 Andrea I. Luppi
Andrea I. Luppi Lennart R. B. Spindler
Lennart R. B. Spindler David K. Menon
David K. Menon Emmanuel A. Stamatakis
Emmanuel A. Stamatakis- 1Division of Anaesthesia, School of Clinical Medicine, University of Cambridge, Cambridge, United Kingdom
- 2Department of Clinical Neurosciences, University of Cambridge, Cambridge, United Kingdom
- 3Wolfson Brain Imaging Centre, University of Cambridge, Cambridge, United Kingdom
“Neural inertia” is the brain’s tendency to resist changes in its arousal state: it is manifested as emergence from anaesthesia occurring at lower drug doses than those required for anaesthetic induction, a phenomenon observed across very different species, from invertebrates to mammals. However, the brain is also subject to another form of inertia, familiar to most people: sleep inertia, the feeling of grogginess, confusion and impaired performance that typically follows awakening. Here, we propose a novel account of neural inertia, as the result of sleep inertia taking place after the artificial sleep induced by anaesthetics. We argue that the orexinergic and noradrenergic systems may be key mechanisms for the control of these transition states, with the orexinergic system exerting a stabilising effect through the noradrenergic system. This effect may be reflected at the macroscale in terms of altered functional anticorrelations between default mode and executive control networks of the human brain. The hypothesised link between neural inertia and sleep inertia could explain why different anaesthetic drugs induce different levels of neural inertia, and why elderly individuals and narcoleptic patients are more susceptible to neural inertia. This novel hypothesis also enables us to generate several empirically testable predictions at both the behavioural and neural levels, with potential implications for clinical practice.
Introduction
Anaesthesia and Sleep
General anaesthesia refers to a pharmacological intervention designed to produce a state of controlled and reversible unconsciousness and unresponsiveness to sensory stimulation. Its discovery is among the greatest in medical history: it allows surgeons to perform millions of life-saving interventions every year, which would be otherwise impossible or extremely distressing.
However, the mechanisms of anaesthetic action in the brain remain incompletely understood—especially since multiple anaesthetic drugs exist, with different pharmacological profiles (Scharf and Kelz, 2013). Nevertheless, anaesthesia is not the only way in which one can become unconscious: the brain exhibits a strong need for periodic unconsciousness in the form of sleep, with the average human spending about a third of their life in this state. A sleep-like state of rapidly reversible physical quiescence, with elevated thresholds to sensory stimulation, has been identified in most species, including even insects (Shaw, 2000) and nematodes (Raizen et al., 2008).
In addition to behavioural similarities with sleep, several anaesthetic drugs generate EEG rhythms that resemble those observed during different stages of sleep: halothane and isoflurane produce a theta rhythm (5–9 Hz) reminiscent of rapid eye movement (REM) sleep (Pang et al., 2009), whereas the GABA-ergic agent propofol and the α2-adrenoreceptor agonist dexmedetomidine induce slow-wave activity (<4 Hz) analogous to what is observed during non-REM (NREM) sleep (Gent and Adamantidis, 2017). Given the behavioural and electrophysiological similarities between sleep and the effects of several anaesthetic agents, the neuronal circuitry underlying sleep may provide critical insights into the mechanisms of anaesthetic action (Karan et al., 2007), with evidence that at least some anaesthetics do in fact intervene on sleep-wake regulating neurons, especially in hypothalamic areas (Franks, 2008; Zecharia et al., 2009; Zhang et al., 2015; Gent and Adamantidis, 2017)—although it should be noted that this similarity is not universal: some other anaesthetics produce desynchronised EEG with little resemblance to sleep EEG, e.g., ketamine, benzodiazepines (Gent and Adamantidis, 2017). The function of sleep is only partly understood, and several different theories have been put forward to explain the existence of this peculiar state (Vyazovskiy, 2015; Joiner, 2016; Krueger et al., 2016), including energy restoration (Berger and Phillips, 1995; Schmidt, 2014) memory consolidation (Abel et al., 2013) and synaptic homeostasis (Tononi and Cirelli, 2014, 2016). Nevertheless, the brain circuits that control sleep are relatively well understood: a wake-promoting and a sleep-promoting system interact in the brain (Figure 1; Saper et al., 2005; Luppi, 2010; Weber and Dan, 2016).
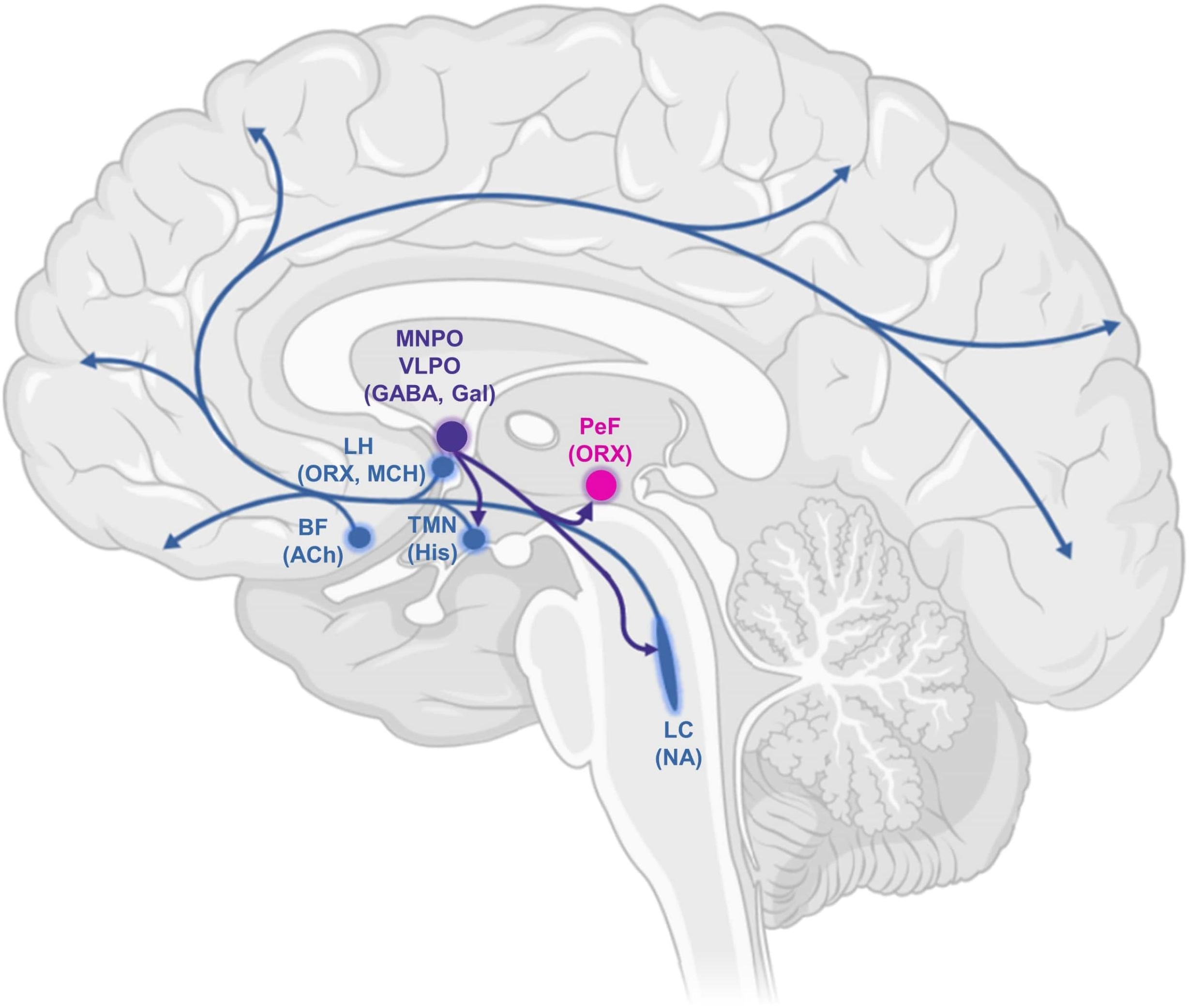
Figure 1. Schematic drawing of some key components of the ascending arousal system, highlighting projections of the ventrolateral preoptic area. This comprises cortical projection neurons originating from the basal forebrain (BF); the recently characterised orexin/hypocretin neurons in the lateral hypothalamus (LH); perifornical orexin neurons (PeF); and several monoaminergic nuclei: the noradrenergic locus coeruleus (LC), the histaminergic tuberomammillary nucleus (TMN) and the ventrolateral preoptic area (VLPO) and median preoptic area (MNPO). Serotonergic and dopaminergic components are not shown. MCH, melanin-concentrating hormone; Gal, galanin; Ach, acetylcholine; ORX, orexin; His, histamine; NA, noradrenaline.
The ascending reticular activating system (Moruzzi and Magoun, 1949) comprises cholinergic, monoaminergic (serotonin, noradrenaline, histamine) and orexinergic nuclei in the brainstem, basal forebrain, and hypothalamus—with wide-ranging projections throughout the entire brain (Luppi, 2010). The hypothalamus also contains key sleep-promoting neuronal populations; in particular, the ventrolateral preoptic area (VLPO) and median preoptic area (MNPO) primarily express the inhibitory neurotransmitters γ-aminobutyric acid (GABA) and galanin, and project to all major hypothalamic and brainstem nuclei of the wake-promoting system (Sherin et al., 1996). Homeostatically arranged, the sleep-active neurons of the preoptic hypothalamus are in turn inhibited by the wake-active nuclei they target, especially those of predominantly noradrenergic and serotonergic transmitter phenotype (Gallopin et al., 2000; Chou et al., 2002). This architecture of mutually inhibitory wake-promoting and sleep-promoting circuits constitutes what is known as a “flip-flop switch” (Saper et al., 2001, 2005, 2010): a bistable system characterised by sharp transitions between its two possible states. Damage to the wake-promoting system causes excessive sleep, while insomnia results from damage to the VLPO (Economo, 1930; Lu et al., 2000). In addition to their sleep-promoting effects, VLPO neurons have also been implicated in the mechanisms of action of anaesthetic drugs (Moore et al., 2012; Zhang et al., 2015). Of note, recent evidence also indicates a common role of hypothalamic neuroendocrine cells of the mouse in sleep generation and general anaesthesia induced by several different anaesthetics, with opto- or chemo-genetic activation of these cells promoting both slow-wave sleep and anaesthesia, and the opposite result obtained by inhibiting them (Jiang-Xie et al., 2019).
Current theories propose that at least some anaesthetic drugs may exert their effect by recruiting the brain’s endogenous mechanisms for the production of unconsciousness (Franks, 2008; Alkire et al., 2009; Scharf and Kelz, 2013; Van Swinderen and Kottler, 2014; but see Vanini et al., 2020, for a recent suggestion that this may not be the case, for isoflurane). This may occur through activation of the sleep-promoting pathways, inhibition of the wake-promoting ones, or both [especially since, given their mutually inhibitory nature, activating one will also result in inhibition of the other (Pace-Schott and Hobson, 2002)].
Neural Inertia and Sleep Inertia
Neural Inertia
“Neural inertia” refers to the brain’s tendency to resist changes in its arousal state: it is manifested as emergence from anaesthesia (recovery of responsiveness, ROR) occurring at lower drug doses than those required for anaesthetic induction (loss of responsiveness, LOR) (Friedman et al., 2010). Thus, for intermediate dosages between those required for ROR and LOR, a given individual may be anaesthetised or awake, depending on their previous state. This “path dependence” (referred to as hysteresis in physics; Figure 2) is in contrast with pharmacokinetic-pharmacodynamic accounts, which assume that anaesthetic state is fully determined by current effect-site concentration of anaesthetic (McKay et al., 2006).
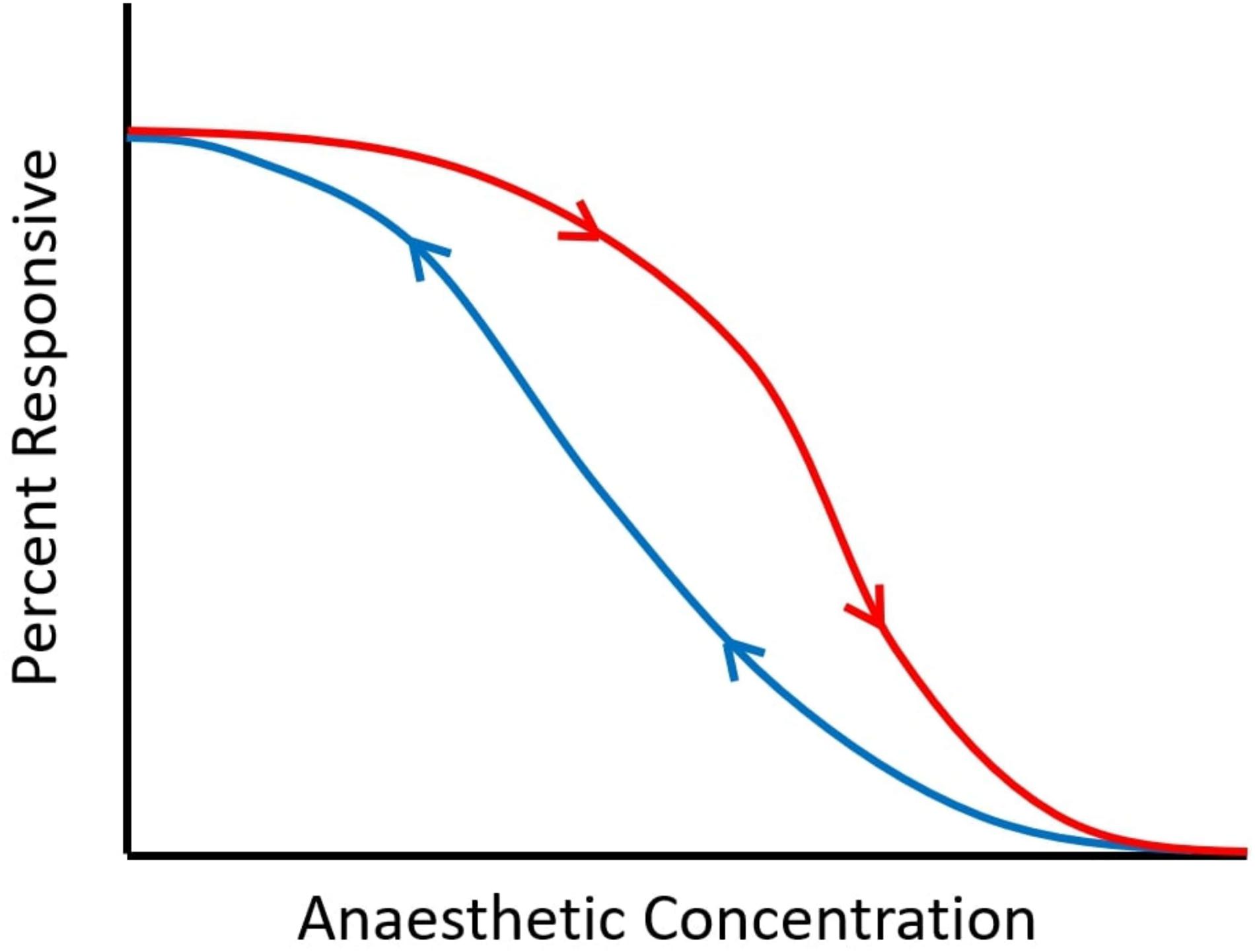
Figure 2. Schematic of neural inertia. As anaesthetic dose is increased, responsiveness is diminished. However, the dose at which a certain proportion of responses is observed is not the same for induction (downward arrow, in red) and emergence (upward arrow, in blue), indicating path-dependence (hysteresis). Between the two curves, subjects may be awake or anaesthetised, depending on whether the drug concentration is being increased or decreased. The wider the gap between the two curves, the greater the hysteresis.
Rather, evidence of hysteresis between anaesthetic induction and emergence obtained in mice and Drosophila led to the proposal that the brain has a tendency to resist transitions in its arousal state, called “neural inertia” (Friedman et al., 2010). Indeed, bistable systems—of which the brain appears to be one, with respect to its sleep-wake states (Saper et al., 2001)—tend to show distinct non-overlapping paths between their states, indicating hysteresis (Chatterjee et al., 2008). Consistent with theoretical work on anaesthesia (Steyn-Ross et al., 2004), this is precisely what Friedman and colleagues observed with regard to anaesthetic induction and emergence in both mammals and invertebrates (Friedman et al., 2010). However, evidence for neural inertia in humans is less clear-cut (Sepúlveda et al., 2019) since it is not possible to measure anaesthetic concentration in the brain in the same way this is commonly done in animal models. Sepulveda and colleagues (Sepúlveda et al., 2018) found that LOR occurred at greater propofol concentrations than ROR, but noted that this result may be alternatively explained by incomplete equilibration between plasma and effect-site concentrations. A different team of researchers (Kuizenga et al., 2018) did not find evidence of neural inertia with propofol, whereas they did observe it with sevoflurane, when combined with the opioid remifentanil. These authors also observed that the choice of marker (behavioural endpoint) with respect to which to compute differences in drug concentration at induction and emergence (e.g., loss and recovery of responsiveness, or EEG features) may also make a difference in investigators’ ability to detect evidence of neural inertia (Kuizenga et al., 2018).
In line with this observation, Warnaby et al. (2017) reported hysteresis for the prevalence of slow-wave EEG activity for both propofol and sevoflurane, with or without addition of opioids; slow-wave persistence was therefore proposed as a marker of neural inertia in humans. While some authors (Colin et al., 2018) criticised this study by arguing that the hysteresis observed by Warnaby and colleagues can be collapsed if a different effect-site equilibration model is assumed, recent modelling work by Proekt and Kelz (2020) demonstrated that—since effect-site concentration is a theoretical construct that cannot be measured directly—it is experimentally impossible to distinguish between an equilibration model that collapses hysteresis and one that does not, even when hysteresis is in part attributable to genuine neuronal dynamics. Therefore, although it is clear that improved methodologies will be required (Proekt and Kelz, 2020), there is reason to believe that humans may also be subject to neural inertia—a postulation consistent with the unequivocal evidence that neural inertia is a widespread phenomenon observed in species as diverse as fruit flies, zebrafish, and rodents (Sepúlveda et al., 2019; Wasilczuk et al., 2020). As Proekt and Kelz observe: “whereas going from the structured to the unstructured state is trivial, the restoration of structure is not generically expected after a dramatic perturbation” (Proekt and Kelz, 2020). Thus, emergence may be an active rather than passive phenomenon, the understanding of which will likely need to invoke specific and distinct neurobiological mechanisms beyond a mere reversal of the induction process.
Sleep Inertia
Transitions in the brain’s arousal state do not occur only after anaesthesia, but also after sleep. Familiar to many people, this state of transition between sleep and wakefulness, characterised by low levels of arousal and vigilance, sleepiness, confusion, and a temporary reduction in performance, is called sleep inertia (SI) (Tassi and Muzet, 2000; Voss, 2010; Trotti, 2017). Sleep inertia dissipates with time awake, with estimates of its typical duration ranging from 20 to 30 min (Dinges et al., 1987; Tassi et al., 1992) to 1–2 h post-awakening (Jewett et al., 1999). Although sleep inertia occurs even in the absence of sleep debt (Akerstedt and Folkard, 1997), its effects are more profound and long-lasting after a period of sleep deprivation (Ferrara and De Gennaro, 2000). Finally, waking up from slow-wave sleep appears to have the most profound negative impact on subsequent vigilance and performance (Dinges, 1990; Bonnet, 1993; Matchock and Mordkoff, 2014).
From a behavioural perspective, sleep inertia affects performance in the same way as sleepiness (Balkin and Badia, 1988). The human electroencephalographic (EEG) signatures of sleep inertia are also analogous to what is observed at increased levels of sleepiness (Voss, 2010). For approximately 10 min post-awakening, EEG is characterised by elevated low-frequency (1–9 Hz) and reduced beta (18–25 Hz) power (Ogilvie and Simons, 1992; Ferrara et al., 2006; Marzano et al., 2011). Analogous results have been obtained in rodents using intracranial recordings during the first 10 min post-sleep: neuronal activity was low upon awakening, with brief periods of neuronal silence (Vyazovskiy et al., 2014). Crucially, such population OFF periods are typically observed not only during sleep, but also after prolonged wake, as revealed by intracranial recordings in rats (Vyazovskiy et al., 2011). Likewise, recordings in monkeys transitioning from wake to sleep show sleep-like patterns of activity in their visual cortex, even while performing a visual task (Pigarev et al., 1997). Thus, across species sleep inertia appears to be the post-sleep counterpart of pre-sleep sleepiness, with both states characterised by similar behavioural changes and EEG signatures, as well as local sleep-like OFF periods.
Neural Inertia as the Effect of Sleep Inertia
Single-gene mutations that increase or decrease neural inertia also affect the sleep-wake cycle, pointing to a connection between anaesthesia, neural inertia and sleep in both invertebrates and mammals (Friedman et al., 2010; Joiner et al., 2013). Here, we propose that neural inertia—the reduction in anaesthetic dose required for emergence compared to induction—may be an effect of the sleep inertia that follows anaesthetic-induced sleep. Specifically, GABA-ergic anaesthetics such as propofol and the inhalational agents sevoflurane, isoflurane, and halothane are believed to induce a state of artificial sleep (Brown et al., 2010; Van Swinderen and Kottler, 2014; but see Vanini et al., 2020). Like natural sleep, this artificial sleep should then be followed by sleep inertia—especially for intravenous drugs such as propofol that induce an artificial sleep characterised by high levels of slow-wave activity (SWA) (Brown et al., 2011; Murphy et al., 2011; Gent and Adamantidis, 2017), since sleep inertia is particularly pronounced upon awakening from slow-wave sleep (Dinges, 1990).
Thus, in the process of emerging from anaesthesia the brain would find itself in the state of sleep inertia, which is behaviourally and neurally equivalent to sleepiness. Since sleepiness is known to increase susceptibility to anaesthesia with propofol, isoflurane, and sevoflurane by lowering the dose that is required for induction, as indicated by rodent studies (Tung et al., 2002; Pal et al., 2011; Scharf and Kelz, 2013), this could explain neural inertia: due to being in a state equivalent to sleepiness, the brain during emergence is more susceptible to anaesthetics than it was at induction, and a smaller dose is sufficient to maintain unconsciousness—producing the hysteresis characteristic of neural inertia.
If this hypothesis is correct, then we predict that neural inertia should be larger when awakening from “recovery sleep” after sleep deprivation, since sleep deprivation increases the sleep inertia that is observed after awakening (Ferrara and De Gennaro, 2000). This is precisely what is observed empirically, with higher neural inertia in previously sleep-deprived animals (Joiner et al., 2013). Moreover, this hypothesis could explain why Friedman and colleagues (Friedman et al., 2010) observed greater neural inertia with halothane than with isoflurane—a result that was recently replicated in mice exposed to equipotent doses of isoflurane, sevoflurane, and halothane, demonstrating that different anaesthetics have different effects on neural inertia, distinct from their potency (Wasilczuk et al., 2020). Specifically, to explain these results we note that unlike isoflurane, halothane does not reduce NREM sleep-debt in rodents (Pick et al., 2011; Scharf and Kelz, 2013). Thus, higher levels of NREM sleep debt would be present upon emergence from halothane than isoflurane, leading to stronger sleep inertia, and hence stronger neural inertia, as observed.
Thus, we have proposed that anaesthesia causes artificial, SWA-rich sleep, which in turn induces sleep inertia. The latter’s effects resemble those of sleepiness, which increases sensitivity to anaesthetics. Therefore, a lower dose of anaesthetic will suffice to keep the brain anaesthetised, resulting in neural inertia at emergence (Figure 3). This hypothesis for the origin of neural inertia could be tested by inducing anaesthesia during the state of sleep inertia, and assessing the prediction that the induction dose will be lower than usual and comparable to the drug level at which emergence typically occurs.
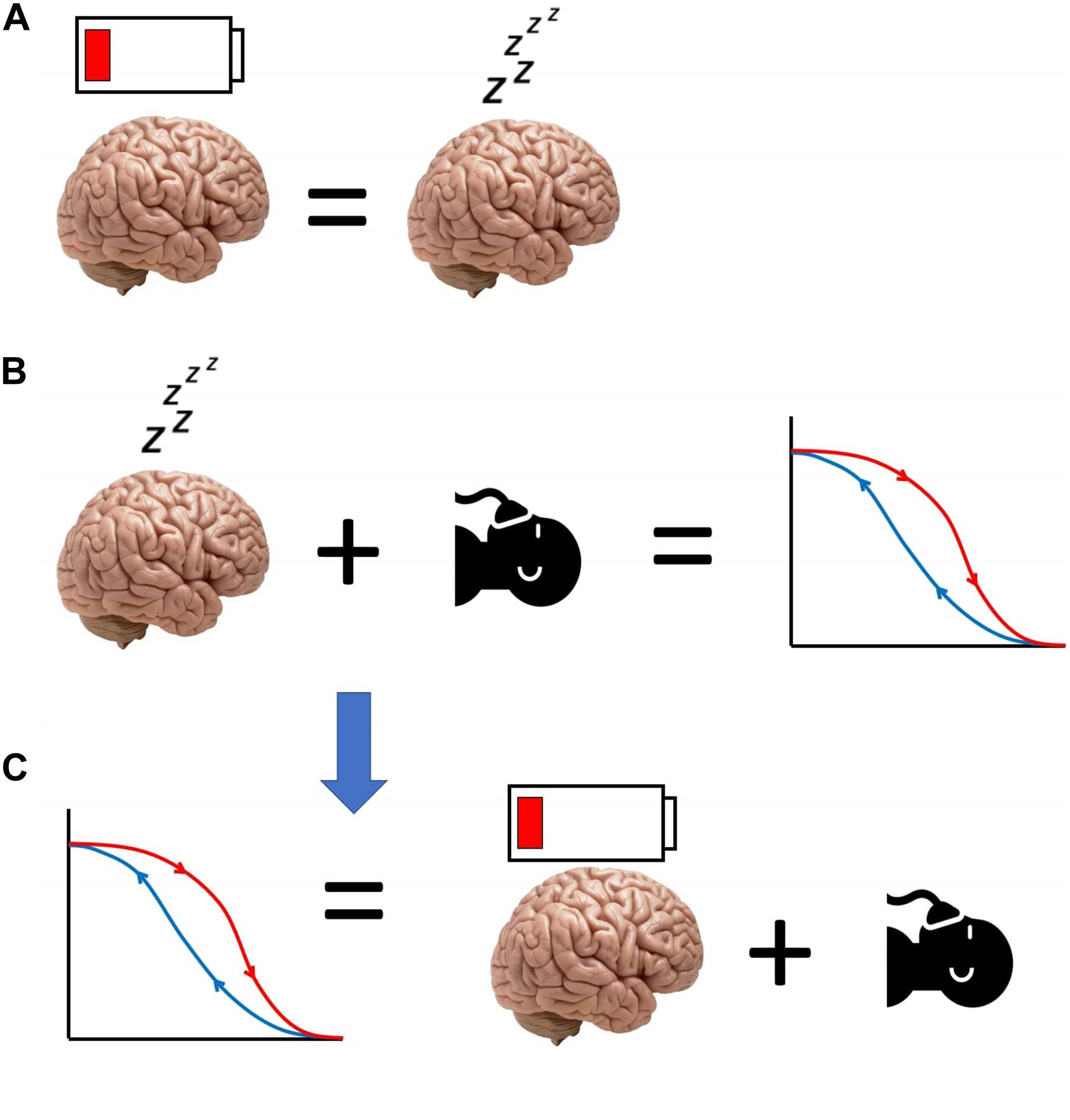
Figure 3. Schematic of our hypothesis equating neural inertia with post-anaesthetic sleep inertia. (A) Sleep inertia (represented by the depleted battery icon over the brain) is neurally and behaviourally equivalent to sleepiness. (B) Sleepiness reduces the need for anaesthetic, and increases post-anaesthetic neural inertia. Therefore, (C) neural inertia may be seen as the manifestation of sleep inertia occurring after anaesthesia, reducing the amount of anaesthetic that is needed for the brain to be unresponsive.
Furthermore, our hypothesis predicts that in the presence of neural inertia, neural activity during emergence should resemble the patterns of sleep-like activity characteristic of sleepiness and sleep inertia—and indeed, there is evidence that slow-wave activity reminiscent of sleep dominates human EEG at the beginning of emergence from anaesthesia, before most patients transition to non-slow-wave activity and subsequent waking (Chander et al., 2014). Additionally, individual measures of susceptibility to sleep inertia could be used to predict individual susceptibility to neural inertia, such as the recently developed Sleep Inertia Questionnaire (Kanady and Harvey, 2015). Indeed, there is already evidence that state-dependent EEG markers at baseline can predict individual susceptibility to anaesthetic induction with propofol (Chennu et al., 2016; Zhang et al., 2020), and future research may seek to determine whether such markers are related to sleep inertia.
We also note that our hypothesis would likely not apply to the dissociative anaesthesia induced by ketamine, whose molecular mechanisms of action and neurophysiological effects at the micro- and macroscale are very different from other known anaesthetics, and do not appear to resemble sleep (Hemmings et al., 2019). Although we are not aware of tests of neural inertia with ketamine, our hypothesis leads us to predict that little should be observed, since sleep does not seem to be involved in the context of dissociative anaesthesia. Testing this prediction in humans is not straightforward, for the same reason that complicates existing attempts to identify neural inertia in humans (Sepúlveda et al., 2019): namely Proekt and Kelz (2020) demonstrated that since effect-site concentration cannot be measured directly, effect-site models could be constructed to collapse hysteresis even when it would actually be attributable to genuine neuronal dynamics. However, the hypothesis is not specific to humans and could be tested in other species for which neural inertia has already been demonstrated with other anaesthetics (Friedman et al., 2010; Joiner et al., 2013; McKinstry-Wu et al., 2019; Wasilczuk et al., 2020), with the prediction being that little hysteresis should be observed. Additionally, we reported above that if neural inertia is due to the increased susceptibility to anaesthetics that occurs during post-anaesthetic sleep inertia, then our hypothesis predicts that higher susceptibility to anaesthesia should be observed during sleep inertia (e.g., as induced by awakening from slow-wave sleep). We expect that ketamine would constitute an exception to this general prediction—which should be testable in humans.
Recently, a modelling study observed that neural inertia is compatible with an account of the brain as a bistable system, stochastically switching between two states (Proekt and Hudson, 2018). If the states are seen as wells in an energy landscape, the system can be conceptualised as transitioning between them whenever noise-driven (stochastic) fluctuations are large enough to overcome the energy differential between the wells. Under conditions of low noise, the system is therefore more likely to remain trapped in whatever state it is currently occupying, and therefore inertia (resistance to state transitions) will be observed (Proekt and Hudson, 2018). It is important to note that our hypothesis of neural inertia as the effects of sleep inertia arising from anaesthetic-induced “artificial sleep” is not incompatible with this account of neural inertia: the two operate at different levels of explanation (Marr, 2010). In fact, if our hypothesis is correct, then it suggests that the account of Proekt and Hudson (2018) could also be invoked to understand sleep inertia.
If corroborated, the hypothesis presented here could have direct relevance for clinical practice: anaesthetists could use tools such as the recently developed Sleep Inertia Questionnaire (Kanady and Harvey, 2015) to evaluate each patient’s individual susceptibility to sleep inertia, which we expect should predict (together with their current amount of sleep debt) their individual likelihood of experiencing neural inertia.
Neuroimaging Evidence: Diminished Anticorrelations in the Inert Human Brain
At the macroscale, there is additional recent evidence to suggest that anaesthesia resembles the state of sleep inertia. Under conditions of normal restfulness, it is well known from functional MRI that the human brain self-organises into distinct sets of brain regions, known as resting-state networks (Yeo et al., 2011; Smith et al., 2012). In particular, a “default mode” network (DMN) of medial frontal and parietal regions, and a set of “task-positive” networks such as the “executive control” network of lateral fronto-parietal regions (FPN) and the “dorsal attention network” (DAN) tend to exhibit anticorrelated patterns of activation (Raichle et al., 2001; Fox et al., 2005) (but note that the DMN can also be recruited by tasks, especially pertaining to self-referential cognition, “mental time travel,” or automated processing (Vatansever et al., 2015a,b, 2017; Buckner and DiNicola, 2019) (Figure 4).
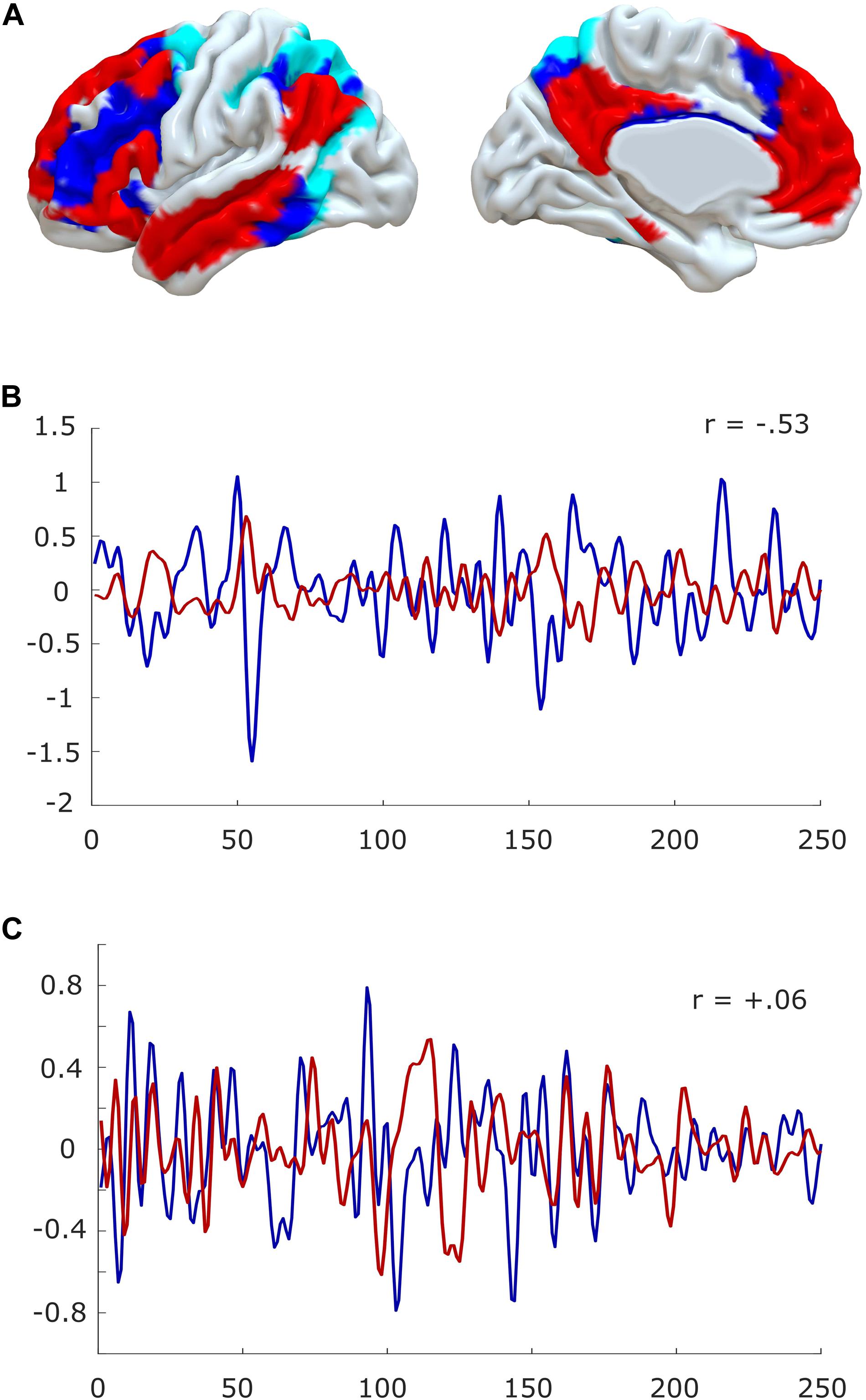
Figure 4. Anticorrelations in the human brain. (A) Surface projection of the default mode network (red) and fronto-parietal (blue)/dorsal attention networks (cyan) onto medial and lateral surfaces of a standard brain (left hemisphere). (B) The timecourses of default mode and fronto-parietal networks are anti-correlated during quiet wakefulness. (C) Anticorrelations are reduced or even abolished in the anaesthetised brain. Data from one representative subject, before and during propofol anaesthesia; for experimental details, see Stamatakis et al. (2010) and Varley et al. (2020).
Intriguingly, recent EEG-fMRI evidence indicates that loss of DMN-FPN/DAN anticorrelations is a neural correlate of sleep inertia itself in humans (Vallat et al., 2018; but see Chen et al., 2020). Indeed, earlier work had also demonstrated, by employing positron emission tomography (PET) that for a short period of time after awakening (5–20 min, compatible with the duration of sleep inertia; Trotti, 2017), there is a gradual increase of cerebral blood flow in heteromodal areas, especially lateral prefrontal cortex (lPFC), a core component of the executive control network (Balkin et al., 2002). Additionally, as previously mentioned, both awakening from deep sleep and previous sleep deprivation intensify subsequent sleep inertia upon awakening. And indeed, a loss of DMN-FPN/DAN anticorrelations is also observed during sleep in humans (Sämann et al., 2011), as well as in the awake but sleep-deprived human brain (De Havas et al., 2012). Thus, sleep inertia and conditions that favour it, share a common neural substrate in the reduction of DMN-FPN/DAN anticorrelations. Conversely, caffeine consumption, perhaps the most widely adopted countermeasure to sleep inertia (Van Dongen et al., 2001) is known to have the opposite effect: it increases the anticorrelations between DMN and FPN/DAN in the human brain (Wong et al., 2012).
This suggests that sleep inertia, at least in the human brain, may correspond to a carry-over of diminished DMN-FPN/DAN anticorrelations. Remarkably, perturbed DMN-FPN/DAN interactions are also one of the most robustly observed neural markers of human loss of consciousness induced by a variety of anaesthetics (Boveroux et al., 2010; Guldenmund et al., 2013; Golkowski et al., 2019; Luppi et al., 2019, 2020; Huang et al., 2020) (Figure 4), and the anticorrelations are even diminished one hour after emergence from sevoflurane anaesthesia (Nir et al., 2020). Thus, we propose that neural inertia may be the effect of anaesthetic-induced sleep inertia, which corresponds to a carry-over of diminished anticorrelations between DMN and FPN/DAN. In other words, we propose that the inert brain is a brain that has lost its characteristic anticorrelations. This specific hypothesis could be empirically tested, since it predicts that humans experiencing higher neural inertia after anaesthesia should exhibit more prominent loss of anticorrelations.
Inertia in the Aging Brain
Intriguingly, the hypothesis presented here may also explain why older adults are more susceptible to neural inertia (Warnaby et al., 2017). Namely, according to the present view, this is because they are more susceptible to sleep inertia. Reduced and fragmented sleep is common among the elderly, and especially patients with Alzheimer’s disease (Bonanni et al., 2005; Guarnieri et al., 2012). Since fragmented sleep tends to increase subsequent slow-wave activity (Bonnet, 1987), awakening from which causes higher levels of sleep inertia (Dinges, 1990), as does sleep deprivation, the elderly should show higher levels of sleep inertia. This is indeed the case (Silva and Duffy, 2008).
Additionally, if the hypothesis proposed here about the link between sleep inertia and neural inertia is correct, these populations should also suffer from higher levels of neural inertia. Again, this is precisely what is observed: rat studies indicate that ageing increases sensitivity to anaesthetics, and prolongs their effect (Chemali et al., 2015); likewise, older humans are also more susceptible to anaesthesia (Kanonidou and Karystianou, 2007). Furthermore, recent evidence indicates that age influences the newly discovered EEG marker of neural inertia in humans, slow wave activity saturation (SWAS): SWAS is more likely to cease abruptly rather than gradually in older patients, predicting their likelihood of post-operative delirium (Warnaby et al., 2017).
Neuroimaging evidence in older adults further supports the link between sleep and neural inertia and loss of anticorrelations between DMN and FPN/DAN: it is well established that aging corresponds to a reduction of anticorrelations between these networks (Keller et al., 2015; Siman-Tov et al., 2017), even in the absence of concomitant psychiatric conditions (Kobuti Ferreira et al., 2015) and more so in those with mild cognitive impairment (Esposito et al., 2018). Thus, older brains are intrinsically more prone to loss of anticorrelations, and suffer from higher sleep inertia and higher neural inertia.
Molecular Mechanisms of Sleep and Neural Inertia
Orexin/Hypocretin
One candidate system for the control of sleep inertia—and hence, we have argued, neural inertia—is the orexinergic system. Located exclusively in the lateral hypothalamus (De Lecea et al., 1998; Sakurai et al., 1998), orexin/hypocretin neurons are wake-active (Lee et al., 2005; de Lecea and Huerta, 2014), and innervate the wake-promoting monoaminergic and cholinergic nuclei (Carter et al., 2012). And indeed, using channelrhodopsin-2 to selectively stimulate orexin neurons promotes awakening from sleep in mice (Adamantidis et al., 2007), and increased wakefulness is reported in rodents after orexin-A administration, either intracerebroventricular or directly into monoaminergic and cholinergic wake-promoting nuclei (Hagan et al., 1999; Sakurai and Mieda, 2011).
Conversely, optogenetic suppression of orexin neurons with archaerhodopsin has sleep-promoting effects in mice (Tsunematsu et al., 2011, 2013); and in humans, orexin blockers are now available as medication against insomnia (Bennett et al., 2014). These effects were confirmed using Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) to chemogenetically activate or silence orexin neurons, resulting in increased wakefulness or sleep in rodents, respectively (Sasaki et al., 2011). Loss of orexin neurons causes the sleep disorder narcolepsy in dogs (Lin et al., 1999) and humans (Nishino et al., 2000; Thannickal et al., 2000), and the same is obtained by selective orexin knock-out in mice (Chemelli et al., 1999; Mochizuki et al., 2004), as well as pharmacological lesions in rats (Gerashchenko et al., 2001). Crucially, narcolepsy is characterised by an unstable and fragmented sleep-wake cycle, and difficulty in becoming awake (i.e., high sleep inertia) (Scammell, 2003). Indeed, sleep inertia is often present in narcoleptic children (Wise, 1998).
Thus, there is ample evidence, in both humans and other animals, that orexin and orexinergic neurons play a crucial role in sleep-wake regulation (Mieda, 2017). A recent computational study indicates that the specific role of orexin may be to stabilise the transitions between sleep and wake (Fulcher et al., 2014). According to the model, a bistable region of state-space exists when the inputs to the sleep-promoting and wake-promoting systems are balanced, and state transitions are easy (Figure 5). By increasing the activity of wake-promoting monoaminergic nuclei upon awakening, orexin pushes the system out of the bistable region, stabilising it. Indeed, simulating orexin loss in the model lowered transition thresholds, resulting in frequent wake-sleep transitions and sleep fragmentation, analogous to what is observed in orexin-deficient narcoleptic patients.
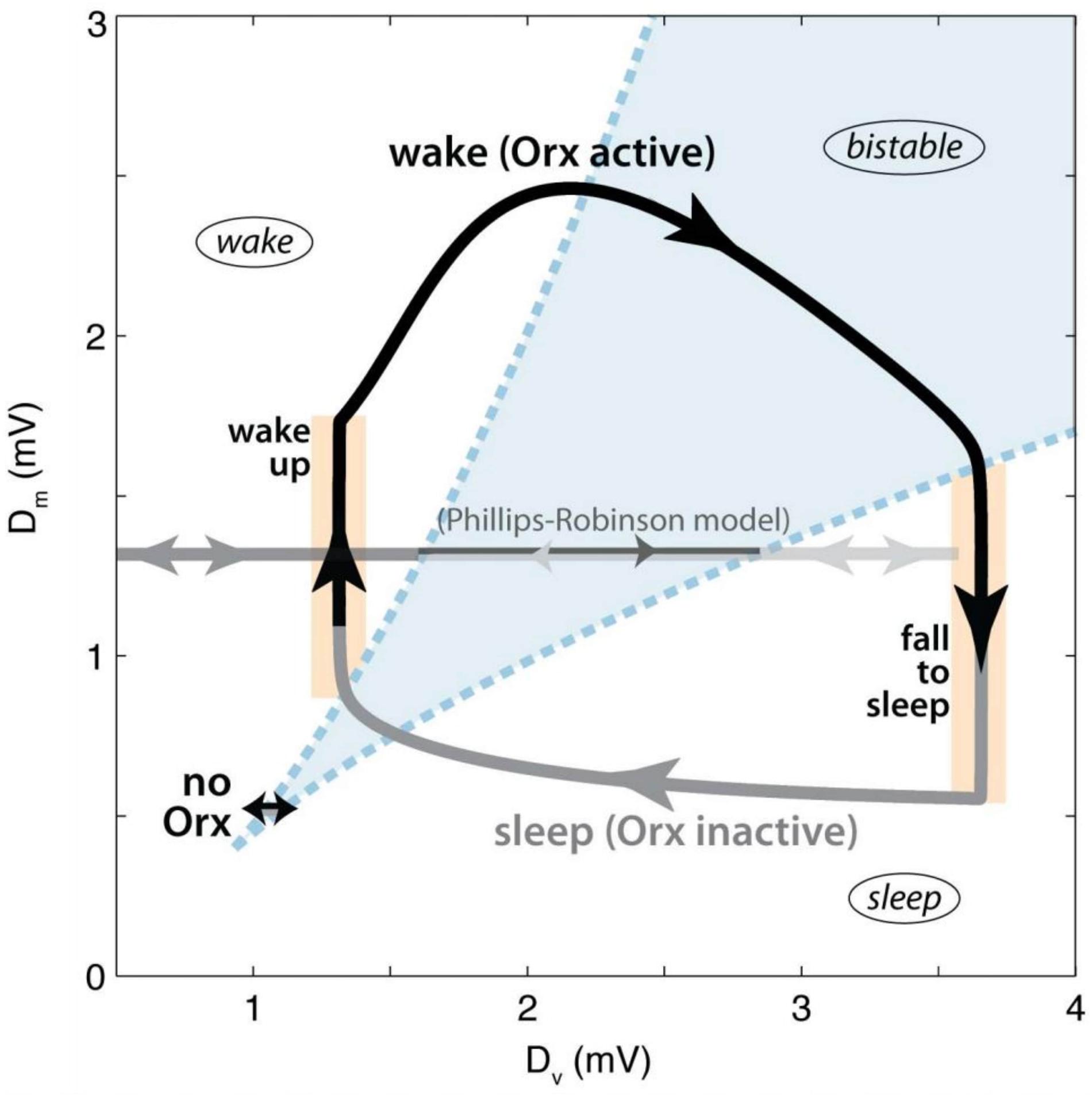
Figure 5. Dynamics of orexin stabilisation of state transitions according to the model of Fulcher et al. (2014). The axes represent the net drives to the wake-promoting (DM) and sleep-promoting (DV) circuits. Regions are labelled, with the bistable region shown in blue. The black arrow represents the waking period, while the grey arrow represents sleep. The trajectory marked “no Orx” represents the dynamics of the model in the absence of orexin input. Figure adapted from Figure 3 of Fulcher et al. (2014), published under CC-BY licence.
Intriguingly, recent neuroimaging work using a variant of functional MRI called MR encephalography, which has high temporal resolution (100 ms), determined that human narcoleptic patients have aberrant interactions between DMN and FPN/DAN, characterised by delayed and monotonic interactions, which the authors interpreted as a compromised ability of task-positive networks to suppress the DMN (Järvelä et al., 2020); Once again, this observation is in line with our proposed macroscale identification of sleep inertia with abnormal anticorrelations between large-scale networks of the brain.
Thus, evidence suggests that low orexin levels lead to high levels of sleep inertia, and its associated neural signatures. According to the hypothesis developed here, such high sleep inertia should be accompanied by high levels of neural inertia. This is indeed the case: case reports suggest high neural inertia in at least some narcoleptic human patients (Mesa et al., 2000; Burrow et al., 2005), confirmed by the increased neural inertia observed in rodents with narcolepsy arising from genetic ablation of orexin neurons (Hara et al., 2001; Kelz et al., 2008). Moreover, orexin is known to be involved in anaesthetic action: the activity of orexin neurons is reduced by propofol, sevoflurane and isoflurane, as indicated by a reduced number of c-Fos-immuno-reactive orexinergic neurons in rodents (Kelz et al., 2008; Zhang et al., 2012; Scharf and Kelz, 2013). Moreover, rodent studies show that reduced activation of orexin neurons during anaesthesia is exacerbated when the anaesthesia is administered under conditions of sleep deprivation (Ran et al., 2015). Conversely, intracerebroventricular administration of orexin-A (though not orexin-B) causes emergence from propofol, isoflurane and sevoflurane anaesthesia in rats (Dong et al., 2009; Shirasaka et al., 2011; Zhang et al., 2012, 2016), and similar results have also been obtained in mice, whereby activation of orexin neurons with DREADDs facilitated emergence from isoflurane anaesthesia (Zhou et al., 2018). Thus, orexin appears to play a major role in anaesthesia and the sleep-wake cycle, with its absence increasing both sleep inertia and neural inertia.
Noradrenaline
The action of orexin neurons is believed to occur mainly through excitation of monoaminergic wake-promoting nuclei, which they innervate (Sakurai and Mieda, 2011). In particular, orexinergic neurons may exert their effects on sleep-wake transitions through the noradrenergic locus coeruleus (LC) (Carter et al., 2012). Orexin neurons send strong excitatory projections to the LC, and the wake-inducing effect of orexin infusion involves activation of the LC (Hagan et al., 1999).
Indeed, the fragmented sleep-wake cycle of narcolepsy was reconsolidated by restoring orexin receptors in the LC of mice, and equivalent results were achieved by chemogenetically activating these neurons with DREADDs (Hasegawa et al., 2014). Furthermore, optogenetic inactivation of LC prevents the arousal-promoting effect of optogenetically activating orexin neurons; conversely, the latter is potentiated by concomitant stimulation of LC neurons (Carter et al., 2012). Thus, there is strong evidence that noradrenergic system activity is one of the primary routes through which orexin neurons perform their regulatory role (De Lecea, 2015).
Specifically supporting a role for noradrenaline in neural inertia, previous work (Friedman et al., 2010) established that genetic deletion of dopamine-ß-hydroxylase (DBH) in mice to remove noradrenergic signalling resulted in hypersensitivity to isoflurane anaesthesia, as well as increased neural inertia. This could be reversed by pharmacologic CNS-specific rescue of adrenergic signalling, achieved by providing the amino acid L-DOPS so that it would be converted into noradrenaline by L-amino acid decarboxylase (Friedman et al., 2010). In humans, Kuizenga et al. (2018) reported evidence of neural inertia when sevoflurane was supplemented with remifentanil, which is believed to influence sleep-wake regulation through adrenergic neurotransmission (McCormick and Bal, 1997; Samuels and Szabadi, 2008).
Indeed, implication of orexin and noradrenaline in neural inertia has been considered before (Sepúlveda et al., 2019; Wasilczuk et al., 2020). Wasilczuk et al. (2020) observed that halothane does not suppress hypothalamic orexinergic neurons and LC noradrenergic neurons (Gompf et al., 2009), whereas isoflurane does suppress them (Kelz et al., 2008). Thus, these authors proposed that this difference may underlie the increased neural inertia induced by halothane compared with isoflurane (Friedman et al., 2010; Wasilczuk et al., 2020) due to non-abolished orexinergic activity. As mentioned above, our own explanation of the same phenomenon is in terms of halothane failing to reduce sleep debt, unlike isoflurane (Pick et al., 2011), thereby producing more sleep inertia (and hence neural inertia, according to our account). These two explanations are not in contrast: indeed, they suggest that a fruitful avenue for future research may be to seek a connection between persistent orexinergic activity and halothane’s failure to discharge sleep debt.
On the other hand, studies providing a direct link between noradrenaline and sleep inertia are presently lacking; nevertheless, several indirect lines of evidence suggest that low levels of noradrenaline may be related to sleep inertia. Behaviourally, noradrenaline is implicated in cognitive functions such as sustained attention and working memory (Chamberlain and Robbins, 2013; Spencer et al., 2015), which are especially vulnerable to sleep deprivation (Goel et al., 2009; Killgore, 2010)—of which sleep inertia is a post-awakening counterpart, we have argued here. Noradrenaline is also increased following consumption of coffee (Papadelis et al., 2003), and caffeine consumption can reverse many of the cognitive adverse effects of clonidine (Smith et al., 2003), which mimics the state of reduced arousal observed as a result of sleep deprivation by reducing turnover of central noradrenaline, by binding to autoreceptors (Nutt and Glue, 1988).
Recently, Bellesi et al. (2016) used in vivo microdialysis to demonstrate decreasing levels of prefrontal noradrenaline in rodents undergoing sleep deprivation, correlating with an increase in low EEG frequencies tracking the need to sleep. Thus, low levels of prefrontal noradrenaline could contribute to explain the cognitive deficits observed during sleepiness induced by prolonged wakefulness. Crucially, noradrenaline restoration to baseline levels post-awakening was slower in prefrontal cortex than in other areas, such as M1—and in humans, prefrontal regions are those that were found to have reduced cerebral blood flow upon awakening in the PET study of Balkin et al. (2002). Thus, evidence suggests that decreased prefrontal noradrenaline could also explain the confusion and cognitive deficits observed during sleep inertia—especially since this state is very similar to sleepiness, as we have shown. This evidence also suggests that, if our hypothesis is correct, then we should expect noradrenaline to modulate the prevalence of anticorrelations between DMN and FPN/DAN in the human brain, since anticorrelations are also enhanced by caffeine and decreased by sleepiness (De Havas et al., 2012), sleep (Sämann et al., 2011), sleep inertia (Vallat et al., 2018), and anaesthesia (Boveroux et al., 2010; Golkowski et al., 2019; Luppi et al., 2019; Huang et al., 2020). Interestingly, recent studies indicate that caffeine infusion can accelerate emergence from isoflurane anaesthesia in both rodents and humans (Fong et al., 2018; Fox et al., 2020), and future research may seek to determine whether this effect corresponds to faster recovery of anticorrelations in the brain after anaesthesia (Nir et al., 2020) and whether it is specifically attributable to caffeine’s action on noradrenergic neuromodulation (Papadelis et al., 2003; Smith et al., 2003).
Indeed, as major wake- and alertness-promoting neurotransmitter, noradrenaline is modulated by both sleep and anaesthesia—just as we should expect if noradrenaline were involved in both sleep and neural inertia, as we propose here. Noradrenaline levels are highest during wake and drop during sleep (Léna et al., 2005) and stimulation of the noradrenergic LC of mice induces waking (Carter et al., 2010, 2013; Berridge et al., 2012); activity of the LC is inhibited by GABA during sleep (Gervasoni et al., 1998), as well as during propofol and isoflurane anaesthesia in mice (Zecharia et al., 2009). Administration of noradrenaline by microinjection into the central medial nucleus of the thalamus accelerates emergence from propofol anaesthesia in rodents, and reverses the local physiological effects of propofol (Fu et al., 2016). Likewise, pharmacogenetic activation of noradrenergic neurons in the LC with virally delivered DREADDs promotes EEG markers of neural arousal and accelerates emergence from isoflurane anaesthesia in rats, an effect that can be prevented by application of noradrenergic antagonists (Vazey and Aston-Jones, 2014). The anaesthetic dexmedetomidine also operates on noradrenergic transmission: as an adrenergic α-2 receptor agonist, it decreases the firing of LC neurons (Nelson et al., 2003), and indeed α-2A receptor activation inhibits noradrenergic LC neurons (Lakhlani et al., 1997).
Although it was originally thought that dexmedetomidine would induce sedation by inhibiting the LC (Sanders and Maze, 2012) thereby removing the noradrenergic inhibition on the sleep-promoting VLPO neurons (Nelson et al., 2003), recent evidence suggests a more intricate picture: acute inhibition of LC neurons does not induce strong sleep in mice (Carter et al., 2010), and LC inhibition is not required for low doses of dexmedetomidine to produce their sedative effects, since knockdown of LC α2A adrenergic receptors in mice does not prevent sedation, even though loss of the righting reflex is still observed at high doses (Zhang et al., 2015). Intriguingly, the same hypothalamic neurons in the mouse are involved in inducing recovery sleep and dexmedetomidine-induced sedation, by locally exciting neurons in the preoptic area (Zhang et al., 2015).
Other studies also indicate a more complicated picture: microdialysis of noradrenaline into rat prefrontal or parietal cortex under constant levels of sevoflurane anaesthesia failed to produce wake-like behaviour—although it did produce wake-like EEG (Pal et al., 2018). Similar failure to awaken rats from continuous sevoflurane anaesthesia was also reported after pharmacological blockade of noradrenaline reuptake (Kenny et al., 2015). Since cholinergic stimulation of prefrontal cortex did induce wake-like behaviour in the rats studies by Pal et al. (2018), this evidence suggests that a full picture will likely need to also take additional neuromodulatory systems into account. Dopamine in particular has been implicated, largely in rodent studies. Lesions to the wake-active dopaminergic ventral tegmental area in the brainstem shorten the induction time of anaesthesia, and lengthen the time taken for recovery—whereas both electrical and optogenetic stimulation of the VTA can reverse the anaesthetic effects of propofol in rats and mice (Solt et al., 2014; Taylor et al., 2016). These contributions of dopaminergic signalling have recently also been extended to a dopaminergic population in the ventral periaqueductal grey (Li et al., 2018; Liu et al., 2020). Given the shared pathways of dopaminergic and noradrenergic transmitter production, it seems plausible that these transmitters and their nuclei in the brainstem may act in-concert to produce wakefulness, and to counter the effects of sleep inertia and neural inertia, as evidenced by their influences on recovery and induction times. Likewise, the recent discovery that hypothalamic neuroendocrine cells are involved in both slow-wave sleep and general anaesthesia induced by multiple classes of anaesthetic drugs (Jiang-Xie et al., 2019) suggests that a fuller understanding of the link between sleep and neural inertia may benefit from taking into account neuroendocrine involvement.
Discussion
Overall, there is converging human and animal evidence that neural inertia strongly resembles sleep inertia, in terms of both behavioural manifestations and microscale and macroscale neural markers. Both phenomena are influenced by orexin neurons, which seem to perform a state-stabilising function via noradrenergic transmission. Loss of orexin neurons in narcolepsy, results in fragmented sleep-wake cycles and increases in both sleep inertia and neural inertia. Therefore, we have argued here that neural inertia may in fact be a manifestation of sleep inertia, as it occurs after the artificial slow-wave sleep induced by anaesthetics. Of note, this hypothesis can account for phenomena as diverse as the higher inertia-inducing properties of halothane vs. isoflurane (Friedman et al., 2010; Wasilczuk et al., 2020), and the increased susceptibility to neural inertia in the elderly and in narcoleptic patients.
If our hypothesis is correct, then it could have implications for clinical practice: by assessing each patient’s individual susceptibility to sleep inertia and current sleep debt, anaesthetists may be able to estimate individual likelihood of their patient experiencing neural inertia. In turn, this may better equip them to counteract adverse effects such as post-anaesthetic delirium (Warnaby et al., 2017; Sepúlveda et al., 2019).
Multiple sources of evidence—behavioural and neurophysiological, in animals and humans—suggest that orexin may play a stabilising effect between states of sleep and wakefulness, possibly (though likely not exclusively) through its effects on locus coeruleus noradrenergic neurons. Together, these neuromodulatory systems may be key in determining sleepiness, sleep inertia and what we have argued is its post-anaesthetic counterpart: neural inertia. Nevertheless, direct evidence explicitly linking all pieces of this puzzle together is still lacking, and even evidence of a link between noradrenergic modulation and sleep inertia is at present only indirect. Further studies explicitly investigating involvement of noradrenaline and other neuromodulators in relation to sleep inertia remain necessary, as a test of the hypothesis presented here.
Of course, the brain is a remarkably complex system. There are other components of the sleep- and wake-promoting circuits beyond orexin and noradrenaline, and they are likely to play some direct or indirect role in the phenomena of sleep inertia and neural inertia, and the stabilisation of arousal states more broadly. All these circuits are intricately interconnected, and changes in one are likely to have multiple repercussions. Indeed, investigating such interactions will be required to further elucidate the hypothesis proposed here. Nevertheless, here we have provided a number of predictions that are testable with current scientific techniques, and we hope that these predictions will stimulate fruitful avenues for further research—whether or not they ultimately support our hypothesis.
Data Availability Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Author Contributions
AL: conceptualisation and writing—original draft. LS and ES: writing—editing. ES: supervision. ES and DM: project administration and funding acquisition. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Gates Cambridge Trust (to AL, grant OPP1144), the Cambridge European Trust (to LS), the Stephen Erskine Fellowship (Queens’ College, Cambridge, to ES), and grants from the National Institute for Health Research (NIHR, United Kingdom), Cambridge Biomedical Research Centre and NIHR Senior Investigator Awards, and the British Oxygen Professorship of the Royal College of Anaesthetists (to DM). DM is a Fellow of the CIFAR Brain, Mind, and Consciousness Programme.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AL is grateful to Dr. Katie Warnaby for helpful discussions on the notion of neural inertia.
References
Abel, T., Havekes, R., Saletin, J. M., and Walker, M. P. (2013). Sleep, plasticity and memory from molecules to whole-brain networks. Curr. Biol. 23, 774–788. doi: 10.1016/j.cub.2013.07.025
Adamantidis, A. R., Zhang, F., Aravanis, A. M., Deisseroth, K., and De Lecea, L. (2007). Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 450, 420–424. doi: 10.1038/nature06310
Akerstedt, T., and Folkard, S. (1997). A three process model of the regulation of alertness and sleepiness sleep arousal and performance. Chronobiol. Int. 14, 115–123.
Alkire, M. T., Hudetz, A. G., and Tononi, G. (2009). Consciousness and anesthesia NIH public access. Science 322, 876–880. doi: 10.1126/science.1149213
Balkin, T. J., and Badia, P. (1988). Relationship between sleep inertia and sleepiness: cumulative effects of four nights of sleep disruption/restriction on performance following abrupt nocturnal awakening. Biol. Psychol. 27, 245–258. doi: 10.1016/0301-0511(88)90034-8
Balkin, T. J., Braun, A. R., Wesensten, N. J., Jeffries, K., Varga, M., Baldwin, P., et al. (2002). The process of awakening: a PET study of regional brain activity patterns mediating the re-establishment of alertness and consciousness. Brain 125, 2308–2319. doi: 10.1093/brain/awf228
Bellesi, M., Tononi, G., Cirelli, C., and Serra, P. A. (2016). Region-Specific dissociation between cortical noradrenaline levels and the sleep/wake cycle. Sleep 39, 143–154. doi: 10.5665/sleep.5336
Bennett, T., Bray, D., and Neville, M. W. (2014). Suvorexant, a dual orexin receptor antagonist for the management of insomnia. P T 39, 264–266.
Berger, R. J., and Phillips, N. H. (1995). Energy conservation and sleep. Behav. Brain Res. 69, 65–73. doi: 10.1016/0166-4328(95)00002-B
Berridge, C. W., Schmeichel, B. E., and España, R. A. (2012). Noradrenergic modulation of wakefulness/arousal. Sleep Med. Rev. 16, 187–197. doi: 10.1016/j.smrv.2011.12.003
Bonanni, E., Maestri, M., Tognoni, G., Fabbrini, M., Nucciarone, B., Manca, M. L., et al. (2005). Daytime sleepiness in mild and moderate Alzheimer’s disease and its relationship with cognitive impairment. J. Sleep Res. 14, 311–317. doi: 10.1111/j.1365-2869.2005.00462.x
Bonnet, M. H. (1987). Sleep restoration as a function of periodic awakening, movement, or electroencephalographic change. Sleep 10, 364–373. doi: 10.1093/sleep/10.4.364
Bonnet, M. H. (1993). Cognitive effects of sleep and sleep fragmentation. Sleep 16, S65–S67. doi: 10.1093/sleep/16.suppl_8.s65
Boveroux, P., Vanhaudenhuyse, A., and Phillips, C. (2010). Breakdown of within- and between-network resting state during propofol-induced loss of consciousness. Anesthesiology 113, 1038–1053.
Brown, E. N., Lydic, R., and Schiff, N. D. (2010). General anesthesia, sleep, and coma. N. Engl. J. Med. 27, 2638–2650.
Brown, E. N., Purdon, P. L., and Van Dort, C. J. (2011). General anesthesia and altered states of arousal: a systems neuroscience analysis. Annu. Rev. Neurosci. 34, 601–628. doi: 10.1146/annurev-neuro-060909-153200
Buckner, R. L., and DiNicola, L. M. (2019). The brain’s default network: updated anatomy, physiology and evolving insights. Nat. Rev. Neurosci. 20, 593–608. doi: 10.1038/s41583-019-0212-7
Burrow, B., Burkle, C., Warner, D. O., and Chini, E. N. (2005). Postoperative outcome of patients with narcolepsy. A retrospective analysis. J. Clin. Anesth. 17, 21–25. doi: 10.1016/j.jclinane.2004.03.007
Carter, M. E., Brill, J., Bonnavion, P., Huguenard, J. R., Huerta, R., and de Lecea, L. (2012). Mechanism for Hypocretin-mediated sleep-to-wake transitions. Proc. Natl. Acad. Sci. U.S.A. 109, E2635–E2644. doi: 10.1073/pnas.1202526109
Carter, M. E., de Lecea, L., and Adamantidis, A. (2013). Functional wiring of hypocretin and LC-NE neurons: implications for arousal. Front. Behav. Neurosci. 7:43. doi: 10.3389/fnbeh.2013.00043
Carter, M. E., Yizhar, O., Chikahisa, S., Nguyen, H., Adamantidis, A., Nishino, S., et al. (2010). Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat. Neurosci. 13, 1526–1535. doi: 10.1038/nn.2682
Chamberlain, S. R., and Robbins, T. W. (2013). Noradrenergic modulation of cognition: therapeutic implications. J. Psychopharmacol. 27, 694–718. doi: 10.1177/0269881113480988
Chander, D., García, P. S., Maccoll, J. N., Illing, S., and Sleigh, J. W. (2014). Electroencephalographic variation during end maintenance and emergence from surgical anesthesia. PLoS One 9:e106291. doi: 10.1371/journal.pone.0106291
Chatterjee, A., Kaznessis, Y. N., and Hu, W. S. (2008). Tweaking biological switches through a better understanding of bistability behavior. Curr. Opin. Biotechnol. 19, 475–481. doi: 10.1016/j.copbio.2008.08.010
Chemali, J. J., Kenny, J. D., Olutola, O., Taylor, N. E., Kimchi, E. Y., Purdon, P. L., et al. (2015). Ageing delays emergence from general anaesthesia in rats by increasing anaesthetic sensitivity in the brain. Br. J. Anaesth. 115, i58–i65. doi: 10.1093/bja/aev112
Chemelli, R. M., Willie, J. T., Sinton, C. M., Elmquist, J. K., Scammell, T., Lee, C., et al. (1999). Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98, 437–451. doi: 10.1016/S0092-8674(00)81973-X
Chen, X., Hsu, C. F., Xu, D., Yu, J., and Lei, X. (2020). Loss of frontal regulator of vigilance during sleep inertia: a simultaneous EEG-fMRI study. Hum. Brain Mapp. 41, 4288–4298. doi: 10.1002/hbm.25125
Chennu, S., O’Connor, S., Adapa, R., Menon, D. K., and Bekinschtein, T. A. (2016). Brain connectivity dissociates responsiveness from drug exposure during propofol-induced transitions of consciousness. PLoS Comput. Biol. 12:e1004669. doi: 10.1371/journal.pcbi.1004669
Chou, T. C., Bjorkum, A. A., Gaus, S. E., Lu, J., Scammell, T. E., and Saper, C. B. (2002). Afferents to the ventrolateral preoptic nucleus. J. Neurosci. 22, 977–990. doi: 10.1523/jneurosci.22-03-00977.2002
Colin, P. J., Kuizenga, M. H., Vereecke, H. E. M., and Struys, M. M. R. F. (2018). Pharmacokinetic pharmacodynamic perspective on the detection of signs of neural inertia in humans. Anesthesiology 129, 373–375. doi: 10.1097/ALN.0000000000002287
De Havas, J. A., Parimal, S., Soon, C. S., and Chee, M. W. L. (2012). Sleep deprivation reduces default mode network connectivity and anti-correlation during rest and task performance. Neuroimage 59, 1745–1751. doi: 10.1016/j.neuroimage.2011.08.026
De Lecea, L. (2015). Optogenetic control of hypocretin (Orexin) neurons and arousal circuits. Curr. Top. Behav. Neurosci. 25, 367–378. doi: 10.1007/7854_2014_364
de Lecea, L., and Huerta, R. (2014). Hypocretin (orexin) regulation of sleep-to-wake transitions. Front. Pharmacol. 5:16. doi: 10.3389/fphar.2014.00016
De Lecea, L., Kilduff, T. S., Peyron, C., Gao, X. B., Foye, P. E., Danielson, P. E., et al. (1998). The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U.S.A. 95, 322–327. doi: 10.1073/pnas.95.1.322
Dinges, D. F. (1990). “Are you awake? Cognitive performance and reverie during the hypnopompic state,” in Sleep and Cognition, eds R. Bootsen, J. F. Kihlstrom, and D. L. Schacter (Washington, DC: American Psychological Association Press), 159–175. doi: 10.1037/10499-012
Dinges, D. F., Orne, M. T., Whitehouse, W. G., and Orne, E. C. (1987). Temporal placement of a nap for alertness: contributions of circadian phase and prior wakefulness. Sleep 10, 313–329. doi: 10.1093/sleep/10.4.313
Dong, H., Niu, J., Su, B., Zhu, Z., Lv, Y., Li, Y., et al. (2009). Activation of orexin signal in basal forebrain facilitates the emergence from sevoflurane anesthesia in rat. Neuropeptides 43, 179–185. doi: 10.1016/j.npep.2009.04.006
Economo, C. V. (1930). Sleep as a problem of localization. J. Nerv. Ment. Dis. 71, 249–259. doi: 10.1097/00005053-193003000-00001
Esposito, R., Cieri, F., Chiacchiaretta, P., Cera, N., Lauriola, M., Di Giannantonio, M., et al. (2018). Modifications in resting state functional anticorrelation between default mode network and dorsal attention network: comparison among young adults, healthy elders and mild cognitive impairment patients. Brain Imaging Behav. 12, 127–141. doi: 10.1007/s11682-017-9686-y
Ferrara, M., Curcio, G., Fratello, F., Moroni, F., Marzano, C., Pellicciari, M. C., et al. (2006). The electroencephalographic substratum of the awakening. Behav. Brain Res. 167, 237–244. doi: 10.1016/j.bbr.2005.09.012
Ferrara, M., and De Gennaro, L. (2000). The Sleep Inertia Phenomenon During the Sleep-Wake Transition: Theoretical and Operational Issues. Available Online at: https://www.researchgate.net/publication/12367344 (accessed October 27, 2020).
Fong, R., Wang, L., Zacny, J. P., Khokhar, S., Apfelbaum, J. L., Fox, A. P., et al. (2018). Caffeine accelerates emergence from isoflurane anesthesia in humans a randomized, double-blind, crossover study. Anesthesiology 129, 912–920. doi: 10.1097/ALN.00000000000002367
Fox, A. P., Wagner, K. R., Towle, V. L., Xie, K. G., and Xie, Z. (2020). Caffeine reverses the unconsciousness produced by light anesthesia in the continued presence of isoflurane in rats. PLoS One 15:e0241818. doi: 10.1371/journal.pone.0241818
Fox, M. D., Snyder, A. Z., Vincent, J. L., Corbetta, M., Van Essen, D. C., and Raichle, M. E. (2005). The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc. Natl. Acad. Sci. U.S.A. 102, 9673–9678. doi: 10.1073/pnas.0504136102
Franks, N. P. (2008). General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal. Nat. Rev. Neurosci. 9, 370–386. doi: 10.1038/nrn2372
Friedman, E. B., Sun, Y., Moore, J. T., Hung, H. T., Meng, Q. C., Perera, P., et al. (2010). A conserved behavioral state barrier impedes transitions between anesthetic-induced unconsciousness and wakefulness: evidence for neural inertia. PLoS One 5:e11903. doi: 10.1371/journal.pone.0011903
Fu, B., Yu, T., Yuan, J., Gong, X., and Zhang, M. (2016). Noradrenergic transmission in the central medial thalamic nucleus modulates the electroencephalographic activity and emergence from propofol anesthesia in rats. J. Neurochem. 140, 862–873. doi: 10.1111/jnc.13939
Fulcher, B. D., Phillips, A. J. K., Postnova, S., and Robinson, P. A. (2014). A physiologically based model of orexinergic stabilization of sleep and wake. PLoS One 9:e91982. doi: 10.1371/journal.pone.0091982
Gallopin, T., Fort, P., Eggermann, E., Cauli, B., Luppi, P. H., Rossier, J., et al. (2000). Identification of sleep-promoting neurons in vitro. Nature 404, 992–995. doi: 10.1038/35010109
Gent, T., and Adamantidis, A. (2017). Anaesthesia and sleep. Clin. Transl. Neurosci. 1:2514183X1772628. doi: 10.1177/2514183x17726281
Gerashchenko, D., Kohls, M. D., Greco, M., Waleh, N. S., Salin-Pascual, R., Kilduff, T. S., et al. (2001). Hypocretin-2-saporin lesions of the lateral hypothalamus produce narcoleptic-like sleep behavior in the rat. J. Neurosci. 21, 7273–7283.
Gervasoni, D., Darracq, L., Fort, P., Soulière, F., Chouvet, G., and Luppi, P. H. (1998). Electrophysiological evidence that noradrenergic neurons of the rat locus coeruleus are tonically inhibited by GABA during sleep. Eur. J. Neurosci. 10, 964–970. doi: 10.1046/j.1460-9568.1998.00106.x
Goel, N., Rao, H., Durmer, J. S., and Dinges, D. F. (2009). Neurocognitive consequences of sleep deprivation. Semin. Neurol. 29, 320–339. doi: 10.1055/s-0029-1237117
Golkowski, D., Larroque, S. K., Vanhaudenhuyse, A., Plenevaux, A., Boly, M., Di Perri, C., et al. (2019). Changes in whole brain dynamics and connectivity patterns during sevoflurane- and propofol-induced unconsciousness identified by functional magnetic resonance imaging. Anesthesiology 130, 898–911. doi: 10.1097/ALN.0000000000002704
Gompf, H., Chen, J., Sun, Y., Yanagisawa, M., Aston-Jones, G., and Kelz, M. B. (2009). Halothane-induced hypnosis is not accompanied by inactivation of orexinergic output in rodents. Anesthesiology 111, 1001–1009. doi: 10.1097/ALN.0b013e3181b764b3
Guarnieri, B., Adorni, F., Musicco, M., Appollonio, I., Bonanni, E., Caffarra, P., et al. (2012). Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement. Geriatr. Cogn. Disord. 33, 50–58. doi: 10.1159/000335363
Guldenmund, P., Demertzi, A., Boveroux, P., Boly, M., Vanhaudenhuyse, A., Bruno, M.-A., et al. (2013). Thalamus, brainstem and salience network connectivity changes during propofol-induced sedation and unconsciousness. Brain Connect. 3, 273–285. doi: 10.1089/brain.2012.0117
Hagan, J. J., Leslie, R. A., Patel, S., Evans, M. L., Wattam, T. A., Holmes, S., et al. (1999). Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc. Natl. Acad. Sci. U.S.A. 96, 10911–10916. doi: 10.1073/pnas.96.19.10911
Hara, J., Beuckmann, C. T., Nambu, T., Willie, J. T., Chemelli, R. M., Sinton, C. M., et al. (2001). Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30, 345–354. doi: 10.1016/S0896-6273(01)00293-8
Hasegawa, E., Yanagisawa, M., Sakurai, T., and Mieda, M. (2014). Orexin neurons suppress narcolepsy via 2 distinct efferent pathways. J. Clin. Invest. 124, 604–616. doi: 10.1172/JCI71017
Hemmings, H. C., Riegelhaupt, P. M., Kelz, M. B., Solt, K., Eckenhoff, R. G., Orser, B. A., et al. (2019). Towards a comprehensive understanding of anesthetic mechanisms of action: a decade of discovery. Trends Pharmacol. Sci. 40, 464–481. doi: 10.1016/j.tips.2019.05.001
Huang, Z., Zhang, J., Wu, J., Mashour, G. A., and Hudetz, A. G. (2020). Temporal circuit of macroscale dynamic brain activity supports human consciousness. Sci. Adv. 6, 87–98. doi: 10.1126/sciadv.aaz0087
Järvelä, M., Raatikainen, V., Kotila, A., Kananen, J., Korhonen, V., Uddin, L., et al. (2020). Lag analysis of fast fMRI reveals delayed information flow between the default mode and other networks in narcolepsy. Cereb. Cortex Commun. 1:tgaa073.
Jewett, M. E., Wyatt, J. K., Ritz-De Cecco, A., Bir Khalsa, S., Dijk, D. J., and Czeisler, C. A. (1999). Time course of sleep inertia dissipation in human performance and alertness. J. Sleep Res. 8, 1–8. doi: 10.1111/j.1365-2869.1999.00128.x
Jiang-Xie, L. F., Yin, L., Zhao, S., Prevosto, V., Han, B. X., Dzirasa, K., et al. (2019). A common neuroendocrine substrate for diverse general anesthetics and sleep. Neuron 102, 1053–1065.e4. doi: 10.1016/j.neuron.2019.03.033
Joiner, W. J. (2016). Unraveling the evolutionary determinants of sleep. Curr. Biol. 26, R1073–R1087. doi: 10.1016/j.cub.2016.08.068
Joiner, W. J., Friedman, E. B., Hung, H.-T., Koh, K., Sowcik, M., Sehgal, A., et al. (2013). Genetic and anatomical basis of the barrier separating wakefulness and anesthetic-induced unresponsiveness. PLoS Genet. 9:e1003605. doi: 10.1371/journal.pgen.1003605
Kanady, J. C., and Harvey, A. G. (2015). Development and validation of the sleep inertia questionnaire (SIQ) and assessment of sleep inertia in analogue and clinical depression. Cognit. Ther. Res. 39, 601–612. doi: 10.1007/s10608-015-9686-4
Karan, S. B., Perlis, M., and Ward, D. (2007). Anesthesia and sleep medicine: an opportunity to be mutually informative? Semin. Anesth. Perioper. Med. Pain 26, 42–48. doi: 10.1053/j.sane.2007.06.002
Keller, J. B., Hedden, T., Thompson, T. W., Anteraper, S. A., Gabrieli, J. D. E., and Whitfield-Gabrieli, S. (2015). Resting-state anticorrelations between medial and lateral prefrontal cortex: association with working memory, aging, and individual differences. Cortex 64, 271–280. doi: 10.1016/j.cortex.2014.12.001
Kelz, M. B., Sun, Y., Chen, J., Cheng Meng, Q., Moore, J. T., Veasey, S. C., et al. (2008). An essential role for orexins in emergence from general anesthesia. Proc. Natl. Acad. Sci. U.S.A. 105, 1309–1314. doi: 10.1073/pnas.0707146105
Kenny, J. D., Taylor, N. E., Brown, E. N., and Solt, K. (2015). Dextroamphetamine (but Not Atomoxetine) induces reanimation from general anesthesia: implications for the roles of dopamine and norepinephrine in active emergence. PLoS One 10:e0131914. doi: 10.1371/journal.pone.0131914
Killgore, W. D. S. (2010). Effects of sleep deprivation on cognition. Prog. Brain Res. 185, 105–129. doi: 10.1016/B978-0-444-53702-7.00007-5
Kobuti Ferreira, L., Carolina Brocanello Regina, A., Kovacevic, N., da Graça Morais Martin, M., Paim Santos, P., de Godoi Carneiro, C., et al. (2015). Aging effects on whole-brain functional connectivity in adults free of cognitive and psychiatric disorders. Cereb. Cortex 26, 3851–3865. doi: 10.1093/cercor/bhv190
Krueger, J. M., Frank, M. G., Wisor, J. P., and Roy, S. (2016). Sleep function: toward elucidating an enigma. Sleep Med. Rev. 28, 42–50. doi: 10.1016/j.smrv.2015.08.005
Kuizenga, M. H., Colin, P. J., Reyntjens, K. M. E. M., Touw, D. J., Nalbat, H., Knotnerus, F. H., et al. (2018). Test of neural inertia in humans during general anaesthesia. Br. J. Anaesth. 120, 525–536. doi: 10.1016/j.bja.2017.11.072
Lakhlani, P. P., Macmillan, L. B., Guo, T. Z., Mccool, B. A., Lovinger, D. M., Maze, M., et al. (1997). Substitution of a mutant α2a-adrenergic receptor via “hit and run” gene targeting reveals the role of this subtype in sedative, analgesic, and anesthetic-sparing responses in vivo. Proc. Natl. Acad. Sci. U.S.A. 94, 9950–9955. doi: 10.1073/pnas.94.18.9950
Lee, M. G., Hassani, O. K., and Jones, B. E. (2005). Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J. Neurosci. 25, 6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005
Léna, I., Parrot, S., Deschaux, O., Muffat-Joly, S., Sauvinet, V., Renaud, B., et al. (2005). Variations in extracellular levels of dopamine, noradrenaline, glutamate, and aspartate across the sleep-wake cycle in the medial prefrontal cortex and nucleus accumbens of freely moving rats. J. Neurosci. Res. 81, 891–899. doi: 10.1002/jnr.20602
Li, J., Yu, T., Shi, F., Zhang, Y., Duan, Z., Fu, B., et al. (2018). Involvement of ventral periaqueductal gray dopaminergic neurons in propofol anesthesia. Neurochem. Res. 43, 838–847. doi: 10.1007/s11064-018-2486-y
Lin, L., Faraco, J., Li, R., Kadotani, H., Rogers, W., Lin, X., et al. (1999). The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98, 365–376. doi: 10.1016/S0092-8674(00)81965-0
Liu, C., Zhou, X., Zhu, Q., Fu, B., Cao, S., Zhang, Y., et al. (2020). Dopamine neurons in the ventral periaqueductal gray modulate isoflurane anesthesia in rats. CNS Neurosci. Ther. 26, 1121–1133. doi: 10.1111/cns.13447
Lu, J., Greco, M. A., Shiromani, P., and Saper, C. B. (2000). Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J. Neurosci. 20, 3830–3842.
Luppi, A. I., Craig, M. M., Pappas, I., Finoia, P., Williams, G. B., Allanson, J., et al. (2019). Consciousness-specific dynamic interactions of brain integration and functional diversity. Nat. Commun. 10:4616. doi: 10.1038/s41467-019-12658-9
Luppi, A. I., Mediano, P. A., Rosas, F. E., Allanson, J., Carhart-Harris, R. L., Williams, G. B., et al. (2020). A synergistic workspace for human consciousness revealed by integrated information decomposition. bioRxiv [preprint] doi: 10.1101/2020.11.25.398081
Luppi, P.-H. H. (2010). Neurochemical aspects of sleep regulation with specific focus on slow-wave sleep. World J. Biol. Psychiatry 11(Suppl. 1), 4–8. doi: 10.3109/15622971003637611
Marr, D. (2010). Vision?: A Computational Investigation into the Human Representation and Processing of Visual Information. MIT Press. Available Online at: http://cognet.mit.edu/book/vision (accessed September 2, 2020).
Marzano, C., Ferrara, M., Mauro, F., Moroni, F., Gorgoni, M., Tempesta, D., et al. (2011). Recalling and forgetting dreams: theta and alpha oscillations during sleep predict subsequent dream recall. J. Neurosci. 31, 6674–6683. doi: 10.1523/JNEUROSCI.0412-11.2011
Matchock, R. L., and Mordkoff, J. T. (2014). Effects of sleep stage and sleep episode length on the alerting, orienting, and conflict components of attention. Exp. Brain Res. 232, 811–820. doi: 10.1007/s00221-013-3790-z
McCormick, D. A., and Bal, T. (1997). SLEEP AND AROUSAL: thalamocortical mechanisms. Annu. Rev. Neurosci. 20, 185–215. doi: 10.1146/annurev.neuro.20.1.185
McKay, I. D. H., Voss, L. J., Sleigh, J. W., Barnard, J. P., and Johannsen, E. K. (2006). Pharmacokinetic-pharmacodynamic modeling the hypnotic effect of sevoflurane using the spectral entropy of the electroencephalogram. Anesth. Analg. 102, 91–97. doi: 10.1213/01.ane.0000184825.65124.24
McKinstry-Wu, A. R., Wasilczuk, A. Z., Harrison, B. A., Bedell, V. M., Sridharan, M. J., Breig, J. J., et al. (2019). Analysis of stochastic fluctuations in responsiveness is a critical step toward personalized anesthesia. Elife 8:e50143. doi: 10.7554/eLife.50143
Mesa, A., Diaz, A. P., and Frosth, M. (2000). Narcolepsy and anesthesia. Anesthesiology 92, 1194–1196. doi: 10.1097/00000542-200004000-00040
Mieda, M. (2017). The roles of orexins in sleep/wake regulation. Neurosci. Res. 118, 56–65. doi: 10.1016/j.neures.2017.03.015
Mochizuki, T., Crocker, A., McCormack, S., Yanagisawa, M., Sakurai, T., and Scammell, T. E. (2004). Behavioral state instability in orexin knock-out mice. J. Neurosci. 24, 6291–6300. doi: 10.1523/JNEUROSCI.0586-04.2004
Moore, J. T., Chen, J., Han, B., Meng, Q. C., Veasey, S. C., Beck, S. G., et al. (2012). Direct activation of sleep-promoting VLPO neurons by volatile anesthetics contributes to anesthetic hypnosis. Curr. Biol. 22, 2008–2016. doi: 10.1016/j.cub.2012.08.042
Moruzzi, G., and Magoun, H. W. (1949). Brain stem reticular formation and activation of the EEG. Electroencephalogr. Clin. Neurophysiol. 1, 455–473. doi: 10.1016/0013-4694(49)90219-9
Murphy, M., Bruno, M.-A., Riedner, B. A., Boveroux, P., Noirhomme, Q., Landsness, E. C., et al. (2011). Propofol anesthesia and sleep: a high-density EEG study. Sleep 34, 283A–291A.
Nelson, L. E., Lu, J., Guo, T., Saper, C. B., Franks, N. P., and Maze, M. (2003). The α2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology 98, 428–436. doi: 10.1097/00000542-200302000-00024
Nir, T., Jacob, Y., Huang, K. H., Schwartz, A. E., Brallier, J. W., Ahn, H., et al. (2020). Resting-state functional connectivity in early postanaesthesia recovery is characterised by globally reduced anticorrelations. Br. J. Anaesth. 125, 529–538. doi: 10.1016/j.bja.2020.06.058
Nishino, S., Ripley, B., Overeem, S., Lammers, G. J., and Mignot, E. (2000). Hypocretin (orexin) deficiency in human narcolepsy. Lancet 355, 39–40. doi: 10.1016/S0140-6736(99)05582-8
Nutt, D. J., and Glue, P. (1988). Aspects of alpha-2-adrenoceptor function in normal volunteers. Hum. Psychopharmacol. Clin. Exp. 3, 235–245. doi: 10.1002/hup.470030403
Ogilvie, R. D., and Simons, I. (1992). “Falling asleep and waking up: a comparison of EEG spectra,” in Sleep, Arousal and Performance, eds R. J. Broughton and R. D. Ogilvie (Boston: Birkhäuser), 73–87.
Pace-Schott, E. F., and Hobson, J. A. (2002). The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat. Rev. Neurosci. 3, 591–605. doi: 10.1038/nrn895
Pal, D., Dean, J. G., Liu, T., Li, D., Watson, C. J., Hudetz, A. G., et al. (2018). Differential role of prefrontal and parietal cortices in controlling level of consciousness. Curr. Biol. 28, 2145–2152.e5 doi: 10.1016/J.CUB.2018.05.025
Pal, D., Lipinski, W. J., Walker, A. J., Turner, A. M., and Mashour, G. A. (2011). State-specific effects of sevoflurane anesthesia on sleep homeostasis: selective recovery of slow wave but not rapid eye movement sleep. Anesthesiology 114, 302–310. doi: 10.1097/ALN.0b013e318204e064
Pang, D. S. J., Robledo, C. J., Carr, D. R., Gent, T. C., Vyssotski, A. L., Caley, A., et al. (2009). An unexpected role for TASK-3 potassium channels in network oscillations with implications for sleep mechanisms and anesthetic action. Proc. Natl. Acad. Sci. U.S.A. 106, 17546–17551. doi: 10.1073/pnas.0907228106
Papadelis, C., Kourtidou-Papadeli, C., Vlachogiannis, E., Skepastianos, P., Bamidis, P., Maglaveras, N., et al. (2003). Effects of mental workload and caffeine on catecholamines and blood pressure compared to performance variations. Brain Cogn. 51, 143–154. doi: 10.1016/S0278-2626(02)00530-4
Pick, J., Chen, Y., Moore, J. T., Sun, Y., Wyner, A. J., Friedman, E. B., et al. (2011). Rapid eye movement sleep debt accrues in mice exposed to volatile anesthetics. Anesthesiology 115, 702–712. doi: 10.1097/ALN.0b013e31822ddd72
Pigarev, I. N., Nothdurft, H. C., and Kastner, S. (1997). Evidence for asynchronous development of sleep in cortical areas. Neuroreport 8, 2557–2560. doi: 10.1097/00001756-199707280-00027
Proekt, A., and Hudson, A. E. (2018). A stochastic basis for neural inertia in emergence from general anaesthesia. Br. J. Anaesth. 121, 86–94. doi: 10.1016/j.bja.2018.02.035
Proekt, A., and Kelz, M. B. (2020). Explaining anaesthetic hysteresis with effect-site equilibration. Br. J. Anaesth. 126, 265–278. doi: 10.1016/j.bja.2020.09.022
Raichle, M. E., MacLeod, A. M., Snyder, A. Z., Powers, W. J., Gusnard, D. A., and Shulman, G. L. (2001). A default mode of brain function. Proc. Natl. Acad. Sci. U.S.A. 98, 676–682. doi: 10.1073/pnas.98.2.676
Raizen, D. M., Zimmerman, J. E., Maycock, M. H., Ta, U. D., You, Y., Sundaram, M. V., et al. (2008). Lethargus is a caenorhabditis elegans sleep-like state. Nature 451, 569–572. doi: 10.1038/nature06535
Ran, M. Z., Wu, W., Li, J. N., Yang, C., Ouyang, P. R., Deng, J., et al. (2015). Reduction of Orexin-A is Responsible for prolonged emergence of the rat subjected to sleep deprivation from isoflurane anesthesia. CNS Neurosci. Ther. 21, 298–300. doi: 10.1111/cns.12380
Sakurai, T., Amemiya, A., Ishii, M., Matsuzaki, I., Chemelli, R. M., Tanaka, H., et al. (1998). Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92, 573–585. doi: 10.1016/S0092-8674(00)80949-6
Sakurai, T., and Mieda, M. (2011). Connectomics of orexin-producing neurons: interface of systems of emotion, energy homeostasis and arousal. Trends Pharmacol. Sci. 32, 451–462. doi: 10.1016/j.tips.2011.03.007
Sämann, P. G., Wehrle, R., Hoehn, D., Spoormaker, V. I., Peters, H., Tully, C., et al. (2011). Development of the brain’s default mode network from wakefulness to slow wave sleep. Cereb. Cortex 21, 2082–2093. doi: 10.1093/cercor/bhq295
Samuels, E., and Szabadi, E. (2008). Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function Part I: principles of functional organisation. Curr. Neuropharmacol. 6, 235–253. doi: 10.2174/157015908785777229
Sanders, R. D., and Maze, M. (2012). Noradrenergic trespass in anesthetic and sedative states. Anesthesiology 117, 945–947. doi: 10.1097/ALN.0b013e3182700c93
Saper, C. B., Chou, T. C., and Scammell, T. E. (2001). The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 24, 726–731. doi: 10.1016/S0166-2236(00)02002-6
Saper, C. B., Fuller, P. M., Pedersen, N. P., Lu, J., and Scammell, T. E. (2010). Sleep state switching. Neuron 68, 1023–1042. doi: 10.1016/j.neuron.2010.11.032
Saper, C. B., Scammell, T. E., and Lu, J. (2005). Hypothalamic regulation of sleep and circadian rhythms. Nature 437, 1257–1263. doi: 10.1038/nature04284
Sasaki, K., Suzuki, M., Mieda, M., Tsujino, N., Roth, B., and Sakurai, T. (2011). Pharmacogenetic modulation of orexin neurons alters sleep/wakefulness states in mice. PLoS One 6:20360. doi: 10.1371/journal.pone.0020360
Scammell, T. E. (2003). The neurobiology, diagnosis, and treatment of narcolepsy. Ann. Neurol. 53, 154–166. doi: 10.1002/ana.10444
Scharf, M. T., and Kelz, M. B. (2013). Sleep and anesthesia interactions: a pharmacological appraisal. Curr. Anesthesiol. Rep. 3, 1–9. doi: 10.1007/s40140-012-0007-0
Schmidt, M. H. (2014). The energy allocation function of sleep: a unifying theory of sleep, torpor, and continuous wakefulness. Neurosci. Biobehav. Rev. 47, 122–153. doi: 10.1016/j.neubiorev.2014.08.001
Sepúlveda, P. O., Carrasco, E., Tapia, L. F., Ramos, M., Cruz, F., Conget, P., et al. (2018). Evidence of hysteresis in propofol pharmacodynamics. Anaesthesia 73, 40–48. doi: 10.1111/anae.14009
Sepúlveda, P. O., Tapia, L. F., and Monsalves, S. (2019). Neural inertia and differences between loss of and recovery from consciousness during total intravenous anaesthesia: a narrative review. Anaesthesia 74, 801–809. doi: 10.1111/anae.14609
Shaw, P. J. (2000). Correlates of sleep and waking in Drosophila melanogaster. Science 287, 1834–1837. doi: 10.1126/science.287.5459.1834
Sherin, J. E., Shiromani, R. W., McCarley, R. W., and Saper, C. B. (1996). Activation of ventrolateral preoptic neurons during sleep. Science 271, 216–219.
Shirasaka, T., Yonaha, T., Onizuka, S., and Tsuneyoshi, I. (2011). Effects of orexin-A on propofol anesthesia in rats. J. Anesth. 25, 65–71. doi: 10.1007/s00540-010-1071-6
Silva, E. J., and Duffy, J. F. (2008). Sleep inertia varies with circadian phase and sleep stage in older adults. Behav. Neurosci. 122, 928–935. doi: 10.1037/0735-7044.122.4.928
Siman-Tov, T., Bosak, N., Sprecher, E., Paz, R., Eran, A., Aharon-Peretz, J., et al. (2017). Early age-related functional connectivity decline in high-order cognitive networks. Front. Aging Neurosci. 8:330. doi: 10.3389/fnagi.2016.00330
Smith, A., Brice, C., Nash, J., Rich, N., and Nutt, D. J. (2003). Caffeine and central noradrenaline: effects on mood, cognitive performance, eye movements and cardiovascular function. J. Psychopharmacol. 17, 283–292. doi: 10.1177/02698811030173010
Smith, S. M., Miller, K. L., Moeller, S., Xu, J., Auerbach, E. J., Woolrich, M. W., et al. (2012). Temporally-independent functional modes of spontaneous brain activity. Proc. Natl. Acad. Sci. U.S.A. 109, 3131–3136. doi: 10.1073/pnas.1121329109
Solt, K., Van Dort, C. J., Chemali, J. J., Taylor, N. E., Kenny, J. D., and Brown, E. N. (2014). Electrical stimulation of the ventral tegmental area induces reanimation from general anesthesia. Anesthesiology 121, 311–319. doi: 10.1097/ALN.0000000000000117
Spencer, R. C., Devilbiss, D. M., and Berridge, C. W. (2015). The cognition-enhancing effects of psychostimulants involve direct action in the prefrontal cortex. Biol. Psychiatry 77, 940–950. doi: 10.1016/j.biopsych.2014.09.013
Stamatakis, E. A., Adapa, R. M., Absalom, A. R., and Menon, D. K. (2010). Changes in resting neural connectivity during propofol sedation. PLoS One 5:e14224. doi: 10.1371/journal.pone.0014224
Steyn-Ross, M. L., Steyn-Ross, D. A., and Sleigh, J. W. (2004). Modelling general anaesthesia as a first-order phase transition in the cortex. Prog. Biophys. Mol. Biol. 85, 369–385. doi: 10.1016/j.pbiomolbio.2004.02.001
Tassi, P., Nicolas, A., Dewasmes, G., Eschenlauer, R., Ehrhart, J., Salame, P., et al. (1992). Effects of noise on sleep inertia as a function of circadian placement of a one-hour nap. Percept. Mot. Skills 75, 291–302. doi: 10.2466/pms.1992.75.1.291
Taylor, N. E., Van Dort, C. J., Kenny, J. D., Pei, J., Guidera, J. A., Vlasov, K. Y., et al. (2016). Optogenetic activation of Dopamine neurons in the ventral tegmental area induces reanimation from general anesthesia. Proc. Natl. Acad. Sci. U.S.A. 113, 12826–12831. doi: 10.1073/pnas.1614340113
Thannickal, T. C., Moore, R. Y., Nienhuis, R., Ramanathan, L., Gulyani, S., Aldrich, M., et al. (2000). Reduced number of hypocretin neurons in human narcolepsy. Neuron 27, 469–474. doi: 10.1016/S0896-6273(00)00058-1
Tononi, G., and Cirelli, C. (2014). Sleep and the price of plasticity: from synaptic and cellular homeostasis to memory consolidation and integration. Neuron 81, 12–34. doi: 10.1016/j.neuron.2013.12.025
Tononi, G., and Cirelli, C. (2016). “Sleep and synaptic down-selection,” in Micro-, Meso-and Macro-Connectomics of the Brain, eds G. Buzsáki and Y. Christen (Cham: Springer), 11–18. doi: 10.1007/978-3-319-27777-6
Trotti, L. M. (2017). Waking up is the hardest thing I do all day: sleep inertia and sleep drunkenness. Sleep Med. Rev. 35, 76–84. doi: 10.1016/j.smrv.2016.08.005
Tsunematsu, T., Kilduff, T. S., Boyden, E. S., Takahashi, S., Tominaga, M., and Yamanaka, A. (2011). Acute optogenetic silencing of orexin/hypocretin neurons induces slow-wave sleep in mice. J. Neurosci. 31, 10529–10539. doi: 10.1523/JNEUROSCI.0784-11.2011
Tsunematsu, T., Tabuchi, S., Tanaka, K. F., Boyden, E. S., Tominaga, M., and Yamanaka, A. (2013). Long-lasting silencing of orexin/hypocretin neurons using archaerhodopsin induces slow-wave sleep in mice. Behav. Brain Res. 255, 64–74. doi: 10.1016/j.bbr.2013.05.021
Tung, A., Szafran, M. J., Bluhm, B., and Mendelson, W. B. (2002). Sleep deprivation potentiates the onset and duration of loss of righting reflex induced by propofol and isoflurane. Anesthesiology 97, 906–911. doi: 10.1097/00000542-200210000-00024
Vallat, R., Meunier, D., Nicolas, A., and Ruby, P. (2018). Hard to wake up? The cerebral correlates of sleep inertia assessed using combined behavioral, EEG and fMRI measures. NeuroImage 184, 266–278. doi: 10.1016/j.neuroimage.2018.09.033
Van Dongen, H. P. A., Price, N. J., Mullington, J. M., Szuba, M. P., Kapoor, S. C., and Dinges, D. F. (2001). Caffeine eliminates psychomotor vigilance deficits from sleep inertia. Sleep 24, 813–819. doi: 10.1093/sleep/24.7.813
Van Swinderen, B., and Kottler, B. (2014). Explaining general anesthesia: a two-step hypothesis linking sleep circuits and the synaptic release machinery. BioEssays 36, 372–381. doi: 10.1002/bies.201300154
Vanini, G., Bassana, M., Mast, M., Mondino, A., Cerda, I., Phyle, M., et al. (2020). Activation of preoptic GABAergic or glutamatergic neurons modulates sleep-wake architecture, but not anesthetic state transitions. Curr. Biol. 30, 779–787.e4. doi: 10.1016/j.cub.2019.12.063
Varley, T. F., Luppi, A. I., Pappas, I., Naci, L., Adapa, R., Owen, A. M., et al. (2020). Consciousness & brain functional complexity in propofol anaesthesia. Sci. Rep. 10:1018. doi: 10.1038/s41598-020-57695-3
Vatansever, D., Menon, D. K., Manktelow, A. E., Sahakian, B. J., and Stamatakis, E. A. (2015a). Default mode network connectivity during task execution. Neuroimage 122, 96–104. doi: 10.1016/j.neuroimage.2015.07.053
Vatansever, D., Menon, D. K., and Stamatakis, E. A. (2017). Default mode contributions to automated information processing. Proc. Natl. Acad. Sci. U.S.A. 114, 12821–12826. doi: 10.1073/pnas.1710521114
Vatansever, D., Menon, X. D. K., Manktelow, A. E., Sahakian, B. J., and Stamatakis, E. A. (2015b). Default mode dynamics for global functional integration. J. Neurosci. 35, 15254–15262. doi: 10.1523/JNEUROSCI.2135-15.2015
Vazey, E. M., and Aston-Jones, G. (2014). Designer receptor manipulations reveal a role of the locus coeruleus noradrenergic system in isoflurane general anesthesia. Proc. Natl. Acad. Sci. U.S.A. 111, 3859–3864. doi: 10.1073/pnas.1310025111
Voss, U. (2010). Changes in EEG Pre and Post Awakening. Amsterdam: Elsevier Inc. doi: 10.1016/S0074-7742(10)93002-X
Vyazovskiy, V. V. (2015). Sleep, recovery, and metaregulation: explaining the benefits of sleep. Nat. Sci. Sleep 7, 171–184. doi: 10.2147/NSS.S54036
Vyazovskiy, V. V., Cui, N., Rodriguez, A. V., Funk, C., Cirelli, C., and Tononi, G. (2014). The dynamics of cortical neuronal activity in the first minutes after spontaneous awakening in rats and mice. Sleep 37, 1337–1347. doi: 10.5665/sleep.3926
Vyazovskiy, V. V., Olcese, U., Hanlon, E. C., Nir, Y., Cirelli, C., and Tononi, G. (2011). Local sleep in awake rats. Nature 472, 443–447. doi: 10.1038/nature10009
Warnaby, C. E., Sleigh, J. W., Hight, D., Jbabdi, S., and Tracey, I. (2017). Investigation of slow-wave activity saturation during surgical anesthesia reveals a signature of neural inertia in humans. Anesthesiology 127, 645–657. doi: 10.1097/ALN.0000000000001759
Wasilczuk, A. Z., Harrison, B. A., Kwasniewska, P., Ku, B., Kelz, M. B., Mckinstry-Wu, A. R., et al. (2020). Resistance to state transitions in responsiveness is differentially modulated by different volatile anaesthetics in male mice. Br. J. Anaesth. 125, 308–320. doi: 10.1016/j.bja.2020.06.007
Weber, F., and Dan, Y. (2016). Circuit-based interrogation of sleep control. Nature 538, 51–59. doi: 10.1038/nature19773
Wong, C. W., Olafsson, V., Tal, O., and Liu, T. T. (2012). Anti-correlated networks, global signal regression, and the effects of caffeine in resting-state functional MRI. Neuroimage 63, 356–364. doi: 10.1016/j.neuroimage.2012.06.035
Yeo, B. T. T., Krienen, F. M., Sepulcre, J., Sabuncu, M. R., Lashkari, D., Hollinshead, M., et al. (2011). The organization of the human cerebral cortex estimated by intrinsic functional connectivity. J. Neurophysiol. 106, 1125–1165. doi: 10.1152/jn.00338.2011
Zecharia, A. Y., Nelson, L. E., Gent, T. C., Schumacher, M., Jurd, R., Rudolph, U., et al. (2009). The involvement of hypothalamic sleep pathways in general anesthesia: testing the hypothesis using the GABAA receptor 3N265M knock-In mouse. J. Neurosci. 29, 2177–2187. doi: 10.1523/JNEUROSCI.4997-08.2009
Zhang, L. N., Li, Z. J., Tong, L., Guo, C., Niu, J. Y., Hou, W. G., et al. (2012). Orexin-a facilitates emergence from propofol anesthesia in the rat. Anesth. Analg. 115, 789–796. doi: 10.1213/ANE.0b013e3182645ea3
Zhang, L. N., Yang, C., Ouyang, P. R., Zhang, Z. C., Ran, M. Z., Tong, L., et al. (2016). Orexin-A facilitates emergence of the rat from isoflurane anesthesia via mediation of the basal forebrain. Neuropeptides 58, 7–14. doi: 10.1016/j.npep.2016.02.003
Zhang, Y., Wang, Y., Yan, F., Song, D., Wang, H., Wang, Q., et al. (2020). Influence of pre-anesthesia dynamic frontal-parietal communication on individual susceptibility to propofol. Clin. Neurophysiol. 131, 2566–2577. doi: 10.1016/j.clinph.2020.07.018
Zhang, Z., Ferretti, V., Güntan, I., Moro, A., Steinberg, E. A., Ye, Z., et al. (2015). Neuronal ensembles sufficient for recovery sleep and the sedative actions of α 2 adrenergic agonists. Nat. Neurosci. 18, 553–561. doi: 10.1038/nn.3957
Keywords: neural inertia, sleep inertia, anaesthesia, orexin, noradrenaline, anticorrelations, aging
Citation: Luppi AI, Spindler LRB, Menon DK and Stamatakis EA (2021) The Inert Brain: Explaining Neural Inertia as Post-anaesthetic Sleep Inertia. Front. Neurosci. 15:643871. doi: 10.3389/fnins.2021.643871
Received: 19 December 2020; Accepted: 05 February 2021;
Published: 02 March 2021.
Edited by:
Xiao Yu, Boston Children’s Hospital and Harvard Medical School, United StatesReviewed by:
Peng Zhong, SRI International, United StatesWei Ba, Imperial College London, United Kingdom
Max Kelz, University of Pennsylvania, United States
Copyright © 2021 Luppi, Spindler, Menon and Stamatakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea I. Luppi, YWw4NTdAY2FtLmFjLnVr