 Rajani Choudhuri
Rajani Choudhuri

- Radiation Biology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA
Purpose: To observe the effect of guggulsterone (GS) on the radiation response in human cancer cell lines. Materials and methods: The radiation response of cancer cells treated with GS was observed by cell survival studies, cell growth assay, NF-κB activity assay, western blotting of some key growth promoting receptors, the DNA repair protein γH2AX, and flow cytometry for DNA analyses. Results: GS inhibited radiation induced NF-κB activation and enhanced radiosensitivity in the pancreatic cell line, PC-Sw. It reduced both cell cycle movement and cell growth. GS reduced ERα protein in MCF7 cells and IGF1-Rβ protein in colon cancer cells and pancreatic cancer cells and inhibited DNA double strand break (DSB) repair following radiation. Conclusion: GS induced radiation sensitization may be due to several different mechanisms including the inhibition of NF-κB activation and reductions in IGF1-Rβ. In addition, GS induced γH2AX formation, primarily in the S-phase, indicates that DNA DSB’s in the S-phase may be another reason for GS induced radiosensitivity. ERα down-regulation in response to GS suggests that it can be of potential use in the treatment of estrogen positive tumors that are resistant to tamoxifen.
Introduction
A major goal in cancer therapy has been to identify drugs that are selectively cytotoxic to tumor cells in order to increase tumor control and lower the toxicity associated with normal tissue damage (Hait and Hambley, 2009; Hambley and Hait, 2009; Ma and Adjei, 2009). Alternatively another approach has been to identify agents, while not curative, may slow or even halt tumor growth over a long period of time, essentially turning cancer into a chronic and manageable disease. For this latter goal there has been renewed interest in natural products that may be less toxic to cells but still have important actions on blocking pro-survival/growth signal transduction pathways. One such natural product is derived from an extract from guggul tree from India containing the compound, guggulsterone (GS; Shishodia et al., 2008). GS is a derivative of the resin of guggul tree Commiphora mukul and has been widely reported as a hypolipidemic agent (Nityanand et al., 1989; Shishodia et al., 2008). Numerous animal and clinical studies have indicated its potential as a therapeutic agent for dyslipidemia (Dixit et al., 1980; Chander et al., 1996). Antagonism of the farnesoid X receptor (FXR) has been suggested as the mechanism for the lipid lowering action of GS (Urizar et al., 2002). It has also been proposed that GS can bind to several steroid receptors at a higher affinity than to FXR (Burris et al., 2005). GS has also been found to activate estrogen receptor alpha (Erα), progesterone receptor (PR), and pregnane X receptor (PXR; Brobst et al., 2004).
Interestingly, GS inhibits activation of NF-κB and decreases the expression of anti-apoptotic, angiogenic, and metastasis promoting proteins (Shishodia and Aggarwal, 2004; Lv et al., 2008; Xiao and Singh, 2008). GS has been also reported to suppress the constitutive activation of NF-κB in tumor cells (Shishodia and Aggarwal, 2004). Activation of pro-survival pathways leading to inhibition of apoptosis can have effects on the radiosensitization of cells (Chautard et al., 2010). One such activator or pro-survival pathways is NF-κB which can be induced by ionizing radiation (IR; Li and Karin, 1998). Given the correlation between NF-κB activation with radiation and inhibition by GS we investigated the effect of GS on the radiosensitivity of human tumor cell lines. GS was found to down-regulate IR-induced activation of NF-κB and enhance the radiosensitivity of four human tumor cell lines. Further, GS was shown to inhibit cell growth and inhibit IR-induced DNA damage repair. These findings warrant further research toward evaluation of GS as IR modifier for potential clinical applications.
Materials and Methods
Reagents
Guggulsterone was obtained from Steraloids, Inc. (Newport, RI, USA) and dissolved in DMSO at a concentration of 25 mM. Mouse monoclonal ER alpha and rabbit polyclonal ER beta antibodies were from LabVision Corp. (Fremont, CA, USA); rabbit polyclonal IGF-1Rβ, p21 antibodies, and PARP-1 (F-2) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA); mouse monoclonal anti-phospho histone H2AX (Ser139), clone JBW301, and rabbit antiserum to histone H2A (acidic patch) were from Upstate Cell Signaling Solutions (Temecula, CA, USA). Mouse anti-actin antibody was purchased from Chemicon Intl. (Temecula, CA, USA). All the HRP linked secondary antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Goat anti-mouse Alexa Fluor 488 secondary antibody was purchased from Invitrogen (Carlsbad, CA, USA).
Cell Survival Studies
HT29 (human colon carcinoma), MCF7 (human breast carcinoma), MIA PaCa-2 (human pancreatic carcinoma), and Bx-PC3 (human pancreatic carcinoma) cells were obtained from American Type Culture Collection (Rockville, MD, USA). PC-Sw (human pancreatic adenocarcinoma) cells were obtained from Dr. William Sindelar (Liebmann et al., 1994). All cell lines were grown in RPMI 1640 supplemented with 10% fetal bovine serum, 100 units/ml penicillin and 50 μg/ml streptomycin. Stock cultures of cells were maintained in exponential growth in an incubator at 37°C in a humidified atmosphere of 95% air and 5% CO2.
For cell survival studies, cells were plated (5 × 105 cells/100 mm culture dishes) and incubated for 16 h at 37°C. GS was added to the exponentially growing cells 24 h prior to IR. A range of IR doses was delivered to cell samples using an Eldorado 8 cobalt-60 teletherapy unit (Theratronics International Ltd. Kanata, ON, Canada) at dose rates of 2.0–2.5 Gy/min. Vehicle control radiation survival curves were conducted in parallel. Twenty-four hours after IR and drug treatment, cells were trypsinized, counted, plated, and incubated for 10–14 days. Colonies were fixed with methanol/acetic acid (3:1) and stained with crystal violet. Colonies with > 50 cells were scored and cell survival determined after correcting for the plating efficiency and for GS cytotoxicity alone. Survival curve data were fit using a linear-quadratic model according to Albright (1987). Survival curves for each cell were repeated 2–3 times. The dose modification factor (DMF) was determined by taking the ratio of IR doses at the 10% survival level (control IR dose divided by the GS-treated IR dose). DMF values > 1 indicate enhancement of radiosensitivity.
Cell Growth Assay
Cells (6 × 104) were plated in 60 mm Petri dishes and incubated overnight. The next day 10, 25, and 50 μM Z-guggulsterone was added to the dishes (triplicate). After 120 h the cells were trypsinized with 0.05% Trypsin–EDTA from Gibco (Carlsbad, CA, USA) and counted.
NF-κB Activity Assay
Nuclear extracts were prepared (Active Motif, Carlsbad, CA, USA) from HT29 and PC-Sw cells that were either (a) irradiated, or (b) treated with 50 or 100 μM GS for 24 h, or (c) pre-treated for 24 h with 50 or 100 μM GS and then irradiated. NF-κB p50 and p65 transcription factor activity assays were carried out using the TransAM™ NF-κB family transcription factor assay kit (Active Motif, Carlsbad, CA, USA).
Western Blotting
Cells (1 × 106) were plated in 100 mm Petri dishes. GS at various concentrations was added to the cells the following day. Twenty-four hours later some of the Petri dishes were irradiated with a single dose of 6 Gy. The next day all the samples were rinsed with cold PBS and the cells lysed in RIPA buffer (Santa Cruz Biotechnology, Inc. Santa Cruz, CA, USA). The samples were centrifuged at 14000 rpm in a refrigerated centrifuge and the supernatant collected. The samples were kept at 4°C if used on the same day or frozen at −70°C for storage. Protein concentration was determined with DC Protein Assay kit (Bio-Rad). Forty micrograms of protein was separated on Tris–Glycine gels (Invitrogen, Carlsbad, CA, USA) and transferred to nitrocellulose membrane using iBlot Dry Blotting System from Invitrogen (Carlsbad, CA, USA). Non-specific protein binding was blocked by incubating the membranes for 1 h in 3% blocking grade non-fat dry milk (Bio-Rad, Hercules, CA, USA) in TBST. The membranes were then left overnight at 4°C in the primary antibody at a dilution of 1:100 for ERα; 1:200 for ERβ, IGF-1Rβ, p21, and 1:5000 for anti-actin. The membranes were washed thrice in TBST and incubated for 1 h in horseradish peroxidase conjugated secondary antibody at a dilution of 1:2000 except for actin where the secondary antibody was used at a dilution of 1:10000. The proteins were then visualized by chemiluminescence (Perkin Elmer, Waltham, MA, USA) using Fluor Chem SP imager (Alpha Innotech, San Leandro, CA, USA).
For the determination of phosphohistone H2AX the cells were harvested and frozen at −70°C. The samples were thawed on ice and incubated on ice for 10 min in four volumes of lysis buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl with 0.5 mM DTT, and 1.5 mM PMSF). The samples were centrifuged at 14000 rpm for 10 min in a refrigerated centrifuge. The supernatant was aspirated and the pellet was incubated on ice with 0.2 M H2SO4 for 4 h. The samples were centrifuged at 14000 rpm for 10 min at 4°C. The supernatant (acid soluble histones) was saved and nine volumes of ice cold acetone were added. The samples were kept overnight at −20°C. After centrifugation at 4°C the pellet was re-suspended in 4 M urea, protein estimated, and 20 μg of acid extracted protein run on 18% gels. Following transfer non-specific binding was blocked by incubating the membranes in 3% milk in TBST for 1 h and incubated overnight at 4°C in 1:10000 anti-phosphohistone H2AX antibody. The membranes were washed in TBST, incubated for 1 h in secondary antibody and visualized by chemiluminescence. The membranes were then stripped using Re-Blot Plus mild antibody stripping solution (Chemicon Int. Temecula, CA, USA) and re-probed with an H2A antibody for equal loading verification. Densitometric analysis of protein bands was accomplished with image analyzer software coupled with the Fluorchem FC800 system (Alpha Innotech, San Leandro, CA, USA). Density values for each protein were normalized to actin or other control protein values. All westerns were repeated 2–3 times and normalized against actin (in the case of Erα, ERβ, IGF-1Rβ, p21). The H2AX experiments were repeated twice and normalized against H2A.
Flow Cytometry
Cell cycle analysis was conducted using 1 × 106 cells plated in 100 mm Petri dishes. The cells were either: (a) treated with GS (50 μM), (b) IR (7 Gy), or (c) treated with GS for 24 h and irradiated. After 24 h of incubation the cells were washed with PBS, trypsinized, fixed in cold 70% ethanol diluted in HBSS and stored at 4°C. Solutions were brought to room temperature and cells were centrifuged at 1000 rpm for 5 min and the supernatant discarded. The pellet was washed once in cold PBS and suspended in 1 ml of 20 μg/ml of propidium iodide solution containing 0.1% Triton X-100 and 500 ng of DNase free RNase. The cells were immediately analyzed for cell cycle changes. All flow experiments were repeated twice.
For γH2AX studies cells (106) were plated in 100 mm Petri dishes and following various treatments were fixed in 70% ethanol diluted in HBSS and stored at 4°C. The cells were then centrifuged at 1000 rpm for 5 min in a refrigerated centrifuge and the supernatant aspirated. The pellets were suspended in 1 ml of cold TST (TBS + 4% FBS + 0.1% Triton X-100) and left on ice for 10 min. After centrifugation the supernatant was discarded and the cells were incubated on a shaker for 2 h at 4°C in 200 μl of 1:500 anti-phosphohistone H2AX antibody diluted in TST. The cells were then rinsed with TST and incubated on a shaker at room temperature for 1 h in 200 μl of 1:200 secondary antibody (Alexa 488 goat anti-mouse IgG). The cells were rinsed with cold TBS and 5 μg/ml of propidium iodide in cold TBS was added to the cells. Both samples for DNA analysis and γH2AX studies were analyzed using a BD FACS Calibur (BD Biosciences, San Jose, CA, USA).
Statistical Methods
Experiments were repeated 2–3 times and means and SD determined. Student’s t-test was used to determine the significance of p-values indicated.
Results
Cell Growth and Cell Cycle Effects
Given the reported activation by GS of the various hormone receptors (Brobst et al., 2004) we initially set out to determine if GS would alter cell growth in the estrogen responsive breast cell line, MCF7. Figure 1A demonstrates that incubating MCF7 cells with 10 μM GS up to 120 h reduced cell growth by approximately 60%. Figures 1B,C shows the effect of various concentrations of GS on two other tumor cell lines, HT29, and PC-Sw cells after 120 h of growth. There was little effect of 10 μM GS on PC-Sw cell growth, however, this concentration did reduce growth in HT29 by 35%. Greater growth inhibition was achieved using 25 and 50 μM GS, such that 50 μM GS incubation reduced growth by greater than 70% in all three cell lines (p < 0.05 for all three cell lines). Clonogenic survival studies indicated that the growth inhibition found was not due to GS toxicity as 50 μM GS incubation showed minimal toxicity on the MCF7, HT29, or the PC-Sw cells (Table 1). Given the growth inhibitory effects of GS, studies were conducted to determine if the GS induced growth inhibition lead to altered cell cycle distribution. As seen in Figure 1D 50 μM GS incubation alone in PC-Sw cells did alter the cell cycle distribution after 24 h incubation (%G1 increased from 34 to 51, %S decreased from 44 to 36, and %G2/M decreased from 22 to 12; Figure 1D). To determine if GS was inhibiting cell cycle movement, PC-Sw cells were irradiated with 6 Gy (with or without GS pre-treatment), after which cell cycle movement was followed for 18 h post-IR exposure. For IR treatment alone cells quickly moved out of G1 (decreased from 20 to 8%) into S and blocked at the G2/M border (increased from 22 to 44%) 6 h post-treatment (Figure 1E). By 12 h cells continued to leave S-phase and block in G2 (Figure 1F), and by 18 h had moved through the G2/M block as G1 increased from 16 to 58% (Figure 1G). In contrast, cells pre-treated for 24 h with 50 μM GS followed by IR showed no cell cycle movement over the entire time course examined [%G1(52% at 3 h – 54% at 18 h), %S(40% at 3 h – 41% at 18 h), %G2/M(8% at 3 h – 5% at 18 h)]. Thus, GS treatment completely blocked cell movement in all phases of the cell cycle following an exposure to IR.
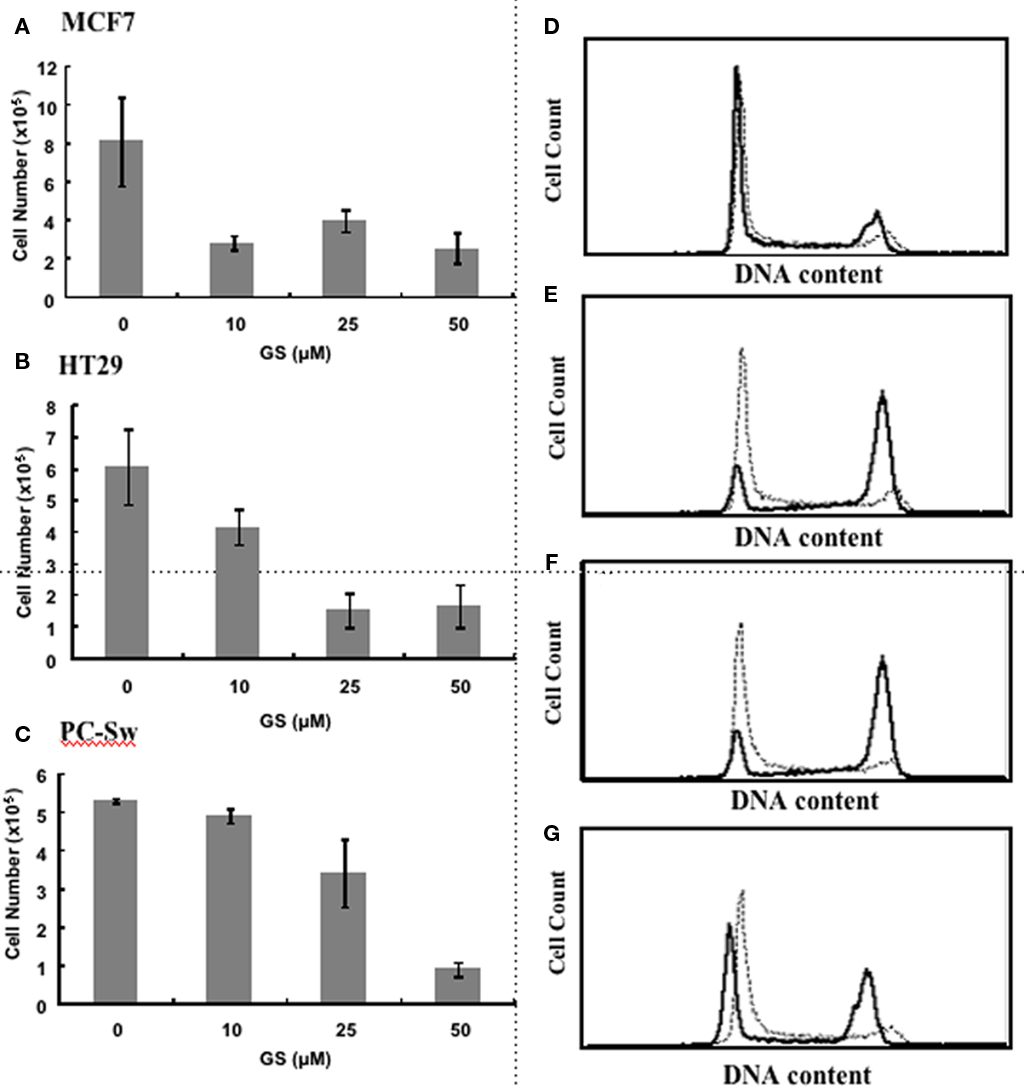
Figure 1. Effect of GS on cell growth and cell cycle movement. Cell growth measurements for (A) MCF7 cells, (B) HT29 cells, and (C) PC-Sw cells incubated with 0, 10, 25, or 50 μM GS over a 5-day period. Cells were plated initially at 2 × 105 and counted after 5 days of growth. Flow cytometry study of PC-Sw cells treated either with 6 Gy of IR or treated with 50 μM GS for 24 h followed by 6 Gy IR. (D) untreated PC-Sw cells (solid line) vs 24 h GS-treated PC-Sw cells (dotted line). (E) 6 h post-IR (solid line) vs 6 h post-IR + 50 μM GS (dotted line) cells, (F) 12 h post-IR (solid line) vs 12 h post-IR + 50 μM GS (dotted line) cells, (G) 18 h post-IR (solid line) vs 18 h post-IR vs 18 h post-IR + 50 μM GS (dotted line) cells.
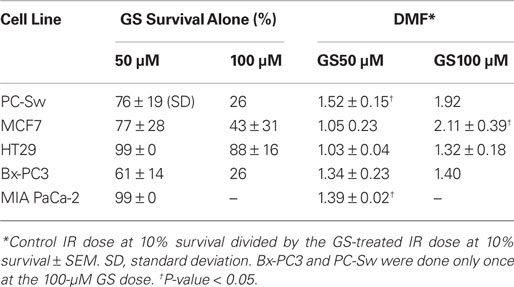
Table 1. Guggulsterone cytotoxicity alone and IR dose modification factors.
Effects on Growth Factor Receptors
Because of the profound effect of GS on the cell growth experiments were conducted to determine what effect GS incubation was having on various growth factor receptors in these cells. Changes in three growth factor receptors, ERα ERβ and IGF-1Rβ were analyzed after 24 h GS incubation alone or in combination with IR in MCF7 cells. IR alone elevated ERα expression, while GS alone at both concentrations substantially down-regulated ERα protein levels (Figure 2A). Likewise, ERα expression was inhibited with the combination of IR and GS (Figure 2A). GS did not significantly affect either ERβ Figure 3A nor IGF-1Rβ levels in the MCF7 cells (Figure 2B) with any of the treatment combinations. Both the HT29 cells and PC-Sw cells tested negative for the Erα receptor (data not shown); however, Figure 2B shows that IGF-1Rβ was down-regulated by 24 h incubation with 50 or 100 μM GS (70–80% reduction). IR treatment alone showed little effect on IGR-1Rβ in these cells, while the combination of IR and GS was similar for GS alone treatment.
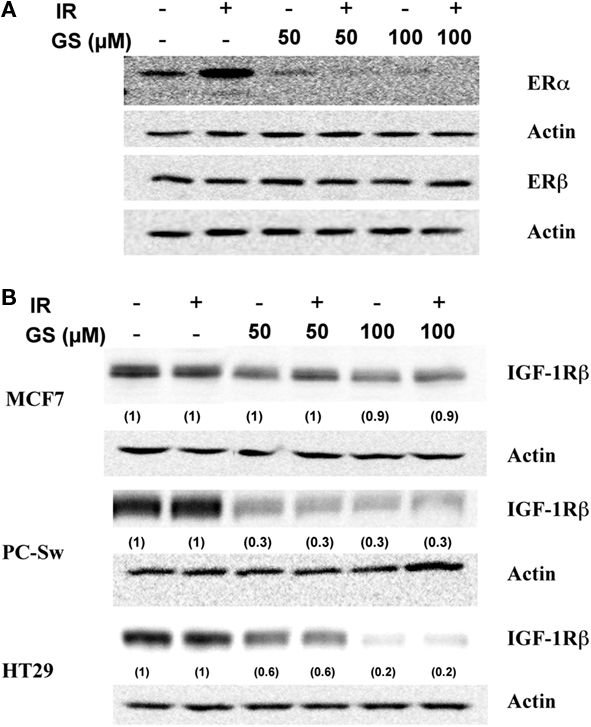
Figure 2. The effect of GS and IR on ERα, ERβ, and IGF-1Rβ levels. (A) MCF7 cells were treated with either 50 or 100 μM GS for 24 h and probed for ERα and ERβ changes. After treatment protein was isolated and processed for Western analysis to determine both ERα and ERβ levels. Blots were stripped and re-probed with actin antibodies. (B) MCF7, HT29, and PC-Sw cells were treated with 50 or 100 μM GS for 24 h and probed for changes in the levels of IGF-1Rβ. After treatment protein was isolated and processed for Western analysis to determine IGF-1Rβ levels. Blots were stripped and re-probed with actin antibodies. Numbers in parenthesis represent normalized protein levels using actin as the loading controls.
P21 Expression
Guggulsterone has been shown to induce the cyclin dependent kinase inhibitor, p21WAF/CIP1, leading to cell cycle blocks (Shishodia et al., 2007). The effects of IR and GS treatment on p21WAF/CIP1 induction were evaluated in two human tumor cell lines differing in their p53 status (MCF7, p53 normal; HT29 p53 mutated). In both cells lines p21WAF/CIP1 was up-regulated by either 50 μM (10.6 ± 0.2-fold for the HT29 and 3.45 ± 2.4-fold for MCF7 cells) or 100 μM GS (Figure 3). IR alone increased p21WAF/CIP1 in MCF7 cells, but not HT29 cells suggesting that GS must increase p21WAF/CIP1 via a p53 and IR-independent mechanism.
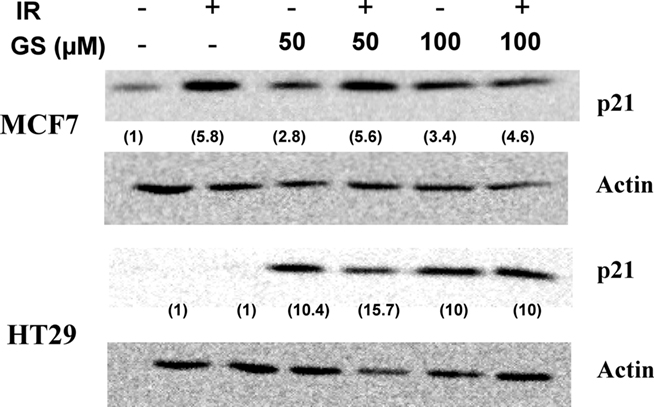
Figure 3. p21WAF/CIP1 induction post-IR and GS exposure on MCF7 or HT29 cells. Cells were treated with either 50 or 100 μM GS alone for 24 h or with 50 or 100 μM GS for 24 h followed by 6 Gy of IR. Protein was extracted after a 24-h post-IR period. Western analysis was done to determine p21WAF/CIP1 levels. Blots were stripped and re-probed with actin antibodies for loading controls. Numbers in parenthesis represent the normalized p21 levels determined using actin as the loading control.
GS-Mediated Radiosensitivity
The ability to interfere with cell cycle movement after IR exposure suggested that GS might alter the radiosensitivity of cells. Pilot studies initially carried out with PC-Sw cells indicated that 50 μM GS incubation for times greater than 4 h increased cytotoxicity after a single 6 Gy IR dose (6 Gy alone = 0.278 (relative survival), 6 Gy + 4 h GS = 0.269, 6 Gy + 8 h GS = 0.152, 6 Gy + 18 h GS = 0.133, 6 Gy + 24 h GS = 0.186). Subsequently, for full dose survival curves, cells were pre-treated for 24 h with GS (50 or 100 μM final concentration) followed by IR. Studies with multiple cancer cell lines (MCF7, HT29, Bx-PC3, DU145) showed enhanced radiosensitization after GS incubation (Figure 4; Table 1). As indicated, 50 μM GS incubation was not cytotoxic alone but toxicity was increased significantly with 100 μm GS incubation (Table 1). GS enhanced the radiosensitivity of the pancreatic cell lines at a concentration of 50 μM; whereas, the other cell lines did not show sensitization with this concentration. GS-mediated enhancement of radiosensitivity was observed in all the cell lines that were pre-treated with 100 μM GS with DMFs ranging from 1.3 to 2.1 (Table 1).
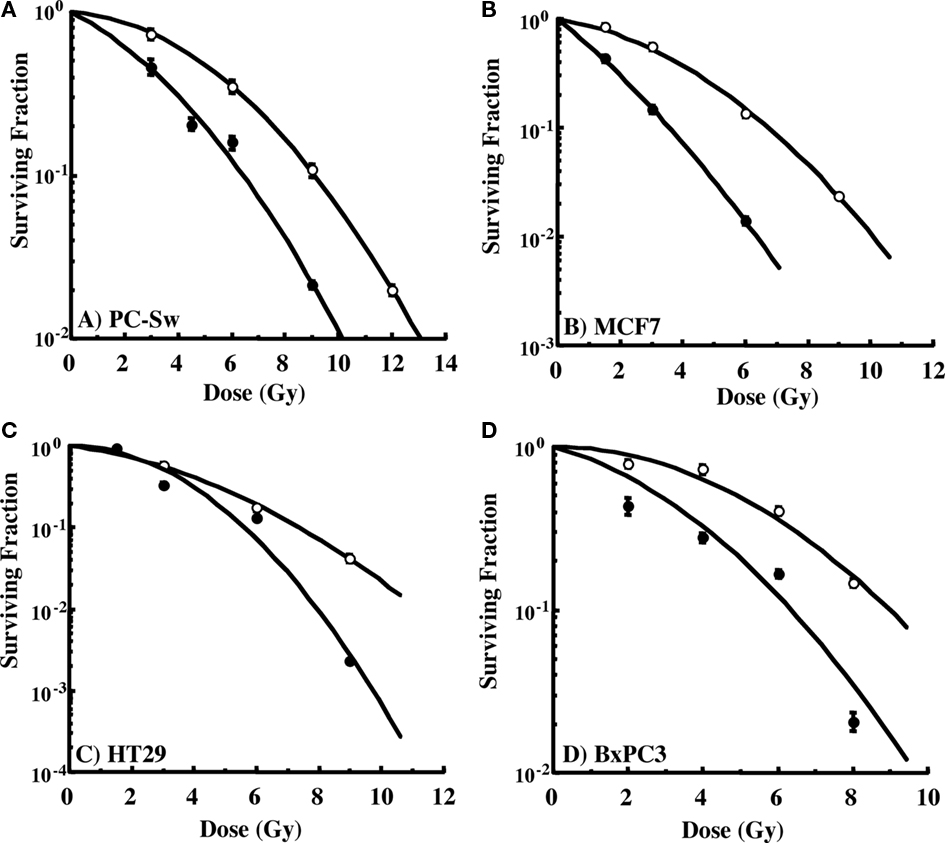
Figure 4. Radiation cell survival curves for cells pre-incubated for 24 h with or without 50 or 100 μM GS and then exposed to varying doses of IR. (A) PC-Sw cells incubated with 50 μM GS for 24 h, (B) MCF7 cells incubated with 100 μM GS (C) HT29 cells incubated with 100 μM GS, (D) BX-PC3 cells incubated with 100 μM GS. GS cytotoxicity data and DMFs are shown in Table 1.
Phosphorylation of H2AX
To assess the effects of GS on IR-induced DNA damage and repair, phosphorylation of the H2AX protein (γH2AX) was evaluated. Cells were pre-treated without or with 50 or 100 μM GS, exposed to 6 Gy, rinsed, and incubated in fresh medium. Figure 5A shows γH2AX levels as a function of time after IR in MCF7, HT29, or PC-Sw cells. All three cell lines exhibited increased γH2AX levels 1 h post-IR (5.7 ± 2.8, 4.8, and 5.4 ± 0.8, for MCF7, HT29, and PC-Sw cells, compared to control intensities, respectively). These levels were reduced at 24 h post-IR indicating repair. Pre-treatment using 50 μM GS followed by IR did not change the kinetics of IR repair in any of the cell lines tested. However, 100 μM GS pre-treatment did inhibit DNA repair in the MCF7 and HT29 cells as the γH2AX remained elevated at 24 h. GS treatment alone also induced γH2AX formation in the MCF7 (2.4 ± 0.4) and HT29 (5.1) cells as did 100 μM GS, while 50 μM GS treatment alone also increased γH2AX expression (2.8 ± 0.8) in the PC-Sw cells. Since GS treatment alone increased γH2AX levels, we examined whether the γH2AX levels were increased uniformly in all parts of the cell cycle of PC-Sw cells by using flow cytometry with simultaneous DNA and γH2AX labeling (Figures 5B,C). Compared to untreated controls, a 24-h 50 μM GS treatment predominantly increased the γH2AX levels in S-phase cells.
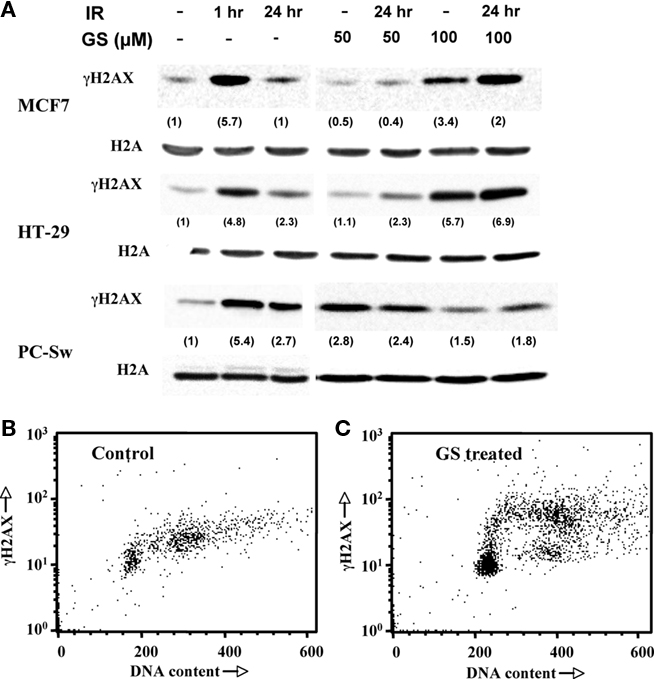
Figure 5. Effect of GS and IR exposure on γH2AX formation in various cell lines. (A) MCF7, HT29, and PC-Sw cells were treated with 6 Gy IR alone, 50 or 100 μM GS for 24 h alone, or 50 or 100 μM GS for 24 h followed by 6 Gy IR exposure. Samples for γH2AX induction were taken at 1 and 24 h. Two parameter flow cytometry histograms of γ-H2AX versus DNA content in control (B) or GS treated (C) PC-Sw cells. Cells were treated with 50 μM GS for 24 h and were then fixed and analyzed using an anti-γH2AX antibody and co-stained with PI for total DNA content.
Additional studies were conducted to determine if the H2AXγ observed was accompanied by the formation of the cleaved PARP fragment. MCF7 cells showed a cleaved PARP fragment after 72 h incubation with 100 μM GS while the HT29 cells showed damage by 48 h with 100 μM incubation (data not shown). HT29 cells showed a small amount of cleaved PARP with 50 μM GS only after 72 h GS incubation (data not shown). Neither the MCF7 nor the HT29 cells showed any cleaved PARP after 24 h GS incubation.
NF-κB Activation
It has been suggested that inactivation of NF-κB can enhance IR sensitization in cell lines (Deorukhkar and Krishnan, 2010). Since GS has been shown to suppress TNF-mediated NF-κB activation (Shishodia and Aggarwal, 2004) we examined if GS could alter NF-κB activation after radiation exposure. HT29 and PC-Sw cells were pre-treated with GS (50 and 100 μM) for 24 h alone, exposed to 10 Gy alone or incubated with GS and irradiated with 10 Gy and analyzed for NF-κB activation 3 h post-IR using a nuclear trans-activation assay. Two different NF-κB subunits (p50 and p65) were evaluated. NF-κB activation after 10 Gy IR was observed in both cell lines since an increase in the levels of the p50 (26% – HT29, 66% PC-Sw) and p65 (73% – HT29, 24% – PC-Sw) subunits was observed as shown in Figure 6. Constitutive NF-κB activity was reduced by GS treatment alone at 50 μM (p65 ∼ 30% for PC-Sw, 8% for HT29; p50 ∼ 19% PC-Sw, 14% for HT29) and 100 μM (p65 ∼ 33% for PC-Sw, 19% for HT29; p50 ∼ 30% for PC-Sw, 20% for HT29), although in some cases not significantly. After IR treatment GS significantly lowered the radiation induced NF-κB activity for p65 (51–52% for PC-Sw and 94–96% for HT29) and p50 (90–95% for PC-Sw and 52–57% for HT29) in both cell lines.
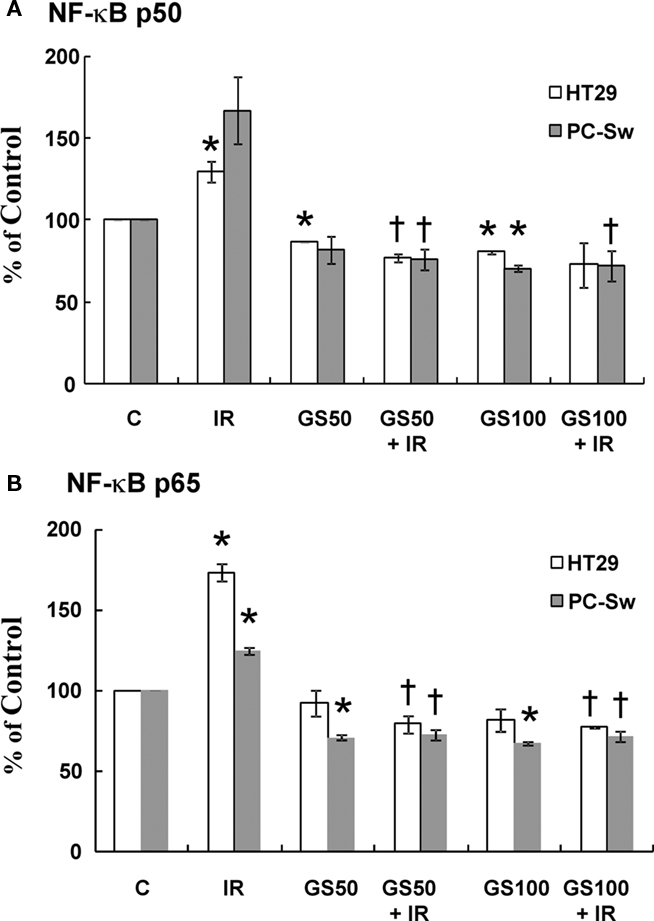
Figure 6. NF-κB p50 and p65 activity in HT29 and PC-Sw cells measured after GS and IR exposure. Cells were pre-treated with GS (50 and 100 μM) for 24 h alone, exposed to 10 Gy alone, or incubated with GS and irradiated with 10 Gy and analyzed for NF-κB activation 3 h post-IR using a nuclear transactivation assay for NF-κB p50 (A) or NF-κB p65 (B). *p values < 0.05 compared to control, †p values compared to IR.
Discussion
Guggulsterone, a plant polyphenol, has several interesting biological properties that may have clinical applications. GS has been evaluated as an anti-hyperlipidemia agent with several clinical trials yielding inconclusive results (Szapary et al., 2003; Nohr et al., 2009). The anti-tumor activity of GS appears to be related to apoptotic mechanisms in a variety of tumor cell lines (Shishodia and Aggarwal, 2004; Samudio et al., 2005; Singh et al., 2005, 2007; Shishodia et al., 2007; An et al., 2009) and also in differentiating 3T3-L1 adipocytes (Yang et al., 2008), implicating consideration of GS in obesity. Our initial interest in GS came from studies showing that GS inhibits the activation of NF-κB a nuclear transcription factor that activates pro-survival pathways (Wang et al., 1996; Shishodia and Aggarwal, 2004). Since IR induces both NF-κB translocation and activation, we examined whether the inhibition of this pro-survival pathway by GS might lead to enhanced radiosensitivity via this mechanism. Support for this mechanism was found in the PC-Sw cells where 50 μM GS incubation inhibited IR-induced NF-κB activation (Figure 6) and increased the radiosensitization in these cells (Figure 4A). However, it is unlikely that this is the only mechanism, since 50 μM GS inhibited IR-induced NF-κB activation in the HT29 cells (Figure 6) but failed to enhance the radiosensitivity in these cells (Figure 4C). As previously reported, GS treatment not only inhibited IR-induced NF-κB activation, it also lowered constitutive NF-κB activation in control untreated cells (Figure 6; Shishodia and Aggarwal, 2004).
Because the GS inhibition of NF-κB activation did not completely correlate with radiation sensitivity we also examined several other potential mechanisms of sensitization. First, given the propensity of GS to interact with several nuclear receptors such as PR, PXR, and ERα we examined if GS could interact with the IGF-1Rβ receptor, a receptor linked with radiation sensitization (Macaulay et al., 2001). Expression of IGF-1Rβ has been shown to confer radioresistance in cancer patients (Rocha et al., 1997). It has also been shown that inactivation of IGF-1Rβ radiosensitizes human lung cancer cells (Cosaceanu et al., 2005). We observed in PC-Sw and HT29 cells, that either 50 μM GS (in PC-Sw cells) or 100 μM GS (in the HT29 cells) incubation did decrease the IGF-1Rβ levels (Figure 2B), and this decrease correlated with the induction of radiosensitization seen in these cells (Figures 4A,C). However, IGF-1Rβ in MCF7 cells showed minimal reduction after 100 μM GS incubation (Figure 2B) although this concentration of GS did increase sensitization to IR (Figure 4B). Hence, it is possible that the GS reduction of IGF-1Rβ may contribute to radiosensitization in certain cell types but in others the effect of GS on different receptors may be more important (such as ERα in MCF7 cells). The mechanism of IGF-1Rβ receptor interaction in the repair of IR damage may be related to its effect on the repair related protein, ATM translocation (Macaulay et al., 2001; Peretz et al., 2001). Whether GS is altering this pathway remains to be determined.
Guggulsterone clearly interacts with steroidal receptors (Bovee et al., 2008) with the greatest activation being on the PR activity (Burris et al., 2005). It has been suggested that the direct interaction of GS with the PXR receptor is partially responsible for the activation of this receptor (Brobst et al., 2004). Our data shows that GS treatment down-regulates the ERα receptor in MCF7 cells. This is important because GS may have potential uses in estrogen positive tumors, which have developed resistance to the anti-estrogen, tamoxifen (Higgins and Stearns, 2009). Whether GS-mediated down-regulation of ERα plays a significant role in enhanced radiosensitivity will require further study. It is interesting to note that tamoxifen and estradiol have been shown to alter radiosensitivity of MCF7 cells suggesting that perturbation of the ER pathway can influence radiation response (Wazer et al., 1989; Paulsen et al., 1996).
Along with the down-regulation of the estrogen receptor GS inhibited cell growth in a variety of human tumor cell lines. Other groups have reported inhibition of cell growth either by MTT assay or 3H-thymidine uptake (Singh et al., 2005, 2007). Growth inhibition was reported to occur because of a G1/S or S-phase block. (Singh et al., 2007) However, Figures 1E–G shows that when PC-Sw cells are irradiated and followed for cell cycle changes that all parts of the cell cycle appear to be affected by GS as almost no movement is observed after GS + IR treatments. Thus, it appears that GS completely abrogates or at least greatly slows down cell cycle movement in each part of the cell cycle, including G1, S, and G2/M. One molecular target responsible for cell cycle blockage with GS incubation was the induction of the CDKI p21WAF1/CIP1 (Figure 3). Importantly, p21WAF1/CIP1 induction also occurred in p53 mutated cells (HT29), which IR did not up-regulate (Figure 3), and hence, suggesting that GS up-regulates p21WAF1/CIP1 by a p53 independent mechanism. Our cell cycle results do not entirely agree with earlier conclusions (Shishodia et al., 2007) that the block was in the S-phase part of the cell cycle in that GS incubation arrested cells in all parts of the cell cycle as clearly no movement was seen from any of the cell cycle compartments following a 6-Gy dose. Shishodia et al. (2007) did find that cdc2 (CDK1) and cyclin D1 were down-regulated, and since these two components of the cell cycle would be expected to impact both G1 and G2/M progression it would seem likely that movement throughout the cell cycle would be impacted.
Guggulsterone was a potent radiosensitizer at 100 μM in all tumor cell lines studied (Figure 4; Table 1) while in several pancreas cell lines GS significantly increased radiosensitization using 50 μM GS (Table 1). Interestingly, this effect was correlated with the induction of γH2AX phosphorylation as 50 μM GS incubation increased γH2AX in the PC-Sw cells but not in the other cell lines (Figure 5). In addition, the other cell lines did show γH2AX phosphorylation when incubated with 100 μM GS (Figure 5). Thus it appears that GS can induce double strand breaks (DSB’s) in cells and this process could also be responsible for the increased radiosensitization observed. One area, which has received increased attention, is the formation of γH2AX foci when cells are exposed to treatments causing replication stress (Peretz et al., 2001; Chanoux et al., 2009). These conditions (usually with drugs) act by inhibiting the rapid repair of DSB’s, which takes place during replication of the DNA during S-phase to relieve topological tension (Nitiss, 2009). Importantly, flow cytometry for γH2AX showed that GS-treated cells had γH2AX induced primarily in S-phase cells suggesting that GS was indeed acting to create sites of DSB’s (Figures 5B,C). Thus, one mechanism of radiosensitization may be the production of DSB’s in mid-to-late S-phase cells as this part of the cell cycle is considered to be the most resistant to IR exposure (Hall and Gaiaccia, 2006).
Another possible advantage of using GS as a clinical radiosensitizer has been reported detailing the selectivity of GS for tumor vs normal cells. Earlier studies have showed that a normal prostate epithelial cell line is resistant to growth inhibition and apoptosis caused by GS when compared to prostate tumor cells (Singh et al., 2005). We examined cleaved PARP after 100 μM GS incubation in both MCF7 and HT29 cells and found that cleaved PARP was observed only after 48 h in HT29 cells and 72 h GS incubation in MCF7 cells. Since radiation studies were done using a 24 h GS incubation, the activation of apoptosis did not appear to be a major pathway for sensitization; however, additional studies are needed to fully examine this mechanism and if GS may interact with normal tissue differently than with the tumor cells examined in this study.
It is interesting to note that the GS concentrations that are effective in bringing about radiosensitization are in the range of the doses, which were used to impact on the inflammatory, apoptotic, and cell cycle pathways (Shishodia and Aggarwal, 2004; Shishodia et al., 2007; An et al., 2009). The ability of GS to modulate ERα and other membrane receptors suggest that it might be a useful drug to alter disease conditions where these receptors are thought to impact. The ability of GS to modulate radiosensitivity in human cancer cell lines warrants further study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the Intramural Research Program of the Center of Cancer Research, National Cancer Institute, National Institutes of Health.
References
Albright, N. (1987). Computer programs for the analysis of cellular survival data. Radiat. Res. 112, 331–340.
An, M. J., Cheon, J. H., Kim, S. W., Kim, E. S., Kim, T. I., and Kim, W. H. (2009). Guggulsterone induces apoptosis in colon cancer cells and inhibits tumor growth in murine colorectal cancer xenografts. Cancer Lett. 279, 93–100.
Bovee, T. F., Schoonen, W. G., Hamers, A. R., Bento, M. J., and Peijnenburg, A. A. (2008). Screening of synthetic and plant-derived compounds for (anti)estrogenic and (anti)androgenic activities. Anal. Bioanal. Chem. 390, 1111–1119.
Brobst, D. E., Ding, X., Creech, K. L., Goodwin, B., Kelley, B., and Staudinger, J. L. (2004). Guggulsterone activates multiple nuclear receptors and induces CYP3A gene expression through the pregnane X receptor. J. Pharmacol. Exp. Ther. 310, 528–535.
Burris, T. P., Montrose, C., Houck, K. A., Osborne, H. E., Bocchinfuso, W. P., Yaden, B. C., Cheng, C. C., Zink, R. W., Barr, R. J., Hepler, C. D., Krishnan, V., Bullock, H. A., Burris, L. L., Galvin, R. J., Bramlett, K., and Stayrook, K. R. (2005). The hypolipidemic natural product guggulsterone is a promiscuous steroid receptor ligand. Mol. Pharmacol. 67, 948–954.
Chander, R., Khanna, A. K., and Kaapor, N. K. (1996). Lipid lowering activity of guggulsterone from Commiphora mukul in hyperlipaemic rats. Phytother. Res. 10, 508–511.
Chanoux, R. A., Yin, B., Urtishak, K. A., Asare, A., Bassing, C. H., and Brown, E. J. (2009). ATR and H2AX cooperate in maintaining genome stability under replication stress. J. Biol. Chem. 284, 5994–6003.
Chautard, E., Loubeau, G., Tchirkov, A., Chassagne, J., Vermot-Desroches, C., Morel, L., and Verrelle, P. (2010). Akt signaling pathway: a target for radiosensitizing human malignant glioma. Neuro Oncol. 12, 434–443.
Cosaceanu, D., Carapancea, M., Castro, J., Ekedahl, J., Kanter, L., Lewensohn, R., and Dricu, A. (2005). Modulation of response to radiation of human lung cancer cells following insulin-like growth factor 1 receptor inactivation. Cancer Lett. 222, 173–181.
Deorukhkar, A., and Krishnan, S. (2010). Targeting inflammatory pathways for tumor radiosensitization. Biochem. Pharmacol. 80, 1904–1914.
Dixit, V. P., Joshi, S., Sinha, R., Bharvava, S. K., and Varma, M. (1980). Hypolipidemic activity of guggal resin (Commiphora mukul) and garlic (Allium sativum Linn.) in dogs (Canis familiaris) and monkeys (Presbytis entellus entellus Dufresne). Biochem. Exp. Biol. 16, 421–424.
Hait, W. N., and Hambley, T. W. (2009). Targeted cancer therapeutics. Cancer Res. 69, 1263–1267; discussion 1267.
Hall, E. J., and Gaiaccia, A. J. (2006). Radiology for the Radiologist. Philadelphia: Lippincott Williams & Wilkins.
Hambley, T. W., and Hait, W. N. (2009). Is anticancer drug development heading in the right direction? Cancer Res. 69, 1259–1262.
Higgins, M. J., and Stearns, V. (2009). Understanding resistance to tamoxifen in hormone receptor-positive breast cancer. Clin. Chem. 55, 1453–1455.
Li, N., and Karin, M. (1998). Ionizing radiation and short wavelength UV activate NF-kappaB through two distinct mechanisms. Proc. Natl. Acad. Sci. U.S.A. 95, 13012–13017.
Liebmann, J., Cook, J. A., Fisher, J., Teague, D., and Mitchell, J. B. (1994). In vitro studies of Taxol as a radiation sensitizer in human tumor cells. J. Natl. Cancer Inst. 86, 441–446.
Lv, N., Song, M. Y., Kim, E. K., Park, J. W., Kwon, K. B., and Park, B. H. (2008). Guggulsterone, a plant sterol, inhibits NF-kappaB activation and protects pancreatic beta cells from cytokine toxicity. Mol. Cell. Endocrinol. 289, 49–59.
Ma, W. W., and Adjei, A. A. (2009). Novel agents on the horizon for cancer therapy. CA Cancer J. Clin. 59, 111–137.
Macaulay, V. M., Salisbury, A. J., Bohula, E. A., Playford, M. P., Smorodinsky, N. I., and Shiloh, Y. (2001). Downregulation of the type 1 insulin-like growth factor receptor in mouse melanoma cells is associated with enhanced radiosensitivity and impaired activation of Atm kinase. Oncogene 20, 4029–4040.
Nitiss, J. L. (2009). DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 9, 327–337.
Nityanand, S., Srivastava, J. S., and Asthana, O. P. (1989). Clinical trials with gugulipid. A new hypolipidaemic agent. J. Assoc. Physicians India 37, 323–328.
Nohr, L. A., Rasmussen, L. B., and Straand, J. (2009). Resin from the mukul myrrh tree, guggul, can it be used for treating hypercholesterolemia? A randomized, controlled study. Complement. Ther. Med. 17, 16–22.
Paulsen, G. H., Strickert, T., Marthinsen, A. B., and Lundgren, S. (1996). Changes in radiation sensitivity and steroid receptor content induced by hormonal agents and ionizing radiation in breast cancer cells in vitro. Acta Oncol. 35, 1011–1019.
Peretz, S., Jensen, R., Baserga, R., and Glazer, P. M. (2001). ATM-dependent expression of the insulin-like growth factor-I receptor in a pathway regulating radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 1676–1681.
Rocha, R. L., Hilsenbeck, S. G., Jackson, J. G., Vandenberg, C. L., Weng, C., Lee, A. V., and Yee, D. (1997). Insulin-like growth factor binding protein-3 and insulin receptor substrate-1 in breast cancer: correlation with clinical parameters and disease-free survival. Clin. Cancer Res. 3, 103–109.
Samudio, I., Konopleva, M., Safe, S., Mcqueen, T., and Andreeff, M. (2005). Guggulsterones induce apoptosis and differentiation in acute myeloid leukemia: identification of isomer-specific antileukemic activities of the pregnadienedione structure. Mol. Cancer Ther. 4, 1982–1992.
Shishodia, S., and Aggarwal, B. B. (2004). Guggulsterone inhibits NF-kappaB and IkappaBalpha kinase activation, suppresses expression of anti-apoptotic gene products, and enhances apoptosis. J. Biol. Chem. 279, 47148–47158.
Shishodia, S., Harikumar, K. B., Dass, S., Ramawat, K. G., and Aggarwal, B. B. (2008). The guggul for chronic diseases: ancient medicine, modern targets. Anticancer Res. 28, 3647–3664.
Shishodia, S., Sethi, G., Ahn, K. S., and Aggarwal, B. B. (2007). Guggulsterone inhibits tumor cell proliferation, induces S-phase arrest, and promotes apoptosis through activation of c-Jun N-terminal kinase, suppression of Akt pathway, and downregulation of antiapoptotic gene products. Biochem. Pharmacol. 74, 118–130.
Singh, S. V., Choi, S., Zeng, Y., Hahm, E. R., and Xiao, D. (2007). Guggulsterone-induced apoptosis in human prostate cancer cells is caused by reactive oxygen intermediate dependent activation of c-Jun NH2-terminal kinase. Cancer Res. 67, 7439–7449.
Singh, S. V., Zeng, Y., Xiao, D., Vogel, V. G., Nelson, J. B., Dhir, R., and Tripathi, Y. B. (2005). Caspase-dependent apoptosis induction by guggulsterone, a constituent of ayurvedic medicinal plant Commiphora mukul, in PC-3 human prostate cancer cells is mediated by Bax and Bak. Mol. Cancer Ther. 4, 1747–1754.
Szapary, P. O., Wolfe, M. L., Bloedon, L. T., Cucchiara, A. J., Dermarderosian, A. H., Cirigliano, M. D., and Rader, D. J. (2003). Guggulipid for the treatment of hypercholesterolemia: a randomized controlled trial. JAMA 290, 765–772.
Urizar, N. L., Liverman, A. B., Dodds, D. T., Silva, F. V., Ordentlich, P., Yan, Y., Gonzalez, F. J., Heyman, R. A., Mangelsdorf, D. J., and Moore, D. D. (2002). A natural product that lowers cholesterol as an antagonist ligand for FXR. Science 296, 1703–1706.
Wang, C. Y., Mayo, M. W., and Baldwin, A. S. Jr. (1996). TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science 274, 784–787.
Wazer, D. E., Tercilla, O. F., Lin, P. S., and Schmidt-Ullrich, R. (1989). Modulation in the radiosensitivity of MCF-7 human breast carcinoma cells by 17B-estradiol and tamoxifen. Br. J. Radiol. 62, 1079–1083.
Xiao, D., and Singh, S. V. (2008). z-Guggulsterone, a constituent of ayurvedic medicinal plant Commiphora mukul, inhibits angiogenesis in vitro and in vivo. Mol. Cancer Ther. 7, 171–180.
Keywords: guggulsterone, radiosensitization, tumor cells, NF-κB activation, cell growth, DNA damage repair
Citation: Choudhuri R, DeGraff W, Gamson J, Mitchell JB and Cook JA (2011) Guggulsterone-mediated enhancement of radiosensitivity in human tumor cell lines. Front. Oncol. 1:19. doi: 10.3389/fonc.2011.00019
Received: 12 May 2011; Accepted: 05 July 2011;
Published online: 21 July 2011.
Edited by:
Zelig Tochner, University of Pennsylvania, USAReviewed by:
Michael L. Freeman, Vanderbilt University School of Medicine, USAJoel S. Greenberger, University of Pittsburgh Medical Center-Shadyside, USA
Melissa Ann Fath, University of Iowa, USA
Copyright: © 2011 Choudhuri, DeGraff, Gamson, Mitchell and Cook. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: James B. Mitchell, Radiation Biology Branch, National Cancer Institute, Building 10, Room B3-B69, 9000 Rockville Pike, Bethesda, MD 20892, USA. e-mail:amJtQGhlbGl4Lm5paC5nb3Y=