


- 1 Department of Radiation Oncology, Warren Alpert Medical School of Brown University and Rhode Island Hospital, Providence, RI, USA
- 2 The Department of Electrical Engineering and Computer Science, School of Engineering, Case Western Reserve University, Cleveland, OH, USA
- 3 Department of Radiation Oncology, Case Western Reserve University, Cleveland, OH, USA
Over the last 7 years, we have focused our experimental and computational research efforts on improving our understanding of the biochemical, molecular, and cellular processing of iododeoxyuridine (IUdR) and ionizing radiation (IR) induced DNA base damage by DNA mismatch repair (MMR). These coordinated research efforts, sponsored by the National Cancer Institute Integrative Cancer Biology Program (ICBP), brought together system scientists with expertise in engineering, mathematics, and complex systems theory and translational cancer researchers with expertise in radiation biology. Our overall goal was to begin to develop computational models of IUdR- and/or IR-induced base damage processing by MMR that may provide new clinical strategies to optimize IUdR-mediated radiosensitization in MMR deficient (MMR−) “damage tolerant” human cancers. Using multiple scales of experimental testing, ranging from purified protein systems to in vitro (cellular) and to in vivo (human tumor xenografts in athymic mice) models, we have begun to integrate and interpolate these experimental data with hybrid stochastic biochemical models of MMR damage processing and probabilistic cell cycle regulation models through a systems biology approach. In this article, we highlight the results and current status of our integration of radiation biology approaches and computational modeling to enhance IUdR-mediated radiosensitization in MMR− damage tolerant cancers.
I Introduction
Scientific and Clinical Rationale for Targeting “CT-and IR-Resistant” MMR Deficient Human Cancers
DNA mismatch repair (MMR) is a highly conserved but complex DNA repair system that ensures genomic stability on several levels including: correcting mismatches generated during DNA replication; blocking genetic recombination events between divergent DNA sequences; and mediating cell death in response to certain DNA damaging agents (Jiricny, 2006). MMR deficiency is principally associated with the autosomal dominant hereditary non-polyposis colorectal cancer (HNPCC) syndrome, resulting from MMR gene mutations including the hMLH1, hMSH2, hMSH6, and hPMS2 genes (Lynch and de la Chapelle, 2003). MMR deficiency is also associated with an increasing number of sporadic microsatellite-instability-high (MSI-H) solid tumors, principally related to promoter methylation of hMLH1 or hMSH2 genes (Peltomaki, 2003). These sporadic MSI-H cancers include several types of GI cancers (gastric, pancreatic, esophageal, colorectal), GYN cancers (endometrial, ovarian), GU cancers (bladder, ureter) as well as non-small cell lung (NSCL) cancers, and high grade primary brain tumors, where MMR deficiency (MSI-H phenotype) is found in up to 10–20% of these common cancers (Peltomaki, 2003). Importantly, MMR deficiency is associated with in vitro/in vivo “damage tolerance” (resistance) to multiple different classes of clinically active chemotherapy drugs (Stojic et al., 2004; Jiricny, 2006; Modrich, 2006; Kinsella, 2009) as well as to other types of DNA damage (stress) including ionizing radiation (IR; Yan et al., 2001, 2009; Brown et al., 2003; Cejka et al., 2004) and hypoxia (Kondo et al., 2001; Mihaylova et al., 2003; Koshiji et al., 2005; Klein and Glazer, 2010). Interestingly, promoter hypermethylation of hMLH1 and hMSH2 genes and subsequent loss of MMR protein expression was found in nearly 50% of NSCL cancers occurring in non-smokers and was associated with a poor prognosis even in early stage lung cancers (Hsu et al., 2005). However, MSI-H localized colon cancers appear to have a better prognosis than MMR proficient (MMR+) tumors following surgery (Lynch and de la Chapelle, 2003; Peltomaki, 2003). These conflicting clinical data underscore the biological complexity of MMR and its translational importance to cancer therapeutics.
The MMR pathway is a multiprotein system that has three sub-processes (Lynch and de la Chapelle, 2003; Jiricny, 2006; Kinsella, 2009). These sub-processes involve: first, mismatch recognition by MutSα (a MSH2/MSH6 dimer) or MutSβ (a MSH2/MSH3 dimer); second, mismatch excision, which is initiated by the binding of MutLα (a MLH1/PMS2 dimer) or MutLβ (a MLH1/MLH3 dimer) to MutSα and the subsequent recruitment of an exonuclease (EXO1) that sequentially removes nucleotides between an adjacent single-strand break (SSB) up to and beyond the mismatch on the daughter DNA strand; and third, resynthesis and ligation initiated by DNA polymerase δ along with at least two other proteins, proliferating cell nuclear antigen (PCNA) and replication protein A (RPA), followed by sealing the nick located in the daughter strand by a DNA ligase.
“Damage tolerance” (drug-resistance) has been demonstrated in MMR deficient (MMR−) human and murine cells to methylating agents such as temozolomide, dacarbazine, and procarbazine; platinum analogs including cisplatinum and carboplatinum; anthracyclines such as adriamycin; and nucleoside analogs such as 6-thioguanine (6-TG), iododeoxyuridine (IUdR), and the fluoropyrimidines [both 5-fluorouracil (5-FU) and 5-fluorodeoxyuridine (FUdR)] (Berry et al., 1999, 2000, 2003; Meyers et al., 2001, 2003; Yan et al., 2003; Jiricny, 2006; Modrich, 2006; Kinsella, 2009). IR resistance in MMR− cells is also found, particularly using low dose rate (LDR)–IR (Yan et al., 2009). Based on biochemical and molecular analyses, MMR proteins (principally the MutSα complex) have been shown to recognize a majority of these chemically modified “mispairs.” However, these chemically or IR modified DNA bases will not be removed during the DNA degradation step of MMR unless they are present in the strand containing the discontinuity (i.e., the daughter strand). In the absence of functional MMR, cells do not “see” these chemically or IR modified bases in their DNA and continue to replicate; accumulating mutations in DNA as a result.
Following recognition of a drug or an IR-induced adduct in DNA, MMR+ cells may undergo a cytotoxic (cell death) response. It is still unclear whether this response is directly caused by the MMR machinery initiating a signaling cascade directing the cells to either repair the mismatch or undergo cell death (general damage sensor model of MMR), or whether this toxicity is indirect due to futile attempts by MMR to repair the DNA containing a drug or IR-induced structural adduct in the template (parent strand) consistent with the futile cycle repair model of MMR (Stojic et al., 2004; Jiricny, 2006; Modrich, 2006). Typically, MMR+ cells display a prolonged G2 cell cycle arrest and a subsequent cytotoxic response following two or more rounds of DNA replication after exposure to drugs such as temozolomide, 6-TG, and the fluoropyrimidines (5-FU, FdUrd; Meyers et al., 2001, 2003; Yan et al., 2003; Stojic et al., 2004; Jiricny, 2006; Kinsella, 2009). These temporal observations may indicate that the MMR proteins recognize altered mismatches resulting from subsequent rounds (>2) of replication of DNA containing the adducts, rather than the initial adducts formed by the incorporation or interaction of these drugs and IR with DNA.
Regardless of the mechanism by which MMR mediates a cytotoxic response in cells to the many different types of drug or IR-induced DNA adducts, deficiency in MMR (MMR−) results in a “damage tolerance” or “drug and IR-resistance” phenotype, which may have direct implications for the success of chemotherapy as well as fluoropyrimidine- and/or platinum-based radiosensitization in the clinic. There are an increasing number of examples in the literature of clinically relevant chemotherapy, radiotherapy, or chemotherapy + radiotherapy “resistance” in MMR− human cancers. For example, in a human glioblastoma multiforme tumor xenograft model, loss of MMR following treatment with procarbazine was found to confer resistance to temozolomide, MNNG, and busulfan, in addition to procarbazine (Friedman et al., 1997). A follow-up clinical study by the same investigators has suggested that analysis of the levels of MLH1 and MSH2 expression prior to chemotherapy with temozolomide may help predict treatment responses in patients with newly diagnosed malignant gliomas (Friedman et al., 1998). In this clinical study, patients with MMR− malignant gliomas showed a markedly reduced response rate and survival compared to patients with MMR+ gliomas. Importantly, the use of radiation therapy and concomitant temozolomide (TMZ) followed by monthly maintenance TMZ is now the standard of care in patients with glioblastoma (Stupp et al., 2005). Indeed, somatic point mutations in hMSH6 are found in up to 30% of recurrent/progressive glioblastomas, which were not present in pre-treatment specimens. Inactivation of hMSH6 was correlated with prior or ongoing TMZ exposure and resulted in enhanced tumor growth and shorter survival (Cahill et al., 2007).
Prior studies in ovarian cancer cell lines suggest that treatment with platinum compounds “selects” for surviving cells with lower MMR protein expression or loss of MMR Brown et al. (1997). A significant decrease in expression of both MSH2 and MLH1 proteins after cisplatinum chemotherapy was found in patients with sporadically arising ovarian tumors (Samimi et al., 2000). Decreased protein expression of MLH1 following adriamycin-based chemotherapy in breast cancer patients was also reported to correlate significantly with a reduced disease-free survival (p = 0.0025), (Mackay et al., 2000) indicating that the resistance that MMR deficiency imparts to many cancer chemotherapy agents is clinically relevant. Analyses of the use of fluoropyrimidine-based adjuvant chemotherapy in patients with MMR− (microsatellite-instability-high, MSI-H) colon and esophageal cancers showed significantly less benefit in disease-free survival in comparison to a significant benefit in patients with MMR+ colon and esophageal cancers. (Kishi et al., 2003; Ribic et al., 2003; Sargent et al., 2010) Additionally, MMR− endometrial and rectal cancers show reduced local control and pathologic response rates following radiation therapy alone (Bilbao et al., 2010) or with 5-fluorouracil-radiation therapy combined modality treatment, (Choi et al., 2007) respectively. In contrast, we have demonstrated that these MMR− “drug and IR resistant” human cancers show enhanced IUdR–DNA incorporation and increased IUdR (IPdR)-mediated radiosensitization using experimental in vitro/in vivo models.(Berry and Kinsella, 2001; Seo et al., 2004, 2005; Kinsella et al., 2007, 2008) The results of these experimental data will be reviewed below.
Scientific and Clinical Rationale for the Use of IUdR and Its Oral Prodrug, IPdR, as Radiosensitizers in MMR Deficient Human Cancers
IUdR is a halogenated thymidine analog and has been recognized as an in vitro/in vivo and potential clinical radiosensitizer for several decades (Kinsella, 1996). Following active nucleoside transport across cell membranes, IUdR is sequentially phosphorylated to IdUTP and incorporated into DNA in competition with thymidine triphosphate (dTTP; Kinsella, 1996). The biochemical mechanisms of cellular radiosensitization are related to the generation of highly reactive free radicals by IR from IUdR–DNA incorporation resulting in enhanced IR-induced DNA strand breaks [both single (SSB) and double strand (DSB)] while also altering IR damage repair (Kinsella, 1996). The extent (%) of thymidine replacement by IUdR in DNA is generally recognized to correlate directly with the extent (%) of radiosensitization. Targeted radiosensitization by IUdR of certain human cancers has been tested based on the higher proliferation rate in these cancers compared to adjacent normal tissues. In our prior in vivo pre-clinical studies using daily oral or continuous intravenous infusions of IUdR for 4–10 days prior to short course IR (four fractions/4 days), we noted enhanced radiosensitization (1.3–1.5× compared to IR alone) but also increased % IUdR–DNA incorporation in both bone marrow and intestine (Kinsella et al., 1996). Clinically, systemic acute toxicities to bone marrow and intestine limited the duration and total dose of continuous intravenous infusions of IUdR during fractionated external beam radiation therapy (Epstein et al., 1994; Schulz et al., 2004).
IPdR is a pyrimidinone nucleoside, which was originally synthesized as an antiviral agent, based on the hypothesis that nucleosides without an amino- or keto-group at position four of the pyrimidine ring would be used as a substrate of viral thymidine kinase (TK) but not mammalian TK (Saif et al., 2007). However, these same investigators subsequently found that IPdR could be efficiently converted to IUdR by an aldehyde oxidase that was mainly localized to rodent liver (Saif et al., 2007). Subsequently, we have demonstrated that an aldehyde oxidase in normal liver tissue in rodents (mice, rats) and non-rodent animals (ferrets, monkeys) rapidly converts IPdR to IUdR in vivo as well as showing similar IPdR metabolism using cytosolic extracts of normal human liver in vitro (Kinsella et al., 1994, 1998). We have also reported that oral IPdR can significantly improve the therapeutic index of IUdR-mediated radiosensitization, compared to continuous infusion IUdR using human colorectal and glioblastoma tumor xenograft models (Kinsella et al., 1994, 1998, 2000a,b). The extent (%) of IUdR–DNA incorporation in small intestine and bone marrow were remarkably reduced with similar or superior % IUdR–DNA incorporation in the human tumor xenografts using oral IPdR given once daily × 14 days compared to the maximum tolerated dose of IUdR as a continuous infusion (Kinsella et al., 1994, 1998, 2000a,b).
As part of our ICBP investigations, we have shown that IUdR (and IPdR) cytotoxicity are affected by both MMR and base excision repair, with both DNA repair systems capable of recognizing G:IU mispairs (Kinsella, 2009). We have also demonstrated that MMR− cells retain higher levels of IUdR–DNA incorporation using isogenic in vitro and in vivo models, which results in enhanced radiosensitization in MMR− vs. MMR+ cancers (Kinsella, 2008). As such, we are now developing IPdR as a potential clinical radiosensitizer for MMR− and other “IR-resistant” cancers in collaboration with the National Cancer Institute. One goal of the development of in silico (computational) models of MMR processing of both IUdR and IR (as detailed below) is to provide insights for clinical trial design to optimize the dosing schedule for orally administered IPdR for IUdR-mediated radiosensitization in MMR− and other IR-resistant human cancers (Kinsella, 2008, 2009).
II Overview and Integration of Biochemical, Molecular, and Cellular Data on MMR Processing of Nucleoside Drug (IUdR vs. 6-TG) Damage and IR Damage
We are interested in studying the differences in biochemical and molecular MMR processing of two different nucleoside analogs (IUdR and 6-TG) as well as the subsequent cell cycle and cell death signaling following MMR processing. Currently, we hypothesize that the differences in MMR processing support both a futile cycle repair model for MMR in 6-TG-DNA processing and a general damage sensor model for MMR in IUdR–DNA processing. These hypotheses are supported by experimental data and mathematical models reported by our group (Yamane et al., 2004, 2007; Yan et al., 2004; Yamane and Kinsella, 2005; Seo et al., 2006; Gurkan et al., 2007a; Zeng et al., 2007; Zeng and Kinsella, 2008) and briefly summarized as follows.
MMR Processing of 6-TG-Induced DNA Base Damage
We have previously shown that MMR+ cells are highly sensitive to cytotoxicity (>2–3 log cell kill) to 6-TG treatment compared to MMR− cells, with MMR+ cells showing an initial prolonged G2/M arrest followed by cell death after several cell replication cycles following 6-TG treatment (Berry et al., 1999, 2000; Yan et al., 2003). 6-TG is known to be incorporated into DNA in human cells in place of dGTP during the first S-phase and a small amount (∼1 per 104 bases) of incorporated 6-TG is rapidly methylated in situ to 6-thiomethylguanine (6-meTG) by endogenous S-adenosylmethionine (Swann et al., 1996). During the next replication cycle (without further 6-TG exposure), 6-meTG on the parental strand can pair with either a cytosine or a thymine and both 6-meTG:C and 6-meTG:T mispairs are recognized by MMR (Swann et al., 1996). According to the futile repair cycle model, MMR then recognizes these mismatches and initiates repair of the daughter strand causing a SSB (Yamane et al., 2004; Yan et al., 2004; Jiricny, 2006). Because 6-meTG in the parental strand is not removed, MMR repeatedly processes this damage at the same site, leading to confined SSB formation in the daughter strand to activate a G2/M checkpoint signaling cascade. Using an isogenic pair of MMR+ and MMR− human colon cancer cell lines, we previously demonstrated that more SSBs (but not DSBs) were produced in the MMR+ tumor cells at later times (after several cell doublings) following 6-TG treatment (Yan et al., 2003). The time course of SSB formation was temporally correlated with the time course of the 6-TG-induced G2/M arrest, which increased later (2–6 days), not earlier, after 6-TG removal in MMR+ cells (Yan et al., 2003). Thus, we argued that the formation of SSBs should be considered as a secondary signal produced by MMR processing after recognition of the primary signal, i.e., 6-meTG:C or 6-meTG:T mispairs. Additionally, we speculated that the continued formation of SSBs provides a link to the subsequent G2/M signaling.
In our follow-up studies of MMR futile cycle processing of 6-TG, (Yamane et al., 2004; Yan et al., 2004) we demonstrated that the ATR/Chk1 pathway is responsible for the delayed (2–3 days) signaling of the MMR-mediated G2 arrest following 6-TG treatment. Using chemical inhibitors and small interfering RNA (siRNA) knockdown of ATR and Chk1, we showed that the damage signaling kinases ATR and Chk1 increase within 1 day following 6-TG treatment and correlate temporally with a G2 arrest (Yan et al., 2004). In contrast, Chk2 (another cell cycle kinase) increased gradually over 2–6 days following 6-TG treatment, which correlated with a later increased tetraploid (4C) G1 arrest. Thus, our simplified current model of MMR processing of 6-TG (Figure 1) suggests that a MMR-mediated 6-TG-induced G2 arrest is ATR/Chk1 dependent but ATM/Chk2 independent. Both apoptotic (type 1) and autophagic (type 2) programmed cell death pathways are activated using p53-dependent and p53- independent mechanisms (Yamane et al., 2004, 2007; Yan et al., 2004; Yamane and Kinsella, 2005; Zeng et al., 2007; Zeng and Kinsella, 2008). More recently, we have shown that 6-TG-induced autophagy by MMR processing is positively regulated by both BNIP3 and mTOR/S6K1 activation (Zeng and Kinsella, 2008, 2010). Additionally, an ATM- and ATR-independent activation of Chk2 appears involved in a later 4C G1 arrest, which blocks cells that escape from the initial G2 checkpoint arrest following 6-TG treatment. We speculate that the formation of a 4C G1 cell population may also contribute to the enhanced cell death by senescence in MMR+ cells following 6-TG treatment.
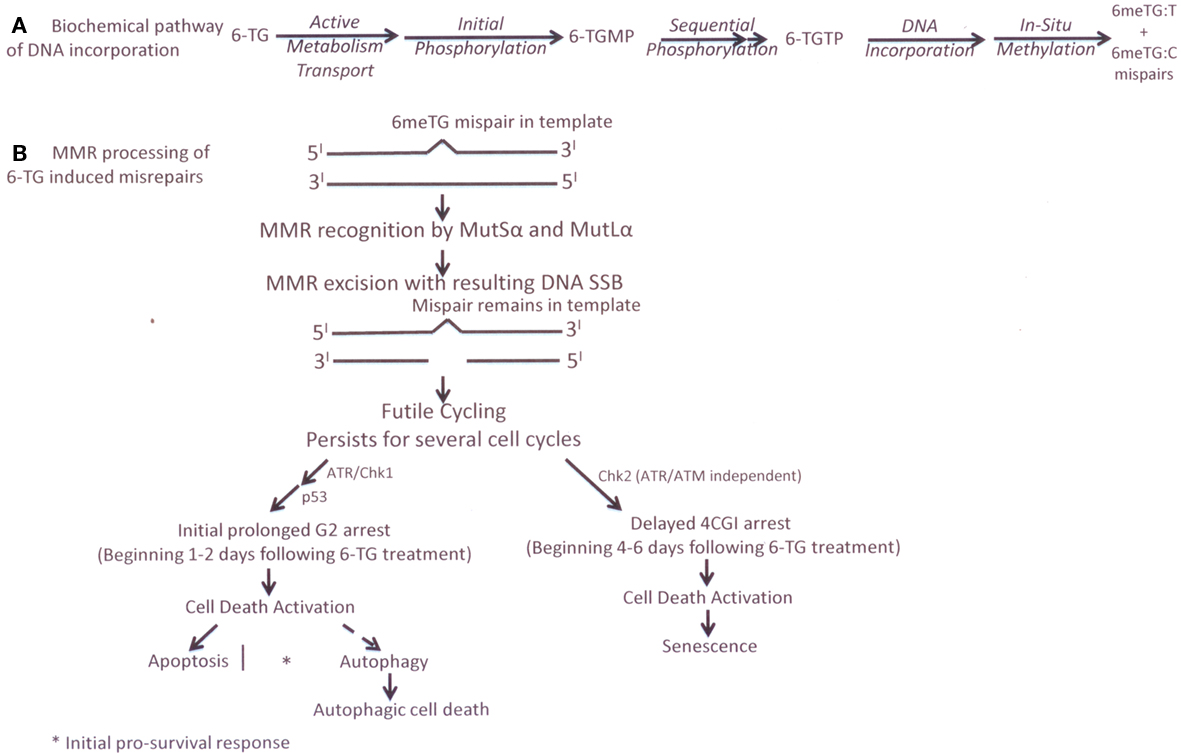
Figure 1. (A) cellular metabolism of 6-TG (B) current experimental biology futile cycle model of MMR processing of 6-TG-induced DNA base damage in MMR* cells.
The time course and the biologic components involved in our current model of the biochemical and molecular processing of 6-TG damage in a MMR+ human colon cancer cell line, based on our experimental data, (Yamane et al., 2004, 2007; Yan et al., 2004; Yamane and Kinsella, 2005; Zeng et al., 2007; Zeng and Kinsella, 2008) are presented in Figure 1.
MMR Processing of IUdR-Induced DNA Base Damage
In contrast to our experimental data on MMR processing of 6-TG (Figure 1), we found that MMR processing of IUdR–DNA damage results in modest cytotoxicity without a significant G2 cell cycle arrest and the time course of repair is reduced, i.e., over the initial 1–2 cell cycles following IUdR exposure, based on in vitro data using various isogenically matched human tumor and murine knock-out cell lines for both MSH2 and MLH1 genes/proteins (Berry et al., 1999, 2000, 2003). Biochemically, IUdR is rapidly converted intracellularly to IdUMP by the rate limiting enzyme, thymidine kinase, and then is sequentially phosphorylated to IdUTP which competes directly with dTTP for DNA incorporation during the first replication following drug exposure (Figure 2; Kinsella, 1996, 2009). Using MMR+/MMR− isogenic matched human tumor and mouse MEF cell lines and clinically relevant IUdR drug concentrations (1–10 μM) for treatment times of <1 cell doubling time, we found a 2–3 fold higher % IUdR–DNA incorporation in the MMR− vs. MMR+ cells immediately following drug exposure with little subsequent cytotoxicity (< <1 log) and no G2 cell cycle delay (Berry et al., 1999, 2000; Seo et al., 2006; Gurkan et al., 2007a). Additionally, the increased % IUdR–DNA incorporation persisted for several cell population doublings following IUdR treatment in MMR− cells without any delayed cytotoxicity compared to isogenic MMR+ cells, consistent with a post-replicative MMR processing which can repair (remove) IUdR–DNA incorporation in MMR+ cells similar to mismatched bases (e.g., G-T).
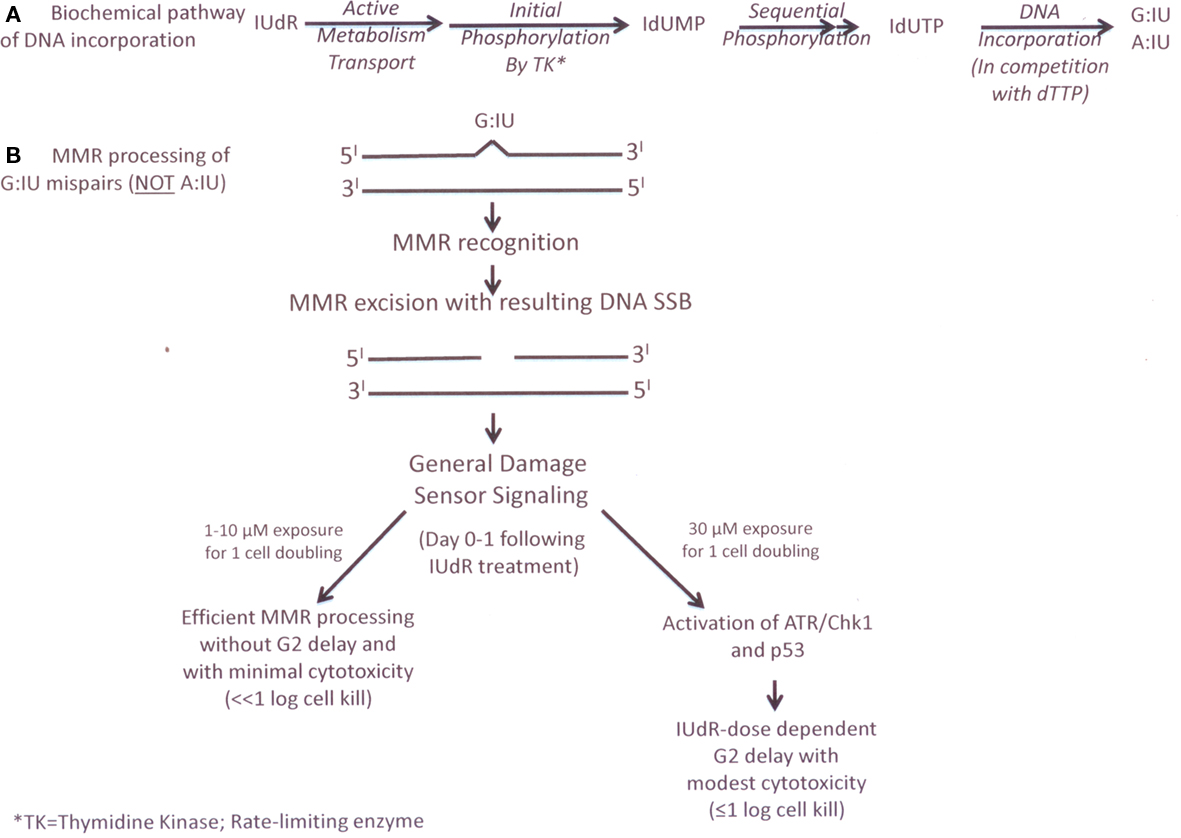
Figure 2. (A) cellular metabolism of IUdR (B) current experimental biology general damage sensor model of MMK processing of lUdR.
These in vitro results were initially surprising because IUdR would not be expected to result in a DNA mismatch following incorporation. Since the iodine atom is substituted at the five position of the pyrimidine ring, IUdR is not different from thymidine in its ability to form hydrogen bonds (Freese, 1959). However, it has been shown previously that mispairs can be generated during replication across DNA containing halogenated thymidine analogs like IUdR, including G:IU and A:IU mispairs (Rydberg, 1977). These data led us to hypothesize a general DNA damage sensor model for MMR processing of IUdR (Figure 2) that differs from futile cycling MMR processing of 6-TG (Figure 1). Additionally, based on the higher levels of IUdR–DNA incorporation in MMR− (e.g., MMR− or MSI-H human cancers) cells compared to MMR+ (e.g., normal proliferating tissues such as bone marrow) cells, we predicted a potential therapeutic gain for IUdR-mediated radiosensitization in MMR− human tumors, (Kinsella, 1996) that we demonstrated in vivo using an athymic mouse model with simultaneously implanted MMR+ and MMR− human colon cancer xenografts (Seo et al., 2004).
In a purified protein study, (Berry et al., 2003) we examined more direct interactions between MutSα and IUdR–DNA incorporation by testing the ability of human MutSα (the hMSH2/hMSH6 dimer involved in initial mismatch recognition) to bind to double-stranded DNA containing the physiologically relevant base mispairs A:IU and G:IU as well the two other possible mispairs, C:IU and T:IU. Using an electrophoretic mobility shift assay (EMSA) analysis, we found MutSα (but not MutSβ) “recognized” the G:IU mispair with high affinity (> than a G:T mispair in the same sequence context; Berry et al., 2003). From a biochemical perspective, of the possible pairings between Watson–Crick bases and IUdR, a G:IU would bear the greatest similarity to a G:T mismatch in DNA. However, the van-der-Waals radius of iodine is just slightly larger than that of the methyl group in the 5 position of thymidine, (Kinsella, 1996) which may cause a greater distortion of the DNA helix containing G:IU than would a G:T mismatch. This may explain the higher affinity of MutSα for G:IU than G:T in the same sequence context as we published (Berry et al., 2003). Using confocal microscopy, we also demonstrated co-localization of IUdR–DNA and MSH2 protein (part of the MutSα heterodimer) in discrete nuclear foci in synchronized MMR+ human tumor cells following IUdR treatment in vitro (Berry et al., 2003). These results were the first demonstration of recognition of IUdR–DNA mispairs by MMR and confirm our prior data showing lower% IUdR–DNA levels in MMR+ vs. MMR− cells during and following IUdR treatment (Berry et al., 1999, 2000).
We also found that MutSα has very little binding affinity for A:IU, C:IU, or T:IU mispairs in the same sequence context, suggesting that MMR is not involved in processing these other possible IU mispairs in DNA (Berry et al., 2003). These protein data are consistent with our previous cellular data, in which we found that, although the levels of IUdR–DNA decrease over several cell population doublings in MMR+ cells, approximately 5% of the % thymidine substitution by IUdR remained (Berry et al., 1999, 2000). Thus, it is likely that IUdR pairs with other bases in addition to G:IU and we speculate that these other pairs (e.g., A:IU) are not recognized nor processed by MMR.
However, we also note some similarity in MMR processing of G:IU mispairs compared to MMR processing of 6-TG-DNA mispairs. In another in vitro study, (Seo et al., 2006) we found that the concomitant or sequential use of chemical inhibitors of ATR (caffeine) and of Chk1 (UCN-01) or the use of MMR+ but ATR− (by siRNA knockdown) cells can alter processing of IUdR–DNA incorporation, resulting in a IUdR-dose-dependent increase in DNA SSB formation (by alkaline comet assay) and a persistence of high% IUdR–DNA levels following IUdR treatment. Nevertheless, using clinically relevant IUdR concentrations (1–10 μM) in these modified cell lines, we still did not observe any significant G2 cell cycle delay nor cell death in the next 2 days following IUdR treatment (Seo et al., 2006). However, exposure to higher IUdR concentrations (>30 μM) results in modest cytotoxicity (by apoptosis and autophagy) and a G2 cell cycle delay. Our simplified current model and time course of this “general damage sensor” MMR processing of IUdR–DNA damage are detailed in Figure 2.
MMR Processing of IR-Induced DNA Base Damage
We have previously reported that the MLH1−, MMR− human colon carcinoma HCT116 cell line and MLH1−, MMRnull mouse embryonic fibroblast EF5 cells show a reduced and shorter G2 arrest after high dose rate (HDR; 2–4 Gy/min) IR compared to their matched MLH1+, MMR+ cells. The MMR− cells also showed a modest reduced clonogenic survival (Davis et al., 1998). In a follow-up publication, we extended our initial findings to two additional matched MLH1− and MLH1+ human tumor cell lines, A2780/CP70 and RKO (Yan et al., 2001). The human ovarian cancer cell model (A2780/CP70) was previously established by Brown et al. (2003) while we established the in vitro human colon cancer model (RKO) using azacytidine, a demethylating agent, to result in increased MLH1 protein expression in RKO (MLH1−, MMR−) cells. These data confirmed our previous finding that the expression of MLH1 protein (establishing a MMR+ phenotype) results in a robust IR-induced G2 arrest, which is higher (up to 80% of cell population) and more sustained (up to two cell population doubling times) compared to the G2 arrest in irradiated genetically matched MLH1−, MMR− cells. Again, only small differences in clonogenic survival were noted using HDR IR.
Because the four MLH1 MMR− cell model systems used by our group in these two reports (Davis et al., 1998; Yan et al., 2001) differ in the cell type (i.e., mouse and different human tumor origins), in the genetic/epigenetic basis of MLH1 deficiency, and in the techniques used to establish MLH1-proficient counterparts (human chromosome 3 transfer or 5-deoxyazacytidine treatment for demethylation), our combined data suggest that the reduced and shorter G2 arrest following IR is a general characteristic of MLH1 MMR deficiency. In our initial study, (Davis et al., 1998) we also examined the role of p53 in this altered G2 arrest by knocking-out p53 function with E6 protein in the MLH1− HCT116 and the MLH1+ HCT116/3–6 cell lines. The presence or absence of the p53 protein did not affect the G2 arrest in the HCT116 cell system. By using two additional MLH1− human tumor cell lines that differ in p53 status (negative protein expression in A2780/CP70 and positive in RKO) in the more recent study, (Yan et al., 2001) we confirmed that the p53 protein does not have a significant impact on the G2 cell cycle response to HDR IR damage in MLH1− and MLH1+ cells. However, p53 activation may be important in MMR processing of certain chemically induced mismatches leading to both apoptotic and autophagic cell deaths as we described (Zeng et al., 2007) and illustrate in Figure 1 for 6-TG treatment.
We also examined a MSH2−, MMR− human endometrial cancer cell system (HEC59) to determine whether MSH2 has an effect on G2 arrest following IR treatment (Yan et al., 2001). We found that the MSH2-deficient HEC59 cells show the classical damage-tolerance response of MMR deficiency to 6-TG treatment as manifested by an increased cell survival and a decreased G2 arrest. We also found that, following HDR IR, the MSH2−, MMR− HEC59 cells exhibited a reduced and a shorter G2 arrest compared to the MMR+ HEC59/2–4 cells. The findings that both MLH1 deficiency and MSH2 deficiency exert the same effect on G2 arrest after HDR IR indicate that defective MMR partially impairs a HDR IR-induced G2 arrest following IR damage. We also showed that there are no differences in the G1 or S-phase cell cycle responses following IR in these isogenic pairs of MMR− and MMR+ cells.
IR is recognized to cause a large variety of DNA damage including DSBs, SSBs, cross-links, and oxidative base damage. A hypothesis that MMR is involved in the repair of IR-induced DNA damage has been proposed by our group and several others (Fritzell et al., 1997; Davis et al., 1998; DeWeese et al., 1998; Zeng et al., 2000; Yan et al., 2001, 2009; Brown et al., 2003; Cejka et al., 2004). These reports suggest that MMR may recognize and process IR-induced DNA mispairs formed during DNA replication that contain oxidative-damaged bases, such as 8-oxoguanine, which can mispair with thymine or adenine. MMR can recognize and remove these mispairs (Chang et al., 2002). Additionally, it was shown that non-cytotoxic levels of H2O2 inactivate both single base-pair mismatch and insertion-deletion loop repair activities of MMR in a dose-dependent fashion and that inactivation is most likely due to oxidative damage (Chang et al., 2002).
In our studies, (Davis et al., 1998; Yan et al., 2001) we also found that the extent and time course of tyrosine15-phosphorylation of cdc2 differs following HDR IR in MMR− vs. MMR+ cells. For up to one cell doubling time post-IR, phospho-Tyr15-cdc2 was increased in both MMR+ and MMR− cell lines, corresponding to the peak G2 arrest. However, the levels of phospho-Tyr15-cdc2 in the MMR− cell lines were lower compared to the MMR+ cell lines, again correlating with the difference in G2 peak between MMR− and MMR+ cells. After approximately 2 cell doubling times post-IR, levels of phospho-Tyr15-cdc2 decreased markedly in the MMR− cell lines, corresponding temporally to the loss of G2 arrest, while the levels of phospho-Tyr15-cdc2 remained high in the MMR+ cell lines. Based on these observations, we speculate the MMR system might help mediate an IR-induced G2 arrest by interacting with other proteins (not p53) that activate the cdc2 signaling pathway or MMR might interact directly with one or more components of the cdc2 signaling pathway. We speculate that MSH2 senses certain IR-induced DNA damage (e.g., oxidative damage) and MLH1 relays this damage signal to other proteins to regulate the cdc2 pathway.
Clonogenic survival following HDR IR (1–4 Gy/min) showed no marked differences between MMR+ and MMR− cells in the murine cells and human tumor cell model systems used in our studies and others (Fritzell et al., 1997; Davis et al., 1998; DeWeese et al., 1998; Zeng et al., 2000; Yan et al., 2001, 2009; Brown et al., 2003; Cejka et al., 2004). However, a much larger survival difference (>1 log cell kill) was found in MMR− vs. MMR+ human tumor cells following LDR IR (1–18 cGy/hr continuously for 24–96 h; Yan et al., 2009). In this LDR study, which mimics IR dose rates used clinically in prostate cancer brachytherapy, we found significant IR-resistance in MMR− cells with a reduced G2/M arrest and enhanced HPRT gene mutation rates. In contrast, MMR+ tumor cells showed enhanced cytotoxicity during and following LDR IR via both apoptotic and autophagic death pathways and a dose rate and total IR dose-dependent G2/M arrest, (Yan et al., 2009) similar to other reports (DeWeese et al., 1998; Zeng et al., 2000).
Interestingly, we also found an increased late-S-phase population in the MLH1+ cells during protracted LDR IR, (Yan et al., 2009) that is not previously reported. This late-S-phase accumulation is not easy to recognize without nocodazole treatment. Indeed, it is only made possible when the nocodazole-trapped mitotic cell population increases significantly, resulting in a shift of the G2/M peak to the right. Previously, Brown et al. (2003) have reported that MMR is required for the S-phase checkpoint following HDR IR. Our data also suggest a role for MLH1 in slowing late-S-phase progression during prolonged LDR IR, (Yan et al., 2009) although the mechanism underlying this late-S-phase accumulation in MLH1+ cells remains to be fully elucidated. We speculate that there may be interplay between MLH1 protein (or MMR) and homologous recombination in the late-S-phase during prolonged LDR IR as the persistent LDR IR leads to continuous generation of DNA damage and homologous recombination is believed to occur mainly in the late-S and G2-phases (Yan et al., 2009).
In our study, we did not find significant changes in the protein levels of many different DNA repair proteins during prolonged LDR IR (Yan et al., 2009). However, we did find that the levels of MLH1 protein and its dimeric partner PMS2 protein were elevated (up to four-fold increase in MLH1) after protracted LDR IR in the MLH1+ cells, whereas the levels of MSH2 protein and its dimeric partner MSH6 protein were concomitantly reduced. These results using the isogenic HCT116 cell lines were confirmed in two additional MLH1+ human cancer cell lines (i.e., HT29 colon cancer and U251 glioblastoma cell lines; Yan et al., 2009). In addition, a similar result is found in HEC59 endometrial cancer cells, (Yan et al., 2009) which are MSH2−, suggesting that functional MMR is not a prerequisite for MLH1 protein accumulation during prolonged LDR IR. Importantly, the increase in MLH1 protein levels seems to result from reduced MLH1 protein degradation.
The other DNA repair protein found to be altered in this LDR IR study was Rad51, which in contrast to MLH1, was found to be progressively decreased in response to prolonged LDR IR (Yan et al., 2009). Rad51, a strand transferase, is a key component in homologous recombination and overexpression of Rad51 results in increased homologous recombination capability. MMR is previously reported to be involved in homologous recombination (Surtees et al., 2004) because mismatches can be produced within the heteroduplex generated by strand exchange during DNA replication to allow bypass of unrepaired damage. During prolonged LDR IR, low levels of continuous DNA damage (including low levels of DSBs) result, as manifested by a sustained elevation of p53 and p21 proteins but without significant γH2AX induction (Yan et al., 2009). Because homologous recombination is one of the major repair pathway of IR-induced double-strand breaks and is most active during late-S-phase, our data showing the inverse correlation of MLH1 and Rad51 protein levels during LDR IR (Yan et al., 2009) lead us to speculate that altered MLH1 protein degradation (with enhanced MutLα levels) affects homologous recombination through reduced expression of Rad51. From a clinical-translational perspective, our data suggest that MLH1-proficiency may decrease cancer susceptibility following prolonged LDR IR, and may also increase the efficacy of localized prolonged LDR IR as used in cancer brachytherapy. A summary of the MMR processing of HDR IR and LDR IR, including the cell cycle and cytotoxic responses, is illustrated in Figure 3.
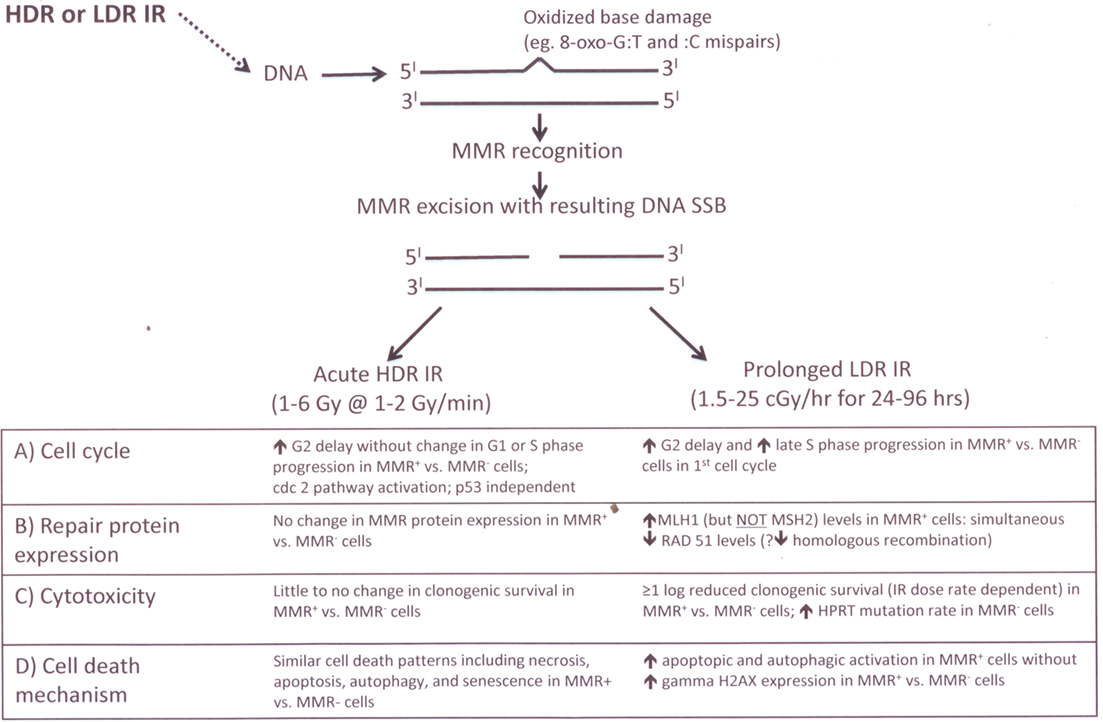
Figure 3. Current stains of experimental biology model of MMR processing of IR-induced base damage following acute IIDR and prolonged LDR IR exposures.
Development of in silico Models for MMR Processing Of IUdR and/or IR Damage and for MMR Effects on Cell Cycle Kinetics Following IUdR and/or IR Damage
Current Status in the Development of an in silico Model of MMR Damage Processing
We are developing computational models to quantitatively analyze the effects of MMR processing on the effectiveness of the treatment strategies where IUdR and/or IR are used. The models are developed at two different levels, at the molecular level and at the population level. The models that are being developed at the molecular level are introduced in this section, and the population level cell cycle models are discussed in Section B. The molecular level models are being developed to study the dynamics of MMR damage processing. We have first modeled the dynamics of the recognition sub-process of the MMR pathway as discussed below. We are currently working on incorporating the dynamics of the excision and repair synthesis sub-processes to this model. The complete model of the MMR process will be used to study the time course of the damage repair dynamics by the MMR pathway. The outputs of the entire MMR pathway model are the amount of damage repaired by the pathway as well as the remaining unrepaired damage in the DNA. This quantitative time course information on the amount of repaired and unrepaired DNA damage will then be used in combination with the cell cycle models discussed in Section B in order to determine the cellular responses under different damage conditions.
The MMR status of the cells is important for determining both the amount of IUdR incorporation and the cell cycle response to different amounts of IUdR incorporation. The models at the molecular level will provide the amount of IUdR incorporation, which will then be used in the cell cycle models to determine the corresponding cell cycle kinetics. The response to IR is dependent on both the amount of IUdR incorporated into the DNA as well as the cell cycle state when the cells are irradiated. The objective is to then optimize this response by using the computational models we are developing both at the molecular level and at the cell population level. The models will provide the time when the maximum difference in the amount of IUdR incorporation into DNA between MMR− and MMR+ cells occurs, with MMR− cells having incorporated more IUdR than MMR+ cells. At the same time, the cell cycle models will provide information on the percentage of cells in the different cell cycle states, which will be used to determine the time when most of the MMR− cells are in G2 phase where they are most sensitive to IR, and when most of the MMR+ cells are in S-phase where they are least sensitive to IR. The main objective of such optimization is to maximize the cell death in MMR− cells by IR while sparing the MMR+ cells.
To better understand the dynamics involved in our observed biochemical and molecular differential MMR processing of 6-TG (Figure 1) vs. IUdR (Figure 2) damage, as well as IR damage (Figure 3), we are applying dynamic systems methods to develop an in silico model of MMR. The MMR process is modeled as a stochastic hybrid system that incorporates both the continuous and discrete dynamic behaviors of MMR (Figure 4), as published in the Proceedings of the IEEE/NIH BISTI Life Science Systems and Applications Workshop, 2007 (Gurkan et al., 2007b). As indicated above, the MMR process involves a finite number of sub-processes, each involving biochemical dynamics for recognition, excision, and repair. Consequently, a stochastic hybrid systems modeling framework is the most appropriate for modeling the DNA MMR pathway because of the discrete- and continuous-state dynamics of each of the MMR sub-processes. The discrete-states in the model are the recognition, excision, and repair synthesis sub-processes, (Hespanha, 2006) that are defined in terms of the dynamics of an interacting multiprotein system that defines the biochemistry of the pathway. For the study of cancer treatment with chemotherapy and/or IR, another important discrete-state needs to be included in this modeling framework, the cell death state. Understanding the conditions that will drive the process to cell death is crucial for effective translational research aimed at developing more effective cancer therapeutics.
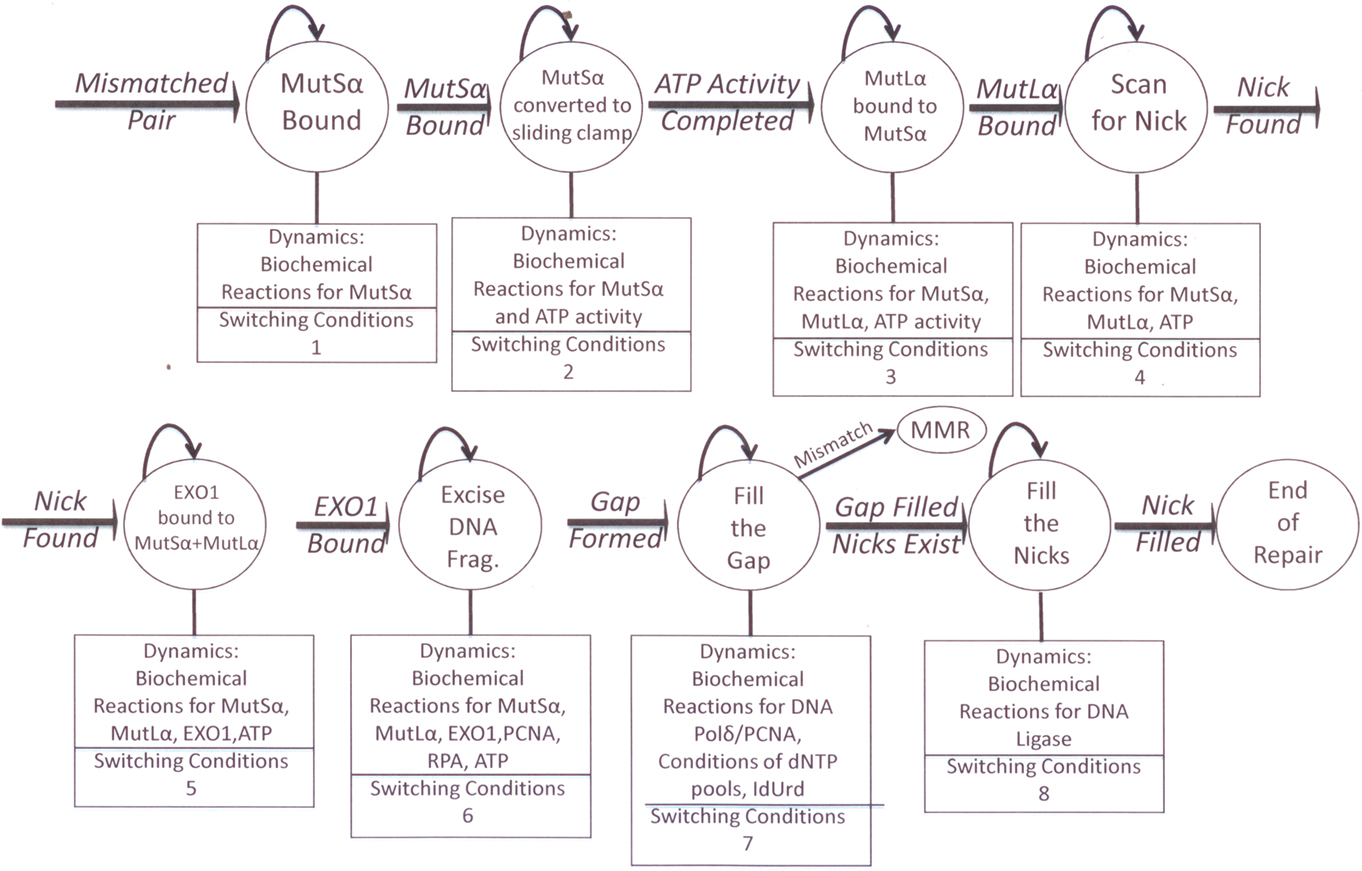
Figure 4. Modeling framework: hybrid model human DNA MMR.
The hybrid model (Figure 4) has the following states: binding of MutS, binding of MutL, excision of the DNA fragment, DNA resynthesis and filling of the nick by ligation together with any fault states that are associated with failure modes that can occur in the process. For example, the fault states can be used to model how MMR+ cells are transformed to MMR− cells, and this will facilitate a more detailed investigation into how various types of MMR defects can be detected and diagnosed using in vitro and in vivo experimental data. Because the process evolves probabilistically, for example the binding of the MutS and MutL proteins are random events, a stochastic hybrid modeling framework is the most appropriate. Further, the uncertainty of enzyme concentrations and activities that define the biochemical reactions of the pathway is best modeled using non-deterministic (probabilistic) parameter sets. In this model, the ATP activity for the binding of MutS and MutL, the excision of the DNA, the resynthesis, and the ligation process have continuous dynamics defined by the biochemical activities of the corresponding enzymes. The transitions between the discrete-states in the MMR process can be modeled as discrete events where the transition rules (switching conditions) are defined by protein concentrations or possibly in terms of the types of conformational changes. The conditions that drive the cells into the cell death state of the hybrid model are also studied because of their importance in cancer therapeutics.
A crucial component in the modeling process is the laboratory data that are necessary for model development. The temporal resolution (sampling) as well as the quantification of the data are critical items that need to be addressed. The main problem in the current cancer biology literature is the lack of appropriate time course data with the required time resolution and level of quantification. This limitation makes it difficult to develop realistic models because the parameters estimated using the available experimental data are generally far from being biologically meaningful.
Another major problem is the multiple and diverse sources of experimental data for the MMR pathway proteins. The activities for the enzymes that are measured often differ by orders of magnitude among different experiments and sources. In order to obtain biologically realistic and meaningful models, it is important that the data are obtained from the same laboratory under the same experimental conditions. Quantification of the experimental data is another problem that needs to be resolved. This problem arises especially when protein concentrations are studied using western blot analysis where experimental data are obtained as relative rather than absolute measurements. These data cannot be used directly for estimating model parameters.
Given the limitations of the existing literature, we have recently begun to study the initial step in MMR processing (i.e., MutSα binding) of G:IU vs. G:T mispairs using a purified protein system as described by the Modrich laboratory (Constantin et al., 2005). To date, we have made modest progress in generating these preliminary experimental data and subsequent computational modeling as described below. To begin to generate the experimental data, we were required to adapt and/or modify several techniques from the literature as follows.
First, as opposed to the construction of a G:T mismatch, nucleotide based mismatches such as G:IU require the use of synthetic oligomers, as IUdR cannot be produced in cells or by PCR methods. Consequently, we developed several more efficient modifications to the method of Wang and Hays (2006) for construction of a G: IU incorporated plasmid substrate. Our modifications include the use of a different combination of endonuclease enzymes, Nhe1/BciV1, and two new redesigned plasmids, pWDAH1A and pWDSH1B (Du, 2011). We reported a high efficiency (up to 90%) of these mismatch substrates and confirmed recognition using a functional assay (Du, 2011).
Second, for MutSα heterodimer purification, we obtained a MutSα expression baculovirus from the Modrich lab and used their purification strategy (Genschel, 2006). Briefly, SF9 insect cells (Invitrogen, Carlsbad, CA, USA) expressing MutSα protein, infected at a MOI (multiplicity of infection) of two were grown in a 1 l flask containing 300 ml of serum-free HyQ SFX media (HyClone, Logan, UT, USA) at 27°C. After 60 h of incubation, cells were collected by centrifugation, and the cell pellet was resuspended Logan, UT pended in media. This was followed by a four-step column purification involving initial passage through a ssDNA-cellulose column (USB, Cleveland, OH, USA), then through a HiTrap Q FF anion exchange column (GE Healthcare, Piscataway, NJ, USA) in an AKTA purifier 10 UPC machine (GE Healthcare), and finally, twice through a Mono Q 5/50 GL column (GE Healthcare). At each step, western blotting and Coomassie Blue staining were used to identify the target proteins. Final purity was estimated to be ≥95% as illustrated in the 1D SDS-PAGE gel silver staining in Figure 5.
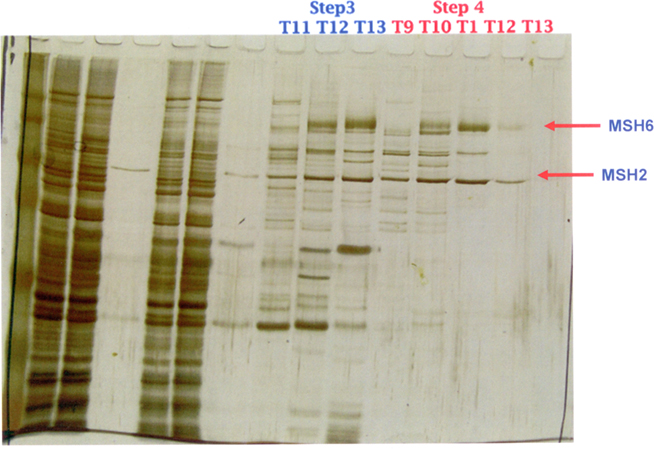
Figure 5. SDS-PAGE Gel of Purification results for MutSa (MSH2 – MSH6).
Third, we used the surface plasmon resonance (SPR) technique to generate our preliminary data on the association and dissociation kinetics of MutSα binding to the dsDNA 120 bp mismatch substrates containing the G:T and G:IU mispairs described above, as well as a G:C control. The application of the principles of SPR to measure the kinetics of bimolecular interactions are well described in the literature, (Myszka, 1997) and the SPR protocol from the Modrich lab was used (Blackwell et al., 2001). Briefly, a Biacore 3000 System and a Sensor Chip SA (GE Healthcare) were used (Blackwell et al., 2001). The 5′ end of the mismatch substrates were biotynlated to facilitate attachment to the sensor surface and MutSα with ATP (1:1) was injected at a flow rate of 10–40 μl/min followed by dissociation time of at least 2 min. The association and dissociation constants, as well as the equilibrium dissociation constants were calculated using Biacore evaluation software and the results are summarized in Table 1.

Table 1. Association and dissociation rate constants (ka and kd), and equilibrium dissociation constant (KD) for substrates that contain G:C, G:T, or G:IU obtained from surface plasmon resonance analysis.
Next, stochastic simulations are used to study the binding of MutSα to mispairs where the rate constants are obtained from the Biacore analysis. The reactions used in the simulations are as follows:
MutSα + DNA (with G:C or G:T or G:IU) ↔ MutSα:DNA.
The association and dissociation rate constants used in the simulations are given in Table 1. The substrate concentration was taken to be 5 nM (12612 molecules for a volume of 4.2 × 10−12l), and the MutSα concentration was 10nM (25224 molecules). The simulation results are shown in Figure 6.
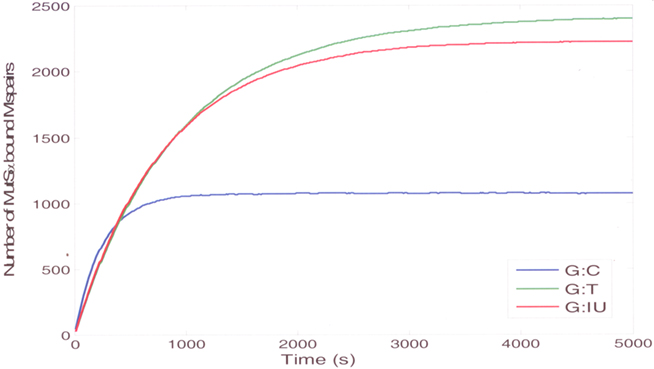
Figure 6. The stochastic simulation results (mean curves) for the binding of MutSa to substrates that contain G:C (blue curve), G:T (green curve), or G:IU (red curve).
We have used the exact stochastic simulation algorithm (SSA) of Gillespie (1976, 1977) originally developed for the analysis of chemical reaction systems where the number of molecules of some of the reactants can become small. When the molecule numbers are large, the SSA results are the same as the deterministic ordinary differential equation (ODE) system solutions. The applicability of the SSA to reactions with small or large numbers of molecules makes it an attractive method to use, because the molecule numbers in the cell can become small, and the ODE modeling approach is not valid. The SSA generates numerical realizations of the stochastic processes defined by a chemical master equation using a Monte Carlo simulation strategy. The algorithm is based on the calculation of the probability that a particular reaction occurs on an infinitesimal time interval. The output of the stochastic simulation is the time profiles of the molecular concentrations of the reactants. For MutSα binding reactions, the output is the number of mispairs bound by MutSα.
Details of the derivations and calculations involved in SSA can be found in detail in prior publications from Gillespie (1976, 1977). We summarize the steps in Table 2, and we have used Matlab® (The MathWorks, Inc., Natick, MA, USA) to implement SSA. The simulations need to be repeated, and the average output curve needs to be calculated from the outputs of individual simulations. We have simulated the binding and unbinding reactions until they reach the steady-state for 200 times, and we have averaged the outputs of these 200 simulations.

Table 2. The steps in the SSA algorithm and the implementation details.
The simulation results agree with the experimental data that show that the binding affinity of MutSα to the normal substrate without mispairs is low, and it is highest for the substrate with G:T mispairs (Figure 6). The binding profile of MutSα to a substrate with a G:IU is very similar to a substrate with G:T mispair. The simulation results show that, at steady-state, the amount of MutSα bound to DNA is highest for DNA containing G:T mispairs indicating that the affinity of MutSα is highest for DNA containing G:T mispairs. The amounts of MutSα bound to DNA containing G:IU mispairs at steady-state is comparable to the amounts in case of G:T mispairs agreeing with the experimental results showing that the affinities in the case of G:T mispairs and G:IU mispairs are comparable. The amount of MutSα bound DNA with G:C as the control at steady-state is almost half of the amounts for DNA with G:T and G:IU mispairs, agreeing with the experimental results indicating low affinity to DNA containing normal G:C pairs.
The stochastic simulations using data obtained from purified protein experiments are used to computationally model the kinetics of the recognition of different mispairs by the DNA MMR pathway. Both the experimental data and the simulation results showed that the MMR system recognizes the damage created by IUdR in the DNA in the form of G:IU mispairs. We are currently working toward the development of a computational model for the dynamics of the complete MMR pathway.
The iterative nature of our systems biology approach to develop in silico models to study MMR processing of drug and/or IR damage is described as follows. The first stage of the model development is parallel to the wet-lab reconstitution of the MMR pathway using a purified protein system. In this stage, the enzymatic steps of the pathway are reconstituted in vitro to study the kinetics of the enzymatic activities. Although the MMR pathway has been reconstituted before in the literature, (Constantin et al., 2005; Zhang et al., 2005) the kinetics of the enzymes were not measured in these experiments. It is crucial to quantify these dynamics for development of the computational model of the MMR process. Although some of these enzyme activities can be found in the literature, not all of these activities are measured in the same experimental setup. It is very important for dynamic system modeling of the MMR process that the data are consistent and are obtained from the same experimental source. Model parameters (e.g., kinetic constants and propensities of reactions) are then directly calculated from the available experimental measurements. The model developed for the reconstitution system is then used to study the MMR process dynamics through a structured set of hypotheses driven in silico experiments.
In the second stage, nuclear extracts are used in the experiments in place of purified proteins. These experiments provide the data that are necessary to analyze the dynamics of the MMR process in cell lines. In the third stage of modeling, data from cellular system experiments are used to further refine the models. Simulations for different initial damage concentrations and different protein concentrations are used to capture actual experimental outputs and to relate cell repair capacity to the amount of damage. These in silico scenarios are tested and verified by wet-lab experiments. Experimental results are used to modify and validate the model through an iterative process, and the final cellular models can then be used to accurately predict the dynamics of repair responses to different treatment strategies.
Current Status in the Development of an in silico Model of MMR Effects on Cell Cycle Kinetics Following IUdR and/or IR Damage
Cell cycle kinetics can be modeled using deterministic and probabilistic computational approaches. Clyde et al. (2006) provide a review of different cell cycle models and illustrate how mathematical modeling can be used to identify new targets for drug and small molecule development in cancer and other diseases of unregulated proliferation. In general, deterministic ODE models of biochemical and molecular reactions are developed using a bottom-up mechanistic approach. However, the continuous evolution of an ever-increasing number of cell cycle regulatory proteins and protein complexes, with both positive and negative feedbacks, complicates the adaptability of these deterministic models to realistic biological environments (Cain and Chau, 1997; Bernard et al., 2003; Chen et al., 2004; Novak and Tyson, 2004; Toettcher et al., 2009). Additionally, these models rely heavily on time sensitive quantitative kinetic data, often requiring multiple repeated measurements over time intervals of seconds to minutes (Chen et al., 2004; Toettcher et al., 2009; Ubezio et al., 2009).
In contrast, probabilistic models developed using a top-down approach to cell cycle dynamics incorporate quantitative measurements from flow cytometry and other related techniques over time intervals of hours to days. We recently published such a top-down approach to assess differences in cell cycle dynamics in MMR− and MMR+ human tumor cell populations with and without IUdR treatment (Gurkan et al., 2007a). In this prior study, we analyzed differences in clonogenic survival,% IUdR–DNA cellular incorporation and cell cycle kinetics by flow cytometry using initially synchronized isogenic MMR+ and MMR− HCT116 human colorectal cancer cell lines, treated with varying concentrations of IUdR (0–30 μM) for up to one cell cycle (cell doubling) time (24 h). We found that initially synchronized MMR− cells have a shortened (2 hour) G2/M phase, even without IUdR treatment, compared to MMR+ cells. The additional damage to IUdR (principally manifested as G:IU mispairs) caused an IUdR-dose-dependent increase in the G2/M arrest in MMR+ cells during the first cell cycle, while IUdR treated MMR− cells showed a late S-phase delay as well. We speculated that the late S-phase delay represented signaling damage recognition of the persistent (unrepaired) G:IU mispairs in MMR− cells by other DNA repair systems, including base excision repair and/or homologous recombination (Taverna et al., 2003; Turner et al., 2006; Kinsella, 2009). However, we found only modest (< <1 log) differences in clonogenic survival in the clinically relevant range of IUdR concentrations (1–10 μM) with lower survival in MMR− cells. In contrast, we have found marked (>1 log) decreased survival in MMR+ cells compared to MMR− cells following treatment with other nucleoside analogs, such as 6-TG and no cell cycle changes (particularly G2/M arrest) until at least the second or third cell cycle following drug treatment (Figure 1; Berry and Kinsella, 2001; Meyers et al., 2001; Yan et al., 2003; Kinsella, 2009).
Using our experimental flow cytometry and IUdR–DNA incorporation data, we developed two computational models (Gurkan et al., 2007a). The first was a stochastic model of the progression of the cell cycle states for one cell cycle in synchronized MMR+ and MMR− HCT116 cell populations (Figure 7). The second model defined the relation between the % of cells in the different cell cycle states and the corresponding% IUdR–DNA incorporation (as a surrogate measure of IUdR-mediated radiosensitization) at any time during the first cell cycle period. In a more recent study (unpublished), we adapt and modify our probabilistic cell cycle model to study the effects for up to two cell cycles of IUdR alone (10 μM), IR alone (5 Gy), and the combined use of IUdR followed by HDR IR. In our experimental design, HDR IR was delivered at three specific time intervals following release from synchronization when the cell populations were becoming asynchronous but a majority were still in G1 (13 h), S (16 h), or G2/M (21 h), respectively. Following IR, cells were sampled hourly using flow cytometry until hour 28 from release. To the best of our knowledge, this study is the most comprehensive experimental/computational analysis of a drug-IR interaction by flow cytometry for 1–2 cell population doublings.
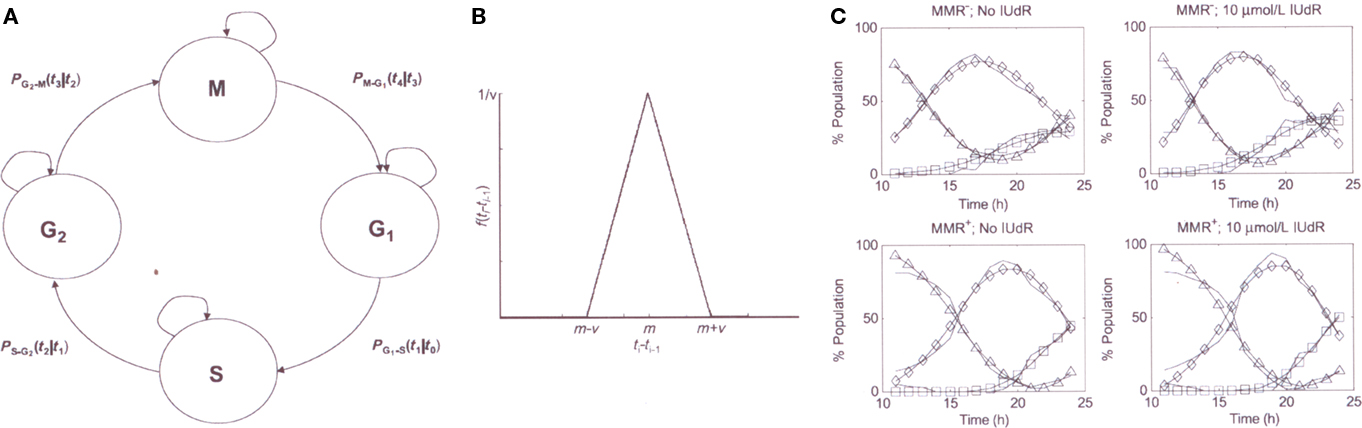
Figure 7. (A) stochastic in silica model of the cell cycle (B) example probability density function (C) cell cycle model outputs compared to experimental data p, experimental data (−); model outputs: GI (Δ), S (♦), and G2 (□) (adapted from reference Gurkan et al., 2007a).
We confirmed our prior observations (Gurkan et al., 2007a) of differences in cell cycle dynamics comparing MMR+ and MMR− cell populations with and without IUdR treatment. Untreated MMR− cells exit G1 into S ∼2 h faster than MMR+ cells during the first and second cell cycles while also demonstrating a shorter G2/M arrest. Following IUdR treatment, MMR+ cells show a more pronounced G2/M arrest in both the first and second cycles, while a more pronounced G1 arrest is found in MMR− cells during the second cell cycle.
A differential cell cycle response is also seen following HDR IR alone. Compared to untreated cells, IR treated MMR− cells show a more pronounced (3×) G2/M arrest in cycle one while also showing a longer (2 h) G1 arrest. In contrast, IR treated MMR+ cells have a similar G1 arrest and only a 2× increase in the G2/M arrest in the first cell cycle compared to untreated MMR+ cells. However, since the G2/M arrest following 5 Gy is so long in the first cell cycle, our sensitivity analyses of IR effects during the second cell cycle are limited by the overall time (28 h) of data sampling. No differences in clonogenic survival were found over a dose range of 2–10 Gy in these isogenic MMR+ and MMR− HCT116 cells, as previously reviewed (Jiricny, 2006; Kinsella, 2009). Future experiments will require lower IR doses (1–3 Gy/min) and possibly shorter sampling intervals over a longer time. We also plan to evaluate the effects of LDR IR on cell cycle kinetics of MMR+ vs. MMR− cells.
In summary, there are significant changes in cell cycle dynamics with IUdR alone, HDR IR alone, and IUdR + IR in both MMR+ and MMR− human tumor cells, based on our flow cytometry data and subsequent probabilistic modeling. Our current plan is to further develop these probabilistic models of IUdR ± IR interactions using data derived from in vivo systems of HCT116 (and other) human tumor cell lines as xenografts in athymic mice which will also allow for assessment of treatment effects (toxicities) on normal tissues (which are MMR+) to calculate a therapeutic index (Gurkan et al., 2007b).
One clear advantage to the in vivo or clinical use of IUdR to target MMR− tumors for radiosensitization is that the drug can also be used to measure tumor cell kinetics (Rodriguez et al., 1994). By measuring the labeling index (LI) using immunohistochemistry and the duration of S-phase (Ts) by flow cytometry at any time point, the potential tumor doubling time (Tpot) can be calculated using the formula: Tpot = Ts/LI × 0.9 (Rodriguez et al., 1994). We have previously hypothesized that for IUdR to be an effective in vivo/clinical radiosensitizer, an IUdR dosing schedule is needed to “label” nearly all (>95%) tumor cells with up to 5% IUdR–DNA cellular incorporation (Rodriguez et al., 1994; Kinsella, 1996). Using HCT116 MMR− tumor xenografts, we found that continuous intravenous IUdR infusions with a steady-state plasma concentration of 1 μM (clinically achievable) resulted in 95% LI with 2% IUdR–DNA incorporation following a 7 day infusion. Serial (daily) measurements of these parameters showed them to have exponential saturation kinetics with a half-life approximating Tpot (Rodriguez et al., 1994). More recently, we used the HCT116 MMR+ and MMR− tumor system as xenografts where MMR− tumors showed 100% LI and 3.5% IUdR–DNA incorporation after a 14 day, once daily, oral dosing of an IUdR prodrug (IPdR), which resulted in a 50% increase in tumor growth delay compared to IR alone, without significant bone marrow or GI toxicities from the prodrug (Seo et al., 2004). There is also limited clinical data on continuous infusion IUdR that can be used in support of our IUdR-IR modeling (Epstein et al., 1994; Schulz et al., 2004). In head and neck cancers, tumor biopsies taken following a 14 day IUdR infusion showed a 65% LI and 3% IUdR–DNA tumor cell incorporation (Epstein et al., 1994). A more recent Phase I clinical trial of continuous infusion IUdR × 28 days showed up to 4–5% IUdR–DNA incorporation in circulation granulocytes as a surrogate for tumor and steady-state plasma levels of 1.5 μM at the maximum tolerated dose (Schulz et al., 2004).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported, in part, by NIH Grant U56 CA112963, NIH Grant R21 CA140901, the DBJ Foundation, and the University Radiation Medicine Foundation.
References
Bernard, S., Pujo-Menjouet, L., and Mackey, M. C. (2003). Analysis of cell kinetics using a cell division marker: mathematical modeling of experimental data. Biophys. J. 84, 3414–3424.
Berry, S. E., Davis, T. W., Schupp, J. E., Hwang, H. S., de Wind, N., and Kinsella, T. J. (2000). Selective radiosensitization of drug-resistant MutS homologue-2 (MSH2) mismatch repair-deficient cells by halogenated thymidine (dThd) analogues: MSH2 mediates dThd analogue DNA levels and the differential cytotoxicity and cell cycle effects of the dThd analogues and 6-thioguanine. Cancer Res. 60, 5773–5780.
Berry, S. E., Garces, C., Hwang, K., Meyers, M., Davis, T. W., Boothman, D. A., and Kinsella, T. J. (1999). The mismatch repair protein, hMLH1, mediates 5-substituted halogenated thymidine analogue cytotoxicity, DNA incorporation, and radiosensitization in human colon cancer cells. Cancer Res. 59, 1840–1845.
Berry, S. E., and Kinsella, T. J. (2001). Targeting DNA mismatch repair for human tumor radiosensitization. Semin. Radiat. Oncol. 11, 300–315.
Berry, S. E., Loh, T., Yan, T., and Kinsella, T. J. (2003). Role of MutSα in the recognition of iododeoxyuridine in DNA. Cancer Res. 63, 5490–5495.
Bilbao, B., Lara, P. C., Ramirez, R., Henriquez-Hernandez, L. A., Rodriguez, G., Falcon, O., Leon, L., Perucho, M., Diaz-Chico, B. N., and Diaz-Chico, J. C. (2010). Microsatellite instability predicts clinical outcome in radiation treated endometrioid endometrial cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 76, 9–13.
Blackwell, L. J., Wang, S., and Modrich, P. (2001). DNA chain length dependence of formation and dynamics of hMutSα-hMutLα-heteroduplex complexes. J. Biol. Chem. 276, 33233–33240.
Brown, K. D., Rathi, A., Kamath, R., Beardsley, D. I., Zhan, Q., Mannino, J. L., and Baskaran, R. (2003). The mismatch repair system is required for S-phase checkpoint activation. Nat. Genet. 33, 80–84.
Brown, R., Hirst, G. L., Gallagher, W. M., McIlwrath, A. J., Margison, G. P., van der Zee, A. G., and Anthoney, D. A. (1997). hMLH1 expression and cellular responses of ovarian tumour cells to treatment with cytotoxic anticancer agents. Oncogene 15, 45–52.
Cahill, D. P., Levine, K. K., Betensky, R. A., Codd, P. J., Romany, C. A., Reavie, L. B., Batchelor, T. T., Futreal, A., Stratton, M. R., Curry, W. T., Iafrate, A. J., and Louis, D. A. (2007). Loss of mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 13, 2038–2045.
Cain, S. J., and Chau, P. C. (1997). Transition probability cell cycle model. Part II-non-balanced growth. J. Theor. Biol. 185, 69–79.
Cejka, P., Stojic, L., Marra, G., and Jiricny, J. (2004). Is mismatch repair really required for ionizing radiation-induced DNA damage signaling? Nat. Genet. 36, 432–433.
Chang, C. L., Marra, G., Chauhan, D. P., Ha, H. T., Chang, D. K., Ricciardiello, L., Randolph, A., Carethers, J. M., and Boland, C. R. (2002). Oxidative stress inactivates the human DNA mismatch repair system. Am. J. Physiol. Cell Physiol. 283, C148–C154.
Chen, K. C., Calzone, L., Csikasz-Nagy, A., Cross, F. R., Novak, B., and Tyson, J. J. (2004). Integrative analysis of cell cycle control in budding yeast. Mol. Biol. Cell 15, 3841–3862.
Choi, M. Y., Lauwers, G. Y., Hur, C., Willett, C. G., and Chung, D. C. (2007). Microsatellite instability is frequently observed in rectal cancer and influenced by neoadjuvant chemoradiation. Int. J. Radiat. Oncol. Biol. Phys. 68, 1584
Clyde, R. G., Brown, J. L., Hupp, T. R., Zhelev, N., and Crawford, J. W. (2006). The role of modeling in identifying drug targets for diseases of the cell cycle. J. R. Soc. Interface 22, 617–627.
Constantin, N., Dzantiev, L., Kadyriv, F. A., and Modrich, P. (2005). Human mismatch repair: reconstitution of a nick-directed bidirectional reaction. J. Biol. Chem. 280, 39752–39761.
Davis, T. W., Wilson-Van Patten, C., Meyers, M., Kunugi, K. A., Cuthill, S., Reznikoff, C., Garces, C., Boland, C. R., Kinsella, T. J., and Boothman, D. A. (1998). Defective expression of the DNA mismatch repair protein, hMLH1, alters the G2/M Cell Cycle checkpoint arrest following ionizing radiation. Cancer Res. 58, 767–778.
DeWeese, T. L., Shipman, J. M., Larrier, N. A., Buckley, N. M., Kidd, L. R., Groopman, J. D., Cutler, R. G., teRiele, H., and Nelson, W. G. (1998). Mouse embryonic stem cells carrying one or two defective MSH2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proc. Natl. Acad. Sci. U.S.A. 95, 11915–11920.
Du, W., and Kinsella, T.J. (2011). A rapid simple DNA mismatch repair substrate construction method. Front. Radiat. Oncol. 1:8. doi: 10.3389/fonc.2011.00008
Epstein, A. H., Lebovics, R. S., Goffman, T., Teague, D., Fuetsch, E. S., Glatstein, E., Okunieff, P., and Cook, J. A. (1994). Treatment of locally advanced cancer of the head and neck with 5′-iododeoxyuridine and hyperfractionated radiation therapy: measurement of cell labeling and thymidine replacement. J. Natl. Cancer Inst. 86, 1775–1780.
Freese, E. (1959). The specific mutagenic effect of base analogues on phage T4. J. Mol. Biol. 1, 87–105.
Friedman, H. S., Johnson, S. P., Dong, Q., Scholds, S. C., Rasheed, A., Bigner, S. H., Ali-Osman, F., Dolan, E., Colvin, O. M., Houghton, P., Germain, G., Drummond, J. T., Keir, S., Marcelli, S., Bigner, D. D., and Modrich, P. (1997). Methylator resistance mediated by mismatch repair deficiency in a glioblastoma multiforme xenograft. Cancer Res. 57, 2933–2936.
Friedman, H. S., McLendon, R. E., Kerby, T., Dugan, M., Bigner, S. H., Henry, A. J., Ashley, D. M., Krischer, J., Lovell, S., Rasheed, K., Marchev, F., Seman, A. J., Cokgor, I., Rich, J., Stewart, E., Colvin, O. M., Provenzale, J. M., Bigner, D. D., Haglund, M. M., Friedman, A. H., and Modrich, P. L. (1998). DNA mismatch repair and O6-alkylguanine-DNA alkyltransferase analysis and response to Temodal in newly diagnosed malignant glioma. J. Clin. Oncol. 16, 3851–3857.
Fritzell, J., Narayanan, L., Baker, S., and Glazer, P. M. (1997). Role of DNA mismatch repair in the cytotoxicity of ionizing radiation. Cancer Res. 57, 5143–5147.
Genschel, J., and Modrich, P. (2006). Analysis of the excision step in human DNA mismatch repair. Meth. Enzymol. 408, 273–284.
Gillespie, D. T. (1976). A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys. 22, 403–434.
Gillespie, D. T. (1977). Exact stochastic simulation of coupled chemical reactions. J. Physical Chem. 81, 2340–2361.
Gurkan, E., Schupp, J. E., Azhar, M. A., Kinsella, T. J., and Loparo, K. A. (2007a). Probabilistic modeling of DNA mismatch repair (MMR) effects on cell cycle dynamics and iododeoxyuridine (IUdR)-DNA incorporation: Implications for targeting MMR-deficient tumors for IUdR-mediated radiosensitization. Cancer Res. 67, 10993–11000.
Gurkan, E., Schupp, J. E., Kinsella, T. J., and Loparo, K. A. (2007b). “A conceptual modeling framework for the study of DNA mismatch repair pathway to improve therapeutic gain in cancer treatment,” in Proc of the IEEE/NIH BISTI 2007 Life Science Systems and Applications Workshop (LISSA 2007), Bethesda, MD.
Hespanha, J. P. (2006). Modeling and analysis of stochastic hybrid systems. Control Theor. Appl. IEE Proc. 153, 520–535.
Hsu, H. S., Wen, C. K., Tang, Y. A., Lin, R. K., Li, W. Y., Hsu, W. H., and Wang, Y. C. (2005). Promoter hypermethylation is the predominant mechanism in hMLH1 and hMSH2 deregulation and is a poor prognostic factor in nonsmoking lung cancer. Clin. Cancer Res. 11, 5410–5416.
Kinsella, T. J. (1996). An approach to the radiosensitization of human tumors. Cancer J. Sci. Am. 2, 184–193.
Kinsella, T. J. (2008). Update on radiosensitization by the halogenated thymidine analogs-molecular mechanisms of drug processing and cell death signaling: implications for further clinical trials. Cancer Biol. Ther. 7, 1567–1569.
Kinsella, T. J. (2009). Coordination of DNA mismatch repair and base excision repair. Processing of chemotherapy and radiation damage for targeting resistant cancers. Clin. Cancer Res. 15, 1853–1859.
Kinsella, T. J., Glennon, M. C., Kunugi, K. A., and Lindstrom, M. J. (1996). In vivo modulation of iododeoxyuridine metabolism and incorporation into cellular DNA by 5′-amino-5′-deoxythymidine in normal mouse tissues and two human colon cancer xenografts. Clin. Cancer Res. 2, 981–989.
Kinsella, T. J., Kinsella, M. T., Hong, S. W., Johnson, J. P., Burback, B., and Tosca, P. J. (2008). Toxicology and pharmacokinetic study of orally administered 5-iodo-2-pyrimidinone-2′-deoxyribose (IPdR) × 28 days in Fischer-344 rats: impact on initial phase I trial design of IPdR-mediated radiosensitization. Cancer Chemother. Pharmacol. 61, 323–334.
Kinsella, T. J., Kinsella, M. T., Seo, Y., and Berk, G. (2007). 5-iodo-2-pyrimidinone-2′-deoxyribose (IPdR)-mediated cytotoxicity and radiosensitization in U87 human glioblastoma xenografts. Int. J. Radiat. Oncol. Biol. Phys. 69, 1254–1261.
Kinsella, T. J., Kunugi, K. A., Vielhuber, K. A., McCulloch, W., Liu, S. H., and Cheng, Y. C. (1994). An in vivo comparison of oral 5-iodo-2′-deoxyuridine and 5-iodo-2-pyrimidinone-2′-deoxyribose toxicity, pharmacokinetics, and DNA incorporation in athymic mouse tissues and the human colon cancer xenograft, HCT116. Cancer Res. 54, 2695–2700.
Kinsella, T. J., Kunugi, K. A., Vielhuber, K. A., Potter, D. M., Fitzsimmons, M. E., and Collins, J. M. (1998). Preclinical evaluation of 5-iodo-2-pyrimidinone-2′-deoxyribose as a prodrug for 5-iodo-2′-deoxyuridine-mediated radiosensitization in mouse and human tissues. Clin. Cancer Res. 4, 99–109.
Kinsella, T. J., Schupp, J. E., Davis, T. W., Berry, S. E., Hwang, H. S., Warren, K., Balis, F., Barnett, J., and Sands, H. (2000a). Preclinical study of the systemic toxicity and pharmacokinetics of 5-iodo-2-pyrimidinone-2′-deoxyribose as a radiosensitizing prodrug in two, non-rodent animal species: implications for phase I study design. Clin. Cancer Res. 6, 3670–3679.
Kinsella, T. J., Vielhuber, K. A., Kunugi, K. A., Schupp, J., Davis, T. W., and Sands, H. (2000b). Preclinical toxicity and efficacy study of a 14-day schedule of oral 5-iodo-2-pyrimidinone-2′-deoxyribose as a prodrug for 5-iodo-2′-deoxyuridine radiosensitization in U251 human glioblastoma xenografts. Clin. Cancer Res. 6, 1468–1475.
Kishi, K., Doki, Y., Yano, M., Yasuda, T., Fujiwara, Y., Takiguchi, S., Kim, S., Higuchi, I., and Monden, M. (2003). Reduced MLH1 expression after chemotherapy is an indicator for poor prognosis in esophageal cancers. Clin. Cancer Res. 9, 4368–4375.
Klein, T. J., and Glazer, P. M. (2010). The tumor microenvironment and DNA repair. Semin. Radiat. Oncol. 20, 282–287.
Kondo, A., Safaei, R., Mishima, M., Niedner, H., Lin, X., and Howell, S. B. (2001). Hypoxia-induced enrichment and mutagenesis of cells that have lost DNA mismatch repair. Cancer Res. 61, 7603–7607.
Koshiji, M., To, K. K., Hammer, S., Kumamoto, K., Harris, A. L., Modrich, P., and Huang, L. E. (2005). HIF-1α induces genetic instability by transcriptionally downregulating MutSα expression. Mol. Cell 17, 793–803.
Lynch, H. T., and de la Chapelle, A. (2003). Hereditary colorectal cancer. N. Engl. J. Med. 348, 919–932.
Mackay, H. J., Cameron, D., Rahilly, M., Mackean, M. J., Paul, J., Kaye, S. B., and Brown, R. (2000). Reduced MLH1 expression in breast tumors after primary chemotherapy predicts disease-free survival. J. Clin. Oncol. 18, 87–93. [Erratum appears in J. Clin. Oncol. 18, 944].
Meyers, M., Hwang, A., Wagner, M. W., Bruening, A. J., Veigl, M. L., Sedwick, W. D., and Boothman, D. A. (2003). A role for DNA mismatch repair in sensing and responding to fluoropyrimidine damage. Oncogene 22, 7376–7388.
Meyers, M., Wagner, M. W., Hwang, H. S., Kinsella, T. J., and Boothman, D. A. (2001). Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 61, 5193–5201.
Mihaylova, V. T., Bindra, R. S., Yuan, J., Campisi, D., Narayanan, L., Jensen, R., Giordano, F., Johnson, R. S., Rockwell, S., and Glazer, P. M. (2003). Decreased expression of the DNA mismatch repair gene MLH1 under hypoxic stress in mammalian cells. Mol. Cell. Biol. 23, 3265–3273.
Myszka, D. G. (1997). Kinetic analysis of macromolecular interactions using surface Plasmon resonance biosensors. Curr. Opin. Biotechnol. 8, 50–57.
Novak, B., and Tyson, J. J. (2004). A model for restriction point control of the mammalian cell cycle. J. Theor. Biol. 230, 563–579.
Peltomaki, P. (2003). Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. Clin. Oncol. 21, 1174–1179.
Ribic, C. M., Sargent, D. J., Moore, M. J., Thibodeau, S. N., French, A. J., Goldberg, R. M., Hamilton, S. R., Laurent-Puig, P., Gryfe, R., Shepard, L. E., Tu, D., Redston, M., and Gallinger, S. (2003). Tumor microsatellite instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N. Engl. J. Med. 349, 247–257.
Rodriguez, R., Ritter, M. A., Fowler, J. F., and Kinsella, T. J. (1994). Kinetics of cell labeling and thymidine replacement after continuous infusion of halogenated pyrimidines in vivo. Int. J. Radiat. Oncol. Biol. Phys. 29, 105–113.
Rydberg, B. (1977). Bromouracil mutagenesis in escherichia coli evidence for involvement of mismatch repair. Mol. Gen. Genet. 152, 19–28.
Saif, M. F., Cheng, T.-C., Berk, G., and Kinsella, T. J. (2007). IPdR: a novel oral radiosensitizer. Expert Opin. Investig. Drugs 16, 1415–1424.
Samimi, G., Fink, D., Varki, N. M., Husain, A., Hoskins, W. J., ALberts, D. S., and Howell, S. B. (2000). Analysis of MLH1 and MSH2 expression in ovarian cancer before and after platinum drug-based chemotherapy. Clin. Cancer Res. 6, 1415–1421.
Sargent, D. J., Marsoni, S., Monges, G., Thibodeau, S. N., Labianca, R., Hamilton, S. R., French, A. J., Kabat, B., Foster, N. R., Torr, V., Bibic, C., Grothey, A., Moore, M., Zaniboni, A., Seitz, J-F., Sinicrope, F., and Gallinger, S. (2010). Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J. Clin. Oncol. 28, 3219–3226.
Schulz, C. A., Mehta, M. P., Badie, B., McGinn, C. J., Robins, H. I., Hayes, L., Chappell, R., Volkman, J., Binger, K., Arzoomanian, R., Simon, K., Alberti, D., Feierabend, C., Tutsch, K. D., Kunugi, K. K., Wilding, G., and Kinsella, T. J. (2004). Continuous 28-day iododeoxyuridine infusion and hyperfractionated accelerate radiotherapy for malignant glioma. Int. J. Radiat. Oncol. Biol. Phys. 59, 1107–1115.
Seo, Y., Yan, T., Schupp, J. E., Colussi, V., Taylor, K. L., and Kinsella, T. J. (2004). Differential radiosensitization in DNA mismatch repair proficient and deficient human colon cancer xenografts with 5-iodo-2-pyrimidinone-2′-deoxyribose. Clin. Cancer Res. 10, 7520–7528.
Seo, Y., Yan, T., Schupp, J. E., Radivoyevitch, T., and Kinsella, T. J. (2005). Schedule-dependent drug effects of oral 5-iodo-2-pyrimidinone-2′-deoxyribose as an in vivo radiosensitizer in U251 human glioblastoma xenografts. Clin. Cancer Res. 11, 7508–7515.
Seo, Y., Yan, T., Schupp, J. E., Yamane, K., Radivoyevitch, T., and Kinsella, T. J. (2006). The interaction between two radiosensitizers: 5-iododeoxyuridine and caffeine. Cancer Res. 66, 490–498.
Stojic, L., Brun, R., and Jiricny, J. (2004). Mismatch repair and DNA damage signaling. DNA Repair (Amst.) 3, 1091–1101.
Stupp, R., Mason, W. P., van den Bent, M. J., Weller, M., Fisher, B., Taphoorn, M. J., Belanger, K., Brandes, A. A., Marosi, C., Bogdahn, U., Curschmann, J., Janzer, R. C., Ludwin, S. K., Gorlia, T., Allgeier, A., Lacombe, D., Cairncross, J. G., Eisenhauer, E., and Mirimanoff, R. O. for the European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups and The National Cancer Institute of Canada Clinical Trials Group. (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996.
Surtees, J. A., Argueso, J. L., and Alani, E. (2004). Mismatch repair proteins: key regulators of genetic recombination. Cytogenet. Genome Res. 107, 146–159.
Swann, P. F., Waters, T. R., Moulton, D. C., Xu, Y. Z., Zheng, Q., Edwards, M., and Mace, R. (1996). Role of post replicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 273, 1109–1111.
Taverna, P., Hwang, H. S., Schupp, J. E., Radivoyevitch, T., Session, N. N., Reddy, G., Zarling, D. A., and Kinsella, T. J. (2003). Inhibition of base excision repair potentiates iododoxyuridine-induced cytotoxicity and radiosensitization. Cancer Res. 63, 838–846.
Toettcher, J. E., Loewer, A., Ostheimer, G. J., Yaffe, M. B., Tidor, B., and Lahav, G. (2009). Distinct mechanisms act in concert to mediate cell cycle arrest. Proc. Natl. Acad. Sci. U.S.A. 106, 785–790.
Turner, D. P., Cortellino, S., Schupp, J. E., Caretti, E., Loh, T., Kinsella, T. J., and Bellacosa, A. (2006). The DNA N-glycosylase MED1 exhibits preference for halogenated pyrimidines and is involved in the cytotoxicity of 5-iododeoxyuridine. Cancer Res. 66, 7686–7693.
Ubezio, P., Lupi, M., Branduardi, D., Cappella, P., Cavallini, E., Colombo, V., Matera, G., Natoli, C., and Tomasoni, D. (2009). Quantitative assessment of the complex dynamics of G1, S, and G2-M checkpoint activities. Cancer Res. 69, 5234–5240.
Wang, H., and Hays, J. B. (2006). Construction of MMR plasmid substrates and analysis of MMR error correction and excision. Methods Mol. Biol. 314, 345–353.
Yamane, K., and Kinsella, T. J. (2005). Casein kinase 2 regulates both apoptosis and the cell cycle following DNA damage induced by 6-thioguanine. Clin. Cancer Res. 11, 2355–2363.
Yamane, K., Schupp, J. E., and Kinsella, T. J. (2007). BRCA1 activates a G2/M cell cycle checkpoint by 6-TG-induced DNA mismatch damage. Cancer Res. 67, 6286–6292.
Yamane, K., Yan, T., Taylor, K., and Kinsella, T. J. (2004). Mismatch repair-mediated G2/M arrest by 6-thioguanine involves the ATR-Chk1 pathway. Biochem. Biophys. Res. Commun. 318, 297–302.
Yan, T., Berry, S. E., Desai, A. B., and Kinsella, T. (2003). DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin. Cancer Res. 9, 2327–2334.
Yan, T., Desai, A. B., Jacobbeger, J. W., Sramroski, R. M., Loh, T., and Kinsella, T. J. (2004). Chk1 and Chk2 are differentially involved in mismatch repair-mediated 6-thioguanine-induced cell cycle checkpoint responses. Mol. Cancer Ther. 9, 1147–1157.
Yan, T., Seo, Y., and Kinsella, T. J. (2009). Differential cellular responses to prolonged low dose rate ionizing radiation in MLH1-proficient and-deficient colorectal cancer HCT116 cells. Clin. Cancer Res. 15, 6912–6920.
Yan, T., Schupp, J. E., Hwang, H. S., Wagner, M. W., Berry, S. E., Strickfaden, S., Veigl, M. L., Sedwick, W. D., Boothman, D. A., and Kinsella, T. J. (2001). Loss of DNA mismatch repair imparts defective cdc2 signaling and G2 arrest responses without altering survival after ionizing radiation. Cancer Res. 61, 8290–8297.
Zeng, M., Narayanan, L., Xu, X., Prolla, T. A., Liskay, R. M., and Glazer, P. M. (2000). Ionizing radiation-induced apoptosis via separate Pms2- and p53-dependent pathways. Cancer Res. 60, 4889–4893.
Zeng, X., and Kinsella, T. J. (2008). Mammalian target of rapamycin and S6 kinase 1 positively regulates 6-thioguanine induced autophagy. Cancer Res. 68, 2384–2390.
Zeng, X., and Kinsella, T. J. (2010). BNIP3 is essential for mediating 6-thioguanine-induced autophagy following DNA mismatch repair processing. Cell Res. 20, 665–675.
Zeng, X., Yan, T., Schupp, J. E., Seo, Y., and Kinsella, T. J. (2007). DNA mismatch repair initiates 6-thioguanine-induced autophagy through p53 activation in human tumor cells. Clin. Cancer Res. 13, 1315–1321.
Keywords: mismatch repair, ionizing radiation, iododeoxyuridine, systems biology
Citation: Kinsella TJ, Gurkan-Cavusoglu E, Du W and Loparo KA (2011) Integration of principles of systems biology and radiation biology: toward development of in silico models to optimize IUdR-mediated radiosensitization of DNA mismatch repair deficient (damage tolerant) human cancers. Front. Oncol. 1:20. doi: 10.3389/fonc.2011.00020
Received: 09 May 2011;
Accepted: 12 July 2011;
Published online: 10 August 2011.
Edited by:
James B. Mitchell, National Institutes of Health, USAReviewed by:
Michael L. Freeman, Vanderbilt University School of Medicine, USAJohn Cook, National Cancer Institute, USA
Copyright: © 2011 Kinsella, Gurkan-Cavusoglu, Du and Loparo. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Timothy J. Kinsella, Department of Radiation Oncology, Rhode Island Hospital, Physicians Office Building, Suite 130, 110 Lockwood Street, Providence, RI 02903, USA. e-mail:dGtpbnNlbGxhQGxpZmVzcGFuLm9yZw==