 Marcello Maugeri-Saccà1
Marcello Maugeri-Saccà1

- 1 Department of Hematology, Oncology and Molecular Medicine, Istituto Superiore di Sanità, Rome, Italy
- 2 Mediterranean Institute of Oncology, Viagrande, Catania, Italy
Recent breakthroughs in translational oncology are opening new perspectives for the treatment of cancer. The advent of targeted therapies has provided the proof-of-concept to selectively turn-off deregulated oncogenic proteins, while the identification and validation of predictive biomarkers of response has allowed to improve, at least in some cases, their performance. Moreover, a subpopulation of tumor-propagating cells has been identified from many solid and hematological tumors. These cells share functional properties of normal stem cells, and are commonly referred to as cancer stem cells (CSCs). It is emerging that CSCs are defended against broadly used anticancer agents by means of different, partly interconnected, mechanisms. However, CSCs rely on specific pathways involved in self-renewal that can be pharmacologically antagonized by experimental molecular targeted agents, some of which have recently entered early phases of clinical development. Here, we discuss the spectrum of pharmacological strategies under clinical or preclinical development for CSCs targeting.
The Cancer Stem Cells Concept
The idea that cancer originates from stem cells traces back to the “embryonal rest theory,” asserting that cancer arises from embryonic remnants persisting in adult tissues. However, the “cancer stem cell model” has captured great interest only in recent years following the isolation of a rare cellular fraction of leukemia-initiating cells with stem cell-like features (Bonnet and Dick, 1997). Ever since, this concept has been corroborated by the isolation of cancer stem-like cells, commonly referred to as cancer stem cells (CSCs), from many solid tumors ranging from highly prevalent cancers (Al-Hajj et al., 2003; Ricci-Vitiani et al., 2007; Eramo et al., 2008) to less common neoplasms such as glioblastoma multiforme (Singh et al., 2003; Galli et al., 2004) and thyroid cancer (Todaro et al., 2010). This new paradigm implies that oncogenesis has its epicenter in a tissue-resident stem cell. Thus, a tumor is hierarchically organized, similar to adult tissues, with a CSC at the top of the pyramid that serves as a precursor of the whole population. The discovery of CSCs has questioned the “clonal evolution model” which, tracing its roots to the Darwinian evolutionary principle, postulated that different mutant clones acquire a survival advantage as a consequence of the natural competition with other clones. However, the hierarchical and clonal evolution models are not mutually exclusive as recently demonstrated by the genetic heterogeneity of cancer propagating cells, which suggests a clonal evolution within the stem cell pool (Anderson et al., 2011). It is conceivable that transformed stem cells maintain, although in a distorted manner, stem cell traits such as defensive ability against chemicals and mechanisms involved in self-renewal. Consistent with this, chemotherapy-induced death stimuli are constrained in a multifaceted way, such as through increased DNA repair ability and high expression of multidrug resistance (MDR) efflux pumps (Eyler and Rich, 2008). Conversely, the pharmacological inhibition of self-renewal-related pathways selectively depletes CSCs in different preclinical models (Bar et al., 2007; Hoey et al., 2009). Moreover, CSCs directly contribute to the tumor-supportive ability of the microenvironment by participating in tumor vasculogenesis through the direct differentiation into vascular cells (El Hallani et al., 2010; Ricci-Vitiani et al., 2010; Wang et al., 2010b; Soda et al., 2011). If many functional properties of CSCs are thought to account for the limited efficacy of chemotherapy, the refinement of knowledge on adult stem cells and their malignant counterparts is revealing unexpected ways for developing innovative anticancer agents (Figure 1).
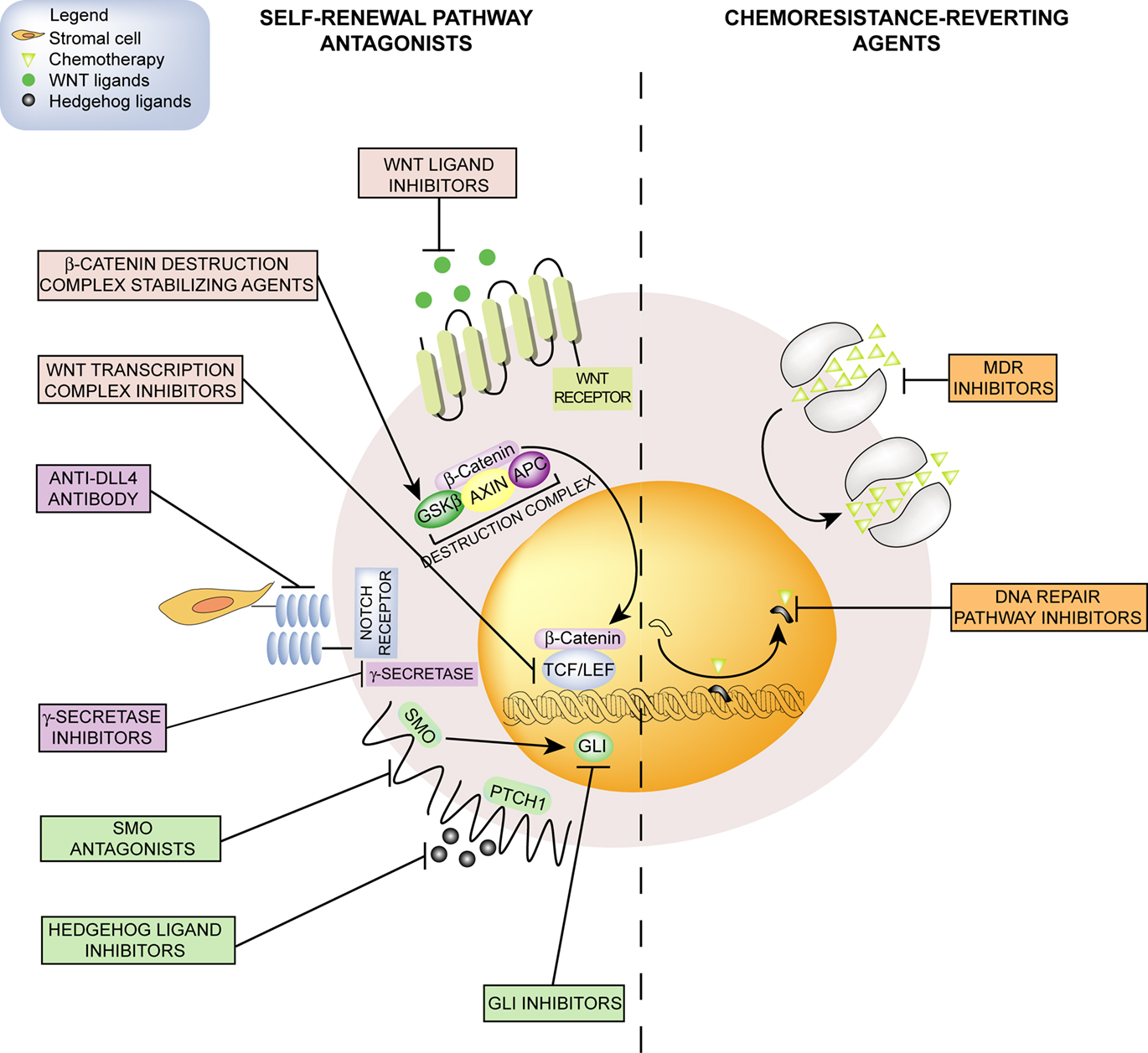
Figure 1. Emerging pharmacological strategies for CSCs targeting include self-renewal pathway antagonists and chemoresistance-reverting agents. Self-renewal-linked signals (left) such as Hedgehog, Notch, and Wnt/β-catenin pathways can be antagonized by molecular targeted agents including ligand-binding molecules, receptor antagonists, or agents inhibiting intracellular effectors. Chemosensitivity (right) could be restored by inhibiting MDR efflux pumps that extrude chemotherapeutic agents of natural origin, or by interfering with the DNA repair machinery that removes DNA-alkylating agent adducts. CSCs, cancer stem cells; GSKβ, glycogen synthase kinase 3β; APC, adenomatosis polyposis coli; TCF/LEF, T-cell transcription factor/lymphoid enhancer-binding factor; DLL4, delta-like ligand 4; SMO, smoothened; PTCH1, patched; GLI, glioma-associated oncogene homolog; MDR, multidrug resistance.
Strategies to Revert Chemoresistance
Adult stem cells maintain tissue function throughout life. To accomplish this function, stem cells are protected from endogenous or exogenous insults to avoid exhausting their replicative function. For instance, evidence indicates that adult stem cells survive cytotoxic injuries and then reconstitute the damaged tissue (Dekaney et al., 2009). Growing evidence indicates that CSCs possess similar stem cell properties that protect them against chemotherapy.
It is known that cancer cells improperly activate DNA repair pathways to overcome chemotherapy-induced cell death (Hoeijmakers, 2001). Key effectors of the DNA damage response machinery have been evaluated in clinical studies to determine the benefit of cancer patients from chemotherapy, even though the predictive value of DNA damage repair-linked biomarkers remains to be addressed (Vilmar and Sørensen, 2009). It has been demonstrated that glioblastoma stem-like cells (GBM-SCs) repair ionizing radiation-induced DNA lesions more readily that differentiated glioma cells through the activation of ataxia telangiectasia mutated (ATM) and checkpoint kinase 1 (Chk1; Bao et al., 2006). Likewise, both colon (Gallmeier et al., 2011) and lung (our unpublished data) CSCs aberrantly exploit the ATR/Chk1 axis to escape chemotherapy cytotoxicity, as demonstrated by the observation that Chk1 inhibition sensitized CSCs to different chemotherapeutic agents inducing mitotic catastrophe. Agents interfering with DNA repair have recently entered clinical development. The molecular background underlying the development of these compounds is a modality of gene–gene interaction known as synthetic lethality. According with this model, the co-occurrence of two events, the first genetic and the second pharmacological, results in the inhibition of two redundant pathways that finally lead to cell death. Poly-ADP ribose polymerase (PARP) inhibitors are the DNA repair-interfering agents at the most advanced stage of clinical development for the treatment of breast (Tutt et al., 2010) and ovarian (Audeh et al., 2010) cancers carrying BRCA1 or BRCA2 germline mutations and triple-negative breast cancer (O’Shaughnessy et al., 2011). The synthetic lethality concept could be also exploited for developing Chk1 inhibitors. When exposed to DNA-damaging agents, p53-deficient cells are unable to undergo G1 arrest and rely on Chk1 to activate cell cycle checkpoints (Zhou and Elledge, 2000). Thus, the pharmacological abrogation of Chk1 could selectively kill cancer cells with p53 defects. Chk1 inhibitors have recently entered clinical trials combined with different antiblastic compounds, although clinical data are not yet available.
DNA repair pathways compete with apoptotic signaling to decide the fate of damaged cells. However, CSCs display a tendency toward an anti-apoptotic state that favors cell survival following chemotherapy (Signore et al., 2011). For instance, it has been demonstrated that interleukin-4 (IL-4) is associated with the overexpression of anti-apoptotic mediators and induces a chemoresistant phenotype in colon CSCs (Todaro et al., 2007). Since IL-4 is overexpressed in many epithelial cancers (Todaro et al., 2008), it is conceivable that other types of CSCs exploit IL-4 as a defensive mechanism.
The combination of differentiation-inducing agents and chemotherapy can cure the majority of patients affected by acute promyelocytic leukemia (Sanz et al., 2004). The use of differentiation therapy may not be limited to leukemia. Recently, a randomized phase II trial demonstrated an increased response rate in non-small cell lung cancer patients when all-trans retinoic acid was associated with platinum-containing therapy (Arrieta et al., 2010). The identification of CSCs has fostered the identification of molecules with pro-differentiation effects. The pro-apoptotic/pro-differentiative bone morphogenetic protein 4 (BMP4) sensitizes colon CSCs to 5-fluorouracil and oxaliplatin, and eradicates CSC-derived tumors in animal models (Lombardo et al., 2011). Likewise, GBM-SCs exposed to BMP4 displayed reduced clonogenic ability coupled with an increased expression of neural differentiation markers (Piccirillo et al., 2006).
Next, both adult stem cells and CSCs may express high levels of MDR pumps (Moitra et al., 2011). This protective system extrudes from cancer cells a broad range of amphiphilic compounds including taxanes, anthracyclines, and vinca alkaloids. The ability to pump out different chemicals is currently exploited for the HOECHST dye efflux assay, a technique used for CSCs isolation that define them as side population (SP). Acute myeloid leukemia SP (Wulf et al., 2001) and neuroblastoma SP (Hirschmann-Jax et al., 2004) are characterized by a greater ability in extruding different chemotherapeutic agents compared with the non-SP. Moreover, long-term exposure of breast cancer cells to doxorubicin resulted in the acquisition of stem-like and chemoresistant properties (Calcagno et al., 2010), as documented by the overexpression of both self-renewal-related and MDR-related genes. Although first and second generation ABC inhibitors failed to demonstrate a clinical benefit, more potent third generation antagonists have been synthesized and are undergoing clinical development (Wu et al., 2008). However, it has been demonstrated that the SP does not always enrich for stem-like cells, at least when evaluating GBM cell lines and primary cells (Broadley et al., 2011), confirming previous findings indicating that chemoresistance of GBM-SCs is independent of the activity of ABC transporters (Eramo et al., 2006).
Quiescence physiologically defends adult stem cells against harmful insults and avoids the exhaustion of their replicative potential (Wilson et al., 2008). Likewise, CSCs usually exhibit a slow proliferation kinetics as demonstrated by label-retaining approaches, which revealed that label-retaining cells meet the operative criteria to be defined CSCs and survive chemotherapy, unlike the non-label-retaining population (Dembinski and Krauss, 2009; Gao et al., 2010). Since chemotherapy is active against rapidly dividing cells, it is conceivable that prolonged exit from cell cycle enables CSCs to survive chemo-radiotherapy. Moreover, it is known that a considerable percentage of patients with apparent organ-confined disease will experience distant recurrence years later radical surgery and adjuvant systemic therapy. Thus, it is conceivable that disseminated cancer cells are able to remain quiescent for years (“tumor dormancy”), thus making CSCs the ideal candidate for explaining this temporal pattern of recurrence. Hematological malignancies are representing the benchmark for the development of anticancer agents forcing dormant cancer cells to proceed through the cell cycle. Recent studies have revealed that some cytokines, such as interferon-alpha and granulocyte colony-stimulating factor, or arsenic trioxide efficiently promote cycling of dormant leukemic stem cells, thus representing a promising way for restoring chemosensitivity (Essers and Trumpp, 2010). Others drugs that could be exploited for inducing exit from quiescence are histone deacetylase inhibitors (HDACis). These compounds act at the epigenetic level producing different effects spanning from apoptosis to differentiation, and the first-in-class HDACi vorinostat has been approved for treating refractory cutaneous T-cell lymphoma (Lane and Chabner, 2009). It has been demonstrated that, unlike imatinib alone, HDACis combined with imatinib induce apoptosis in quiescent chronic myelogenous leukemia stem cells (Zhang et al., 2010). However, it is important to underline that chemotherapy and molecular targeted agents are significantly more active in hematological malignancies than in solid tumors. In this latter case, pharmacological strategies for breaking tumor dormancy should be carefully evaluated, especially when developed in the adjuvant setting in the attempt to eradicate minimal residual disease. Paradoxically, maintaining disseminated cancer cells in a quiescent state may represent an alternative way for achieving long-lasting recurrence-free interval in solid tumors, the priority goal in the adjuvant setting.
Targeting Self-Renewal Pathways
The aberrant activation of self-renewal-linked signals is thought to be the main determinant of CSCs fate. The Hedgehog (Hh), Notch, and Wnt/β-catenin are the most studied and characterized pathways.
Rationale and Strategies for Targeting the Hedgehog Pathway
The Hh pathway plays a crucial role during mammalian development and becomes later silenced in adult tissues (Ingham and McMahon, 2001). The interest on Hh in cancer biology comes from evidences demonstrating the inappropriate reactivation of its signaling in many tumors (Merchant and Matsui, 2010). In the absence of ligand stimulation the transmembrane-spanning receptor patched (PTCH1) maintains the pathway in the “off” state by repressing the activity of the serpentine receptor smoothened (SMO). Upon ligand binding, PTCH1 inhibition on SMO is relieved, allowing SMO to engage downstream effectors consisting in glioma-associated oncogene homolog (GLI) transcription factors. Target genes are involved in many cellular functions such as proliferation, survival, metastatization, and pathway auto-regulation.
The tumor-enhancing activity of aberrant Hh signaling occurs through different and tumor-specific modalities. Among these, the mutation-driven manner is the best characterized, resulting from the constitutive activation of the transduction machinery as the consequence of loss- or gain-of-function mutations in negative or positive controllers, respectively (Xie et al., 1997, 1998). Germline mutations in PTCH1 underpin the Gorlin syndrome (Hahn et al., 1996), a condition associated with the predisposition to develop multiple basal cell carcinoma. Moreover, many sporadic forms of basal cell carcinoma are characterized by a comparable mutation pattern (Daya-Grosjean and Couvé-Privat, 2005). Likewise, PTCH1 knockout mice revealed that aberrant Hh pathway activation causes unrestricted proliferation of cerebellar stem cells that results in the onset of medulloblastoma (Yang et al., 2008). Consistent with this, genome-wide expression profiles unveiled that up to one-third of medulloblastoma patients carry somatic mutations in SMO, PTCH1, or downstream effectors (Thompson et al., 2006). While activating/inactivating mutations sustain aberrant Hh activation in a niche of tumors, a dual paracrine model is probably the main modality of Hh activation in many cancers (Dierks et al., 2007; Yauch et al., 2008). Accordingly, the overproduction of Hh ligands by cancer cells activates the stromal compartment that responds with the production of growth factors. Alternatively, ligands secreted by stromal cells exert their activity on recipient cancer cells. To further complicate this picture, pathway trans-activation via the PI3K/AKT axis (Riobo et al., 2006), KRAS (Ji et al., 2007), and TGF-β (Dennler et al., 2007) has been documented.
Increasing evidence is connecting the Hh pathway with CSCs. The control of the self-renewal by Hh has been documented in chronic myeloid leukemia (Zhao et al., 2009) and, through the modulation of Bmi-1, in breast CSCs (Liu et al., 2006). In multiple myeloma a marked asymmetry has been reported in Hh pathway components distribution and sensitivity to pathway inhibition between B-cell-like progenitors and mature myelomatous plasma cells (Peacock et al., 2007). Moreover, Hh pathway inhibition significantly hampers GBM-SCs clonogenicity (Bar et al., 2007) and preferentially depletes pancreatic CSCs (Feldmann et al., 2007).
The first evidence suggesting that Hh signaling could be pharmacologically antagonized came from the identification of the teratogenic steroidal alkaloid cyclopamine (Chen et al., 2002). Subsequently, high-throughput screening of small-molecule libraries led to the identification of several SMO antagonists (Frank-Kamenetsky et al., 2002). The first-in-class oral SMO inhibitor GDC-0449 was initially tested in a phase I study enrolling 33 patients with locally advanced or metastatic basal cell carcinoma, demonstrating good activity and acceptable safety profile (Von Hoff et al., 2009). Moreover, a massive tumor regression has been reported in an adult patient with a plurimetastatic PTCH-mutant medulloblastoma, even though disease restaging revealed tumor progression associated with the onset of a resistance-conferring SMO mutation (Rudin et al., 2009). Despite this encouraging activity in tumors carrying mutations in Hh pathway effectors, a comparable efficacy did not emerge against tumors with wild-type Hh components. In fact, both a phase II randomized study comparing chemotherapy with or without GDC-0449 in metastatic colorectal cancer patients (Berlin et al., 2010) and a placebo-controlled trial with GDC-0449 as maintenance therapy in advanced ovarian cancer patients (Kaye et al., 2010) failed to reach the primary endpoint. It is worth considering that Hh pathway, when unaffected by somatic mutations, is activated in a paracrine manner and, therefore, its inhibition could lead to cytostatic, rather than cytotoxic, effects. Thus, it is arguable that the evaluation of parameters of rapid tumor response in proof-of-concept studies might underestimate the benefit from this class of drugs when evaluated in patients whose tumors display wild-type Hh effectors. Based on this assumption, the pharmacological inhibition of Hh could offer greater opportunities as adjuvant therapies in order to prevent distant recurrence. An attractive hypothesis could be that while during early phases of the natural history of tumors (i.e., the adjuvant setting) cancer cells may rely on a tumor-supportive microenvironment, later phases (i.e., the metastatic setting) could be biologically characterized by the self-sufficiency of cancer cells. Such acquired ability of cancer cells to thrive in a microenvironment-independent manner could result in insensitivity to molecular targeted agents acting by depriving cancer cells of paracrine-acting stimuli. Moreover, GDC-0449 pharmacokinetics is influenced by several factors, including solubility-limited absorption, slow rate of metabolic elimination and interaction with plasma proteins. In particular, both total level and genetic variants of the plasma protein alpha-1-acid glycoprotein (AAG) seem to account for the interindividual variability of GDC-0449 bioavailability (Graham et al., 2011), thus suggesting that alternative schedules should be investigated in clinical trials. The disappointing results from the studies mentioned above raise the need for alternative strategies of Hh inhibition. Since Hh transduction machinery converges on the GLI transcription factors, small-molecule inhibitors of GLI proteins (GANT61 and GANT58; Lauth et al., 2007) or, although less specific, indirect inhibitors of GLI activity (PI3K/AKT inhibitors) could allow a deeper inhibition of Hh signaling. Alternatively, ligand inhibition through neutralizing antibodies (5E1 antibody) or ligand-binding molecules (robotnikinin) have been proposed to avoid blocking Hh pathway in the whole spectrum of its developmental functions (Yauch et al., 2008; Stanton et al., 2009), which is of utmost importance when treating young patients. In summary, while Hh inhibition is emerging as a new opportunity in cancer therapy, early clinical data are conflicting and mirror the heterogeneity of mechanisms sustaining pathway activation. Thus, the tumor-dependent activity of Hh signaling has to be fully dissected for optimal clinical testing of Hh pathway inhibitors.
Rationale and Strategies for Targeting the Notch Pathway
Notch is a short-range-acting communication system that exerts its function via cell-to-cell contact. The developmental Notch pathway has been implicated in various pro-tumorigenic activities spanning from cell survival to motility (Koch and Radtke, 2007). In mammals, the pathway is composed by four transmembrane receptors (Notch 1–4) and five ligands [delta-like ligand (DLL) 1, 3, 4, Jagged 1, and 2]. The pairing of Notch ligand-receptor leads to conformational changes in the receptor that undergoes two sequential cleavages operated by the “A disintegrin and metallopeptidase” 10 and 17 and gamma-secretase enzymes. The resulting Notch intracellular domain (Nic) is released and translocates to the nucleus where it interacts with co-activators to finally generate the Notch transcriptional complex. Moreover, Notch communicates with other oncogenic signals including the NFkB pathway, the hypoxia sensor HIF1α and the estrogen receptor alpha (Pannuti et al., 2010). The four Notch paralogs are also endowed of tumor-suppressive properties. Notch 2 expression correlates with better prognosis in breast cancer (Parr et al., 2004), while in mesothelioma counteracts the pro-survival effects of Notch 1 (Graziani et al., 2008).
Breast cancer represents a benchmark for studying the influence of Notch pathway on CSCs. Consistent with this, a significant reduction of mammosphere-forming efficiency has been achieved with a gamma-secretase inhibitor (GSI) or a Notch 4-neutralizing antibody in ductal carcinoma in situ of the breast (Farnie et al., 2007). Since a similar effect was seen with the EGFR tyrosine kinase inhibitor gefitinib, it is conceivable that the dual inhibition of Notch and EGFR may exert a synergistic effect. The HER2 gene is amplified in approximately 20% of human breast cancers and, more recently, has been implicated in breast CSCs maintenance and expansion (Cicalese et al., 2009; Magnifico et al., 2009). Evidence connecting Notch with HER2 comes from the demonstration that the HER2 promoter contains Notch-binding sequences, while Notch signaling activation has been documented in HER2-overexpressing cells (Korkaya and Wicha, 2009). Notably, Notch inhibition through small interfering RNA or a GSI impaired mammosphere formation and determined HER2 down-regulation (Magnifico et al., 2009). Finally, human breast cancer mammospheres have been shown to acquire an hypoxia-resistant phenotype following Notch 3 induction by IL-6 (Sansone et al., 2007). Since cancer cells cultured under hypoxic condition gain stem-like properties (Li et al., 2009), it is conceivable that Notch pathway and the hypoxia-sensing machinery dynamically cooperate in controlling the CSC pool. Accordingly, GBM-SCs are characterized by high Notch activity that confers resistance to ionizing radiation, whereas Notch inhibition depleted GBM-SCs and reverted radioresistance (Fan et al., 2010; Wang et al., 2010a). GBM-SCs exposed to the differentiation-inducing agent retinoic acid underwent both growth arrest and expression of lineage-specific differentiation markers (Ying et al., 2011). Consistently, microarray analysis of retinoic acid-treated cells revealed a molecular profile characterized by the down-regulation of Notch components, while the constitutive activation of Nic rescued GBM-SCs from retinoic acid-induced differentiation and subsequent depletion. Finally, the Notch pathway has been also implicated in colon cancer. In this model, the inhibition of the DLL4 combined with irinotecan has been associated with a reduction of KRAS-mutant colon CSCs frequency (Fischer et al., 2011). Given that KRAS mutations are detectable in approximately 40% of colorectal tumors and confer resistance to anti-EGFR therapy, Notch antagonists should be exploited for improving the management of the large segment of patients unsuitable for cetuximab and panitumumab.
The inhibition of Notch pathway is undergoing early clinical trials aimed at determining the safety profile and optimal schedule of GSIs. Since hormone receptor-positive breast cancer relies on Notch activation when hormonal signaling are pharmacologically inhibited (Rizzo et al., 2008), different clinical trials are evaluating the combination of GSIs with anti-estrogen therapy. Moreover, a phase I study combining the GSI RO4929097 with carboplatin and paclitaxel as neoadjuvant therapy in stage II and III triple-negative breast cancer patients is currently enrolling. Triple-negative breast cancers harbor CSC-like characteristics and are deficient in DNA repair (Perou, 2011). Thus, this study will provide valuable information given the potential synergism between GSIs and DNA-damaging agents. Beside GSIs, anti-DLL4 agents are in phase I clinical development. Even though safety data are still unavailable, antibody-mediated inhibition of Notch pathway has been connected in preclinical models with the onset of vascular tumors and liver histopathological alterations such as sinusoidal dilation and centrilobular hepatocyte atrophy (Li et al., 2010; Yan et al., 2010). Liver gene expression of DLL4-treated mice revealed a significant up-regulation of endothelium-specific genes, indicating that DLL4 signaling is crucial for maintaining endothelial cells in a quiescent state. Similar alterations were noted with a Notch 1-specific inhibitory antibody and the GSI dibenzazepine, thus indicating that these changes represent a class effect of DLL4 inhibition. In summary, optimal clinical development of Notch antagonists must take into account the complexity of the pathway. In particular, the opposite effects of Notch receptors observed in different tumor types raise the need for more selective modalities of Notch inhibition.
Rationale and Strategies for Targeting the Wnt Pathway
The canonical Wnt/β-catenin signaling is by far the best characterized among Wnt pathways. This signaling is mainly regulated at the level of β-catenin, a protein maintained at low cytoplasmic concentration by a destruction complex. Upon activation of the Wnt pathway, the inhibition of the β-catenin destruction complex allows β-catenin to translocate to the nucleus, leading to the expression of target genes involved in several cellular processes encompassing proliferation, motility and stem cell maintenance (Moon et al., 2002).
The role of the Wnt pathway in cancer has been recognized since the Vogelstein’s “adenoma to carcinoma sequence,” which identified mutations in adenomatosis polyposis coli (APC) gene as a critical event during colorectal carcinogenesis. The most direct evidence linking the Wnt pathway with CSCs comes from a recent study demonstrating that high Wnt activity identifies the colon CSC population (Vermeulen et al., 2010). Moreover, myofibroblast-secreted factors instructed differentiated colon cancer cells to activate the β-catenin-dependent transcription, leading to the gain of a stem-like phenotype. Although in a less direct way, the increased nuclear level of β-catenin that characterizes the switch from chronic phase to blast crisis in chronic myelogenous leukemia suggests the influence of Wnt on leukemia stem cells (Jamieson et al., 2004).
A number of Wnt modulators or inhibitors have been identified (de Sousa et al., 2011). COX-2 inhibitors such as celecoxib and rofecoxib seem to exert modulatory activity on Wnt through a reduced production of prostaglandin E2, a molecule able to prevent the degradation of β-catenin. c-MET inhibitors have been also indicated as indirect Wnt antagonists. The rationale underlying the anti-Wnt activity of these compound comes from the mitigation of the activity of c-MET downstream machinery, which relieves the inhibition on glycogen synthase kinase 3β, a key enzyme of the destruction complex. Finally, high-throughput drug screening has allowed the identification of a variety of Wnt inhibitors acting at various level of the pathway, including compounds stabilizing key components of the destruction complex, transcription factor antagonists and molecules inhibiting Wnt secretion. However, the majority of direct Wnt antagonists are in preclinical development, and only few of these have recently entered phase I dose-finding studies.
Conclusion
Mounting evidence points to CSCs as responsible for tumor generation and treatment failure. Notwithstanding, the CSCs concept is still object of controversies. For instance, in some tumors a distinct repertoire of cell surface markers identifies cellular subsets that share stem-like features (Eramo et al., 2010). Moreover, the tumorigenic population in melanoma seems to be rather frequent (>25%) and reversible (Quintana et al., 2010). Regardless of current controversies on CSCs, a deeper understanding of molecular mechanisms governing the biology of tumorigenic cells should be considered a priority for optimal development of innovative anticancer drugs. It is now clear that traditional cancer cell lines generate tumors that markedly differ from the human disease, whereas the use of CSC-based tumor xenografts allows more reliable testing of anticancer agents by generating tumors that closely resemble parental human cancers (Baiocchi et al., 2010). It is conceivable that pharmacological inhibition of paracrine-acting pathways results in cytostatic effects correlated with the disruption of the tumor-microenvironment interplay. Thus, self-renewal pathway antagonists could be more effective in targeting microscopic residual disease rather than bulk tumors. As a result, clinical development of these compounds should be carried out in appropriate patient populations such as the adjuvant or maintenance setting. Taking into account the complex modalities of activation of these pathways, the identification of predictive biomarkers remains a challenge, ideally requiring biomarker measurement within a microenvironmental context. It is also worth considering that the therapeutic potential of self-renewal pathway antagonists could be counterbalanced by potential serious adverse events due to interference with crucial mechanisms of tissue homeostasis. Therefore, a deeper understanding of adult stem cell biology is required to determine a therapeutic window for anti-CSC agents. Finally, CSC-related endpoints should be included in proof-of-principle clinical trials. To this end, the neoadjuvant setting offers the opportunity to evaluate the CSC pool, whose qualitative and quantitative modifications could be correlated with long-term outcomes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Giuseppe Loreto for technical assistance.
References
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., and Clarke, M. F. (2003). Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 3983–3988.
Anderson, K., Lutz, C., van Delft, F. W., Bateman, C. M., Guo, Y., Colman, S. M., Kempski, H., Moorman, A. V., Titley, I., Swansbury, J., Kearney, L., Enver, T., and Greaves, M. (2011). Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469, 356–361.
Arrieta, O., González-De la Rosa, C. H., Aréchaga-Ocampo, E., Villanueva-Rodríguez, G., Cerón-Lizárraga, T. L., Martínez-Barrera, L., Vázquez-Manríquez, M. E., Ríos-Trejo, M. A., Alvarez-Avitia, M. A., Hernández-Pedro, N., Rojas-Marín, C., and De la Garza, J. (2010). Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 28, 3463–3471.
Audeh, M. W., Carmichael, J., Penson, R. T., Friedlander, M., Powell, B., Bell-McGuinn, K. M., Scott, C., Weitzel, J. N., Oaknin, A., Loman, N., Lu, K., Schmutzler, R. K., Matulonis, U., Wickens, M., and Tutt, A. (2010). Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376, 245–251.
Baiocchi, M., Biffoni, M., Ricci-Vitiani, L., Pilozzi, E., and De Maria, R. (2010). New models for cancer research: human cancer stem cell xenografts. Curr. Opin. Pharmacol. 10, 380–384.
Bao, S., Wu, Q., McLendon, R. E., Hao, Y., Shi, Q., Hjelmeland, A. B., Dewhirst, M. W., Bigner, D. D., and Rich, J. N. (2006). Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760.
Bar, E. E., Chaudhry, A., Lin, A., Fan, X., Schreck, K., Matsui, W., Piccirillo, S., Vescovi, A. L., DiMeco, F., Olivi, A., and Eberhart, C. G. (2007). Cyclopamine-mediated Hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 25, 2524–2533.
Berlin, J., Bendell, J., Hart, L. L., Firdaus, I., Gore, I., Hermann, R. C., Mackey, H., Yauch, B., Graham, R. A., and Bray, G. L. (2010). A phase 2, randomized, double-blind, placebo-controlled study of hedgehog pathway inhibitor (HPI) GDC-0449 in patients with previously untreated metastatic colorectal cancer (MCRC). Ann. Oncol. 21(Suppl. 8), Abstract LBA21.
Bonnet, D., and Dick, J. E. (1997). Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, 730–737.
Broadley, K. W., Hunn, M. K., Farrand, K. J., Price, K. M., Grasso, C., Miller, R. J., Hermans, I. F., and McConnell, M. J. (2011). Side population is not necessary or sufficient for a cancer stem cell phenotype in glioblastoma multiforme. Stem Cells 29, 452–461.
Calcagno, A. M., Salcido, C. D., Gillet, J. P., Wu, C. P., Fostel, J. M., Mumau, M. D., Gottesman, M. M., Varticovski, L., and Ambudkar, S. V. (2010). Prolonged drug selection of breast cancer cells and enrichment of cancer stem cell characteristics. J. Natl. Cancer Inst. 102, 1637–1652.
Chen, J. K., Taipale, J., Cooper, M. K., and Beachy, P. A. (2002). Inhibition of hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev. 16, 2743–2748.
Cicalese, A., Bonizzi, G., Pasi, C. E., Faretta, M., Ronzoni, S., Giulini, B., Brisken, C., Minucci, S., Di Fiore, P. P., and Pelicci, P. G. (2009). The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell 138, 1083–1095.
Daya-Grosjean, L., and Couvé-Privat, S. (2005). Sonic hedgehog signaling in basal cell carcinomas. Cancer Lett. 225, 181–192.
Dekaney, C. M., Gulati, A. S., Garrison, A. P., Helmrath, M. A., and Henning, S. J. (2009). Regeneration of intestinal stem/progenitor cells following doxorubicin treatment of mice. Am. J. Physiol. Gastrointest. Liver Physiol. 297, 461–470.
Dembinski, J. L., and Krauss, S. (2009). Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 26, 611–623.
Dennler, S., André, J., Alexaki, I., Li, A., Magnaldo, T., ten Dijke, P., Wang, X. J., Verrecchia, F., and Mauviel, A. (2007). Induction of sonic Hedgehog mediators by transforming growth factor-β: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 67, 6981–6696.
de Sousa, E. M., Vermeulen, L., Richel, D., and Medema, J. P. (2011). Targeting Wnt signaling in colon cancer stem cells. Clin. Cancer Res. 17, 647–653.
Dierks, C., Grbic, J., Zirlik, K., Beigi, R., Englund, N. P., Guo, G. R., Veelken, H., Engelhardt, M., Mertelsmann, R., Kelleher, J. F., Schultz, P., and Warmuth, M. (2007). Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 13, 944–951.
El Hallani, S., Boisselier, B., Peglion, F., Rousseau, A., Colin, C., Idbaih, A., Marie, Y., Mokhtari, K., Thomas, J. L., Eichmann, A., Delattre, J. Y., Maniotis, A. J., and Sanson, M. (2010). A new alternative mechanism in glioblastoma vascularization: tubular vasculogenic mimicry. Brain 133, 973–982.
Eramo, A., Haas, T. L., and De Maria, R. (2010). Lung cancer stem cells: tools and targets to fight lung cancer. Oncogene 29, 4625–4635.
Eramo, A., Lotti, F., Sette, G., Pilozzi, E., Biffoni, M., Di Virgilio, A., Conticello, C., Ruco, L., Peschle, C., and De Maria, R. (2008). Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 15, 504–514.
Eramo, A., Ricci-Vitiani, L., Zeuner, A., Pallini, R., Lotti, F., Sette, G., Pilozzi, E., Larocca, L. M., Peschle, C., and De Maria, R. (2006). Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 13, 1238–1241.
Essers, M. A., and Trumpp, A. (2010). Targeting leukemic stem cells by breaking their dormancy. Mol. Oncol. 4, 443–450.
Eyler, C. E., and Rich, J. N. (2008). Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 26, 2839–2845.
Fan, X., Khaki, L., Zhu, T. S., Soules, M. E., Talsma, C. E., Gul, N., Koh, C., Zhang, J., Li, Y. M., Maciaczyk, J., Nikkhah, G., Dimeco, F., Piccirillo, S., Vescovi, A. L., and Eberhart, C. G. (2010). Notch pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28, 5–16.
Farnie, G., Clarke, R. B., Spence, K., Pinnock, N., Brennan, K., Anderson, N. G., and Bundred, N. J. (2007). Novel cell culture technique for primary ductal carcinoma in situ: role of Notch and epidermal growth factor receptor signaling pathways. J. Natl. Cancer Inst. 99, 616–627.
Feldmann, G., Dhara, S., Fendrich, V., Bedja, D., Beaty, R., Mullendore, M., Karikari, C., Alvarez, H., Iacobuzio-Donahue, C., Jimeno, A., Gabrielson, K. L., Matsui, W., and Maitra, A. (2007). Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: a new paradigm for combination therapy in solid cancers. Cancer Res. 67, 2187–2196.
Fischer, M., Yen, W. C., Kapoun, A. M., Wang, M., O’Young, G., Lewicki, J., Gurney, A., and Hoey, T. (2011). Anti-DLL4 inhibits growth and reduces tumor-initiating cell frequency in colorectal tumors with oncogenic KRAS mutations. Cancer Res. 71, 1520–1525.
Frank-Kamenetsky, M., Zhang, X. M., Bottega, S., Guicherit, O., Wichterle, H., Dudek, H., Bumcrot, D., Wang, F. Y., Jones, S., Shulok, J., Rubin, L. L., and Porter, J. A. (2002). Small-molecule modulators of hedgehog signaling: identification and characterization of smoothened agonists and antagonists. J. Biol. 1, 10.
Galli, R., Binda, E., Orfanelli, U., Cipelletti, B., Gritti, A., De Vitis, S., Fiocco, R., Foroni, C., Dimeco, F., and Vescovi, A. (2004). Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 64, 7011–7021.
Gallmeier, E., Hermann, P. C., Mueller, M. T., Machado, J. G., Ziesch, A., De Toni, E. N., Palagyi, A., Eisen, C., Ellwart, J. W., Rivera, J., Rubio-Viqueira, B., Hidalgo, M., Bunz, F., Göke, B., and Heeschen, C. (2011). Inhibition of ataxia telangiectasia- and Rad3-related function abrogates the in vitro and in vivo tumorigenicity of human colon cancer cells through depletion of the CD133(+) tumor-initiating cell fraction. Stem Cells 29, 418–429.
Gao, M. Q., Choi, Y. P., Kang, S., Youn, J. H., and Cho, N. H. (2010). CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene 29, 2672–2680.
Graham, R. A., Lum, B. L., Cheeti, S., Jin, J. Y., Jorga, K., Von Hoff, D. D., Rudin, C. M., Reddy, J. C., Low, J. A., and Lorusso, P. M. (2011). Pharmacokinetics of Hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors: the role of alpha-1-acid glycoprotein binding. Clin. Cancer Res. 17, 2512–2520.
Graziani, I., Eliasz, S., De Marco, M. A., Chen, Y., Pass, H. I., De May, R. M., Strack, P. R., Miele, L., and Bocchetta, M. (2008). Opposite effects of Notch-1 and Notch-2 on mesothelioma cell survival under hypoxia are exerted through the Akt pathway. Cancer Res. 68, 9678–9685.
Hahn, H., Wicking, C., Zaphiropoulous, P. G., Gailani, M. R., Shanley, S., Chidambaram, A., Vorechovsky, I., Holmberg, E., Unden, A. B., Gillies, S., Negus, K., Smyth, I., Pressman, C., Leffell, D. J., Gerrard, B., Goldstein, A. M., Dean, M., Toftgard, R., Chenevix-Trench, G., Wainwright, B., and Bale, A. E. (1996). Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 85, 841–851.
Hirschmann-Jax, C., Foster, A. E., Wulf, G. G., Nuchtern, J. G., Jax, T. W., Gobel, U., Goodell, M. A., and Brenner, M. K. (2004). A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. U.S.A. 101, 14228–14233.
Hoeijmakers, J. H. (2001). Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374.
Hoey, T., Yen, W. C., Axelrod, F., Basi, J., Donigian, L., Dylla, S., Fitch-Bruhns, M., Lazetic, S., Park, I. K., Sato, A., Satyal, S., Wang, X., Clarke, M. F., Lewicki, J., and Gurney, A. (2009). DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 5, 168–177.
Ingham, P. W., and McMahon, A. P. (2001). Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 15, 3059–3087.
Jamieson, C. H., Weissman, I. L., and Passegue, E. (2004). Chronic versus acute myelogenous leukemia: a question of self-renewal. Cancer Cell 6, 531–533.
Ji, Z., Mei, F. C., Xie, J., and Cheng, X. (2007). Oncogenic KRAS activates Hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 282, 14048–14055.
Kaye, S. B., Fehrenbacher, L., Holloway, R., Horowitz, N., Karlan, B., Amit, A., Slomovitz, B., Chang, I., Yauch, R. L., and Reddy, J. C. (2010). A phase 2, randomized, placebo-controlled study of Hedgehog (HH) pathway inhibitor GDC-0449 as maintenance therapy in patients with ovarian cancer in 2nd or 3rd complete remission (CR). Ann. Oncol. 21(Suppl. 8), Abstract LBA25.
Koch, U., and Radtke, F. (2007). Notch and cancer: a double-edged sword. Cell. Mol. Life Sci. 64, 2746–2762.
Korkaya, H., and Wicha, M. S. (2009). HER-2, notch, and breast cancer stem cells: targeting an axis of evil. Clin. Cancer Res. 15, 1845–1847.
Lane, A. A., and Chabner, B. A. (2009). Histone deacetylase inhibitors in cancer therapy. J. Clin. Oncol. 27, 5459–5468.
Lauth, M., Bergström, A., Shimokawa, T., and Toftgård, R. (2007). Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. U.S.A. 104, 8455–8460.
Li, J. L., Jubb, A. M., and Harris, A. L. (2010). Targeting DLL4 in tumors shows preclinical activity but potentially significant toxicity. Future Oncol. 6, 1099–1103.
Li, Z., Bao, S., Wu, Q., Wang, H., Eyler, C., Sathornsumetee, S., Shi, Q., Cao, Y., Lathia, J., McLendon, R. E., Hjelmeland, A. B., and Rich, J. N. (2009). Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513.
Liu, S., Dontu, G., Mantle, I. D., Patel, S., Ahn, N. S., Jackson, K. W., Suri, P., and Wicha, M. S. (2006). Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 66, 6063–6071.
Lombardo, Y., Scopelliti, A., Cammareri, P., Todaro, M., Iovino, F., Ricci-Vitiani, L., Gulotta, G., Dieli, F., De Maria, R., and Stassi, G. (2011). Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology 140, 297–309.
Magnifico, A., Albano, L., Campaner, S., Delia, D., Castiglioni, F., Gasparini, P., Sozzi, G., Fontanella, E., Menard, S., and Tagliabue, E. (2009). Tumour-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are trastuzumab sensitive. Clin. Cancer Res. 15, 2010–2021.
Merchant, A. A., and Matsui, W. (2010). Targeting Hedgehog – a cancer stem cell pathway. Clin. Cancer Res. 16, 3130–3140.
Moitra, K., Lou, H., and Dean, M. (2011). Multidrug efflux pumps and cancer stem cells: insights into multidrug resistance and therapeutic development. Clin. Pharmacol. Ther. 89, 491–502.
Moon, R. T., Bowerman, B., Boutros, M., and Perrimon, N. (2002). The promise and perils of Wnt signaling through β-catenin. Science 296, 1644–1646.
O’Shaughnessy, J., Osborne, C., Pippen, J. E., Yoffe, M., Patt, D., Rocha, C., Koo, I. C., Sherman, B. M., and Bradley, C. (2011). Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 364, 205–214.
Pannuti, A., Foreman, K., Rizzo, P., Osipo, C., Golde, T., Osborne, B., and Miele, L. (2010). Targeting Notch to target cancer stem cells. Clin. Cancer Res. 16, 3141–3152.
Parr, C., Watkins, G., and Jiang, W. G. (2004). The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int. J. Mol. Med. 14, 779–786.
Peacock, C. D., Wang, Q., Gesell, G. S., Corcoran-Schwartz, I. M., Jones, E., Kim, J., Devereux, W. L., Rhodes, J. T., Huff, C. A., Beachy, P. A., Watkins, D. N., and Matsui, W. (2007). Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. U.S.A. 104, 4048–4053.
Perou, C. M. (2011). Molecular stratification of triple-negative breast cancers. Oncologist 16(Suppl.), 61–70.
Piccirillo, S. G., Reynolds, B. A., Zanetti, N., Lamorte, G., Binda, E., Broggi, G., Brem, H., Olivi, A., Dimeco, F., and Vescovi, A. L. (2006). Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 444, 761–765.
Quintana, E., Shackleton, M., Foster, H. R., Fullen, D. R., Sabel, M. S., Johnson, T. M., and Morrison, S. J. (2010). Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 18, 510–523.
Ricci-Vitiani, L., Lombardi, D. G., Pilozzi, E., Biffoni, M., Todaro, M., Peschle, C., and De Maria, R. (2007). Identification and expansion of human colon-cancer-initiating cells. Nature 445, 111–115.
Ricci-Vitiani, L., Pallini, R., Biffoni, M., Todaro, M., Invernici, G., Cenci, T., Maira, G., Parati, E. A., Stassi, G., Larocca, L. M., and De Maria, R. (2010). Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468, 824–828.
Riobo, N. A., Lu, K., Ai, X., Haines, G. M., and Emerson, C. P. Jr. (2006). Phosphoinositide 3-kinase and AKT are essential for sonic Hedgehog signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 4505–4510.
Rizzo, P., Miao, H., D’Souza, G., Osipo, C., Song, L. L., Yun, J., Zhao, H., Mascarenhas, J., Wyatt, D., Antico, G., Hao, L., Yao, K., Rajan, P., Hicks, C., Siziopikou, K., Selvaggi, S., Bashir, A., Bhandari, D., Marchese, A., Lendahl, U., Qin, J. Z., Tonetti, D. A., Albain, K., Nickoloff, B. J., and Miele, L. (2008). Cross-talk between notch and the estrogen receptor in breast cancer suggests novel therapeutic approaches. Cancer Res. 68, 5226–5235.
Rudin, C. M., Hann, C. L., Laterra, J., Yauch, R. L., Callahan, C. A., Fu, L., Holcomb, T., Stinson, J., Gould, S. E., Coleman, B., LoRusso, P. M., Von Hoff, D. D., de Sauvage, F. J., and Low, J. A. (2009). Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 36, 1173–1178.
Sansone, P., Storci, G., Tavolari, S., Guarnieri, T., Giovannini, C., Taffurelli, M., Ceccarelli, C., Santini, D., Paterini, P., Marcu, K. B., Chieco, P., and Bonafè, M. (2007). IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Invest. 117, 3988–4002.
Sanz, M. A., Martin, G., Gonzalez, M., León, A., Rayón, C., Rivas, C., Colomer, D., Amutio, E., Capote, F. J., Milone, G. A., De La Serna, J., Román, J., Barragán, E., Bergua, J., Escoda, L., Parody, R., Negri, S., Calasanz, M. J., and Bolufer, P. (2004). Risk-adapted treatment of acute promyelocytic leukemia with all-trans-retinoic acid and anthracycline monochemotherapy: a multicenter study by the PETHEMA group. Blood 103, 1237–1243.
Signore, M., Ricci-Vitiani, L., and De Maria, R. (2011). Targeting apoptosis pathways in cancer stem cells. Cancer Lett. doi: 10.1016/j.canlet.2011.01.013 [Epub ahead of print].
Singh, S. K., Clarke, I. D., Terasaki, M., Bonn, V. E., Hawkins, C., Squire, J., and Dirks, P. B. (2003). Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828.
Soda, Y., Marumoto, T., Friedmann-Morvinski, D., Soda, M., Liu, F., Michiue, H., Pastorino, S., Yang, M., Hoffman, R. M., Kesari, S., and Verma, I. M. (2011). Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 108, 4274–4280.
Stanton, B. Z., Peng, L. F., Maloof, N., Nakai, K., Wang, X., Duffner, J. L., Taveras, K. M., Hyman, J. M., Lee, S. W., Koehler, A. N., Chen, J. K., Fox, J. L., Mandinova, A., and Schreiber, S. L. (2009). A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat. Chem. Biol. 5, 154–156.
Thompson, M. C., Fuller, C., Hogg, T. L., Dalton, J., Finkelstein, D., Lau, C. C., Chintagumpala, M., Adesina, A., Ashley, D. M., Kellie, S. J., Taylor, M. D., Curran, T., Gajjar, A., and Gilbertson, R. J. (2006). Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J. Clin. Oncol. 24, 1924–1931.
Todaro, M., Iovino, F., Eterno, V., Cammareri, P., Gambara, G., Espina, V., Gulotta, G., Dieli, F., Giordano, S., De Maria, R., and Stassi, G. (2010). Tumorigenic and metastatic activity of human thyroid cancer stem cells. Cancer Res. 70, 8874–8885.
Todaro, M., Lombardo, Y., Francipane, M. G., Alea, M. P., Cammareri, P., Iovino, F., Di Stefano, A. B., Di Bernardo, C., Agrusa, A., Condorelli, G., Walczak, H., and Stassi, G. (2008). Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived interleukin-4. Cell Death Differ. 15, 762–772.
Todaro, M., Perez Alea, M., Di Stefano, A. B., Cammareri, P., Vermeulen, L., Iovino, F., Tripodo, C., Russo, A., Gulotta, G., Medema, J. P., and Stassi, G. (2007). Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 1, 389–402.
Tutt, A., Robson, M., Garber, J. E., Domchek, S. M., Audeh, M. W., Weitzel, J. N., Friedlander, M., Arun, B., Loman, N., Schmutzler, R. K., Wardley, A., Mitchell, G., Earl, H., Wickens, M., and Carmichael, J. (2010). Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376, 235–244.
Vermeulen, L., De Sousa E Melo, F., van der Heijden, M., Cameron, K., de Jong, J. H., Borovski, T., Tuynman, JB., Todaro, M., Merz, C., Rodermond, H., Sprick, M. R., Kemper, K., Richel, D. J., Stassi, G., and Medema, J. P. (2010). Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 12, 468–476.
Vilmar, A., and Sørensen, J. B. (2009). Excision repair cross-complementation group 1 (ERCC1) in platinum-based treatment of non-small cell lung cancer with special emphasis on carboplatin: a review of current literature. Lung Cancer 64, 131–139.
Von Hoff, D. D., LoRusso, P. M., Rudin, C. M., Reddy, J. C., Yauch, R. L., Tibes, R., Weiss, G. J., Borad, M. J., Hann, C. L., Brahmer, J. R., Mackey, H. M., Lum, B. L., Darbonne, W. C., Marsters, J. C. Jr., de Sauvage, F. J., and Low, J. A. (2009). Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 361, 1164–1172.
Wang, J., Wakeman, T. P., Lathia, J. D., Hjelmeland, A. B., Wang, X. F., White, R. R., Rich, J. N., and Sullenger, B. A. (2010a). Notch promotes radioresistance of glioma stem cells. Stem Cells 28, 17–28.
Wang, R., Chadalavada, K., Wilshire, J., Kowalik, U., Hovinga, K. E., Geber, A., Fligelman, B., Leversha, M., Brennan, C., and Tabar, V. (2010b). Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468, 829–833.
Wilson, A., Laurenti, E., Oser, G., van der Wath, R. C., Blanco-Bose, W., Jaworski, M., Offner, S., Dunant, C. F., Eshkind, L., Bockamp, E., Lió, P., Macdonald, H. R., and Trumpp, A. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129.
Wu, C. P., Calcagno, A. M., and Ambudkar, S. V. (2008). Reversal of ABC drug transporter-mediated multidrug resistance in cancer cells: evaluation of current strategies. Curr. Mol. Pharmacol. 1, 93–105.
Wulf, G. G., Wang, R. Y., Kuehnle, I., Weidner, D., Marini, F., Brenner, M. K., Andreeff, M., and Goodell, M. A. (2001). A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood 98, 1166–1173.
Xie, J., Johnson, R. L., Zhang, X., Bare, J. W., Waldman, F. M., Cogen, P. H., Menon, A. G., Warren, R. S., Chen, L. C., Scott, M. P., and Epstein, E. H. Jr. (1997). Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. 57, 2369–2372.
Xie, J., Murone, M., Luoh, S. M., Ryan, A., Gu, Q., Zhang, C., Bonifas, J. M., Lam, C. W., Hynes, M., Goddard, A., Rosenthal, A., Epstein, E. H. Jr., and de Sauvage, F. J. (1998). Activating smoothened mutations in sporadic basal-cell carcinoma. Nature 391, 90–92.
Yan, M., Callahan, C. A., Beyer, J. C., Allamneni, K. P., Zhang, G., Ridgway, J. B., Niessen, K., and Plowman, G. D. (2010). Chronic DLL4 blockade induces vascular neoplasms. Nature 463, E6–E7.
Yang, Z. J., Ellis, T., Markant, S. L., Read, T. A., Kessler, J. D., Bourboulas, M., Schüller, U., Machold, R., Fishell, G., Rowitch, D. H., Wainwright, B. J., and Wechsler-Reya, R. J. (2008). Medulloblastoma can be initiated by deletion of patched in lineage-restricted progenitors or stem cells. Cancer Cell 14, 135–145.
Yauch, R. L., Gould, S. E., Scales, S. J., Tang, T., Tian, H., Ahn, C. P., Marshall, D., Fu, L., Januario, T., Kallop, D., Nannini-Pepe, M., Kotkow, K., Marsters, J. C., Rubin, L. L., and de Sauvage, F. J. (2008). A paracrine requirement for hedgehog signaling in cancer. Nature 455, 406–410.
Ying, M., Wang, S., Sang, Y., Sun, P., Lal, B., Goodwin, C. R., Guerrero-Cazares, H., Quinones-Hinojosa, A., Laterra, J., and Xia, S. (2011). Regulation of glioblastoma stem cells by retinoic acid: role for Notch pathway inhibition. Oncogene. doi: 10.1038/onc.2011.58 [Epub ahead of print].
Zhang, B., Strauss, A. C., Chu, S., Li, M., Ho, Y., Shiang, K. D., Snyder, D. S., Huettner, C. S., Shultz, L., Holyoake, T., and Bhatia, R. (2010). Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell 17, 427–442.
Zhao, C., Chen, A., Jamieson, C. H., Fereshteh, M., Abrahamsson, A., Blum, J., Kwon, H. Y., Kim, J., Chute, J. P., Rizzieri, D., Munchhof, M., VanArsdale, T., Beachy, P. A., and Reya, T. (2009). Hedgehog signaling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776–779.
Keywords: cancer stem cells, chemoresistance, self-renewal pathways, molecular targeted agents
Citation: Maugeri-Saccà M, Zeuner A and De Maria R (2011) Therapeutic targeting of cancer stem cells. Front. Oncol. 1:10. doi: 10.3389/fonc.2011.00010
Received: 18 April 2011; Paper pending published: 10 May 2011;
Accepted: 09 June 2011; Published online: 17 June 2011.
Edited by:
Silvia Giordano, University of Torino, ItalyReviewed by:
Justin Lathia, Lerner Research Institute, USASaverio Minucci, European Institute of Oncology, Italy
Copyright: © 2011 Maugeri-Saccà, Zeuner and De Maria. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Ruggero De Maria, Department of Hematology, Oncology and Molecular Medicine, Istituto Superiore di Sanità, Viale Regina Elena 299, 00161 Rome, Italy. e-mail:cnVnZ2Vyby5kZW1hcmlhQGlzcy5pdA==