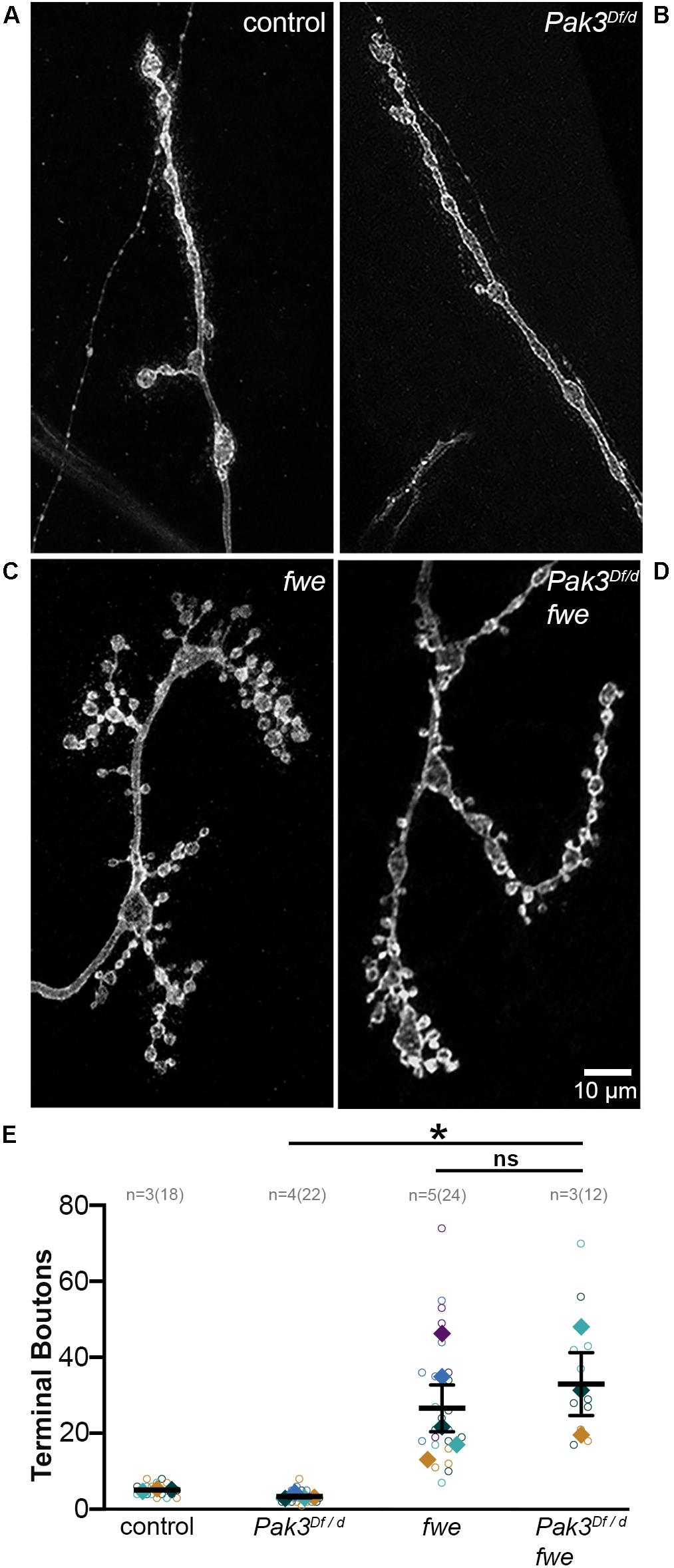
Figure 4. Pak3 loss of function does not rescue all supernumerary bouton mutants. (A–D) Anti-HRP staining shows neuronal morphology at the NMJ. (A) Wild type synaptic boutons are arrayed linearly with only a few branches and (B) Pak3 loss of function mutants, Pak3Df/Pak3d02472, show no differences from wild type. (C) Loss of function mutations in flower (fweDB56/fweDB25) result in extreme branching of the neuronal arbor and small bouton size, similar to spastin mutant morphology. (D) Loss of Pak3 function does not rescue the supernumerary bouton phenotype in fwe mutants, indicating that Pak3 loss is not a generalized mechanism to reduce extra synaptic branching. (E) Individual terminal bouton numbers (open circles) and larval averages (solid diamonds) are shown for each of the genotypes in (A–D). Significant differences are calculated by one-way ANOVA and Sidak’s multiple comparisons test. * denotes 0.01 < p ≤ 0.05.
Discussion
Through an unbiased, forward genetic screen for modifiers of Spastin activity, we previously showed that the actin regulator Pak3 is required for the manifestation of morphological and functional synaptic defects due to spastin loss (Ozdowski et al., 2011). Here, we have determined that Pak3 functions in larval subperineurial glia cells, where it promotes projections that extend into the NMJ synaptic arbor. The SPG projections are highly dependent on Pak3, whether in wild type animals, where these glia serve supportive roles in synaptic development (e.g., Fuentes-Medel et al., 2009, 2012), or in spastin mutants, where the projections are twice as frequent and synapses are defective.
The ability of Pak3 loss to suppress spastin mutant defects suggests that Pak3-dependent glial projections are a critical aspect of SPG toxicity when spastin is lost. Excess glial projections could, for example, reflect phagocytic behavior gone awry, given that wild type projections positively regulate synaptic arbor morphology through this mechanism (Fuentes-Medel et al., 2009). Indeed, these phagocytic engulfment events, mediated by the Draper receptor, have been shown to play both neuroprotective and neurodegenerative roles in the CNS, depending on context (reviewed in Logan, 2017; Hilu-Dadia and Kurant, 2019). Whether SPG projections in spastin mutants have analogous functions remains to be determined, however, particularly given the lack of correlation we observed between synaptic bouton and SPG morphologies at individual NMJs in both wild type and mutant animals. Even with rapid dissection, it is possible that our fixed preparations fail to accurately capture the distribution of these dynamic glial structures and their precise relationship to associated synaptic boutons. Additionally, glial projections may represent only a subset of Pak3-mediated changes within SPG cells contributing to the spastin mutant phenotype. Finally, while Pak3 in SPG is required for the spastin mutant phenotype, other cell types may also be involved. We do not yet know whether spastin loss affects perineurial glia behavior, for example, or the underlying muscle or adjacent trachea, all of which have been shown to interact with the presynaptic arbor (Fuentes-Medel et al., 2009; Brink et al., 2012; Chen et al., 2019).
In addition to defining the contribution of these additional cell types, future experiments will be important to determine whether Pak3 is altered in response to spastin loss within the same SPG cells, or if changes in Pak3 are induced cell non-autonomously. Our earlier results used tissue-specific rescue to demonstrate that spastin is primarily required in neurons (Sherwood et al., 2004; Du et al., 2010), and expression studies in mouse and human also support this neuron-specific requirement (Ma et al., 2006). If so, this argues for a model in which spastin-deficient neurons send a signal that leads to Pak3 upregulation in SPG, and subsequent deleterious effects on synaptic boutons. Given that our neuron-specific rescue of the spastin phenotype was incomplete, however, we cannot rule out roles for Spastin in other cells (Sherwood et al., 2004). Spastin RNAi has been ineffective in our hands, and neither antibodies nor promoter-GAL4 lines have convincingly revealed its endogenous expression pattern in Drosophila. However, Spastin expression has been shown via Western blots in normal human primary cerebellar astrocytes, glioblastoma cells, and rodent reactive astrocytes, consistent with possible roles in non-neuronal cell types and specifically glia (Ma et al., 2006; Draberova et al., 2011; Havlicek et al., 2014).
Although they remain the “support cells” of the nervous system, the idea that glia can also induce neuronal toxicity, particularly in the context of other defects, is increasingly evident. In flies, the requirement for Pak3 to elicit neuronal dysfunction during spastin loss bears striking parallels to the glial-derived, prodegenerative signaling molecule Eiger (the mammalian TNF-α ortholog; Keller et al., 2011). Neither protein is required for normal synapse development; rather, it is their glial deployment in mutant contexts (Pak3 in spastin mutants, Eiger in ankyrin mutants), that promotes neuronal dysfunction. Our inability to generate excess glial projections or significantly alter bouton number by overexpressing Pak3 in wild type animals supports, additionally, that activation of these glial pathways is only damaging to neurons in specific mutant contexts. Conversely, as seen with the fwe experiment, absence of these proteins does not serve as a generalized mechanism by which even potentially related synaptic defects can be rescued.
Dramatic changes in glial morphology and behavior, from non-reactive to reactive states, are also observed in mammalian nervous systems in response to damage (reviewed in Liddelow and Barres, 2017). Ischemic versus inflammatory insults induce distinct forms of reactive astrocytes; while ischemic damage leads to “A2” reactive astrocytes that support repair and survival, neuroinflammatory insults induce “A1” reactive astrocytes that are toxic to synapses. These A1 reactive astrocytes are now thought to play causal roles in the progression of several neurodegenerative diseases including ALS, Alzheimer’s, Parkinson’s, and Huntington’s. Our results suggest that AD-HSP is also a candidate for this list, and that putative reactive glial behavior could be mediated by excess Pak activity. Drosophila Pak3 bears considerable amino acid identity and functional correspondence to the vertebrate group 1 Paks (Mentzel and Raabe, 2005), which are extensively studied for their roles in several cancers and actively targeted in drug development (reviewed in Rane and Minden, 2019). Should evidence for a Pak-mediated reactive glial mechanism be revealed in AD-HSP, the group I Paks will be promising targets for therapeutic intervention.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
EO and NS contributed to the conception and experimental design. JW, SE, and HA generated and characterized genetic tools, including the efficacy of GAL4 drivers and RNAi lines, and obtained pilot data on bouton and glial phenotypes. EO performed dissections and immunofluorescence with anti-Pak3, -Slit, -Elav, and -GFP, and quantified adult eclosion rates, boutons for the fwe mutant experiment, and glial projections. EO, NS, and HA performed genetic crosses, larval dissections, immunofluorescence, acquired confocal images, and performed statistics on numeric data for the tissue-specific RNAi experiments. NS wrote the first draft of the manuscript. EO compiled the figures and figure legends and contributed to manuscript revision. All authors read and approved the written work.
Funding
This work was enabled by generous funding from the Shepard Broad Foundation, Raquelle de la Rocha, and the GT-Hele Fund. Publication costs were supported by the Duke University Libraries’ Compact for Open Access Publishing Equity (COPE) Fund to remove cost barriers in making Duke research open.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Buzz Baum, Sami Bahri, Marc Freeman, Vanessa Auld, Hugo Bellen, Mark Metzstein, and Donald Fox for generously sharing reagents and fly stocks, as well as to Nicole Fox, Jaeda Coutinho-Budd, and Eric Monson for helpful discussions. We thank former lab members, particularly Charlene Chen, Caitlin Cristante, and Daniel Ren for their assistance with the fwe mutant experiments. Additional stocks were obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537), National Institute of Genetics (NIG-FLY, Japan), Kyoto Drosophila Genomics and Genetics Resources (DGGR, Kyoto Institute of Technology), and the Vienna Drosophila Resource Center (VDRC, www.vdrc.at). Monoclonal antibodies were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology. Duke’s Light Microscopy Core Facility, the D. Sherwood lab, and E. Spana provided valuable microscopy support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2020.00912/full#supplementary-material
FIGURE S1 | Ubiquitous or glial-specific Pak3RNAi expression in wild type versus spastin mutant backgrounds suppresses the effects of spastin mutation. From left to right: Loss of spastin in Controls induces a nearly twofold increase in total bouton number (left two genotypes), and this effect is not altered by the presence of the Pak3RNAi transgene (next two genotypes). Ubiquitous Pak3RNAi expression makes larval synapses resistant to spastin removal, as bouton numbers in wild type (left; e,sqh > Pak3[RNAi]) and spastin mutant (right; e,sqh > Pak3[RNAi],spas) backgrounds are not significantly different. Neither neuron- nor muscle-specific Pak3RNAi expression suppress supernumerary boutons in spastin mutants. These results are consistent with those of Figure 2. The effects of tracheal Pak3RNAi expression are inconclusive, as the btl-GAL4 driver alone may suppress the spastin mutant phenotype independent of Pak3 (see Figure 2). Statistical significance is determined by Student’s t-test. P-values are denoted as ns for p > 0.05, * for 0.01 < p ≤ 0.05, ** for 0.001 < p ≤ 0.01, and *** for p ≤ 0.001.
FIGURE S2 | Expression of two different Pak3 RNAi transgenes, and use of a different pan-neuronal driver, independently support alleviation the spastin mutant phenotype by glial, and not neuronal, Pak3 knockdown. Simultaneous pan-neuronal expression of the Pak3GL0028 and Pak3v39844 RNAi transgenes (“Pak3[2xRNAi]”) does not alleviate supernumerary boutons of spastin mutants (p = 0.32 compared to paired control). The neuronal driver used in these experiments is elavC155-GAL4 rather than nsyb-GAL4; Dcr2 was also expressed to increase RNAi efficacy. In contrast, pan-glial expression of these Pak3 RNAi transgenes has the same effect as Pak3NIG.14895R–2 RNAi expression (from Figure 2 and Supplementary Figure S1), reducing terminal bouton number in spastin mutants to wild type levels (p = 1.3 × 10–3; Student’s t-test).
References
Allison, R., Edgar, J. R., and Reid, E. (2019). Spastin MIT domain disease-associated mutations disrupt lysosomal function. Front. Neurosci. 13:1179. doi: 10.3389/fnins.2019.01179
Asano, Y., Jimenez-Dalmaroni, A., Liverpool, T. B., Marchetti, M. C., Giomi, L., Kiger, A., et al. (2009). Pak3 inhibits local actin filament formation to regulate global cell polarity. HFSP J. 3, 194–203. doi: 10.2976/1.3100548
Baek, S. H., Cho, H. W., Kwon, Y. C., Lee, J. H., Kim, M. J., Lee, H., et al. (2012). Requirement for Pak3 in Rac1-induced organization of actin and myosin during Drosophila larval wound healing. FEBS Lett. 586, 772–777. doi: 10.1016/j.febslet.2012.01.061
Bahri, S., Wang, S., Conder, R., Choy, J., Vlachos, S., Dong, K., et al. (2010). The leading edge during dorsal closure as a model for epithelial plasticity: pak is required for recruitment of the Scribble complex and septate junction formation. Development 137, 2023–2032. doi: 10.1242/dev.045088
Brink, D. L., Gilbert, M., Xie, X., Petley-Ragan, L., and Auld, V. J. (2012). Glial processes at the Drosophila larval neuromuscular junction match synaptic growth. PLoS One 7:e37876. doi: 10.1371/journal.pone.0037876
Chang, C. L., Weigel, A. V., Ioannou, M. S., Pasolli, H. A., Xu, C. S., Peale, D. R., et al. (2019). Spastin tethers lipid droplets to peroxisomes and directs fatty acid trafficking through ESCRT-III. J. Cell. Biol. 218, 2583–2599. doi: 10.1083/jcb.201902061
Chen, P. Y., Tsai, Y. W., Cheng, Y. J., Giangrande, A., and Chien, C. T. (2019). Glial response to hypoxia in mutants of NPAS1/3 homolog Trachealess through Wg signaling to modulate synaptic bouton organization. PLoS Genet. 15:e1007980. doi: 10.1371/journal.pgen.1007980
Collins, C. A., and DiAntonio, A. (2007). Synaptic development: insights from Drosophila. Curr. Opin. Neurobiol. 17, 35–42. doi: 10.1016/j.conb.2007.01.001
Dickman, D. K., Lu, Z., Meinertzhagen, I. A., and Schwarz, T. L. (2006). Altered synaptic development and active zone spacing in endocytosis mutants. Curr. Biol. 16, 591–598. doi: 10.1016/j.cub.2006.02.058
Draberova, E., Vinopal, S., Morfini, G., Liu, P. S., Sladkova, V., Sulimenko, T., et al. (2011). Microtubule-severing ATPase spastin in glioblastoma: increased expression in human glioblastoma cell lines and inverse roles in cell motility and proliferation. J. Neuropathol. Exp. Neurol. 70, 811–826. doi: 10.1097/NEN.0b013e31822c256d
Du, F., Ozdowski, E. F., Kotowski, I. K., Marchuk, D. A., and Sherwood, N. T. (2010). Functional conservation of human Spastin in a Drosophila model of autosomal dominant-hereditary spastic paraplegia. Hum. Mol. Genet. 19, 1883–1896. doi: 10.1093/hmg/ddq064
Duan, R., Jin, P., Luo, F., Zhang, G., Anderson, N., and Chen, E. H. (2012). Group I PAKs function downstream of Rac to promote podosome invasion during myoblast fusion in vivo. J. Cell. Biol. 199, 169–185. doi: 10.1083/jcb.201204065
Felix, M., Chayengia, M., Ghosh, R., Sharma, A., and Prasad, M. (2015). Pak3 regulates apical-basal polarity in migrating border cells during Drosophila oogenesis. Development 142, 3692–3703. doi: 10.1242/dev.125682
Franke, J. D., Montague, R. A., and Kiehart, D. P. (2005). Nonmuscle myosin II generates forces that transmit tension and drive contraction in multiple tissues during dorsal closure. Curr. Biol. 15, 2208–2221. doi: 10.1016/j.cub.2005.11.064
Freeman, M. R. (2015). Drosophila central nervous system glia. Cold Spring Harb Perspect Biol 7:a020552. doi: 10.1101/cshperspect.a020552
Fuentes-Medel, Y., Ashley, J., Barria, R., Maloney, R., Freeman, M., and Budnik, V. (2012). Integration of a retrograde signal during synapse formation by glia-secreted TGF-beta ligand. Curr. Biol. 22, 1831–1838. doi: 10.1016/j.cub.2012.07.063
Fuentes-Medel, Y., Logan, M. A., Ashley, J., Ataman, B., Budnik, V., and Freeman, M. R. (2009). Glia and muscle sculpt neuromuscular arbors by engulfing destabilized synaptic boutons and shed presynaptic debris. PLoS Biol. 7:e1000184. doi: 10.1371/journal.pbio.1000184
Havlicek, S., Kohl, Z., Mishra, H. K., Prots, I., Eberhardt, E., Denguir, N., et al. (2014). Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 23, 2527–2541. doi: 10.1093/hmg/ddt644
Hazan, J., Fonknechten, N., Mavel, D., Paternotte, C., Samson, D., Artiguenave, F., et al. (1999). Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 23, 296–303. doi: 10.1038/15472
Hilu-Dadia, R., and Kurant, E. (2019). Glial phagocytosis in developing and mature Drosophila CNS: tight regulation for a healthy brain. Curr. Opin. Immunol. 62, 62–68. doi: 10.1016/j.coi.2019.11.010
Jenett, A., Rubin, G. M., Ngo, T. T., Shepherd, D., Murphy, C., Dionne, H., et al. (2012). A GAL4-driver line resource for Drosophila neurobiology. Cell. Rep. 2, 991–1001. doi: 10.1016/j.celrep.2012.09.011
Jeong, B., Kim, T. H., Kim, D. S., Shin, W. H., Lee, J. R., Kim, N. S., et al. (2019). Spastin contributes to neural development through the regulation of microtubule dynamics in the primary cilia of neural stem cells. Neuroscience 411, 76–85. doi: 10.1016/j.neuroscience.2019.05.024
Kasher, P. R., De Vos, K. J., Wharton, S. B., Manser, C., Bennett, E. J., Bingley, M., et al. (2009). Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem. 110, 34–44. doi: 10.1111/j.1471-4159.2009.06104.x
Keller, L. C., Cheng, L., Locke, C. J., Muller, M., Fetter, R. D., and Davis, G. W. (2011). Glial-derived prodegenerative signaling in the Drosophila neuromuscular system. Neuron 72, 760–775. doi: 10.1016/j.neuron.2011.09.031
Kumar, R., Sanawar, R., Li, X., and Li, F. (2017). Structure, biochemistry, and biology of PAK kinases. Gene 605, 20–31. doi: 10.1016/j.gene.2016.12.014
Kuo, Y. W., Trottier, O., Mahamdeh, M., and Howard, J. (2019). Spastin is a dual-function enzyme that severs microtubules and promotes their regrowth to increase the number and mass of microtubules. Proc. Natl. Acad. Sci. U.S.A. 116, 5533–5541. doi: 10.1073/pnas.1818824116
Lee, T., and Luo, L. (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461. doi: 10.1016/s0896-6273(00)80701-1
Liddelow, S. A., and Barres, B. A. (2017). Reactive astrocytes: production, function, and therapeutic potential. Immunity 46, 957–967. doi: 10.1016/j.immuni.2017.06.006
Logan, M. A. (2017). Glial contributions to neuronal health and disease: new insights from Drosophila. Curr. Opin. Neurobiol. 47, 162–167. doi: 10.1016/j.conb.2017.10.008
Lord, S. J., Velle, K. B., Mullins, R. D., and Fritz-Laylin, L. K. (2020). SuperPlots: communicating reproducibility and variability in cell biology. J. Cell. Biol. 219:e202001064. doi: 10.1083/jcb.202001064
Lumb, J. H., Connell, J. W., Allison, R., and Reid, E. (2012). The AAA ATPase spastin links microtubule severing to membrane modelling. Biochim. Biophys. Acta 1823, 192–197. doi: 10.1016/j.bbamcr.2011.08.010
Ma, D. L., Chia, S. C., Tang, Y. C., Chang, M. L., Probst, A., Burgunder, J. M., et al. (2006). Spastin in the human and mouse central nervous system with special reference to its expression in the hippocampus of mouse pilocarpine model of status epilepticus and temporal lobe epilepsy. Neurochem. Int. 49, 651–664. doi: 10.1016/j.neuint.2006.05.008
Mentzel, B., and Raabe, T. (2005). Phylogenetic and structural analysis of the Drosophila melanogaster p21-activated kinase DmPAK3. Gene 349, 25–33. doi: 10.1016/j.gene.2004.12.030
Oliva, C., Soldano, A., Mora, N., De Geest, N., Claeys, A., Erfurth, M. L., et al. (2016). Regulation of Drosophila brain wiring by neuropil interactions via a slit-robo-RPTP signaling complex. Dev. Cell. 39, 267–278. doi: 10.1016/j.devcel.2016.09.028
Ozdowski, E. F., Gayle, S., Bao, H., Zhang, B., and Sherwood, N. T. (2011). Loss of Drosophila melanogaster p21-activated kinase 3 suppresses defects in synapse structure and function caused by spastin mutations. Genetics 189, 123–135. doi: 10.1534/genetics.111.130831
Papadopoulos, C., Orso, G., Mancuso, G., Herholz, M., Gumeni, S., Tadepalle, N., et al. (2015). Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 11:e1005149. doi: 10.1371/journal.pgen.1005149
Park, S. H., Zhu, P. P., Parker, R. L., and Blackstone, C. (2010). Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest. 120, 1097–1110. doi: 10.1172/JCI40979
Rane, C. K., and Minden, A. (2019). P21 activated kinase signaling in cancer. Semin. Cancer Biol. 54, 40–49. doi: 10.1016/j.semcancer.2018.01.006
Ribeiro, S. A., D’Ambrosio, M. V., and Vale, R. D. (2014). Induction of focal adhesions and motility in Drosophila S2 cells. Mol. Biol. Cell. 25, 3861–3869. doi: 10.1091/mbc.E14-04-0863
Semenova, G., and Chernoff, J. (2017). Targeting PAK1. Biochem. Soc. Trans. 45, 79–88. doi: 10.1042/BST20160134
Sepp, K. J., Schulte, J., and Auld, V. J. (2000). Developmental dynamics of peripheral glia in Drosophila melanogaster. Glia 30, 122–133. doi: 10.1002/(sici)1098-1136(200004)30:2<122::aid-glia2>3.0.co;2-b
Sepp, K. J., Schulte, J., and Auld, V. J. (2001). Peripheral glia direct axon guidance across the CNS/PNS transition zone. Dev. Biol. 238, 47–63. doi: 10.1006/dbio.2001.0411
Sherwood, N. T., Sun, Q., Xue, M., Zhang, B., and Zinn, K. (2004). Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2:e429. doi: 10.1371/journal.pbio.0020429
Shiga, Y., Tanaka-Matakatsu, M., and Hayashi, S. (1996). A nuclear GFP / beta-galactosidase fusion protein as a marker for morphogenesis in living Drosophila. Dev. Growth Diff. 38, 99–106. doi: 10.1046/j.1440-169x.1996.00012.x
Stone, M. C., Rao, K., Gheres, K. W., Kim, S., Tao, J., La Rochelle, C., et al. (2012). Normal spastin gene dosage is specifically required for axon regeneration. Cell. Rep. 2, 1340–1350. doi: 10.1016/j.celrep.2012.09.032
Tarrade, A., Fassier, C., Courageot, S., Charvin, D., Vitte, J., Peris, L., et al. (2006). A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum. Mol. Genet. 15, 3544–3558. doi: 10.1093/hmg/ddl431
Trotta, N., Orso, G., Rossetto, M. G., Daga, A., and Broadie, K. (2004). The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr. Biol. 14, 1135–1147. doi: 10.1016/j.cub.2004.06.058
Vemu, A., Szczesna, E., Zehr, E. A., Spector, J. O., Grigorieff, N., Deaconescu, A. M., et al. (2018). Severing enzymes amplify microtubule arrays through lattice GTP-tubulin incorporation. Science 361:eaau1504. doi: 10.1126/science.aau1504
Wood, J. D., Landers, J. A., Bingley, M., McDermott, C. J., Thomas-McArthur, V., Gleadall, L. J., et al. (2006). The microtubule-severing protein Spastin is essential for axon outgrowth in the zebrafish embryo. Hum. Mol. Genet. 15, 2763–2771. doi: 10.1093/hmg/ddl212
Yao, C. K., Lin, Y. Q., Ly, C. V., Ohyama, T., Haueter, C. M., Moiseenkova-Bell, V. Y., et al. (2009). A synaptic vesicle-associated Ca2+ channel promotes endocytosis and couples exocytosis to endocytosis. Cell 138, 947–960. doi: 10.1016/j.cell.2009.06.033
Yao, C. K., Liu, Y. T., Lee, I. C., Wang, Y. T., and Wu, P. Y. (2017). A Ca2+ channel differentially regulates Clathrin-mediated and activity-dependent bulk endocytosis. PLoS Biol. 15:e2000931. doi: 10.1371/journal.pbio.2000931
Keywords: Spastin, Pak3, Autosomal Dominant Hereditary Spastic Paraplegia, p21-activated kinase, subperineurial glia, microtubule severing proteins, reactive glia
Citation: Ozdowski EF, Wentzell JS, Engert SM, Abbott H and Sherwood NT (2020) Suppression of spastin Mutant Phenotypes by Pak3 Loss Implicates a Role for Reactive Glia in AD-HSP. Front. Neurosci. 14:912. doi: 10.3389/fnins.2020.00912
Received: 31 March 2020; Accepted: 06 August 2020;
Published: 04 September 2020.
Edited by:
Cahir Joseph O’Kane, University of Cambridge, United KingdomReviewed by:
Yong Qing Zhang, Chinese Academy of Sciences (CAS), ChinaJ. Troy Littleton, Massachusetts Institute of Technology, United States
Mihaela Serpe, National Institutes of Health (NIH), United States
Copyright © 2020 Ozdowski, Wentzell, Engert, Abbott and Sherwood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina T. Sherwood, bmluYS5zaGVyd29vZEBkdWtlLmVkdQ==