 Zhongshan Shi
Zhongshan Shi Zhigang Lei
Zhigang Lei Fan Wu
Fan Wu Luoxing Xia
Luoxing Xia Yiwen Ruan
Yiwen Ruan Zao C. Xu
Zao C. Xu- 1Department of Anatomy and Cell Biology, Indiana University School of Medicine, Indianapolis, IN, United States
- 2Guangdong-Hongkong-Macau Institute for CNS Regeneration, Jinan University, Guangzhou, China
- 3Department of Neurology, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, China
- 4Third Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 5Jiangsu Province Co-innovation Center of Neuroregeneration, Nantong University, Nantong, China
Seizures are among the most common neurological sequelae of stroke, and diabetes notably increases the incidence of post-ischemic seizures. Recent studies have indicated that Sestrin3 (SESN3) is a regulator of a proconvulsant gene network in human epileptic hippocampus. But the association of SESN3 and post-ischemic seizures in diabetes remains unclear. The present study aimed to reveal the involvement of SESN3 in seizures following transient cerebral ischemia in diabetes. Diabetes was induced in adult male mice and rats via intraperitoneal injection of streptozotocin (STZ). Forebrain ischemia (15 min) was induced by bilateral common carotid artery occlusion, the 2-vessel occlusion (2VO) in mice and 4-vessel occlusion (4VO) in rats. Our results showed that 59% of the diabetic wild-type mice developed seizures after ischemia while no seizures were observed in non-diabetic mice. Although no apparent cell death was detected in the hippocampus of seizure mice within 24 h after the ischemic insult, the expression of SESN3 was significantly increased in seizure diabetic mice after ischemia. The post-ischemic seizure incidence significantly decreased in SESN3 knockout mice. Furthermore, all diabetic rats suffered from post-ischemic seizures and non-diabetic rats have no seizures. Electrophysiological recording showed an increased excitatory synaptic transmission and intrinsic membrane excitability in dentate granule cells of the rat hippocampus, together with decreased IA currents and Kv4.2 expression levels. The above results suggest that SESN3 up-regulation may contribute to neuronal hyperexcitability and seizure generation in diabetic animals after ischemia. Further studies are needed to explore the molecular mechanism of SESN3 in seizure generation after ischemia in diabetic conditions.
Introduction
Stroke is the major cause of death in the world. Seizures/epilepsy is a common complication after stroke (Bladin et al., 2000; Li et al., 2010). It has been reported by the American Diabetes Association that diabetic patients have a 2–4 fold higher risk of inducing a stroke. Hyperglycemia in people with diabetes will not only damage the central nervous system, but also increase the possibility of seizures following stroke (Ottman et al., 2011). Therefore, to reveal the molecular mechanism underlying post-ischemic epilepsy in diabetic animals may bring out a new clue in treatment of these patients.
Sestrins are a family of stress-inducible proteins, consisting of Sestrin1 (SESN1), Sestrin2 (SESN2) and Sestrin3 (SESN3). Sestrin-mediated inhibition of mTORC1 occurs via AMPK-mediated activation of the TSC1/2 complex (Budanov and Karin, 2008). The Sesn1 and Sesn2 genes are under transcriptional control of p53, while SESN3 is transcriptionally regulated by the AKT/FOXO axis, through FOXO1/FOXO3a-mediated gene expression (Budanov et al., 2004, 2010). SESN3 is a strong genetic risk factor that regulates proconvulsive cytokines and genes (Johnson et al., 2015) and plays a key role in metabolic homeostasis (Lee et al., 2012). It has reported that SESN3 regulates human hippocampal epilepsy (Johnson et al., 2015). However, the role of SESN3 in diabetes after stress, such post-ischemia seizures, remains unclear.
Hippocampus is a critical structure as an epileptic focus and epileptogenic zone. Increase of neuronal excitability is a widely recognized cause of seizures/epilepsy generation. It is conceivable that both membrane excitability and excitatory synaptic transmission are enhanced in hippocampal neurons in diabetic animals after ischemia, which results in seizures and epilepsy (Luhmann and Heinemann, 1992; Congar et al., 1995). In epileptic animals, inhibitory basket cells in the dentate gyrus receive less excitatory synaptic drive, and the transmission failure at basket cell-to- granule cell is increased (Zhang and Buckmaster, 2009). A-type potassium currents (IA currents) are voltage-dependent, transient outward K+ currents that quickly activate at subthreshold membrane potentials and rapidly inactivate. Considerable evidence has shown that IA currents play a pivotal role in the regulation of neuronal excitability (Carrasquillo et al., 2012). It has been reported that Kv4.2 knockout mice increased susceptibility to convulsant stimulation and Kv4.2 truncation mutation was seen in a patient with temporal lobe epilepsy (Singh et al., 2006; Barnwell et al., 2009). Our previous study has shown that traumatic brain injury (TBI) causes a downregulation of IA in hippocampal neurons, which is associated with the hyperexcitability in the post-traumatic hippocampus and leads to an enhanced seizure susceptibility (Lei et al., 2012). And the reduction of IA channels in hippocampus is involved in the development of post-ischemic seizures and epilepsy after ischemia (Lei et al., 2014, 2016).
To explore the role of SESN3 in hippocampal hyperexcitability and post-ischemia seizures occurring in diabetic animals, the present study first examined changes in expression of SESN3 and neuronal excitability after ischemia in diabetic animals. Secondly, we employed SESN3 knockout (KO) mice to investigate the casual effect of SESN3 on post-ischemic seizures. Our results showed that SESN3 up-regulation played an important role in hippocampal hyperexcitability and contributed to post-ischemia epileptogenesis in diabetic animals, through down-regulation of Kv4.2-mediated IA currents.
Materials and Methods
Animals
Experimental protocols were approved by the Institutional Animals Care and Use Committee of the Indiana University School of Medicine in accordance with the care and use of laboratory animals. Male mice of C57B16/J (25–30 g, 5–7 weeks old) and male Wistar rats (150–200 g, 5–7 weeks old) were used in the present study. The mice were randomly divided into 4 experimental groups: Control (CTL, n = 10); ischemia (IS, n = 10); diabetes (STZ, n = 10); and diabetes with ischemia (STZ-IS, n = 27). SESN3 KO mice (n = 7) were provided by Dr. Charlie Dong, Department of Biochemistry and Molecular Biology, Indiana University School of Medicine (Tao et al., 2015). Rats were randomly divided into 4 groups: Control (CTL, n = 10); ischemia (IS, n = 10); diabetes (STZ, n = 10) and diabetes with ischemia (STZ-IS, n = 17).
Diabetic Animal Model
Streptozotocin (STZ)-induced diabetes is a well-established animal model (Malhotra et al., 1981). To induce hyperglycemia, mice were intraperitoneally injected with STZ dissolved in 0.1 M citrate buffer (pH4.5, prepared just before use), final concentration of 7.5 mg/ml. Mice were fasted for 4 h before STZ injection (150 mg/kg) and had access to water freely. Rats were fasted overnight and STZ (50 mg/kg) was injected (i. p.) for two consecutive days. Blood was collected from the saphenous vein. Blood glucose level (BGL) was measured by One Touch Ultra glucometer 6 days after STZ injection. The animals with a BGL of ≥300 mg/dL were considered diabetic (Srinivasan et al., 2005). Transient global ischemia was performed at 1 week after STZ injection.
Transient Forebrain Ischemia Model
Mice were initially anesthetized with 5% isoflurane in a chamber. After shaving hair around the neck, mice were placed on a surgery board and anesthetized with 2% isoflurane via a nasal mask. Then the bilateral common carotid arteries were separated and clipped for 15 min. During clipping, the rectal temperature was maintained at 37°C with a heating lamp via a feedback system. Then the wound was stitched and the mice injected with buprenorphine SR to relieve the pain.
Transient global ischemia in rats was induced by transient occlusion of bilateral common carotid arteries and permanent occlusion of both vertebral arteries (4-VO) as described in our previous studies (Ruan et al., 2009). On the first day, the adult male Wistar rats were anesthetized with 2% isoflurane via a nasal mask. Both vertebral arteries were electrocauterized. A length of silicone tubing (0.025″ I.D., 0.047″ O.D.) was placed loosely around each common carotid artery and passed through two holes in a small teflon button before being tied in a loop. A suture line was tied at the end of the loop. The incision was closed with wound clips. The rats were then allowed to recover from anesthesia overnight. The next day, the carotid arteries were occluded in the lightly anesthetized rats. The 2% lidocaine-HCl (Sparhawk Laboratories., Lenexa, KS, United States) was applied as a local anesthetic in the region of incision. Under light anesthesia, the ventral neck suture was removed. The silicone tubing was then threaded and drawn through a 2-cm plastic cylinder, compressing the artery against the Teflon button. During occlusion, the isoflurane was removed and the rectal temperature was maintained at 37°C with a heating lamp via a feedback system, and the completeness of global ischemia was confirmed by testing the loss of righting and pupil reflexes. After the termination of 15 min occlusion, the silicone tubing, teflon buttons and sutures were removed and the incision was closed. The rats were injected with buprenorphine SR to relieve the pain. Rats were then allowed to recover in a standard cage.
Behavioral (i.e., convulsive) seizures include symptoms on a scale from III to V using the Racine scale (Racine, 1972). A class III seizure was characterized by forelimb clonus, an erect tail and lordotic posturing. A class IV was characterized by continued forelimb clonus and rearing on hindlimbs. Animals showing all of these behaviors in combination with a fall were defined as having a class V seizure (Lei et al., 2014, 2016).
Histochemistry Staining and Immunohistochemical Staining
HE Staining
Mice were sacrificed immediately after seizure onset. The animals were deeply anesthetized using 5% isoflurane and perfused with phosphate buffered saline (PBS, 0.01 mol/L, pH 7.4) for about 3 min, followed by 4% paraformaldehyde in PBS for about 10 min. After 1 day of post fixation, 40-μm coronal sections containing the hippocampus were cut with a Vibratome (VT1000; Leica, Nussloch, Germany). The sections were stained with hematoxylin and eosin (H&E, Thermo Fisher Scientific, Pittsburgh, PA, United States).
Immunohistochemical Staining
Rats were deeply anesthetized, perfused with PBS and fixed with 4% paraformaldehyde in PBS (Lei et al., 2012). After being postfixed overnight, six sets of coronal sections of the hippocampus were cut (40 μm) with a Vibratome (Technical Products International) and collected in PBS. The sections from all groups were stained together in each immunohistochemical step. The sections were incubated for 30 min in 0.3% H2O2 (Sigma) to quench the endogenous peroxidase activity. Subsequently, the sections were blocked and permeabilized in permeabilization solution (5% goat or horse serum, 0.1% Triton X-100 in PBS) for 1 h at room temperature before they were incubated with rabbit polyclonal anti-SESN3 (1:100; Abcam, Cambridge, MA, United States) in permeabilization solution overnight at 4°C. After being washed, the sections were incubated with biotinylated goat anti-rabbit IgG (1:200; Vector) in blocking solution (5% horse or goat serum in PBS) for 1 h at room temperature. After three washes, the sections were processed with Vectastain ABC Kits (HRP, Vector) and visualized by using 0.05% 3,3’-diaminobenzidine (DAB, sigma) in PBS containing 0.015% H2O2 for 1–2 min. The sections were then mounted onto slides, air dried, dehydrated in graded series of ethanols, infiltrated in xylene, and embedded in paraffin before they were examined with a microscope (BX50; Olympus). Images were acquired with a digital camera coupled to control software (DP70-BSW; Olympus) at 4 and 20 X magnification. The conditions for catching images was controlled constantly throughout all sections.
Western Blotting
Mice were deeply anesthetized with 5% isoflurane and perfused with cold saline. The brain was removed quickly and the hippocampus was dissected and placed in an EP tube with EDTA-RIPA buffer (Boston BioProducts, Worcester, MA, United States). The tissue was homogenized by smashing and ultrasonication. Insoluble cell debris was removed by centrifugation at 13,000 rpm for 30 min at 4°C and the resulting supernatants were collected for analysis. Protein concentration was determined by BCA assay with a commercial Kit (Bio-Rad, Hercules, CA, United States) and then adjusted to the same concentration among different samples followed by being boiled with 2X sodium dodecyl sulfate gel-loading buffer (Invitrogen, Carlsbad, CA, United States). Then sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out. Following being concentrated in 5% stacking gel, equal amount of protein (30 μg) was separated in 10% SDS-PAGE gels and transferred to 0.2 μm PVDF membrane. Then the membrane was blocked in 3% BSA in TBST for 2 h at room temperature (RT) followed by being incubated in primary antibody overnight at 4°C. We used rabbit polyclonal anti-Kv4.2 (1:1000; Chemicon, Temecula, CA, United States), rabbit polyclonal anti-SESN3 (1:1000; Abcam, Cambridge, MA, United States), mouse monoclonal anti-GAPDH (1:10000; Thermo Fisher Scientific, Rockford, IL, United States), or mouse monoclonal anti-β-actin antibodies (1:20000; Sigma). After rinsed in TBST 3 times for 10 min each, the membranes were incubated in the HRP-conjugated secondary antibodies (Vector, Burlingame, CA, United States) for 1 h at room temperature. After incubation, membranes were rinsed three times for 10 min with TBST again. Following a final rinse, the signals were detected with enhanced chemiluminescence (Amersham, Piscataway, NJ, United States) and visualized by exposing to X-ray films (Fuji, Tokyo, Japan). Band densitometry analysis of membrane was performed by using scanned images of non-saturated immunoblot films in NIH Image J 1.37.
Real-Time RT-PCR
Total RNA was prepared using an Absolutely RNA RT-PCR Miniprep kit (Stratagene, Cedar Creek, TX, United States). Reverse transcription of 1-μg total RNA to cDNA was performed using the StrataScript qPCR cDNA synthesis kit (Stratagene). Real-time quantitative polymerase chain reaction amplification was performed in a Stratagene MX 3005P Thermal Cycler (La Jolla, CA, United States) using RT2 SYBR Green qPCR Master Mix. Primers for SYBR Green-based real-time PCR were purchased from SuperArray Bioscience (Frederick, MD, United States). The relative amount of target mRNA was calculated using the comparative cycle threshold (Ct) method and normalizing each target gene with Ct of housekeeping gene, GAPDH.
Electrophysiological Recording
Electrophysiological techniques used in this study have been established in our lab for years (Li et al., 2009; Lei et al., 2012). Rats were be deeply anesthetized with 5% isoflurane and perfused transcardially with cold sucrose solution. The brain was quickly removed and immersed in ice-cold artificial cerebrospinal fluid (ACSF). Brain slices containing hippocampus (300-μm thickness) was cut using a vibratome (Leica VT1000). Hippocampal slices were incubated in ACSF (continuously equilibrated with 95% O2-5% CO2) for >1 h at room temperature (∼ 24°C) before being transferred to the recording chamber. The ACSF was comprised of the following (in mM): NaCl 130; KCl 3; CaCl2 2; MgCl2 2; NaH2PO4 1.25; NaHCO3 26; Glucose 10. Recording electrodes were prepared from borosilicate glass (Warner Instruments, Hamden, CT, United States) using a horizontal electrode puller (Sutter Instruments, Novato, CA, United States) to obtain a tip resistance of 3–5 MΩ. Neurons were visualized with an infrared-DIC microscope (Olympus BX50WI) through a CCD camera. Recordings were made using Axopatch 200B (Axon Instrument). Capacitive transients were reduced by adjusting the capacitance compensation of the amplifier. Signals were filtered at 2 KHz and digitized at a sampling rate of 5 KHz using data acquisition program Axograph 4.6 (Axon Instruments). The data were stored in a Macintosh computer for offline analysis.
Whole-cell current-clamp recording were used to study the membrane properties. The internal solution contained (in mM): KMeSO4 120, KCl 12, MgCl2 1, EGTA 1, CaCl2 0.2, HEPES 10, and Mg-ATP 2, pH 7.3, 290–295 mOsm/L. Depolarizing current pulses (800 ms, 20–180 pA) were applied to evoke either a single firing or repetitive firings. The membrane input resistance (Rin) was derived from the linear portion of the current-voltage curve. The spike latency was measured from the onset of the current injection to the peak of the first action potential. The spike threshold will be measured at the beginning of the upstroke of the first action potential (AP).
To study synaptic transmission, the membrane potential was held at -80 mV. The internal solution contained (in mM): CsCl 43, CsMeSO4 92, TEA 5, EGTA 2, MgCl2 1, HEPES 10, and Mg-ATP 4. For miniature analysis, an episode of 5–10 min was collected for miniature excitatory postsynaptic currents (mEPSCs). Tetrodotoxin (TTX, 1 μM) was applied during mEPSCs recording. Bicuculline (BIC) was used to block GABAA receptors at a concentration of 30 μM. The amplitude and frequency of the synaptic events were measured. EPSCs above a threshold (2.5 SDs above baseline noise) was automatically detected by a sliding template algorithm and manually checked off-line.
To study IA currents, the internal solution was the same as that used for measuring the membrane properties. The voltage-dependent outward potassium currents were evoked by voltage steps (from −90 mV to +60 mV in 10 mV increments, 400 ms) following a 300 ms hyperpolarizing pulse of −130 mV in the presence of TTX (1 μM) and CdCl2 (300 μM). IA currents were isolated by subtracting the currents evoked after depolarized pre-pulses (0 mV, 100 ms) from those evoked without depolarized pre-pulses.
Statistics
Student t-test was used to compare two groups of data. Paired t-test was used to compare the same values before and after treatment. The post-ischemia seizure rates in different experimental groups were compared with the Chi-square test. One-way analysis of variance (ANOVA) was used for multiple-group comparisons followed by post hoc analysis with Fisher’s PLSD. All statistical analyses were performed via StatView 5.0 software (Abacus Concepts). The difference was considered significant when P < 0.05.
Results
The Post-ischemic Seizure Incidence Significantly Increased in Diabetic Mice
Seven days after STZ injection, the blood glucose levels of animals were significantly elevated compared with those before injection (Before: 119.4 ± 5.9 mg/dL; 7 days after: 450.1 ± 25.6 mg/dL, n = 14, P < 0.01, Figure 1A). After STZ injection, the body weight of these mice decreased markedly (Before: 26.3 ± 0.8 g; 7 days after: 22.8 ± 0.7 g, n = 19, P < 0.01, Figure 1B). Furthermore, seven cages housing the diabetic mice (100%) were found wet with urine after STZ injection while the cages housing the control mice (0%) were in normal condition (P < 0.01, Figure 1C). These data indicate that the mice showed typical diabetic symptoms after STZ injection. Although both the control and diabetic mice were subjected to 15 min ischemia, no seizures were observed in 10 control mice after ischemia whereas 16 out of 27 (59.2%) diabetic mice exhibited post-ischemia seizures (P < 0.01, Chi-square test, Figure 1D). The time of first onset of seizures in mice varied from 0.6 to 22.8 h following ischemia (Median = 5.7 ± 2.1 h, n = 12). In our preliminary experiment, we found that the frequence of post-ischemia seizures increased and finally developed into status epilepticus (SE) before the mice died within 12 h after the first onset of seizures. SE is a very severe type of seizure, starting with forelimbs and hind limbs rigidly pushed away from the body, followed by violent shaking, vibrating, and jumping (Lei et al., 2014, 2016). Therefore, we sacrificed mice immediately after the first onset of seizures (Supplementary Videos).
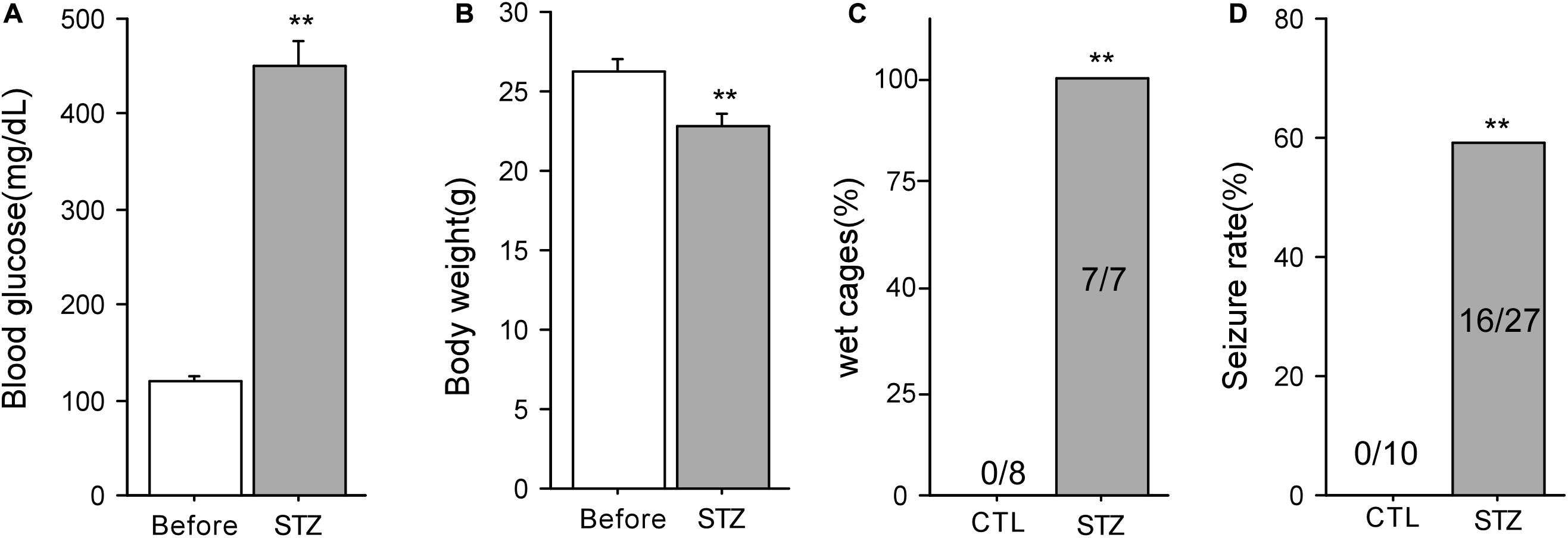
Figure 1. Diabetic animal model in mice and post-ischemic seizures. (A) Blood glucose levels of mice before and 7 days after STZ injection (n = 14). (B) The body weight of mice before and 7 days after STZ injection (n = 19). (C) The percentage of wet cages holding control and diabetic mice. (D) 59% of the mice with diabetes developed seizures following 15-min ischemia, whereas none of the mice with normoglycemia had seizures after ischemia. **P < 0.01 vs. control groups.
No Apparent Cell Death Is Observed Right After Post-ischemic Seizures in Diabetic Mice
In order to investigate whether the post-stroke seizure in diabetic mice was attributable to neuronal death, we examined the morphology of hippocampal neurons following ischemia using H&E staining (Figure 2). Ischemic neuronal injury is determined by the eosinophilic cytoplasm, nuclear pyknosis, and shrinkage of the cell body in H&E sections. The results showed that there was no obvious neuronal damage in the CA1 area, CA3 area, and dentate gyrus (DG) among the control, ischemic, diabetic, and diabetic-ischemic mice before they were sacrified.
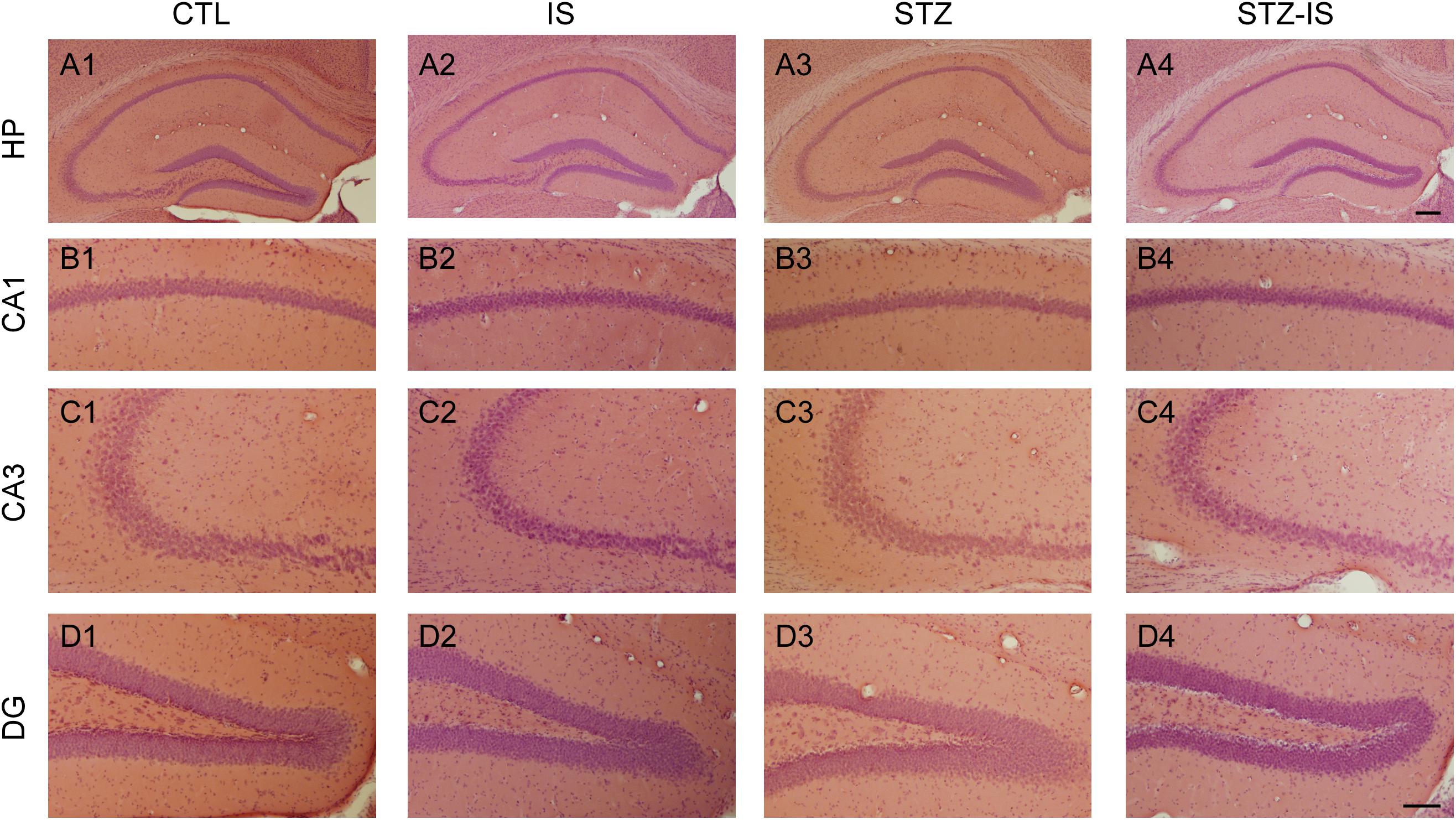
Figure 2. Histology of the hippocampus in different experimental conditions. Representative photographs show H&E staining of brain sections of CA1, CA3, and DG in control (CTL), ischemia (IS), diabetes and diabetes-ischemia (STZ) mice. No apparent cell death was observed in the hippocampus 12 h after ischemia in normoglycemia and diabetic mice. The scale bar of (A4) (200 μm) is equal to (A1–A3). And the scale bar of (D4) (100 μm) is equal to (B1–D3).
Higher Expression of SESN3 in Post-ischemia Seizures in Diabetic Animals
Sestrin3 positively regulates epilepsy gene network that contains a specialized, highly expressed transcriptional module encoding proconvulsive cytokines and Toll-like receptor signaling genes in human hippocampal epilepsy (Johnson et al., 2015). In order to reveal whether SESN3 is related to post-ischemia seizures or diabetes, we detected the expression of SESN3 in each group of animals. As shown in Figure 3A, compared with the control (100 ± 8.4%), there were no significant changes in the expression of SESN3 protein in the ischemia group (110.7 ± 4.7%), or diabetic group, both P > 0.05. However, the expression of SESN3 protein increased significantly in diabetic ischemia group (144.5 ± 23.6% vs. 100 ± 8.4%, P < 0.05).
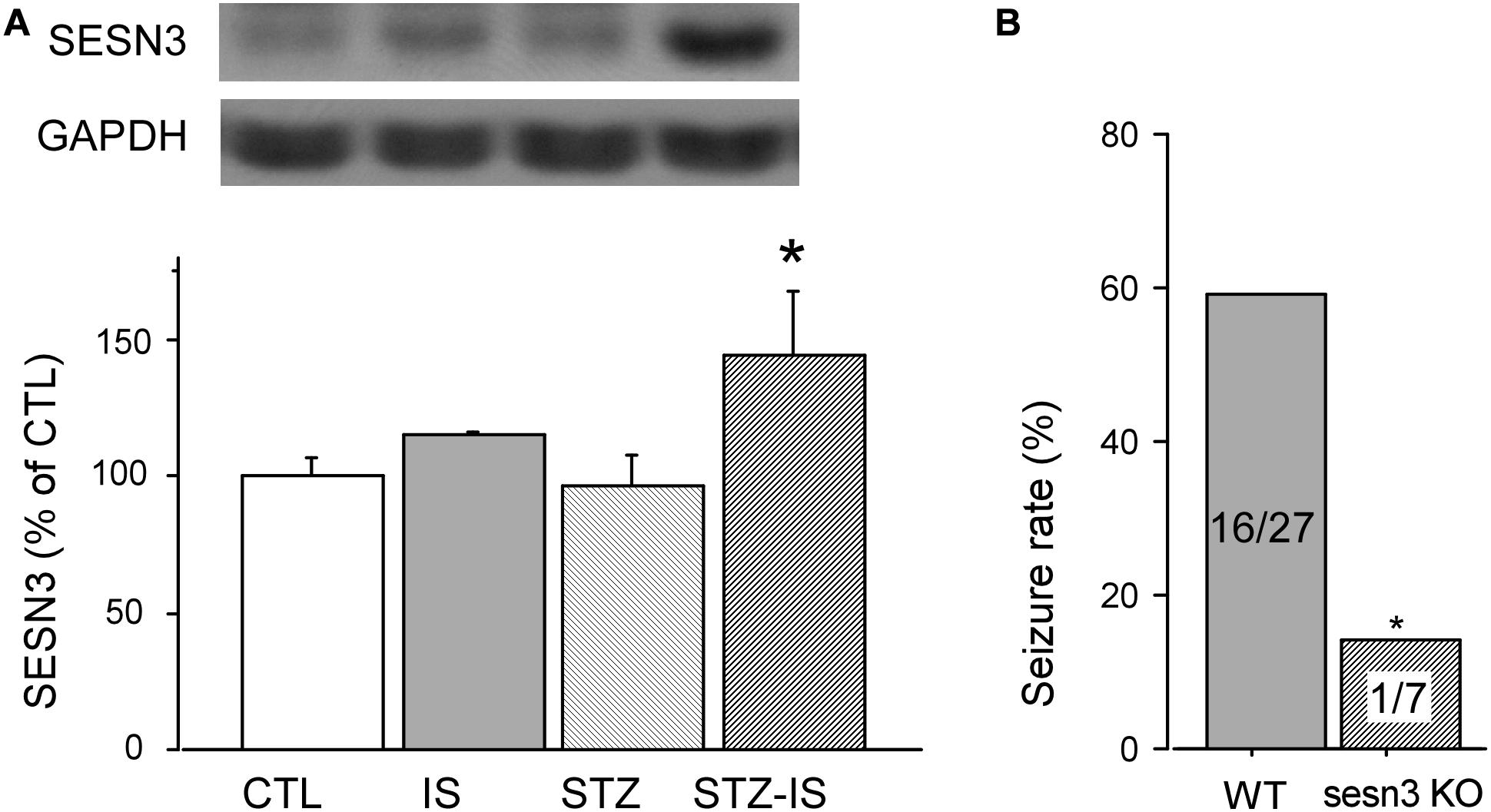
Figure 3. Western blot results showing SESN3 protein levels increase in diabetic mice after 15-min ischemia. (A) The expression of SESN3 increased significantly in diabetic ischemia group (*P < 0.05 vs. the control group). (B) Compared to wild type diabetic mice following ischemia, the post-ischemic seizure incidence significantly decreased in diabetic sesn3 KO mice, *P < 0.05 vs. the control group.
The above results suggest that the expression of SESN3 is related to ischemic diabetes-induced seizures. To prove this, we employed sesns3 KO mice to establish sesns KO ischemic mice. The diabetes of sesn3 KO mice was also produced by injection of STZ. The blood glucose level was similar to the wild type mice which was 120.7 ± 6.1 mg/dL before injection of STZ and reached 455.9 ± 30.8 mg/dL (n = 10) 7 days following injection of STZ. After the mice underwent 15 min global ischemia, the seizure incidence was significantly decreased to 14.2% compared to WT ischemic diabetes mice (WT-STZ-IS) (n = 7, P < 0.05, Figure 3B). The above results demonstrate the critical role of SESN3 in post-ischemia seizures in diabetic mice.
Abnormal Electrical Activity Was Induced in Post-ischemic Seizures Under Diabetic Conditions
To investigate mechanisms of post-ischemic seizures in diabetic animals, the excitability of hippocampal neurons of rats was examined among different groups. Diabetic rats were induced by injection of streptozotocin (STZ) (50 mg/kg, i.p.) each day for two consecutive days. The blood glucose levels were significantly increased in diabetic rats (422 ± 16 mg/dL, n = 10) at 1 week after STZ injection when compared with the controls (117 ± 5 mg/dL, n = 10), P < 0.01. The post-ischemia seizure rate was significantly increased in diabetic rats as compared with the controls (100% in diabetes vs. 0% in control, P < 0.05). To reveal the changes in SESN3 expression of diabetic animals after ischemia, the mRNA and protein levels of SESN3 were compared in different experimental groups. Diabetic rats were sacrificed shortly after the onset of seizures following ischemia. As shown in Figure 4A, quantitative PCR data indicated that SESN3 mRNA expression was 43.8% higher in diabetic animals after ischemia than that of the controls (STZ-ischemia: 143.8 ± 6.0% of control, P < 0.05). Western blotting data showed that SESN3 protein in hippocampus increased by 21.3% in diabetic rats and by 34.4% in diabetic rats after ischemia when compared with the controls (Figure 4B). Immunohistochemical studies showed that intensity of SESN3 staining in the hippocampus was increased in diabetic rats and it was strongest in the CA3 region of the hippocampus in diabetic-ischemic rats (Figure 4C).
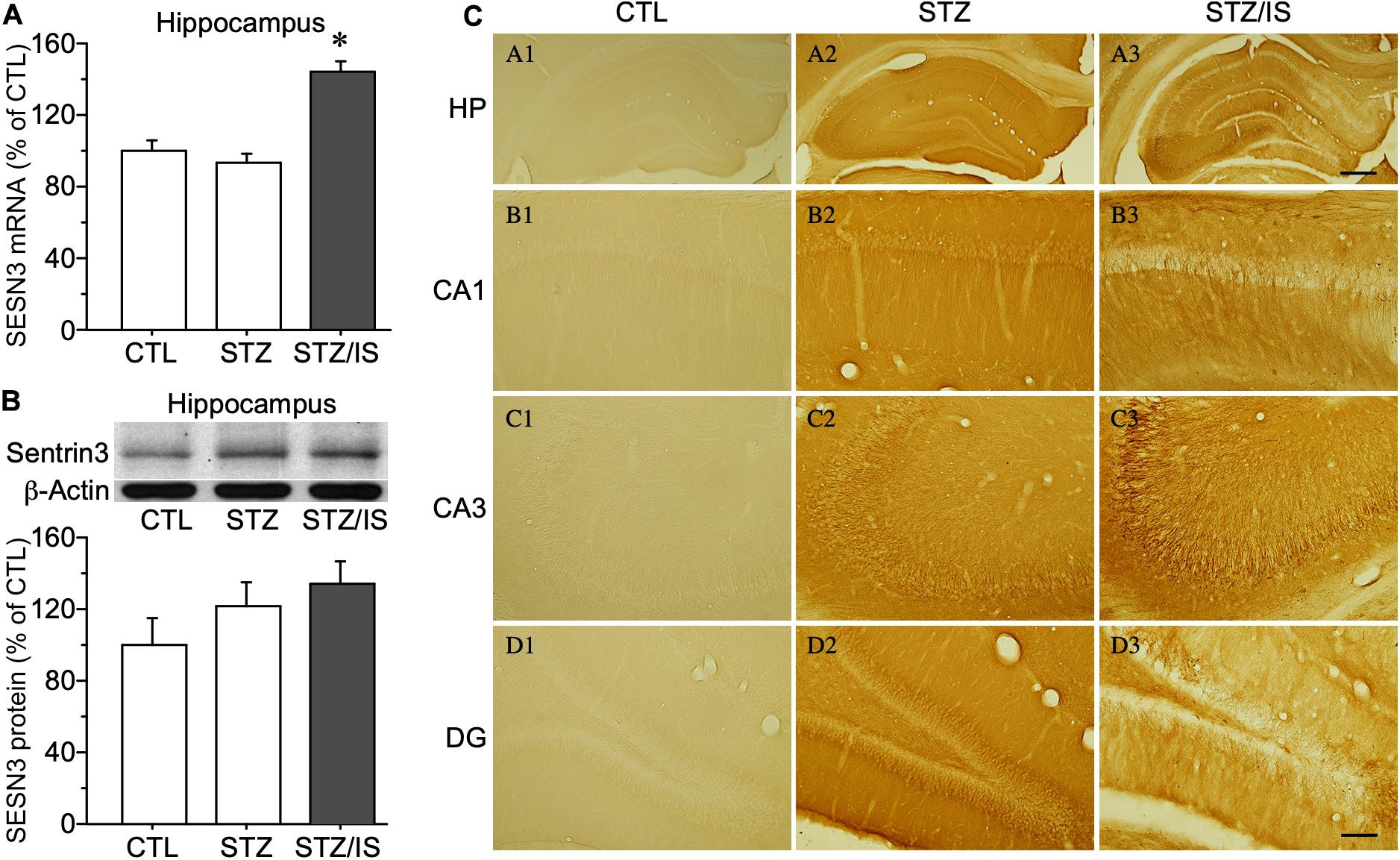
Figure 4. Quantitative PCR and Western blot results showing changes in sesn3 mRNA and protein levels in different experimental groups (A) Sesn3 mRNA levels in the hippocampus increased in diabetic rats after 15-min ischemia, *P < 0.05 vs. CTL and STZ groups. (B) The SESN3 protein levels in the hippocampus also appeared an increasing trend in diabetic rats after ischemia. (C) The intensity of immunostaining of SESN3 in the hippocampus is higher in diabetes after ischemia, especially in the CA3 region (C3). The scale bar of (A3) (500 μm) is equal to (A1–A3). And the scale bar of (D3) (100 μm) is equal to (B1–D3).
Whole-cell recording of dentate granule cells of diabetic rats was performed. The animals were sacrificed immediately after onset seizure and transverse brain slices at 300 μm were prepared. mEPSCs of dentate granule cells were compared in each groups. As shown in Figure 5A, in comparison with the control rats, the frequency of mEPSCs was significantly increased in diabetic animals after ischemia (control: 0.74 ± 0.10 Hz, n = 9 vs. STZ-ischemia: 3.35 ± 0.68 Hz, n = 7, P < 0.01). Furthermore, the membrane properties of these neurons were investigated (Tables 1, 2). The resting membrane potential was depolarized in diabetic animals with ischemia (Control: −89. 3 ± 0.9 mV; STZ-ischemia: −77.2 ± 4.0 mV, P < 0.05), the input resistance was also increased (Control: 248.1 ± 37.8 MΩ; STZ-ischemia: 338.5 ± 38.0 MΩ, P < 0.05). The latency of first spike was significantly shortened (Figure 5B, Control: 0.467 ± 0.079 s; STZ-ischemia: 0.046 ± 0.024 s, P < 0.01). These data suggest that the hyperexcitability of granule cells after ischemia may be related to the onset of seizure.
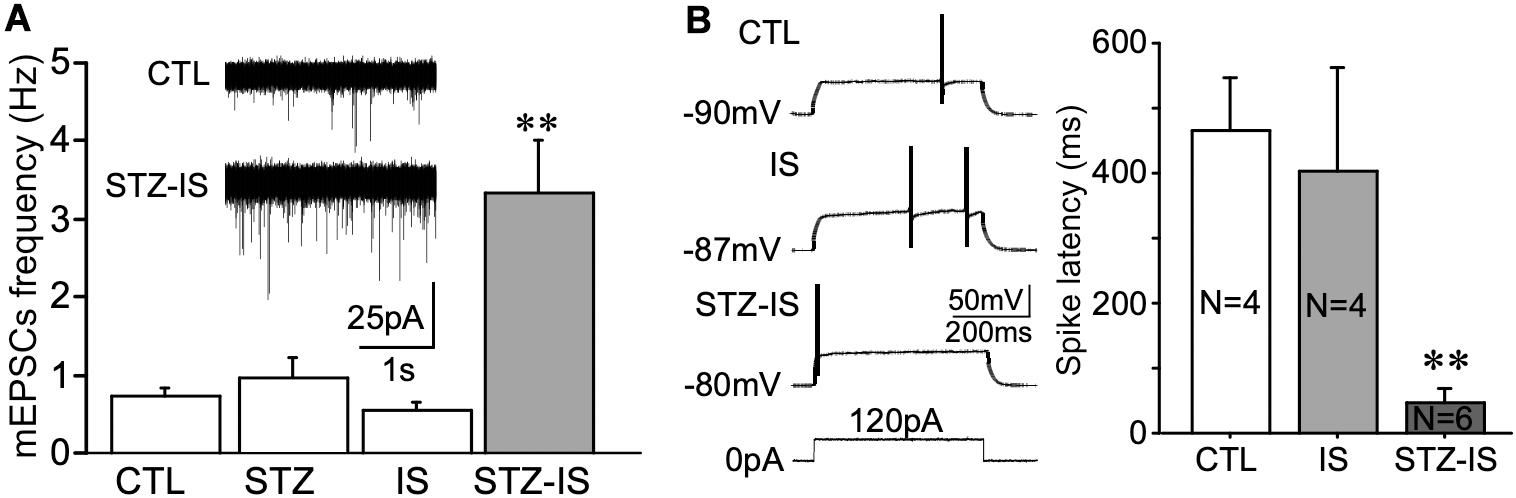
Figure 5. Electrophysiological recording displaying changes in neuronal excitability in different experimental groups. (A) The frequency of mEPSCs in granule cells significantly increases after ischemia in diabetic rats (**P < 0.01 vs. CTL). (B) The latency of the first spike evoked by depolarizing current pulse in granule cells of STZ/IS animals was significantly shorter than the control ones (**P < 0.01), suggesting that the membrane excitability was increased after ischemia in diabetic rats.

Table 1. Passive membrane properties of granule cells after ischemia in diabetic rats.

Table 2. Active membrane properties of granule cells after ischemia in diabetic rats.
IA currents play an important role in controlling neuronal excitability and therefore may contribute to seizure generation after ischemia. Kv4.2 is the major subunit of IA channels. Western blotting showed a significant reduction of total Kv4.2 protein levels in the hippocampus of seizure animals (47.1 ± 9.7% of control, n = 3, P < 0.01; Figure 6A). IA was recorded from dentate granule cells of hippocampal slices. The current density of IA was reduced after 15-min ischemia in diabetic animals (Figure 6B). These data suggest that IA currents of granule cells in diabetic rats are decreased after ischemia and contribute to the increase of excitability.
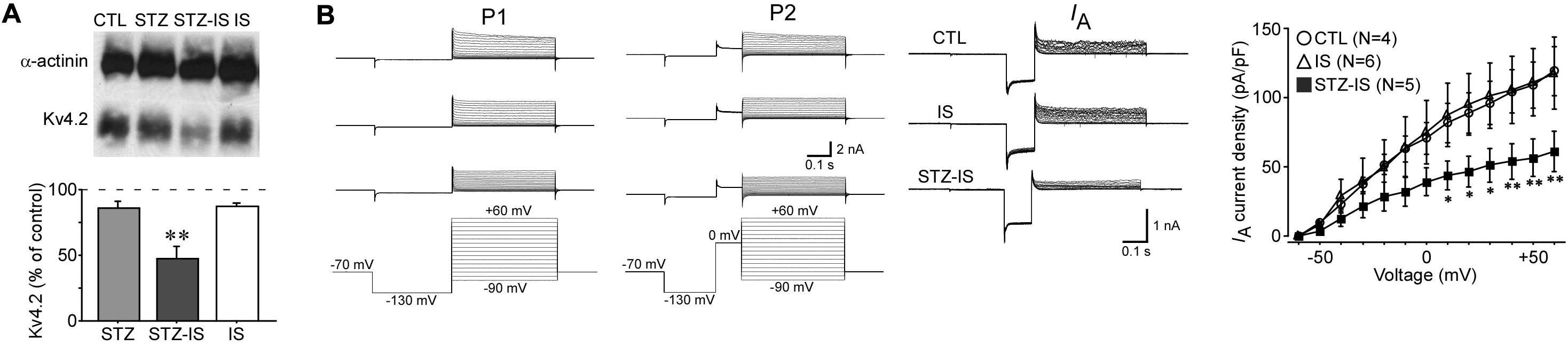
Figure 6. Electrophysiological recording showing changes in neuronal inhibition in different experimental groups. (A) Western blotting showing a reduction of total Kv4.2 protein levels in seizure rats after 15-min diabetic ischemia (**P < 0.01 vs. control group). (B) IA currents in granule cells were reduced in diabetic rats after ischemia (*P < 0.05 vs. control group).
Discussion
Epilepsy has long-term devastating effects on stroke victims and a financial burden on society. It is well known that diabetic patients have a higher incidence of stroke and worse post-stroke seizures as compared to the non-diabetic patients (Krakow et al., 2010; Adelow et al., 2011). However, the mechanisms of post-stroke seizure/epilepsy in patients with diabetes are not fully understood and no effective treatment is currently available to those patients. The current treatment of seizures following brain injury is symptomatic and could not prevent the recurrent epilepsy (Temkin et al., 1990; Temkin, 2009).
Our studies and others have shown that transient global ischemia induces selective neuronal death in vulnerable area in the CA1 pyramidal neurons of the hippocampus, cortical pyramidal neurons and spiny neurons of the striatum, rather than causes infarct area of the cerebral cortex (Kirino, 1982; Pulsinelli et al., 1982; Ruan et al., 2006; Shi et al., 2017). Delayed neuronal death usually occurs in the hippocampus at 2–3 days after transient global ischemia. That is why we did not detect the cell death in the hippocampus at 12 h after ischemia. In this case, the incidence of seizures after ischemia seems not related to the CA1 neural death of the hippocampus.
Recent studies found that sestrin 3 is a regulator of a proconvulsant gene network in human epileptic hippocampus and seizures are reduced by inhibition of high rates of glucose metabolism via lactate dehydrogenase (LDH) (Johnson et al., 2015; Sada et al., 2015). SESN3 has been implicated in the regulation of multiple pathways including AMPK, mTORC1, and mTORC2 axes, which in turn regulates hepatic insulin signal transduction and glucose metabolism (Tao et al., 2015). Therefore, it is conceivable that SESN3 is associated with the seizures following ischemia in diabetes.
To investigate the role of SESN3 in post-ischemia seizures under diabetic condition, we established a mouse model of post-ischemia seizures. Compared with the rat model we used before (Xia et al., 2016), it is useful to use knockout mice to establish the causal relationship of SESN3 in post-ischemic seizures. First, we executed STZ injection to induce diabetes in mice. At 7 days after injection, the increased blood glucose, the reduced body weight and the enhanced amount of urine indicate the diabetic status. The present study showed that there was a trend of increased SESN3 protein level and a significant increase in SESN3 mRNA level in STZ-IS group, which is positive associated with post-ischemic seizures. This suggests that SESN3 gene plays important role in post-ischemic seizures.
Global cerebral ischemia occurs when the blood supply to the entire brain or a large part of the brain is disrupted, resulting in the tissue deprived of oxygen and glucose that damages the brain. It is well known that diabetes exacerbates ischemic brain damage (Tureyen et al., 2011; Wei et al., 2013). We examined the brain damage in our mouse model. It is well known that transient global ischemia mainly damages the CA1 pyramidal neurons of hippocampus rather than the CA3 neurons, and this kind of cell death was delayed until 3 days after ischemia and reperfusion (Kirino, 1982; Arai et al., 1986). In the present study, our HE staining showed that there was no obvious cell death both in CA1 and CA3 area of hippocampus less than 24 h after ischemia in wild type mice with normal blood glucose, which was consistent with the delayed neuronal death. We also did not observe neuronal death in the hippocampus when the mouse brains were immediately fixed at the onset of post-ischemic seizures in diabetic animals. However, if the mouse brains were fixed after the occurrence of seizures, the CA1 neurons remained intact while some neuronal death was observed in the CA3 and hilus areas (Lei et al., 2014, 2016). The above observation suggests that the neuronal death in the CA3 and hilus areas may not be a result of ischemia, but may be related to the seizure activities.
Increase of neuronal excitability is a widely recognized cause of seizures/epilepsy generation. Hippocampus is a critical structure as an epileptic focus and epileptogenic zone, and thus as a target of resections for surgical treatment of temporal lobe epilepsy. It is possible that the both membrane excitability and excitatory synaptic transmission are enhanced in hippocampal neurons in diabetic animals after ischemia, which results in seizures and epilepsy. A long-term hyperexcitability and seizure-like activities have been recorded in cortical and hippocampal slices prepared from animals after global ischemia (Luhmann and Heinemann, 1992; Congar et al., 1995). Recently, combining in vivo EEG and in vitro voltage-clamp recording, Epsztein et al. have shown that the seizure threshold in CA3 pyramidal neurons is reduced after cerebral ischemia (Epsztein et al., 2006). Intrinsic membrane properties play an important role in neuronal excitability and therefore the alteration of membrane properties after ischemia is closely associated with seizure generation. On the other hand, Congar et al. (1995) have shown that the resting membrane potential of CA3 neurons shift to more positive levels after ischemia. In addition, in epileptic animals, the inhibitory basket cells in dentate gyrus receive less excitatory synaptic drive, the transmission failure to granule cells is increased (Lowery et al., 2009). Consistent with the above observations, the frequency of miniature inhibitory postsynaptic currents (mIPSCs) in granule cells is reduced in TLE or kainate induced seizure (Shao and Dudek, 2005). As we know there is no evidence showing glia cells express SESN3 by far. Therefore, it is possible that the both membrane excitability and excitatory synaptic transmission are enhanced in hippocampal neurons in diabetic animals after ischemia, which results in seizures and epilepsy.
In recent years, accumulating evidence suggests the involvement of inflammatory and immune processes in the etiopathogenesis of seizures (Vezzani and Granata, 2005). Inflammatory responses induced by stroke are associated with acute symptomatic seizures and a high risk of developing epilepsy (Bartfai et al., 2007; Vezzani et al., 2011). It is conceivable that up-regulation of sestrins in hippocampus could lead to seizure generation in diabetes after ischemia. However, how sestrins regulate inflammation in the brain and their role in epileptogenesis in diabetes are not clear. Recent studies have shown that glucose regulates the mTOR pathway through a Sestrin-dependent pathway (Ben-Sahra et al., 2013). Several preclinical and some clinical studies have indicated that the hyperactivation of the mTOR signaling pathway appears involved in acquiring epilepsy (Griffith and Wong, 2018). Here, we found an increased neuronal excitability, together with decreased IA currents and Kv4.2 in the hippocampus. It is well-known that IA currents are important gatekeepers of excitability and synchronicity in the CNS. Somadendritic IA currents are mainly mediated by the potassium channel Kv4.2. IA currents and Kv4.2 protein levels are decreased in rodent models of epilepsy, suggesting that the downregulation of IA currents is a pathological mechanism in epilepsy (Lei et al., 2012, 2014). So far, the mechanisms that lead to this downregulation of IA currents and Kv4.2 protein levels are still unclear. activation of mTORC1 signaling increases the abundance of sestrins which prevents ROS accumulation via inhibition of mTOR (Lee et al., 2010). It has been reported that mTOR activity was increased in animals with epilepsy (Ostendorf and Wong, 2015; Citraro et al., 2016). Evidence showed that mTOR dysregulation could potentially contribute to hyperexcitability and recurrent seizures by regulating Kv4.2-mediated IA currents (Yao et al., 2013; Nguyen and Anderson, 2018). We speculate that the SESN3-regulated mTOR pathway might cause the downregulation of Kv4.2 and IA currents, resulting in neuronal hyperexcitability and the development of post-ischemic seizures under diabetic condition.
We found that the frequency of mEPSCs was significantly increased in diabetic animals after ischemia, and no significant changes was observed in the amplitude of mEPSCs. This finding suggest that presynaptic changes may alter firing properties of postsynaptic DG cells. NMDA receptors are assembled from the NR1 subunit and at least one type of NR2 subunit, mainly NR2A and/or NR2B. NR2A-containing NMDA receptors are believed to be located predominantly at synaptic sites, whereas NR2B-containing NMDA receptors are located predominantly extrasynaptically. Distinct roles of NR2A- versus NR2B-containing or synaptic versus extrasynaptic NMDA receptors have been extensively studied in synaptic plasticity and neurodegenerative diseases (Hardingham et al., 2002). Our previous study have shown that NR2B-containing NMDA receptor-mediated downregulation of Kv4.2 potassium channels (Lei et al., 2010).
Dentate granule cells in the hippocampus survive after ischemic insult but are implicated in seizure generation (Pun et al., 2012). The abnormal hyperexcitability of granule cells results in seizure activities (Young et al., 2009). Our results suggest that the hyperexcitability of granule cells after ischemia in diabetes may be related to the onset of seizure by downregulation of IA currents.
In summary, the present study demonstrates that seizure incidence increased in diabetic animals after ischemia together with an up-regulation of SESN3 expression. The seizure rate of sesn3 KO mice was significantly reduced after ischemia compared with the wild type mice. The excitability of hippocampal neurons was increased and Kv4.2-mediated IA currents was decreased in these animals. These results suggest that up-regulation of SESN3 contributes to post-ischemia seizures in the diabetic condition.
However, there are some shortcomings of the study, such as due to time point of sacrifice changes can be cause or consequence of seizures, lack of non-diabetic sesn3 KO group, and lack of EEG for quantification of seizure activity. More precise experimental design is necessary for the further study.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animals Care and Use Committee of the Indiana University School of Medicine.
Author Contributions
ZX and YR conceived and designed the experiments. ZS and ZL performed the experiments. ZS, ZL, FW, and LX analyzed the data. ZS, ZL, ZX, and YR wrote, revised, and commented on the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by AHA 13SDG17140056 (to ZL), the ISCBIR grant 19966 (to ZX), and 14GRNT20410061 and ISDH-19966 (to ZX). This work was also supported by the National Natural Science Foundation of China (30971530), the 111 Project, Ministry of Education and State Administration of Foreign Experts, China (B14036), and the Key Basic Study and Functional Product Research of Wolfberry Grant from Ningxia Province.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Drs. X. Charlie Dong, Xiwen Xiong, and Yang Zhang (Department of Biochemistry and Molecular Biology, Indiana University School of Medicine) for providing the Sesn3 KO mice and technical support. The authors also thank Dr. Greg Campbell, the professor of Department of Cell and Developmental Biology, University College London, for reviewing the manuscript and proofreading of English grammar.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2020.591207/full#supplementary-material
References
Adelow, C., Andersson, T., Ahlbom, A., and Tomson, T. (2011). Prior hospitalization for stroke, diabetes, myocardial infarction, and subsequent risk of unprovoked seizures. Epilepsia 52, 301–307. doi: 10.1111/j.1528-1167.2010.02757.x
Arai, H., Passonneau, J. V., and Lust, W. D. (1986). Energy metabolism in delayed neuronal death of CA1 neurons of the hippocampus following transient ischemia in the gerbil. Metab. Brain Dis. 1, 263–278.
Barnwell, L. F., Lugo, J. N., Lee, W. L., Willis, S. E., Gertz, S. J., Hrachovy, R. A., et al. (2009). Kv4.2 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia 50, 1741–1751. doi: 10.1111/j.1528-1167.2009.02086.x
Bartfai, T., Sanchez-Alavez, M., Andell-Jonsson, S., Schultzberg, M., Vezzani, A., Danielsson, E., et al. (2007). Interleukin-1 system in CNS stress: seizures, fever, and neurotrauma. Ann. N. Y. Acad. Sci. 1113, 173–177. doi: 10.1196/annals.1391.022
Ben-Sahra, I., Dirat, B., Laurent, K., Puissant, A., Auberger, P., Budanov, A., et al. (2013). Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ. 20, 611–619. doi: 10.1038/cdd.2012.157
Bladin, C. F., Alexandrov, A. V., Bellavance, A., Bornstein, N., Chambers, B., Cote, R., et al. (2000). Seizures after stroke: a prospective multicenter study. Arch. Neurol. 57, 1617–1622.
Budanov, A. V., and Karin, M. (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460. doi: 10.1016/j.cell.2008.06.028
Budanov, A. V., Lee, J. H., and Karin, M. (2010). Stressin’ Sestrins take an aging fight. EMBO Mol. Med. 2, 388–400. doi: 10.1002/emmm.201000097
Budanov, A. V., Sablina, A. A., Feinstein, E., Koonin, E. V., and Chumakov, P. M. (2004). Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 304, 596–600. doi: 10.1126/science.1095569
Carrasquillo, Y., Burkhalter, A., and Nerbonne, J. M. (2012). A-type K+ channels encoded by Kv4.2, Kv4.3 and Kv1.4 differentially regulate intrinsic excitability of cortical pyramidal neurons. J. Physiol. 590(Pt 16), 3877–3890. doi: 10.1113/jphysiol.2012.229013
Citraro, R., Leo, A., Constanti, A., Russo, E., and De Sarro, G. (2016). mTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol. Res. 107, 333–343. doi: 10.1016/j.phrs.2016.03.039
Congar, P., Khazipov, R., and Ben-Ari, Y. (1995). Direct demonstration of functional disconnection by anoxia of inhibitory interneurons from excitatory inputs in rat hippocampus. J. Neurophysiol. 73, 421–426. doi: 10.1152/jn.1995.73.1.421
Epsztein, J., Milh, M., Bihi, R. I., Jorquera, I., Ben-Ari, Y., Represa, A., et al. (2006). Ongoing epileptiform activity in the post-ischemic hippocampus is associated with a permanent shift of the excitatory-inhibitory synaptic balance in CA3 pyramidal neurons. J. Neurosci. 26, 7082–7092. doi: 10.1523/jneurosci.1666-06.2006
Griffith, J. L., and Wong, M. (2018). The mTOR pathway in treatment of epilepsy: a clinical update. Future Neurol. 13, 49–58. doi: 10.2217/fnl-2018-0001
Hardingham, G. E., Fukunaga, Y., and Bading, H. (2002). Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414. doi: 10.1038/nn835
Johnson, M. R., Behmoaras, J., Bottolo, L., Krishnan, M. L., Pernhorst, K., Santoscoy, P. L. M., et al. (2015). Systems genetics identifies Sestrin 3 as a regulator of a proconvulsant gene network in human epileptic hippocampus. Nat. Commun. 6:6031. doi: 10.1038/ncomms7031
Kirino, T. (1982). Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 239, 57–69. doi: 10.1016/0006-8993(82)90833-2
Krakow, K., Sitzer, M., Rosenow, F., Steinmetz, H., Foerch, C., and Arbeitsgruppe Schlaganfall, H. (2010). Predictors of acute poststroke seizures. Cerebrovasc. Dis. 30, 584–589. doi: 10.1159/000319777
Lee, J. H., Budanov, A. V., Park, E. J., Birse, R., Kim, T. E., Perkins, G. A., et al. (2010). Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 327, 1223–1228. doi: 10.1126/science.1182228
Lee, J. H., Budanov, A. V., Talukdar, S., Park, E. J., Park, H. L., Park, H. W., et al. (2012). Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 16, 311–321. doi: 10.1016/j.cmet.2012.08.004
Lei, Z., Deng, P., Li, J., and Xu, Z. C. (2012). Alterations of A-type potassium channels in hippocampal neurons after traumatic brain injury. J, Neurotrauma 29, 235–245. doi: 10.1089/neu.2010.1537
Lei, Z., Deng, P., Li, Y., and Xu, Z. C. (2010). Downregulation of Kv4.2 channels mediated by NR2B-containing NMDA receptors in cultured hippocampal neurons. Neuroscience 165, 350–362. doi: 10.1016/j.neuroscience.2009.10.041
Lei, Z., Zhang, H., Liang, Y., Cui, Q., Xu, Z., and Xu, Z. C. (2014). Reduced expression of IA channels is associated with postischemic seizures in hyperglycemic rats. J. Neurosci. Res. 92, 1775–1784. doi: 10.1002/jnr.23445
Lei, Z., Zhang, H., Liang, Y., and Xu, Z. C. (2016). Reduced expression of IA channels is associated with post-ischemic seizures. Epilepsy Res. 124, 40–48. doi: 10.1016/j.eplepsyres.2016.05.008
Li, Y., Blanco, G. D., Lei, Z., and Xu, Z. C. (2010). Increased GAD expression in the striatum after transient cerebral ischemia. Mol. Cell. Neurosci. 45, 370–377. doi: 10.1016/j.mcn.2010.07.010
Li, Y., Lei, Z., and Xu, Z. C. (2009). Enhancement of inhibitory synaptic transmission in large aspiny neurons after transient cerebral ischemia. Neuroscience 159, 670–681. doi: 10.1016/j.neuroscience.2008.12.046
Lowery, R. L., Zhang, Y., Kelly, E. A., Lamantia, C. E., Harvey, B. K., and Majewska, A. K. (2009). Rapid, long-term labeling of cells in the developing and adult rodent visual cortex using double-stranded adeno-associated viral vectors. Dev. Neurobiol. 69, 674–688. doi: 10.1002/dneu.20735
Luhmann, H. J., and Heinemann, U. (1992). Hypoxia-induced functional alterations in adult rat neocortex. J. Neurophysiol. 67, 798–811. doi: 10.1152/jn.1992.67.4.798
Malhotra, A., Penpargkul, S., Fein, F. S., Sonnenblick, E. H., and Scheuer, J. (1981). The effect of streptozotocin-induced diabetes in rats on cardiac contractile proteins. Circ. Res. 49, 1243–1250. doi: 10.1161/01.res.49.6.1243
Nguyen, L. H., and Anderson, A. E. (2018). mTOR-dependent alterations of Kv1.1 subunit expression in the neuronal subset-specific Pten knockout mouse model of cortical dysplasia with epilepsy. Sci. Rep. 8:3568. doi: 10.1038/s41598-018-21656-8
Ostendorf, A. P., and Wong, M. (2015). mTOR inhibition in epilepsy: rationale and clinical perspectives. CNS Drugs 29, 91–99. doi: 10.1007/s40263-014-0223-x
Ottman, R., Lipton, R. B., Ettinger, A. B., Cramer, J. A., Reed, M. L., Morrison, A., et al. (2011). Comorbidities of epilepsy: results from the Epilepsy Comorbidities and Health (EPIC) survey. Epilepsia 52, 308–315. doi: 10.1111/j.1528-1167.2010.02927.x
Pulsinelli, W. A., Brierley, J. B., and Plum, F. (1982). Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 11, 491–498. doi: 10.1002/ana.410110509
Pun, R. Y., Rolle, I. J., Lasarge, C. L., Hosford, B. E., Rosen, J. M., Uhl, J. D., et al. (2012). Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron 75, 1022–1034. doi: 10.1016/j.neuron.2012.08.002
Racine, R. J. (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294.
Ruan, Y. W., Lei, Z., Fan, Y., Zou, B., and Xu, Z. C. (2009). Diversity and fluctuation of spine morphology in CA1 pyramidal neurons after transient global ischemia. J. Neurosci. Res. 87, 61–68. doi: 10.1002/jnr.21816
Ruan, Y. W., Zou, B., Fan, Y., Li, Y., Lin, N., Zeng, Y. S., et al. (2006). Dendritic plasticity of CA1 pyramidal neurons after transient global ischemia. Neuroscience 140, 191–201. doi: 10.1016/j.neuroscience.2006.01.039
Sada, N., Lee, S., Katsu, T., Otsuki, T., and Inoue, T. (2015). Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367. doi: 10.1126/science.aaa1299
Shao, L. R., and Dudek, F. E. (2005). Detection of increased local excitatory circuits in the hippocampus during epileptogenesis using focal flash photolysis of caged glutamate. Epilepsia 46(Suppl. 5), 100–106. doi: 10.1111/j.1528-1167.2005.01018.x
Shi, Z., Zhu, L., Li, T., Tang, X., Xiang, Y., Han, X., et al. (2017). Neuroprotective mechanisms of Lycium barbarum polysaccharides against ischemic insults by regulating NR2B and NR2A containing NMDA receptor signaling pathways. Front. Cell. Neurosci. 11:288. doi: 10.3389/fncel.2017.00288
Singh, B., Ogiwara, I., Kaneda, M., Tokonami, N., Mazaki, E., Baba, K., et al. (2006). A Kv4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol. Dis. 24, 245–253.
Srinivasan, K., Viswanad, B., Asrat, L., Kaul, C. L., and Ramarao, P. (2005). Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol. Res. 52, 313–320. doi: 10.1016/j.phrs.2005.05.004
Tao, R., Xiong, X., Liangpunsakul, S., and Dong, X. C. (2015). Sestrin 3 protein enhances hepatic insulin sensitivity by direct activation of the mTORC2-Akt signaling. Diabetes 64, 1211–1223. doi: 10.2337/db14-0539
Temkin, N. R. (2009). Preventing and treating posttraumatic seizures: the human experience. Epilepsia 50(Suppl. 2), 10–13. doi: 10.1111/j.1528-1167.2008.02005.x
Temkin, N. R., Dikmen, S. S., Wilensky, A. J., Keihm, J., Chabal, S., and Winn, H. R. (1990). A randomized, double-blind study of phenytoin for the prevention of post-traumatic seizures. N. Engl. J. Med. 323, 497–502. doi: 10.1056/NEJM199008233230801
Tureyen, K., Bowen, K., Liang, J., Dempsey, R. J., and Vemuganti, R. (2011). Exacerbated brain damage, edema and inflammation in type-2 diabetic mice subjected to focal ischemia. J. Neurochem. 116, 499–507. doi: 10.1111/j.1471-4159.2010.07127.x
Vezzani, A., French, J., Bartfai, T., and Baram, T. Z. (2011). The role of inflammation in epilepsy. Nat. Rev. Neurol. 7, 31–40. doi: 10.1038/nrneurol.2010.178
Vezzani, A., and Granata, T. (2005). Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46, 1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x
Wei, N., Yu, S. P., Gu, X. H., Chen, D. D., Whalin, M. K., Xu, G. L., et al. (2013). The involvement of autophagy pathway in exaggerated ischemic brain damage in diabetic mice. CNS Neurosci. Ther. 19, 753–763. doi: 10.1111/cns.12123
Xia, L., Lei, Z., Shi, Z., Guo, D., Su, H., Ruan, Y., et al. (2016). Enhanced autophagy signaling in diabetic rats with ischemia-induced seizures. Brain Res. 1643, 18–26. doi: 10.1016/j.brainres.2016.04.054
Yao, J. J., Sun, J., Zhao, Q. R., Wang, C. Y., and Mei, Y. A. (2013). Neuregulin-1/ErbB4 signaling regulates Kv4.2-mediated transient outward K+ current through the Akt/mTOR pathway. Am. J. Physiol. Cell Physiol. 305, C197–C206. doi: 10.1152/ajpcell.00041.2013
Young, C. C., Stegen, M., Bernard, R., Müller, M., Bischofberger, J., Veh, R. W., et al. (2009). Upregulation of inward rectifier K+ (Kir2) channels in dentate gyrus granule cells in temporal lobe epilepsy. J. Physiol. 587(Pt 17), 4213–4233. doi: 10.1113/jphysiol.2009.170746
Keywords: epilepsy, stroke, hyperglycemia, excitability, SESN3
Citation: Shi Z, Lei Z, Wu F, Xia L, Ruan Y and Xu ZC (2021) Increased Sestrin3 Contributes to Post-ischemic Seizures in the Diabetic Condition. Front. Neurosci. 14:591207. doi: 10.3389/fnins.2020.591207
Received: 04 August 2020; Accepted: 14 December 2020;
Published: 15 January 2021.
Edited by:
Mohammad Badruzzaman Khan, Augusta University, United StatesReviewed by:
Chengwen Zhou, Vanderbilt University, United StatesHamid Gholami Pourbadie, Pasteur Institute of Iran (PII), Iran
Copyright © 2021 Shi, Lei, Wu, Xia, Ruan and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zao C. Xu, emN4dUBpdXB1aS5lZHU=; Yiwen Ruan, dHlpd2VuQGpudS5lZHUuY24=; eWl3ZW5ydWFuQHlhaG9vLmNvbQ==
†These authors have contributed equally to this work