

- 1 Department of Radiation Oncology, Case Integrative Cancer Biology Program, Case Western Reserve University, Cleveland, OH, USA
- 2 Department of Radiation Oncology, Rhode Island Hospital, Warren Alpert Medical School of Brown University, Providence, RI, USA
A more flexible and higher-yielding in vitro DNA mismatch repair (MMR) substrate construction method, which was developed initially by Wang and Hays, is described for the construction of a nucleotide-based chemical mismatch (G/IU) and a G/T mismatch. Our modifications use the combination of two endonuclease enzymes (NheI and BciVI) and two new redesigned plasmids (pWDAH1A and pWDSH1B). In our modified methodology, plasmids are initially digested with the nicking endonucleases, followed by the streptavidin treatment. The mismatch-containing oligo is then annealed to the gap DNA and finally ligated to produce a mismatch-containing DNA substrate. We report a high efficiency (up to 90%) of these mismatch substrates and confirm recognition using a functional assay. These modifications, coupled with the use of the redesigned plasmids, can be applied for the construction of other types of chemically induced mismatches as well as insertion-deletion loops for future in vitro studies of MMR processing by our group and others.
Introduction
DNA mismatches may arise from endogenous replication errors or from exogenous sources including occupational and therapeutic chemical and ionizing radiation exposures. These mismatches may be repaired during scheduled or unscheduled DNA synthesis by the DNA mismatch repair (MMR) pathway (for reviews, see references Kunkel and Erie, 2005; Iyer et al., 2006; Modrich, 2006; Li, 2008), as well as by other DNA repair pathways such as base excision repair (BER; Kinsella, 2009). Failure to correct these mismatches by MMR may significantly increase the incidence of cancer in patients with genetic defects (e.g., hereditary non-polyposis colon cancer; Bellizzi and Frankel, 2009). Additionally, epigenetic silencing by promoter methylation of MMR genes (particularly MLH1 and MSH2) is associated with sporadic cancers of gastrointestinal, gynecological and genitourinary origin, which are characterized by a microsatellite instability-high (MSI-H) phenotype (Bellizzi and Frankel, 2009). MMR-deficient cancers are reported to be resistant, or more accurately, damage-tolerant, to cytotoxicity by various classes of chemotherapeutic agents including nucleoside analogs and to certain types of ionizing radiation (Li, 2008; Kinsella, 2009; Yan et al., 2009). Consequently, a better understanding of how MMR processes non-classical DNA mismatches may be helpful in designing cancer prevention strategies as well as in developing novel cancer therapeutics for MMR-deficient cancers.
Our laboratory initially reported that incorporation of the halogenated thymidine analog, iododeoxyuridine (IUdR) causes specific DNA mismatches, particularly G/IU, which are efficiently processed by MMR (Berry and Kinsella, 2001; Berry et al., 2003). In fact, MMR-deficient cancer cells and human tumor xenografts retain higher levels of IUdR–DNA incorporation compared to MMR-proficient cells. However, the increased percentage IUdR–DNA incorporation in MMR-deficient cells/tumors does not result in excessive cytotoxicity per se, but does enhance cell killing with subsequent exposure to ionizing radiation. As such, the use of IUdR or its prodrug IPdR, during radiation therapy has been used in pre-clinical studies to “target” MMR-deficient cancers while sparing normal (MMR-proficient) tissues (Berry and Kinsella, 2001; Berry et al., 2003; Seo et al., 2004). Other types of nucleoside analogs, such as the fluoropyrimidine, 5-fluorouracil (5-FU) and the purine analog, 6-thioguanine (6-TG), which are used clinically as cancer chemotherapeutic agents, are also processed by MMR but result in marked cytotoxicity in MMR-proficient cells compared to IUdR whereas MMR-deficient tumor cells show clear damage-tolerance in pre-clinical and clinical studies to 5-FU and 6-TG (Berry and Kinsella, 2001; Meyers et al., 2001; Berry et al., 2003; Li, 2008; Kinsella, 2009).
To begin to better understand the observed cellular differences in MMR processing of specific DNA mismatches resulting from treatment with different nucleoside analogs (IUdR, 5-FU, 6-TG) compared to endogenous mismatches like G/T, we initially plan to use using a purified protein system as described by the Modrich and Li laboratories (Zhang et al., 2005; Modrich, 2006; York and Modrich, 2006). However, as opposed to the construction of a G/T mismatch, these nucleotide based mismatches such as G/IU require the use of synthetic oligomers, as IUdR cannot be produced in cells or by a PCR methodology. As part of our preliminary investigations into MMR processing of specific nucleoside based mismatches, we developed several more efficient modifications to the method of Wang and Hays (2006) for the construction of a G/IU incorporated plasmid substrate. Our modifications include the use of a different combination of endonuclease enzymes, NheI/BciVI, and two new redesigned plasmids, pWDAH1A and pWDSH1B, in the construction of supercoiled G/IU versus G/T mismatch-containing DNA. Together, the advancements to the in vitro MMR assay described here provide an opportunity to more rapidly and efficiently investigate the biochemical mechanisms that drive MMR processing of specific DNA mismatches.
Materials and Methods
Plasmids and Enzymes
The original plasmids pSYAH1A and pSYSH1B were kindly provided by York and Modrich(2006; Duke University). These pUC19Y-derived plasmids (originally from Dr. Hays’ lab) were used to construct both pSYAH1A and pSYSH1B, the only difference of which is they share the same insertion fragment, but are inserted into the parental plasmid in opposite directions. All enzymes, including Nt.BbvCI, Nt.BstNBI, and T4 DNA ligase, were obtained from New England Biolab (NEB, Boston, MA, USA), except where otherwise noted. Oligonucleotides (oligos) and biotinylated oligos were synthesized by Invitrogen (Carlsbad, CA, USA), while the IUdR-containing oligo was from Operon (Huntsville, AL, USA). Streptavidin beads were purchased from Invitrogen and the streptavidin column was from GE Healthcare (Piscataway, NJ, USA).
Preparation of Plasmid DNA
To construct DNA substrates, we redesigned the insertion sequence of the plasmid pSYAH1A so that the newly designed plasmids have overlapping restrictive sites of NheI and a latent BciVI site. The source plasmid pSYAH1A was digested using SalI and AatII, and the purified linear double-stranded (ds)DNA was annealed to the following sequences: 5′-CGAGTCGATCCGT CGACACCTGCTAGCATCCTTAAGCTTGATCAGAGTCGATCCG-3′ and 5′-TCGACGG ATCGACTCTGATCAAGCTTAAGGATCTAGCAGGTGTC GAC GGAT CGACTCGACGT-3′. This new plasmid is named pWDAH1A, which has two Nt.BstNBI recognition sites in the sense strand, parting from a fragment of 43 nucleotides. The mismatch of either “T” or “IU” will be introduced by replacing this fragment with a synthetic segment. For the pWDSH1B plasmid, we used the following sequences as insertions: 5′-TCGACGAGTCGATCCGAGCTCACCTGC TAGCATCCTTAAGCTTGATCAGAGTCGATCCGACGT-3′ and 5′-CGGATCGACTCTGA TCAAGCTTAAG GATGCTAGCAGGTGAGCTCGGATCGACTCG-3′. The two plasmids share the same but inverted sequence.
The annealed oligos and SalI/AatII digested linearized DNA were ligated by T4 DNA ligase at room temperature and then grown in bacterial supercompetent XL10 cells (Stratagene, La Jolla, CA, USA) for 16 h following the manufacturer’s manual. The plasmids were extracted using a Marligen Miniprep kit (Marligen, Rockville, MD, USA). Purified plasmids were fully sequenced and confirmed in both strands.
For large-scale preparation, pWDAH1A plasmid-containing bacteria XL10 cells were grown in approximately 1000 ml of LB broth (USB, Cleveland, OH, USA). High purity and high quality plasmids were harvested according to the manufacturer’s manual.
Generation of Gap DNA
For a typical digestion reaction, 400 μg of plasmid DNA were used and 400 units of Nt.BstNBI were added in the reaction containing NEBuffer 3 buffer, and digestion was performed at 55°C for 4–6 h. At least 90% of supercoiled plasmid DNA was transformed into nick DNA. Agarose gel electrophoresis was used to confirm the digestion. Digestion was stopped by denaturation at 90°C for 10 min, followed by gradually cooling down to room temperature, in the presence of 20-fold excess of a biotinylated 43-mer oligo (Biotin-GATCGACTCTGATCAAGCTTAAGGATGCTAGCAGGT GTCGACG), which is complementary to the fragment between the two nicks. The 43-nt fragment released under the denaturating conditions was annealed to the excess biotinylated oligos to form dsDNA to facilitate removal. These biotinylated oligos (either single-stranded or double-stranded) were removed by centrifugation using a Amicon Ultra-4 50K column (Millipore, Billerica, MA, USA). Most of the excess oligos were removed by initial centrifugation and the remaining trace oligos were removed by subsequent passage through a streptavidin column. The passage through the column also concentrated the crude gap DNA. DNA concentrations were monitored using a NanoDrop 1000 spectrophotometer (NanoDrop, Wilmington, DE, USA) and were confirmed by agarose gel electrophoresis. The removal of the 43-nt fragment inhibited recognition reaction of NheI, as the anti-sense strand is now single-stranded. Our calculation based on the comparison before and after filtration indicated that 90% of gap DNA is recovered.
However, contamination of the remaining 10% of impurity in the gap DNA is too much and unacceptable. Consequently, any residual impurity DNA (mostly nicked DNA) is initially ligated, in the presence of NEBuffer 4 buffer, ligase and ATP, into supercoiled DNA. The supercoiled DNA is then separated using CsCl density-gradient ultracentrifugation and the target band is recovered using SSC (Saline Sodium Citrate) saturated isopropanol to extract Ethidium Bromide. Briefly, for 1 ml the ligated products, 1 g of CsCl is added to an Ultra-clear tube (Beckman, Palo Alto, CA, USA) and a SW50.1 rotor (Beckman) is used in the Beckman L7 ultracentrifuge for 48 h at a speed of 50000 rpm. The band is identified under UV light and recovered.
Ligation of Nicked dsDNA with Mismatch(s)
5′-end phosphorylated 43-nt oligos are added to the gap DNA at 1:1 ratio to produce a mismatched or matched DNA. The annealing reaction is performed in the presence of NEBuffer 4 buffer at 90°C for 30 s and then cooled at a rate of 0.1°C/s using a MJ Research Thermal Cycler (now Bio-Rad, Hercules, CA, USA). Then, 1 mM of ATP and 100 units of T4 DNA ligase in 500 μl reaction volume are used for the ligation reaction and kept overnight at room temperature.
Under this condition, we found a ligation efficiency of up to 90%, as judged by agarose gel electrophoresis, significantly higher than the prior method, which reported an efficiency of 50% (Wang and Hays, 2006). The ligation products are then digested by NheI to check whether it could cleave the mismatch-containing DNA recognition sites. This ligation reaction produced a mixture of circular dsDNA without nick (ligated) and circular dsDNA with nick (un-ligated). To further purify the mismatched DNA, CsCl density-gradient ultracentrifugation was applied again, as described above.
Identification and Confirmation of Mismatched DNA
Both strands of the constructed mismatch DNA are sequenced to confirm the presence of the mismatch. Note that in this case, the mismatched nucleotides in the template will pair with a cytosine in the daughter strand. The purified ligated DNA construct with the mismatch is then digested using Nt.BbvCI (to produce a nick in the anti-sense strand approximately 200-nt 5′ to the mismatch) or Nb.BbvCI (to produce a nick in the sense strand, 200-nt 3′ to the mismatch).
Nuclear extract and DNA substrate are incubated to assess mismatch correction. The nuclear extract is obtained from Active Motif (Carlsbad, CA, USA). The DNA substrate is mixed with nuclear extract in the presence of the following buffer solution: 20 mM Tris/HCl (pH 7.6), 1.5 mM ATP, 1 mM glutathione, 100 μM dNTP, 5 mM MgCl2, and 50 μg/ml BSA. After incubation at 37°C for 20 min, proteinase K buffer (10 mM Tris/HCl, 5 mM EDTA, 0.5% SDS) is added and the incubation is continued for 60 min at 37°C. DNA is precipitated by sequential phenol and ethanol exposures. The DNA is next dissolved in nuclease-free water and subjected to digestion using NheI. Since NheI cannot cleave a G/T or G/IU mismatch, any additional band that appeared indicated that the DNA substrate has been repaired by the nuclear extract.
Results and Discussion
Two Newly Designed Plasmids
The thymidine analog IUdR, an effective radiosensitizer, is known to be incorporated into genomic DNA after administration and can activate MMR processing. Our specific interest is not only to understand how MMR works, but also to learn how MMR processes exogenous chemically induced analogs (York and Modrich, 2006), compared to endogenous mismatches. The most widely used strategy to study endogenous MMR is to use a T/G mismatch and a combination of HindIII/XhoI recognition sites (Fang and Modrich, 1993; Wang and Hays, 2000, 2001; Constantin et al., 2005; Zhang et al., 2005) in a purified protein system (Constantin et al., 2005; Zhang et al., 2005) or with nuclear extracts (Wang and Hays, 2001, 2006). However, the T/G and HindIII/XhoI combination is not suitable to study MMR processing of our G/IU mismatch, as the presence of unnatural IUdR makes the repaired nucleotide un-identifiable by XhoI. We thus sought to develop a new restriction combination.
To construct a heteroduplex with a G/IU mismatch, we redesigned two plasmids (pWDAH1A and pWDSH1B plasmid) for subsequent DNA mismatch substrate construction (Figure 1). The pWDAH1A plasmid has two Nt.BstNBI sites both in the anti-sense strand, whereas pWDSH1B has two identical inverted nick sites in the sense strand. Both plasmids have NheI and a latent overlapping BciVI recognition site (Figure 1). These plasmids are sequenced and confirmed in both directions. Our preliminary data indicate that NheI cannot recognize and cleave the newly designed G/T and G/IU combination (data not shown), which means this combination is applicable to our experiment.
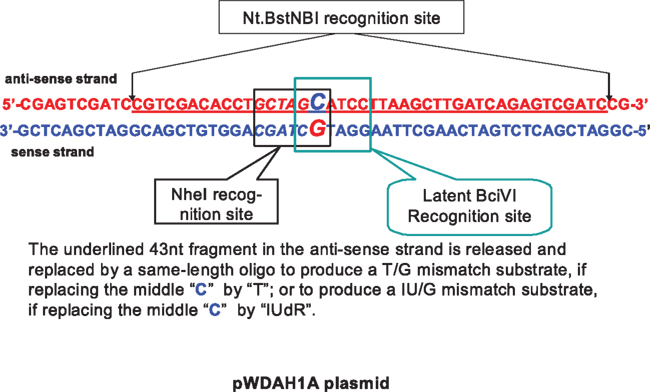
Figure 1. Sequences of the newly designed plasmid, containing a NheI site and a latent BciVI recognition site. The 43-nt fragment between two Nt. BstNBI sites in the anti-sense strand can be replaced by a new mismatch-containing oligo to produce a mismatch substrate.
The overall approach is illustrated in Figure 2. Briefly, Nt.BstNBI digestion is first used to produce a dual-nick DNA; the products are then centrifuged and bound to a streptavidin column to obtain gap DNA. The mismatch-introducing oligo is annealed with the gap DNA followed by ligation to yield the mismatch-containing heteroduplex DNA. Following CsCl gradient ultracentrifugation, the DNA substrates are nicked with the nicking nucleases.
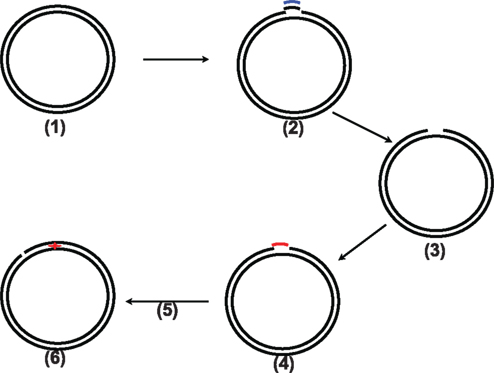
Figure 2. Illustration of the whole procedure of this new method. (1) the original pWDAH1A/SH1B plasmid; (2) Nt. BstNBI digestion to produce two nicks followed by addition of 20× biotinylated supplementary oligo; (3) two rounds of treatment with streptavidin beads and column to remove the biotinylated oligo to generate gap DNA; (4) the mismatch-containing DNA oligo added to anneal with the gap DNA, followed by a ligation reaction; (5) CsCl density ultracentrifuge, DNA recovery, and nick generation using Nt.BbvCI or Nb.BbvCI; (6) mismatch-containing DNA substrate (G/T mismatch or G/IU).
Production of Gap DNA
To generate the gap DNA, we used a simple strategy to remove the short 43-nt oligo between two Nt.BstNBI recognition sites (Figure 2, steps 2 and 3). A slow cool-down following heat denaturation facilitates the pairing of a biotinylated complementary oligo with the oligo released to form dsDNA. Centrifugation using Amicon centrifugal filter units (MW cut-off 50K) from Millipore removed most of the oligos (Figure 3, lane 4), while the remaining small amount of the oligo can be removed by passage through a streptavidin column to acquire as high efficiency as possible(Figure 3, lane 5).
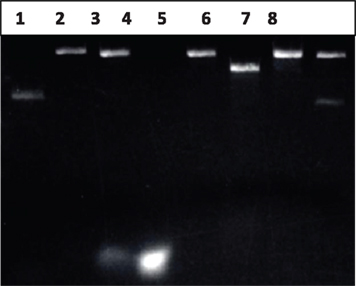
Figure 3. Identification of gap DNA. Lane 1: 200 ng of pWDAH1A plasmid; Lane 2: Nt. BstNBI-digested pWDAH1A plasmid (Nick DNA), 200 ng; Lane 3: Nt.BstNBI-digested pWDAH1A plasmid + biotinylated Rem-seq oligo; Lane 4: flow-through of mixture of lane 3; Lane 5: gap DNA, 200 ng; Lane 6: NheI-digested pWDAH1A plasmid DNA (linear DNA); Lane 7: NheI digestion of 200 ng gap DNA; Lane 8: ligation of gap DNA. Two hundred nanograms of DNA loaded each lane.
However, gap DNA remains only 90% pure, as there is small amount of single-nick DNA or undigested DNA in mixture (estimation from lanes 7 and 8 of Figure 3 indicates that approximately 10% is impurity). To assess the purity of gap DNA, the ligation reaction and NheI-digested reaction are performed. As shown in Figure 3, lanes 7 and 8, compared with the original undigested DNA (lane 1), most of original plasmid has been transformed into gap DNA, while NheI digestion of DNA found some impurity. Those approximately 10% of impurities should represent incompletely digested plasmid. The less pure gap DNA is then ligated. The formation of the nicked DNA by the ligation reaction into supercoiling DNA makes it easier to separate from gap DNA by our CsCl density-gradient ultracentrifugation method described above.
Generation of the Mismatched Nick DNA
The mismatch (T/G or IU/G) containing oligo is annealed with gap DNA, the oligo is inserted into the gap and ligated in the presence of 1 mM ATP. Our agarose gel electrophoresis indicated that the ligation efficiency is high (up to 90%), as shown in Figure 4, lane 4 (G/T mismatch) and lane 5(G/IU), where the nicked DNA binds with more Ethidium Bromide dye when compared with the supercoiled DNA. The ligated circular closed DNA is subjected to CsCl density-gradient centrifugation again.
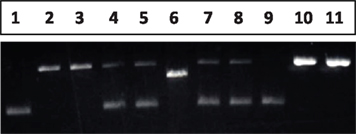
Figure 4. Generation and identification of the mismatch DNA substrate. Lane 1: pWDAH1A plasmid (supercoiled DNA); Lane 2, 3: gap DNA obtained after CsCl gradient ultracentrifuge + oligo (containing “T” or “IUdR”) heteroduplex, without ligation; Lane 4, 5: ligation of lane 2 and 3, gap DNA + mismatch-introducing oligo to produce G/T mismatch; Lane 6: NheI digestion of pWDAH1A plasmid; Lane 7, 8: NheI digestion of G/T- or G/IU-containing DNA; Lane 9: CsCl-method recovered mismatch DNA (G/T); Lane 10, 11: Nt. BbvCI-digested nick G/T or G/IU mismatch DNA as a DNA substrate.
The recovered DNA is then sequenced for confirmation of the mismatch. As shown in Figure 5, sequencing data confirmed the presence of a G/T mismatch. Note that the nucleotides of the original G/T mismatch will be exhibited as A/C in the sequencing reaction. For G/IU mismatch, there is a difference. When the template (sense strand) is “G,” the newly polymerized strand in the sequencing reaction is shown as a “C” as expected; however, when the template in the anti-sense strand contains IU, DNA polymerase would select a “G” in the G/T mismatch, to match the non-naturally occurring IU. Hence, it will show a “G/C.” However, these results do not suggest that this is a paired match DNA, as our digestion analysis indicates that NheI is unable to cleave the DNA, implying the presence of the mismatch (Figure 4, lane 8). To our knowledge, it is the first available sequencing data containing IUdR in the template strand and the first report that DNA polymerase will select “C” as a complementary nucleotide.
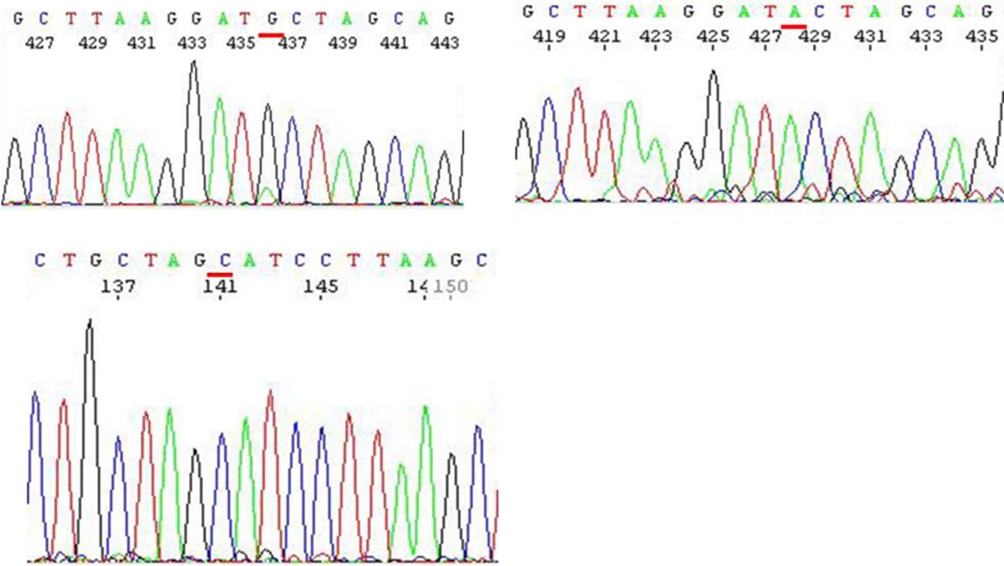
Figure 5. Sequencing data of a G/T mismatch DNA and a G/IU mismatch DNA substrate. Upper left: G/IU DNA, template – anti-sense strand; Upper Right: T/G DNA, template – anti-sense strand; Lower left: G/IU DNA, template – sense strand.
The closed circular DNA is digested by either Nt.BbvCI (in the anti-sense strand) or Nb.BbvCI (in the sense strand) to introduce a nick which mimics the template strand. The two plasmids make construction of four DNA substrates are available for MMR analysis.
Confirmation of Mismatch DNA Substrate
To further confirm that the new construct serves as a DNA substrate in the MMR study, we performed a functional assay using the MMR-proficient HeLa S3 nuclear extract (product of Active Motif). As shown in Figure 6, the nuclear extract data demonstrate that a mismatch DNA substrate is recognized and repaired such that the mismatched “T” or “IU” in the daughter strand is removed and replaced by a “C,” which restores the NheI recognition site, clearly indicating the success of our substrate constructions.
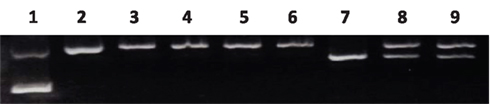
Figure 6. Confirmation of the mismatch DNA substrate. Lane 1: supercoiled DNA; Lane 2: Nick DNA control (Nt. BbvCI-digested); Lane 3, 4: G/T and G/IU nick DNA substrate; Lane 5, 6: NheI digestion of lane 3, 4; Lane 7: NheI digestion of pWDAH1A plasmid, linear; Lane 8: G/T nick DNA substrate + NE + proteinase K digestion, DNA extraction using ethanol + NheI digestion; Lane 9: G/IU nick DNA substrate + NE + proteinase K digestion, DNA extraction using ethanol + NheI digestion.
Toward our overall goal of improved cancer therapeutics for “damage-tolerant” MMR-deficient tumors (Berry and Kinsella, 2001; Kinsella, 2009), we have developed a probabilistic cell cycle model of MMR (Gurkan et al., 2007a). Using this model, we experimentally and computationally verified that the cell cycle dynamics for MMR-deficient and -proficient cells are different, with and without IUdR treatment. Eventually, we plan to integrate both the probabilistic cell cycle MMR model and the stochastic hybrid MMR drug processing model to “optimize” the timing and duration of drug (IUdR) and ionizing radiation when the % IUdR–DNA incorporation is maximally different between MMR-deficient cancers and MMR-proficient normal tissues so as to improve the therapeutic index of IUdR-mediated radiosensitization (Gurkan et al., 2007b).
Conclusion
The data shown here demonstrate that a faster, higher-yielding DNA mismatch substrate construction method is established successfully. These modifications, together with the redesigned plasmids, will be applied for the construction of other types of chemically induced mismatches as well as insertion-deletion loops (IDL) for future in vitro studies of MMR processing by our group and others.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported, in part, by NIH Grant U56 CA112963, the DBJ Foundation, and the University Radiation Medicine Foundation.
References
Bellizzi, A. M., and Frankel, W. L. (2009). Colorectal cancer due to deficiency in DNA mismatch repair function: a review. Adv. Anat. Pathol. 16, 405–417.
Berry, S. E., and Kinsella, T. J. (2001). Targeting DNA mismatch repair for radiosensitization. Semin. Radiat. Oncol. 11, 300–315.
Berry, S. E., Loh, T., Yan, T., and Kinsella, T. J. (2003). Role of MutSalpha in the recognition of iododeoxyuridine in DNA. Cancer Res. 63, 5490–5495.
Constantin, N., Dzantiev, L., Kadyrov, F. A., and Modrich, P. (2005). Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J. Biol. Chem. 280, 39752–39761.
Fang, W. H., and Modrich, P. (1993). Human strand-specific mismatch repair occurs by a bidirectional mechanism similar to that of the bacterial reaction. J. Biol. Chem. 1268, 11838–11844.
Gurkan, E., Schupp, J. E., Aziz, M. A., Kinsella, T. J., and Loparo, K. A. (2007a). Probabilistic modeling of DNA mismatch repair effects on cell cycle dynamics and iododeoxyuridine-DNA incorporation. Cancer Res. 67, 10993–11000.
Gurkan, E., Schupp, J. E., Kinsella, T. J., and Loparo, K. A. (2007b). “A conceptual modeling framework for the study of DNA mismatch repair pathway to improve therapeutic gain in cancer treatment,” in IEEE/NIH BISTI 2007 Life Science Systems and Applications Workshop (LISSA 2007), Bethesda, MD.
Iyer, R. R., Pluciennik, A., Burdett, V., and Modrich, P. (2006). DNA mismatch repair: functions and mechanisms. Chem. Rev. 106, 302–323.
Kinsella, T. J. (2009). Coordination of DNA mismatch repair and base excision repair processing of chemotherapy and radiation damage for targeting resistant cancers. Clin. Cancer Res. 15, 1853–1859.
Meyers, M., Wagner, M. W., Hwang, H.-S., Kinsella, T. J., and Boothman, D. A. (2001). Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 61, 5193–5201.
Seo, Y., Yan, T., Schupp, J. E., Colussi, V., Taylor, K. L., and Kinsella, T. J. (2004). Differential radiosensitization in DNA mismatch repair-proficient and’deficient human colon cancer xenografts with 5-iodo-2-pyrimidinone-2′-deoxyribose. Clin. Cancer Res. 10, 7520–8.
Wang, H., and Hays, J. B. (2000). Preparation of DNA substrates for in vitro mismatch repair. Mol. Biotechnol. 15, 97–104.
Wang, H., and Hays, J. B. (2001). Simple and rapid preparation of gapped plasmid DNA for incorporation of oligomers containing specific DNA lesions. Mol. Biotechnol. 19, 133–140.
Wang, H., and Hays, J. B. (2006). Construction of MMR plasmid substrates and analysis of MMR error correction and excision. Methods Mol. Biol. 314, 345–353.
Yan, T., Seo, Y., and Kinsella, T. J. (2009). Differential cellular responses to prolonged LDR-IR in MLH1-proficient and MLH1-deficient colorectal cancer HCT116 cells. Clin. Cancer Res. 15, 6912–6920.
York, S. J., and Modrich, P. (2006). Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. J. Biol. Chem. 281, 22674–22683.
Keywords: mismatch repair, mismatch substrates construction, iododeoxyuridine
Citation: Du W and Kinsella TJ (2011) A rapid, simple DNA mismatch repair substrate construction method. Front. Oncol. 1:8. doi: 10.3389/fonc.2011.00008
Received: 16 April 2011;
Accepted: 20 May 2011;
Published online: 06 June 2011.
Edited by:
Anatoly Dritschilo, Georgetown University School of Medicine, USAReviewed by:
Sean Collins, Georgetown University Hospital, USAJoel S. Greenberger, University of Pittsburgh Medical Center-Shadyside, USA
Copyright: © 2011 Du and Kinsella. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Timothy J. Kinsella, Department of Radiation Oncology, Rhode Island Hospital, Physicians Office Building, Suite 130, 110 Lockwood Street, Providence, RI 02903, USA. e-mail:dGtpbnNlbGxhQGxpZmVzcGFuLm9yZw==