 Amélie Poulin-Brière
Amélie Poulin-Brière Edris Rezaei
Edris Rezaei Silvia Pozzi
Silvia Pozzi- 1Department of Psychiatry and Neuroscience, Laval University, Quebec, QC, Canada
- 2Cellular and Molecular Neuroscience Division, CERVO Brain Research Centre, Quebec, QC, Canada
Amyotrophic Lateral Sclerosis (ALS) is a mid-life onset neurodegenerative disease that manifests its symptomatology with motor impairments and cognitive deficits overlapping with Frontotemporal Lobar Degeneration (FTLD). The etiology of ALS remains elusive, with various mechanisms and cellular targets implicated, and no treatment can reverse or stop the progression of the pathology. Therapeutic interventions based on passive immunization are gaining attention for neurodegenerative diseases, and FDA recently approved the first antibody-based approach for Alzheimer's disease. The present systematic review of the literature aims to highlight the efforts made over the past years at developing antibody-based strategies to cure ALS. Thirty-one original research papers have been selected where the therapeutic efficacy of antibodies were investigated and described in patients and animal models of ALS. Antibody-based interventions analyzed, target both extracellular molecules implicated in the pathology and intracellular pathogenic proteins known to drive the disease, such as SOD1, TDP-43 or C9ORF72 repeats expansions. The potentials and limitations of these therapeutic interventions have been described and discussed in the present review.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease that primarily affects motor neurons (MN) of the motor cortex and spinal tract. Ninety percent of ALS cases have sporadic origin (sporadic ALS, sALS), whereas the remaining 10% develop the disease due to inherited gene mutations (familial ALS, fALS). The clinical manifestations of these two groups of ALS are indistinguishable, suggesting a convergence of varying pathways onto one unambiguous outcome, the degeneration of motor neurons and loss of muscles functions. Although several efforts have been made to decipher the pathogenic mechanisms of ALS, its etiology remains elusive, with various mechanisms and cellular targets suggested.
To date, there is no effective treatment for ALS. Patients are currently treated with Riluzole and Edaravone, two drugs approved by the Food and Drug Administration (FDA), that increase the survival up to 14 months (Miller et al., 2012) and slow the disease progression (Abe et al., 2017). Different therapeutic strategies have been tested in animal models with promising results. Kinase inhibitors such as Masitinib (tyrosine kinase inhibitor) or Fasudil (rho kinase inhibitor) increase survival and slow the disease progression in SOD1G93A mice (Takata et al., 2013; Trias et al., 2016). Gene-therapy based strategies are now becoming more available and specifically tested for ALS conditions (Amado and Davidson, 2021). Antisense oligonucleotides (ASO) against SOD1, C9ORF72 repeats, ATXN2 and FUS demonstrate beneficial effects in animal models. Gene editing, using the CRISPR/Cas9 technology, has been implemented for SOD1 and C9ORF72 repeats expansions with promising results in patients-derived cells and animal models. Clinical trials are currently underway to test the efficacy of these approaches in ALS patients (Amado and Davidson, 2021; Yang et al., 2021).
In recent years, there has been increasing interest in the use of monoclonal antibodies to treat neurodegenerative disorders (Freskgård and Urich, 2017; Mortada et al., 2021), with the goal of targeting misfolded intra- or extra-cellular proteins, such as amyloid beta peptide, tau, or alpha-synuclein (Valera et al., 2016). Very recently, the U.S. FDA has approved Aducanumab, a recombinant monoclonal antibody against amyloid beta plaques, for the treatment of Alzheimer's disease patients (Nimmo et al., 2021). Antibodies show a considerable number of advantages when used for therapeutic purposes (Elgundi et al., 2017; Slastnikova et al., 2018; Regazzi et al., 2020). They possess a long half-life, and, due to their nature, they can efficiently target proteins in their physiological state, after post-translational modifications or in a misfolded conformation, with high specificity and affinity. In addition, they can be conjugated to effector molecules and engineered to bind multiple targets. Finally, antibodies can be improved to interact with specific intracellular or extracellular proteins, and can be fragmented to nanobodies for efficient cellular penetration.
Multiple efforts have been made in the past 15 years to develop and test antibodies for therapeutic purposes in ALS. The present review aims to collect and discuss scientific papers where the therapeutic effect of an antibody-based approach was evaluated for ALS patients or models, describing both the potentials of the therapeutic interventions and their limitations.
Methods
Research Question and Objectives
In the present review, we aim at providing an overview of the therapeutic interventions based on the delivery of antibodies (passive immunization approaches) that have been developed and tested in patients and/or models of ALS. Here, we summarize the main publications, based on original and experimental data, that proposed therapeutic interventions with a focus on target, antibody used, outcomes and limitations.
Data Source and Search Strategy
The present review was developed following the PRISMA protocol (Liberati et al., 2009; Moher et al., 2009) with no restrictions on journal or period of publication. The data collection was conducted in May 2021, therefore all papers published after this period were not considered. The search was performed in PubMed, Web of Science, Scopus and Embase databases, using the following main keywords: “amyotrophic lateral sclerosis,” “ALS” and “antibod*,” that allowed the inclusion of both “antibody” and “antibodies” wordings.
Screening Process and Eligibility Criteria
All the publications found were imported in the citation manager Endnote (Endnote, citation manager software, version X7 for Windows, Clarivate Analytics, Philadelphia) for further screening. We first eliminated duplicates, and then performed an “exclusion on format” analysis, aimed at removing all studies that were not original articles based on empirical data, thus only research articles, clinical trials and case reports were further considered. We proceeded to an “exclusion on language” and kept all the studies that were in English, French or Italian as authors could further select and analyze the papers in these languages. Using Endnote we performed an “exclusion on keywords.” Titles, abstracts and keywords were screened for the presence of population keywords: “amyotrophic lateral sclerosis” and “ALS,” keeping only papers focusing on ALS, and the presence of intervention keywords: “therap*,” “therapeutic,” “treatment,” “antibod*” and “immunoglobulin.” Selected studies were analyzed using the software Covidence (Covidence, Systematic Review Software, Melbourne, retrieved from https://www.covidence.org). Titles and abstracts of manuscripts were fully screened to assess their relevance for our review and excluded if they were not describing a therapeutic intervention based on the development or evaluation of an antibody-based approach. A full-text screening was then performed on the four following criteria of eligibility: nature, finality, method, and availability (Lawarée et al., 2020). Based on nature criteria, all non-empirical publications such as reviews were excluded. Review papers were not included in the final analysis but considered only as key references to understand the overall portrait of the topic analyzed, and to verify the presence of an already published work on the same subject. Studies, where the objective was not to describe or to test a therapeutic intervention based on antibodies, were excluded on finality criteria. Method criteria were used to exclude publications lacking details on the experimental plan and scientific data. Papers not available online or requiring a paid registration not provided by our institution (Université Laval) were excluded based on the availability criteria.
Data Extraction
Selected papers were compiled in one table (now divided in Tables 1, 2) where the main bibliographic information (first author and year of publication) and the details about the study (targeted protein, type of antibody used, antibody clone, targeted epitope, model used and treatment details) where listed together with observed outcomes and limitations, according to the SALSA framework (Grant and Booth, 2009).
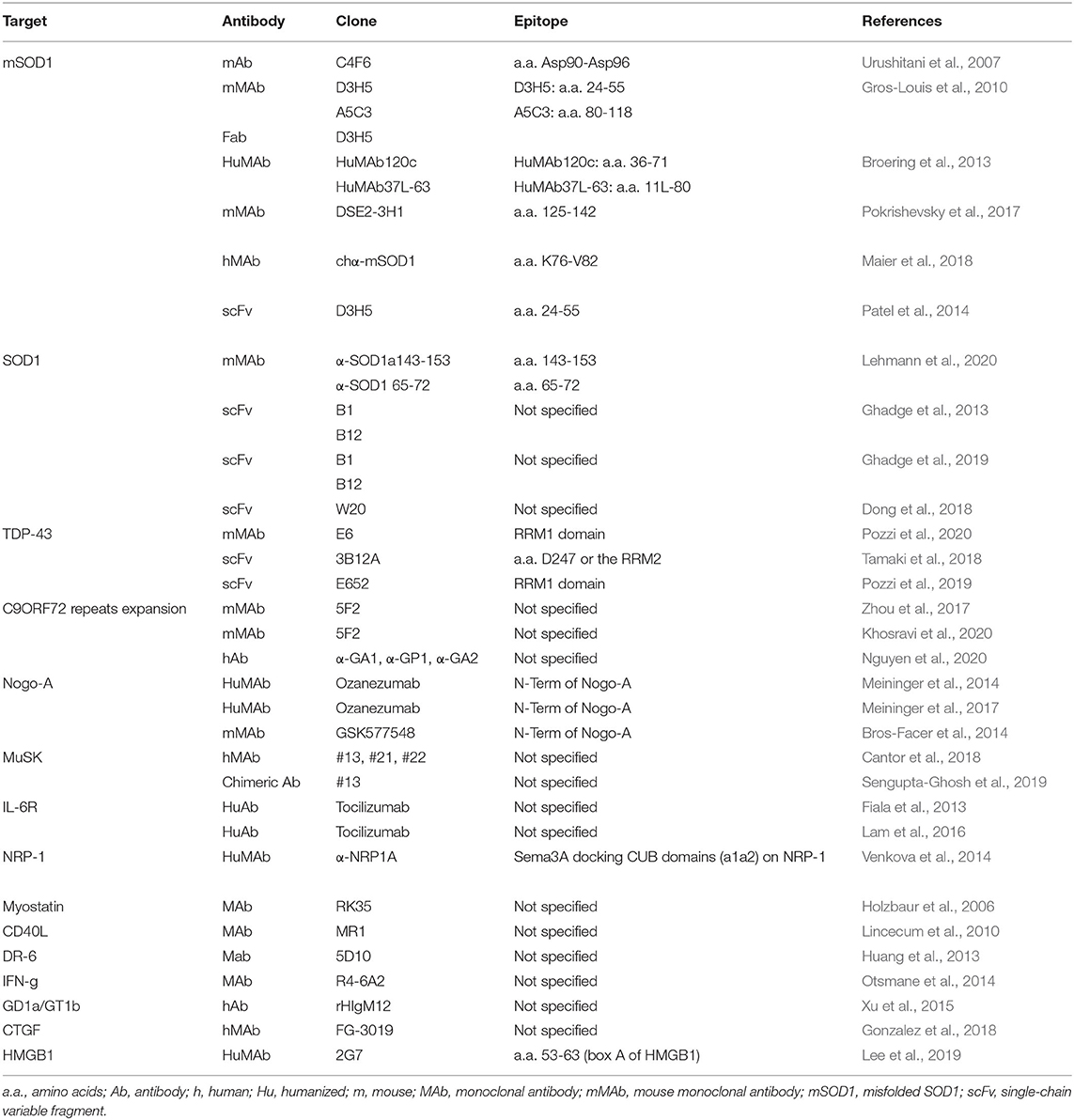
Table 1. Summary of the main characteristics of the antibody-based interventions identified in the 31 original articles analyzed in this work.
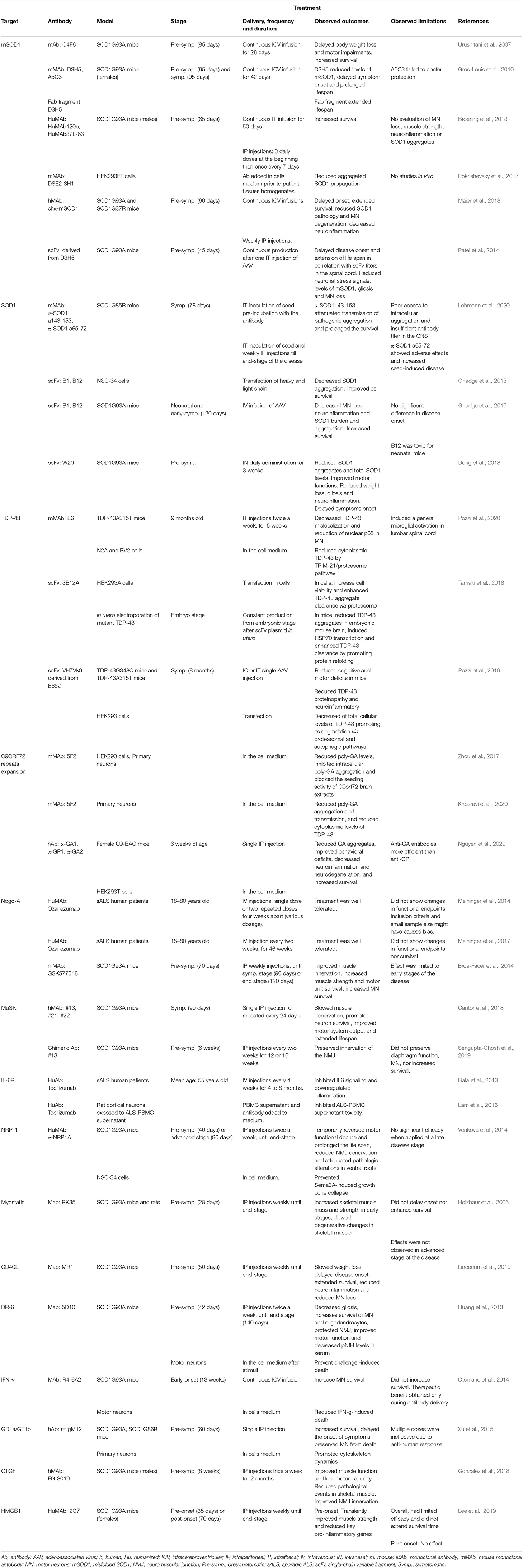
Table 2. Summary of the main characteristics of treatments tested in the 31 original articles analyzed in this work.
Data Analysis
A thematic summary technique was used for data synthesis. We defined “targeted protein” as a specific thematic category to classify the selected publications and grouped single manuscript studies under the “other targeted proteins” category. We revised and discussed the outcomes of the studies under each category, evaluating the type of antibody employed, the specific epitope bound, and the models used for the study.
Results
Description of the Selected Papers and Identification of Thematic Categories
The systematic review conduced for antibody-based therapeutic interventions for ALS resulted in the final selection of 31 papers listed in Tables 1, 2.
As shown in Figure 1, we primarily found a total of 5,329 publications to screen. 1,710 papers were excluded as duplicates, none after screening for “exclusion on format” criteria and 100 based on “exclusion on language.” A total of 2,717 publications were eliminated after “exclusion on intervention keyworks” in title, abstract and keywords session. N = 836 were eliminated for “population” and n = 1,881 for “intervention” keywords, leaving 802 articles for further analysis with Covidence software. The title and abstract of the remaining papers were fully screened (Supplementary Table 1), leading to exclusion of n = 675 manuscripts of which the subject was not relevant for our review despite the presence of keywords, leaving 127 papers for further screening (Supplementary Table 2). The final full text analysis based on nature, finality method and availability criteria led to the exclusion of 96 total studies. N = 59 studies were excluded based on nature, n = 25 based on finality, n = 3 based on method and n = 9 based on availability of a full text to analyze, leaving a total of 31 studies for the present review.
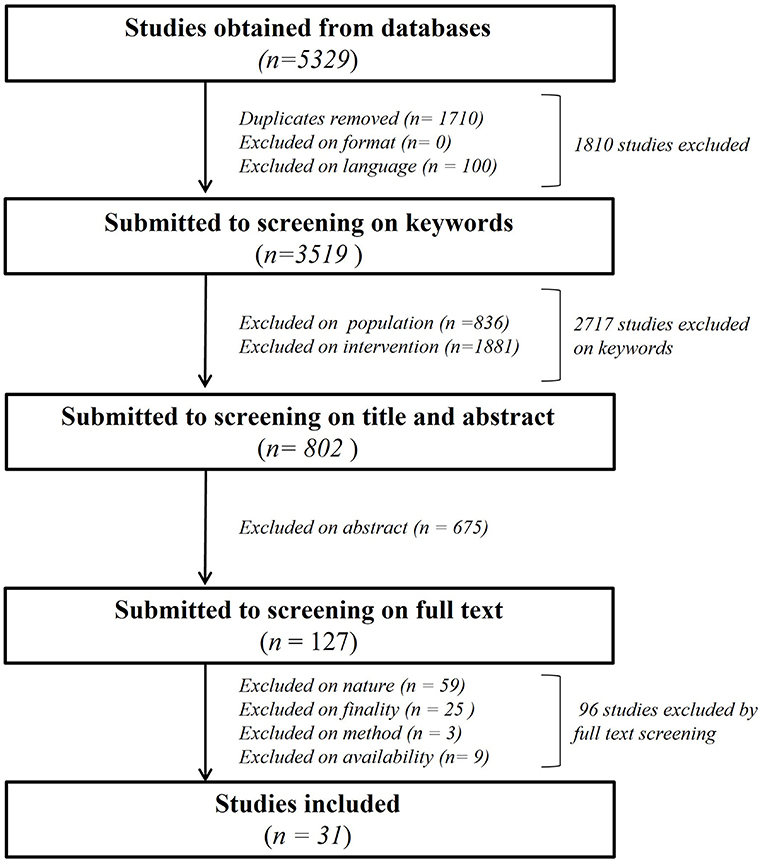
Figure 1. Flowchart of the systematic literature analysis describing all the steps of the manuscript's selection leading to the 31 analyzed paper.
The selected papers were analyzed by year of publication as shown in Figure 2. The first paper describing an antibody-based therapy for ALS targeted myostatin and was published in 2006, followed by a paper targeting SOD1 that was published at the beginning of 2007. The majority of the remaining selected papers were published between 2010 and 2020. We identified the categories for the thematic summary and applied them to the selected papers. As summarized in Figure 2, SOD1 is presently the main target of antibody-based approaches described in 10 papers; antibodies against TDP-43 and C9ORF72 repeats were discussed in 3 papers each; 3 papers described antibodies against Nogo-A; 2 papers described antibodies against IL-6 receptor and Musk; and a total of 8 manuscripts targeted respectively NRP-1, myostatin, CD40 ligand, DR-6 receptor, INF-γ, GD1a, CTGF and HMGB1 that were then grouped in the thematic category of “other targeted proteins.”
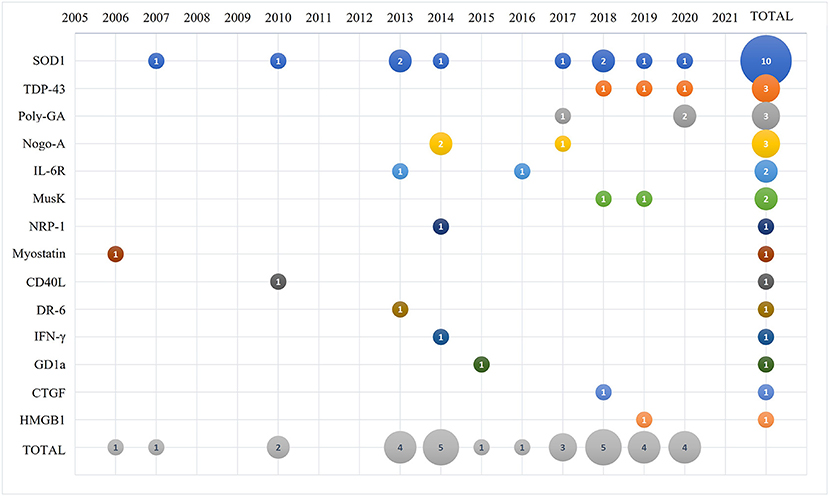
Figure 2. Bubble plot of the 31 selected and analyzed papers describing the number of manuscripts published (number in the bubble) per selected target (y-axis) and time (x-axis).
Antibody-Based Interventions for SOD1
Superoxide dismutase 1 (SOD1) have been unequivocally implicated in ALS. SOD1 mutation accounts for 2–5% of ALS cases (Gros-Louis et al., 2010). Moreover, SOD1 intracellular aggregation, which causes specific degeneration of motor neurons (Gurney et al., 1994), has been observed in sporadic ALS cases without SOD1 mutation (Bosco et al., 2010; Forsberg et al., 2010; Guareschi et al., 2012; Pokrishevsky et al., 2012; Paré et al., 2018). From the 31 studies included, 10 studies were focused on the protein SOD1. These studies were published between 2007 and 2020. Five of them used full-length monoclonal antibodies (MAb), three with mouse and two with human origin. One used a full-length antibody purified from antisera of actively immunized mice, whereas the other four studies used single-chain variable fragments (scFv).
Full Length Antibodies Against SOD1
The first passive immunization approach against misfolded SOD1 was studied and proposed by Urushitani et al. (2007). The aim of the study was to test active and passive immunization protocols aimed at reducing the burden of extracellular SOD1 mutants in the nervous tissue of mouse models of ALS. An antibody specific for human misfolded SOD1 (a.a. Asp90–Asp96), named C4F6, was purified from C57BL/6 mice inoculated with recombinant human G93ASOD1 and administered to SOD1G93A mice by intraventricular infusion for 28 days starting from the pre-symptomatic stage (85 days of age). Disease progression was monitored using a temporal profile of the body weight and hindlimb reflex score and the passive immunization approach demonstrated to significantly delay body weight loss, hindlimb reflex impairment, and to increase the life span of treated mice. By highlighting the significant effect of passive immunization vs. active immunization, this paper opened the door for antibody-based approaches against SOD1 for ALS treatment (Urushitani et al., 2007).
In 2010, Gros-Louis et al. investigated passive immunization of mutant SOD1G93A female mice with D3H5 or A5C3, two mMAb targeting misfolded SOD1 (mSOD1) (Gros-Louis et al., 2010). The antibodies were obtained from supernatant of hybridoma cell lines derived from mice injected with the metal free apo-form of recombinant SOD1 with G93A mutation as antigen. A5C3 antibody targets amino acids (a.a.) 80-118, while D3H5 targets a.a. 24-55 (Rotunno and Bosco, 2013). After intracerebroventricular (ICV) infusion for 42 days, D3H5 antibody succeeded in delaying symptoms onset and mortality of ALS mice and in reducing mSOD1 levels by 23% in spinal cord of treated mice compared with a group infused with control mouse IgG. The treatment showed interesting, although not significant, reduction in motor neuron loss, attenuation of astrocytosis, and increased number of innervated neuromuscular junctions (NMJ). Authors also tested the Fab fragment of D3H5, which demonstrated to confer some protection by extending the lifespan by 6 days. On the other hand, A5C3 failed to confer any protection (Gros-Louis et al., 2010).
Eleven humanized MAbs directed against human SOD1, five of which binding only to unfolded or misfolded hSOD1, were developed by Broering et al. in 2013 (Broering et al., 2013). During exploratory experiments, male mice expressing SOD1G93A were treated with the antibodies in the cerebrospinal fluid (CSF) by lumbar intrathecal (IT) injections with osmotic pump for 50 days, starting from a pre-symptomatic stage of the disease (65 days of age). Although not significant, due to small group size, antibodies showed no toxicity and tended to increase survival in SOD1G93A mice. Experiments were then repeated in a larger cohort of mice with the two antibodies that yielded the best results in terms of survival, i.e., HuMAb120c and HuMAb37L-63, confirming their therapeutic effect. These two antibodies recognize linear epitopes and bind to a.a. 36-71 present on the zinc-binding loop (Ab120c) and to 11L-80 epitope (Ab37L-63), not accessible in properly folded hSOD1. HuMAb37L-63 was also tested by intraperitoneal (IP) administration. This delivery method yielded higher levels of antibody in the nervous tissue but same survival time as IT injections. No further analyses on motor neuron loss, muscle strength, neuroinflammation or SOD1 aggregates were performed in this study (Broering et al., 2013).
Few years later, Pokrieshevsky et al. conducted a study using a mouse monoclonal antibody (mMAb) specific to misfolded SOD1 (DSE2-3H1), binding to a.a. 125-142 of extended electrostatic loop of full-length SOD1 protein, demonstrated to confer prion-like properties to the protein (Rotunno and Bosco, 2013; Pokrishevsky et al., 2017). In HEK293FT cells transfected with chimeric SOD1-GFP protein and exposed to hSOD1-fALS spinal cord tissue homogenates, the group demonstrated the ability of misfolded SOD1, present in the fALS tissues, to trigger the aggregation of the reporter protein. Interestingly, the treatment with DSE2-3H1antibody for 30–45 min prior to the exposure of tissues homogenates successfully attenuated the aggregation of SOD1-GFP protein (Pokrishevsky et al., 2017).
In 2018, Maier et al. developed a recombinant human monoclonal antibody (hMAb), specific to human misfolded SOD1 (Maier et al., 2018) and, to avoid mouse anti-human responses upon chronic use, a chimeric version was used in ALS mice models. The antibody was designed to specifically target a.a. K76-V82 in the IV loop of native SOD1. ICV infusion with minipumps from a pre-symptomatic stage (60 days of age) until the end stage of disease ameliorated motor symptoms and reduced SOD1 pathology in SOD1G93A mice. Gait abnormalities were significantly improved, body weight loss was attenuated, and median survival increased compared to vehicle-treated control littermates. Histological analysis of spinal cords revealed a trend toward a decrease in neuroinflammation and a significant reduction in SOD1 aggregate amount by 51%. When administered IP as a single dose, the antibody was able to penetrate the central nervous system (CNS) and bind to misfolded SOD1. It improved the survival of SOD1G93A mice and their motor performance, even at low doses. It led to 66% reduction in misfolded SOD1 and 25% reduction in SOD1 aggregates together with a reduction of 37% in microgliosis and 43% in astrogliosis. In slowly progressing SOD1G37R mice, when injected IP, the chimeric antibody delayed disease onset, ameliorated motor impairments and increased survival. Mice treated with chimeric α-mSOD1 displayed higher muscular strength, with 20% increase in weight of the hindlimb muscles, and preservation of 50% of motor neurons (Maier et al., 2018).
More recently, Lehmann et al. investigated the effect of disordered SOD1-selective antibodies on the SOD1 transmission of aggregation properties. In their model, intrathecal inoculation of hSOD1 aggregates isolated from the CNS of ALS mice model exacerbated pathology and SOD1 aggregation in SOD1G85R mice (Lehmann et al., 2020). Authors developed two mMAbs specific to epitopes exposed only in aggregated forms of human SOD1 (hSOD1), i.e., a.a. 143-153 of C-terminus of hSOD1 (α-SOD1143-153) and a.a. 65-72 (α-SOD165-72). Antibodies were pre-incubated with the aggregate strains and further injected into mice, or directly tested by weekly IP injections before inoculation of aggregate strains. In both cases, Ab administration or Ab-preincubated strains, were delivered from early symptomatic stage (78 days of age) until end-stage of the disease. Both pre-incubation with aggregates and IP injections of α-SOD1143-153 attenuated transmission of pathogenic aggregation and prolonged the survival of transgenic mice by up to 47%. α-SOD165-72 antibody, which displayed lower affinity for hSOD1, showed adverse effects and aggravated seed-induced ALS-like disease. Moreover, α-SOD1143-153 was found in association with aggregated hSOD1 in spinal cord homogenates. However, the administration of antibodies in non-inoculated hSOD1G85R mice did not improve survival nor mitigated spontaneously evolving aggregation, suggesting that the antibody effect is specific to seed-induced ALS-like disease (Lehmann et al., 2020).
Nanobodies Against SOD1
The first nanobodies against SOD1 were generated by Ghadge et al. (2013), who developed two human-derived single chain antibodies (scFvs) named B1 and B12, binding to all three WT, G93A and A4V SOD1 forms (Ghadge et al., 2013). NSC-34 cells were co-transfected with the scFv antibody and A4VSOD1-YFP to induce SOD1 aggregation. After 48 h of A4VSOD1-YFP transfection, ~24% of YFP expressing cells had bright focal punctuate areas of fluorescence that were significantly decreased following co-transfection with B1 or B12 scFv. Clones B1 and B12 also improved cell survival following expression of either A4V or G93A mutations compared to control groups with empty vector or a negative control scFv (Ghadge et al., 2013). In 2019, the same group subcloned B1 and B12 into an AAV9 virus and injected it intravenously (IV) in neonatal and early symptomatic (120 days old) SOD1G93A mice. B1 and B12 decreased motor neuron loss, microgliosis, astrogliosis, and SOD1 amount and aggregation, as shown by reduction of percentage of anti-SOD1 positive area in spinal cord sections. No significant difference in the disease onset was observed, but the α-SOD1 treatment significantly increased survival compared to control mice, with the longest survival measured after AAV:scFvB1 delivery into neonatal SOD1G93A mice. On the contrary, AAV:scFvB12 was toxic for neonatal mice (Ghadge et al., 2019).
Concomitantly to the first single chain antibody, Patel et al. produced in 2014 a scFv antibody derived from the monoclonal antibody D3H5, previously tested by Gros-Louis et al. (2010) (Patel et al., 2014). After a single intrathecal injection of AAV2/1:D3H5scFv in young SOD1G93A mice (45 days of age), AAV-mediated expression of the scFv delayed disease onset and extended life span by up to 28%. Mice survival was directly correlated with the amount of scFv antibody present in the spinal cord. The treatment attenuated neuronal stress pathways such as GAP-43 and ATF3 upregulation, reduced gliosis and levels of misfolded SOD1 in the spinal cord of SOD1G93A mice as well as decreased motor neuron loss (Patel et al., 2014).
Finally, in 2018, Dong et al. produced a human scFv antibody, named W20, targeting toxic SOD1 oligomers, which was delivered intranasally (IN) every day for 3 weeks in SOD1G93A mice before the onset of symptoms (Dong et al., 2018). Authors observed a significant reduction of SOD1 aggregates in the brain stem, and reduced SOD1 levels in the spinal cord lysates of SOD1G93A mice. Oligomeric forms of the protein were reduced in W20-treated mice showing a significant 37.6 and 22.3% of reductions in fibrillar SOD1 and prefibrillar oligomers respectively, compared to vehicle-treated SOD1G93A mice. Treated mice displayed improved rotarod performance, significantly increased muscle strength, reduced hindlimb clasping, reduced weight loss, and markedly reduced gliosis and neuroinflammation, together with a slight delay in symptoms onset (Dong et al., 2018).
Antibody-Based Interventions for TDP-43
TDP-43 proteinopathy is a hallmark of ALS (Neumann et al., 2006), characterized by cytoplasmic mislocalization and aggregation of the TAR DNA binding protein 43 (TDP-43), which loses its physiological properties, leading to neuronal death. Out of the 31 papers included in the analysis, 3 studies targeted TDP-43 protein. These studies were published between 2018 and 2020. One of them used a monoclonal full-length antibody (MAb), whereas the other two studies used single chain antibodies (scFvs).
Full Length Antibodies Against TDP-43
Pozzi et al. produced 8 monoclonal antibodies directed mainly against the RNA recognition motif 1 (RRM1) domain of TDP-43 (Pozzi et al., 2019), known to be sensitive to TDP-43 proteinopathy (Chang et al., 2013; Shodai et al., 2013) and involved in the interaction between TDP-43 and p65 NFκB (Swarup et al., 2011). From those 8 clones, only three of them, named C10, G8 and E6, were able to clearly detect TDP-43 with a visible preference for the cytoplasmic protein. Among the three full-length monoclonal antibodies, the E6 clone was the most effective at interfering with the binding of TDP-43 to p65 (Pozzi et al., 2019). Authors tested the E6 full-length antibody both in vitro and in vivo, where E6 showed no toxicity (Pozzi et al., 2020). In immortalized neuronal cells, E6 reduced levels of cytoplasmic TDP-43 by 33% compared to control antibody, inducing the TRIM-21 and the lysosome-dependent mechanisms of degradation. E6 full-length antibody also reduced NFκB activation in neuronal cells and in microglial cells challenged with LPS, due to either a reduction of the cytoplasmic TDP-43 or the inhibition of TDP-43/p65 interaction (Pozzi et al., 2020). Repeated intrathecal injections of E6 antibody showed to be safe and feasible for symptomatic TDP-43A315T mice, and the antibody demonstrated a broad diffusion throughout the length of the spinal cord together with motor neuron and microglia uptake. Five-weeks of repeated injections of E6 full-length antibody in TDP-43A315T mice resulted in mitigation of cytoplasmic TDP-43 mislocalization and reduction of nuclear p65 in motor neurons; however, E6 treatment induced a general microglial activation in lumbar spinal cord, suggested to be associated with the Fc domain of the antibody (Pozzi et al., 2020).
Nanobodies Against TDP-43
In 2018, Tamaki et al. produced two nanobodies derived from 3B12A MAb directed against the a.a. D247 of the RRM2 domain masked in the physiological state (Tamaki et al., 2018). Authors showed a specific binding of the scFv antibodies to the mislocalized and aggregated form of the protein. The two produced scFvs contained a PEST sequence, which induces proteasome-mediated proteolysis, and were genetically engineered with a chaperone-mediated autophagy (CMA) sequence or a proteasome localization signal (CL1) to enhance their degradation. In transfected HEK293A cell, proteasome-mediated degradation of aggregated TDP-43 was accelerated, enhancing TDP-43 aggregates clearance and cell viability. TDP-43 aggregates were also reduced in brains of an embryonic mouse model where the plasmids encoding for the scFv antibodies were in utero electroporated together with mutant TDP-43 plasmids to induce the pathology. Authors correlated the neuroprotection observed in mice to the transcriptional induction of HSP70, suggesting that HSP70 was involved in TDP-43 clearance by promoting protein refolding (Tamaki et al., 2018).
Few months later, Pozzi et al. produced two scFvs antibodies derived from E6 MAb (Pozzi et al., 2020), named VH1Vk9 and VH7Vk9, binding to the RRM1 domain of TDP-43 protein (Pozzi et al., 2019). The scFvs were designed with a secretion signal peptide, which enabled them to target distant cells. ScFv antibodies transfected in HEK293 cells induced a decrease in total cellular levels of TDP-43 by enhancing the polyubiquitination signaling for proteasomal and autophagic degradation. In the same cells, VH1Vk9 and VH7Vk9 reduced TDP-43 mislocalization and aggregation induced by ethacrynic acid (EA) treatment, a model of TDP-43 proteinopathy. Both VH1Vk9 and VH7Vk9 were able to disrupt the protein-protein interaction between TDP-43 and p65 and to reduce LPS-induced activation of microglial cells. As VH7Vk9 was more efficient than VH1Vk9 in reducing NFkB activity and TDP-43 proteinopathy, in vivo experiments were conducted by delivering the selected scFv through a viral vector. AAV2/9:VH7Vk9 expression reduced the LPS-induced microglial activation by almost 50% in vivo. In symptomatic TDP-43G348C mice, a single intracortical (IC) injection of AAV2/9:VH7Vk9 mitigated cognitive impairment, neuroinflammation and TDP-43 pathology as early as 2 months after injection. Moreover, in symptomatic TDP-43A315T mice, injected IT with AAV2/9:VH7Vk9, the VH7Vk9 scFv antibody improved motor performance and neuromuscular junction innervation, together with reduced neuroinflammation and TDP-43 cytoplasmic accumulation (Pozzi et al., 2019).
Antibody-Based Interventions for C9ORF72 Repeats Expansion
In 2011, a hexanucleotide GGGGCC (G4C2)n repeat expansion in the C9orf72 gene was found to be implicated in frontotemporal lobar degeneration and amyotrophic lateral sclerosis as the most common known genetic cause (DeJesus-Hernandez et al., 2011; Renton et al., 2011; Gijselinck et al., 2012). These repeats are transcribed into repetitive RNA, which forms sense and antisense RNA (Ash et al., 2013). Despite being within a non-coding region of C9orf72, these repetitive RNAs are translated into five aggregating dipeptide repeat proteins (DPRs) named poly-GA, poly-GP, poly-PA, and poly-PR, forming neuronal inclusions, and sequestering several RNA binding proteins (Mori et al., 2013). Out of 31 studies included, 3 studies focused on the poly-GA protein. These studies were published between 2017 and 2020. Two of them used a mouse monoclonal full-length antibody, whereas the last one used a human recombinant full-length antibody.
In 2017, Zhou et al. studied the spread of intraneuronal pathology of dipeptide repeat proteins (DPRs) and investigated the potential of an antibody therapy to inhibit this phenomenon (Zhou et al., 2017). First, using co-culture systems of primary or immortalized cells, they demonstrated that poly-GA, poly-GP and poly-PA repeat species can be transmitted between cells and cause seed aggregation in recipient cells. They also observed that exposing cells to brain extracts from ALS patients with C9orf72 repeat expansion increased poly-GA, -GR and -GP levels and aggregation, suggesting a vicious cycle. In a second set of experiments, authors tested a mouse monoclonal IgG2A antibody against poly-GA DPRs, named 5F2, produced through immunizing mice with insoluble recombinant poly-GA repeats (Mackenzie et al., 2013). In GA175GFP-transfected HEK293 cells, 3-day treatment with 5F2 anti-GA antibody was able to reduce poly-GA levels and aggregation compared to isotype control treated cells. The effect was confirmed in primary neurons treated for 6 days with 1mg/ml antibody. Finally, preincubation of C9orf72 patients brain extracts with the antibody prevented the seeding of aggregates in (G4C2)80-expressing HEK293 cells (Zhou et al., 2017).
Few years later, Khosravi et al. used co-culture systems of primary neurons to demonstrate that poly-GA aggregation can induce TDP-43 cytoplasmic mislocalization and aggregation in poly-GA expressing cells and in neighboring cells by inhibition of proteasome function (Khosravi et al., 2020). Treatment with the 5F2 anti-GA antibody ameliorated the TDP-43 pathology in both donor and recipient cells. Authors corroborated these results by immunodepleting poly-GA with 5F2 antibody from media of DRPs-expressing cells, establishing that immunodepletion prevented poly-GA uptake in recipient cells and thus reduced TDP-43 pathology compared to depletion with control IgG (Khosravi et al., 2020).
In 2020, Nguyen et al. purified three high affinity human recombinant antibodies against GA and GP C9orf72 RAN proteins, respectively named α-GA1, α-GP1, and α-GA2 (Nguyen et al., 2020). Antibodies were previously obtained from libraries of memory B cells of healthy individuals (Sevigny et al., 2016). The antibodies were able to recognize aggregated GA or GP RAN proteins in both HEK293 cells and C9-BAC mouse tissues, a mouse model of ALS/FTLD containing 500–750 GA-repeats (Liu et al., 2016), as well as in human cerebellar samples with C9orf72 expansions. In C9orf72 patient-derived motor neurons (iMNs), treatment with α-GA1 and α-GA2, but not α-GP1 nor control antibody, reduced GA levels. To understand the mechanisms involved in the increased poly-GA clearance, authors conducted a proximity ligation assay (PLA), demonstrating that TRIM21 protein interacts with the Fc region of α-GA1 enhancing the clearance of poly-GA/anti-GA1 complex. Moreover, they showed that treatment with α-GA1 antibody released proteosomal and autophagy proteins sequestered in poly-GA aggregates, resulting in a rescue of proteasomal activity and increased protein clearance. Anti-GA1, but not anti-GA2 nor anti-GP1, was able to rescue cultured cells from DRPs induced toxicity, suggesting a therapeutic potential for α-GA1. In C9-BAC mice, α-GA1, α-GA2, α-GP2 (30 mg/kg) or PBS was administered by weekly IP injections from 6 to 43 weeks of age. Murine chimeric derivatives were used to avoid mouse anti-human responses. The three antibodies were able to penetrate the CNS and reduced both soluble and aggregated RAN proteins. Both anti-GA antibodies improved survival of C9-BAC mice and reduced gait abnormalities as well as anxiety-like behavior, whereas the α-GP1 antibody did not. Authors hypothesized that a higher reduction of poly-GP was needed for behavioral improvements. Anti-GA1 treatment, which showed higher efficacy in rescuing the phenotype, probably due to its increased affinity for longer GA-repeats (60 and 120), was also able to significantly reduce inflammation (astrocytosis) in motor cortex, and to increase neuronal survival in spinal cord. Interestingly, using an aglycosylated α-GA1 antibody, authors demonstrated, both in cells and mice, that a fully glycosylated Fc region of the α-GA1 antibody is required for α-GA1 cellular uptake and for its therapeutic effects such as reduction of GA-aggregates, neurons preservation, and improvement of behavioral performance (Nguyen et al., 2020).
Antibody-Based Interventions for the Neurite Outgrowth Inhibitor A
Nogo is a myelin-associated neurite outgrowth inhibitor in the CNS (Caroni and Schwab, 1988), which has three protein products: Nogo-A (1,163 amino acids), Nogo-B (360 amino acids) and Nogo-C (199 amino acids) (Chen et al., 2000; Prinjha et al., 2000). Nogo-A was shown to increase in SOD1G86R transgenic mice at the pre-symptomatic stage, while Nogo-C levels were decreased (Dupuis et al., 2002; Jokic et al., 2006). Moreover, altered expression of Nogo proteins in the post-mortem and biopsy samples from diagnosed ALS patients (brachial plexus nerve and deltoid muscle) was reported (Jokic et al., 2006). Nogo-A expression has been linked to axonal denervation (Jokic et al., 2006). Knock-out of Nogo-A in ALS mice increased the number of healthy motor neurons in soleus muscle and the mice survival, whereas overexpression of Nogo-A in muscles gave rise to the disassembly of the neuromuscular junction (Jokic et al., 2006). Out of 31 studies included, 3 studies focused on the Nogo-A protein. These studies were published between 2014 and 2017. One study used a mouse antibody whereas the other two used a humanized antibody.
Ozanezumab (GSK1223249), a humanized monoclonal antibody (HuMAb) targeting the N- terminus of Nogo A was generated by Schnell and Schwab by immunization of mice in 1990 (Schnell and Schwab, 1990). The Fc portion of the antibody includes two amino acid substitutions at positions 235 and 237, taking away the ability of monoclonal antibodies to recruit immune effector cells and complement. This humanized anti-NogoA antibody was first used in 2014 by Meininger et al. for a phase I study in sporadic ALS patients where safety, pharmacokinetics (PK), and functional effects of Ozanezumab were assessed (Meininger et al., 2014). The trial was performed as a randomized study, in which Ozanezumab was compared to placebo in a double-blind manner. Two groups of patients received respectively (1) a single dose (SD) of Ozanezumab (0.01, 0.1, 1, 5, or 15 mg/kg) or (2) two repeated doses (RD) of Ozanezumab (0.5, 2.5, or 15 mg/kg). In both cases the antibody was administered intravenously (IV). In total, 18 patients (18–80 years of age) received placebo, and 53 patients (age matched) received Ozanezumab. A clear co-localization between Nogo-A and Ozanezumab was observed in skeletal muscle biopsies by IHC staining and the percentage of co-localization correlated with the amount of Ozanezumab injected. Ozanezumab was well tolerated both as SD (0.01–15 mg/kg) and RD (0.5–15 mg/kg). No adverse events (AEs) were reported with increasing doses of Ozanezumab although the incidence of AEs was numerically higher in patients receiving a repeated injection of the two higher doses, but the proportion of AEs and serious adverse events (SAEs) was similar between treated and placebo groups. Generally, SAEs and deaths were not related to the treatment, but doubts were raised for one cardiac non-serious AE (mild sinus tachycardia). Although the study was not designed to obtain relevant observations in functional endpoints, a positive trend in some clinical measurements such as ALS Functional Rating Scale (ALSFRS-R), slow vital capacity (SVC), and manual muscle testing (MMT) was observed with the highest dose (Meininger et al., 2014).
The results of this study allowed the authors to start a Phase II randomized and double-blinded clinical trial, aimed at investigating Ozanezumab efficacy and safety in both familial and sporadic ALS patients (Meininger et al., 2017), who were allowed to continue taking riluzole during the study. Patients were divided into two groups that received either placebo (n = 151) or 15 mg/kg of Ozanezumab (n = 152) delivered intravenously every 2 weeks for 46 weeks. Assessments were performed at 48 and 60 weeks after treatment. The study was a combined analysis of the ALS Functional Rating Scale-Revised (ALSFRS-R) and overall survival. Even though Ozanezumab was generally well tolerated, and that reported AEs were similar between treatment and placebo groups, no difference was observed in scores nor survival rates at 48 weeks. No efficacy was therefore observed for Ozanezumab, suggesting a poor reliability of anti-Nogo-A treatments for ALS patients (Meininger et al., 2017).
In 2014, concomitantly with the first clinical trial of Ozanezumab, Bros-Facer et al. studied the effect of an anti-Nogo-A antibody on the disease phenotype and progression in the SOD1G93A mice (Bros-Facer et al., 2014). In this study, a murine antibody (GSK577548: the murine parental antibody for Ozanezumab) was generated against Nogo-A. Authors treated SOD1G93A mice by weekly IP injections starting from 70 days of age (pre-symptomatic stage) until 90 (late symptomatic stage) or 120 days of age (near to end-stage). After confirming the presence of the antibody in muscles and its co-localization with Nogo-A, authors observed a reduction of Nogo-A levels, as well as CHRNA1 and MuSK, two markers of neuromuscular dysfunction, in 90 days old anti-Nogo-A treated mice compared to control group. No differences in expression were observed between the groups at 120 days. The innervation of two muscles (slow-twitch soleus muscle and fast-twitch extensor digitorum longus (EDL) muscle) was analyzed by staining for silver cholinesterase showing a clear decrease in denervation in both muscles of antibody-treated SOD1G93A mice at 90 days of age. This improvement in innervation led to an increased muscle force and motor unit survival. Functional physiological analysis of the muscle contractile ability and staining for succinate dehydrogenase (SDH), an oxidative enzyme that increases in fast twitch fibers during disease progression, showed that treatment with the antibody prevented pathological alterations. Similarly, Nissl staining of lumbar spinal cord sections showed 21% increase in motor neuron survival of treated mice. Although encouraging results were observed in treated mice, improvements were mainly observed at 90 days of age and not maintained at end-stage of the disease (120 days of age), concluding that treatment with anti-Nogo-A antibody can significantly improve neuromuscular function only in early disease stages of SOD1G93A mouse model (Bros-Facer et al., 2014).
Antibody-Based Interventions for the Muscle-Specific Kinase
In ALS animal models, loss of neuromuscular synapses occurs prior to the loss of motor neurons (Alhindi et al., 2021). Although defects in the MuSK signaling pathway are not associated with ALS, studies show that overexpression of MuSK stabilizes neuromuscular synapses in SOD1G93A ALS mouse model, preventing denervation and slowing down motor dysfunction (Pérez-García and Burden, 2012). Moreover, stimulation of the MuSK receptor tyrosine-kinase activity with Agrin leads to anchoring and enhanced expression of critical post-synaptic proteins (Zhang et al., 2011; Burden et al., 2013). Out of 31 studies included, two studies focused on the MuSK protein. These studies were published between 2018 and 2019. Both used a humanized full-length antibody.
In 1997, 21 single-chain antibodies were isolated by phage display with the aim to modulate MuSk functions. Four of these scFvs, named #13, #21, #22 and #2, were highly specific for MuSk. Among them, #13 and #22 were able to activate the kinase and induce AChRs clustering in the cultured myotube cell lines (Xie et al., 1997). In 2018 Cantor et al. generated human IgG1 antibodies from single chain antibodies #21, #13 and #22 that recognize mouse MuSK (Cantor et al., 2018) with the aim to determine whether increasing MuSK activity with a pharmacological approach would preserve neuromuscular synapses in SOD1G93A mice and slow down the loss of motor innervation. Unlike agrin, the agonist antibodies stimulated MuSK in a Lrp4-independent manner, binding directly to the Fz-like domain of MuSK, which dimerizes and stimulates its phosphorylation. Preliminary experiments in C2 mouse muscle cells and WT mice confirmed that MuSK antibodies engaged with and stimulated MuSK and were well tolerated by the mice. A single IP injection of 2 mg/kg #13 antibody was sufficient to saturate MuSK at the synapse and increased its phosphorylation. Authors administered 10 mg/kg of the engineered antibody #13, or control human IgG1, by a single IP injection into male and female SOD1G93A mice after their disease onset (90 days of age). Twenty days after the single injection, they analyzed diaphragm muscle innervation and observed up to 2.7-fold increase in fully innervated synapses, and a 3.7-fold decrease of fully denervated synapses. Chronic administration with 10 mg/kg of a reverse chimera MuSK agonist antibody (murine IgG2A backbone lacking effector functions) every 24 days, decreased spinal cord motor neuron loss and maintained the number of innervated diaphragm neuromuscular synapses stable for 50 days more than control antibody, where neuromuscular junctions continue to denervate. Rescuing motor neurons and synaptic junctions resulted in an improved motor system output in #13 antibody treated mice, observed with the Compound Muscle Action Potential (CMAP) test. Treatment also extended lifespan of SOD1G93A mice by 7 days in females and 10 days in males (Cantor et al., 2018).
In 2019, Sengupta-Ghosh et al. (2019) tested MuSK agonist antibody #13 (on murine IgG2A backbone) by chronic IP administration of 10 or 100 mg/kg of antibody every 2 weeks in male and female SOD1G93A mice. Unlike Cantor et al. (2018), treatment started at 6 weeks of age, prior to disease onset, until 18 weeks of age (for histology analysis), or 22 weeks of age (for the functional analysis). Authors showed that treatment was able to preserve innervation of the NMJ in the diaphragm, as previously demonstrated. However, they discovered that it was insufficient to provide any functional benefit such as preservation of respiratory or diaphragm function (as measured by plethysmography or the CMAP test) nor was it able to alter the disease progression. Moreover, no improvement of motor neuron loss in the spinal cord and brainstem, no reduction of body weight loss, and no extension of mice survival were observed (Sengupta-Ghosh et al., 2019).
Antibody-Based Interventions for IL-6 Receptor
Sporadic ALS patients show chronic peripheral and central nervous system inflammation characterized by infiltration in the spinal cord of inflammatory macrophages, IL-17A-positive T cells, and mast cells (Henkel et al., 2004; Fiala et al., 2010). Studies show that inflammatory macrophages play an important role in motor neuron death in the ALS conditions. Indeed, 19% of motor neurons in post-mortem ALS spinal cords, both healthy-appearing and apoptotic neurons, exhibit evidence of phagocytosis by interleukin-6 (IL-6)- and tumor necrosis factor-α (TNFα)-positive macrophages (Liu et al., 2012). Moreover, IL-6 signaling has proved toxic for mouse brain in a dose-dependent manner (Campbell et al., 2014).
Out of 31 studies included, 2 studies focused on the IL-6 receptor (IL-6R). These studies were published between 2006 and 2013. Both used a recombinant humanized monoclonal antibody named Tocilizumab.
In 2012, Mizwicki et al. provided evidence for attenuation of inflammatory activation in the peripheral blood mononuclear cells (PBMCs) and in the CSF of ALS patients through inhibition of IL-6R by treatment with Tocilizumab, a humanized antibody (HuAb) against IL-6R (Mizwicki et al., 2012). In 2013, Fiala et al. studied the in vivo effects of Tocilizumab infusion therapy in sporadic ALS patients (Fiala et al., 2013). Eleven patients with sporadic ALS (mean age 55 years) were selected for the study. Ten patients provided peripheral blood specimens. First, authors measured baseline mRNA expression of genes implicated in inflammation by RT-PCR and divided the patients according to the level of inflammation into group 1 (5 patients with high inflammation) and group 2 (5 patients with low inflammation) (Fiala et al., 2010; Mizwicki et al., 2012). Antibody infusion was performed every 4 weeks from 4 to 8 months, with doses increasing from 4 to 8 mg/kg. Blood samples were taken before and 1 h after each infusion. Levels of inflammatory genes were measured in blood PBMC (peripheral blood mononuclear cells) together with serum cytokines and compared to the previously analyzed baseline levels. Results showed that Tocilizumab infusions downregulated inflammatory genes (in particular IL-1 and IL-6) both in PBMC and serum of group 1 (high inflammatory baseline) whereas group 2 patients (low inflammatory baseline) experienced an up-regulation of IL-1 and IL-6 mRNAs levels together with a strong release of IL-6 in serum. Interestingly, the progression of neurological decline (measured by ALSFRS-R) was attenuated in three patients that adhered to the therapy. Although the study was not a controlled double-blind trial, authors were able to conclude that infusions of Tocilizumab might be beneficial for sALS patients by normalizing IL-1 and IL-6 expression, but that the effects are individual as well as time- and dose-dependent (Fiala et al., 2013).
In 2016, Lam et al. investigated the effect of Tocilizumab in rat primary cortical neurons exposed to an ALS patients' PBMC secreted proteins. The investigation of the antibody treatment was part of a bigger study on the transcriptional and epigenetic differences in PBMCs from two twins, one of whom being diagnosed for ALS (Lam et al., 2016). Although no genetic differences were found in hematopoietic cells of these twins, authors reported a higher abundance of CD14-positive monocytes and lower presence of T- and NK-cells in the ALS-twin compared to her healthy sibling. RNA sequencing of PBMCs supported these findings and highlighted significant changes in chemokines and metalloproteases genes produced by ALS PBMC. Moreover, PBMC form the ALS-twin spontaneously produced high concentrations of IL-6, IL-1 and TNF-α, whereas the PBMC of the healthy twin had the same levels only after stimulation with SOD1. Having observed a higher production of IL-6, authors treated cultured rat primary cortical neurons with PBMC supernatants for 6 h (1:100 ratio of supernatant to medium) and demonstrated that PBMC supernatant from the ALS-twin, but not that of the healthy twin, was toxic for neurons and increased their degeneration. The level of neuronal degeneration and death was strongly reduced by treating cells with Tocilizumab for 2 h, but not with anti-IL-1 or anti-TNF-α antibodies (Lam et al., 2016).
Antibody-Based Interventions for Other Proteins
Finally, out of the 31 manuscripts analyzed, 8 papers described antibody interventions directed against other proteins, namely NRP-1, myostatin, CD40L, DR-6, IFN-γ, GD1a, CTGF and HMGB1. These studies were published between 2006 and 2019 and all used monoclonal antibodies. Four of them used mouse monoclonal Ab, and the other four used human or humanized monoclonal Ab. All these antibodies were tested in SOD1G93A mice and administered by IP injections, except for α-IFN-γ, which was administered ICV.
Neuropilin-1
Semaphorin 3A (Sema3A), a destabilizing factor for axonal innervation of neuromuscular junction, triggers distal axonopathy and muscle denervation through its receptor, Neuropilin-1 (NRP-1). Both proteins were upregulated in pre-symptomatic ALS mouse models and ALS patients (Körner et al., 2016; Maimon et al., 2018).
In 2014, Venkova et al. tested a humanized monoclonal antibody specific for the Sema3A docking CUB domains (a1a2) of NRP-1 (α-NRP-1A), provided by Genentech Inc (Venkova et al., 2014). In immortalized motoneuron-like cultured cells, α-NRP-1A antibody prevented Sema3A-induced growth cone collapse. In mice, α-NRP-1 antibody was administered by IP injections twice a week until end stage. When administered to pre-symptomatic (40 days of age) SOD1G93A mice, α-NRP-1A delayed and temporarily reversed motor functional decline, as assessed by rotarod task, and prolonged the lifespan. Moreover, it also reduced NMJ denervation and increased percentage of large-caliber axons in ventral roots. Despite these protective effects in pre-symptomatic treated mice, when administered at advanced stages of the disease (90 days of age) the antibody did not show any significant improvement (Venkova et al., 2014).
Myostatin
Myostatin is a member of the TGF-β superfamily. It regulates muscle development and homeostasis, by inhibiting both the proliferation and differentiation of satellite cells and myoblasts (McCroskery et al., 2003), and negatively regulates muscle regeneration after injury (McCroskery et al., 2005).
In 2006, Holzbauer et al. tested a myostatin/GDF-8 neutralizing antibody (RK35), previously generated from hybridoma cells obtained from mice immunized with recombinant myostatin (Whittemore et al., 2003), in both mouse and rat models expressing SOD1G93A (Holzbaur et al., 2006). The antibody was administered weekly, starting at pre-symptomatic stage (28 days of age) until sacrifice or end stage of the disease. In both mouse and rat models, inhibition of myostatin resulted in increased muscle mass and strength, which was maintained during the early stages (56–90 days of age) of disease but lost by end-stage. Myostatin inhibition slowed down degenerative changes in skeletal muscles (limb and diaphragm muscles) but did not delay the appearance of gait abnormalities or limb paralysis, nor did it extend the survival of diseased mice or rats. Changes observed in early stages were lost in late stages of the disease, except for the increase in diaphragm muscle mass and function that was maintained until the end stage in RK35-treated animals (Holzbaur et al., 2006).
CD40 Ligand
T-cell infiltration and activation has been observed during the disease progression in central and peripheral nervous system tissues of SOD1G93A mice (Lincecum et al., 2010). This phenomenon is mediated by the engagement of CD40L with its receptor (Roy et al., 1993).
In 2010, Lincecum et al. investigated the effect of a mouse monoclonal antibody, named MR1, directed against the mouse CD40 ligand (Lincecum et al., 2010). The antibody was derived from mouse hybridoma cells obtained after immunization of mice with IP injections of activated ThO (D1.6) (Noelle et al., 1992a,b). MR1R1 antibody was administered weekly starting at pre-symptomatic stage (50 days of age) until end stage. Treatment slowed weight loss, delayed disease onset, and extended survival of mice. Moreover, it lowered glial fibrillary acidic protein (GFAP) and Mac-2 staining, two markers glia activation, and reduced the loss of motor neurons (Lincecum et al., 2010).
Death Receptor 6
Death Receptor 6 (DR-6) is a tumor necrosis factor receptor which, after oligomerization, activates different pathways that can lead to neuronal death (Nikolaev et al., 2009).
In 2013 Huang et al. observed elevated levels of DR-6 in spinal cords of SOD1G93A mice and post-mortem tissues of ALS patients and investigated the effect of an anti-DR-6 antibody named 5D10 (Huang et al., 2013). The antibody was obtained from mouse hybridoma cells derived from mice after immunization with the recombinant protein. In vitro, the antibody promoted motor neuron survival, via activation of Akt phosphorylation and inhibition of the caspase 3 signaling pathways, following sodium arsenite treatment, growth factor withdrawal, or co-culture with SOD1G93A astrocytes. In vivo, IP delivery twice a week of 5D10 starting from asymptomatic stage (42 days of age) until 140 days of age promoted motor neuron survival, decreased astrocytes activation, and induced oligodendrocyte proliferation in the spinal cord and protected NMJ in gastrocnemius muscle from denervation. Moreover, 5D10-treated mice showed decreased pNfH levels in serum and 79% increase in hindlimb grip strength, suggesting improved motor function (Huang et al., 2013).
Interferon Gamma
IFN-γ is a pro-inflammatory cytokine whose levels were found increased in spinal cord of ALS mice and patients (Aebischer et al., 2011).
Otsmane et al. tested a rat IgG1 antibody against mouse IFN-γ, named R4-6A2, purified from hybridoma cell lines (Otsmane et al., 2014). In vitro, treatment with α-IFN-γ protected cultured mouse motor neurons from IFN-γ-induced death. The antibody was then delivered in the CSF of SOD1G93A mice by osmotic pumps ICV implanted, starting at early-onset of disease (13 weeks of age). In treated mice, authors observed a significant increase of surviving motor neurons but no changes in mice survival. The beneficial effect was observed only for the period during which the antibody was present in the CNS (Otsmane et al., 2014).
G1a and GT1b Gangliosides Complex
Gangliosides are glycosphingolipids that play important roles in the modulation of membrane proteins and ion channels, in cell signaling and in communication among cells (Sipione et al., 2020). GD1a and GT1b are enriched on the membrane of neurons and facilitate the interactions between oligodendrocytes and axons, promoting long-term axonal stability. Changes in the ganglioside profile have been observed in ALS patients and animal models (Rapport et al., 1985; Dodge et al., 2015).
Xu et al. tested a recombinant natural neurite-promoting human IgM antibody, named rHIgM12, that binds to the gangliosides complex GD1a and GT1b on the surface of neurons. In primary neurons, rHIgM12 promotes the cytoskeleton dynamics by decreasing tubulin acetylation and increasing its tyrosination (Xu et al., 2015). A single IP injection at pre-symptomatic stage (60 days of age) allowed the antibody to cross the blood brain barrier and reach the CNS. Authors reported increased survival in SOD1G93A and SOD1G86R mice (10 and 8 days, respectively), a 16-day delay in the onset of neurological deficits, as measured by nocturnal activity, and a 5-day delay of weight loss. Moreover, mice treated with rHIgM12 displayed fewer degenerating spinal cord axons and more NeuN-positive anterior horn neurons. Multiple doses of rHIgM12 in animal models were not effective due to the development of a strong anti-human antibody response that inactivated circulating rHIgM12 (Xu et al., 2015).
Connective Tissue Growth Factor (CTGF)
Connective tissue growth factor (CTGF/CCN2) is a matricellular protein, found up-regulated in spinal cord tissues of ALS patients (Spliet et al., 2003) and in muscles of symptomatic SOD1G93A mice (Gonzalez et al., 2017). Its involvement in ALS is still unclear although its functions in skeletal muscles (Morales et al., 2011) might be relevant for the pathology.
In 2018, Gonzalez et al. tested a human monoclonal IgG antibody against CTGF/CCN2, named FG-3019, obtained from FibroGen. Inc. (Gonzalez et al., 2018). The antibody was administered by IP injection to male mice at pre-symptomatic stage (8 weeks old), three times a week for 2 months. FG-3019 antibody delayed body weight loss and improved muscle function and locomotor capacity, as shown by longer hanging time, increased muscle strength and longer traveled distances at faster speed. Antibody reduced fibrosis and improved skeletal muscle architecture as demonstrated by reduced interstitial space and myofiber atrophy. Moreover it reduced NMJ denervation and myelin degeneration in the sciatic nerve (Gonzalez et al., 2018).
High Motility Group Box 1
High motility group box 1 (HMGB1) is an ubiquitously expressed nuclear protein but also an extracellular mediator of RAGE and TLR4 activation, two proteins implicated in the neuroinflammatory pathways observed in ALS (Brites and Vaz, 2014; Lee et al., 2015). Interestingly, HMGB1 has been shown to translocate from the nucleus to cytoplasm in ALS patients and animal models (Lo Coco et al., 2007; Casula et al., 2011), suggesting a potential pathogenic role for HMGB1 in ALS.
In 2019 Lee et al. tested a humanized IgG2b antibody targeting the extracellular damage-associated molecular pattern (DAMP) form of HMGB1 (Lee et al., 2019). The anti-HMGB1 (clone 2G7), purified from hybridomas, was administered to SOD1G93A female mice every week until end stage of disease. Administration started from 35 days of age (pre-onset) for prevention or from 70 days (post-onset) for treatment. Pre-onset treatment transiently improved muscle strength assessed by grip test and reduced key pro-inflammatory genes in spinal cord, whereas post-onset treatment had no effect. Treatment at none of the two time points extended the survival time nor did it affect neuroinflammation (Lee et al., 2019).
Discussion
Antibody-based therapies are gaining relevance for the treatment of neurodegenerative diseases (Mortada et al., 2021) and the FDA approval of a passive immunization approach for Alzheimer's disease demonstrates the feasibility of these interventions for neurodegenerative conditions. According to our results, over the last 15 years, 31 original papers have been published demonstrating the potential of antibody-treatments for ALS.
The antibody-based strategies target both intracellular and extracellular proteins demonstrating the adaptability of immunoglobulins for therapeutic purposes. More than fifty percent of the revised paper indeed have been generated against extracellular proteins or membrane receptors with the aim of inhibiting the downstream effects of receptor activation. Interestingly, the other portion targeted intracellular proteins, known to cause ALS either in their pathological (misfolded or mutant SOD1, C9ORF72 repeats) or physiological (TDP-43) forms. Targeting a toxic intracellular protein as SOD1, TDP-43 or C9ORF72 repeats in ALS is an essential requirement, as these proteins are presently the main triggers of the pathology. Full-length antibodies possess two main advantages as therapeutic intracellular strategies. First, they can be specifically up-taken by neurons and microglial cells through the clathrin-dependent Fcγ receptor endocytosis (Peress et al., 1993; Congdon et al., 2013), two cell populations relevant to the disease; and second, once inside the cells, they activate physiological processes that induce the clearance of targeted proteins such as the TRIM-21/proteasome (McEwan et al., 2017) or the endosomes/lysosomes degradative machinery (Gu et al., 2013). On the other hand, the presence of the Fc domain may facilitate the activation of inflammatory processes, which can be avoided by using full-length-derived single chain antibodies or modified Fc domains, as demonstrated by some of the analyzed approaches.
Antibodies are big molecules and might pose difficulties in penetrating the CNS due to the natural defense structure of the blood-brain barrier (BBB). The use of single chain antibodies could overcome this issue since they are smaller in size and possess higher cellular penetration capacity. Moreover, scFv antibodies have the potential to enhance the target protein degradation, and the fact that they can be easily engineered increases their future therapeutic potential. On the other hand, it is worth mentioning that the delivery of a single chain antibody through viral vectors might present further limitations in controlling and modulating the treatment in case of adverse effects. Despite the limitations posed by BBB, most of the antibody-based interventions identified through the present systematic literature review used a systemic delivery of the immunoglobulin with efficient target engagement and therapeutic effect. The use of a systemic rather than localized delivery represents a huge advantage for patients since therapeutic interventions localized in the CNS might be riskier and more dangerous for them. That being said, systemic delivery might increase the risk of side effects as some of the targeted proteins are expressed also outside the CNS. This subject has been poorly investigated in the reviewed papers but appear necessary to characterize therapeutic interventions. Future studies should consider evaluating the presence and effect of antibodies delivered systemically also outside the CNS. Moreover, novel strategies should be considered to improve the bioavailability of a systemically delivered Ab into the CNS. It is worth mentioning that no intervention analyzed has used local muscular delivery. The neuromuscular junction is a crucial structure in the context of ALS as it represents the contact surface between degenerating motor neurons and atrophic muscles. This synaptic structure appears to be underestimated as a delivery hub and future therapeutic interventions should also consider this administration strategy to target both motor neurons and muscle cells or molecules involved in NMJ degeneration.
Another relevant issue to point out following the analysis of the selected literature is the use of ALS models. Although not all targeting SOD1, most of the analyzed studies used the mutant SOD1 model. This model is so far the most utilized as it perfectly recapitulates the symptomatology and pathological events occurring in ALS but, unfortunately, it also presents some limitations. First, it is highly heterogeneous. The different number of transgene copies, the various levels of mutant protein expressed, or the genetic background of the animals (Bendotti et al., 2020) make the comparison of the obtained results and their reproducibility highly difficult. Second, it represents a very small portion of familial ALS cases therefore, its use can limit the observed efficiency of the therapeutic intervention to few forms of ALS excluding many other ALS cases. It is, therefore, necessary to implement protocols where the target and the therapeutic efficiency are studied in other familial-derived ALS models and to increase scientific efforts toward developing models for sporadic ALS that represent the 90% of all ALS cases.
Most of the analyzed papers describe preclinical trial or proof of principle studies in animal- or patients-derived cells. Although the antigen presence was validated also in ALS patient tissues, a relevant discrepancy emerges when considering that the antigen expression is evaluated in postmortem tissues and then targeted with a therapy in animal models at the pre-symptomatic stage. As sporadic ALS patients are diagnosed only years after the beginning of their symptoms, therapeutic strategies for ALS should be investigated with the use of animal models only after the onset of symptoms to have a more translational approach for patients. Moreover, as described by some of the mentioned papers, a treatment starting at the pre-symptomatic stage often proves less effective when delivered at the symptomatic stage, corroborating the need to evaluate therapeutic efficacy when the pathology has already manifested itself. For this reason, the identification of early and prognostic biomarkers in ALS patients may give rise to new therapeutic strategies with a more precise intervention window and a better evaluation of the therapeutic efficacy.
Conclusions
In conclusion, with this review we systematically searched and described scientific efforts made in the past years in the production and evaluation of therapeutic interventions with passive immunization in patients and models of amyotrophic lateral sclerosis. Although some improvements still need to be made in order to increase the efficiency in ALS patients, the therapeutic interventions analyzed in this review showcase the strong potential of antibodies as a therapeutic approach for ALS. Owing to the specificity of these molecules and their potential to block/inhibit downstream pathological events or increase the clearance of the pathogenic proteins, passive immunization is on its way to become a therapeutic intervention for this disease.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
SP and AP-B designed the study. AP-B and ER performed the literature research and analysis. AP-B, ER, and SP wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work has been funded by the ALS Canada and Vincent Bourque Foundations. SP is the recipient of the 2020 ALS Career Transition Award from ALS Canada and Vincent Bourque Foundations and of the 2019 Marlene Reimer Brain Star of the year award CIHR-INMHA (ICT-171454).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Authors would like to kindly thank Dr. Jean-Pierre Julien for critical discussion on the manuscript, and Dr. Gohar Fakhfouri for English language revision.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2021.790114/full#supplementary-material
Abbreviations
a.a., amino acids; AAV, adeno-associated virus; Ab, antibody; Abs, antibodies; AChRs, acetylcholine receptors; AEs, adverse events; ALS, amyotrophic lateral sclerosis; ASO, antisense oligonucleotides; BBB, blood-brain barrier; C9ORF72, chromosome 9 open reading frame 72; CD40, cluster of differentiation 40; CMAP, compound muscle action potential; CNS, central nervous system; CSF, cerebrospinal fluid; CTGF, connective tissue growth factor; DPRs, dipeptide repeat proteins; DR-6, death receptor 6; ECM, extracellular matrix; EDL, extensor digitorum longus; Fab, fragment antigen-binding; fALS, familial ALS; Fc, Fragment crystallizable; FDA, Food and Drug Administration; FRS-R, Functional Rating Scale-Revised; FTLD, frontotemporal lobar degeneration; GFAP, glial fibrillary acidic protein; GFP, green fluorescent protein; HEK, human embryonic kidney; HMGB1, high motility group box 1; HSP, heat shock protein; IC, intracortical; ICV, intracerebroventricular; IgG/M, Immunoglobulin G/M; IHC, immunohistochemistry; IL-6, interleukin 6; IN, intranasal; INF-γ, interferon gamma; IP, intraperitoneal; IT, intrathecal; IV, intravenous; LPS, lipopolysaccharide; MAb, monoclonal antibody; mMAb, mouse monoclonal antibody; MMT, manual muscle testing; MN, motor neurons; mSOD1, misfolded SOD1 protein; MuSK, muscle-specific kinase; NFκB, nuclear factor-kappa B; NK, natural killer; NMJ, neuromuscular junctions; Nogo, neurite outgrowth inhibitor; NRP-1, neuropilin-1; PBMC, peripheral blood mononuclear cell; PK, pharmacokinetics; RD, repeated dose; RRM1, RNA recognition motif 1; RT-PCR, reverse transcriptase polymerase chain reaction; SAEs, serious adverse events; sALS, sporadic ALS; scFv, single-chain variable fragment; SD, single dose; Sema3A, semaphorin 3A; SOD1, superoxide dismutase 1; SVC, slow vital capacity; TDP-43, TAR DNA-binding protein 43; TGF-β, transforming growth factor beta; TNF-α, tumor necrosis factor α; TRIM-21, tripartite motif containing-21; WT, wild type.
References
Abe, K., Aoki, M., Tsuji, S., Itoyama, Y., Sobue, G., Togo, M., et al. (2017). Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 16, 505–512. doi: 10.1016/S1474-4422(17)30115-1
Aebischer, J., Cassina, P., Otsmane, B., Moumen, A., Seilhean, D., Meininger, V., et al. (2011). IFNγ triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ. 18, 754–768. doi: 10.1038/cdd.2010.143
Alhindi, A., Boehm, I., and Chaytow, H. (2021). Small junction, big problems: neuromuscular junction pathology in mouse models of amyotrophic lateral sclerosis (ALS). J. Anat. 2021:13463. doi: 10.1111/joa.13463
Amado, D. A., and Davidson, B. L. (2021). Gene therapy for ALS: A review. Mol. Ther. doi: 10.1016/j.ymthe.2021.04.008
Ash, P. E. A., Bieniek, K. F., Gendron, T. F., Caulfield, T., Lin, W.-L., DeJesus-Hernandez, M., et al. (2013). Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77, 639–646. doi: 10.1016/j.neuron.2013.02.004
Bendotti, C., Bonetto, V., Pupillo, E., Logroscino, G., Al-Chalabi, A., Lunetta, C., et al. (2020). Focus on the heterogeneity of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 8421, 1–11. doi: 10.1080/21678421.2020.1779298
Bosco, D. A., Morfini, G., Karabacak, N. M., Song, Y., Gros-Louis, F., Pasinelli, P., et al. (2010). Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 13, 1396–1403. doi: 10.1038/nn.2660
Brites, D., and Vaz, A. R. (2014). Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front. Cell. Neurosci. 8:117. doi: 10.3389/fncel.2014.00117
Broering, T. J., Wang, H., Boatright, N. K., Wang, Y., Baptista, K., Shayan, G., et al. (2013). Identification of human monoclonal antibodies specific for human SOD1 recognizing distinct epitopes and forms of SOD1. PLoS ONE 8:e61210. doi: 10.1371/journal.pone.0061210
Bros-Facer, V., Krull, D., Taylor, A., Dick, J. R. T., Bates, S. A., Cleveland, M. S., et al. (2014). Treatment with an antibody directed against Nogo-A delays disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 23, 4187–4200. doi: 10.1093/hmg/ddu136
Burden, S. J., Yumoto, N., and Zhang, W. (2013). The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harb. Perspect. Biol. 5, a009167–a009167. doi: 10.1101/cshperspect.a009167
Campbell, I. L., Erta, M., Lim, S. L., Frausto, R., May, U., Rose-John, S., et al. (2014). Trans-signaling is a dominant mechanism for the pathogenic actions of Interleukin-6 in the brain. J. Neurosci. 34, 2503–2513. doi: 10.1523/JNEUROSCI.2830-13.2014
Cantor, S., Zhang, W., Delestrée, N., Remédio, L., Mentis, G. Z., and Burden, S. J. (2018). Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody. Elife 7:e34375. doi: 10.7554/eLife.34375
Caroni, P., and Schwab, M. E. (1988). Antibody against myelin associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron 1, 85–96. doi: 10.1016/0896-6273(88)90212-7
Casula, M., Iyer, A. M., Spliet, W. G. M., Anink, J. J., Steentjes, K., Sta, M., et al. (2011). Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience 179, 233–243. doi: 10.1016/j.neuroscience.2011.02.001
Chang, C., Chiang, M., Toh, E. K.-W., Chang, C.-F., and Huang, T. (2013). Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 587, 575–582. doi: 10.1016/j.febslet.2013.01.038
Chen, M. S., Huber, A. B., van der Haar, M. E., Frank, M., Schnell, L., Spillmann, A. A., et al. (2000). Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403, 434–439. doi: 10.1038/35000219
Congdon, E. E., Gu, J., Sait, H. B. R., and Sigurdsson, E. M. (2013). Antibody uptake into neurons occurs primarily via clathrin-dependent Fcα receptor endocytosis and is a prerequisite for acute tau protein clearance. J. Biol. Chem. 288, 35452–35465. doi: 10.1074/jbc.M113.491001
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Dodge, J. C., Treleaven, C. M., Pacheco, J., Cooper, S., Bao, C., Abraham, M., et al. (2015). Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 112, 8100–8105. doi: 10.1073/pnas.1508767112
Dong, Q., Zhu, J., Liu, S., Yu, X., and Liu, R. (2018). An oligomer-specific antibody improved motor function and attenuated neuropathology in the SOD1-G93A transgenic mouse model of ALS. Int. Immunopharmacol. 65, 413–421. doi: 10.1016/j.intimp.2018.10.032
Dupuis, L., Gonzalez de Aguilar, J.-L., di Scala, F., Rene, F., de Tapia, M., Pradat, P.-F., et al. (2002). Nogo provides a molecular marker for diagnosis of amyotrophic lateral sclerosis. Neurobiol. Dis. 10, 358–365. doi: 10.1006/nbdi.2002.0522
Elgundi, Z., Reslan, M., Cruz, E., Sifniotis, V., and Kayser, V. (2017). The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 122, 2–19. doi: 10.1016/j.addr.2016.11.004
Fiala, M., Chattopadhay, M., La Cava, A., Tse, E., Liu, G., Lourenco, E., et al. (2010). IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J. Neuroinflammation 7, 76. doi: 10.1186/1742-2094-7-76
Fiala, M., Mizwicki, M. T., Weitzman, R., Magpantay, L., and Nishimoto, N. (2013). Tocilizumab infusion therapy normalizes inflammation in sporadic ALS patients. Am. J. Neurodegener. Dis. 2, 129–39. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/23844337
Forsberg, K., Jonsson, P. A., Andersen, P. M., Bergemalm, D., Graffmo, K. S., Hultdin, M., et al. (2010). Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS ONE 5:e11552. doi: 10.1371/journal.pone.0011552
Freskgård, P. O., and Urich, E. (2017). Antibody therapies in CNS diseases. Neuropharmacology 120, 38–55. doi: 10.1016/j.neuropharm.2016.03.014
Ghadge, G. D., Kay, B. K., Drigotas, C., and Roos, R. P. (2019). Single chain variable fragment antibodies directed against SOD1 ameliorate disease in mutant SOD1 transgenic mice. Neurobiol. Dis. 121, 131–137. doi: 10.1016/j.nbd.2018.08.021
Ghadge, G. D., Pavlovic, J. D., Koduvayur, S. P., Kay, B. K., and Roos, R. P. (2013). Single chain variable fragment antibodies block aggregation and toxicity induced by familial ALS-linked mutant forms of SOD1. Neurobiol. Dis. 56, 74–78. doi: 10.1016/j.nbd.2013.04.007
Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. doi: 10.1016/S1474-4422(11)70261-7
Gonzalez, D., Contreras, O., Rebolledo, D. L., Espinoza, J. P., van Zundert, B., and Brandan, E. (2017). ALS skeletal muscle shows enhanced TGF-β signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PLoS ONE 12:e0177649. doi: 10.1371/journal.pone.0177649
Gonzalez, D., Rebolledo, D. L., Correa, L. M., Court, F. A., Cerpa, W., Lipson, K. E., et al. (2018). The inhibition of CTGF/CCN2 activity improves muscle and locomotor function in a murine ALS model. Hum. Mol. Genet. 27, 2913–2926. doi: 10.1093/hmg/ddy204
Grant, M. J., and Booth, A. (2009). A typology of reviews: an analysis of 14 review types and associated methodologies. Heal. Inf. Libr. J. 26, 91–108. doi: 10.1111/j.1471-1842.2009.00848.x
Gros-Louis, F., Soucy, G., Larivière, R., and Julien, J. P. (2010). Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J. Neurochem. 113, 1188–1199. doi: 10.1111/j.1471-4159.2010.06683.x
Gu, J., Congdon, E. E., and Sigurdsson, E. M. (2013). Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce Tau protein pathology. J. Biol. Chem. 288, 33081–33095. doi: 10.1074/jbc.M113.494922
Guareschi, S., Cova, E., Cereda, C., Ceroni, M., Donetti, E., Bosco, D. A., et al. (2012). An over-oxidized form of superoxide dismutase found in sporadic amyotrophic lateral sclerosis with bulbar onset shares a toxic mechanism with mutant SOD1. Proc. Natl. Acad. Sci. USA 109, 5074–5079. doi: 10.1073/pnas.1115402109
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Henkel, J. S., Engelhardt, J. I., Siklós, L., Simpson, E. P., Kim, S. H., Pan, T., et al. (2004). Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 55, 221–235. doi: 10.1002/ana.10805
Holzbaur, E. L. F., Howland, D. S., Weber, N., Wallace, K., She, Y., Kwak, S., et al. (2006). Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol. Dis. 23, 697–707. doi: 10.1016/j.nbd.2006.05.009
Huang, G., Lee, X., Bian, Y., Shao, Z., Sheng, G., Pepinsky, R. B., et al. (2013). Death receptor 6 (DR6) antagonist antibody is neuroprotective in the mouse SOD1G93A model of amyotrophic lateral sclerosis. Cell Death Dis. 4, e841–e841. doi: 10.1038/cddis.2013.378
Jokic, N., Gonzalez de Aguilar, J., Dimou, L., Lin, S., Fergani, A., Ruegg, M. A., et al. (2006). The neurite outgrowth inhibitor Nogo-A promotes denervation in an amyotrophic lateral sclerosis model. EMBO Rep. 7, 1162–1167. doi: 10.1038/sj.embor.7400826
Khosravi, B., LaClair, K. D., Riemenschneider, H., Zhou, Q., Frottin, F., Mareljic, N., et al. (2020). Cell-to-cell transmission of C9orf72 poly-(Gly-Ala) triggers key features of ALS / FTD. EMBO J. 39. doi: 10.15252/embj.2019102811
Körner, S., Böselt, S., Wichmann, K., Thau-Habermann, N., Zapf, A., Knippenberg, S., et al. (2016). The axon guidance protein semaphorin 3A is increased in the motor cortex of patients with amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 75, 326–333. doi: 10.1093/jnen/nlw003
Lam, L., Chin, L., Halder, R. C., Sagong, B., Famenini, S., Sayre, J., et al. (2016). Epigenetic changes in T-cell and monocyte signatures and production of neurotoxic cytokines in ALS patients. FASEB J. 30, 3461–3473. doi: 10.1096/fj.201600259RR
Lawarée, J., Jacob, S., and Ouimet, M. (2020). A scoping review of knowledge syntheses in the field of evaluation across four decades of practice. Eval. Program Plann. 79, 101761. doi: 10.1016/j.evalprogplan.2019.101761
Lee, J. D., Liu, N., Levin, S. C., Ottosson, L., Andersson, U., Harris, H. E., et al. (2019). Therapeutic blockade of HMGB1 reduces early motor deficits, but not survival in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 16, 45. doi: 10.1186/s12974-019-1435-2
Lee, J. Y., Lee, J. D., Phipps, S., Noakes, P. G., and Woodruff, T. M. (2015). Absence of toll-like receptor 4 (TLR4) extends survival in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 12, 90. doi: 10.1186/s12974-015-0310-z
Lehmann, M., Marklund, M., Bolender, A.-L., Bidhendi, E. E., Zetterström, P., Andersen, P. M., et al. (2020). Aggregate-selective antibody attenuates seeded aggregation but not spontaneously evolving disease in SOD1 ALS model mice. Acta Neuropathol. Commun. 8, 161. doi: 10.1186/s40478-020-01032-2
Liberati, A., Altman, D. G., Tetzlaff, J., Mulrow, C., Gotzsche, P. C., Ioannidis, J. P. A., et al. (2009). The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: explanation and elaboration. BMJ 339, b2700–b2700. doi: 10.1136/bmj.b2700
Lincecum, J. M., Vieira, F. G., Wang, M. Z., Thompson, K., De Zutter, G. S., Kidd, J., et al. (2010). From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat. Genet. 42, 392–399. doi: 10.1038/ng.557
Liu, G., Fiala, M., Mizwicki, M. T., Sayre, J., Magpantay, L., Siani, A., et al. (2012). Neuronal phagocytosis by inflammatory macrophages in ALS spinal cord: inhibition of inflammation by resolvin D1. Am. J. Neurodegener. Dis. 1, 60–74. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/22787561
Liu, Y., Pattamatta, A., Zu, T., Reid, T., Bardhi, O., Borchelt, D. R., et al. (2016). C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 90, 521–534. doi: 10.1016/j.neuron.2016.04.005
Lo Coco, D., Veglianese, P., Allievi, E., and Bendotti, C. (2007). Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of ALS. Neurosci. Lett. 412, 73–77. doi: 10.1016/j.neulet.2006.10.063
Mackenzie, I. R., Arzberger, T., Kremmer, E., Troost, D., Lorenzl, S., Mori, K., et al. (2013). Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol. 126, 859–879. doi: 10.1007/s00401-013-1181-y
Maier, M., Welt, T., Wirth, F., Montrasio, F., Preisig, D., McAfoose, J., et al. (2018). A human-derived antibody targets misfolded SOD1 and ameliorates motor symptoms in mouse models of amyotrophic lateral sclerosis. Sci. Transl. Med. 10. doi: 10.1126/scitranslmed.aah3924
Maimon, R., Ionescu, A., Bonnie, A., Sweetat, S., Wald-Altman, S., Inbar, S., et al. (2018). miR126-5p downregulation facilitates axon degeneration and NMJ disruption via a non-cell-autonomous mechanism in ALS. J. Neurosci. 38, 5478–5494. doi: 10.1523/JNEUROSCI.3037-17.2018
McCroskery, S., Thomas, M., Maxwell, L., Sharma, M., and Kambadur, R. (2003). Myostatin negatively regulates satellite cell activation and self-renewal. J. Cell Biol. 162, 1135–1147. doi: 10.1083/jcb.200207056
McCroskery, S., Thomas, M., Platt, L., Hennebry, A., Nishimura, T., McLeay, L., et al. (2005). Improved muscle healing through enhanced regeneration and reduced fibrosis in myostatin-null mice. J. Cell Sci. 118, 3531–3541. doi: 10.1242/jcs.02482
McEwan, W. A., Falcon, B., Vaysburd, M., Clift, D., Oblak, A. L., Ghetti, B., et al. (2017). Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation. Proc. Natl. Acad. Sci. U. S. A. 114, 574–579. doi: 10.1073/pnas.1607215114
Meininger, V., Genge, A., van den Berg, L. H., Robberecht, W., Ludolph, A., Chio, A., et al. (2017). Safety and efficacy of ozanezumab in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 16, 208–216. doi: 10.1016/S1474-4422(16)30399-4
Meininger, V., Pradat, P.-F., Corse, A., Al-Sarraj, S., Rix Brooks, B., Caress, J. B., et al. (2014). Safety, pharmacokinetic, and functional effects of the nogo-a monoclonal antibody in amyotrophic lateral sclerosis: a randomized, first-in-human clinical trial. PLoS ONE 9:e97803. doi: 10.1371/journal.pone.0097803
Miller, R. G., Mitchell, J. D., and Moore, D. H. (2012). Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012:CD001447. doi: 10.1002/14651858.CD001447.pub3
Mizwicki, M. T., Fiala, M., Magpantay, L., Aziz, N., Sayre, J., Liu, G., et al. (2012). Tocilizumab attenuates inflammation in ALS patients through inhibition of IL6 receptor signaling. Am. J. Neurodegener. Dis. 1, 305–15. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/23383400 (accessed Nov 21, 2012).
Moher, D., Liberati, A., Tetzlaff, J., and Altman, D. G. (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 6:e1000097. doi: 10.1371/journal.pmed.1000097
Morales, M. G., Cabello-Verrugio, C., Santander, C., Cabrera, D., Goldschmeding, R., and Brandan, E. (2011). CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J. Pathol. 225, 490–501. doi: 10.1002/path.2952
Mori, K., Lammich, S., Mackenzie, I. R. A., Forné, I., Zilow, S., Kretzschmar, H., et al. (2013). hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 125, 413–423. doi: 10.1007/s00401-013-1088-7
Mortada, I., Farah, R., Nabha, S., Ojcius, D. M., Fares, Y., Almawi, W. Y., et al. (2021). Immunotherapies for neurodegenerative diseases. Front. Neurol. 12:654739. doi: 10.3389/fneur.2021.654739
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. doi: 10.1126/science.1134108
Nguyen, L., Montrasio, F., Pattamatta, A., Tusi, S. K., Bardhi, O., Meyer, K. D., et al. (2020). Antibody therapy targeting RAN proteins rescues C9 ALS/FTD phenotypes in C9orf72 mouse model. Neuron 105, 645–662.e11. doi: 10.1016/j.neuron.2019.11.007
Nikolaev, A., McLaughlin, T., O'Leary, D. D. M., and Tessier-Lavigne, M. (2009). APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457, 981–989. doi: 10.1038/nature07767
Nimmo, J. T., Kelly, L., Verma, A., Carare, R. O., Nicoll, J. A. R., and Dodart, J.-C. (2021). Amyloid-β and α-synuclein immunotherapy: from experimental studies to clinical trials. Front. Neurosci. 15:733857. doi: 10.3389/fnins.2021.733857
Noelle, R. J., Ledbetter, J. A., and Aruffo, A. (1992a). CD40 and its ligand, an essential ligand-receptor pair for thymus-dependent B-cell activation. Immunol. Today 13, 431–433. doi: 10.1016/0167-5699(92)90068-I
Noelle, R. J., Roy, M., Shepherd, D. M., Stamenkovic, I., Ledbetter, J. A., and Aruffo, A. (1992b). A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc. Natl. Acad. Sci. 89, 6550–6554. doi: 10.1073/pnas.89.14.6550
Otsmane, B., Aebischer, J., Moumen, A., and Raoul, C. (2014). Cerebrospinal fluid-targeted delivery of neutralizing anti-IFNγ antibody delays motor decline in an ALS mouse model. Neuroreport 25, 49–54. doi: 10.1097/WNR.0000000000000043
Paré, B., Lehmann, M., Beaudin, M., Nordström, U., Saikali, S., Julien, J.-P., et al. (2018). Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 8, 14223. doi: 10.1038/s41598-018-31773-z
Patel, P., Kriz, J., Gravel, M., Soucy, G., Bareil, C., Gravel, C., et al. (2014). Adeno-associated virus-mediated delivery of a recombinant single-chain antibody against misfolded superoxide dismutase for treatment of amyotrophic lateral sclerosis. Mol. Ther. 22, 498–510. doi: 10.1038/mt.2013.239
Peress, N. S., Fleit, H. B., Perillo, E., Kuljis, R., and Pezzullo, C. (1993). Identification of FcγRI, II and III on normal human brain ramified microglia and on microglia in senile plaques in Alzheimer's disease. J. Neuroimmunol. 48, 71–79. doi: 10.1016/0165-5728(93)90060-C
Pérez-García, M. J., and Burden, S. J. (2012). Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep. 2, 497–502. doi: 10.1016/j.celrep.2012.08.004
Pokrishevsky, E., Grad, L. I., Yousefi, M., Wang, J., Mackenzie, I. R., and Cashman, N. R. (2012). Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS ONE 7:e35050. doi: 10.1371/journal.pone.0035050
Pokrishevsky, E., Hong, R. H., Mackenzie, I. R., and Cashman, N. R. (2017). Spinal cord homogenates from SOD1 familial amyotrophic lateral sclerosis induce SOD1 aggregation in living cells. PLoS ONE 12:e0184384. doi: 10.1371/journal.pone.0184384
Pozzi, S., Codron, P., Soucy, G., Renaud, L., Cordeau, P. J., Dutta, K., et al. (2020). Monoclonal full-length antibody against TAR DNA binding protein 43 reduces related proteinopathy in neurons. JCI Insight 5:e140420. doi: 10.1172/jci.insight.140420
Pozzi, S., Thammisetty, S. S., Codron, P., Rahimian, R., Plourde, K. V., Soucy, G., et al. (2019). Virus-mediated delivery of antibody targeting TAR DNA-binding protein-43 mitigates associated neuropathology. J. Clin. Invest. 129, 1581–1595. doi: 10.1172/JCI123931
Prinjha, R., Moore, S. E., Vinson, M., Blake, S., Morrow, R., Christie, G., et al. (2000). Inhibitor of neurite outgrowth in humans. Nature 403, 383–384. doi: 10.1038/35000287
Rapport, M. M., Donnenfeld, H., Brunner, W., Hungund, B., and Bartfeld, H. (1985). Ganglioside patterns in amyotrophic lateral sclerosis brain regions. Ann. Neurol. 18, 60–67. doi: 10.1002/ana.410180111
Regazzi, M., Golay, J., and Molinaro, M. (2020). Monoclonal antibody monitoring: clinically relevant aspects, a systematic critical review. Ther. Drug Monit. 42, 45–56. doi: 10.1097/FTD.0000000000000681
Renton, A. E., Majounie, E., Waite, A., Simón-Sánchez, J., Rollinson, S., Gibbs, J. R., et al. (2011). A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268. doi: 10.1016/j.neuron.2011.09.010
Rotunno, M. S., and Bosco, D. A. (2013). An emerging role for misfolded wild-type SOD1 in sporadic ALS pathogenesis. Front. Cell. Neurosci. 7:253. doi: 10.3389/fncel.2013.00253
Roy, M., Waldschmidt, T., Aruffo, A., Ledbetter, J. A., and Noelle, R. J. (1993). The regulation of the expression of gp39, the CD40 ligand, on normal and cloned CD4+ T cells. J. Immunol. 151, 2497–510. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/8103067
Schnell, L., and Schwab, M. E. (1990). Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature 343, 269–272. doi: 10.1038/343269a0
Sengupta-Ghosh, A., Dominguez, S. L., Xie, L., Barck, K. H., Jiang, Z., Earr, T., et al. (2019). Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1G93A mouse model of ALS. Neurobiol. Dis. 124, 340–352. doi: 10.1016/j.nbd.2018.12.002
Sevigny, J., Chiao, P., Bussière, T., Weinreb, P. H., Williams, L., Maier, M., et al. (2016). The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537, 50–56. doi: 10.1038/nature19323
Shodai, A., Morimura, T., Ido, A., Uchida, T., Ayaki, T., Takahashi, R., et al. (2013). Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 288, 14886–14905. doi: 10.1074/jbc.M113.451849
Sipione, S., Monyror, J., Galleguillos, D., Steinberg, N., and Kadam, V. (2020). Gangliosides in the brain: physiology, pathophysiology and therapeutic applications. Front. Neurosci. 14:1004. doi: 10.3389/fnins.2020.572965
Slastnikova, T. A., Ulasov, A. V., Rosenkranz, A. A., and Sobolev, A. S. (2018). Targeted intracellular delivery of antibodies: the state of the art. Front. Pharmacol. 9:1208. doi: 10.3389/fphar.2018.01208
Spliet, W. G. M., Aronica, E., Ramkema, M., Aten, J., and Troost, D. (2003). Increased expression of connective tissue growth factor in amyotrophic lateral sclerosis human spinal cord. Acta Neuropathol. 106, 449–457. doi: 10.1007/s00401-003-0741-y
Swarup, V., Phaneuf, D., Dupré, N., Petri, S., Strong, M., Kriz, J., et al. (2011). Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB-mediated pathogenic pathways. J. Exp. Med. 208, 2429–2447. doi: 10.1084/jem.20111313
Takata, M., Tanaka, H., Kimura, M., Nagahara, Y., Tanaka, K., Kawasaki, K., et al. (2013). Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br. J. Pharmacol. 170, 341–351. doi: 10.1111/bph.12277
Tamaki, Y., Shodai, A., Morimura, T., Hikiami, R., Minamiyama, S., Ayaki, T., et al. (2018). Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci. Rep. 8, 1–16. doi: 10.1038/s41598-018-24463-3
Trias, E., Ibarburu, S., Barreto-Núñez, R., Babdor, J., Maciel, T. T., Guillo, M., et al. (2016). Post-paralysis tyrosine kinase inhibition with masitinib abrogates neuroinflammation and slows disease progression in inherited amyotrophic lateral sclerosis. J. Neuroinflammation 13:177. doi: 10.1186/s12974-016-0620-9
Urushitani, M., Ezzi, S. A., and Julien, J. P. (2007). Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U. S. A. 104, 2495–2500. doi: 10.1073/pnas.0606201104
Valera, E., Spencer, B., and Masliah, E. (2016). Immunotherapeutic approaches targeting amyloid-β, α-Synuclein, and Tau for the treatment of neurodegenerative disorders. Neurotherapeutics 13, 179–189. doi: 10.1007/s13311-015-0397-z
Venkova, K., Christov, A., Kamaluddin, Z., Kobalka, P., Siddiqui, S., and Hensley, K. (2014). Semaphorin 3A signaling through neuropilin-1 is an early trigger for distal axonopathy in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 73, 702–713. doi: 10.1097/NEN.0000000000000086
Whittemore, L.-A., Song, K., Li, X., Aghajanian, J., Davies, M., Girgenrath, S., et al. (2003). Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem. Biophys. Res. Commun. 300, 965–971. doi: 10.1016/S0006-291X(02)02953-4
Xie, M.-H., Yuan, J., Adams, C., and Gurney, A. (1997). Direct demonstration of MuSK involvement in acetylcholine receptor clustering through identification of agonist ScFv. Nat. Biotechnol. 15, 768–771. doi: 10.1038/nbt0897-768
Xu, X., Denic, A., Jordan, L. R., Wittenberg, N. J., Warrington, A. E., Wootla, B., et al. (2015). A natural human IgM that binds to gangliosides is therapeutic in murine models of amyotrophic lateral sclerosis. Dis. Model. Mech. 8, 831–842. doi: 10.1242/dmm.020727
Yang, X., Ji, Y., Wang, W., Zhang, L., Chen, Z., Yu, M., et al. (2021). Amyotrophic lateral sclerosis: molecular mechanisms, biomarkers, and therapeutic strategies. Antioxidants 10:1012. doi: 10.3390/antiox10071012
Zhang, W., Coldefy, A.-S., Hubbard, S. R., and Burden, S. J. (2011). Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J. Biol. Chem. 286, 40624–40630. doi: 10.1074/jbc.M111.279307
Keywords: amyotrophic lateral sclerosis, antibody-based therapy, systematic review, knowledge synthesis, passive immunization
Citation: Poulin-Brière A, Rezaei E and Pozzi S (2021) Antibody-Based Therapeutic Interventions for Amyotrophic Lateral Sclerosis: A Systematic Literature Review. Front. Neurosci. 15:790114. doi: 10.3389/fnins.2021.790114
Received: 06 October 2021; Accepted: 08 November 2021;
Published: 29 November 2021.
Edited by:
Justin John Yerbury, University of Wollongong, AustraliaReviewed by:
Makoto Urushitani, Shiga University of Medical Science, JapanSerena Stanga, University of Turin, Italy
Copyright © 2021 Poulin-Brière, Rezaei and Pozzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Pozzi, c2lsdmlhLnBvenppQGNlcnZvLnVsYXZhbC5jYQ== orcid.org/0000-0002-1500-8158