
 Julia M. Fraile
Julia M. Fraile
- Departamento de Bioquímica y Biología Molecular, Facultad de Medicina, Instituto Universitario de Oncología, Universidad de Oviedo, Oviedo, Spain
The “oncogene addiction” concept refers to the dependence of cancer cells on the function of the oncogenes responsible for their transformed phenotype, while the term “non-oncogene addiction” has been introduced to define the exacerbated necessity of the normal function of non-mutated genes. In this Perspective, we focus on the importance of proteolytic enzymes to maintain the viability of cancer cells and hypothesize that most, if not all, tumors present “addiction” to a number of proteolytic activities, which in turn may represent valuable targets of anti-cancer therapies, even without being mutated or over-expressed by the malignant cells.
Progression from normalcy to malignancy results from the acquisition of several fundamental changes in cellular behavior, which in turn derive from the accumulation of a large number of genetic and epigenetic changes (Galluzzi et al., 2011; Hanahan and Weinberg, 2011). A few of the genetic alterations present in a cancer cell are essential for the development of the cancer phenotype (driver mutations) and are accompanied by a much larger number of incidental passenger mutations that do not contribute directly to oncogenesis but still may have an impact on the biology of the tumor cells (Haber and Settleman, 2007). The heterogeneous arrangement of molecular abnormalities present in cancer cells gives rise to a complex and largely unpredictable scenario of interwoven alterations in signaling and metabolic pathways (Wood et al., 2007). The multiple genetic interactions taking place at the cancer genome are responsible for the biological resilience of tumor cells, but also for specific weaknesses which, if appropriately exploited, could provide valuable targets of therapeutic intervention (Ashworth et al., 2011).
Cancer therapy is based on the increased susceptibility of tumor cells and tissues to different agents. Drugs traditionally used for cancer treatment present anti-tumor effect due to their preferential action against rapidly proliferating cells. Despite the large amount of resources invested in the development of novel drugs and in the optimization of administration schemes, these approaches are limited by the lack of specificity, which often results in unacceptable toxicity, and the evolution of the cancer cell population to eventually become resistant to the treatment.
Oncogene Addiction
In line with the multiple efforts to develop more efficient and selective therapies based on targeting tumor-specific traits, the concept of “oncogene addiction” was introduced to emphasize the apparent dependency of cancer cells on one or a few genes for the maintenance of the malignant phenotype (Weinstein, 2002; Weinstein and Joe, 2006, 2008). Important support for this concept derived from studies with genetically modified animal models. Thus, Ron DePinho and colleagues reported that oncogenic H-Ras is essential for tumor maintenance in a doxycycline-inducible H-RasV12G mouse melanoma model null for the tumor suppressor INK4a, as downregulation of the oncogene resulted in tumor regression (Chin et al., 1999). Moreover, Harold Varmus and coworkers developed transgenic mice expressing constitutively active K-Ras under the control of doxycycline in type II pneumocytes. The induction of the oncogene led to development of lung adenocarcinomas, which underwent apoptotic regression upon removal of the inductor, demonstrating that the function of this oncogene is necessary to maintain the viability of tumor cells even in the absence of key tumor suppressors such as p53 or Ink4A (Fisher et al., 2001). Similar approaches revealed the role of myc in the maintenance of tumors initiated by this oncogene in transgenic animals. In this regard, Felsher and Bishop (1999) reported that a myc transgene expressed in hematopoietic cells caused malignant T cell leukemias and acute myeloid leukemias, while switching off oncogene expression caused differentiation, proliferative arrest, and apoptosis of the leukemia cells. The role of myc in the maintenance of myc-driven skin premalignant lesions as well as invasive pancreatic tumors in transgenic mice was also reported (Pelengaris et al., 1999, 2002). Finally, tetracycline-regulated expression of a BCR–ABL transgene allowed demonstrating its role in the maintenance of leukemia initiated by this fusion oncogene in mice (Huettner et al., 2000).
However, the best validation of the oncogene addiction concept came from the success obtained with a few anti-cancer agents directed against the proteins encoded by the driver oncogenes of particular cancer types. Even though the number of such molecularly targeted therapies is low and their clinical benefits still limited, these therapies have improved the survival of patients suffering some previously intractable cancers (Haber et al., 2011). Well-known cases of oncogenes clinically targeted with either humanized monoclonal antibodies or small-molecule inhibitors include HER2 in breast cancer (Eisenhauer, 2001), EGFR and ALK in non-small cell lung cancer (Kwak et al., 2010; Maemondo et al., 2010), BCR–ABL in chronic myeloid leukemia (Peggs and Mackinnon, 2003), KIT in gastrointestinal stromal tumor (Heinrich et al., 2003), or B-RAF in metastatic melanoma (Flaherty et al., 2010), among others.
These results strongly support the notion that oncogenic alterations are required both for tumor establishment and maintenance and therefore, at least in theory, represent optimal anti-cancer drug targets (Sharma and Settleman, 2007). Despite the tremendous significance of this concept, the complexity of the mutational landscape of human cancers limits its practical application, as large-scale sequencing approaches have revealed that most tumors harbor diverse combination of low-frequency mutations rather than one or a few frequently mutated oncogenes as drivers of the neoplastic phenotype (Wood et al., 2007; Diamandis et al., 2010). Besides, some of the most prevalent tumorigenic genetic alterations do not yield easily targetable products. This is the case for most loss of function mutations in tumor suppressor genes, but also for many oncogenes such as the members of the Ras family, in which activating mutations do not involve the acquisition of a new or exacerbated catalytic activity, making oncogene addiction difficult to exploit therapeutically.
Non-Oncogene Addiction
A growing body of evidence has demonstrated that there are many proteins which, even without undergoing activating mutations, are critical for tumor development and maintenance. This fact led to the introduction of the concept of “non-oncogene addiction” to define the exacerbated dependence of tumors on the normal function of certain genes which are not mutated oncogenes or tumor suppressors (Solimini et al., 2007). It has been proposed that non-oncogene addiction is based on the increased cellular stresses experienced by tumor cells as a side effect of the transformed phenotype, such as DNA damage and replication, metabolic or oxidative stress, and protein misfolding, making cancer cells more dependent on the respective stress support systems. Consequently, non-oncogene addiction provides a wide spectrum of potential therapeutic targets, whose inhibition could reverse the oncogenic phenotype, affecting the viability of normal cells at a lower degree. This goal can be achieved either by inhibiting the activity of the stress support mechanisms (stress sensitization) or through exacerbation of the existing oncogenic stress (stress overload; Solimini et al., 2007; Luo et al., 2009b). Tumor-intrinsic non-oncogene addiction factors act in a cell-autonomous fashion, while tumor-extrinsic factors are required to provide stromal and vascular support to the tumor. The first group includes proteins involved in protection against DNA damage and replicative stress, mitotic stress, proteotoxic stress, metabolic stress, oxidative stress, nutrient stress, hypoxia, and immune response modulation. Tumor-extrinsic factors include proteins regulating angiogenesis and stromal support.
Current cancer therapies often exploit non-oncogene addiction-related phenomena to selectively kill cancer cells. For example, DNA-damaging agents and ionizing radiation take advantage of the genetic instability of cancer cells and their defects in DNA damage response to overload this stress defense system. Analogously, the taxanes used for the treatment of breast and ovarian cancers cause an overload of mitotic stress by interfering with proper spindle–kinetochore attachment (Jordan and Wilson, 2004). However, the concept of non-oncogene addiction did not play a significant role in the development of these therapies. Recently, the direct involvement of several proteins in this phenomenon has been reported. In this regard, one of the best examples of non-oncogene addiction factors is represented by HSF1 (heat shock factor 1), an evolutionary conserved transcription factor that coordinates the heat shock response in eukaryotes and protects the cell against protein misfolding and aggregation. HSF1 is required for tumor initiation and maintenance in a variety of cancer models, and in consequence it has been proposed to represent a promising drug target (Dai et al., 2007). Also involved in cell protection against unfolded protein stress, heat shock protein 70 (HSP70) has been shown to be inhibited by small molecules with anti-cancer effects in preclinical models (Galluzzi et al., 2009; Leu et al., 2009), further illustrating the relevance of protein homeostasis for the development of new anti-cancer therapies. Also, the transcription factor interferon regulatory factor 4 (IRF4), which maintains the expression of the MYC oncogene through an autoregulatory circuit, has been reported to be essential for myeloma cells independently of their transforming oncogenic mechanisms, constituting another paradigmatic example of non-oncogene addiction factor (Shaffer et al., 2008).
Synthetic Lethality in Cancer
The potential of non-oncogene addiction as basis of anti-cancer therapies relies on the identification of the putative factors involved in this process. Thus, investigating the genetic interactions taking place in cancer biology both in vivo and in experimental settings can result in the identification of new therapeutic targets and strategies. This idea has fueled a number of synthetic lethality screens. The concept of synthetic lethality derives from classical genetic studies and refers to the interaction among two genes in which alterations in any of them are compatible with cell or organism viability, while the combination of defects in both genes causes lethality or fitness impairment (Kaelin, 2005; Chan and Giaccia, 2011). Synthetic lethal targeting of cancer cells provides a genotype-selective approach as only the cancer cells with alterations in specific genes or pathways are sensitive to the therapy and, at the same time, it allows to exploit cancer alterations that are difficult to target directly. Consequently, a number of studies have used synthetic lethal screens for identifying new anti-cancer drugs. Some of these studies have compared the activity of collections of chemical compounds on isogenic cell lines with and without specific genetic alterations. Alternatively, genetic screens of synthetic lethality can be performed using siRNA or shRNA libraries to identify genes whose knockdown is preferentially harmful to the cells carrying a defined molecular defect. So far, the most successful application of the synthetic lethality concept in anti-cancer research has led to the discovery and development of poly(ADP-ribose) polymerase (PARP) inhibitors to treat cancers with BRCA1 or BRCA2 mutations (Bryant et al., 2005; Farmer et al., 2005; Rehman et al., 2010). Other studies have pursued the identification of compounds and genes synthetically lethal with genetic alterations in other tumor suppressors such as VHL or PTEN as well as in oncogenes such as RAS or MYC, leading to the identification of several non-oncogene addiction factors differentially essential for the cancer cells carrying these particular alterations (Chan and Giaccia, 2011).
Protease Addiction
Proteolytic enzymes play a large variety of critical roles in the regulation and execution of most physiological processes and pathological conditions (Lopez-Otin and Bond, 2008) acting frequently through a complex crosstalk with other important regulators of cell biology such as protein kinases and phosphatases (Lopez-Otin and Hunter, 2010). Consequently, the potential of tumor proteases as targets of anti-cancer therapies has been frequently explored (Overall and Lopez-Otin, 2002). The success of some of these attempts was limited by the overwhelming complexity of the human degradome (the repertoire of proteolytic enzymes, substrates, and inhibitors) and by the fact that some proteases are important players in the natural anti-cancer defenses rather than being cancer-promoting enzymes (Lopez-Otin and Matrisian, 2007). However, a wide diversity of proteases are involved in physiological mechanisms such as those responsible for the protection against most if not all the above discussed cancer-associated stresses. Moreover, the mechanisms of action of proteases have been the subject of innumerable studies over many decades and are probably the best known among all enzymatic activities, facilitating the development of specific inhibitors (Turk, 2006). Consequently, we hypothesize that most if not all tumors present “addiction” to a number of proteolytic activities, which in turn may represent valuable targets of anti-cancer therapies, even without being mutated or over-expressed by the malignant cells (Figure 1). Below we discuss briefly a few examples supporting this proposal.
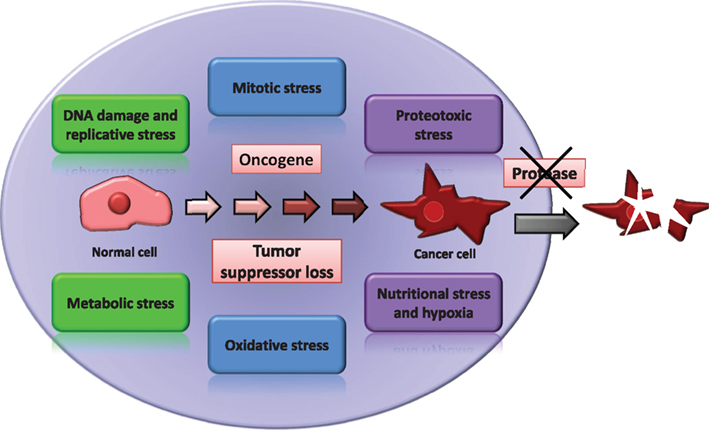
Figure 1. Protease addiction in cancer. Cancer cells are under different forms of cellular stress as a side effect of the transformed phenotype. We hypothesize that for most cases of cancer, it will be possible to find one or more proteases specifically required for the maintenance of cancer cell viability.
Proteotoxic Stress and Proteasome Addiction in Cancer
Aneuploidy, copy-number variations and transcriptional alterations in cancer cells alter the dosage balance of components in protein complexes, causing proteotoxic stress and making cancer cells especially dependent on chaperone pathways as mentioned above, as well as on the cell machinery responsible for protein turnover. The responsibility for the degradation of unfolded proteins relies mainly in the ubiquitin–proteasome system, and consequently the proteolytic enzymes acting in this system are potential candidates for pharmacological targeting (Bedford et al., 2011). The therapeutic potential of this degradative pathway has been demonstrated by the development of bortezomib, an inhibitor of the 26S proteasome used for the treatment of myeloma and mantle cell lymphoma. Bortezomib is a peptide boronic acid that inhibits the proteasome by binding to its chymotrypsin-like site. Bortezomib induces apoptosis of cancer cells through mechanisms that probably include stabilization of proapoptotic proteins, inhibition of NF-κB function and interference with the unfolded protein response, causing endoplasmic reticulum stress. Besides, some of the anti-cancer effects of bortezomib seem to be due to alterations of the microenvironment and angiogenesis inhibition (Adams, 2004; Navon and Ciechanover, 2009). Remarkably, bortezomib shows additive effects with other therapies in several experimental models, chemosensitizing cancer cells to radiation therapy or to drugs such as doxorubicin, irinotecan, gemcitabine, cyclophosphamide, or cisplatin (Adams, 2004), supporting the synergistic effect of the stress overload and stress sensitization anti-cancer approaches discussed above. The turnover of cancer-relevant proteins in the proteasome can also be targeted indirectly through the inhibition of E3 ubiquitin ligases. In this regard, the inhibition of members of the cullin–RING subfamily has shown promise against cancer (Soucy et al., 2009). Moreover, the possibility of specifically targeting the activity of individual E3 enzymes opens new avenues in this field (Aghajan et al., 2010; Orlicky et al., 2010), widening the possibilities of exploiting proteolytic addiction for cancer treatment.
Taspase Addiction in Cancer
Threonine aspartase 1 (taspase 1) is a threonine endoprotease that mediates the cleavage of intracellular proteins after aspartate residues (Hsieh et al., 2003). Taspase 1 was first identified as the enzyme responsible for the cleavage of the mixed lineage leukemia (MLL) protein, a proteolytic event which is crucial for MLL stabilization and, in consequence, for proper HOX gene expression and MLL-mediated tumorigenesis (Hsieh et al., 2003; Takeda et al., 2006). Taspase 1 is also over-expressed in solid tumors and acts on other regulatory proteins such as transcription factor IIA (TFIIA) or DNA polymerase zeta (DPOLZ), among other substrates (Zhou et al., 2006; Knauer et al., 2011). Interestingly, taspase 1 has been reported to be required for in vitro transformation of mouse embryonic fibroblasts by diverse combinations of oncogene pairs (Takeda et al., 2006), but also to maintain the transformed phenotype of these cells (Chen et al., 2010). Moreover, taspase 1 knockdown inhibited the proliferation of several human cancer cell lines and enhanced apoptosis, exhibiting a synergistic effect with chemotherapeutic agents at least in part through downregulation of the antiapoptotic protein MCL-1. These findings prompted Chen et al. (2010) to propose taspase 1 as a non-oncogene addiction protease with potential as a target of anti-cancer therapies.
Synthetic Lethality of Proteases with Oncogenes and Tumor Suppressor Genes
Considering the overwhelming diversity of functions played by proteases, it seems reasonable to expect that one or more proteolytic enzymes will be involved in synthetic lethality interactions with most oncogenes and tumor suppressor genes (Figure 2). In our opinion, the results derived from several large-scale synthetic lethality screens carried out with some of the most relevant oncogenes support this hypothesis. The RNAi-based genome-wide screen performed by Stephen Elledge’s group with KRAS provides one of the best examples in this regard (Luo et al., 2009a). These authors discovered a set of genes whose silencing was preferentially lethal for cancer cells carrying activating KRAS mutations due to their exacerbated mitotic stress. The Ras synthetic lethal factors identified in this study included the Polo-like kinase PLK1, the anaphase promoting complex/cyclosome (an E3 ubiquitin ligase that promotes the degradation of mitotic proteins) and, remarkably, the COP9 signalosome and the proteasome, among others. In agreement with the results of their RNAi screen, KRAS mutant cells are more sensitive to proteasome inhibitors such as bortezomib, providing pharmacological evidence of the protease addiction of these cancer cells (Luo et al., 2009a). Interestingly, Ras oncogenes also seem to involve another type of protease addiction: autophagy addiction, as cancer cells with activating mutations in these genes show hyperactivation of this degradative system, whereas downregulation of autophagy proteins causes growth impairment in a subset of these cells (Guo et al., 2011).
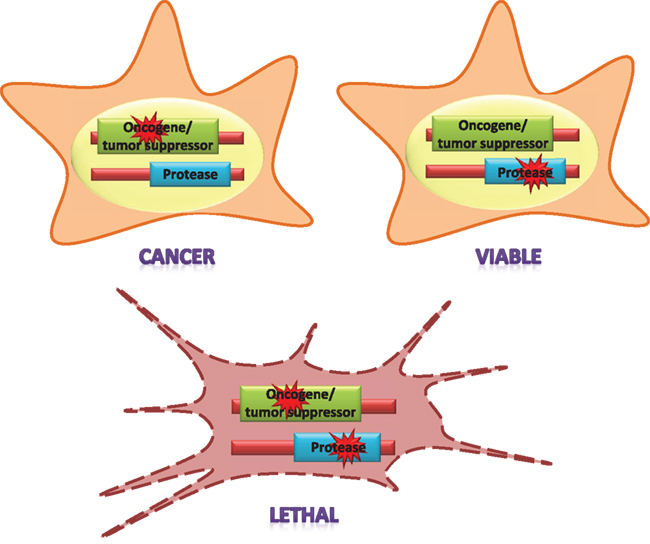
Figure 2. The concept of synthetic lethality applied to the interactions of proteases and cancer genes. Mutations in oncogenes and tumor suppressor genes act as drivers of neoplastic transformation. A protease is “synthetic lethal” with an oncogene or a tumor suppressor gene if its function is specifically indispensable for the survival of cells with mutations in those genes. Thus, a mutation in the protease-encoding gene or the pharmacological inhibition of the enzyme is tolerable for normal cells, but not for cancer cells.
Other synthetic lethal screens with different oncogenes have been based on the use of non-genome-wide RNAi libraries, with a strong bias toward kinases among “cancer-relevant genes” and without representation of most members of the human degradome. Nevertheless, additional examples of synthetic lethality of proteases with oncogenes have been found over the last few years. Thus, Bivona et al. (2011) carried out a screen with a shRNA library targeting approximately 2,000 cancer-related genes to identify proteins whose ablation rendered lung cancer cells sensitive to EGFR inhibition. Interestingly, besides components of the FAS and NF-kB pathway, the list of proteins identified in this study includes PSMD14 (also known as POH1/RPN11), a deubiquitinating enzyme component of the proteasome that has been involved in regulation of the ErbB2 receptor (Liu et al., 2009).
Besides the above discussed role of the ubiquitin–proteasome system in the protection of the cell against proteotoxic stress, this proteolytic system plays a critical role in the regulation of the cell cycle through the rapid turnover of regulatory proteins. In consequence, diverse components of this system are likely to be involved in non-oncogene addiction and synthetic lethality phenomena in different types of tumors. An interesting example of this possibility is represented by ubiquitin specific protease 2 (USP2), a deubiquitinating enzyme from the USP family of cysteine proteases reported to stabilize cyclin D1. Cyclin D1 is an important regulator of the G1/S phase transition and is activated by chromosomal translocations or over-expressed in several cancer types. Shan et al. (2009) reported that USP2 specifically deubiquitinates and stabilizes cyclin D1, whereas knockdown of this protease causes cyclin D1 degradation and growth arrest in cancer cells dependent on this cell cycle regulator. Thus, the resulting non-oncogene addiction of these cancer cells to USP2 derives from its synthetic lethality with cyclin D1 and it could provide the rational to develop inhibitory compounds with anti-cancer potential.
The identification of druggable proteins involved in synthetic lethality interactions with tumor suppressors can be of great potential for anti-cancer therapy, as brilliantly demonstrated by the development of PARP inhibitors to treat BRCA1/2-related cancers (Rehman et al., 2010). This possibility has been explored in a number of RNAi-based screens, but the identification of proteases selectively required for the maintenance of cancer cells with mutations in particular tumor suppressors has been hampered by the limited RNAi libraries used in most of these studies (Bommi-Reddy et al., 2008; Baldwin et al., 2010). It seems reasonable to assume that a more comprehensive exploration of the human degradome will allow the identification of targetable proteolytic enzymes. Remarkably, the work based on the results of a RNAi screen with a library targeting 2,287 genes (Lovejoy et al., 2009) led to the finding that ubiquitin specific protease 11 (USP11) participates in repair of DNA double-strand breaks via homologous recombination. Silencing of this protease causes spontaneous DNA damage repair activation and hypersensitivity to PARP inhibition, ionizing radiation and other sources of genotoxic stress (Wiltshire et al., 2010). The synthetic lethal interaction between USP11 and PARP uncovered by this work illustrates the potential of proteases as critical players in maintaining cancer cell viability and highlights the special relevance of the ubiquitin–proteasome system in this regard.
Conclusion and Perspectives
Proteases have been regarded too often as demolition machines lacking the required finesse to be good candidate targets of anti-cancer treatments. Fortunately, this view is changing dramatically. The extraordinary diversity of mechanisms of action used by the different proteases constituting the human degradome and the disparity of structural features underlying their functions, represent formidable challenges for the development of protease inhibitors but also provide a unique opportunity for synthesizing exquisitely selective compounds of potential anti-cancer interest. According to the “cancer protease addiction” hypothesis that we put forward in this Perspective, it should be possible for any given tumor type to find proteolytic activities that are essential for cancer cell proliferation or survival, or whose inhibition could act synergistically with other therapies. We have discussed here a few examples of degradome members that represent good examples of protease addiction in different neoplastic contexts (Figure 3), but we have not attempted to provide a comprehensive coverage of this topic. Thus, we have not discussed some conspicuous examples, such as gamma-secretase, a proteolytic complex required for Notch maturation and consequently a likely non-oncogene addiction factor in neoplasias depending on this protein. Interestingly, gamma-secretase inhibitors have been reported to present a synergistic effect with glucocorticoids in leukemia cells (Real et al., 2009) and selectively target cancer stem cells in some neoplasias (Kondratyev et al., 2011), illustrating the diversity of the biological repercussions of targeting protease addiction for cancer treatment. Currently available data point to the ubiquitin–proteasome system as the most promising source of druggable proteases for cancer treatment. The successful development of therapies based on proteasome inhibitors provides strong support to this idea and represents the most fruitful application of the non-oncogene addiction concept. The functions played by ubiquitin ligases and deubiquitinases parallel those of protein kinases and phosphatases in biological regulation by posttranslational protein modification, but constitute a considerably more complex system in structure, mechanism, and regulation. These enzymes participate in functions far beyond the destruction of unnecessary or damaged protein molecules, and their potential as cancer targets remains largely unexplored. Hopefully, this first attempt to summarize the current information supporting our proposal that protease addiction is a widespread process in cancer, may facilitate the building of a conceptual framework which can be helpful for translating this hypothesis into clinical benefits for cancer patients.
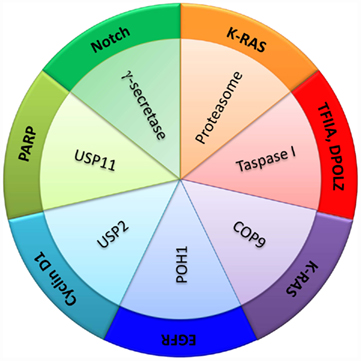
Figure 3. Functional interactions between proteases and cancer genes in the context of protease addiction processes in cancer. Several examples of proteolytic activities are shown in relation with different proteins involved in cancer development and survival, as discussed in the main text.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Our work is supported by grants from Ministerio de Ciencia e Innovación-Spain, Fundación “M. Botín,” PCTI-FICYT Asturias, and European Union (FP7 MicroEnviMet). The Instituto Universitario de Oncología is supported by Obra Social Cajastur and Acción Transversal del Cáncer-RTICC.
References
Adams, J. (2004). The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5, 417–421.
Aghajan, M., Jonai, N., Flick, K., Fu, F., Luo, M., Cai, X., Ouni, I., Pierce, N., Tang, X., Lomenick, B., Damoiseaux, R., Hao, R., Del Moral, P. M., Verma, R., Li, Y., Li, C., Houk, K. N., Jung, M. E., Zheng, N., Huang, L., Deshaies, R. J., Kaiser, P., and Huang, J. (2010). Chemical genetics screen for enhancers of rapamycin identifies a specific inhibitor of an SCF family E3 ubiquitin ligase. Nat. Biotechnol. 28, 738–742.
Ashworth, A., Lord, C. J., and Reis-Filho, J. S. (2011). Genetic interactions in cancer progression and treatment. Cell 145, 30–38.
Baldwin, A., Grueneberg, D. A., Hellner, K., Sawyer, J., Grace, M., Li, W., Harlow, E., and Munger, K. (2010). Kinase requirements in human cells: V. Synthetic lethal interactions between p53 and the protein kinases SGK2 and PAK3. Proc. Natl. Acad. Sci. U.S.A. 107, 12463–12468.
Bedford, L., Lowe, J., Dick, L. R., Mayer, R. J., and Brownell, J. E. (2011). Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat. Rev. Drug Discov. 10, 29–46.
Bivona, T. G., Hieronymus, H., Parker, J., Chang, K., Taron, M., Rosell, R., Moonsamy, P., Dahlman, K., Miller, V. A., Costa, C., Hannon, G., and Sawyers, C. L. (2011). FAS and NF-kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 471, 523–526.
Bommi-Reddy, A., Almeciga, I., Sawyer, J., Geisen, C., Li, W., Harlow, E., Kaelin, W. G. Jr., and Grueneberg, D. A. (2008). Kinase requirements in human cells: III. Altered kinase requirements in VHL-/- cancer cells detected in a pilot synthetic lethal screen. Proc. Natl. Acad. Sci. U.S.A. 105, 16484–16489.
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., Kyle, S., Meuth, M., Curtin, N. J., and Helleday, T. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917.
Chan, D. A., and Giaccia, A. J. (2011). Harnessing synthetic lethal interactions in anticancer drug discovery. Nat. Rev. Drug Discov. 10, 351–364.
Chen, D. Y., Liu, H., Takeda, S., Tu, H. C., Sasagawa, S., Van Tine, B. A., Lu, D., Cheng, E. H., and Hsieh, J. J. (2010). Taspase1 functions as a non-oncogene addiction protease that coordinates cancer cell proliferation and apoptosis. Cancer Res. 70, 5358–5367.
Chin, L., Tam, A., Pomerantz, J., Wong, M., Holash, J., Bardeesy, N., Shen, Q., O’Hagan, R., Pantginis, J., Zhou, H., Horner, J. W. II, Cordon-Cardo, C., Yancopoulos, G. D., and DePinho, R. A. (1999). Essential role for oncogenic Ras in tumour maintenance. Nature 400, 468–472.
Dai, C., Whitesell, L., Rogers, A. B., and Lindquist, S. (2007). Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 130, 1005–1018.
Diamandis, E. P., Hudson, T., Kallioniemi, O., Liu, E. T., and Lopez-Otin, C. (2010). Cancer genomes. Clin. Chem. 56, 1660–1664.
Eisenhauer, E. A. (2001). From the molecule to the clinic – inhibiting HER2 to treat breast cancer. N. Engl. J. Med. 344, 841–842.
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., Santarosa, M., Dillon, K. J., Hickson, I., Knights, C., Martin, N. M., Jackson, S. P., Smith, G. C., and Ashworth, A. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921.
Felsher, D. W., and Bishop, J. M. (1999). Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 4, 199–207.
Fisher, G. H., Wellen, S. L., Klimstra, D., Lenczowski, J. M., Tichelaar, J. W., Lizak, M. J., Whitsett, J. A., Koretsky, A., and Varmus, H. E. (2001). Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 15, 3249–3262.
Flaherty, K. T., Puzanov, I., Kim, K. B., Ribas, A., McArthur, G. A., Sosman, J. A., O’Dwyer, P. J., Lee, R. J., Grippo, J. F., Nolop, K., and Chapman, P. B. (2010). Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819.
Galluzzi, L., Giordanetto, F., and Kroemer, G. (2009). Targeting HSP70 for cancer therapy. Mol. Cell 36, 176–177.
Galluzzi, L., Vitale, I., and Kroemer, G. (2011). Past, present, and future of molecular and cellular oncology. Front. Oncol. 1:1. doi: 10.3389/fonc.2011.00001
Guo, J. Y., Chen, H. Y., Mathew, R., Fan, J., Strohecker, A. M., Karsli-Uzunbas, G., Kamphorst, J. J., Chen, G., Lemons, J. M., Karantza, V., Coller, H. A., Dipaola, R. S., Gelinas, C., Rabinowitz, J. D., and White, E. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Heinrich, M. C., Corless, C. L., Demetri, G. D., Blanke, C. D., von Mehren, M., Joensuu, H., McGreevey, L. S., Chen, C. J., Van den Abbeele, A. D., Druker, B. J., Kiese, B., Eisenberg, B., Roberts, P. J., Singer, S., Fletcher, C. D., Silberman, S., Dimitrijevic, S., and Fletcher, J. A. (2003). Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 21, 4342–4349.
Hsieh, J. J., Cheng, E. H., and Korsmeyer, S. J. (2003). Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell 115, 293–303.
Huettner, C. S., Zhang, P., Van Etten, R. A., and Tenen, D. G. (2000). Reversibility of acute B-cell leukaemia induced by BCR-ABL1. Nat. Genet. 24, 57–60.
Jordan, M. A., and Wilson, L. (2004). Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265.
Kaelin, W. G. Jr. (2005). The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 5, 689–698.
Knauer, S. K., Fetz, V., Rabenstein, J., Friedl, S., Hofmann, B., Sabiani, S., Schröder, E., Kunst, L., Proschak, E., Thines, E., Kindler, T., Schneider, G., Marschalek, R., Stauber, R. H., and Bier, C. (2011). Bioassays to monitor taspase1 function for the identification of pharmacogenetic inhibitors. PLoS ONE 6, e18253. doi: 10.1371/journal.pone.0018253
Kondratyev, M., Kreso, A., Hallett, R. M., Girgis-Gabardo, A., Barcelon, M. E., Ilieva, D., Ware, C., Majumder, P. K., and Hassell, J. A. (2011). Gamma-secretase inhibitors target tumor-initiating cells in a mouse model of ERBB2 breast cancer. Oncogene. doi: 10.1038/onc.2011.212. [Epub ahead of print].
Kwak, E. L., Bang, Y. J., Camidge, D. R., Shaw, A. T., Solomon, B., Maki, R. G., Ou, S. H., Dezube, B. J., Jänne, P. A., Costa, D. B., Varella-Garcia, M., Kim, W. H., Lynch, T. J., Fidias, P., Stubbs, H., Engelman, J. A., Sequist, L. V., Tan, W., Gandhi, L., Mino-Kenudson, M., Wei, G. C., Shreeve, S. M., Ratain, M. J., Settleman, J., Christensen, J. G., Haber, D. A., Wilner, K., Salgia, R., Shapiro, G. I., Clark, J. W., and Iafrate, A. J. (2010). Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 363, 1693–1703.
Leu, J. I., Pimkina, J., Frank, A., Murphy, M. E., and George, D. L. (2009). A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 36, 15–27.
Liu, H., Buus, R., Clague, M. J., and Urbe, S. (2009). Regulation of ErbB2 receptor status by the proteasomal DUB POH1. PLoS ONE 4, e5544. doi: 10.1371/journal.pone.0005544
Lopez-Otin, C., and Bond, J. S. (2008). Proteases: multifunctional enzymes in life and disease. J. Biol. Chem. 283, 30433–30437.
Lopez-Otin, C., and Hunter, T. (2010). The regulatory crosstalk between kinases and proteases in cancer. Nat. Rev. Cancer 10, 278–292.
Lopez-Otin, C., and Matrisian, L. M. (2007). Emerging roles of proteases in tumour suppression. Nat. Rev. Cancer 7, 800–808.
Lovejoy, C. A., Xu, X., Bansbach, C. E., Glick, G. G., Zhao, R., Ye, F., Sirbu, B. M., Titus, L. C., Shyr, Y., and Cortez, D. (2009). Functional genomic screens identify CINP as a genome maintenance protein. Proc. Natl. Acad. Sci. U.S.A. 106, 19304–19309.
Luo, J., Emanuele, M. J., Li, D., Creighton, C. J., Schlabach, M. R., Westbrook, T. F., Wong, K. K., and Elledge, S. J. (2009a). A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137, 835–848.
Luo, J., Solimini, N. L., and Elledge, S. J. (2009b). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837.
Maemondo, M., Inoue, A., Kobayashi, K., Sugawara, S., Oizumi, S., Isobe, H., Gemma, A., Harada, M., Yoshizawa, H., Kinoshita, I., Fujita, Y., Okinaga, S., Hirano, H., Yoshimori, K., Harada, T., Ogura, T., Ando, M., Miyazawa, H., Tanaka, T., Saijo, Y., Hagiwara, K., Morita, S., Nukiwa, T., and North-East Japan Study Group. (2010). Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 362, 2380–2388.
Navon, A., and Ciechanover, A. (2009). The 26 S proteasome: from basic mechanisms to drug targeting. J. Biol. Chem. 284, 33713–33718.
Orlicky, S., Tang, X., Neduva, V., Elowe, N., Brown, E. D., Sicheri, F., and Tyers, M. (2010). An allosteric inhibitor of substrate recognition by the SCF(Cdc4) ubiquitin ligase. Nat. Biotechnol. 28, 733–737.
Overall, C. M., and Lopez-Otin, C. (2002). Strategies for MMP inhibition in cancer: innovations for the post-trial era. Nat. Rev. Cancer 2, 657–672.
Peggs, K., and Mackinnon, S. (2003). Imatinib mesylate – the new gold standard for treatment of chronic myeloid leukemia. N. Engl. J. Med. 348, 1048–1050.
Pelengaris, S., Khan, M., and Evan, G. I. (2002). Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 109, 321–334.
Pelengaris, S., Littlewood, T., Khan, M., Elia, G., and Evan, G. (1999). Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol. Cell 3, 565–577.
Real, P. J., Tosello, V., Palomero, T., Castillo, M., Hernando, E., De Stanchina, E., Sulis, M. L., Barnes, K., Sawai, C., Homminga, I., Meijerink, J., Aifantis, I., Basso, G., Cordon-Cardo, C., Ai, W., and Ferrando, A. (2009). Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat. Med. 15, 50–58.
Rehman, F. L., Lord, C. J., and Ashworth, A. (2010). Synthetic lethal approaches to breast cancer therapy. Nat. Rev. Clin. Oncol. 7, 718–724.
Shaffer, A. L., Emre, N. C., Lamy, L., Ngo, V. N., Wright, G., Xiao, W., Powell, J., Dave, S., Yu, X., Zhao, H., Zeng, Y., Chen, B., Epstein, J., and Staudt, L. M. (2008). IRF4 addiction in multiple myeloma. Nature 454, 226–231.
Shan, J., Zhao, W., and Gu, W. (2009). Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol. Cell 36, 469–476.
Sharma, S. V., and Settleman, J. (2007). Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev. 21, 3214–3231.
Solimini, N. L., Luo, J., and Elledge, S. J. (2007). Non-oncogene addiction and the stress phenotype of cancer cells. Cell 130, 986–988.
Soucy, T. A., Smith, P. G., Milhollen, M. A., Berger, A. J., Gavin, J. M., Adhikari, S., Brownell, J. E., Burke, K. E., Cardin, D. P., Critchley, S., Cullis, C. A., Doucette, A., Garnsey, J. J., Gaulin, J. L., Gershman, R. E., Lublinsky, A. R., McDonald, A., Mizutani, H., Narayanan, U., Olhava, E. J., Peluso, S., Rezaei, M., Sintchak, M. D., Talreja, T., Thomas, M. P., Traore, T., Vyskocil, S., Weatherhead, G. S., Yu, J., Zhang, J., Dick, L. R., Claiborne, C. F., Rolfe, M., Bolen, J. B., and Langston, S. P. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736.
Takeda, S., Chen, D. Y., Westergard, T. D., Fisher, J. K., Rubens, J. A., Sasagawa, S., Kan, J. T., Korsmeyer, S. J., Cheng, E. H., and Hsieh, J. J. (2006). Proteolysis of MLL family proteins is essential for taspase1-orchestrated cell cycle progression. Genes Dev. 20, 2397–2409.
Turk, B. (2006). Targeting proteases: successes, failures and future prospects. Nat. Rev. Drug Discov. 5, 785–799.
Weinstein, I. B. (2002). Cancer. Addiction to oncogenes – the Achilles heal of cancer. Science 297, 63–64.
Weinstein, I. B., and Joe, A. (2008). Oncogene addiction. Cancer Res. 68, 3077–3080; discussion 3080.
Weinstein, I. B., and Joe, A. K. (2006). Mechanisms of disease: oncogene addiction – a rationale for molecular targeting in cancer therapy. Nat. Clin. Pract. Oncol. 3, 448–457.
Wiltshire, T. D., Lovejoy, C. A., Wang, T., Xia, F., O’Connor, M. J., and Cortez, D. (2010). Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J. Biol. Chem. 285, 14565–14571.
Wood, L. D., Parsons, D. W., Jones, S., Lin, J., Sjöblom, T., Leary, R. J., Shen, D., Boca, S. M., Barber, T., Ptak, J., Silliman, N., Szabo, S., Dezso, Z., Ustyanksky, V., Nikolskaya, T., Nikolsky, Y., Karchin, R., Wilson, P. A., Kaminker, J. S., Zhang, Z., Croshaw, R., Willis, J., Dawson, D., Shipitsin, M., Willson, J. K., Sukumar, S., Polyak, K., Park, B. H., Pethiyagoda, C. L., Pant, P. V., Ballinger, D. G., Sparks, A. B., Hartigan, J., Smith, D. R., Suh, E., Papadopoulos, N., Buckhaults, P., Markowitz, S. D., Parmigiani, G., Kinzler, K. W., Velculescu, V. E., and Vogelstein, B. (2007). The genomic landscapes of human breast and colorectal cancers. Science 318, 1108–1113.
Keywords: degradome, proteasome, deubiquitinating enzymes, autophagy
Citation: Freije JMP, Fraile JM and López-Otín C (2011) Protease addiction and synthetic lethality in cancer. Front. Oncol. 1:25. doi: 10.3389/fonc.2011.00025
Received: 06 July 2011;
Paper pending published: 20 July 2011;
Accepted: 17 August 2011;
Published online: 05 September 2011.
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Alessandro Rimessi, University of Ferrara, ItalyValerio Donato, New York University Medical Center, USA
Vilma A. Sardao, Center for Neuroscience and Cell Biology, Portugal
Copyright: © 2011 Freije, Fraile and López-Otín. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: José M. P. Freije and Carlos López-Otín, Departamento de Bioquímica y Biología Molecular, Facultad de Medicina, Universidad de Oviedo, 33006-Oviedo, Oviedo, Spain. e-mail:am1wZkB1bmlvdmkuZXM=;Y2xvQHVuaW92aS5lcw==