

- 1 Department of Radiation Oncology, Case Integrative Cancer Biology Program, Case Western Reserve University, Cleveland, OH, USA
- 2 Department of Radiation Oncology, Rhode Island Hospital, Warren Alpert Medical School of Brown University, Providence, RI, USA
Autophagy, a highly regulated cell “self-eating” pathway, is controlled by the action of over 34 autophagy-related proteins (collectively termed Atgs). Although they are fundamentally different processes, autophagy and apoptosis (type I programmed cell death), under certain circumstances, can be regulated by common signaling mediators. Current cancer therapies including chemotherapy and ionizing radiation are known to induce autophagy within tumor cells. However, autophagy plays a dual role of either pro-cell survival or pro-cell death in response to these cancer treatments, depending on the cellular context and the nature of the treatment. We review the current basic and translational cancer research literature on how autophagy impacts tumor cell survival (“to live”) and death (“not to live”) following treatment as well as the role of chemical mediators of autophagy.
Introduction
An apoptotic response in tumors cells has been shown to play a major role in determining the cytotoxicity in response to DNA damaging agents such as ionizing radiation and several different classes of chemotherapeutic drugs (Brown and Wouters, 1999). Recently, macroautophagy (hereafter called autophagy; type II programmed cell death), has also been reported to mediate cytotoxicity to these anti-cancer regimens (Zeng and Kinsella, 2007, 2010).
Autophagy is an evolutionarily conserved lysosomal pathway for degrading cytoplasmic proteins and organelles in eukaryotic cells (Dunn, 1990). The main physiologic function of autophagy is to supervise and maintain cellular protein and organelle quality control, thereby preventing the accumulation of unfolded and aggregated proteins so as to maintain intracellular metabolic homeostasis (Dunn, 1990). Autophagy is involved in many aspects of human health and disease, including cancer. Accumulating evidence suggests that autophagy is a double-edged sword in tumorigenesis, functioning as both a tumor suppressor and a protector of cancer cell survival (Shintani and Klionsky, 2004).
A better understanding of autophagy regulation and its impact on treatment outcomes will potentially allow the identification of novel therapeutic targets in cancer. In this review, we summarize the current knowledge about autophagy machinery, crosstalk between apoptosis and autophagy, and the impact of autophagy on the anti-cancer treatment efficacy of ionizing radiation and chemotherapy in human cancers.
Autophagy Machinery
Autophagy is a ubiquitous cellular pathway in eukaryotic cells that entails the degradation of intracellular components through the lysosomal machinery (Dunn, 1990; Shintani and Klionsky, 2004). This cellular self-consumption process is characterized by sequestration of bulk cytoplasm, long-lived proteins, and cellular organelles in double-membrane vesicles, called autophagosomes. Autophagosomes form from the elongation of small membrane structures called isolation membranes. These membranes can be derived from the endoplasmic reticulum, trans-golgi network, mitochondrial outer membrane, or the plasma membrane. Autophagosomes then fuse with lysosomes, forming autolysosomes where its contents are degraded by lysosomal enzymes (Figure 1; Shintani and Klionsky, 2004).
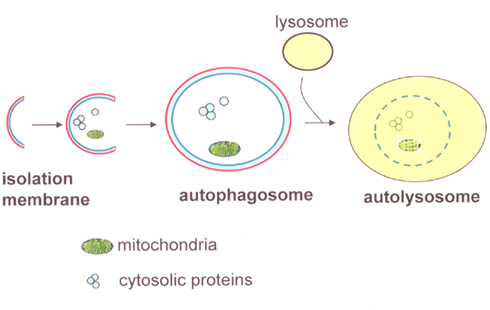
Figure 1. Diagram of autophagy components. The initial phase of autophagy is involved in the formation of isolation membranes. After engulfing part of the cytoplasmic components, such as long-lived proteins and organelles, the isolation membrane elongates and closes to become autophagosomes. Autophagosomes then fuse with lysosomes and mature into autolysosomes, resulting in the degradation of its contents.
Autophagy is a highly regulated process. The process starts with the activation of a Unc-51-like kinase (ULK) complex composed of ULK1/2, Atg13, FIP200, and Atg101 (He and Klionsky, 2009; Yang and Klionsky, 2010). This complex is regulated by mTOR which senses nutrient levels in the environment. Under high-nutrient conditions, mTOR phosphorylates ULK1/2 and then inhibits autophagy; whereas mTOR dissociates from the ULK complex during periods of nutrient deprivation and activates autophagy (Figure 2A). The activation and accumulation of the ULK complex result in the development of the isolation membranes (He and Klionsky, 2009; Yang and Klionsky, 2010). The subsequent development of autophagosomes is dependent on a class III phosphatidylinositol-3 kinase (PI3K), also known as Vps34, complex. The complex containing Beclin1–UVRAG (UV radiation resistance associated gene) and Beclin1–Ambra1 activates autophagy, while the Beclin1–Rubicon complex negatively regulates class III PI3K activity and inhibits autophagy (Yang and Klionsky, 2010). The class III PI3K complex localizes to the isolation membranes and recruits further Atgs like Atg3, Atg5, Atg7, and LC3 [microtubule-associated protein light chain 3], to allow for elongation of the isolation membranes and completion of the autophagosomes (Figure 2B). Mediated by the lysosomal membrane protein LAMP-2 and the small GTPase Rab7, autophagosomes fuse with lysosomes, leading to the breakdown of proteins and organelles by lysosomal enzymes (Yang and Klionsky, 2010; Amaravadi et al., 2011).
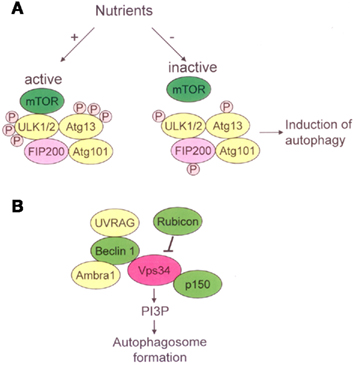
Figure 2. Molecular pathway of activation of autophagy. (A) In presence of nutrients, active mTOR inhibits ULK1/2 – Atg13-FIP200-Atg101 complex through phosphorylating ULK1/2 and Atg13. In response to nutrient deprivation, mTOR dissociates from the ULK complex, resulting in dephosphorylation of ULK1/2 and Atg13 and subsequent induction of autophagy. (B) The class III PI3K complex contains Vps34, p150, Beclin1, UVRAG, and Ambra1. Rubicon negatively regulates class III PI3K activity and inhibits autophagy. The class III PI3K complex localizes to the isolation membranes and produces phosphatidylinositol-3-phosphate (PI3P), allowing for elongation of the isolation membranes and completion of the autophagosomes.
Crosstalk between Apoptosis and Autophagy
Apoptosis, a form of programmed cell death (type I), has been well characterized. It involves the activation of catabolic enzymes – in particular proteases – in signaling cascades, which leads to characteristic changes such as altered nuclear morphology including chromatin condensation and fragmentation, cell shrinkage, plasma membrane blebbing, and apoptotic body formation (Williams, 1991). Although autophagy and apoptosis are markedly different processes, several signaling mediators, as discussed below, regulate both autophagic and apoptotic machinery.
Common Signal Transduction Mediators
Reactive oxygen species not only trigger apoptosis but also stimulate the proteolytic activity of Atg4 and then activate autophagy (Scherz-Shouval et al., 2007). Ceramide is a prominent inducer of apoptosis and also triggers autophagic cell death in malignant glioma cells via activation of BNIP3 (Lavieu et al., 2006). An increase in the cytosolic free Ca2+ concentration activates pro-apoptotic signals while it also potently induces autophagy through activating calmodulin-dependent kinase (Rizzuto and Pozzan, 2006; Hoyer-Hansen et al., 2007). The tumor suppressor p53 is a classic mediator of apoptosis (Vousden and Lane, 2007). However, accumulating evidence indicates that p53 stimulates autophagy through inhibition of mTOR or activation of damage-regulated autophagy modulator (DRAM; Crighton et al., 2006; Zeng et al., 2007; Zeng and Kinsella, 2008).
Dual Role of Atg5 in Autophagy and Apoptosis
Atg5 (autophagy-related-5) also represents a point of crosstalk between autophagic and apoptotic pathways. Atg5 is required for autophagy. It conjugates to Atg12 and associates with isolation membranes to form autophagosomes (Mizushima et al., 1998). Interestingly, Atg5 also plays in an important role in the apoptotic process. The role of Atg5 in apoptosis is mediated by the proteolytic cleavage of Atg5. Upon lethal stress, the 33-kDa full-length Atg5 protein is cleaved by Calpains to remove the C terminus, generating a 24-kDa fragment. This fragment loses its autophagy-inducing activity but acquires a pro-apoptotic function. The truncated Atg5 fragment undergoes translocation from the cytosol to the mitochondria, where it associates with the anti-apoptotic molecule Bcl-xL and then triggers apoptosis (Yousefi et al., 2006).
Autophagy Functions as a Pro-Cell Survival Mechanism in Mediating Radiotherapy and Chemotherapy Damage
Substantial studies have shown that inhibition of autophagy sensitizes cancer cells to DNA damaging anti-cancer reagents. Inhibition of autophagy by 3-methyladenine (3-MA) and Atg7 siRNA enhances 5-FU induced cytotoxicity in human colorectal cancer cells (Li et al., 2010). Autophagy suppression also enhances the therapeutic efficacy of cisplatin and 5-FU in esophageal and colon cancer cells, respectively (Liu et al., 2009; Li et al., 2010). siRNA-mediated silencing of autophagy-related genes such as Beclin 1, Atg3, and Atg4b sensitizes resistant cancer cells to ionizing radiation (Apel et al., 2008). These studies suggest that autophagy may provide a mechanism through which cancer cells acquire resistance to radiotherapy and chemotherapy.
Cancer stem cells, a sub-population of cells with abilities to give rise to all cell types found in a particular cancer sample, have been shown to be highly resistant to ionizing radiation and other anti-cancer treatments (Pajonk et al., 2010). It has been proposed that such therapy resistance may be due to increased autophagy in the “stem cell” sub-population. For example, in malignant gliomas, the cancer stem cells, identified by CD133 positivity, express higher levels of autophagy-related proteins including LC3, Atg5, and Atg12. Higher levels of autophagy are induced in CD133+ cancer stem cells than in CD133− cells following ionizing radiation (Lomonaco et al., 2009). Furthermore, glioma cells treated with autophagy inhibitors exhibit more extensive DNA double-strand breaks than cells treated with radiation alone (Ito et al., 2005). These data suggest that cancer stem cells with increased autophagy activity may provide one of the mechanisms of the resistance to chemotherapy and ionizing radiation.
To reverse the pro-cell survival mechanism exerted by autophagy, hydroxychloroquine (HCQ) or chloroquine (CQ) are being studied in clinical trials as autophagy inhibitors to enhance tumor cell killing when combined with chemotherapy and radiotherapy. HCQ and CQ are widely used as anti-malarial and anti-rheumatic agents. They disrupt lysosome acidification, thereby blocking the maturation of autophagosomes (Bellodi et al., 2009). For example, in patients with glioblastoma, a phase III clinical trial combining radiation and carmustine with either daily CQ or placebo demonstrated a median survival of 24 months in the CQ arm compared with 11 months in the placebo arm (Sotelo et al., 2006). However, the results of this study were not statistically significant, probably due to a small sample size, but it does provide evidence for the safety of adding CQ to current anti-cancer regimens and suggests that CQ has the potential to improve patient survival. Besides CQ and HCQ, there are other drugs and compounds being investigated in pre-clinical stages as autophagy inhibitors. Verteporfin, a benzoporphyrin derivative, has been shown to inhibit autophagosome formation (Donohue et al., 2011). Another small molecule, lucanthone, currently used as an anti-schistosome agent, inhibits autophagy by impairing autophagic degradation (Carew et al., 2011). Thus, there appears to be several classes of autophagy inhibitors with the potential to sensitize cancer cells to certain conventional chemotherapeutic drugs and to ionizing radiation.
Autophagy Functions as a Pro-Cell Death Mechanism in Radiotherapy and Chemotherapy
In contrast to its pro-survival role as discussed above, autophagy has also been shown to function in type II programmed cell death in other cancer cell lines following similar treatments with chemotherapy or ionizing radiation. Stimulation of autophagy by RAD001, a mTOR inhibitor, was shown to produce a synergistic killing effect on papillary thyroid cancer cells by doxorubicin (Fujiwara et al., 2007). Akt inhibition induces autophagy and enhances cell death when combined with chemotherapy as well (Degtyarev et al., 2008). Induction of autophagy in papillary thyroid cancer through mTOR inhibition has also been shown to increase cytotoxicity to ionizing radiation (Lin et al., 2010). Furthermore, it has been reported that vitamin D or a vitamin D analog (compound EB 1089) sensitizes breast cancer cells to ionizing radiation by promoting autophagic cell death (Gewirtz et al., 2009). These data indicate that autophagy also functions as a mechanism of pro-cell death, depending on the cellular context.
One of the explanations that certain types of cancer cells undergo autophagic cell death is that the apoptosis pathway is inactivated in these cells. Substantial evidence suggests that apoptosis is often disabled in cancer cells due to frequent mutations in apoptosis-inducing genes such as p53 and bax (Mrozek et al., 2003). Thus, it is not surprising that tumor cells under extreme stress often die by other mechanisms such as type II autophagic cell death. Therefore, induction of autophagic cell death may be an ideal approach in those cancers that are intrinsically resistant to apoptosis following anti-cancer therapies (e.g., chemotherapy, radiation).
To utilize the role of autophagy as a pro-cell death mechanism, multiple pharmacologic agents are now available for pre-clinical or clinical testing to enhance the treatment efficacy of chemotherapy or ionizing radiation. The most commonly used autophagy-inducers are the mTOR inhibitors. As a currently approved drug for the treatment of renal cell carcinoma, Everolimus, a mTOR inhibitor, induces autophagy and enhances sensitivity to ionizing radiation in prostate cancer and non-small-cell lung cancer (NSCLC) cell lines and xenografts (Cao et al., 2006; Kim et al., 2008). Multiple clinical trials with mTOR inhibitors are under way to investigate the role of induction of autophagy in anti-tumor activity. It has also been shown that the poly(ADP-ribose) polymerase-1 (PARP1) inhibitor ABT-888 induces autophagy and sensitize H460 NSCLC cells to ionizing radiation (Albert et al., 2007). Other small molecules such as bortezomib (a proteasome inhibitors), imatinib (a tyrosine kinase inhibitor), and tamoxifen (an antiestrogen) have been reported to induce autophagy in human cancer cells as well (Janku et al., 2011). These data suggest that autophagy-inducers hold great potential for improving the treatment of cancers following chemotherapy and ionizing radiation in the near future.
Concluding Remarks
The study of autophagy is a very exciting and highly promising area of cancer research. There has been much recent progress in our understanding the pathways that control autophagy. Further exploration of these pathways holds great potential for improving the treatment efficacy of chemotherapy and ionizing radiation. However, despite this potential, one of the most difficult questions remains to be answered: whether autophagy should be inhibited or stimulated to improve clinical outcomes? As illustrated in Figure 3, the impact of autophagy on chemotherapy and radiotherapy mediated tumor cytotoxicity (to live or not to live) seems to depend on the cellular context. Thus, gene expression signatures may be identified as biomarkers to help make such decisions in the future.
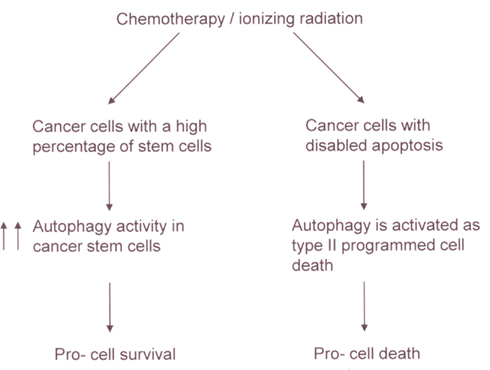
Figure 3. A model for the impact of autophagy on chemotherapy and radiotherapy mediated tumor cytotoxicity. In a tumor with a high percentage of cancer stem cells, chemotherapy, or ionizing radiation can increase autophagy activity in the stem cells, leading to the resistance of the tumor to the treatments. In contrast, in cancer cells with disabled apoptosis, autophagy may be activated as a type II programmed cell death in response to chemotherapy or ionizing radiation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Supported, in part, by NIH Grant U56 CA112963, the DBJ Foundation, and the University Radiation Medicine Foundation.
References
Albert, J. M., Cao, C., Kim, K. W., Willey, C. D., Geng, L., Xiao, D., Wang, H., Sandler, A., Johnson, D. H., Colevas, A. D., Low, J., Rothenberg, M. L., and Lu, B. (2007). Inhibition of poly(ADP-ribose) polymerase enhances cell death and improves tumor growth delay in irradiated lung cancer models. Clin. Cancer Res. 13, 3033–3042.
Amaravadi, R. K., Lippincott-Schwartz, J., Yin, X. M., Weiss, W. A., Takebe, N., Timmer, W., Dipaola, R. S., Lotze, M. T., and White, E. (2011). Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 17, 654–666.
Apel, A., Herr, I., Schwarz, H., Rodemann, H. P., and Mayer, A. (2008). Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res. 68, 1485–1494.
Bellodi, C., Lidonnici, M. R., Hamilton, A., Helgason, G. V., Soliera, A. R., Ronchetti, M., Galavotti, S., Young, K. W., Selmi, T., Yacobi, R., Van Etten, R. A., Donato, N., Hunter, A., Dinsdale, D., Tirro, E., Vigneri, P., Nicotera, P., Dyer, M. J., Holyoake, T., Salomoni, P., and Calabretta, B. (2009). Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Invest. 119, 1109–1123.
Brown, J. M., and Wouters, B. G. (1999). Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 59, 1391–1399.
Cao, C., Subhawong, T., Albert, J. M., Kim, K. W., Geng, L., Sekhar, K. R., Gi, Y. J., and Lu, B. (2006). Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 66, 10040–10047.
Carew, J. S., Espitia, C. M., Esquivel, J. A. II, Mahalingam, D., Kelly, K. R., Reddy, G., Giles, F. J., and Nawrocki, S. T. (2011). Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J. Biol. Chem. 286, 6602–6613.
Crighton, D., Wilkinson, S., O’prey, J., Syed, N., Smith, P., Harrison, P. R., Gasco, M., Garrone, O., Crook, T., and Ryan, K. M. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134.
Degtyarev, M., De Maziere, A., Orr, C., Lin, J., Lee, B. B., Tien, J. Y., Prior, W. W., Van Dijk, S., Wu, H., Gray, D. C., Davis, D. P., Stern, H. M., Murray, L. J., Hoeflich, K. P., Klumperman, J., Friedman, L. S., and Lin, K. (2008). Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol. 183, 101–116.
Donohue, E., Tovey, A., Vogl, A. W., Arns, S., Sternberg, E., Young, R. N., and Roberge, M. (2011). Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J. Biol. Chem. 286, 7290–7300.
Dunn, W. A. Jr. (1990). Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J. Cell Biol. 110, 1923–1933.
Fujiwara, K., Iwado, E., Mills, G. B., Sawaya, R., Kondo, S., and Kondo, Y. (2007). Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int. J. Oncol. 31, 753–760.
Gewirtz, D. A., Hilliker, M. L., and Wilson, E. N. (2009). Promotion of autophagy as a mechanism for radiation sensitization of breast tumor cells. Radiother. Oncol. 92, 323–328.
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93.
Hoyer-Hansen, M., Bastholm, L., Szyniarowski, P., Campanella, M., Szabadkai, G., Farkas, T., Bianchi, K., Fehrenbacher, N., Elling, F., Rizzuto, R., Mathiasen, I. S., and Jaattela, M. (2007). Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 25, 193–205.
Ito, H., Daido, S., Kanzawa, T., Kondo, S., and Kondo, Y. (2005). Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 26, 1401–1410.
Janku, F., Mcconkey, D. J., Hong, D. S., and Kurzrock, R. (2011). Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. doi: 10.1038/nrclinonc.2011.71
Kim, K. W., Hwang, M., Moretti, L., Jaboin, J. J., Cha, Y. I., and Lu, B. (2008). Autophagy upregulation by inhibitors of caspase-3 and mTOR enhances radiotherapy in a mouse model of lung cancer. Autophagy 4, 659–668.
Lavieu, G., Scarlatti, F., Sala, G., Carpentier, S., Levade, T., Ghidoni, R., Botti, J., and Codogno, P. (2006). Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J. Biol. Chem. 281, 8518–8527.
Li, J., Hou, N., Faried, A., Tsutsumi, S., and Kuwano, H. (2010). Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo models. Eur. J. Cancer 46, 1900–1909.
Lin, C. I., Whang, E. E., Donner, D. B., Du, J., Lorch, J., He, F., Jiang, X., Price, B. D., Moore, F. D. Jr., and Ruan, D. T. (2010). Autophagy induction with RAD001 enhances chemosensitivity and radiosensitivity through Met inhibition in papillary thyroid cancer. Mol. Cancer Res. 8, 1217–1226.
Liu, D., Yang, Y., Liu, Q., and Wang, J. (2009). Inhibition of autophagy by 3-MA potentiates cisplatin-induced apoptosis in esophageal squamous cell carcinoma cells. Med. Oncol. 28, 105–111.
Lomonaco, S. L., Finniss, S., Xiang, C., Decarvalho, A., Umansky, F., Kalkanis, S. N., Mikkelsen, T., and Brodie, C. (2009). The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int. J. Cancer 125, 717–722.
Mizushima, N., Sugita, H., Yoshimori, T., and Ohsumi, Y. (1998). A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 273, 33889–33892.
Mrozek, A., Petrowsky, H., Sturm, I., Kraus, J., Hermann, S., Hauptmann, S., Lorenz, M., Dorken, B., and Daniel, P. T. (2003). Combined p53/Bax mutation results in extremely poor prognosis in gastric carcinoma with low microsatellite instability. Cell Death Differ. 10, 461–467.
Pajonk, F., Vlashi, E., and McBride, W. H. (2010). Radiation resistance of cancer stem cells: the 4 R’s of radiobiology revisited. Stem Cells 28, 639–648.
Rizzuto, R., and Pozzan, T. (2006). Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 86, 369–408.
Scherz-Shouval, R., Shvets, E., Fass, E., Shorer, H., Gil, L., and Elazar, Z. (2007). Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 26, 1749–1760.
Shintani, T., and Klionsky, D. J. (2004). Autophagy in health and disease: a double-edged sword. Science 306, 990–995.
Sotelo, J., Briceno, E., and Lopez-Gonzalez, M. A. (2006). Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial. Ann. Intern. Med. 144, 337–343.
Vousden, K. H., and Lane, D. P. (2007). p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283.
Yang, Z., and Klionsky, D. J. (2010). Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131.
Yousefi, S., Perozzo, R., Schmid, I., Ziemiecki, A., Schaffner, T., Scapozza, L., Brunner, T., and Simon, H. U. (2006). Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 8, 1124–1132.
Zeng, X., and Kinsella, T. J. (2007). A novel role for DNA mismatch repair and the autophagic processing of chemotherapy drugs in human tumor cells. Autophagy 3, 368–370.
Zeng, X., Yan, T., Schupp, J. E., Seo, Y., and Kinsella, T. J. (2007). DNA mismatch repair initiates 6-thioguanine-induced autophagy through p53 activation in human tumor cells. Clin. Cancer Res. 13, 1315–1321.
Zeng, X., and Kinsella, T. J. (2008). Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine induced autophagy. Cancer Res. 68, 2384–2390.
Keywords: autophagy, apoptosis, chemotherapy, radiotherapy
Citation: Zeng X and Kinsella TJ (2011) Impact of autophagy on chemotherapy and radiotherapy mediated tumor cytotoxicity: “to live or not to live”. Front. Oncol. 1:30. doi: 10.3389/fonc.2011.00030
Received: 21 July 2011;
Paper pending published: 23 August 2011;
Accepted: 13 September 2011;
Published online: 03 October 2011.
Edited by:
James B. Mitchell, National Institutes of Health, USAReviewed by:
Mira Jung, Georgetown University, USAChristopher Schultz, Medical College of Wisconsin, USA
Copyright: © 2011 Zeng and Kinsella. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Timothy James Kinsella, Department of Radiation Oncology, Rhode Island Hospital, Physicians Office Building, Suite 130, 110 Lockwood Street, Providence, RI 02903, USA. e-mail:dGtpbnNlbGxhQGxpZmVzcGFuLm9yZw==