



- 1 Department of Oncological Sciences, Mount Sinai School of Medicine, New York, NY, USA
- 2 Department of Dermatology, Mount Sinai School of Medicine, New York, NY, USA
- 3 The Tisch Cancer Institute, Mount Sinai Medical Center, New York, NY, USA
The global incidence of melanoma has dramatically increased during the recent decades, yet the advancement of primary and adjuvant therapies has not kept a similar pace. The development of melanoma is often centered on cellular signaling that hyper-activates survival pathways, while inducing a concomitant blockade to cell death. Aberrations in cell death signaling not only promote tumor survival and enhanced metastatic potential, but also create resistance to anti-tumor strategies. Chemotherapeutic agents target melanoma tumor cells by inducing a form of cell death called apoptosis, which is governed by the BCL-2 family of proteins. The BCL-2 family is comprised of anti-apoptotic proteins (e.g., BCL-2, BCL-xL, and MCL-1) and pro-apoptotic proteins (e.g., BAK, BAX, and BIM), and their coordinated regulation and function are essential for optimal responses to chemotherapeutics. Here we will discuss what is currently known about the mechanisms of BCL-2 family function with a focus on the signaling pathways that maintain melanoma tumor cell survival. Importantly, we will critically evaluate the literature regarding how chemotherapeutic strategies directly impact on BCL-2 family function and offer several suggestions for future regimens to target melanoma and enhance patient survival.
Introduction
Skin cancer is one of the leading causes of death worldwide. Presently in the United States, it is estimated that one in five people will develop some form of skin cancer due to a combination of family history, environment, and genetic factors (see the American Cancer Society for more information, http://www.cancer.org/Cancer/SkinCancer-Melanoma/Detailed Guide/melanoma-skin-cancer-risk-factors). DNA damage caused by ultraviolet irradiation is thought to be the main environmental culprit behind the development of this disease. Likewise, genetic disruption of genes necessary for the appropriate balances between survival, proliferation, and cell death are frequently associated with the development and persistence of cells that result in skin cancer. These cancerous cells may also develop mechanisms to evade the immune system resulting in unimpeded cell proliferation, survival, and metastases. Malignant melanoma is the deadliest form of skin cancer, and it arises from the uncontrollable cell division of the basal layer melanin-producing cells called melanocytes (for in-depth review, see Ibrahim and Haluska, 2009). Melanoma commonly involves the skin, but it can occur at other sites such as the eyes, ears, and gastrointestinal tract. While melanoma only accounts for 5% of skin cancers, it is responsible for the majority of skin cancer deaths (Jemal et al., 2009). The prognosis of melanoma is favorable when limited to the skin and wide excision is curative. However, the prognosis of metastatic melanoma is very poor with a 5-year survival rate of less than 20%. Presently, the most used anti-tumor strategy for stage IV metastatic melanoma is dacarbazine (DITC), which as a single treatment is not very effective, and is associated with <20% response rate (Tsao et al., 2004). Adjuvant and combination treatments are also used; for example, cisplatin, vinblastine, and DTIC (the regimen is referred to as “CVD”), in combination with interferon alpha-2α (IFNα) and interleukin-2 (IL-2), but none have proven to be significantly effective, yet they demonstrate substantial toxicity (Legha et al., 1989, 1996; Phan et al., 2001). In the majority of cases where a response does occur, there is often recurrence of the cancer within months. A better understanding of the pathways that are activated to promote melanoma, and targeted during treatment, will hopefully reveal novel therapeutic opportunities. Toward the end of our discussion, we will propose that manipulating the BCL-2 family and the mitochondrial pathway may offer a potential therapeutic inroad to treatment success.
The Mitochondrial Pathway of Apoptosis
In response to chemotherapeutics, cells attempt to eliminate the damage or recover from it by engaging stress response pathways (e.g., the p53 pathway), but often the damage is overwhelming, and the treated cells induce a form of programmed cell death known as apoptosis (Green and Evan, 2002). Apoptotic signaling can initiate from outside the cell via plasma membrane receptors (referred to as the “extrinsic pathway”; e.g., CD95/FAS or tumor necrosis factor receptor, TNFR), or through stress that originates from within the cell (referred to as the “intrinsic” or “mitochondrial pathway”; e.g., macromolecular damage; Guicciardi and Gores, 2009; Chipuk et al., 2010). The extrinsic apoptotic pathway is engaged when pro-apoptotic ligands such as TNFα or CD95L/FASL trigger death receptor signaling which directly leads to the activation of caspases, which are the cysteine–aspartic proteases responsible for inducing the apoptotic phenotype (e.g., DNA laddering, loss of plasma membrane asymmetry, and cellular blebbing; Pop and Salvesen, 2009). The intrinsic pathway is triggered by intracellular stress signals such as DNA damage, oncogenes, hypoxia, and growth factor withdrawal (Chipuk et al., 2010). These cellular stressors transcriptionally and post-transcriptionally regulate the B cell CLL/lymphoma-2 (BCL-2) family of proteins, which are responsible for mitochondrial integrity, subsequent caspase activation, and apoptosis. While these two general pathways are distinct, there are situations of cross-talk where the extrinsic pathway promotes apoptosis through the intrinsic pathway and vice versa. In these situations, the BCL-2 family of proteins is also crucial control points of cellular fate. For the most part, cancer cells utilize and respond to chemotherapeutic agents by regulating the mitochondrial pathway of apoptosis, so our discussions will be focused on these signaling mechanisms (Figure 1).
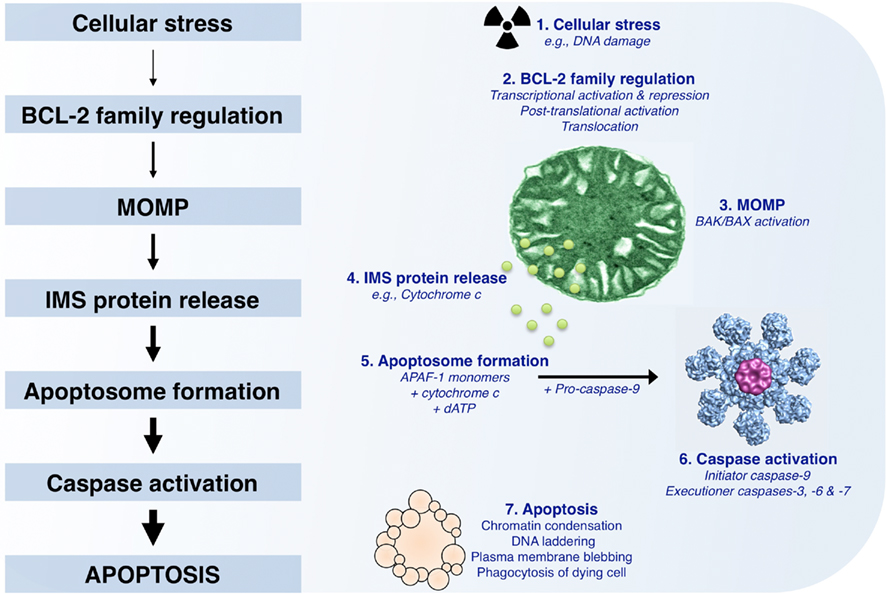
Figure 1. The mitochondrial pathway of apoptosis. Following cellular stress such as DNA damage, the BCL-2 family of proteins is regulated by transcriptional (increased or decreased) and post-translational (e.g., phosphorylation, cleavage, and relocalization) mechanisms. If the cellular stress is irreparable, the culmination of pro-apoptotic signaling will lead to mitochondrial outer membrane permeabilization (MOMP), which allows for the release of soluble intermembrane space proteins, including cytochrome c. APAF-1, cytochrome c, and dATP then coordinate to promote APAF-1 oligomerization and recruitment of pro-caspase-9; this complex is referred to as the “apoptosome.” The apoptosome allows for pro-caspase-9 dimerization and activation, and the subsequent cleavage and activation of executioner caspases (caspase-3, -6, and -7). These caspases promote the characteristic phenotypes of apoptosis, e.g., chromatin condensation, DNA laddering, and plasma membrane blebbing, by cleaving numerous intracellular substrates. Within the body, apoptotic cells are rapidly cleared by phagocytosis to ensure tissue homeostasis.
Once the apoptotic program has been initiated by compromising the outer mitochondrial membrane (OMM), pro-apoptotic factors are released from the mitochondrial intermembrane space (IMS), including cytochrome c (Figure 1; Liu et al., 1996). Monomeric apoptotic protease activating factor 1 (APAF-1) cooperates with cytochrome c and dATP to form the oligomeric caspase activation platform referred to as the “apoptosome” (Li et al., 1997; Zou et al., 1997). The apoptosome recruits, dimerizes, and activates pro-caspase-9, which then promotes the downstream function of pro-caspases-3, -6, and -7 (Figure 1). Often, this step in the mitochondrial pathway is referred to as “the point of no return,” as mitochondrial function and ATP generation gradually wane, and proteolytic cleavage of caspase substrates ensues. Caspase activity is responsible for the morphological phenotypes of apoptosis, including DNA laddering, loss of plasma membrane asymmetry, and phagocytosis (Figure 1; Logue and Martin, 2008). Regulation of the mitochondrial pathway of apoptosis plays a crucial role in the development and treatment of melanoma; and it has been shown that defects in this pathway can result in resistance to tumor suppressor pathways and treatments (for an in-depth review, see Soengas and Lowe, 2003).
The BCL-2 Family of Proteins
The involvement of mitochondria in apoptosis was first suggested when caspase activity resulted from Xenopus oocyte extract co-incubation with purified mitochondria (Newmeyer et al., 1994). This activity was blocked by the addition of BCL-2, suggesting that BCL-2 could prevent mitochondrial engagement of the cytosol. Since then, the BCL-2 family has grown to include almost 20 members that are divided into two functional classes of proteins: anti-apoptotic and pro-apoptotic (Figure 2). Most cells express a variety of anti-apoptotic and pro-apoptotic BCL-2 proteins, and through the regulation of their interactions command survival or commitment to apoptosis (for an in-depth review, see Chipuk and Green, 2008).
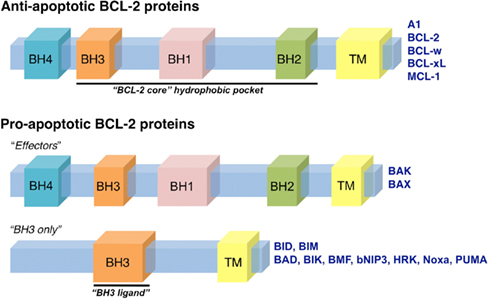
Figure 2. The BCL-2 family of proteins. The BCL-2 family of proteins is divided into anti-apoptotic and pro-apoptotic members. The anti-apoptotic members include: A1, BCL-2, BCL-w, BCL-xL, and MCL-1, all of which contain four BCL-2 homology domains (termed BH1-4). The pro-apoptotic proteins are subdivided into “effector” and “BH3-only” members. The effector proteins, BAK and BAX, also contain BH1-4; while the BH3-only proteins contain only one domain, a BH3, that is required for interactions with anti-apoptotic and effector proteins. The BH3-only proteins include: BAD, BID, BIK, BIM, BMF, bNIP3, HRK, Noxa, and PUMA. The BH1-3 regions form the conserved “BCL-2 core” structural unit, which includes a hydrophobic groove that binds to BH3 domains. Many of the BCL-2 family proteins also contain a transmembrane domain (TM).
Anti-apoptotic BCL-2 proteins are comprised of four BCL-2 homology domains (BH1-4) and are generally integrated within the OMM, but may be present in other membranes like the endoplasmic reticulum (Petros et al., 2004). BCL-2, BCL-xL, and myeloid cell leukemia 1 (MCL-1) are the major members of the anti-apoptotic BCL-2 repertoire that function to preserve OMM integrity by directly binding and inhibiting the pro-apoptotic BCL-2 proteins (Chen et al., 2005; Willis et al., 2005, 2007; Figure 2).
The pro-apoptotic BCL-2 members are divided into effectors (which also contain BH1-4) and the BH3-only proteins (Figure 2). The effector proteins BAK (BCL-2 antagonist killer 1) and BAX (BCL-2 associated x protein) homo-oligomerize into proteolipid pores within the OMM and are required to promote MOMP and cytochrome c release (Lindsten et al., 2000; Wei et al., 2001). However, these effectors require an activation step, upon which they oligomerize and gain the capacity to permeabilize membranes composed of mitochondrial lipids (Kuwana et al., 2002). Activation of BAK and BAX occurs through interaction with so-called “direct activators” (see below), or by physico-chemical effects of detergents, mild heat, or elevated pH (Hsu and Youle, 1997; Khaled et al., 2001; Letai et al., 2002; Pagliari et al., 2005). The BH3-only proteins act to regulate both the anti-apoptotics and effectors (Figures 2 and 3). Two of these, BID (BH3 interacting domain death agonist) and BIM (BCL-2 interacting mediator of cell death), are direct activators of BAK and BAX, acting via their BH3 domains to induce oligomerization and the permeabilization function of BAK and BAX (Wei et al., 2000; Kuwana et al., 2002, 2005; Letai et al., 2002). The process by which BAK and BAX permeabilize the OMM is commonly referred to as “mitochondrial outer membrane permeabilization,” or MOMP.
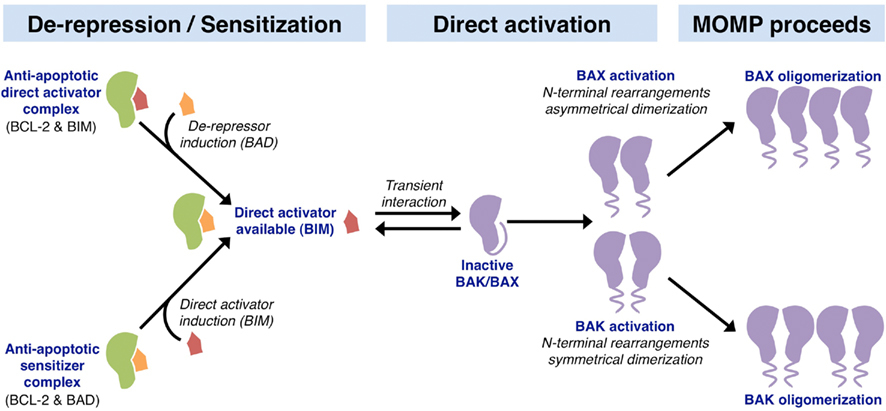
Figure 3. Mechanisms of action within the BCL-2 family leading to MOMP. The upstream involvement of the BCL-2 family in apoptosis can be divided into two parallel activities: de-repression and sensitization. The de-repression model begins at cellular status quo with anti-apoptotic BCL-2 proteins binding and sequestering BH3-only direct activators (e.g., BCL-2 binds BIM). In response to stress, a de-repressor BH3-only protein is induced (e.g., BAD), which can then displace the direct activator and liberate it to activate BAK and BAX. The sensitization model involves the preemptive binding and inactivation of anti-apoptotic proteins by sensitizer BH3-only proteins (e.g., BCL-2 binds BAD). This sensitization prevents the inhibition of direct activator BH3-only proteins induced following apoptotic stimulation (e.g., BIM), thus lowering the threshold level of direct activator necessary to activate the effectors BAK and BAX. Unbound direct activator BH3-only proteins function in the direct activation of BAK and BAX through a transient interaction. These interactions lead to conformational changes involving N-terminal rearrangement, which exposes the BH3 domain of BAK and BAX. Activated BAK monomers pair symmetrically, and oligomerize as sets of dimers to permeabilize the outer mitochondrial membrane leading to MOMP. In contrast, activated BAX monomers pair asymmetrically to oligomerize and promote MOMP.
The activation of BAK and BAX leading to MOMP is complex and incompletely understood. Recent in vitro studies show that activated BID and BAX bind to one another only in association with membranes, and this precedes BAX oligomerization, which is followed by membrane permeabilization (Lovell et al., 2008). The accepted viewpoint is that the expression, stability, and activation of the BH3-only proteins and their interactions with the anti-apoptotic and effector proteins sufficiently links pro-apoptotic signal transduction to MOMP(Chipuk et al., 2010). Broadly speaking, regulation of the BH3-only proteins at the transcriptional and translational levels determines the OMM integrity. When an apoptotic signal is initiated, the anti-apoptotic BCL-2 proteins are inhibited by the sensitizer/de-repressor BH3-only proteins (e.g., BAD, BMF, Hrk, Noxa, and PUMA), which allows for direct activator-induced BAK and BAX activation to promote MOMP, and subsequent apoptosis (Figure 3). Non-BCL-2 family direct activator proteins are also described; several reports suggest a direct activator function for cytosolic p53, MAP-1, and ASC (Tan et al., 2001; Chipuk et al., 2004; Ohtsuka et al., 2004). Furthermore, controversy exists regarding the role of PUMA (p53 upregulated modulator of apoptosis) in mediating BAK and BAX activation, as conflicting results suggest that PUMA is either a de-repressor/sensitizer or direct activator BH3-only protein (Chipuk et al., 2005; Kuwana et al., 2005; Kim et al., 2009). Finally, the steps leading to BAK and BAX activation have been unveiled through studies that focused on structural consequences of BIM and BAK/BAX interactions; these structural details are discussed in Figure 3 (Dewson et al., 2008, 2009; Gavathiotis et al., 2008, 2010). In terms of chemotherapeutic success, pushing a cell to undergo MOMP is suggested to enhance clinical outcomes, and this is highlighted by the development of small molecule inhibitors to the anti-apoptotic BCL-2 proteins, which lower the cellular threshold for MOMP and apoptosis (Letai, 2008). We will discuss these in greater detail later.
Melanoma and the BCL-2 Family
Melanoma is notorious for chemoresistance, and there continues to be tremendous research focused on identifying the molecular mechanisms to explain this phenotype (for an in-depth review, see Hocker et al., 2008). Unlike many other cancers, melanoma rarely displays mutations within the tumor suppressor p53 pathway, which does afford the opportunity to pharmacologically regulate the p53 pathway through chemotherapy-induced DNA damage and stress-responses, and we will discuss this throughout (Hocker and Tsao, 2007). However, mutations in N-RAS [neuroblastoma RAS viral (v-ras) oncogene homolog] and B-RAF (v-raf murine sarcoma viral oncogene homolog B1) are common events in melanoma, which are observed in approximately 20 and 70% of patients, respectively (Shukla et al., 1989; Davies et al., 2002; Reifenberger et al., 2004; Edlundh-Rose et al., 2006). Additionally, the loss of PTEN (phosphatase and tensin homolog) expression, activation of the AKT/PI3K (phosphatidylinositol 3-kinases) pathway, and loss of CDKN2A (cyclin-dependent kinase inhibitor 2A) often synergize with B-RAF mutations leading to the progression from benign nevus, primary tumor, to metastatic disease (Haluska and Hodi, 1998; Kim, 2010; Figure 4).
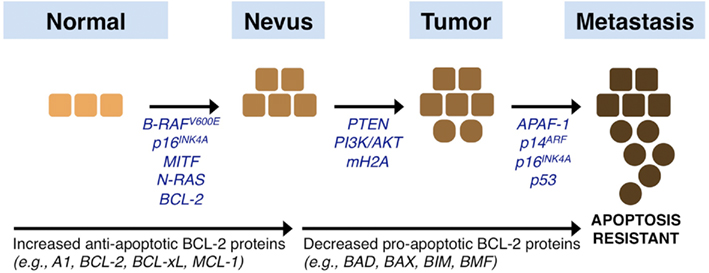
Figure 4. Melanoma signaling networks, progression, and influences by the BCL-2 family. Melanoma originates from melanocytes in the skin that acquire a series of changes that promote the progression from a normal phenotype to a nevus, primary tumor, and eventual metastatic disease. Epigenetically silenced, mutated, or deleted genes that promote disease progression are listed and discussed throughout the text. In brief, common mutations observed early in melanoma development are in the genes encoding N-RAS and B-RAF, and these are often complemented by mutations in the melanocyte regulators such as MITF and BCL-2. Subsequent changes to PTEN, CDKN2A, and mH2A along with activating mutations of the AKT/PI3K pathway synergize with early mutations. Subsequently, alterations in pro-apoptotic sensitivity upstream (e.g., p53) and downstream (e.g., APAF-1) of the mitochondrial pathway of apoptosis are suggested. In parallel to these events, the expression and function of the BCL-2 family is altered to establish marked resistance to pro-apoptotic stimulation: there is increased expression of anti-apoptotic members, and coordinated downregulation of pro-apoptotic members.
Activating mutations in N-RAS and B-RAF lead to positive influences on the cell cycle, mainly by promoting mitosis and preventing cells from engaging their repair and stress machineries, which leads to increased stress and accumulated mutations. Studies also suggest that the expression of AKT/PI3K is directly correlated with melanoma progression; and PTEN expression is subsequently lost by genetic and epigenetic mechanisms to further promote AKT/PI3K survival pathways (Steck et al., 1997; Tsao et al., 1998, 2000; Zhou et al., 2000; Stahl et al., 2004; Karbowniczek et al., 2008). Indeed, a recent mouse model of combined B-RAF V600E and PTEN loss provides in vivo evidence that these two pathways are necessary and sufficient to promote metastatic melanoma formation (Dankort et al., 2009). To counteract the proliferative signals generated from the above circumstances, cells can engage inhibitors to the cell cycle, for example CDKN2A, which encodes for the proteins p16INK4A (cyclin-dependent kinase inhibitor 2A) and p14ARF (alternative reading frame); yet CDKN2A is also mutated, genetically lost, or epigenetically silenced in a significant percentage of melanomas (Castellano and Parmiani, 1999). There are numerous other genes that are targeted during the transition between primary tumor and metastasis; more recently, the histone variant macroH2A was shown to suppress tumor progression of malignant melanoma (Kapoor et al., 2010). In brief, the sum of these mutations leads to an aggressive disease that is highly resistant to chemotherapeutics (Figure 4).
For this discussion, we are focused on understanding how melanomas with significant alterations in N-RAS, B-RAF, and the other pathways listed above fail to engage apoptosis despite aberrations in cellular signaling leading to unregulated proliferation (Figure 4). How are the cellular safeguards against uncontrolled proliferation silenced to prevent apoptosis? More importantly, can we pharmacologically regulate these aberrant pathways to produce better responses in the clinic? The answers to both of these questions point in the direction of the BCL-2 family of proteins. Here, we suggest that this family of anti- and pro-apoptotic members may be what allows for melanoma development, survival, and chemoresistance, yet it offers a pharmacologically tractable Achilles’ heel for successful combination/adjuvant therapies to treat malignant melanoma. In general, there are two approaches to block apoptosis that is normally engaged following deregulated cellular proliferation: the cell may decrease the expression of pro-apoptotic BCL-2 proteins (e.g., BAX or BIM); or the cell can over-express anti-apoptotic BCL-2 proteins (e.g., BCL-2 or MCL-1). In either case, the resulting phenotype is the failure to promote BAK/BAX activation, MOMP, and apoptosis following deregulated cellular proliferation cues or chemotherapeutic treatment. This ultimately leads to metastatic disease with poor prognosis and decreased long-term survival despite chemotherapeutic intervention.
Returning to the pathways that promote melanoma development and chemoresistance, it is widely regarded that B-RAF V600E is the most commonly observed mutation in patients (Davies et al., 2002; Pollock and Meltzer, 2002). This gain of function mutation is a valine to glutamic acid substitution at codon 600, which results in a hyper-activated kinase and subsequent marked increases in MAPK/ERK signaling (Wan et al., 2004). Increased B-RAF V600E activity is demonstrated to promote BIM phosphorylation, leading to its proteasome-mediated degradation in several melanoma models (Cartlidge et al., 2008; Sheridan et al., 2008; Boisvert-Adamo et al., 2009; Goldstein et al., 2009). The B-RAF V600E mutation is observed very early in melanoma development starting at the benign nevus stage. Therefore, reducing BIM levels and all apoptotic pathways that BIM directly regulates (e.g., cytokine/growth factor withdrawal) likely leads to marked resistance to apoptosis (Bouillet et al., 1999). Likewise, since BIM is a direct activator of BAX and BAK, chemotherapies that utilize a BIM-dependent mechanism will likely not be successful (Letai et al., 2002; Kuwana et al., 2005; Dai et al., 2008). Multiple other pro-apoptotic BCL-2 proteins are also targeted by the B-RAF V600E mutation. Several studies suggest that excessive signaling through B-RAF and MAPK promotes BAD (BCL-2 antagonist of cell death) phosphorylation and inactivation, and decreased BMF (BCL-2 modifying factor) and PUMA expression, and all these events lead to abrogated BAK/BAX activation, MOMP, and apoptosis (Cartlidge et al., 2008; Sheridan et al., 2008; Boisvert-Adamo et al., 2009; Goldstein et al., 2009; Keuling et al., 2010; Shao and Aplin, 2010).
There is also a growing literature connecting RAS and B-RAF mutations to the induction of autophagy. Autophagy is a catabolic process which promotes the degradation of cytoplasm and organelles; and this plays a key role in tumorigenesis as it often helps cells cope with stress (for in-depth review, see Kondo et al., 2005). Activating mutations in RAS and B-RAF promote autophagy, which can restrict tumor cell growth (Chen and Karantza-Wadsworth, 2009; Maddodi et al., 2010; Elgendy et al., 2011). In contrast, some tumor cells display an addiction to autophagy, which can promote glucose metabolism and mitochondrial function, leading to enhanced tumorigenesis (Guo et al., 2011; Lock et al., 2011). Interestingly, anti-apoptotic BCL-2 proteins function at the intersection between autophagy and apoptosis by directly regulating Beclin-1 activity, a key protein that is required for the initiation of the formation of the autophagy machinery. Therefore, integrating the mechanistic interplay between the pathways that promote melanoma development, autophagy, and the cell death machinery is of critical importance to understanding this disease and potential therapeutic strategies (Levine et al., 2008; Chipuk et al., 2010). Importantly, the regulation of anti-apoptotic BCL-2 proteins by BH3-only proteins and BH3 mimetics (discussed later) impacts not only on apoptosis, but also autophagy, and subsequent changes to stress signaling, proteostasis, and metabolism; yet we will focus our discussion on cellular commitment to apoptosis.
The role of the anti-apoptotic BCL-2 proteins in melanoma is highlighted by the phenotype of the bcl-2 knockout mouse which displays dramatic graying of the hair because of diminished follicular melanocyte survival (Yamamura et al., 1996). In support of this phenotype, microphthalmia associated transcription factor (MITF), a key transcription factor necessary for melanocyte development and survival, directly induces bcl-2 promoter activity (McGill et al., 2002). Elevated levels of BCL-2 and MITF can promote chemoresistance and are associated with melanoma progression; furthermore, antisense against BCL-2 or MITF promotes a decrease in melanoma cell survival and sensitivity to chemotherapy (Jansen et al., 1998; Gautschi et al., 2001; McGill et al., 2002). This leads us to ask why melanocytes are solely addicted to BCL-2? Perhaps this cell type does not express other anti-apoptotic BCL-2 proteins, so small increases in BH3-only protein activity may promote premature MOMP and apoptosis, whereas this normally tolerated level of BH3-only protein activity would be actively sequestered by BCL-2 to allow for stress resolution. Such a limited repertoire of anti-apoptotic reserve would not appear to favor chemoresistance. However, the malignant transformation and chemoresistance seen in melanomas has also been attributed to increased levels in A1 (BCL-2 related gene A1), BCL-xL, and MCL-1, allowing for a rather substantial shift in the apoptotic threshold (Tang et al., 1998; Leiter et al., 2000; Zhang and Rosdahl, 2006; Placzek et al., 2010; Zhuang et al., 2010). The upregulation of these anti-apoptotic proteins during the transition from nevus to melanoma may indicate their active role in melanoma progression (Wong et al., 2008). Several additional transcription factors such as ETS-1 (v-ets erythroblastosis virus E26 oncogene homolog 1), and the chromatin remodeling factor DEK, promote MCL-1 expression, and may present novel mechanisms (and perhaps chemotherapeutic strategies) as to the mechanism of melanoma survival and chemoresistance (Khodadoust et al., 2009; Dong et al., 2011).
From both in vitro and in vivo melanoma model systems, several lines of evidence suggest that BCL-2 over-expression correlates with a malignant phenotype and a higher metastatic potential (Grover and Wilson, 1996; Takaoka et al., 1997; Xie et al., 1997; Leiter et al., 2000; Utikal et al., 2002; Zhang and Rosdahl, 2006). This seems logical, as a cell expressing more anti-apoptotic proteins should resist apoptosis despite cues to die following events from benign status to malignancy and metastasis. However, there are conflicting results in the literature that describe decreased BCL-2 expression during melanoma progression (Saenz-Santamaria et al., 1994; Ramsay et al., 1995; Tron et al., 1995; Tang et al., 1998; Korabiowska et al., 1999; Sviatoha et al., 2002). Similarly, BCL-2 is described to be constitutively expressed in melanocytes, nevi, and melanoma, with little change in expression levels (Cerroni et al., 1995; Collins and White, 1995; Morales-Ducret et al., 1995; Plettenberg et al., 1995; Miracco et al., 1998; Selzer et al., 1998; Loggini et al., 2001; Gradilone et al., 2003). The observations in both these scenarios can be reconciled if we consider that a cell may have the opportunity to express alternative anti-apoptotic BCL-2 members, such as BCL-w, BCL-xL, MCL-1, or A1. Another possibility is downregulation of the pro-apoptotic effectors BAK and BAX, which is also associated with tumor progression and decreased patient survival (Fecker et al., 2006; Tchernev and Orfanos, 2007). The balance between pro-apoptotic and anti-apoptotic BCL-2 members could also be considered to control apoptosis. For example, the BAX/BCL-2 ratio in chemotherapy-sensitive cells is relatively higher than in cells displaying chemoresistance (Raisova et al., 2001). However, great caution should be employed with monitoring only a few proteins, as MOMP and apoptosis proceed when multiple BH3-only proteins collaborate to promote BAK/BAX activation. Understanding the dynamic and total protein·protein interactions within the BCL-2 family is more indicative of apoptotic sensitivity and chemotherapeutic responses. This has been recently applied to tumors of lymphoid origin, and should be developed to better delineate which patients have the best chance of chemotherapeutic responses (Deng et al., 2007; Ryan et al., 2010).
Returning to our original questions, it appears that melanoma has developed numerous strategies to block apoptosis despite heightened cellular proliferation cues: the loss of BH3-only protein function, along with collateral increases in anti-apoptotic BCL-2 protein expression and diversity. The marked increase in anti-apoptotic BCL-2 proteins suggests that these cells can engage a pro-apoptotic response, and that the increased BCL-2 expression favors a selection process where increased BCL-2 (or other anti-apoptotic members) confers a survival advantage. Strategies that promote BH3-only protein function, while also decreasing the functional anti-apoptotic BCL-2 repertoire, will likely yield the best clinical responses; indeed, we will discuss the results of such strategies in the following sections.
Melanoma Treatment and the BCL-2 Family
Standard treatments for patients diagnosed with stage IV metastatic malignant melanoma are DTIC and IL-2. The FDA approved DTIC in 1975, and it remains the most commonly used single agent chemotherapeutic for metastatic melanoma. DTIC is an alkylating agent, which prevents DNA replication and subsequently blocks proliferation. Unfortunately, the response rate is less than 20% and resistance nearly always occurs (Stein and Brownell, 2008). Other chemotherapeutic agents are currently being studied to improve late stage melanoma treatment but few are proving to be more effective than DTIC. For example, temozolomide, a imidazotetrazine-derivative of DTIC, was tested in clinical trials but did not improve the overall survival of patients compared to DTIC (Patel et al., 2011). In addition to the standard combination chemotherapy regimens [e.g., CVD (cisplatin, vinblastine, DTIC), Dartmouth (DTIC, cisplatin, carmustine, and tamoxifen), and BOLD (bleomycin, vincristine, lomustine, and DTIC)], a number of novel adjuvant therapies are currently being evaluated to improve the efficiency of DTIC-based treatments (Seigler et al., 1980; Legha et al., 1989; Lattanzi et al., 1995). In particular, Ipilimumab, a human IgG1 monoclonal antibody that recognizes cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), enhances T-cell activation, proliferation, and attack of cancer cells (Melero et al., 2007; O’Day et al., 2007; Fong and Small, 2008; Robert and Ghiringhelli, 2009; Hodi et al., 2010). The combination of DTIC and Ipilimumab was recently shown to be associated with a significant increase in survival among patients with untreated metastatic melanoma (Robert et al., 2011).
The mechanism of action for DTIC-based therapies is to kill tumor cells by DNA damage, and these treatments likely signal through the p53 pathway to induce apoptosis (Igney and Krammer, 2002; Vousden and Lane, 2007). Fortunately, p53 is rarely targeted in melanoma, so the pro-apoptotic arm of the p53 pathway has the potential to be engaged. DTIC also directly impacts on the BCL-2 family from several angles: there is downregulation of anti-apoptotic proteins, and coordinated upregulation of BH3-only proteins and effector molecules, like BIM and BAX, respectively (Weber et al., 2009; Jiang et al., 2010b). Importantly, DTIC has been studied in multiple cellular models of melanoma, and the consistent observation is that DTIC promotes apoptosis through a marked regulation of the BCL-2 family leading to MOMP. But what accounts for the low response rate of melanoma patients to DTIC? Can the threshold for BAK/BAX activation, MOMP, and apoptosis be lowered to increase DTIC responses in patients? There is minimal information regarding how combination chemotherapy regimens (e.g., CVD) impact on the BCL-2 family. It is therefore essential to better understand the mechanisms of action to design appropriate co-administered adjuvant therapies. A non-exhaustive list of several chemotherapeutic drugs (some discussed below) and their impact on the BCL-2 family in common melanoma cell lines is provided in Table 1.
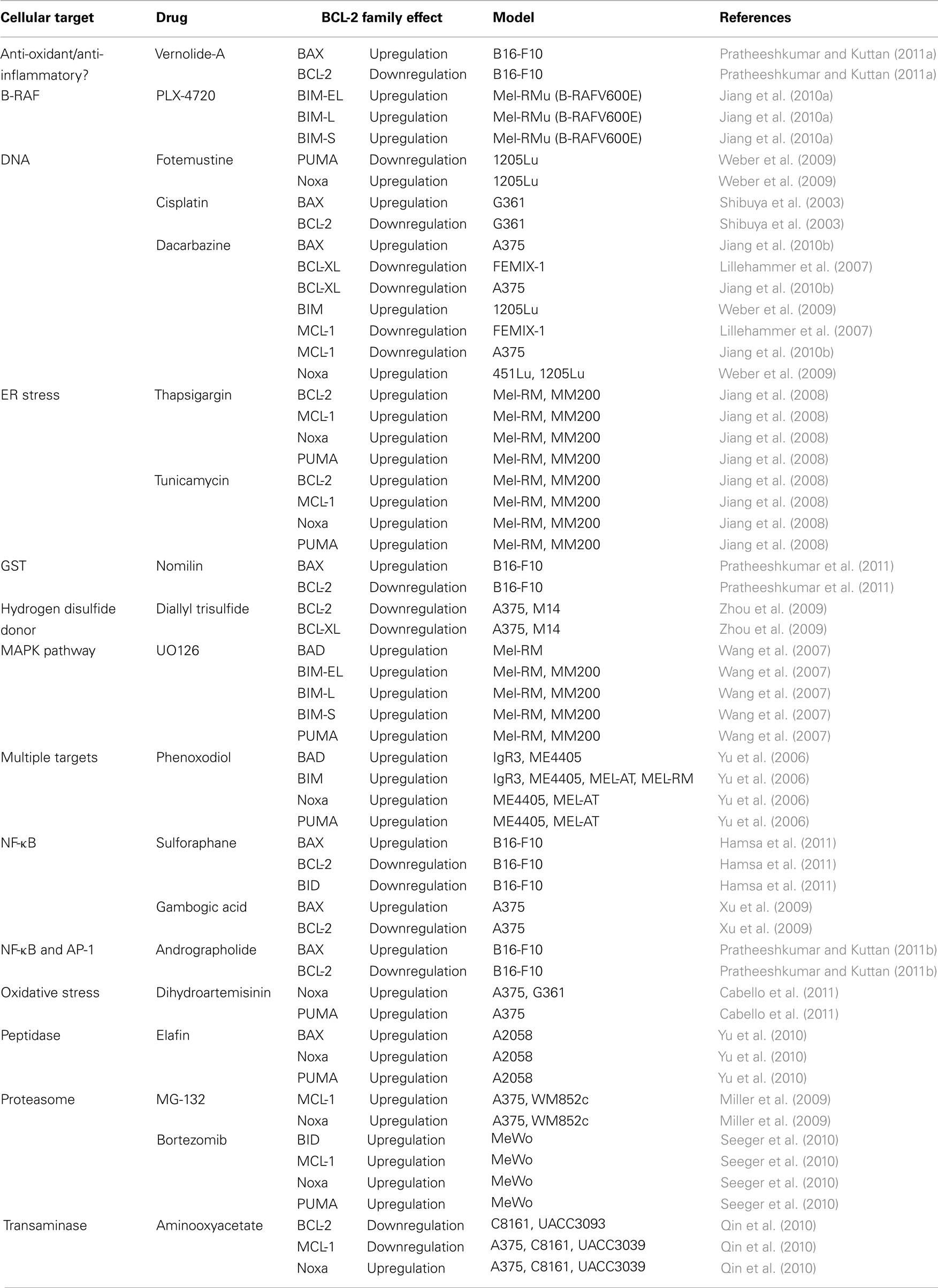
Table 1. A list of chemotherapeutics that induce changes to the BCL-2 family in various melanoma cell lines.
In terms of a targeted therapeutic approach, the frequency of the B-RAF V600E mutation in melanoma presents a novel and unique drug target (Davies et al., 2002; Sheridan et al., 2008). Early studies with B-RAF V600E mutated melanoma lines suggest that silencing B-RAF V600E expression leads to greater sensitivity to apoptosis, which is likely mediated by the combined stabilization and function of BIM to promote BAK/BAX activation (Jiang et al., 2010a). In recent years, treatment of melanoma patients harboring the B-RAF V600E mutation with small molecules (e.g., PLX-4720, PLX-4032) that specifically inhibit the B-RAF V600E kinase has been successful through Phase III clinical trials, and appears to offer greater response rates and increased overall survival compared to DTIC (Flaherty et al., 2010).
However, melanoma cells treated with these drugs have a means of becoming resistant to declining levels of B-RAF signaling. For instance, if PTEN expression or function is lost, there are suggestions in the literature that melanoma cell lines can develop resistance to PLX-4720 induced death due to decreased BIM (Paraiso et al., 2011). The loss of PTEN results in constitutive activation of the PI3K/AKT pathway, which has been described in melanoma suggesting B-RAF inhibitors will only work for a subset of patients carrying the B-RAF V600E mutation and functional PTEN (VanBrocklin et al., 2009). Dual requirements for the PI3K/AKT pathway and constitutive MAPK signaling are also supported by combination treatments such as Sorafenib [small molecular inhibitor of several tyrosine protein kinases (VEGFR and PDGFR) and B-RAF] and nanoliposomal ceramide, which have been shown to synergistically decrease PI3K/AKT and MAPK signaling leading to increased sensitivity to the mitochondrial pathway of apoptosis in vitro (Tran et al., 2008). Furthermore, several MEK pathway inhibitors, such as UO126 and CI-1040, have also been shown to cause upregulation of pro-apoptotic BCL-2 protein function like BIM, BMF, Noxa, and PUMA, and occasionally can lead to the downregulation of anti-apoptotic proteins like BCL-2 and MCL-1 (Wang et al., 2007; VanBrocklin et al., 2009). Perhaps combinations of B-RAF and MEK inhibitors can promote more robust apoptosis due to dual regulations of numerous anti- and pro-apoptotic BCL-2 members (Wang et al., 2007).
From our discussion, we suggest that melanoma likely develops because of altered BCL-2 family function, which allows for survival and proliferation despite signals to die. Likewise, literature supports that chemotherapies engage apoptosis by directly promoting the function of pro-apoptotic BCL-2 members. If so, are there other drugs that can be used in combination with DTIC to alter the balance toward death, perhaps by decreasing anti-apoptotic BCL-2 protein expression, or by inhibiting their function by inducing BH3-only proteins? Indeed, there is already an abundant literature to show that many drugs can directly regulate the BCL-2 family to favor turning on the mitochondrial pathway of apoptosis (Table 1). Numerous DNA damaging agents, such as cisplatin, directly impinge on the mitochondrial pathway of apoptosis by lowering anti-apoptotic BCL-2 protein expression, while also increasing effector levels, and presumably, activation and MOMP (Shibuya et al., 2003). Endoplasmic reticulum stress induced by tunicamycin or thapsigargin can also induce apoptosis of melanoma cells, which has been shown to directly up-regulate PUMA expression (Jiang et al., 2008). Proteasome inhibitors such as bortezomib have been shown to induce BID, Noxa, and PUMA in melanoma cells (Nikiforov et al., 2007; Seeger et al., 2010). Drugs with diverse mechanisms of action including dihydroartemisinin (promotes oxidative stress), ADI-PEG20 (arginine deiminase), and aminooxyacetate (transaminase inhibitor) appear to directly regulate Noxa expression, which has been shown to directly impact on cellular sensitivity to both intrinsic and extrinsic forms of apoptosis (Qin et al., 2010; You et al., 2010; Cabello et al., 2011). The overall outcome of such studies suggests that the BCL-2 family determines the chemotherapeutic response to either engage apoptosis or continue with survival. Indeed, several drugs are suggested to promote apoptosis via BIM, which nicely parallels existing clinical data that shows melanoma progression and poor survival rates are associated with decreased BIM expression (Cartlidge et al., 2008; Dai et al., 2008). Are there strategies to sensitize melanoma cells to chemotherapeutics by using this information? In the following section, we provide evidence that depleting cellular anti-apoptotic BCL-2 function may be the optimal combination therapy to promote chemotherapeutic responses despite over-expressed anti-apoptotic BCL-2 members or decreased pro-apoptotic BCL-2 engagement.
BCL-2 Family Inhibitors and Melanoma Treatment Responses
As mentioned, the BCL-2 family of proteins can influence both the progression and chemotherapeutic responses of melanoma. While conventional chemotherapeutic regimens can modulate the levels of individual anti- and/or pro-apoptotic BCL-2 proteins, changes in one or two proteins will likely induce only minor effects in the pro- and anti-apoptotic balance. Furthermore, since anti-apoptotic proteins are numerous, there are multiple members with the potential to inhibit pro-apoptotic signaling leading to chemoresistance. While there are notions to suggest that loss of BID/BIM or BAK/BAX can lead to cancer development and/or chemoresistance, most data suggest that these proteins are not commonly targeted, which affords the over-expressed anti-apoptotic members a unique opportunity for pharmacological regulation (Letai, 2008). With this understanding, directly targeting the anti-apoptotic BCL-2 proteins, and indirectly the pro-apoptotic machinery they control, is an attractive tool with potential to generate new therapies, as well as complement existing treatments. In this endeavor, the greatest successes have come through the identification of small molecules that mimic the function of BH3-only proteins to bind within the hydrophobic pocket of the anti-apoptotic BCL-2 proteins. These small molecules, referred to as “BH3 mimetics,” functionally neutralize several anti-apoptotic proteins, thus inhibiting their ability to sequester pro-apoptotic proteins. This strategy lowers the cellular threshold leading to BAK/BAX activation, MOMP, and subsequent apoptosis.
The notion that regulating BCL-2 function in melanoma could yield enhanced responses to chemotherapeutics arose from a study that showed bcl-2 antisense improved treatment responses and tumor burden in a xenograft mouse model of human disease (Jansen et al., 1998). However, the use of bcl-2 antisense adjuvant therapy (Oblimersen sodium) with dacarbazine failed to meet expectations in several trials (conducted by Genta Incorporated), although there continues to be some controversy as to whether or not bcl-2 antisense is beneficial (Jansen et al., 2000; Bedikian et al., 2006). Independent of those results, pharmacological regulation of anti-apoptotic BCL-2 proteins continues to be of tremendous interest. A BH3 mimetic that has been studied with considerable success in melanoma is the Abbott compound ABT-737 (and the bio-available form ABT-263). Published in 2005, ABT-737 was identified using a structural activity relationship by nuclear magnetic resonance (SAR by NRM) screen for compounds that could bind within the hydrophobic groove of BCL-xL (Oltersdorf et al., 2005). Upon its discovery, ABT-737 was determined to have high affinity for the anti-apoptotic BCL-2 proteins BCL-2, BCL-w, and BCL-xL, but not A1 or MCL-1. ABT-737 was immediately cast as a promising new drug candidate since it could engage multiple anti-apoptotic BCL-2 family members, and it demonstrated minimal toxicity as a single agent. However, the inability of ABT-737 to inhibit MCL-1 (or A1, however, A1 has restricted expression so the influence in cancer is far less than MCL-1), which is commonly over-expressed in melanomas, highlighted one weakness for using ABT-737 in melanoma, and suggested a possible mechanism by which ABT-737 resistance may be conferred (Yecies et al., 2010). Indeed, melanoma survival and sensitivity to chemotherapeutics appears to be mediated by a combination of anti-apoptotic BCL-2 members as preclinical studies using antisense oligonucleotides against BCL-2, BCL-xL, or MCL-1 sensitized to chemotherapy-induced apoptosis (Gautschi et al., 2001; Del Bufalo et al., 2003; Thallinger et al., 2003).
To better understand their combined influence on cellular survival, a recent study examined the result of reduced MCL-1 expression in combination with ABT-737 treatment in melanoma cells (Keuling et al., 2009). Indeed, the results showed an increased induction of apoptosis, and opened up the possibility for pro-apoptotic synergy between ABT-737 and classical chemotherapeutic treatments that decrease MCL-1 levels. Intriguingly, such combination regimens have already yielded a number of successful studies. For example, ABT-737 in combination with the standard clinically approved anti-melanoma drug, dacarbazine, promoted apoptosis through the induction of Noxa, which likely synergizes with ABT-737 to fully inhibit the anti-apoptotic repertoire (Weber et al., 2009). In a similar scenario, ABT-737 synergized with the proteasome inhibitor MG-132, which was also suggested to induce Noxa expression and inhibition of MCL-1 function leading to increased rates of apoptosis (Fernandez et al., 2005; Qin et al., 2005; Miller et al., 2009). Alternatively, because of frequent mutations in the MAPK pathway in melanoma, ABT-737 has also been studied in combination with MEK and p38 inhibitors, both of which synergize to enhance the apoptotic machinery (Cragg et al., 2008; Keuling et al., 2010).
TW-37 is another BH3 mimetic, but it has a broader range of targets including BCL-2, BCL-xL, and MCL-1(Verhaegen et al., 2006). Due to the ability of TW-37 to inhibit multiple members, in particular MCL-1, it was proposed that TW-37 could synergize with chemotherapeutics to promote apoptosis independently of Noxa regulation. Indeed, when melanoma lines were treated with TW-37 and a MEK inhibitor (U0126), significant apoptosis resulted. Interestingly, the observed synergy did not occur in normal melanocytes, suggesting that the mechanism of action was specific to tumor cells. Curiously, the observed apoptosis was also dependent upon the p53 pathway, which is functionally intact in the majority of melanoma cell lines and patients.
Obatoclax is also an inhibitor to the anti-apoptotic BCL-2 family proteins that has been studied in melanoma. First described in 2007, Obatoclax is believed to inhibit all of the anti-apoptotic BCL-2 proteins, including MCL-1, and thus shows less resistance to treatment than ABT-737 (Nguyen et al., 2007). While Obatoclax has been shown to induce apoptosis as a single agent treatment in other types of cancer, such efficacy has not been observed in melanoma, leading to the study of combination treatments (Trudel et al., 2007). When combined with the endoplasmic reticulum stressors tunicamycin or thapsigargin, increased apoptosis was observed (Jiang et al., 2009). This synergy is likely due to Obatoclax functioning like a sensitizer BH3-only protein to inhibit the anti-apoptotic BCL-2 proteins, lowering the cellular threshold for BAK/BAX activation, and allowing the mitochondrial apoptotic machinery to be engaged when signaled. However, it should be noted that despite its confirmed inhibiting interaction with the anti-apoptotic BCL-2 proteins, albeit at a higher concentrations than ABT-737, the mechanism by which obatoclax elicits a response is not definitive, as studies have suggested alternative targets than the BCL-2 family (Konopleva et al., 2008).
While the above drugs target the anti-apoptotic BCL-2 proteins, none of them have proven to be as effective at binding the anti-apoptotics proteins as the BH3-only proteins themselves. Given the importance of the BH3 domain – anti-apoptotic interaction, which lowers the cellular threshold of apoptosis, short chemically synthesized peptides mimicking the different BH3-only proteins have been generated and tested for function (Letai et al., 2002). Indeed, BH3 domain peptides from different BH3-only proteins revealed the distinction between direct activator and sensitizer/de-repressor BH3-only proteins (Figure 3). The BID and BIM BH3 domain peptides promote BAK/BAX-dependent MOMP and cytochrome c release, whereas the BAD BH3 promotes MOMP by inhibiting BCL-2, BCL-xL, and BCL-w (and revealing BID/BIM activity, see Figure 3), and not by direct BAK/BAX interactions (Chen et al., 2005; Kuwana et al., 2005). One issue that arose with using synthetic peptides was the loss of α-helicity that is required for protein-protein interactions between the BH3-only proteins and the anti-apoptotics. To address this issue, non-natural amino acids containing olefin-bearing tethers were introduced into the BH3 domain sequence, effectively “stapling” the peptide into an α-helical form (Walensky et al., 2004, 2006). These stapled BH3 domains are referred to as “stabilized α-helix of BCL-2 domains” (SAHBs), and these may serve as a new tool for designing cancer therapeutics to target the BCL-2 family and enhance endogenous BH3-only activity. For example, the BID BH3 SAHB was shown to induce apoptosis in leukemia cells as well as inhibit growth of human leukemia xenografts in vivo (Walensky et al., 2004). Recently, a MCL-1 BH3 SAHB was developed which inhibits MCL-1 with very high affinity, and indeed has also been shown to sensitize cancer cells to apoptosis (n.b., the MCL-1 SAHB uniquely binds to the MCL-1 hydrophobic groove; Stewart et al., 2010). The identification of an exclusive and potent MCL-1 inhibitor is important, given the lack of potent inhibitors that target MCL-1 and the emergence of MCL-1 as a chemoresistance factor in a broad range of human cancers. These developments will hopefully serve as an additional platform to discover small molecule regulators of the BCL-2 family. Presently, BH3 domain peptides have not been examined in the context of melanoma treatment; efforts to elucidate their potential impact would be useful for understanding both the biology of melanoma, and hopefully its treatment. Of note, BH3 mimetics also promote autophagy, alone or in combination with B-RAF inhibition; and this may be a contributing mechanism with therapeutic potential in metastatic melanoma (Armstrong et al., 2011; Malik et al., 2011a,b).
Perspectives
Throughout our discussions, we focused on applying the mechanisms of action for the BCL-2 family to the regulation of melanoma tumorigenesis, apoptosis, and treatment. As the BCL-2 family defines the balance between life and death in normal and cancer cells, it is reasonable to seek a full understanding of the cellular mechanisms that control BCL-2 family function to establish how stressed cells undergo apoptosis, and how cancer cells resist pro-apoptotic signals to ensure survival. Here we suggest that multiple anti-apoptotic BCL-2 proteins ensure survival, and that melanocytes undergoing stress during tumorigenesis and metastasis increase the expression and/or function of these pro-survival members. Likewise, chemotherapeutics (single agents and combinations) must promote the inhibition of these pro-survival members by a combination of BH3-only protein functions to engage BAK/BAX activation and achieve desired clinical outcomes.
While we have focused on the direct targeting of the BCL-2 family to promote MOMP, it is important to mention that there are also pharmacological opportunities to promote apoptosis both upstream and downstream of mitochondria. In particular, the development of XIAP (X-linked inhibitor of apoptosis protein) inhibitors is critical as the increased expression of XIAP is positively correlated with melanoma thickness, tumor progression, and chemoresistance (Chawla-Sarkar et al., 2004; Kluger et al., 2007; Emanuel et al., 2008; Hiscutt et al., 2010). XIAP is a potent inhibitor to the apoptotic caspases, so relieving this apoptotic brake is predicted to lower the threshold leading to the cell death phenotype (Eckelman et al., 2006). Indeed, the development of SMAC (second mitochondria-derived activator of caspases, the cellular XIAP inhibitor) mimetics to target XIAP is shown to induce apoptosis in numerous melanoma cell lines (Vucic et al., 2002; Zobel et al., 2006). There are also numerous additional pathways that offer opportunities to enhance tumor cell killing though MOMP and apoptosis. For example, epigenetic mechanisms are implicated in melanoma survival as miR-149 and miR-193b enhance Mcl-1 expression in nevi, patient tumor samples, and melanoma cell lines (Chen et al., 2011; Jin et al., 2011). Histone deacetylase (HDAC) inhibitors sensitize melanoma cells to chemotherapy through a number of mechanisms, including suppression of the RAS/MAPK signaling pathway and enhanced DNA damage signaling leading to apoptosis (Peltonen et al., 2005; Kobayashi et al., 2006; Munshi et al., 2006). Furthermore, the role of chromatin modifications may offer collateral means of pharmacological intervention as the requirement for SWI/SNF complexes in MITF regulated melanocyte-specific gene expression pathways directly impact on BCL-2 expression, and likely sensitivity to chemotherapeutics (de la Serna et al., 2006; Keenen et al., 2010; Vachtenheim et al., 2010). Determining the appropriate combination treatments for each patient based on his/her individual tumor status will likely generate the most effective treatments.
Fortunately, the cellular signaling pathways that intersect between the survival and apoptosis cascades are numerous (e.g., B-RAF mutation and BCL-2 over-expression; proteasome function and MCL-1 over-expression) and this affords the pharmacological opportunity to explore multiple combination strategies to cure melanoma. In the future, enhanced animal models of human disease will likely shed light upon the mechanisms that drive melanocyte tumorigenesis and metastasis. The combined B-RAF V600E/PTEN mouse model mentioned earlier is perhaps the ideal model system to genetically evaluate the role of the BCL-2 family in melanoma progression (Dankort et al., 2009). As further models are developed, they should be evaluated along side the standard models of BCL-2 family function (e.g., bak−/− bax−/−, bid−/−bim−/−, puma−/−, melanocyte-specific bcl-2 and mcl-1 transgenics, etc…) to fully appreciate the influence of the BCL-2 family in mediating melanoma development and treatment. Likewise, a better understanding of the pathways (e.g., mitochondrial dynamics, structural studies, lipid environment etc…) that regulate the BCL-2 family itself will certainly yield novel target strategies to obtain better clinical outcomes. The future shows significant promise to develop more specific and bio-available small molecule inhibitors to the anti-apoptotic BCL-2 proteins, and perhaps drugs that directly promote BAK/BAX activation; this will be another significant milestone in adjuvant-based chemotherapeutic strategies targeting the mitochondrial pathway of apoptosis. The hope is that as we further dissect the mechanisms of the BCL-2 family and apoptotic pathways, novel therapeutic targets and combination strategies will emerge and provide significant benefits to those afflicted with melanoma.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank members of the Chipuk Laboratory for useful discussions and feedback on this review; and the Department of Dermatology at MSSM for its continued support, especially Dr. Mark G. Lebwohl. Dr. Christopher W. Akey (Boston University) for the apoptosome image. We apologize for not discussing and citing all the relevant literature due to space restrictions. The laboratory is funded by NIH R01CA157740 and is supported in part by Research Grant 5-FY11-74 from the March of Dimes Foundation.
References
Armstrong, J. L., Corazzari, M., Martin, S., Pagliarini, V., Falasca, L., Hill, D. S., Ellis, N., Al Sabah, S., Redfern, C. P., Fimia, G. M., Piacentini, M., and Lovat, P. E. (2011). Oncogenic B-RAF signaling in melanoma impairs the therapeutic advantage of autophagy inhibition. Clin. Cancer Res. 17, 2216–2226.
Bedikian, A. Y., Millward, M., Pehamberger, H., Conry, R., Gore, M., Trefzer, U., Pavlick, A. C., DeConti, R., Hersh, E. M., Hersey, P., Kirkwood, J. M., Haluska, F. G., and Oblimersen Melanoma Study Group. (2006). Bcl-2 antisense (oblimersen sodium) plus dacarbazine in patients with advanced melanoma: the Oblimersen Melanoma Study Group. J. Clin. Oncol. 24, 4738–4745.
Boisvert-Adamo, K., Longmate, W., Abel, E. V., and Aplin, A. E. (2009). Mcl-1 is required for melanoma cell resistance to anoikis. Mol. Cancer Res. 7, 549–556.
Bouillet, P., Metcalf, D., Huang, D. C., Tarlinton, D. M., Kay, T. W., Kontgen, F., Adams, J. M., and Strasser, A. (1999). Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286, 1735–1738.
Cabello, C. M., Lamore, S. D., Bair, W. B. III, Qiao, S., Azimian, S., Lesson, J. L., and Wondrak, G. T. (2011). The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Invest. New Drugs 1–13.
Cartlidge, R. A., Thomas, G. R., Cagnol, S., Jong, K. A., Molton, S. A., Finch, A. J., and McMahon, M. (2008). Oncogenic BRAF(V600E) inhibits BIM expression to promote melanoma cell survival. Pigment Cell Melanoma Res. 21, 534–544.
Castellano, M., and Parmiani, G. (1999). Genes involved in melanoma: an overview of INK4a and other loci. Melanoma Res. 9, 421–432.
Cerroni, L., Soyer, H. P., and Kerl, H. (1995). bcl-2 protein expression in cutaneous malignant melanoma and benign melanocytic nevi. Am. J. Dermatopathol. 17, 7–11.
Chawla-Sarkar, M., Bae, S. I., Reu, F. J., Jacobs, B. S., Lindner, D. J., and Borden, E. C. (2004). Downregulation of Bcl-2, FLIP or IAPs (XIAP and survivin) by siRNAs sensitizes resistant melanoma cells to Apo2L/TRAIL-induced apoptosis. Cell Death Differ. 11, 915–923.
Chen, J., Zhang, X., Lentz, C., Abi-Daoud, M., Pare, G. C., Yang, X., Feilotter, H. E., and Tron, V. A. (2011). miR-193b regulates Mcl-1 in melanoma. Am. J. Pathol. PMID: 21893020. [Epub ahead of print].
Chen, L., Willis, S. N., Wei, A., Smith, B. J., Fletcher, J. I., Hinds, M. G., Colman, P. M., Day, C. L., Adams, J. M., and Huang, D. C. (2005). Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell 17, 393–403.
Chen, N., and Karantza-Wadsworth, V. (2009). Role and regulation of autophagy in cancer. Biochim. Biophys. Acta 1793, 1516–1523.
Chipuk, J. E., Bouchier-Hayes, L., Kuwana, T., Newmeyer, D. D., and Green, D. R. (2005). PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309, 1732–1735.
Chipuk, J. E., and Green, D. R. (2008). How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 18, 157–164.
Chipuk, J. E., Kuwana, T., Bouchier-Hayes, L., Droin, N. M., Newmeyer, D. D., Schuler, M., and Green, D. R. (2004). Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303, 1010–1014.
Chipuk, J. E., Moldoveanu, T., Llambi, F., Parsons, M. J., and Green, D. R. (2010). The BCL-2 family reunion. Mol. Cell 37, 299–310.
Collins, K. A., and White, W. L. (1995). Intercellular adhesion molecule 1 (ICAM-1) and bcl-2 are differentially expressed in early evolving malignant melanoma. Am. J. Dermatopathol. 17, 429–438.
Cragg, M. S., Jansen, E. S., Cook, M., Harris, C., Strasser, A., and Scott, C. L. (2008). Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J. Clin. Invest. 118, 3651–3659.
Dai, D. L., Wang, Y., Liu, M., Martinka, M., and Li, G. (2008). Bim expression is reduced in human cutaneous melanomas. J. Invest. Dermatol. 128, 403–407.
Dankort, D., Curley, D. P., Cartlidge, R. A., Nelson, B., Karnezis, A. N., Damsky, W. E. Jr., You, M. J., DePinho, R. A., McMahon, M., and Bosenberg, M. (2009). Braf (V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 41, 544–552.
Davies, H., Bignell, G. R., Cox, C., Stephens, P., Edkins, S., Clegg, S., Teague, J., Woffendin, H., Garnett, M. J., Bottomley, W., Davis, N., Dicks, E., Ewing, R., Floyd, Y., Gray, K., Hall, S., Hawes, R., Hughes, J., Kosmidou, V., Menzies, A., Mould, C., Parker, A., Stevens, C., Watt, S., Hooper, S., Wilson, R., Jayatilake, H., Gusterson, B. A., Cooper, C., Shipley, J., Hargrave, D., Pritchard-Jones, K., Maitland, N., Chenevix-Trench, G., Riggins, G. J., Bigner, D. D., Palmieri, G., Cossu, A., Flanagan, A., Nicholson, A., Ho, J. W., Leung, S. Y., Yuen, S. T., Weber, B. L., Seigler, H. F., Darrow, T. L., Paterson, H., Marais, R., Marshall, C. J., Wooster, R., Stratton, M. R., and Futreal, P. A. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954.
de la Serna, I. L., Ohkawa, Y., Higashi, C., Dutta, C., Osias, J., Kommajosyula, N., Tachibana, T., and Imbalzano, A. N. (2006). The microphthalmia-associated transcription factor requires SWI/SNF enzymes to activate melanocyte-specific genes. J. Biol. Chem. 281, 20233–20241.
Del Bufalo, D., Trisciuoglio, D., Scarsella, M., Zangemeister-Wittke, U., and Zupi, G. (2003). Treatment of melanoma cells with a bcl-2/bcl-xL antisense oligonucleotide induces antiangiogenic activity. Oncogene 22, 8441–8447.
Deng, J., Carlson, N., Takeyama, K., Dal Cin, P., Shipp, M., and Letai, A. (2007). BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12, 171–185.
Dewson, G., Kratina, T., Czabotar, P., Day, C. L., Adams, J. M., and Kluck, R. M. (2009). Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol. Cell 36, 696–703.
Dewson, G., Kratina, T., Sim, H. W., Puthalakath, H., Adams, J. M., Colman, P. M., and Kluck, R. M. (2008). To trigger apoptosis, Bak exposes its BH3 domain and homodimerizes via BH3: groove interactions. Mol. Cell 30, 369–380.
Dong, L., Jiang, C. C., Thorne, R. F., Croft, A., Yang, F., Liu, H., de Bock, C. E., Hersey, P., and Zhang, X. D. (2011). Ets-1 mediates upregulation of Mcl-1 downstream of XBP-1 in human melanoma cells upon ER stress. Oncogene 30, 3716–3726.
Eckelman, B. P., Salvesen, G. S., and Scott, F. L. (2006). Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 7, 988–994.
Edlundh-Rose, E., Egyhazi, S., Omholt, K., Mansson-Brahme, E., Platz, A., Hansson, J., and Lundeberg, J. (2006). NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: a study based on mutation screening by pyrosequencing. Melanoma Res. 16, 471–478.
Elgendy, M., Sheridan, C., Brumatti, G., and Martin, S. J. (2011). Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell 42, 23–35.
Emanuel, P. O., Phelps, R. G., Mudgil, A., Shafir, M., and Burstein, D. E. (2008). Immunohistochemical detection of XIAP in melanoma. J. Cutan. Pathol. 35, 292–297.
Fecker, L. F., Geilen, C. C., Tchernev, G., Trefzer, U., Assaf, C., Kurbanov, B. M., Schwarz, C., Daniel, P. T., and Eberle, J. (2006). Loss of proapoptotic Bcl-2-related multidomain proteins in primary melanomas is associated with poor prognosis. J. Invest. Dermatol. 126, 1366–1371.
Fernandez, Y., Verhaegen, M., Miller, T. P., Rush, J. L., Steiner, P., Opipari, A. W. Jr., Lowe, S. W., and Soengas, M. S. (2005). Differential regulation of noxa in normal melanocytes and melanoma cells by proteasome inhibition: therapeutic implications. Cancer Res. 65, 6294–6304.
Flaherty, K. T., Puzanov, I., Kim, K. B., Ribas, A., McArthur, G. A., Sosman, J. A., O’Dwyer, P. J., Lee, R. J., Grippo, J. F., Nolop, K., and Chapman, P. B. (2010). Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819.
Fong, L., and Small, E. J. (2008). Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J. Clin. Oncol. 26, 5275–5283.
Gautschi, O., Tschopp, S., Olie, R. A., Leech, S. H., Simoes-Wust, A. P., Ziegler, A., Baumann, B., Odermatt, B., Hall, J., Stahel, R. A., and Zangemeister-Wittke, U. (2001). Activity of a novel bcl-2/bcl-xL-bispecific antisense oligonucleotide against tumors of diverse histologic origins. J. Natl. Cancer Inst. 93, 463–471.
Gavathiotis, E., Reyna, D. E., Davis, M. L., Bird, G. H., and Walensky, L. D. (2010). BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol. Cell 40, 481–492.
Gavathiotis, E., Suzuki, M., Davis, M. L., Pitter, K., Bird, G. H., Katz, S. G., Tu, H. C., Kim, H., Cheng, E. H., Tjandra, N., and Walensky, L. D. (2008). BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081.
Goldstein, N. B., Johannes, W. U., Gadeliya, A. V., Green, M. R., Fujita, M., Norris, D. A., and Shellman, Y. G. (2009). Active N-Ras and B-Raf inhibit anoikis by downregulating Bim expression in melanocytic cells. J. Invest. Dermatol. 129, 432–437.
Gradilone, A., Gazzaniga, P., Ribuffo, D., Scarpa, S., Cigna, E., Vasaturo, F., Bottoni, U., Innocenzi, D., Calvieri, S., Scuderi, N., Frati, L., and Aglianò, A. M. (2003). Survivin, bcl-2, bax, and bcl-X gene expression in sentinel lymph nodes from melanoma patients. J. Clin. Oncol. 21, 306–312.
Grover, R., and Wilson, G. D. (1996). Bcl-2 expression in malignant melanoma and its prognostic significance. Eur. J. Surg. Oncol. 22, 347–349.
Guicciardi, M. E., and Gores, G. J. (2009). Life and death by death receptors. FASEB J. 23, 1625–1637.
Guo, J. Y., Chen, H. Y., Mathew, R., Fan, J., Strohecker, A. M., Karsli-Uzunbas, G., Kamphorst, J. J., Chen, G., Lemons, J. M., Karantza, V., Coller, H. A., Dipaola, R. S., Gelinas, C., Rabinowitz, J. D., and White, E. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470.
Haluska, F. G., and Hodi, F. S. (1998). Molecular genetics of familial cutaneous melanoma. J. Clin. Oncol. 16, 670–682.
Hamsa, T. P., Thejass, P., and Kuttan, G. (2011). Induction of apoptosis by sulforaphane in highly metastatic B16F-10 melanoma cells. Drug Chem. Toxicol. 34, 332–340.
Hiscutt, E. L., Hill, D. S., Martin, S., Kerr, R., Harbottle, A., Birch-Machin, M., Redfern, C. P., Fulda, S., Armstrong, J. L., and Lovat, P. E. (2010). Targeting X-linked inhibitor of apoptosis protein to increase the efficacy of endoplasmic reticulum stress-induced apoptosis for melanoma therapy. J. Invest. Dermatol. 130, 2250–2258.
Hocker, T., and Tsao, H. (2007). Ultraviolet radiation and melanoma: a systematic review and analysis of reported sequence variants. Hum. Mutat. 28, 578–588.
Hocker, T. L., Singh, M. K., and Tsao, H. (2008). Melanoma genetics and therapeutic approaches in the 21st century: moving from the benchside to the bedside. J. Invest. Dermatol. 128, 2575–2595.
Hodi, F. S., O’Day, S. J., McDermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., Gonzalez, R., Robert, C., Schadendorf, D., Hassel, J. C., Akerley, W., van den Eertwegh, A. J., Lutzky, J., Lorigan, P., Vaubel, J. M., Linette, G. P., Hogg, D., Ottensmeier, C. H., Lebbé, C., Peschel, C., Quirt, I., Clark, J. I., Wolchok, J. D., Weber, J. S., Tian, J., Yellin, M. J., Nichol, G. M., Hoos, A., and Urba, W. J. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723.
Hsu, Y. T., and Youle, R. J. (1997). Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272, 13829–13834.
Ibrahim, N., and Haluska, F. G. (2009). Molecular pathogenesis of cutaneous melanocytic neoplasms. Annu. Rev. Pathol. 4, 551–579.
Igney, F. H., and Krammer, P. H. (2002). Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer 2, 277–288.
Jansen, B., Schlagbauer-Wadl, H., Brown, B. D., Bryan, R. N., van Elsas, A., Muller, M., Wolff, K., Eichler, H. G., and Pehamberger, H. (1998). bcl-2 antisense therapy chemosensitizes human melanoma in SCID mice. Nat. Med. 4, 232–234.
Jansen, B., Wacheck, V., Heere-Ress, E., Schlagbauer-Wadl, H., Hoeller, C., Lucas, T., Hoermann, M., Hollenstein, U., Wolff, K., and Pehamberger, H. (2000). Chemosensitisation of malignant melanoma by BCL2 antisense therapy. Lancet 356, 1728–1733.
Jemal, A., Siegel, R., Ward, E., Hao, Y., Xu, J., and Thun, M. J. (2009). Cancer statistics, 2009. CA Cancer J. Clin. 59, 225–249.
Jiang, C. C., Lai, F., Tay, K. H., Croft, A., Rizos, H., Becker, T. M., Yang, F., Liu, H., Thorne, R. F., Hersey, P., and Zhang, X. D. (2010a). Apoptosis of human melanoma cells induced by inhibition of B-RAFV600E involves preferential splicing of bimS. Cell Death Dis. 1, e69.
Jiang, G., Liu, Y. Q., Wei, Z. P., Pei, D. S., Mao, L. J., and Zheng, J. N. (2010b). Enhanced anti-tumor activity by the combination of a conditionally replicating adenovirus mediated interleukin-24 and dacarbazine against melanoma cells via induction of apoptosis. Cancer Lett. 294, 220–228.
Jiang, C. C., Lucas, K., Avery-Kiejda, K. A., Wade, M., deBock, C. E., Thorne, R. F., Allen, J., Hersey, P., and Zhang, X. D. (2008). Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res. 68, 6708–6717.
Jiang, C. C., Wroblewski, D., Yang, F., Hersey, P., and Zhang, X. D. (2009). Human melanoma cells under endoplasmic reticulum stress are more susceptible to apoptosis induced by the BH3 mimetic obatoclax. Neoplasia 11, 945–955.
Jin, L., Hu, W. L., Jiang, C. C., Wang, J. X., Han, C. C., Chu, P., Zhang, L. J., Thorne, R. F., Wilmott, J., Scolyer, R. A., Hersey, P., Zhang, X. D., and Wu, M. (2011). MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc. Natl. Acad. Sci. U.S.A. 108, 15840–15845.
Kapoor, A., Goldberg, M. S., Cumberland, L. K., Ratnakumar, K., Segura, M. F., Emanuel, P. O., Menendez, S., Vardabasso, C., Leroy, G., Vidal, C. I., Polsky, D., Osman, I., Garcia, B. A., Hernando, E., and Bernstein, E. (2010). The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 468, 1105–1109.
Karbowniczek, M., Spittle, C. S., Morrison, T., Wu, H., and Henske, E. P. (2008). mTOR is activated in the majority of malignant melanomas. J. Invest. Dermatol. 128, 980–987.
Keenen, B., Qi, H., Saladi, S. V., Yeung, M., and de la Serna, I. L. (2010). Heterogeneous SWI/SNF chromatin remodeling complexes promote expression of microphthalmia-associated transcription factor target genes in melanoma. Oncogene 29, 81–92.
Keuling, A. M., Andrew, S. E., and Tron, V. A. (2010). Inhibition of p38 MAPK enhances ABT-737-induced cell death in melanoma cell lines: novel regulation of PUMA. Pigment Cell Melanoma Res. 23, 430–440.
Keuling, A. M., Felton, K. E., Parker, A. A., Akbari, M., Andrew, S. E., and Tron, V. A. (2009). RNA silencing of Mcl-1 enhances ABT-737-mediated apoptosis in melanoma: role for a caspase-8-dependent pathway. PLoS ONE 4, e6651. doi: 10.1371/journal.pone.0006651
Khaled, A. R., Reynolds, D. A., Young, H. A., Thompson, C. B., Muegge, K., and Durum, S. K. (2001). Interleukin-3 withdrawal induces an early increase in mitochondrial membrane potential unrelated to the Bcl-2 family. Roles of intracellular pH, ADP transport, and F(0)F(1)-ATPase. J. Biol. Chem. 276, 6453–6462.
Khodadoust, M. S., Verhaegen, M., Kappes, F., Riveiro-Falkenbach, E., Cigudosa, J. C., Kim, D. S., Chinnaiyan, A. M., Markovitz, D. M., and Soengas, M. S. (2009). Melanoma proliferation and chemoresistance controlled by the DEK oncogene. Cancer Res. 69, 6405–6413.
Kim, H., Tu, H. C., Ren, D., Takeuchi, O., Jeffers, J. R., Zambetti, G. P., Hsieh, J. J., and Cheng, E. H. (2009). Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36, 487–499.
Kim, M. (2010). Cooperative interactions of PTEN deficiency and RAS activation in melanoma metastasis. Small Gtpases 1, 161–164.
Kluger, H. M., McCarthy, M. M., Alvero, A. B., Sznol, M., Ariyan, S., Camp, R. L., Rimm, D. L., and Mor, G. (2007). The X-linked inhibitor of apoptosis protein (XIAP) is up-regulated in metastatic melanoma, and XIAP cleavage by phenoxodiol is associated with carboplatin sensitization. J. Transl. Med. 5, 6.
Kobayashi, Y., Ohtsuki, M., Murakami, T., Kobayashi, T., Sutheesophon, K., Kitayama, H., Kano, Y., Kusano, E., Nakagawa, H., and Furukawa, Y. (2006). Histone deacetylase inhibitor FK228 suppresses the Ras-MAP kinase signaling pathway by upregulating Rap1 and induces apoptosis in malignant melanoma. Oncogene 25, 512–524.
Kondo, Y., Kanzawa, T., Sawaya, R., and Kondo, S. (2005). The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 5, 726–734.
Konopleva, M., Watt, J., Contractor, R., Tsao, T., Harris, D., Estrov, Z., Bornmann, W., Kantarjian, H., Viallet, J., Samudio, I., and Andreeff, M. (2008). Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res. 68, 3413–3420.
Korabiowska, M., Brinck, U., Ruschenburg, I., Schlott, T., Droese, M., and Stachura, J. (1999). Bcl2 and Bax expression in naevi and melanomas and their relation to ploidy status and proliferation. Pol. J. Pathol. 50, 17–21.
Kuwana, T., Bouchier-Hayes, L., Chipuk, J. E., Bonzon, C., Sullivan, B. A., Green, D. R., and Newmeyer, D. D. (2005). BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 17, 525–535.
Kuwana, T., Mackey, M. R., Perkins, G., Ellisman, M. H., Latterich, M., Schneiter, R., Green, D. R., and Newmeyer, D. D. (2002). Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342.
Lattanzi, S. C., Tosteson, T., Chertoff, J., Maurer, L. H., O’Donnell, J., LeMarbre, P. J., Mott, L., DelPrete, S. A., Forcier, R. J., and Ernstoff, M. S. (1995). Dacarbazine, cisplatin and carmustine, with or without tamoxifen, for metastatic melanoma: 5-year follow-up. Melanoma Res. 5, 365–369.
Legha, S. S., Ring, S., Bedikian, A., Plager, C., Eton, O., Buzaid, A. C., and Papadopoulos, N. (1996). Treatment of metastatic melanoma with combined chemotherapy containing cisplatin, vinblastine and dacarbazine (CVD) and biotherapy using interleukin-2 and interferon-alpha. Ann. Oncol. 7, 827–835.
Legha, S. S., Ring, S., Papadopoulos, N., Plager, C., Chawla, S., and Benjamin, R. (1989). A prospective evaluation of a triple-drug regimen containing cisplatin, vinblastine, and dacarbazine (CVD) for metastatic melanoma. Cancer 64, 2024–2029.
Leiter, U., Schmid, R. M., Kaskel, P., Peter, R. U., and Krahn, G. (2000). Antiapoptotic bcl-2 and bcl-xL in advanced malignant melanoma. Arch. Dermatol. Res. 292, 225–232.
Letai, A., Bassik, M. C., Walensky, L. D., Sorcinelli, M. D., Weiler, S., and Korsmeyer, S. J. (2002). Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2, 183–192.
Letai, A. G. (2008). Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat. Rev. Cancer 8, 121–132.
Levine, B., Sinha, S., and Kroemer, G. (2008). Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 4, 600–606.
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S. M., Ahmad, M., Alnemri, E. S., and Wang, X. (1997). Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489.
Lillehammer, T., Engesaeter, B. O., Prasmickaite, L., Maelandsmo, G. M., Fodstad, O., and Engebraaten, O. (2007). Combined treatment with Ad-hTRAIL and DTIC or SAHA is associated with increased mitochondrial-mediated apoptosis in human melanoma cell lines. J. Gene Med. 9, 440–451.
Lindsten, T., Ross, A. J., King, A., Zong, W. X., Rathmell, J. C., Shiels, H. A., Ulrich, E., Waymire, K. G., Mahar, P., Frauwirth, K., Chen, Y., Wei, M., Eng, V. M., Adelman, D. M., Simon, M.C., Ma, A., Golden, J. A., Evan, G., Korsmeyer, S. J., MacGregor, G. R., and Thompson, C. B. (2000). The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 6, 1389–1399.
Liu, X., Kim, C. N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157.
Lock, R., Roy, S., Kenific, C. M., Su, J. S., Salas, E., Ronen, S. M., and Debnath, J. (2011). Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 22, 165–178.
Loggini, B., Rinaldi, I., Pingitore, R., Cristofani, R., Castagna, M., and Barachini, P. (2001). Immunohistochemical study of 49 cutaneous melanomas: p53, PCNA, Bcl-2 expression and multidrug resistance. Tumori 87, 179–186.
Logue, S. E., and Martin, S. J. (2008). Caspase activation cascades in apoptosis. Biochem. Soc. Trans. 36, 1–9.
Lovell, J. F., Billen, L. P., Bindner, S., Shamas-Din, A., Fradin, C., Leber, B., and Andrews, D. W. (2008). Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084.
Maddodi, N., Huang, W., Havighurst, T., Kim, K., Longley, B. J., and Setaluri, V. (2010). Induction of autophagy and inhibition of melanoma growth in vitro and in vivo by hyperactivation of oncogenic BRAF. J. Invest. Dermatol. 130, 1657–1667.
Malik, S. A., Orhon, I., Morselli, E., Criollo, A., Shen, S., Marino, G., Benyounes, A., Benit, P., Rustin, P., Maiuri, M. C., and Kroemer, G. (2011a). BH3 mimetics activate multiple pro-autophagic pathways. Oncogene 30, 3918–3929.
Malik, S. A., Shen, S., Marino, G., BenYounes, A., Maiuri, M. C., and Kroemer, G. (2011b). BH3 mimetics reveal the network properties of autophagy-regulatory signaling cascades. Autophagy 7, 914–916.
McGill, G. G., Horstmann, M., Widlund, H. R., Du, J., Motyckova, G., Nishimura, E. K., Lin, Y. L., Ramaswamy, S., Avery, W., Ding, H. F., Jordan, S. A., Jackson, I. J., Korsmeyer, S. J., Golub, T. R., and Fisher, D E. (2002). Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 109, 707–718.
Melero, I., Hervas-Stubbs, S., Glennie, M., Pardoll, D. M., and Chen, L. (2007). Immunostimulatory monoclonal antibodies for cancer therapy. Nat. Rev. Cancer 7, 95–106.
Miller, L. A., Goldstein, N. B., Johannes, W. U., Walton, C. H., Fujita, M., Norris, D. A., and Shellman, Y. G. (2009). BH3 mimetic ABT-737 and a proteasome inhibitor synergistically kill melanomas through Noxa-dependent apoptosis. J. Invest. Dermatol. 129, 964–971.
Miracco, C., Santopietro, R., Biagioli, M., Lazzi, S., Nyongo, A., Vatti, R., and Luzi, P. (1998). Different patterns of cell proliferation and death and oncogene expression in cutaneous malignant melanoma. J. Cutan. Pathol. 25, 244–251.
Morales-Ducret, C. R., van de Rijn, M., and Smoller, B. R. (1995). bcl-2 expression in melanocytic nevi. Insights into the biology of dermal maturation. Arch. Dermatol. 131, 915–918.
Munshi, A., Tanaka, T., Hobbs, M. L., Tucker, S. L., Richon, V. M., and Meyn, R. E. (2006). Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol. Cancer Ther. 5, 1967–1974.
Newmeyer, D. D., Farschon, D. M., and Reed, J. C. (1994). Cell-free apoptosis in Xenopus egg extracts: inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell 79, 353–364.
Nguyen, M., Marcellus, R. C., Roulston, A., Watson, M., Serfass, L., Murthy Madiraju, S. R., Goulet, D., Viallet, J., Belec, L., Billot, X., Acoca, S., Purisima, E., Wiegmans, A., Cluse, L., Johnstone, R. W., Beauparlant, P., and Shore, G. C. (2007). Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. U.S.A. 104, 19512–19517.
Nikiforov, M. A., Riblett, M., Tang, W. H., Gratchouck, V., Zhuang, D., Fernandez, Y., Verhaegen, M., Varambally, S., Chinnaiyan, A. M., Jakubowiak, A. J., and Soengas, M. S. (2007). Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc. Natl. Acad. Sci. U.S.A. 104, 19488–19493.
O’Day, S. J., Hamid, O., and Urba, W. J. (2007). Targeting cytotoxic T-lymphocyte antigen-4 (CTLA-4): a novel strategy for the treatment of melanoma and other malignancies. Cancer 110, 2614–2627.
Ohtsuka, T., Ryu, H., Minamishima, Y. A., Macip, S., Sagara, J., Nakayama, K. I., Aaronson, S. A., and Lee, S. W. (2004). ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat. Cell Biol. 6, 121–128.
Oltersdorf, T., Elmore, S. W., Shoemaker, A. R., Armstrong, R. C., Augeri, D. J., Belli, B. A., Bruncko, M., Deckwerth, T. L., Dinges, J., Hajduk, P. J., Joseph, M. K., Kitada, S., Korsmeyer, S. J., Kunzer, A. R., Letai, A., Li, C., Mitten, M. J., Nettesheim, D. G., Ng, S., Nimmer, P. M., O’Connor, J. M., Oleksijew, A., Petros, A. M., Reed, J. C., Shen, W., Tahir, S. K., Thompson, C. B., Tomaselli, K. J., Wang, B., Wendt, M. D., Zhang, H., Fesik, S. W., and Rosenberg, S. H. (2005). An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681.
Pagliari, L. J., Kuwana, T., Bonzon, C., Newmeyer, D. D., Tu, S., Beere, H. M., and Green, D. R. (2005). The multidomain proapoptotic molecules Bax and Bak are directly activated by heat. Proc. Natl. Acad. Sci. U.S.A. 102, 17975–17980.
Paraiso, K. H., Xiang, Y., Rebecca, V. W., Abel, E. V., Chen, Y. A., Munko, A. C., Wood, E., Fedorenko, I. V., Sondak, V. K., Anderson, A. R., Ribas, A., Palma, M. D., Nathanson, K. L., Koomen, J. M., Messina, J. L., and Smalley, K. S. (2011). PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 71, 2750–2760.
Patel, P. M., Suciu, S., Mortier, L., Kruit, W. H., Robert, C., Schadendorf, D., Trefzer, U., Punt, C. J., Dummer, R., Davidson, N., Becker, J., Conry, R., Thompson, J. A., Hwu, W. J., Engelen, K., Agarwala, S. S., Keilholz, U., Eggermont, A. M., Spatz, A., and EORTC Melanoma Group. (2011). Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur. J. Cancer 47, 1476–1483.
Peltonen, K., Kiviharju, T. M., Jarvinen, P. M., Ra, R., and Laiho, M. (2005). Melanoma cell lines are susceptible to histone deacetylase inhibitor TSA provoked cell cycle arrest and apoptosis. Pigment Cell Res. 18, 196–202.
Petros, A. M., Olejniczak, E. T., and Fesik, S. W. (2004). Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta 1644, 83–94.
Phan, G. Q., Attia, P., Steinberg, S. M., White, D. E., and Rosenberg, S. A. (2001). Factors associated with response to high-dose interleukin-2 in patients with metastatic melanoma. J. Clin. Oncol. 19, 3477–3482.
Placzek, W. J., Wei, J., Kitada, S., Zhai, D., Reed, J. C., and Pellecchia, M. (2010). A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 1, e40.
Plettenberg, A., Ballaun, C., Pammer, J., Mildner, M., Strunk, D., Weninger, W., and Tschachler, E. (1995). Human melanocytes and melanoma cells constitutively express the Bcl-2 proto-oncogene in situ and in cell culture. Am. J. Pathol. 146, 651–659.
Pollock, P. M., and Meltzer, P. S. (2002). A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell 2, 5–7.
Pop, C., and Salvesen, G. S. (2009). Human caspases: activation, specificity, and regulation. J. Biol. Chem. 284, 21777–21781.
Pratheeshkumar, P., and Kuttan, G. (2011a). Vernolide-A, a sesquiterpene lactone from Vernonia cinerea, induces apoptosis in B16F-10 melanoma cells by modulating p53 and caspase-3 gene expressions and regulating NF-κB-mediated bcl-2 activation. Drug Chem. Toxicol. 34, 261–270.
Pratheeshkumar, P., and Kuttan, G. (2011b). Effect of vernolide-A, a sesquiterpene lactone from Vernonia cinerea L., on cell-mediated immune response in B16F-10 metastatic melanoma-bearing mice. Immunopharmacol. Immunotoxicol. 33, 533–538.
Pratheeshkumar, P., Raphael, T. J., and Kuttan, G. (2011). Nomilin inhibits metastasis via induction of apoptosis and regulates the activation of transcription factors and the cytokine profile in B16F-10 cells. Integr Cancer Ther. PMID: 21665879. [Epub ahead of print].
Qin, J. Z., Xin, H., and Nickoloff, B. J. (2010). Targeting glutamine metabolism sensitizes melanoma cells to TRAIL-induced death. Biochem. Biophys. Res. Commun. 398, 146–152.
Qin, J. Z., Ziffra, J., Stennett, L., Bodner, B., Bonish, B. K., Chaturvedi, V., Bennett, F., Pollock, P. M., Trent, J. M., Hendrix, M. J., Rizzo, P., Miele, L., and Nickoloff, B. J. (2005). Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 65, 6282–6293.
Raisova, M., Hossini, A. M., Eberle, J., Riebeling, C., Wieder, T., Sturm, I., Daniel, P. T., Orfanos, C. E., and Geilen, C. C. (2001). The Bax/Bcl-2 ratio determines the susceptibility of human melanoma cells to CD95/Fas-mediated apoptosis. J. Invest. Dermatol. 117, 333–340.
Ramsay, J. A., From, L., and Kahn, H. J. (1995). bcl-2 protein expression in melanocytic neoplasms of the skin. Mod. Pathol. 8, 150–154.
Reifenberger, J., Knobbe, C. B., Sterzinger, A. A., Blaschke, B., Schulte, K. W., Ruzicka, T., and Reifenberger, G. (2004). Frequent alterations of Ras signaling pathway genes in sporadic malignant melanomas. Int. J. Cancer 109, 377–384.
Robert, C., and Ghiringhelli, F. (2009). What is the role of cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma? Oncologist 14, 848–861.
Robert, C., Thomas, L., Bondarenko, I., O’Day, S., M, D. J., Garbe, C., Lebbe, C., Baurain, J. F., Testori, A., Grob, J. J., Davidson, N., Richards, J., Maio, M., Hauschild, A., Miller, W. H. Jr., Gascon, P., Lotem, M., Harmankaya, K., Ibrahim, R., Francis, S., Chen, T. T., Humphrey, R., Hoos, A., and Wolchok, J. D. (2011). Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N. Engl. J. Med. 364, 2517–2526.
Ryan, J. A., Brunelle, J. K., and Letai, A. (2010). Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Natl. Acad. Sci. U.S.A. 107, 12895–12900.
Saenz-Santamaria, M. C., Reed, J. A., McNutt, N. S., and Shea, C. R. (1994). Immunohistochemical expression of BCL-2 in melanomas and intradermal nevi. J. Cutan. Pathol. 21, 393–397.
Seeger, J. M., Schmidt, P., Brinkmann, K., Hombach, A. A., Coutelle, O., Zigrino, P., Wagner-Stippich, D., Mauch, C., Abken, H., Kronke, M., and Kashkar, H. (2010). The proteasome inhibitor bortezomib sensitizes melanoma cells toward adoptive CTL attack. Cancer Res. 70, 1825–1834.
Seigler, H. F., Lucas, V. S. Jr., Pickett, N. J., and Huang, A. T. (1980). DTIC, CCNU, bleomycin and vincristine (BOLD) in metastatic melanoma. Cancer 46, 2346–2348.
Selzer, E., Schlagbauer-Wadl, H., Okamoto, I., Pehamberger, H., Potter, R., and Jansen, B. (1998). Expression of Bcl-2 family members in human melanocytes, in melanoma metastases and in melanoma cell lines. Melanoma Res. 8, 197–203.
Shao, Y., and Aplin, A. E. (2010). Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 70, 6670–6681.
Sheridan, C., Brumatti, G., and Martin, S. J. (2008). Oncogenic B-RafV600E inhibits apoptosis and promotes ERK-dependent inactivation of Bad and Bim. J. Biol. Chem. 283, 22128–22135.
Shibuya, H., Kato, Y., Saito, M., Isobe, T., Tsuboi, R., Koga, M., Toyota, H., and Mizuguchi, J. (2003). Induction of apoptosis and/or necrosis following exposure to antitumour agents in a melanoma cell line, probably through modulation of Bcl-2 family proteins. Melanoma Res. 13, 457–464.
Shukla, V. K., Hughes, D. C., Hughes, L. E., McCormick, F., and Padua, R. A. (1989). ras mutations in human melanotic lesions: K-ras activation is a frequent and early event in melanoma development. Oncogene Res. 5, 121–127.
Soengas, M. S., and Lowe, S. W. (2003). Apoptosis and melanoma chemoresistance. Oncogene 22, 3138–3151.
Stahl, J. M., Sharma, A., Cheung, M., Zimmerman, M., Cheng, J. Q., Bosenberg, M. W., Kester, M., Sandirasegarane, L., and Robertson, G. P. (2004). Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 64, 7002–7010.
Steck, P. A., Pershouse, M. A., Jasser, S. A., Yung, W. K., Lin, H., Ligon, A. H., Langford, L. A., Baumgard, M. L., Hattier, T., Davis, T., Frye, C., Hu, R., Swedlund, B., Teng, D. H., and Tavtigian, S. V. (1997). Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362.
Stein, J. A., and Brownell, I. (2008). Treatment approaches for advanced cutaneous melanoma. J. Drugs Dermatol. 7, 175–179.
Stewart, M. L., Fire, E., Keating, A. E., and Walensky, L. D. (2010). The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat. Chem. Biol. 6, 595–601.
Sviatoha, V., Rundgren, A., Tani, E., Hansson, J., Kleina, R., and Skoog, L. (2002). Expression of CD40, CD44, bcl-2 antigens and rate of cell proliferation on fine needle aspirates from metastatic melanoma. Cytopathology 13, 11–21.
Takaoka, A., Adachi, M., Okuda, H., Sato, S., Yawata, A., Hinoda, Y., Takayama, S., Reed, J. C., and Imai, K. (1997). Anti-cell death activity promotes pulmonary metastasis of melanoma cells. Oncogene 14, 2971–2977.
Tan, K. O., Tan, K. M., Chan, S. L., Yee, K. S., Bevort, M., Ang, K. C., and Yu, V. C. (2001). MAP-1, a novel proapoptotic protein containing a BH3-like motif that associates with Bax through its Bcl-2 homology domains. J. Biol. Chem. 276, 2802–2807.
Tang, L., Tron, V. A., Reed, J. C., Mah, K. J., Krajewska, M., Li, G., Zhou, X., Ho, V. C., and Trotter, M. J. (1998). Expression of apoptosis regulators in cutaneous malignant melanoma. Clin. Cancer Res. 4, 1865–1871.
Tchernev, G., and Orfanos, C. E. (2007). Downregulation of cell cycle modulators p21, p27, p53, Rb and proapoptotic Bcl-2-related proteins Bax and Bak in cutaneous melanoma is associated with worse patient prognosis: preliminary findings. J. Cutan. Pathol. 34, 247–256.
Thallinger, C., Wolschek, M. F., Wacheck, V., Maierhofer, H., Gunsberg, P., Polterauer, P., Pehamberger, H., Monia, B. P., Selzer, E., Wolff, K., and Jansen, B. (2003). Mcl-1 antisense therapy chemosensitizes human melanoma in a SCID mouse xenotransplantation model. J. Invest. Dermatol. 120, 1081–1086.
Tran, M. A., Smith, C. D., Kester, M., and Robertson, G. P. (2008). Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin. Cancer Res. 14, 3571–3581.
Tron, V. A., Krajewski, S., Klein-Parker, H., Li, G., Ho, V. C., and Reed, J. C. (1995). Immunohistochemical analysis of Bcl-2 protein regulation in cutaneous melanoma. Am. J. Pathol. 146, 643–650.
Trudel, S., Li, Z. H., Rauw, J., Tiedemann, R. E., Wen, X. Y., and Stewart, A. K. (2007). Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood 109, 5430–5438.
Tsao, H., Atkins, M. B., and Sober, A. J. (2004). Management of cutaneous melanoma. N. Engl. J. Med. 351, 998–1012.
Tsao, H., Zhang, X., Benoit, E., and Haluska, F. G. (1998). Identification of PTEN/MMAC1 alterations in uncultured melanomas and melanoma cell lines. Oncogene 16, 3397–3402.
Tsao, H., Zhang, X., Fowlkes, K., and Haluska, F. G. (2000). Relative reciprocity of NRAS and PTEN/MMAC1 alterations in cutaneous melanoma cell lines. Cancer Res. 60, 1800–1804.
Utikal, J., Leiter, U., Udart, M., Kaskel, P., Peter, R. U., and Krahn, G. M. (2002). Expression of c-myc and bcl-2 in primary and advanced cutaneous melanoma. Cancer Invest. 20, 914–921.
Vachtenheim, J., Ondrusova, L., and Borovansky, J. (2010). SWI/SNF chromatin remodeling complex is critical for the expression of microphthalmia-associated transcription factor in melanoma cells. Biochem. Biophys. Res. Commun. 392, 454–459.
VanBrocklin, M. W., Verhaegen, M., Soengas, M. S., and Holmen, S. L. (2009). Mitogen-activated protein kinase inhibition induces translocation of Bmf to promote apoptosis in melanoma. Cancer Res. 69, 1985–1994.
Verhaegen, M., Bauer, J. A., Martin de la Vega, C., Wang, G., Wolter, K. G., Brenner, J. C., Nikolovska-Coleska, Z., Bengtson, A., Nair, R., Elder, J. T., Van Brocklin, M., Carey, T. E., Bradford, C. R., Wang, S., and Soengas, M. S. (2006). A novel BH3 mimetic reveals a mitogen-activated protein kinase-dependent mechanism of melanoma cell death controlled by p53 and reactive oxygen species. Cancer Res. 66, 11348–11359.
Vousden, K. H., and Lane, D. P. (2007). p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283.
Vucic, D., Deshayes, K., Ackerly, H., Pisabarro, M. T., Kadkhodayan, S., Fairbrother, W. J., and Dixit, V. M. (2002). SMAC negatively regulates the anti-apoptotic activity of melanoma inhibitor of apoptosis (ML-IAP). J. Biol. Chem. 277, 12275–12279.
Walensky, L. D., Kung, A. L., Escher, I., Malia, T. J., Barbuto, S., Wright, R. D., Wagner, G., Verdine, G. L., and Korsmeyer, S. J. (2004). Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466–1470.
Walensky, L. D., Pitter, K., Morash, J., Oh, K. J., Barbuto, S., Fisher, J., Smith, E., Verdine, G. L., and Korsmeyer, S. J. (2006). A stapled BID BH3 helix directly binds and activates BAX. Mol. Cell 24, 199–210.
Wan, P. T., Garnett, M. J., Roe, S. M., Lee, S., Niculescu-Duvaz, D., Good, V. M., Jones, C. M., Marshall, C. J., Springer, C. J., Barford, D., Marais, R., and Cancer Genome Project. (2004). Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867.
Wang, Y. F., Jiang, C. C., Kiejda, K. A., Gillespie, S., Zhang, X. D., and Hersey, P. (2007). Apoptosis induction in human melanoma cells by inhibition of MEK is caspase-independent and mediated by the Bcl-2 family members PUMA, Bim, and Mcl-1. Clin. Cancer Res. 13, 4934–4942.
Weber, A., Kirejczyk, Z., Potthoff, S., Ploner, C., and Hacker, G. (2009). Endogenous noxa determines the strong proapoptotic synergism of the BH3-mimetic ABT-737 with chemotherapeutic agents in human melanoma cells. Transl. Oncol. 2, 73–83.
Wei, M. C., Lindsten, T., Mootha, V. K., Weiler, S., Gross, A., Ashiya, M., Thompson, C. B., and Korsmeyer, S. J. (2000). tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14, 2060–2071.
Wei, M. C., Zong, W. X., Cheng, E. H., Lindsten, T., Panoutsakopoulou, V., Ross, A. J., Roth, K. A., MacGregor, G. R., Thompson, C. B., and Korsmeyer, S. J. (2001). Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292, 727–730.
Willis, S. N., Chen, L., Dewson, G., Wei, A., Naik, E., Fletcher, J. I., Adams, J. M., and Huang, D. C. (2005). Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 19, 1294–1305.
Willis, S. N., Fletcher, J. I., Kaufmann, T., van Delft, M. F., Chen, L., Czabotar, P. E., Ierino, H., Lee, E. F., Fairlie, W. D., Bouillet, P., Strasser, A., Kluck, R. M., Adams, J. M., and Huang, D. C. (2007). Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315, 856–859.
Wong, R. P., Khosravi, S., Martinka, M., and Li, G. (2008). Myeloid leukemia-1 expression in benign and malignant melanocytic lesions. Oncol. Rep. 19, 933–937.
Xie, K., Wang, Y., Huang, S., Xu, L., Bielenberg, D., Salas, T., McConkey, D. J., Jiang, W., and Fidler, I. J. (1997). Nitric oxide-mediated apoptosis of K-1735 melanoma cells is associated with downregulation of Bcl-2. Oncogene 15, 771–779.
Xu, X., Liu, Y., Wang, L., He, J., Zhang, H., Chen, X., Li, Y., Yang, J., and Tao, J. (2009). Gambogic acid induces apoptosis by regulating the expression of Bax and Bcl-2 and enhancing caspase-3 activity in human malignant melanoma A375 cells. Int. J. Dermatol. 48, 186–192.
Yamamura, K., Kamada, S., Ito, S., Nakagawa, K., Ichihashi, M., and Tsujimoto, Y. (1996). Accelerated disappearance of melanocytes in bcl-2-deficient mice. Cancer Res. 56, 3546–3550.
Yecies, D., Carlson, N. E., Deng, J., and Letai, A. (2010). Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 115, 3304–3313.
You, M., Savaraj, N., Wangpaichitr, M., Wu, C., Kuo, M. T., Varona-Santos, J., Nguyen, D. M., and Feun, L. (2010). The combination of ADI-PEG20 and TRAIL effectively increases cell death in melanoma cell lines. Biochem. Biophys. Res. Commun. 394, 760–766.
Yu, F., Watts, R. N., Zhang, X. D., Borrow, J. M., and Hersey, P. (2006). Involvement of BH3-only proapoptotic proteins in mitochondrial-dependent phenoxodiol-induced apoptosis of human melanoma cells. Anticancer Drugs 17, 1151–1161.
Yu, K. S., Lee, Y., Kim, C. M., Park, E. C., Choi, J., Lim, D. S., Chung, Y. H., and Koh, S. S. (2010). The protease inhibitor, elafin, induces p53-dependent apoptosis in human melanoma cells. Int. J. Cancer 127, 1308–1320.
Zhang, H., and Rosdahl, I. (2006). Bcl-xL and bcl-2 proteins in melanoma progression and UVB-induced apoptosis. Int. J. Oncol. 28, 661–666.
Zhou, C., Mao, X. P., Guo, Q., and Zeng, F. Q. (2009). Diallyl trisulphide-induced apoptosis in human melanoma cells involves downregulation of Bcl-2 and Bcl-xL expression and activation of caspases. Clin. Exp. Dermatol. 34, e537–e543.
Zhou, X. P., Gimm, O., Hampel, H., Niemann, T., Walker, M. J., and Eng, C. (2000). Epigenetic PTEN silencing in malignant melanomas without PTEN mutation. Am. J. Pathol. 157, 1123–1128.
Zhuang, L., Scolyer, R. A., Murali, R., McCarthy, S. W., Zhang, X. D., Thompson, J. F., and Hersey, P. (2010). Lactate dehydrogenase 5 expression in melanoma increases with disease progression and is associated with expression of Bcl-XL and Mcl-1, but not Bcl-2 proteins. Mod. Pathol. 23, 45–53.
Zobel, K., Wang, L., Varfolomeev, E., Franklin, M. C., Elliott, L. O., Wallweber, H. J., Okawa, D. C., Flygare, J. A., Vucic, D., Fairbrother, W. J., and Deshayes, K. (2006). Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem. Biol. 1, 525–533.
Keywords: apoptosis, BCL-2 family, cancer, chemotherapy, melanoma, mitochondria
Citation: Anvekar RA, Asciolla JJ, Missert DJ and Chipuk JE (2011) Born to be alive: a role for the BCL-2 family in melanoma tumor cell survival, apoptosis, and treatment. Front. Oncol. 1:34. doi: 10.3389/fonc.2011.00034
Received: 05 August 2011;
Accepted: 22 September 2011;
Published online: 13 October 2011.
Edited by:
Vassiliki Karantza, UMDNJ-Robert Wood Johnson Medical School, USAReviewed by:
Daniel C. Christian Hoessli, International Center for Chemical and Biological Sciences, SwitzerlandTheocharis Panaretakis, Karolinska Institutet, Sweden
Copyright: © 2011 Anvekar, Asciolla, Missert and Chipuk. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Jerry E. Chipuk, Mount Sinai School of Medicine, One Gustave L. Levy Place, Box 1130, New York, NY 10029, USA. e-mail:amVycnkuY2hpcHVrQG1zc20uZWR1
†Rina A. Anvekar and James J. Asciolla and Derek J. Missert have contributed equally to this work.