 Nilo Riva1,2,3*
Nilo Riva1,2,3* Laura Pozzi1Tommaso Russo1,2Giovanni Battista Pipitone4Paride Schito1,2
Laura Pozzi1Tommaso Russo1,2Giovanni Battista Pipitone4Paride Schito1,2 Teuta Domi1
Teuta Domi1 Federica Agosta5,6
Federica Agosta5,6 Angelo Quattrini1Paola Carrera4†
Angelo Quattrini1Paola Carrera4† Massimo Filippi2,3,5,6,7†
Massimo Filippi2,3,5,6,7†
- 1Neuropathology Unit, Division of Neuroscience, Institute of Experimental Neurology (INSPE), San Raffaele Scientific Institute, Milan, Italy
- 2Neurology Unit, San Raffaele Scientific Institute, Milan, Italy
- 3Neurorehabilitation Unit, San Raffaele Scientific Institute, Milan, Italy
- 4Unit of Genomics for Human Disease Diagnosis, San Raffaele Scientific Institute, Milan, Italy
- 5Neuroimaging Research Unit, Division of Neuroscience, Institute of Experimental Neurology (INSPE), San Raffaele Scientific Institute, Milan, Italy
- 6Vita-Salute San Raffaele University, Milan, Italy
- 7Neurophysiology Service, San Raffaele Scientific Institute, Milan, Italy
Introduction: In the last few years, different studies highlighted a significant enrichment of NEK1 loss of function (LoF) variants in amyotrophic lateral sclerosis (ALS), and an additional role for the p.Arg261His missense variant in the disease susceptibility. Several other missense variants have been described so far, whose pathogenic relevance remains however unclear since many of them have been reported in both patients and controls. This study aimed to investigate the presence of NEK1 variants and their correlation with phenotype in a cohort of Italian patients with ALS.
Methods: We sequenced a cohort of 350 unrelated Italian patients with ALS by next-generation sequencing (NGS) and then we analyzed the clinical features of NEK1 carriers.
Results: We detected 20 different NEK1 rare variants (four LoF and 16 missense) in 33 unrelated patients with sporadic ALS (sALS). The four LoF variants (two frameshift and two splice-site variants) were all novel. The p.Arg261His missense variant was enriched in the patients’ cohort (p < 0.001). Excluding this variant from counting, the difference in the frequency of NEK1 rare missense variants between patients and controls was not statistically significant. NEK1 carriers had a higher frequency of flail arm (FA) phenotype compared with the other patients of the cohort (29.2% vs. 6.4%). Nine NEK1 carriers (37.5%) also harbored variants in other ALS-related genes.
Conclusion: This study confirms that NEK1 LoF and p.Arg261. His missense variants are associated with ALS in an Italian ALS cohort and suggests a correlation between the presence of NEK1 variants and FA phenotype.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder characterized by progressive loss of both upper and lower motor neurons, muscular weakness, and atrophy, leading to death within 2–5 years after diagnosis (Riva et al., 2016). It is largely recognized that the clinical spectrum of ALS is widely heterogeneous, with the definition of distinct subphenotypes, characterized by a different involvement of spinal, bulbar, and upper or lower motor neurons (Wijesekera et al., 2009; Chio et al., 2011). In particular, flail arm, flail leg, and pure lower motor neuron are phenotypes where predominant or selective lower motor neurons dysfunction occurs (Schito et al., 2020). About 10% of ALS cases are classified as familial (fALS), whereas the remaining 90% appear to be sporadic (sALS); in about two-thirds of fALS and up to 10% of sALS patients mutations in one or more specific ALS-related genes may be identified (Riva et al., 2016). To date, mutations in at least 30 genes have been implicated in ALS pathogenesis, and more than 120 genes have been proposed as potentially related to the disease. However, only a few of them, i.e., the C9orf72 repeat expansion, SOD1, FUS, TARDBP, and TBK1 genes, which account for up to 63% of fALS and 11% of sALS cases, have been clearly demonstrated to be implicated in the disease (Renton et al., 2014; Veldink, 2017).
The never in mitosis A (NIMA)-related kinase 1 (NEK1) is a widely studied serine/threonine kinase belonging to a family that shares significant homology with Aspergillus nidulans NIMA proteins. NEK1 plays a key role in several cellular functions, such as cell cycle progression, cilia regulation, DNA damage response (DDR), and mitochondrial membrane permeability (Peres de Oliveira et al., 2020). In addition, alterations in its expression have been correlated with human diseases like polycystic kidney disease and short-rib polydactyly syndrome (Type Majewski), Mohr syndrome, and Wilms tumor (Peres de Oliveira et al., 2020). Recently, large-scale whole-exome sequencing gene burden analysis studies highlighted a significant enrichment of NEK1 loss of function (LoF) variants in ALS (Brenner et al., 2016; Kenna et al., 2016), and an additional role for the p.Arg261His missense variant for disease susceptibility (Nguyen et al., 2018). Several other missense variants have been described so far; however, their pathogenic relevance remains to be established, since many of them have been reported in both the patients with ALS and control cases (Kenna et al., 2016; Nguyen et al., 2018). In this context, a previous study demonstrated that NEK1 LoF variants may induce the DNA damage accumulation in motor neurons derived from patients with ALS (Higelin et al., 2018). Other well-established ALS genes, such as C9orf72 and FUS, were previously related to DDR mechanisms, a finding supporting the hypothesis that NEK1 may play a pathogenic role in ALS (Peres de Oliveira et al., 2020; Riancho et al., 2020). According to previous literature, NEK1 variants do not affect the age at onset or survival in patients with ALS; while no clear genotype-phenotype correlation was reported, interestingly, a recent article described an association between NEK1 LoF variants and hand involvement at onset in a cohort of Taiwanese patients with ALS (Tsai et al., 2020).
This study aimed to further investigate the presence and impact of NEK1 variants and to explore potential genotype-phenotype correlations in a cohort of Italian patients with ALS.
Materials and Methods
Participants
In this study, we sequenced a cohort of 350 unrelated Italian patients with ALS (200 men and 150 women). Patients were enrolled at the Department of Neurology, San Raffaele Hospital, between 2014 and 2020. El Escorial revised criteria (Brooks et al., 2000) were used for ALS diagnostic categorization; no strict inclusion or exclusion criteria were adopted for patient selection (Brooks et al., 2000). Phenotypic classification of patients was assessed as previously described (Chio et al., 2011; Schito et al., 2020). We performed neuropsychological screening with Edinburgh Cognitive and Behavioral ALS Screen (Abrahams et al., 2014), followed by a further neuropsychological evaluation according to the diagnostic criteria for the behavioral variant of frontotemporal dementia and the ALSFTD consensus criteria (Rascovsky et al., 2011; Strong et al., 2017; Falzone et al., 2020).
The mean age of onset of our cohort was 57.4 ± 12.4 years; 296 patients (84.6%) had spinal onset ALS while 54 (15.4%) had bulbar onset ALS; twenty-nine patients (8.3%) had fALS. All patients were of Caucasian ethnicity, except three of Hispanic origin and the other three who were Africans. A cohort of 380 non-neurological unrelated Italian patients, for whom next-generation sequencing (NGS) exome sequencing data were available from our in-house database, was selected as the control group. This cohort ware screened to exclude the presence of neurological diseases or comorbidities.
Standard Protocol Approvals, Registrations, and Patient Consents
This study was conducted according to Helsinki criteria and received approval from the Local Ethics Committee of the San Raffaele Hospital; blood samples were collected for diagnostic purposes and stored in our tissue bank, after informed consent, both for ALS and control cohorts.
Genetic Analysis
Targeted NGS, using TruSeq Neurodegeneration Panel by Illumina (San Diego, CA, United States) was performed for NEK1 mutational analysis, following the procedure of the manufacturer. Single variants reported in the FASTQ and VCF output file were analyzed with Illumina Variant Studio V3.0 software1 and visualized via Integrative Genome Viewer software.2 The C9orf72 repeat expansion was also analyzed in all patients, using both amplicon-length and repeat-primed polymerase chain reactions, as described before (Agosta et al., 2017). All the identified NEK1 variants found in patients with ALS and patients’ relatives were confirmed by Sanger sequencing (Pozzi et al., 2017). The clinical significance of reported variants was assessed based on the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). The Amyotrophic Lateral Sclerosis Online (ALSoD) database3 was used to select the 33 ALS-related genes to evaluate the oligogenicity of NEK1 carriers. To exclude the presence of duplicates and to confirm that patients with ALS analyzed were unrelated, a KING tool analysis4 was performed, including a bcftools and plink2-based script (Manichaikul et al., 2010). The same analytical and validated in-house pipeline, and filtering criteria, were adopted for both the patients with ALS and control subjects (for detailed procedures and variants filtering criteria, see Supplementary Material).
Statistical Analysis
Descriptive statistics are reported as count and percentage, for categorical variables, or mean and SD, for continuous variables. NEK1 variants classified as likely benign were excluded from statistical analyses and counting, both in patients with ALS and control cases. Kaplan–Meier univariate analysis was carried out to determine the effect of NEK1 variants on age at onset and survival (defined as the time from symptoms onset to death/tracheostomy). Follow-up data were censored on February 2021. The Fisher’s exact test and binary logistic regression analysis, adjusted for sex and age at onset, were used to explore differences in the frequency of patients carrying NEK1 variants compared with controls, and the influence of NEK1 variants on phenotype (Brenner et al., 2016; Black et al., 2017; Naruse et al., 2019; Tsai et al., 2020). Statistical significance was set at p-value < 0.05. All analyses were performed using the SPSS 22.0 software (Technologies Incorporation, Chicago, Illinois, United States).
Results
We conducted an NGS analysis of 350 Italian patients with ALS. Overall, we detected and confirmed 20 different NEK1 rare variants in 33 unrelated patients, all presenting with sALS. Four of these variants were LoF and 16 were missense; all of these variants were found in heterozygosity (Table 1). After excluding the variants classified as likely benign according to ACMG classification, both in ALS and control cohorts, we identified a total of 14 different rare variants (four LoF and 10 missense) in 24 patients and five missense in controls (Table 1 and Supplementary Table 3). Eight of the identified variants are novel, while the others have a minor allele frequency (MAF) < 0.0005. The only one variant with a higher MAF is the p.Arg261His (global MAF: 0.00385 and population MAF: 0.00398), which has already been reported as an ALS risk factor (Kenna et al., 2016; Nguyen et al., 2018; Lattante et al., 2021). Our cohort of patients with ALS was enriched for NEK1 variants compared with controls (24/350, i.e., 6.86% and 6/380, i.e., 1.58%; p < 0.001).
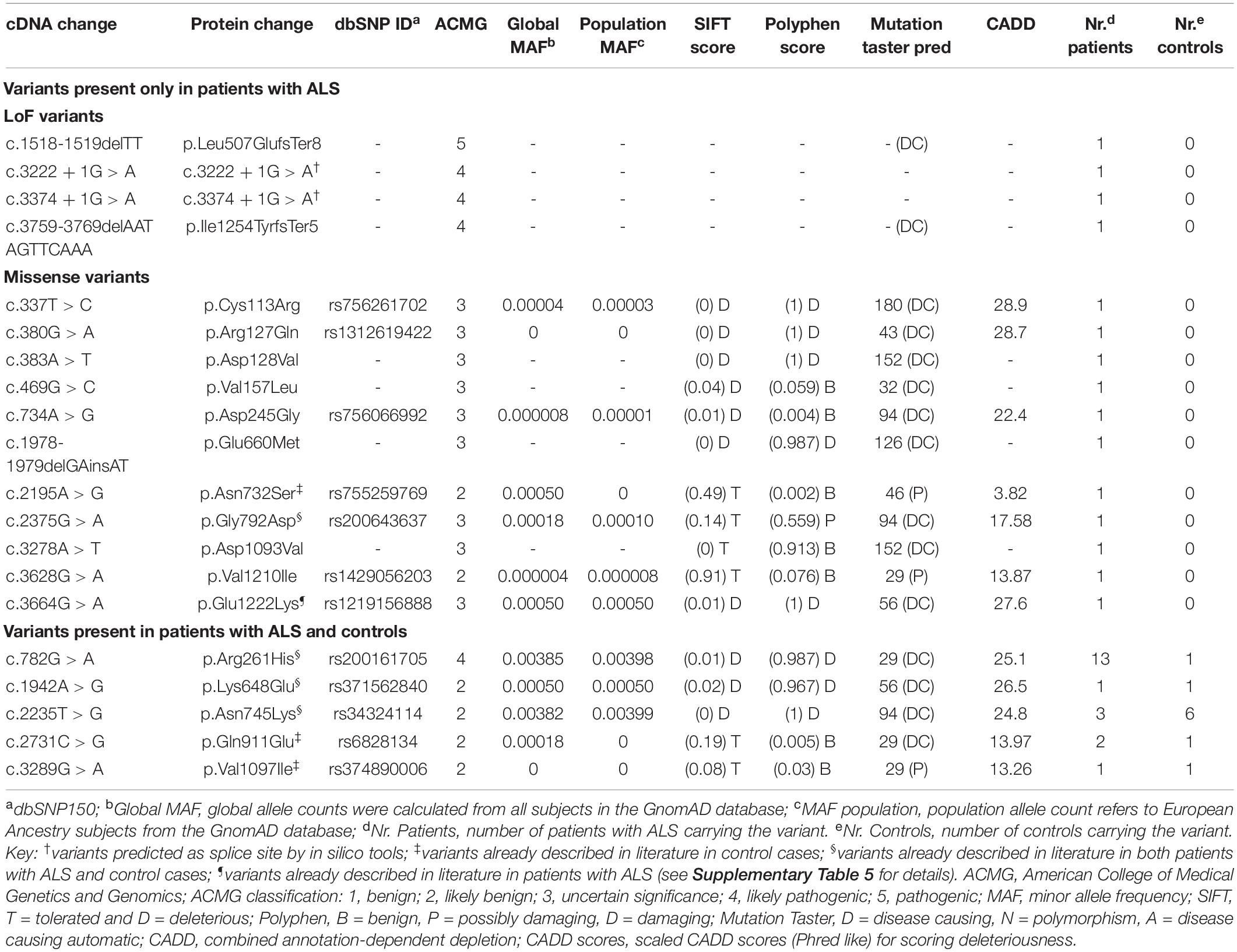
Table 1. All the NEK1 variants found in our NGS analysis.
LoF variants were found in 1.14% (4/350) of patients and none (0/380) in control individuals, in line with previous literature (Brenner et al., 2016; Kenna et al., 2016; Nguyen et al., 2018; Shu et al., 2018; Naruse et al., 2019; Tsai et al., 2020; Lattante et al., 2021), even though this difference did not reach statistical significance. In addition, we also observed a higher frequency of NEK1 missense variants in patients vs. controls (21/350, i.e., 6.0% and 6/380, i.e., 1.58%; p = 0.0015). However, this enrichment was driven by the high prevalence of the p.Arg261His, found in 13 patients with ALS and only one control (p < 0.001). Excluding this latter from counting, the difference between the two groups was not statistically significant (2.57% patients with ALS and 1.32% controls), in line with previous studies (Brenner et al., 2016; Kenna et al., 2016; Nguyen et al., 2018; Shu et al., 2018; Naruse et al., 2019; Tsai et al., 2020; Lattante et al., 2021).
Patients with ALS carrying NEK1 variants did not differ for sex distribution, age at onset, or survival from the other patients of the cohort. However, a binary logistic regression analysis, adjusted for sex and age at onset, revealed a significantly higher risk for NEK1 carriers to manifest the flail arm phenotype (OR 5.34, 95% CI 1.93–14.74). Indeed, we found a significant enrichment of patients presenting with a flail arm phenotype among the NEK1 carriers group (7/24, i.e., 29.2% vs. 21/326, i.e., 6.4%, p = 0.0013).
NEK1 Loss of Function Variants
The four NEK1 LoF were absent from all the public genomic databases, including dbSNP, and also from our in-house control cohort (Table 1). They include two frameshift variants (p.Leu507GlufsTer8, p.Ile1254TyrfsTer5) and the two c.3222 + 1G > A and c.3374 + 1G > A variants, which are predicted to alter the normal splice sites from in silico tools (Supplementary Table 2).
The p.Leu507GlufsTer8 is predicted to cause the transcript degradation through nonsense-mediated decay, while the p.Ile1254TyrfsTer5, being at the C-terminal of the protein, could likely lead to the formation of a truncated protein. These frameshift variants were identified in two unrelated male patients with sALS, both presenting with a flail arm phenotype (Table 2).
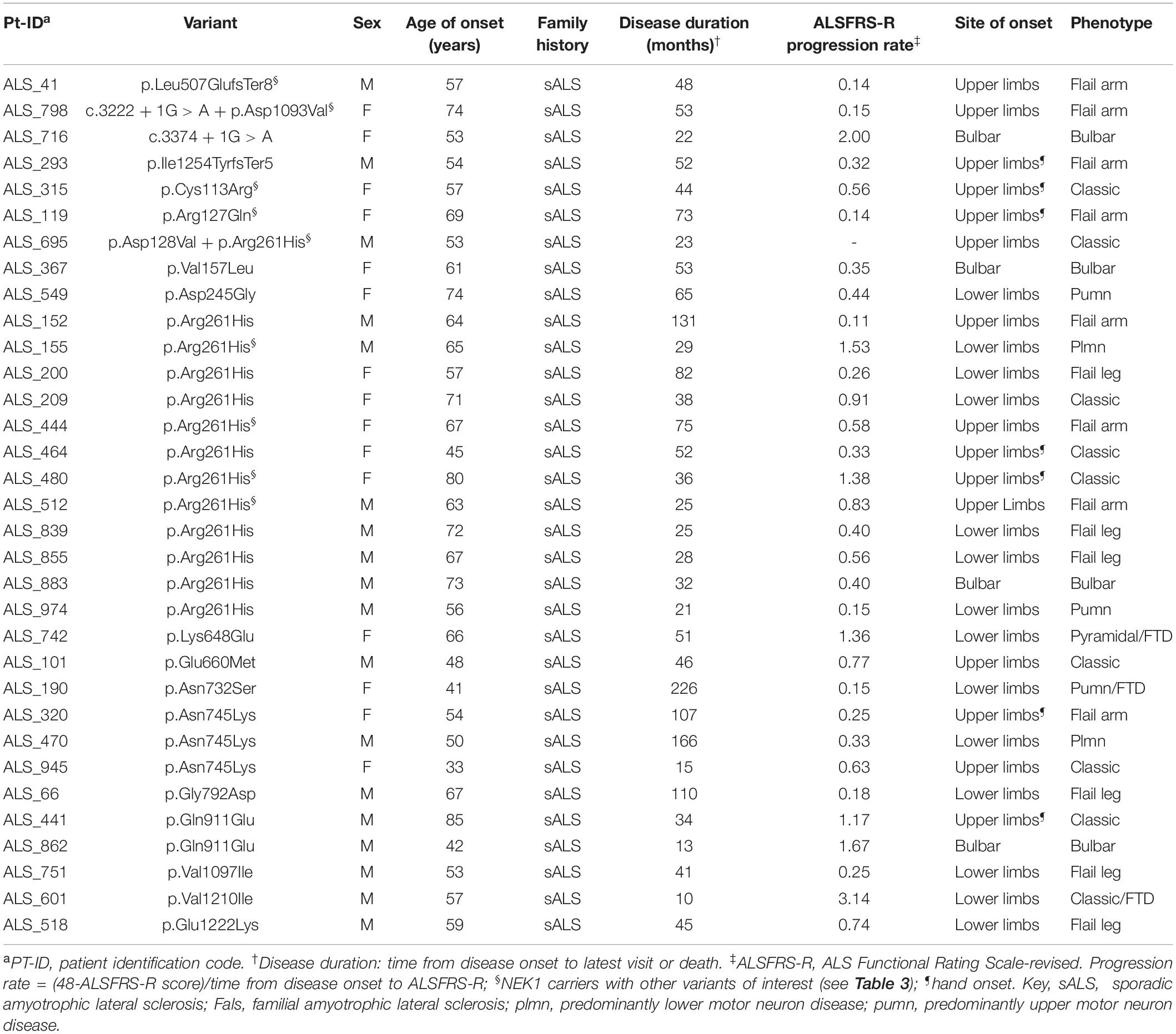
Table 2. Demographic and clinical characteristics of patients with NEK1 variants.
The c.3222 + 1G > A splice site was found in a female patient with sALS also presenting with a flail arm phenotype. Interestingly, this patient also carried a novel NEK1 missense variant (Tables 1, 2). The c.3374 + 1G > A variant was identified in a female patient with sALS presenting with a bulbar phenotype (Table 2); this variant was also detected in her unaffected brother, whose DNA was available for testing (Family #1 in Supplementary Figure 1).
NEK1 Missense Variants
Overall, we found 10 different missense variants in 22 unrelated patients with ALS (Table 1). The most frequent missense variant identified in this study was the p.Arg261His, previously described as ALS-risk factor (Kenna et al., 2016; Nguyen et al., 2018; Lattante et al., 2021), that we found in 13 patients with ALS and one control (3.71 and 0.26%, respectively). Nine missense variants were classified as variants of uncertain significance (VUS), according to the ACMG classification. Among them, the p.Asp128Val, p.Val157Leu, p.Glu660Met, and p.Asp1093Val were novel, neither reported in the literature or dbSNPs database nor found in our in-house controls. The two patients with the novel p.Asp128Val and p.Asp1093Val were both found to carry another NEK1 variant (p.Arg261His and c.3222 + 1G > A, respectively) (Table 3). These patients had a classic and a flail arm phenotype, respectively. The p.Glu660Met variant was the result of the change of two adjacent bases (c.1978-1979delGAinsAT). The other five missense variants classified as VUS were found only in the ALS cohort; among them, the p.Glu1222Lys was previously described in a patient with ALS (Lattante et al., 2021), the p.Gly792Asp in both patients with ALS and controls (Kenna et al., 2016), whereas the others were only reported in dbSNPs database. The other six missense variants were considered as likely benign (Table 1).
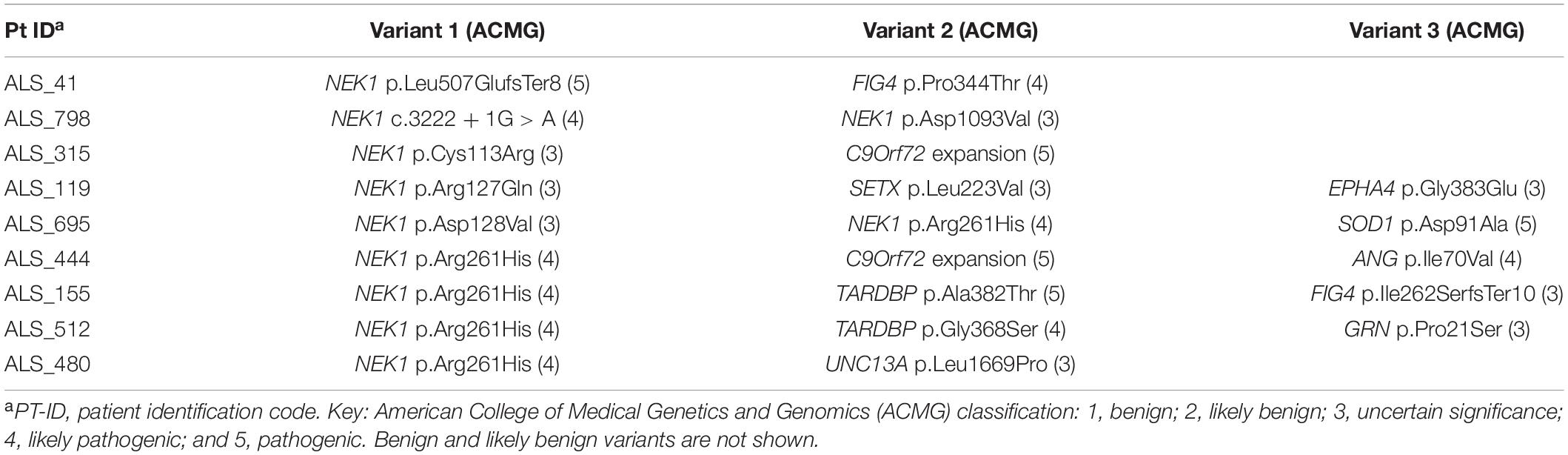
Table 3. List of NEK1 carriers with other variants of interest.
NEK1 Polygenic Carriers
We next investigated whether the NEK1 patients with ALS also carried other variants of interest among other 33 ALS genes (Supplementary Methods). We found that nine patients (37.5%) were oligogenic for other ALS genes (Table 3); in particular, two carried the pathological expansion of the C9orf72 gene and one of them carried also the p.Ile70Val missense in the ANG gene, which was classified as presumed pathogenic (Greenway et al., 2006; Crabtree et al., 2007; Paubel et al., 2008). The patient carrying the NEK1 p.Cys113Arg missense, classified as VUS, also harbored the C9orf72 expansion, while her unaffected mother and brother carried only the NEK1 variant (Family #2 in Supplementary Figure 1). This patient had hand onset and a classic ALS phenotype (Table 2). The other four patients with the C9orf72 expansion carried likely benign NEK1 missense variants and were excluded from counting. Notably, the two unaffected sisters of the patient in Family #3 carried the C9orf72 expansion but were wild type for the NEK1 p.Asn732Ser likely benign variant, identified in their affected sister (Supplementary Figure 1).
Two p.Arg261His NEK1 carriers were also heterozygous for a TARDBP pathogenic variant (p.Ala382Thr and p.Gly368Ser, respectively). The unaffected brother of the patient carrying both the p.Arg261His NEK1 risk factor and the likely pathogenic p.Gly368Ser TARDBP variant was found to carry the TARDBP variant but not the NEK1 missense (Family #4 in Supplementary Figure 1). The patient had a flail arm phenotype (Table 2).
One patient carried also the SOD1 p.Asp91Ala pathogenic variant; notably, he was found to carry the p.Arg261His risk factor and another NEK1 missense variant (p.Asp128Val) (Table 3).
Another patient, presenting with a flail arm phenotype, carried two different NEK1 variants: the c.3222 + 1G > A LoF and the novel p.Asp1093Val missense variant (Table 2).
The other three patients carried a VUS in EPHA4, FIG4, SETX, and UNC13A ALS-related genes (Table 3).
NEK1 polygenic carriers did not show relevant differences in age of onset or survival compared with ALS patients carrying a single NEK1 variant (data not shown).
Discussion
This study describes the genetic variability of the NEK1 gene in a cohort of 350 Italian patients with ALS. Indeed, we identified four LoF and 16 missense variants in 33 unrelated patients with sALS. We also characterized the clinical features of the patients carrying NEK1 variants, highlighting a potential association with the flail arm phenotype.
NEK1 LoF variants have been previously proposed to play a pathogenic role in ALS, since several studies described a significantly higher frequency in both patients with fALS and sALS compared with controls, with a prevalence ranging from 0.4 to 1.8% (Brenner et al., 2016; Kenna et al., 2016; Nguyen et al., 2018; Shu et al., 2018; Naruse et al., 2019; Tsai et al., 2020; Lattante et al., 2021). A previous study also demonstrated that LoF variants lead to NEK1 haploinsufficiency, resulting in DDR impairment (Higelin et al., 2018), a mechanism already associated with other ALS-related genes (Riancho et al., 2020). In our ALS cohort, LoF frequency was 1.1%. However, NEK1 LoF variants should not be considered rare: indeed, despite being less frequent than variants in C9orf72, SOD1, FUS, TARDBP, and TBK1, their prevalence is higher compared with other ALS-related genes (Tsai et al., 2020).
In addition to LoF, we also observed an apparently enriched fraction of NEK1 missense variants in patients with ALS with an overall frequency of 6.0% compared with 1.58% of our in-house controls. Previous studies reported, instead, similar percentages of missense variants in both the ALS and control cases, ranging from 1.1 to 3.7%, suggesting that, collectively, missense variants might not affect ALS susceptibility (Supplementary Tables 4, 5; Brenner et al., 2016; Kenna et al., 2016; Black et al., 2017; Gratten et al., 2017; Nguyen et al., 2018; Shu et al., 2018; Naruse et al., 2019; Tsai et al., 2020; Lattante et al., 2021). The enrichment we observed is explained by the higher prevalence of the p.Arg261His missense in our ALS cohort, which was 10 times more frequent than in controls. The p.Arg261His is the only NEK1 missense variant already reported to be related to an increased risk for ALS (Kenna et al., 2016; Black et al., 2017; Nguyen et al., 2018; Lattante et al., 2021). Nevertheless, previous studies showed p.Arg261His frequency in patients with ALS ranging from 0.6 to 1.7% (Kenna et al., 2016; Nguyen et al., 2018; Lattante et al., 2021), while our data reported a higher frequency of 3.7%. Regarding other NEK1 missense variants, further studies are required to assess their role in ALS pathogenesis.
We then explored whether NEK1 variants in patients with ALS might have an association with specific phenotypic features. Notably, 29.2% of NEK1 carriers presented with a flail arm phenotype, a percentage significantly higher compared with non-NEK1 carriers of our ALS cohort, and with the 11% prevalence of this phenotype reported by previous studies (Wijesekera et al., 2009; Chio et al., 2011; Schito et al., 2020). Among patients with ALS carrying NEK1 LoF variants, three presented with a flail arm phenotype, while the patient whose variant was shared with her unaffected brother had instead a bulbar phenotype. Interestingly, a recent report correlated the presence of a NEK1 LoF variant with a hand-onset ALS in a Taiwanese ALS cohort, reporting it in all LoF carriers (Tsai et al., 2020). Nevertheless, other studies described NEK1 LoF carriers with the same clinical presentation, even if the authors did not directly point out such correlation (Brenner et al., 2016; Shu et al., 2018; Naruse et al., 2019).
Nowadays it is well recognized that ALS and frontotemporal dementia (FTD) share common clinical and genetic features and could represent a pathological continuum (Liscic et al., 2020). In our study, none of the NEK1 LoF carriers had cognitive impairment; moreover, we found three patients carrying a NEK1 likely benign missense variant that presented also with FTD, but all of them carried also the C9orf72 expansion. Intriguingly, none of the patients with ALS carrying NEK1 LoF variants described up to date had cognitive impairment, except for one case (Lattante et al., 2021), whereas C9orf72 association to ALS-FTD is well known (Hodges, 2012).
It has already been proposed that ALS could be a complex multistep pathogenic process, in which the disease presentation results from the combination of different factors (Al-Chalabi et al., 2014; Scarlino et al., 2020). This “multiple-hit hypothesis” could account for the phenotypic heterogeneity observed even in patients harboring the same gene mutation, and for the reduced penetrance of NEK1 variants (Nguyen et al., 2018; Scarlino et al., 2020). Our results are in line with this oligogenic hypothesis since more than one-third of NEK1 carriers had variants in other ALS-related genes. From pedigree analysis, we found that in two different families the patients with ALS carried both the C9orf72 expansion and a NEK1 missense variant, whereas the unaffected relatives carried a variant in only one of these two genes. Similarly, in another family, both the patient and his unaffected brother carried a TARDBP mutation, but only the patient was oligogenic for a NEK1 missense variant. Taken together, these observations support the already proposed hypothesis that NEK1 variants may confer a significant susceptibility to ALS, even though they may be not sufficient per se for disease development, possibly acting as a phenotypic modifier (Nguyen et al., 2018; Lattante et al., 2021). Although several studies already suggested that additional variants may affect ALS disease severity (Pang et al., 2017; Ross et al., 2020), in our cohort the concomitant presence of a NEK1 variant with variants in other ALS-related genes did not seem to significantly influence the disease onset or progression rate.
We acknowledge that this study has some limitations. First, our sample size is relatively small and this cannot be considered a population-based study. However, we were able to provide detailed phenotype information of patients with ALS to test potential genotype-phenotype correlations. Second, it is difficult to determine the potential pathogenic role for the missense variants. Indeed, specific functional studies are needed to evaluate whether the single amino acid change could affect the whole protein function. In our study, the lack of biological material from NEK1 carriers prevented us to perform exhaustive segregation analysis and functional studies at the transcript or protein levels. However, we analyzed also a non-neurological age-matched cohort of controls.
Conclusion
In conclusion, this study supports NEK1’s contribution to ALS pathogenesis in the Italian population. We confirmed previous findings that NEK1 LoF variants, and the p.Arg261His missense variant, are significantly enriched in patients with ALS. Notably, our data also suggest an association of the flail arm phenotype with the presence of NEK1 variants. We also found a relevant number of NEK1 carriers with variants in other ALS-related genes, supporting the hypothesis that NEK1 variants might concur to ALS pathogenesis in an oligogenic model of disease, possibly acting as a phenotypic modifier.
Data Availability Statement
The data presented in the study are deposited in the NCBI SRA Sequence Read Archive Depository, accession number PRJNA817104.
Ethics Statement
The studies involving human participants were reviewed and approved by the Local Ethics Committee of the San Raffaele Hospital. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
NR, PC, and AQ conceived and designed the study. NR, LP, TR, GP PS, TD, FA, PC, and AQ contributed to data collection, analysis, and interpretation and generation of the tables. NR, LP, and TR did the statistical analysis. NR, TR, and PS were involved in patient selection and clinical data collection. MF, PC, and AQ supervised the study. LP and TR wrote the first draft of the article. All authors actively contributed to the writing, critically reviewing of the article for important intellectual content, and approved the final version and contributed to data interpretation.
Funding
This study was supported by the Giovanni Marazzina Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2022.833051/full#supplementary-material
Footnotes
- ^ http://variantstudio.software.illumina.com/
- ^ http://www.broadinstitute.org/software/igv/
- ^ https://alsod.ac.uk/
- ^ https://kingrelatedness.com
References
Abrahams, S., Newton, J., Niven, E., Foley, J., and Bak, T. H. (2014). Screening for cognition and behaviour changes in ALS. Amyotroph. Lateral Scler. Frontotemporal Degener. 15, 9–14. doi: 10.3109/21678421.2013.805784
Agosta, F., Ferraro, P. M., Riva, N., Spinelli, E. G., Domi, T., Carrera, P., et al. (2017). Structural and functional brain signatures of C9orf72 in motor neuron disease. Neurobiol. Aging 57, 206–219. doi: 10.1016/j.neurobiolaging.2017.05.024
Al-Chalabi, A., Calvo, A., Chio, A., Colville, S., Ellis, C. M., Hardiman, O., et al. (2014). Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol. 13, 1108–1113. doi: 10.1016/S1474-4422(14)70219-4
Black, H. A., Leighton, D. J., Cleary, E. M., Rose, E., Stephenson, L., Colville, S., et al. (2017). Genetic epidemiology of motor neuron disease-associated variants in the Scottish population. Neurobiol. Aging 51, 178.e11–178.e20. doi: 10.1016/j.neurobiolaging.2017.04.019
Brenner, D., Muller, K., Wieland, T., Weydt, P., Bohm, S., Lule, D., et al. (2016). NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 139, (Pt 5):e28. doi: 10.1093/brain/aww033
Brooks, B. R., Miller, R. G., Swash, M., and Munsat, T. L. (2000). El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299. doi: 10.1080/146608200300079536
Chio, A., Calvo, A., Moglia, C., Mazzini, L., and Mora, G. (2011). Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J. Neurol. Neurosurg. Psychiatry 82, 740–746. doi: 10.1136/jnnp.2010.235952
Crabtree, B., Thiyagarajan, N., Prior, S. H., Wilson, P., Iyer, S., Ferns, T., et al. (2007). Characterization of human angiogenin variants implicated in amyotrophic lateral sclerosis. Biochemistry 46, 11810–11818. doi: 10.1021/bi701333h
Falzone, Y. M., Domi, T., Agosta, F., Pozzi, L., Schito, P., Fazio, R., et al. (2020). Serum phosphorylated neurofilament heavy-chain levels reflect phenotypic heterogeneity and are an independent predictor of survival in motor neuron disease. J. Neurol. 267, 2272–2280. doi: 10.1007/s00415-020-09838-9
Gratten, J., Zhao, Q. Y., Benyamin, B., Garton, F., He, J., Leo, P. J., et al. (2017). Whole-exome sequencing in amyotrophic lateral sclerosis suggests NEK1 is a risk gene in Chinese. Genome Med. 9:97. doi: 10.1186/s13073-017-0487-0
Greenway, M. J., Andersen, P. M., Russ, C., Ennis, S., Cashman, S., Donaghy, C., et al. (2006). ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat. Genet. 38, 411–413. doi: 10.1038/ng1742
Higelin, J., Catanese, A., Semelink-Sedlacek, L. L., Oeztuerk, S., Lutz, A. K., Bausinger, J., et al. (2018). NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 30, 150–162. doi: 10.1016/j.scr.2018.06.005
Hodges, J. (2012). Familial frontotemporal dementia and amyotrophic lateral sclerosis associated with the C9ORF72 hexanucleotide repeat. Brain 135(Pt 3), 652–655. doi: 10.1093/brain/aws033
Kenna, K. P., van Doormaal, P. T., Dekker, A. M., Ticozzi, N., Kenna, B. J., Diekstra, F. P., et al. (2016). NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 48, 1037–1042. doi: 10.1038/ng.3626
Lattante, S., Doronzio, P. N., Conte, A., Marangi, G., Martello, F., Bisogni, G., et al. (2021). Novel variants and cellular studies on patients’ primary fibroblasts support a role for NEK1 missense variants in ALS pathogenesis. Hum. Mol. Genet. 30, 65–71. doi: 10.1093/hmg/ddab015
Liscic, R. M., Alberici, A., Cairns, N. J., Romano, M., and Buratti, E. (2020). From basic research to the clinic: innovative therapies for ALS and FTD in the pipeline. Mol. Neurodegener. 15:31. doi: 10.1186/s13024-020-00373-9
Manichaikul, A., Mychaleckyj, J. C., Rich, S. S., Daly, K., Sale, M., and Chen, W. M. (2010). Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873. doi: 10.1093/bioinformatics/btq559
Naruse, H., Ishiura, H., Mitsui, J., Takahashi, Y., Matsukawa, T., Tanaka, M., et al. (2019). Burden of rare variants in causative genes for amyotrophic lateral sclerosis (ALS) accelerates age at onset of ALS. J. Neurol. Neurosurg. Psychiatry 90, 537–542. doi: 10.1136/jnnp-2018-318568
Nguyen, H. P., Van Mossevelde, S., Dillen, L., De Bleecker, J. L., Moisse, M., Van Damme, P., et al. (2018). NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol. Aging 61, 255.e1–255.e7. doi: 10.1016/j.neurobiolaging.2017.08.021
Pang, S. Y. Y., Hsu, J. S., Teo, K. C., Li, Y., Kung, M. H. W., Cheah, K. S. E., et al. (2017). Burden of rare variants in ALS genes influences survival in familial and sporadic ALS. Neurobiol. Aging 58, 238.e9–238.e15. doi: 10.1016/j.neurobiolaging.2017.06.007
Paubel, A., Violette, J., Amy, M., Praline, J., Meininger, V., Camu, W., et al. (2008). Mutations of the ANG gene in French patients with sporadic amyotrophic lateral sclerosis. Arch. Neurol. 65, 1333–1336. doi: 10.1001/archneur.65.10.1333
Peres de Oliveira, A., Kazuo Issayama, L., Betim Pavan, I. C., Riback Silva, F., Diniz Melo-Hanchuk, T., Moreira Simabuco, F., et al. (2020). Checking NEKs: overcoming a Bottleneck in Human Diseases. Molecules 25:1778. doi: 10.3390/molecules25081778
Pozzi, L., Valenza, F., Mosca, L., Dal Mas, A., Domi, T., Romano, A., et al. (2017). TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: genetic and functional characterisation. J. Neurol. Neurosurg. Psychiatry 88, 869–875. doi: 10.1136/jnnp-2017-316174
Rascovsky, K., Hodges, J. R., Knopman, D., Mendez, M. F., Kramer, J. H., Neuhaus, J., et al. (2011). Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134, 2456–2477. doi: 10.1093/brain/awr179
Renton, A. E., Chio, A., and Traynor, B. J. (2014). State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 17, 17–23. doi: 10.1038/nn.3584
Riancho, J., Castanedo-Vazquez, D., Gil-Bea, F., Tapia, O., Arozamena, J., Duran-Vian, C., et al. (2020). ALS-derived fibroblasts exhibit reduced proliferation rate, cytoplasmic TDP-43 aggregation and a higher susceptibility to DNA damage. J. Neurol. 267, 1291–1299. doi: 10.1007/s00415-020-09704-8
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Riva, N., Agosta, F., Lunetta, C., Filippi, M., and Quattrini, A. (2016). Recent advances in amyotrophic lateral sclerosis. J. Neurol. 263, 1241–1254. doi: 10.1007/s00415-016-8091-6
Ross, J. P., Leblond, C. S., Laurent, S. B., Spiegelman, D., Dionne-Laporte, A., Camu, W., et al. (2020). Oligogenicity, C9orf72 expansion, and variant severity in ALS. Neurogenetics 21, 227–242. doi: 10.1007/s10048-020-00612-7
Scarlino, S., Domi, T., Pozzi, L., Romano, A., Pipitone, G. B., Falzone, Y. M., et al. (2020). Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: insights from a Next Generation Sequencing Study of an Italian ALS Cohort. Int. J. Mol. Sci. 21:3346. doi: 10.3390/ijms21093346
Schito, P., Ceccardi, G., Calvo, A., Falzone, Y. M., Moglia, C., Lunetta, C., et al. (2020). Clinical features and outcomes of the flail arm and flail leg and pure lower motor neuron MND variants: a multicentre Italian study. J. Neurol. Neurosurg. Psychiatry 91, 1001–1003. doi: 10.1136/jnnp-2020-323542
Shu, S., Lei, X., Liu, F., Cui, B., Liu, Q., Ding, Q., et al. (2018). Mutation screening of NEK1 in Chinese ALS patients. Neurobiol. Aging 71, 267.e1–267.e4. doi: 10.1016/j.neurobiolaging.2018.06.022
Strong, M. J., Abrahams, S., Goldstein, L. H., Woolley, S., Mclaughlin, P., Snowden, J., et al. (2017). Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 153–174. doi: 10.1080/21678421.2016.1267768
Tsai, Y. S., Lin, K. P., Jih, K. Y., Tsai, P. C., Liao, Y. C., and Lee, Y. C. (2020). Hand-onset weakness is a common feature of ALS patients with a NEK1 loss-of-function variant. Ann. Clin. Transl. Neurol. 7, 965–971. doi: 10.1002/acn3.51064
Veldink, J. H. (2017). ALS genetic epidemiology ‘How simplex is the genetic epidemiology of ALS?’. J. Neurol. Neurosurg. Psychiatry 88:537. doi: 10.1136/jnnp-2016-315469
Keywords: motor neuron disease, frontotemporal dementia, neuromuscular diseases, neurodegenerative disorders, genetics, never in mitosis a (NIMA)-related kinase 1, flail arm, ALS phenotype
Citation: Riva N, Pozzi L, Russo T, Pipitone GB, Schito P, Domi T, Agosta F, Quattrini A, Carrera P and Filippi M (2022) NEK1 Variants in a Cohort of Italian Patients With Amyotrophic Lateral Sclerosis. Front. Neurosci. 16:833051. doi: 10.3389/fnins.2022.833051
Received: 13 December 2021; Accepted: 02 March 2022;
Published: 14 April 2022.
Edited by:
Daniele Ghezzi, Carlo Besta Neurological Institute Foundation (IRCCS), ItalyReviewed by:
Dongsheng Fan, Peking University Third Hospital, ChinaSilvia Peverelli, Italian Auxological Institute (IRCCS), Italy
Copyright © 2022 Riva, Pozzi, Russo, Pipitone, Schito, Domi, Agosta, Quattrini, Carrera and Filippi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nilo Riva, cml2YS5uaWxvQGhzci5pdA==
†These authors share senior authorship