 Karla Ramirez1,2†
Karla Ramirez1,2† Rosa Fernández1*†
Rosa Fernández1*† Sarah Collet3
Sarah Collet3 Meltem Kiyar4
Meltem Kiyar4 Enrique Delgado-Zayas1
Enrique Delgado-Zayas1 Esther Gómez-Gil5Tibbert Van Den Eynde3Guy T’Sjoen3Antonio Guillamon6
Esther Gómez-Gil5Tibbert Van Den Eynde3Guy T’Sjoen3Antonio Guillamon6 Sven C. Mueller4
Sven C. Mueller4 Eduardo Pásaro1
Eduardo Pásaro1- 1Laboratory of Psychobiology, Department of Psychology, Institute Advanced Scientific Research Center (CICA), University of A Coruña, A Coruña, Spain
- 2Laboratory of Neurophysiology, Center for Biophysics and Biochemistry, Venezuelan Institute for Scientific Research (IVIC), Caracas, Venezuela
- 3Department of Endocrinology, Ghent University, Ghent, Belgium
- 4Department of Experimental Clinical and Health Psychology, Ghent University, Ghent, Belgium
- 5Department of Psychiatry, Hospital Clínic, Barcelona, Spain
- 6Department of Psychobiology, Faculty of Psychology, National University of Distance Education (UNED), Madrid, Spain
Introduction: The main objective was to carry out a global DNA methylation analysis in a population with gender incongruence before gender-affirming hormone treatment (GAHT), in comparison to a cisgender population.
Methods: A global CpG (cytosine-phosphate-guanine) methylation analysis was performed on blood from 16 transgender people before GAHT vs. 16 cisgender people using the Illumina© Infinium Human Methylation 850k BeadChip, after bisulfite conversion. Changes in the DNA methylome in cisgender vs. transgender populations were analyzed with the Partek® Genomics Suite program by a 2-way ANOVA test comparing populations by group and their sex assigned at birth.
Results: The principal components analysis (PCA) showed that both populations (cis and trans) differ in the degree of global CpG methylation prior to GAHT. The 2-way ANOVA test showed 71,515 CpGs that passed the criterion FDR p < 0.05. Subsequently, in male assigned at birth population we found 87 CpGs that passed both criteria (FDR p < 0.05; fold change ≥ ± 2) of which 22 were located in islands. The most significant CpGs were related to genes: WDR45B, SLC6A20, NHLH1, PLEKHA5, UBALD1, SLC37A1, ARL6IP1, GRASP, and NCOA6. Regarding the female assigned at birth populations, we found 2 CpGs that passed both criteria (FDR p < 0.05; fold change ≥ ± 2), but none were located in islands. One of these CpGs, related to the MPPED2 gene, is shared by both, trans men and trans women. The enrichment analysis showed that these genes are involved in functions such as negative regulation of gene expression (GO:0010629), central nervous system development (GO:0007417), brain development (GO:0007420), ribonucleotide binding (GO:0032553), and RNA binding (GO:0003723), among others.
Strengths and Limitations: It is the first time that a global CpG methylation analysis has been carried out in a population with gender incongruence before GAHT. A prospective study before/during GAHT would provide a better understanding of the influence of epigenetics in this process.
Conclusion: The main finding of this study is that the cis and trans populations have different global CpG methylation profiles prior to GAHT. Therefore, our results suggest that epigenetics may be involved in the etiology of gender incongruence.
Introduction
Sexual development in mammals begins at conception, when the sex chromosome pair is determined as XX or XY. Later, the biological sex will imply differences in gonadal development, hormonal environment, sexual behavior, as well as other physical and behavioral differences. The current hypothesis about brain sexual development points to the existence of a complex “mosaic” model in the mammalian brain with a diversity of mechanisms involved, that allows a variable degree of masculinization/feminization within the brain (Joel et al., 2020).
But in humans, it is possible to differentiate between sex and gender. Whereas, gender identity could be defined as one’s innermost concept of self as male, female, a blend of both or neither (American Psychological Association, 2012; Gómez-Gil, 2019) that could be coincident or not, with the sex assigned at birth. According to this concordance between sex and gender, we can differentiate into “cisgender” or “transgender” people, respectively (Polderman et al., 2018). Gender incongruence (GI) as per International Classification of Diseases ICD-11 (World Health Organization [WHO], 2018) is characterized by a pronounced and persistent incongruence between the individual’s experience of gender and their sex assigned at birth.
According to the latest research, the origin of GI is complex and multifactorial. It might be associated with neurodevelopmental processes of the brain as well as genetic and epigenetic factors. With regards to the neuroanatomy, whereas post-mortem histological studies showed feminization of the central region of the bed nucleus of the stria terminalis in trans women (Zhou et al., 1995), more recent structural MRI studies indicated different brain phenotypes in trans women, trans men, cis women, and cis men (Guillamon et al., 2016; Kreukels and Guillamon, 2016; Nota et al., 2017; Baldinger-Melich et al., 2020; Mueller et al., 2021).
Paralleling the brain structural research, studies have been searching for a genetic component of GI. Some authors found variations in the DNA sequence of the androgen (AR) and estrogen (ERs, α and β) (Henningsson et al., 2005; Hare et al., 2009; Fernández et al., 2014a, b, 2016, 2018, 2020a; Cortés-Cortés et al., 2017; Foreman et al., 2019) that could hypothetically modulate the sensitivity of the nuclear steroid receptors. Furthermore, since AR and ER (α and β) are, at the same time, hormonal receptors and transcription factors, the modulation of gene expression via activation of AR and ERs by their ligands and coactivators, could be another presumptive mechanism underlying GI (Fernández et al., 2021; Ramírez et al., 2021).
Epigenetics offers an interesting complement to genetic studies because it reflects the interconnection between genes and the environment and could be a mechanism underlying GI given its sensitivity to environmental stimuli. Moreover, it could be possible to detect the capacity of the GAHT to modify gene expression and their stability over time.
DNA methylation (DNAm), which is the most stable and widely studied epigenetic modification to date, involves the covalent addition of a methyl group to cytosine residues adjacent to guanine in DNA (CpG sites) (Bird, 1986) and is associated with changes in gene transcription when they are located in gene promoter regions (Suzuki and Bird, 2008). Using DNA methylation analysis, epigenetic regulation has been shown to be critical in the control of sexual differentiation of the brain (McCarthy and Nugent, 2015; Nugent et al., 2015; Joel and McCarthy, 2017; Li et al., 2017; Joel et al., 2020; McCarthy, 2020). Thus, inhibition of DNA methylation in developing mice brains induces aberrant neurobehavioral profiles and disrupts sexually dimorphic neurobehavioral phenotypes in adulthood (Li et al., 2017; McCarthy, 2020). Furthermore, the sex difference in maternal anogenital licking of male compared to female pups produces a different methylation of the estrogen receptor α promoter in the preoptic area (Kurian et al., 2010).
Previous studies carried out in our laboratory in people with GI have found that certain environmental factors such as GAHT modify the methylation profile of the promoters of the ERα (Aranda et al., 2017; Fernández et al., 2020b), the AR and the ERβ (Aranda et al., 2017). Aranda et al. (2017) found no differences in the DNA methylation of the ERα in trans women, while DNA methylation was increased in trans men at 6 and 12 months of GAHT. The AR showed a significant increase of methylation in trans women after 12 months of estrogen supplementation. With respect to the ER α promoter, before the hormone treatment, trans men showed a lower methylation level with respect to both cis men and women, whereas trans women reached an intermediate methylation level with respect to the cis groups. However, after 6 months of GAHT, trans men showed a methylation increase, and both transgender groups reached a midway methylation level between cis men and cis women (Fernández et al., 2020b). Thus, both studies suggest that epigenetic changes in the sex steroid receptor promoters might be associated to GAHT. In fact, 6 months of GAHT was sufficient to modulate epigenetic changes at the estrogen and androgen receptor promoter regions. Yet, these prior studies focused exclusively on the AR and ER receptors and, to our knowledge, a global CpG analysis has not been performed to date in people with GI.
Therefore, taking into account our previous analyses and to achieve a broader perspective of the influence of epigenetics in GI, our main objective of this study was to carry out a global CpG methylation analysis in a transgender population before GAHT and cisgender comparisons, assessed by ethnicity, geographical origin and sex.
Subjects and Methods
Study Participants and Experimental Design
We analyzed sixteen Flemish Belgian transgender people (9 trans men and 7 trans women) before GAHT, and sixteen Flemish Belgian cisgender people (8 cis men and 8 cis women). The population was recruited at the Center for Sexology and Gender, Dept. of Endocrinology at the Ghent University Hospital (Belgium).
To obtain a homogeneous population avoiding stratification (Michels et al., 2013), the following inclusion and exclusion criteria were applied: for transgender people were the presence of GI according to ICD-11 (World Health Organization [WHO], 2018), identification with the other sex (male or female); and no prior history of hormonal treatment.
The exclusion criteria were the presence of psychiatric, neurological and hormonal diseases, and major medical condition. To the cisgender population the same exclusion criteria were applied. The mean age of the cisgender group at the beginning of the investigation study was 27.75 (SD ± 7.6)years and 34.1 (SD ± 14.0) years for the transgender group. Written informed consent was obtained from all participants after a full explanation of the procedures. The study was approved by the Ethical Committees of Gent University Hospital and UNED.
Genomic DNA Methylation Analysis
Genomic DNAs were extracted from peripheral blood samples using the DNeasy Blood and Tissue Kit from Qiagen following the manufacturer’s protocol, and an aliquot of 1 μg DNA per subject was processed for bisulfite conversion (Zymo Research EZ Methylation Kit), according to the manufacturer’s instructions.
DNA methylome was analyzed using the Illumina© Infinium Human Methylation 850k BeadChip array (Illumina, San Diego, CA, United States) that assesses 862,927 cytosine–phosphate–guanine (CpG) sites throughout the genome, covering 99% of RefSeq genes, 95% of CpG islands and high coverage of enhancer regions. In this study we selected the CpGs located in islands because they often coincide with promoters (Illingworth and Bird, 2009), and methylation modification of the promoter regions has the capacity to modify gene expression (Maurano et al., 2015) because methylation of the promoters prevents the binding of RNA polymerases and/or other diverse transcriptional factors to the promoter region, thereby inhibiting DNA transcription (Kang et al., 2019).
Beadchips were scanned with the Illumina iScan SQ system, and image intensities were extracted with the Genome Studio (2011.1). DNA quality checks, data normalization, and statistical filters were performed with the Partek® Genomics Suite® v7.19.1018 Methylation Module. Probes from the X and Y chromosomes were excluded from the study, and probes based on detection P > 0.05 were also filtered to exclude low-quality probes. Analysis of differentially methylated loci in humans and mice often excludes probes on the X and Y chromosomes because of the difficulties caused by the inactivation of one X chromosome in female samples.
Functional normalization, NOOB background correction and dye correction were applied. Principal component analysis (PCA) was performed to visualize clusters in the methylation data, and as a quality control procedure (Figure 1).
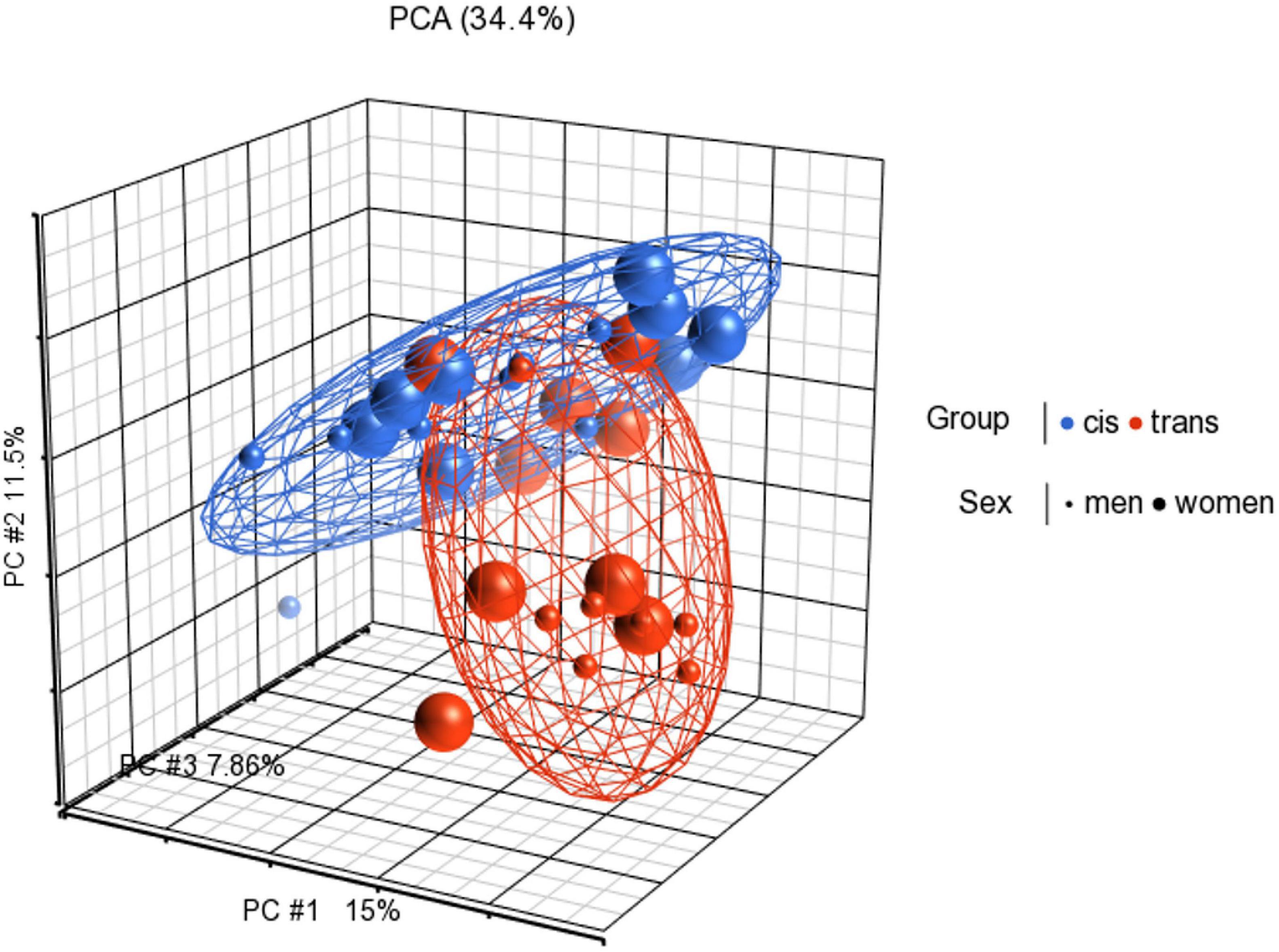
Figure 1. Principal components analysis (PCA) in 3D showing methylation profiles of the study samples. Each sample is represented by a dot, the axes are the first three PCs, the percentages indicate the fraction of variance explained by each PC. The number at the top is the variance explained by the first three PCs. The samples are colored according to the levels of the variable “group” (blue for cisgender individuals and red for transgender individuals), and sized according to the levels of the variable “sex assigned at birth” (small for men and big for women).
The raw methylation score for each probe was represented as methylation beta (β), in which β = intensity of the methylated allele (M)/intensity of the unmethylated allele (U) + intensity of the methylated allele (M) + 100. β-values range from 0 (unmethylated) to 1 (fully methylated) and can be broadly interpreted as the percentage of CpG methylation (Bibikova et al., 2011; Moran et al., 2016). Subsequently β-values were converted to M-values using the following equation: M-value = log2(β/(1 −β)). An M-value close to 0 for a CpG site indicates a similar intensity of the methylated and unmethylated probes, which means the CpG site is about half-methylated. Positive M-values mean that more molecules are methylated than unmethylated, while negative M-values mean that more molecules are unmethylated than methylated. As discussed by Du et al. (2010), the β-value has a more intuitive biological interpretation, but the M-value is more statistically valid for the differential analysis of methylation levels. Because we were performing differential methylation analysis, Partek Genomics Suite automatically created the M-values to use for statistical analysis. Distribution of M-values across the samples was inspected by a box-and-whiskers plot and the distribution of beta-values by a histogram.
Differential methylation analyses (mean M variation, ΔM) aimed to evaluate methylation differences between the studied groups. Individual probes were then filtered based on Illumina detection P < 0.05 value, and a false discovery rate correction (FDR) p < 0.05 and a fold change ≥ ± 2 were applied.
All analyses were done by the Partek® Genomics Suite® software, version 7.0. The human reference genome (GRCh37/hg19 assembly) was used to determine the location and features of the gene region using the UCSC Genome Browser (Kent et al., 2002).
Statistical Analysis
To detect the differential methylation in global CpGs that varies across all samples we performed a 2-way ANOVA test comparing cisgender vs. transgender individuals by their sex assigned at birth. Then, we added two contrast interaction terms to find those genes that specifically change in each group: we contrasted cis men vs. trans people with male sex assigned at birth (trans women), and cis women vs. trans people with female sex assigned at birth (trans men). For each contrast, a P-value, Beta difference (Δβ), and M difference (ΔM) were generated. Hierarchical cluster analysis of the significant CpGs was carried out with the Heatmap function in the Partek® Genomics Suite® 7.0 (Figure 2). P-values were calculated using false discovery rate correction for multiple comparisons, FDR p < 0.05; corrected, two-tailed, and fold change ≥ ± 2).
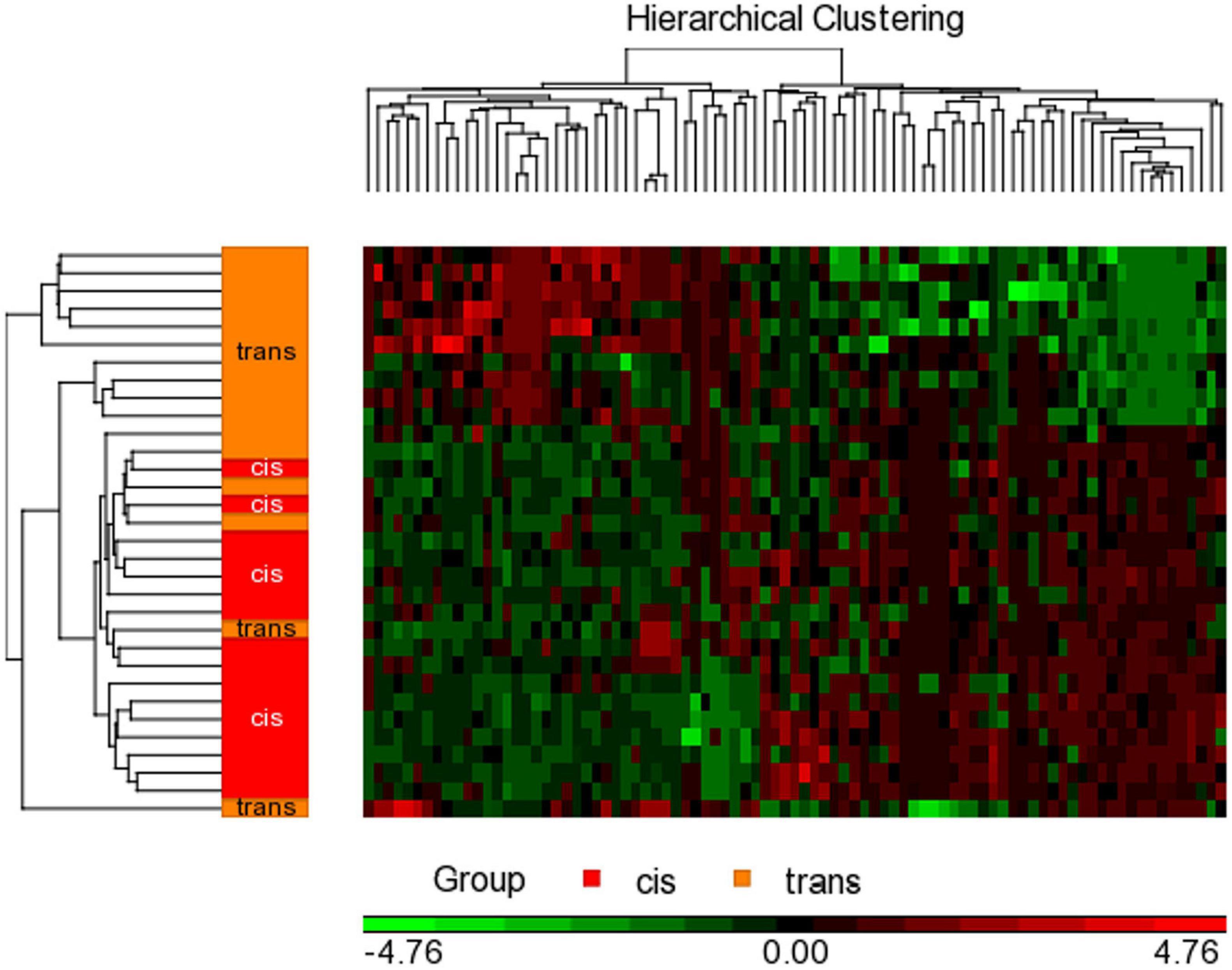
Figure 2. Hierarchical clustering with heat map invoked on the list of significant CpGs FDR p < 0.05 and a fold change ≥ 2.0 in male assigned at birth populations. The experimental groups are rows, while the CpGs from the cisgender vs. transgender spreadsheet are columns. CpGs with higher methylation are colored red, CpGs with lower methylation are colored green. Samples of transgender people are colored orange and samples of cisgender people are colored red in the dendrogram on the left-hand side of the heat map.
Controls Applied to Exclude Genes Associated With Smoking and Age
The most robustly validated findings to date with DNA methylation studies have been the association between DNA methylation in blood and smoking. The genes that have shown the strongest associations to smoking status are: AHRR, 2q37.1, 6p21.33, F2RL3, GPR15, GFI1, CYP1A1, MYO1G, and CNTNAP2 (Flanagan, 2015). We have used this knowledge to create a list of genes related to smoking status that was checked from our list of genes related to our variable of interest. This has been done because methylation alterations are detectable in blood DNA even in ex-smokers who stopped smoking up to 10–20 years before (Flanagan, 2015).
Furthermore, several genes appear consistently associated with age, including ELOVL2, CCDC102B, OTUD7A, and FHL2. Therefore, we have used this list of genes to exclude them from our study, prior to the enrichment analysis (Flanagan, 2015).
Functional and Regulatory Enrichment Analysis
The distribution of significant CpGs differentially methylated in females and males was examined across functional and regulatory annotations. CpG findings were mapped to known genes for enrichment of Gene Ontology (GO) classifications. The GO analysis and pathway enrichment analysis were carried with the Partek® Pathway program and the WebGestalt (WEB-based Gene SeT AnaLysis Toolkit)1 (Liao et al., 2019) using the Genomes (KEGG) and the Panther databases. The GO ontology includes three independent divisions: biological process (BP), molecular function (MF) and cellular component (CC). The biological process can be defined as a biological objective to which the gene or gene product contributes. The molecular function is defined as the biochemical activity of a gene product, while the cellular component refers to the place in the cell where the gene product is active (Draghici, 2012).
Results
Analysis of 2-Way ANOVA Test
When we compared the DNAm of transgender and cisgender populations by the variable sex assigned at birth, we found a baseline of 71,515 CpGs that passed the criterion FDR p < 0.05. Furthermore, 28.5% were in islands. About a third of these positions (32.3%) were hypomethylated while 67.66% were hypermethylated in cis men. In cis women, 27.05% of the CpGs were hypomethylated, while 72.95% were hypermethylated. These statistically significant CpGs were distributed among all autosomes.
Analysis of Cis Men vs. Trans Women
Subsequently, when we specifically contrasted the methylome in people who were male assigned at birth, we found 87 CpGs that passed both criteria (FDR p < 0.05; fold change ≥ ± 2), of which 22 CpGs were located in islands: 14 were hypomethylated while 8 were hypermethylated in the cis population. Table 1 lists the 22 CpG islands in populations assigned male at birth that passed both criteria. The most significant CpG islands were related to genes: WDR45B, SLC6A20, UBALD1, GRASP, NHLH1, PLEKHA5, SLC37A1, NCOA6, and ARL6IP1 (Figure 3).
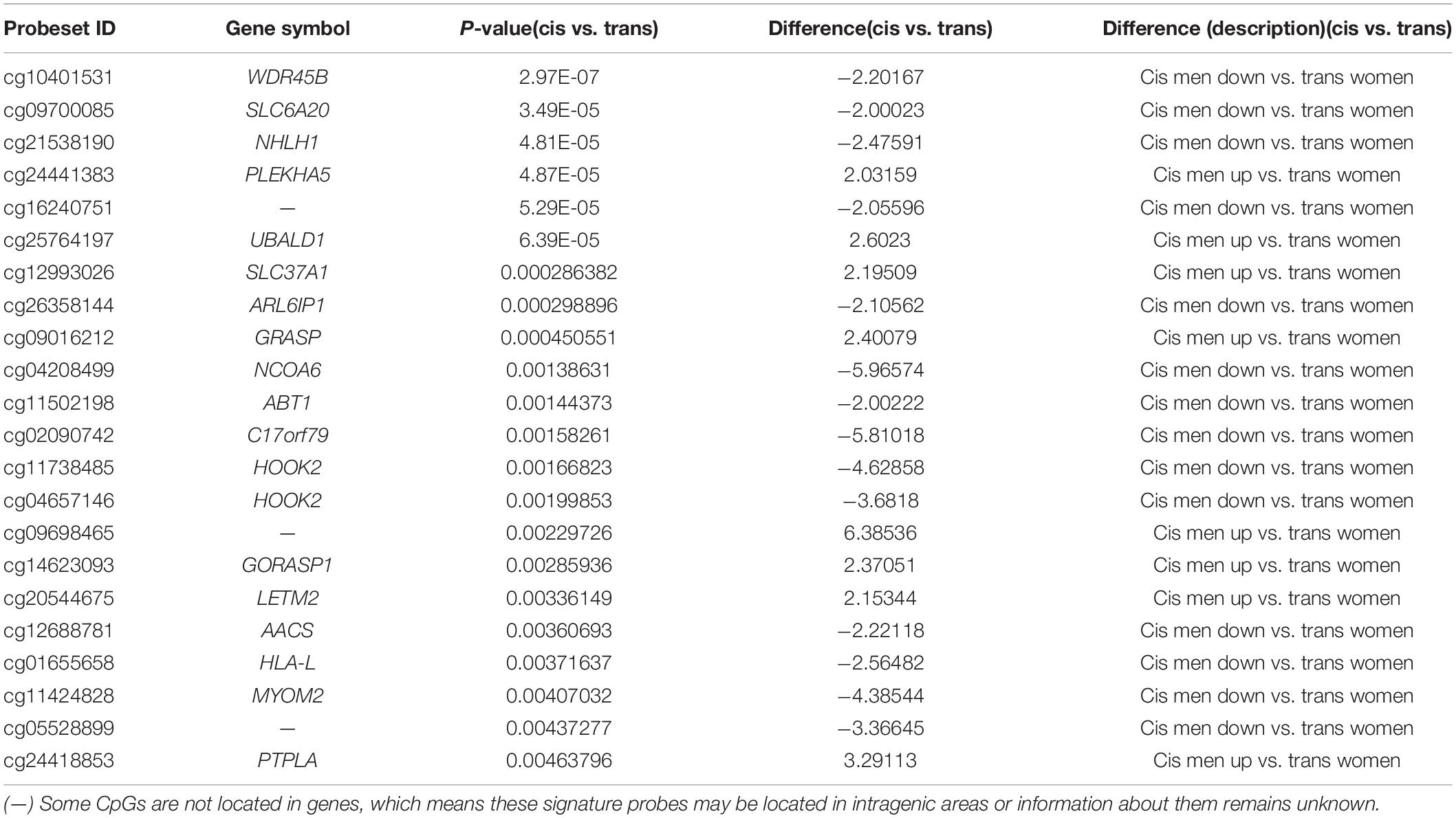
Table 1. The 22 CpG islands that passed statistical correction (FDR p < 0.05; fold change ≥ 2.0), in the population assigned male at birth.
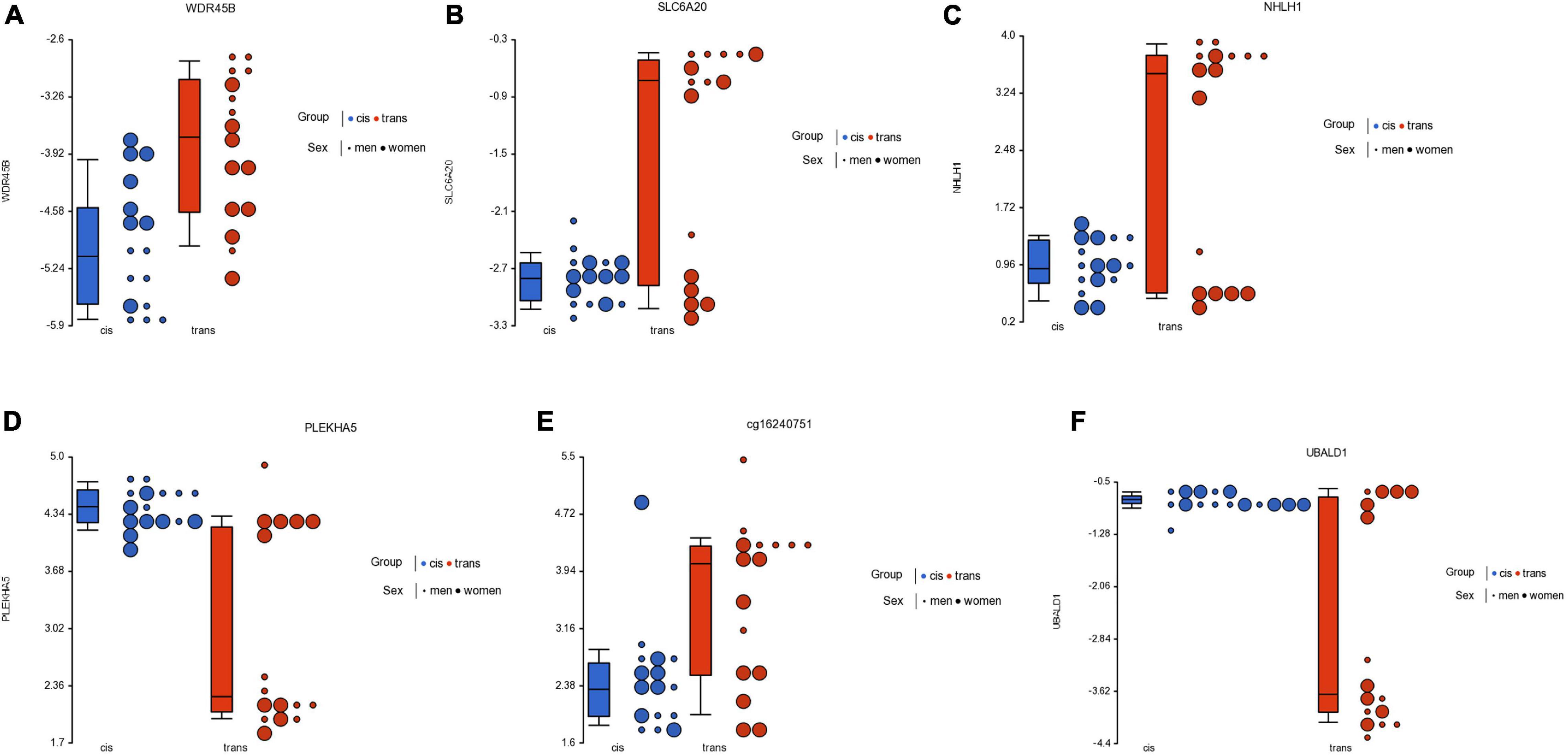
Figure 3. Dot plot showing M-value data for genes WDR45B (A), SLC6A20 (B), NHLH1 (C), PLEKHA5 (D), one intergenic locus (cg16240751) (E), and UBALD1 (F), for cisgender vs. transgender populations by their sex assigned at birth. Each sample is represented by a dot, which corresponds to the overall degree of methylation (M-value data). The samples are colored according to the levels of the variable “group” (blue for cisgender population and red for transgender population), and sized according to the levels of the variable “sex assigned at birth” (big for women and small for men). The middle line is the median, the box represents the upper and the lower quartile, while the whiskers correspond to the 90th and 10th percentiles of the data.
Analysis of Cis Women vs. Trans Men
With respect to the population with female sex assigned at birth, we found 70 CpGs that passed the criterion FDR p < 0.05, of which 2 CpGs also passed the criterion fold change ≥ ± 2 (Table 2), but none were in islands. Table 2 lists the 2 CpGs that passed both criteria. The two significant CpGs were cg23944405 related to gene MPPED2, and cg16149820 that may be in intragenic areas or information about it remains unknown.

Table 2. The 2 CpGs that passed statistical correction (FDR p < 0.05; fold change ≥ 2.0) in the population assigned female at birth.
Functional and Regulatory Enrichment Analysis
Once the significant CpGs had been selected, and prior to making the enrichment analysis, we excluded the list of genes involved in age and smoking. Subsequently, the enrichment analysis was done with the Partek® Gateway program and the WebGestalt. The results of the enrichment tests yielded significant over-representation for the categories of biological process, cellular component, and molecular function ontologies. Among the main molecular functions, we can highlight: negative regulation of gene expression (GO:0010629), central nervous system development (GO:0007417), brain development (GO:0007420), purine nucleotide binding (GO:0017076), ribonucleotide binding (GO:0032553), RNA binding (GO:0003723), and ATP binding (GO:0005524), among others (Table 3).
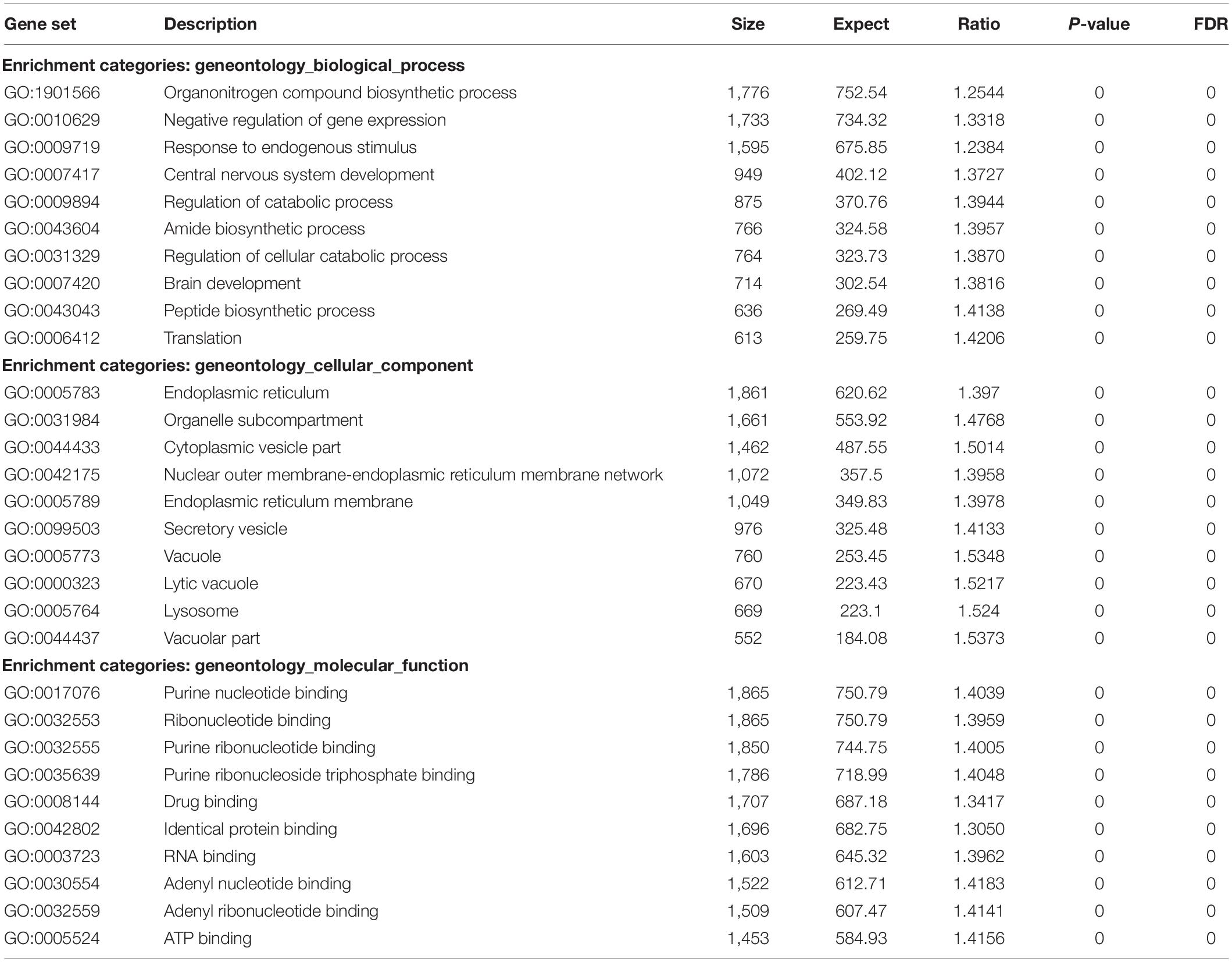
Table 3. The results of the enrichment analysis for the categories of biological process, cellular component, and molecular function ontologies.
Discussion
The main finding of this study is that the cis and trans populations have different global CpG methylation profiles, prior to GAHT. The PCA analysis showed that the spatial representation of the global methylation of these populations clearly differs between them. When comparing male sex assigned at birth individuals (cis men vs. trans women), 22 CpGs with significant methylation were located in islands. However, with respect to female assigned at birth individuals, significant changes of methylation in only 2 CpGs were found, and none were in islands. Furthermore, one of these CpGs, related to the MPPED2 gene, is shared by both, trans men and trans women. Among the most statistically significant CpGs, we found that at least four of these genes were clearly involved in brain development and neurogenesis. These genes are SLC6A20, PLEKHA5, NHLH1, and MPPED2. Overall, our results suggest that these genes could be involved in brain development, and that epigenetic factors play a role in a differential development that might be related to GI.
When comparing cis men vs. trans women 87 CpGs passed statistical correction (FDR p < 0.05; fold change ≥ ± 2), of which 22 CpGs were located in islands: 14 were hypomethylated and 8 were hypermethylated in the cis population. In this study we have considered CpGs islands because they often coincide with promoter areas, and they have the capacity to modify gene expression (Maurano et al., 2015).
The most significant CpGs in trans women were related to genes WDR45B, SLC6A20, NHLH1, PLEKHA5, UBALD1, SLC37A1, ARL6IP1, GRASP, NCOA6, ABT1, and C17orf79 (Table 1 and Figure 3). Among the most statistically significant CpGs, at least four of these genes were involved in brain development and neurogenesis (WDR45B, SLC6A20, NHLH1, and PLEKHA5) and three were related to transcriptional functions (NHLH1, NCOA6, and ABT1). Furthermore, the gene C17orf79 is related to chromatin organization and its activation stimulates the transcription of the AR. Finally, another two genes were related to glutamate synapses (ARL6IP1 and GRASP).
When we analyzed specifically the functions of each gene, we found that WDR45B is a component of the autophagy machinery that controls the major intracellular degradation process by which cytoplasmic materials are packaged into autophagosomes and delivered to lysosomes for degradation. Experiments with knockout (KO) mice exhibit many swollen axons and show cerebellar atrophy (Ji et al., 2020). On the other hand, the gene SLC6A20 synthetizes an amino acid transporter as proline and is a regulator of brain glycine levels. Recent studies have reported that this gene is highly expressed in various brain regions and is also highly expressed in astrocytes and microglia, but only modestly expressed in glutamate or minimally in GABAergic neurons (Bae et al., 2021). This could suggest that SLC6A20 proteins act as the regulator of both proline and glycine homeostasis in the brain.
The gene NHLH1 is involved in neurogenesis that encodes a helix-loop-helix (HLH) protein that belongs to a family of transcription factors, some of which have been shown to play an important role in the growth and development of a wide variety of tissues. This protein is mainly expressed in the brain, specifically in the cerebellum. Ware et al. (2016) proposed that NHLH1 is a neuronal marker. Its function might be regulating the expression of specific neuronal genes at the level of the first neurons, establishing the early axon scaffold tracts.
On the other hand, PLEKHA5 is related to cell migration and cell to cell interactions and might also be a mediator of the brain homing phenotype (Eisele et al., 2015). Yamada et al. (2012) demonstrated that this gene may play an important role in mouse brain development. We also found differences in the methylation profile of the UBALD1 gene, but its function is still unknown, however, it was associated with IL-8 secretion and NF-kappa-B signaling (Frenkel et al., 2019).
With respect to gene NCOA6, the protein encoded by this gene is a transcriptional coactivator that can interact with nuclear hormone receptors to enhance their transcriptional activator functions. It is a nuclear receptor coactivator that directly binds nuclear receptors such as for steroids (glucocorticoid receptors GR and ERs) and stimulates transcriptional activities in a hormone-dependent fashion (Eyster, 2016). Gene ontology annotations related to this gene include chromatin binding and transcription coactivator activity. Besides that, previous DNA analysis of polymorphisms related to SRC-1 and SRC-2 coactivators have pointed out their possible implication in the process of brain dimorphism (Fernández et al., 2021).
A further point in relation to this subject is that studies in mice suggest that the protein encoded by the gene ABT1 is likely to activate basal transcription from class II promoters by interaction with the class II promoter DNA. GO annotations related to this gene include transcription coactivator activity, DNA binding, RNA binding, transcription coactivator activity, or regulation of transcription by RNA polymerase II among others.
On the other hand, when we compared cis women vs. trans men, we found significant methylation in only 2 CpGs, and none were in islands. The Venn analysis showed that one of the significant CpGs was shared by both trans groups. Thus, the cg23944405, located in the MPPED2 gene (Metallophosphoesterase Domain Containing 2) showed statistically significant changes in methylation in trans men and trans women. This gene is expressed in most human tissues, also in the brain, both in cis men and cis women, and is expressed predominantly in fetal brains. Furthermore, Liguori et al. (2012) characterized MPPED2 expression in human tissues of neuronal origin, and demonstrated that MPPED2 expression is modulated during development, attributing to this gene an important role in the processes of neuronal differentiation that occur at the embryonic stage during CNS development. This gene has also been associated with altered inflammation and adverse clinical outcomes in severe blunt trauma (Schimunek et al., 2019). Furthermore, the functional importance of MPPED2 regulation is related to cell cycle inhibition as it induces apoptosis and differentiation of neuronal precursors (Liguori et al., 2012).
Cg23944405 related to the MPPED2 gene is hypermethylated in both trans populations (Figure 4). But this CpG is not located in an island (Table 2), so we cannot conclude that the hypermethylation in the transgender population was related to a low gene expression. Nevertheless, overall, we must point out that low metallophosphoesterase activity (in vitro) may play a role in the development of the CNS.
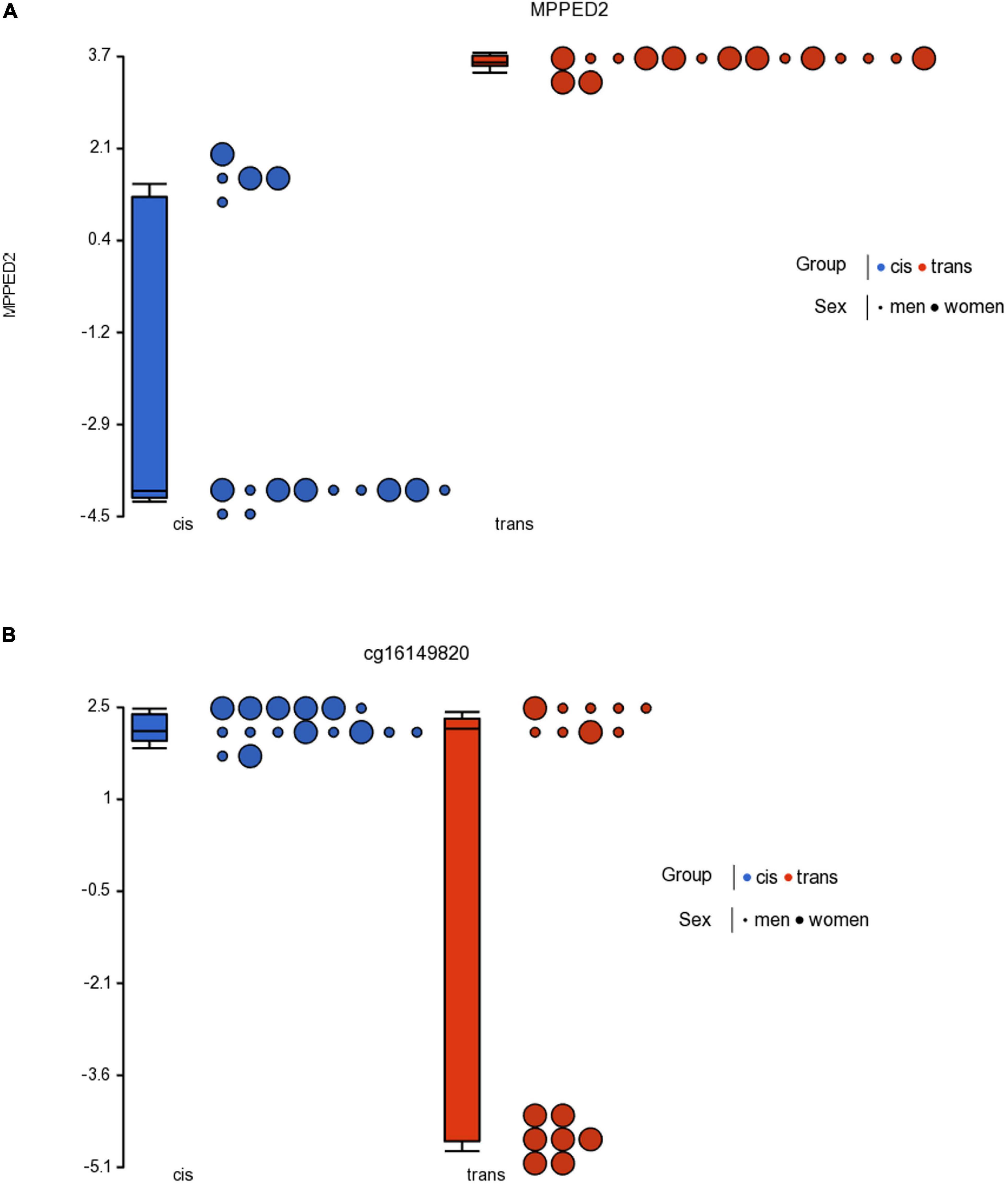
Figure 4. Dot plot showing M-value data for cg23944405 (A) in the MPPED2 gene and cg16149820 (B) for cisgender vs. transgender populations by sex assigned at birth. Each sample is represented by a dot. The samples are colored according to the levels of the variable “group” (blue for cisgender population and red for transgender population), and sized according to the levels of the variable “sex assigned at birth” (big for women and small for men). The middle line is the median, the box represents the upper and the lower quartile, while the whiskers correspond to the 90th and 10th percentiles of the data.
Our previous studies on the genetic basis of GI pointed to the existence of DNA sequences that modulate the sensitivity of the estrogen and androgen receptors in the trans population. Furthermore, we must remember that these nuclear receptors (ER and AR) are at the same time transcription factors, that modulate gene expression. Furthermore, the direct induction of gene expression through the activation of estrogen receptors and the androgen receptor is the presumed route for masculinization of the brain (Sato et al., 2004; Kudwa et al., 2006).
Now, with the present investigation, another small step is taken to increase our knowledge about GI. The results obtained here tell us that also epigenetics also plays an important role in the etiology of GI. Specifically, the differential methylation of essential genes in brain neurodevelopment such as SLC6A20, PLEKHA5, NHLH1, and MPPED2, are also involved in the etiology of GI. These are genes that play an important role in brain neurodevelopment, gene expression, and neuronal migration, which makes it possible to consider the existence of characteristic methylation profiles in the trans population.
In summary, our data reaffirm the hypothesis of a complex origin of GI, as the result of a combination of multiple factors such as hormones, hormone receptors, genetics and now also epigenetics.
Limitations
A potential limitation is that the methylation data was generated for only 32 participants. To make our study more robust, it would be advantageous to repeat this in a larger sample size or validate the conclusions with a new analysis from another trans population with similar characteristics. Also to make it even stronger it would be advantageous to do a longitudinal study that we are also collecting.
Another limitation of our study is that other factors with a known influence on the DNA methylome exist that must be taken into account. For example: sleep profile (Lahtinen et al., 2019), active/sedentary lifestyle (Voisin et al., 2015), nutritional habits (Kadayifci et al., 2018), or life adversity (Cecil et al., 2020).
The effect of sleep deprivation on transcriptome and methylome has previously been studied both in experimental animal models (Lahtinen et al., 2019). Sleep deprivation induces notable changes in the brain transcriptome of rats, affecting protein synthesis, synaptic plasticity, and metabolism (Cirelli and Tononi, 2000; Cirelli et al., 2004).
Moreover, nutrition is another important factor which plays a direct role in DNA methylation (Kadayifci et al., 2018). It is believed that nutrition affects the epigenetic regulation of DNA methylation by altering the substrates and cofactors that are necessary for DNA methylation, and also by changing the activity of enzymes that regulate the one-carbon cycle, and has a role in DNA demethylation activity too.
On the other hand, multiple studies in animals and also in humans have supported a link between early adversity and DNA methylation. The first piece of evidence for the impact of early adversity on the epigenome stemmed from research in animals. In a series of seminal studies based on rodents, Weaver et al. (2004, 2005) found that variations in maternal care during the first week of life led to long-term changes in the pup’s epigenetic regulation of the glucocorticoid receptor gene (Nr3c1), a gene crucially implicated in HPA axis function. These epigenetic changes stably altered Nr3c1 expression, resulting in variations in the density of glucocorticoid receptors in the brain as well as inter-individual differences in the pup’s physiological and behavioral responses to future stressors (Turecki and Meaney, 2016; Cecil et al., 2020).
Since these and other factors have not been taken into account in our study, together with the small sample size, we believe that our study constitutes a preliminary analysis of the influence of epigenetics on gender incongruence.
Conclusion
In conclusion, we have identified two global CpG methylation profiles in cis and trans populations, prior to gender affirming hormonal therapy. These epigenetic changes in DNAm were associated with several genes related to crucial processes during development. Moreover, these methylation data, along with our previous genetic data, support the hypothesis that GI is a complex multifactorial trait, involving intricate interactions between sex steroids, sex steroid receptors, genetics and epigenetics. This supports the view that combining genetic and epigenetic approaches in parallel may be a successful approach to understanding the mechanisms underlying brain dimorphism. Furthermore, this hypothesis is consistent with the current complex “mosaic” model of the masculinization/feminization of the brain (Joel, 2021).
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, GSE173382.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethical Committees of Gent University Hospital and UNED. The participants provided their written informed consent to participate in this study.
Author Contributions
AG, EP, RF, and SM contributed to the conception and design of the study. SC and MK recruited the population, involved in the study, and collected data from participants. SC and TV performed DNA extractions. RF and KR performed the statistical analysis and wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by grants: Xunta de Galicia ED431 B 019/02 (EP), Ministerio de Ciencia, Innovación y Universidades: PGC2018-094919-B-C21 (AG) and PGC2018-094919-B-C22 (RF and EP), and BOF interdisciplinary project (IOP003-18) of Ghent University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to everyone who directly or indirectly contributed to the study.
Footnotes
References
American Psychological Association (2012). Guidelines for psychological practice with lesbian, gay, and bisexual clients. Am. Psychol. 67, 10–42. doi: 10.1037/a0024659
Aranda, G., Fernández-Rebollo, E., Pradas-Juni, M., Hanzu, F. A., Kalko, S. G., Halperin, I., et al. (2017). Effects of sex steroids on the pattern of methylation and expression of the promoter region of estrogen and androgen receptors in people with gender dysphoria under cross-sex hormone treatment. J. Steroid Biochem. Mol. Biol. 172, 20–28. doi: 10.1016/j.jsbmb.2017.05.010
Bae, M., Roh, J. D., Kim, Y., Kim, S. S., Han, H. M., Yang, E., et al. (2021). SLC6A20 transporter: a novel regulator of brain glycine homeostasis and NMDAR function. EMBO Mol. Med. 13:e12632. doi: 10.15252/emmm.202012632
Baldinger-Melich, P., Urquijo Castro, M. F., Seiger, R., Ruef, A., Dwyer, D. B., Kranz, G. S., et al. (2020). Sex matters: a multivariate pattern analysis of sex- and gender-related neuroanatomical differences in cis- and transgender individuals using structural magnetic resonance imaging. Cereb. Cortex 30, 1345–1356. doi: 10.1093/cercor/bhz170
Bibikova, M., Barnes, B., Tsan, C., Ho, V., Klotzle, B., Le, J. M., et al. (2011). High density DNA methylation array with single CpG site resolution. Genomics 98, 288–295. doi: 10.1016/j.ygeno.2011.07.007
Bird, A. P. (1986). CpG-rich islands and the function of DNA methylation. Nature 321, 209–213. doi: 10.1038/321209a0
Cecil, C. A. M., Zhang, Y., and Nolte, T. (2020). Childhood maltreatment and DNA methylation: a systematic review. Neurosci. Biobehav. Rev. 112, 392–409. doi: 10.1016/j.neubiorev.2020.02.019
Cirelli, C., Gutierrez, C. M., and Tononi, G. (2004). Extensive and divergent effects of sleep and wakefulness on brain gene expression. Neuron 41, 35–43. doi: 10.1016/s0896-6273(03)00814-6
Cirelli, C., and Tononi, G. (2000). Gene expression in the brain across the sleep-waking cycle. Brain Res. 885, 303–321. doi: 10.1016/s0006-8993(00)03008-0
Cortés-Cortés, J., Fernández, R., Teijeiro, N., Gómez-Gil, E., Esteva, I., Almaraz, M. C., et al. (2017). Genotypes and haplotypes of the estrogen receptor α gene (ESR1) are associated with Female-to-Male gender dysphoria. J. Sex. Med. 14, 464–472. doi: 10.1016/j.jsxm.2016.12.234
Draghici, S. (2012). Statistics and Data Analysis for Microarrays Using R and Bioconductor, 2nd Edn. London, UK: CRC Press, Taylor & Francis Group.
Du, P., Zhang, X., Huang, C.-C., Jafari, N., Kibbe, W. A., Hou, L., et al. (2010). Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11:587. doi: 10.1186/1471-2105-11-587
Eisele, S. C., Gill, C. M., Shankar, G. M., and Brastianos, P. K. (2015). PLEKHA5: a key to unlock the blood-brain barrier? Clin. Cancer Res. 21, 1978–1980. doi: 10.1158/1078-0432.CCR-14-2604
Eyster, K. M. (2016). The estrogen receptors: an overview from different perspectives. Methods Mol. Biol. 1366, 1–10. doi: 10.1007/978-1-4939-3127-9
Fernández, R., Cortés-Cortés, J., Gómez-Gil, E., Esteva de Antonio, I., Almaraz, M. C., Guillamón, A., et al. (2016). The CYP17-MspA1 rs743572 polymorphism is not associated with gender dysphoria. Genes Genomics 38, 1145–1150. doi: 10.1007/s13258-016-0456-9
Fernández, R., Delgado-Zayas, E., Ramírez, K., Cortés-Cortés, J., Gómez-Gil, E., Esteva, I., et al. (2020a). Analysis of four polymorphisms located at the promoter of the estrogen receptor alpha ESR1 gene in a population with gender incongruence. Sex. Med. 8, 490–500. doi: 10.1016/j.esxm.2020.04.002
Fernández, R., Esteva, I., Gómez-Gil, E., Rumbo, T., Almaraz, M. C., Roda, E., et al. (2014b). The (CA)n polymorphism of ERβ gene is associated with FtM transsexualism. J. Sex. Med. 11, 720–728. doi: 10.1111/jsm.12398
Fernández, R., Esteva, I., Gómez-Gil, E., Rumbo, T., Almaraz, M. C., Roda, E., et al. (2014a). Association study of ERβ, AR, and CYP19A1 genes and MtF transsexualism. J. Sex. Med. 11, 2986–2994. doi: 10.1111/jsm.12673
Fernández, R., Guillamon, A., Cortés-Cortés, J., Gómez-Gil, E., Jácome, A., Esteva, I., et al. (2018). Molecular basis of gender dysphoria: androgen and estrogen receptor interaction. Psychoneuroendocrinology 98, 161–167. doi: 10.1016/j.psyneuen.2018.07.032
Fernández, R., Ramírez, K., Delgado-Zayas, E., Gómez-Gil, E., Esteva, I., Guillamon, A., et al. (2021). “An analysis of the role of estrogens and the receptor coactivators in the basis of gender incongruence,” in Psychoneuroendocrinology, ed. D. I. Kostoglou-Athanassiou (London: IntechOpen), 1–22.
Fernández, R., Ramírez, K., Gómez-Gil, E., Cortés-Cortés, J., Mora, M., Aranda, G., et al. (2020b). Gender-affirming hormone therapy modifies the CpG methylation pattern of the ESR1 gene promoter after six months of treatment in transmen. J. Sex. Med. 17, 1795–1806. doi: 10.1016/j.jsxm.2020.05.027
Flanagan, J. M. (2015). Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol. Biol. 1238, 51–63. doi: 10.1007/978-1-4939-1804-1_3
Foreman, M., Hare, L., York, K., Balakrishnan, K., Sánchez, F. J., Harte, F., et al. (2019). Genetic link between gender dysphoria and sex hormone signaling. J. Clin. Endocrinol. Metab. 104, 390–396. doi: 10.1210/jc.2018-01105
Frenkel, S., Bernstein, C. N., Sargent, M., Kuang, Q., Jiang, W., Wei, J., et al. (2019). Genome-wide analysis identifies rare copy number variations associated with inflammatory bowel disease. PLoS One 14:e0217846. doi: 10.1371/journal.pone.0217846
Gómez-Gil (2019). “Disforia de género,” in Manual de Sexología Clínica, (Editorial Médica Panamericana), eds Castelo-Branco and Molero Rodríguez: Barcelona. 400.
Guillamon, A., Junqué, C., and Gómez-Gil, E. (2016). A review of the status of brain structure research in transsexualism. Arch. Sex. Behav. 45, 1615–1648. doi: 10.1007/s10508-016-0768-5
Hare, L., Bernard, P., Sánchez, F. J. J., Baird, P. N. N., Vilain, E., Kennedy, T., et al. (2009). Androgen receptor repeat length polymorphism associated with male-to-female transsexualism. Biol. Psychiatry 65, 93–96. doi: 10.1016/j.biopsych.2008.08.033
Henningsson, S., Westberg, L., Nilsson, S., Lundstrom, B., Ekselius, L., Bodlund, O., et al. (2005). Sex steroid-related genes and male-to-female transsexualism. Psychoneuroendocrinology 30, 657–664. doi: 10.1016/j.psyneuen.2005.02.006
Illingworth, R. S., and Bird, A. P. (2009). CpG islands–’a rough guide’. FEBS Lett. 583, 1713–1720. doi: 10.1016/j.febslet.2009.04.012
Ji, C., Zhao, H., Li, D., Sun, H., Hao, J., Chen, R., et al. (2020). Role of Wdr45b in maintaining neural autophagy and cognitive function. Autophagy 16, 615–625. doi: 10.1080/15548627.2019.1632621
Joel, D. (2021). Beyond the binary: rethinking sex and the brain. Neurosci. Biobehav. Rev. 122, 165–175. doi: 10.1016/j.neubiorev.2020.11.018
Joel, D., Garcia-Falgueras, A., and Swaab, D. (2020). The complex relationships between sex and the brain. Neuroscientist 26, 156–169. doi: 10.1177/1073858419867298
Joel, D., and McCarthy, M. M. (2017). Incorporating sex as a biological variable in neuropsychiatric research: where are we now and where should we be? Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 42, 379–385. doi: 10.1038/npp.2016.79
Kadayifci, F. Z., Zheng, S., and Pan, Y.-X. (2018). Molecular mechanisms underlying the link between diet and DNA methylation. Int. J. Mol. Sci. 19:4055. doi: 10.3390/ijms19124055
Kang, J. G., Park, J. S., Ko, J.-H., and Kim, Y.-S. (2019). Regulation of gene expression by altered promoter methylation using a CRISPR/Cas9-mediated epigenetic editing system. Sci. Rep. 9:11960. doi: 10.1038/s41598-019-48130-3
Kent, W. J., Sugnet, C. W., Furey, T. S., Roskin, K. M., Pringle, T. H., Zahler, A. M., et al. (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006. doi: 10.1101/gr.229102
Kreukels, B. P. C., and Guillamon, A. (2016). Neuroimaging studies in people with gender incongruence. Int. Rev. Psychiatry 28, 120–128. doi: 10.3109/09540261.2015.1113163
Kudwa, A. E., Michopoulos, V., Gatewood, J. D., and Rissman, E. F. (2006). Roles of estrogen receptors α and β in differentiation of mouse sexual behavior. Neuroscience 138, 921–928. doi: 10.1016/j.neuroscience.2005.10.018
Kurian, J. R., Olesen, K. M., and Auger, A. P. (2010). Sex differences in epigenetic regulation of the estrogen receptor-α promoter within the developing preoptic area. Endocrinology 151, 2297–2305. doi: 10.1210/en.2009-0649
Lahtinen, A., Puttonen, S., Vanttola, P., Viitasalo, K., Sulkava, S., Pervjakova, N., et al. (2019). A distinctive DNA methylation pattern in insufficient sleep. Sci. Rep. 9:1193. doi: 10.1038/s41598-018-38009-0
Li, Y., Ma, Q., Dasgupta, C., Halavi, S., Hartman, R. E., Xiao, D., et al. (2017). Inhibition of DNA methylation in the developing rat brain disrupts sexually dimorphic neurobehavioral phenotypes in adulthood. Mol. Neurobiol. 54, 3988–3999. doi: 10.1007/s12035-016-9957-4
Liao, Y., Wang, J., Jaehnig, E. J., Shi, Z., and Zhang, B. (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 47, W199–W205. doi: 10.1093/nar/gkz401
Liguori, L., Andolfo, I., de Antonellis, P., Aglio, V., di Dato, V., Marino, N., et al. (2012). The metallophosphodiesterase Mpped2 impairs tumorigenesis in neuroblastoma. Cell Cycle 11, 569–581. doi: 10.4161/cc.11.3.19063
Maurano, M. T., Wang, H., John, S., Shafer, A., Canfield, T., Lee, K., et al. (2015). Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 12, 1184–1195. doi: 10.1016/j.celrep.2015.07.024
McCarthy, M. M. (2020). “Origins of sex differentiation of brain and behavior,” in Developmental Neuroendocrinology, eds S. Wray and S. Blackshaw (Cham: Springer International Publishing), 393–412.
McCarthy, M. M., and Nugent, B. M. (2015). At the frontier of epigenetics of brain sex differences. Front. Behav. Neurosci. 9:221. doi: 10.3389/fnbeh.2015.00221
Michels, K. B., Binder, A. M., Dedeurwaerder, S., Epstein, C. B., Greally, J. M., Gut, I., et al. (2013). Recommendations for the design and analysis of epigenome-wide association studies. Nat. Methods 10, 949–955. doi: 10.1038/nmeth.2632
Moran, S., Arribas, C., and Esteller, M. (2016). Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 8, 389–399. doi: 10.2217/epi.15.114
Mueller, S. C., Guillamon, A., Zubiaurre-Elorza, L., Junque, C., Gomez-Gil, E., Uribe, C., et al. (2021). The neuroanatomy of transgender identity: mega-analytic findings from the ENIGMA transgender persons working group. J. Sex. Med. 18, 1122–1129. doi: 10.1016/j.jsxm.2021.03.079
Nota, N. M., Burke, S. M., den Heijer, M., Soleman, R. S., Lambalk, C. B., Cohen-Kettenis, P. T., et al. (2017). Brain sexual differentiation and effects of cross-sex hormone therapy in transpeople: a resting-state functional magnetic resonance study. Neurophysiol. Clin. 47, 361–370. doi: 10.1016/j.neucli.2017.09.001
Nugent, B. M., Wright, C. L., Shetty, A. C., Hodes, G. E., Lenz, K. M., Mahurkar, A., et al. (2015). Brain feminization requires active repression of masculinization via DNA methylation. Nat. Neurosci. 18, 690–697. doi: 10.1038/nn.3988
Polderman, T. J. C., Kreukels, B. P. C., Irwig, M. S., Beach, L., Chan, Y.-M., Derks, E. M., et al. (2018). The biological contributions to gender identity and gender diversity: bringing data to the table. Behav. Genet. 48, 95–108. doi: 10.1007/s10519-018-9889-z
Ramírez, K., Fernández, R., Delgado-Zayas, E., Gómez-Gil, E., Esteva, I., Guillamón, A., et al. (2021). Implications of the estrogen receptor coactivators SRC1 and SRC2 in the biological basis of gender incongruence. Sex. Med. 9:100368. doi: 10.1016/j.esxm.2021.100368
Sato, T., Matsumoto, T., Kawano, H., Watanabe, T., Uematsu, Y., Sekine, K., et al. (2004). Brain masculinization requires androgen receptor function. Proc. Natl. Acad. Sci. U.S.A. 101, 1673–1678. doi: 10.1073/pnas.0305303101
Schimunek, L., Namas, R. A., Yin, J., Barclay, D., Liu, D., El-Dehaibi, F., et al. (2019). MPPED2 polymorphism is associated with altered systemic inflammation and adverse trauma outcomes. Front. Genet. 10:1115. doi: 10.3389/fgene.2019.01115
Suzuki, M. M., and Bird, A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476. doi: 10.1038/nrg2341
Turecki, G., and Meaney, M. J. (2016). Effects of the social environment and stress on glucocorticoid receptor gene methylation: a systematic review. Biol. Psychiatry 79, 87–96. doi: 10.1016/j.biopsych.2014.11.022
Voisin, S., Eynon, N., Yan, X., and Bishop, D. J. (2015). Exercise training and DNA methylation in humans. Acta Physiol. (Oxf). 213, 39–59. doi: 10.1111/apha.12414
Ware, M., Hamdi-Rozé, H., Le Friec, J., David, V., and Dupé, V. (2016). Regulation of downstream neuronal genes by proneural transcription factors during initial neurogenesis in the vertebrate brain. Neural Dev. 11:22. doi: 10.1186/s13064-016-0077-7
Weaver, I. C. G., Cervoni, N., Champagne, F. A., D’Alessio, A. C., Sharma, S., Seckl, J. R., et al. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7, 847–854. doi: 10.1038/nn1276
Weaver, I. C. G., Champagne, F. A., Brown, S. E., Dymov, S., Sharma, S., Meaney, M. J., et al. (2005). Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J. Neurosci. 25, 11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005
World Health Organization (WHO) (2018). The ICD-11. International Classification of Diseases for Mortality and Morbidity Statistics Eleventh Revision. Geneva: World Health Organization.
Yamada, K., Nomura, N., Yamano, A., Yamada, Y., and Wakamatsu, N. (2012). Identification and characterization of splicing variants of PLEKHA5 (Plekha5) during brain development. Gene 492, 270–275. doi: 10.1016/j.gene.2011.10.018
Keywords: DNA methylation, epigenetics, gender dysphoria, gender identity, gender incongruence
Citation: Ramirez K, Fernández R, Collet S, Kiyar M, Delgado-Zayas E, Gómez-Gil E, Van Den Eynde T, T’Sjoen G, Guillamon A, Mueller SC and Pásaro E (2021) Epigenetics Is Implicated in the Basis of Gender Incongruence: An Epigenome-Wide Association Analysis. Front. Neurosci. 15:701017. doi: 10.3389/fnins.2021.701017
Received: 27 April 2021; Accepted: 30 July 2021;
Published: 19 August 2021.
Edited by:
Mitsuhiro Kawata, Kyoto Prefectural University of Medicine, JapanReviewed by:
Arturo Ortega, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoDaniela Grassi, Autonomous University of Madrid, Spain
Copyright © 2021 Ramirez, Fernández, Collet, Kiyar, Delgado-Zayas, Gómez-Gil, Van Den Eynde, T’Sjoen, Guillamon, Mueller and Pásaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Fernández, cm9zYS5mZXJuYW5kZXpAdWRjLmVz
†These authors have contributed equally to this work and share first authorship