 Ignacio Pérez de Castro1*
Ignacio Pérez de Castro1* Cristina Aguirre-Portolés1
Cristina Aguirre-Portolés1 Benedicte Martin2
Benedicte Martin2 Gonzalo Fernández-Miranda1
Gonzalo Fernández-Miranda1 Andrea Klotzbucher3 Michael H. G. Kubbutat3 Diego Megías4
Andrea Klotzbucher3 Michael H. G. Kubbutat3 Diego Megías4 Yannick Arlot-Bonnemains2†
Yannick Arlot-Bonnemains2† Marcos Malumbres1*†
Marcos Malumbres1*†- 1 Molecular Oncology Programme, Cell Division and Cancer Group, Centro Nacional de Investigaciones Oncológicas, Madrid, Spain
- 2 CNRS-UMR 6061, Institut de Génétique et Développement de Rennes, IFR 140 GFAS, Faculté de Médecine, Université Rennes 1, Rennes, France
- 3 ProQinase GmBH, Freiburg, Germany
- 4 Confocal Microscopy Core Unit, Centro Nacional de Investigaciones Oncológicas, Madrid, Spain
Aurora-A is a serine/threonine kinase that plays critical roles in centrosome maturation, spindle dynamics, and chromosome orientation and it is frequently over-expressed in human cancers. In this work, we show that Aurora-A interacts with the SUMO-conjugating enzyme UBC9 and co-localizes with SUMO1 in mitotic cells. Aurora-A can be SUMOylated in vitro and in vivo. Mutation of the highly conserved SUMOylation residue lysine 249 significantly disrupts Aurora-A SUMOylation and mitotic defects characterized by defective and multipolar spindles ensue. The Aurora-AK249R mutant has normal kinase activity but displays altered dynamics at the mitotic spindle. In addition, ectopic expression of the Aurora-AK249R mutant results in a significant increase in susceptibility to malignant transformation induced by the Ras oncogene. These data suggest that modification by SUMO residues may control Aurora-A function at the spindle and that deficiency of SUMOylation of this kinase may have important implications for tumor development.
Introduction
Aurora-A is a serine/threonine kinase with important functions in the biology of centrosomes and chromosomes. This protein belongs to the Aurora kinase family composed of Aurora-A, -B, and -C in mammals (Carmena and Earnshaw, 2003). Despite their sequence similarity, the three mammalian Aurora family members differ in their expression patterns, sub-cellular localization, timing of activity, and roles in cancer development and progression. Dysfunction of these kinases has been associated with defects in the chromosome content of cells that contribute to genetic instability. Among the three Aurora kinases, over-expression of Aurora-A in human cancer was first described in primary breast and colon tumor cells (Sen et al., 1997; Bischoff et al., 1998). Over-expression of this protein induces abnormal centrosome duplication and the subsequent formation of abnormal spindle structures, leading to abnormal chromosome segregation (Zhou et al., 1998; Meraldi et al., 2002). Aurora-A has also been found to be amplified in a subset of human tumors, and was localized to an amplicon associated with poor prognosis in patients with breast and colon tumors (20q13; Bischoff et al., 1998; Royce et al., 2004). Many subsequent studies have identified other tumor types, including pancreatic, ovarian bladder, gastric, and thyroid tumors, in which Aurora-A is over-expressed (Miyoshi et al., 2001; Sakakura et al., 2001; Sen et al., 2002; Gritsko et al., 2003; Li et al., 2003; Moreno-Bueno et al., 2003). It has also been demonstrated that some Aurora-A variants confer low-penetrance susceptibility to tumor development and progression in mices and humans (Ewart-Toland et al., 2003). All these results have aroused interest in Aurora-A as a putative oncogene and as an attractive anti-tumoral therapeutic target (Katayama et al., 2003; Perez De Castro et al., 2008; Lens et al., 2010).
Aurora-A is mainly localized on duplicated centrosomes from the end of the S phase until the beginning of the following G1 phase (Marumoto et al., 2005). Its protein levels seem to be cell cycle regulated as it is synthesized and activated at the end of S phase with a peak in the G2/M phase, and, it is degraded via the ubiquitin/proteasome pathway as cells enter into the next G1 phase (Castro et al., 2002; Taguchi et al., 2002). The kinase activity of the Aurora-A protein is regulated by phosphorylation at several residues including Ser 51, Thr 288, and Ser 342 (Littlepage et al., 2002; Eyers et al., 2003; Kitajima et al., 2007). Degradation of the protein requires the presence of specific amino acids in the C terminal sequence (destruction box – RxxLxD) and the dephosphorylation of Ser 51 (Arlot-Bonnemains et al., 2001; Kitajima et al., 2007). Whether Aurora-A is subject to other post-translational modifications is unknown at present.
Among the post-translational modifications that regulate protein behavior, SUMOylation is characterized by its highly dynamic and reversible nature. Similar to ubiquitination, SUMOylation modulates protein function through post-translational covalent attachment of Small Ubiquitin-related MOdifiers (SUMOs) to lysine residues within targeted proteins. Although invertebrates express a single SUMO protein, three paralogs exist in vertebrates: SUMO1, SUMO2, and SUMO3. SUMO conjugation, which often requires the consensus sequence ψKxE/D (where ψ is a large hydrophobic residue and x is any amino acid), occurs in a stepwise enzymatic cascade (Melchior, 2000; Hay, 2005). The covalent attachment of SUMO to lysine residues is catalyzed by three types of enzymes, a SUMO-activating E1 enzyme (Aos1/Uba2), a SUMO-conjugating E2 enzyme (UBC9), and a SUMO E3 ligase. SUMOylation has been shown to play a crucial role in regulating the activity of numerous cellular processes, including nuclear transport, genome integrity, signal transduction, and transcriptional regulation (Hay, 2005; Ulrich, 2008). An increasing number of studies have demonstrated that SUMOylation is essential for regulating chromosome segregation and progression through mitosis (Dasso, 2008). In yeast and Drosophila, SUMOylation has been shown to be critical for chromosome condensation, sister chromatid cohesion, kinetochore function, and mitotic spindle elongation (Watts, 2007). Multiple studies have confirmed the essential role of SUMO modification in progression through mitosis in vertebrates (Dawlaty et al., 2008; Zhang et al., 2008b; Klein et al., 2009). More recently, we demonstrated that SUMOylation of Aurora-B at Lysine 207 controls its proper localization and activity at centromeres (Fernandez-Miranda et al., 2010). Because this residue resides in a conserved SUMOylation motif in all Aurora kinases we decided to analyze the possibility that SUMO modification is also involved in the regulation of Aurora-A function.
In this manuscript, we report that Aurora-A interacts with the SUMOylation machinery. Aurora-A interacts with the SUMO-conjugating enzyme UBC9 and binds SUMO1 both in vitro and in vivo. Mutation of the predicted SUMO-binding site, Lys249, significantly disrupts Aurora-A SUMOylation and results in several aberrations during mitosis that are accompanied by defective bipolar spindle formation. These defects have consequences for the susceptibility of mutant cells to malignant transformation by the Ras oncogene, suggesting a possible link between SUMO modification and the role of Aurora-A in cancer.
Materials and Methods
Yeast Two-Hybrid, in vitro SUMOylation and Pull-Down Assays
The full-length human Aurora-A sequence was subcloned and expressed as fusion protein with DNA-binding domain (pGBT9) with a selection medium Trp. The human fetal kidney MATCHMAKER cDNA Library was cDNA protein expressed as fusion with activation domain cloned in pACT2 vector library with a selection medium Leu. Yeast strain CG1945 was transformed by means of pairwise combination of both two-hybrid vectors and grown on a medium without leucine or tryptophan. Galactosidase activities were assayed using 5-bromo-4-chloro-3-indolyl b-D-galactopyranoside staining on filter replicates according to Breeden and Nasmyth (1985). Both pACT2 and pGBT9 vectors without insert were used as negative controls. The human UBC9 cDNA was obtained from MRC Geneservice (#14227-h18), then subcloned into a pGEX-4T-1 vector (Amersham Biosciences, #27-4580-01) in order to produce the recombinant GST–UBC9 full-length protein. All constructs were sequenced in full (Genome Express).
Aurora-A(His)6 and GST-UBC9 recombinant proteins were prepared as previously described (Smith and Johnson, 1988; Roghi et al., 1998). Histidine-tagged proteins and proteins bound to histidine-tagged proteins as well as GST–UBC9 and GST–UBC9 bound proteins were isolated by affinity chromatography on Nickel–NTA–agarose and GST–4B agarose, respectively. Proteins from pull-downs were separated on 12.5% SDS polyacrylamide gels and submitted to western blot using the appropriated antibodies against Aurora-A (Abcam, mouse monoclonal 35C1 clone; 1/200) or GST (SIGMA, mouse monoclonal, #G1160, 1/50000).
The mouse Aurora-A cDNA was cloned into pENTR–DTOPO vector using TOPO technology (Invitrogen) and transferred to a destiny vector coexpressing EGFP or V5 tag by a LR recombination reaction of the Gateway system (Invitrogen). Mouse Aurora-A mutants (Aurora-AK153M and Aurora-AK249R) were prepared using the Quick-Change site-directed mutagenesis kit (Stratagene) and were verified by DNA sequencing.
In vitro SUMOylation of recombinant Aurora-A protein was tested using Active Motif SUMO link (Active Motif, #40120) and following manufacturer’s conditions. Products from the in vitro reaction were separated on 12% SDS-PAGE gels stained with Coomassie and transferred to nitrocellulose membranes for the further detection of Aurora-A and SUMO1 proteins using the corresponding antibodies (35C1 for Aurora-A at 1/200; Active Motif, rabbit anti-SUMO1 at 1/4000). The reactions were performed in the presence of SUMO1 or a SUMO1 non-conjugatable mutant.
Sequence Analysis
For protein domain analysis we first used the eukaryotic linear motif (ELM1) algorithm. To further confirm the presence of SUMO motifs we used the specific SUMO prediction software SUMOplot™ Analysis Program2.
Cell Culture, Transfection, and Cellular Assays
HEK293, U2OS, and HeLa human cells were maintained in DMEM medium supplemented with 10% fetal bovine serum and antibiotics and were grown at 37°C in a humidified 5% CO2 atmosphere. NIH-3T3 mouse cells were maintained in the same conditions with 10% calf serum. In the focus-formation assays NIH-3T3 cells were transfected with 10 μg of V5-Aurora-A plasmids and 1 μg of H-Ras G12V following standard calcium phosphate transfection protocol, maintained in culture during 3 weeks and then stained with crystal violet solution for foci quantification. Cell cycle distribution of cell lines were determined by flow cytometry after DNA staining with propidium iodide (Sigma) and analyzed on a FACSCanto flow cytometer (Becton Dickinson). Data were processed using FACSDiva software (Becton Dickinson).
To obtain HeLa cells stably expressing EGFP-tagged Aurora-A variants, cells were transfected with EGFP–Aurora-A expressing vectors that also confer resistance to G418. To select stable clones, G418 (1 mg/ml) was added to the cultures for 10 days. G418-resistant cells were then individually seeded in 96-well microplates. EGFP-positive colonies were expanded and Aurora-A exogenous expression established by immunofluorescence and immunoblotting (Figure A2 in Appendix).
For the taxol sensitive assay HeLa clones expressing EGFP–Aurora-A variants in a stable fashion were treated with 150 nM taxol for 48 h. The percentage of apoptotic cells was then measured using propidium iodide (PI) staining and flow cytometry to quantify the PI positive (dead) population.
The microtubule re-growth assay was performed using HeLa cells stably expressing EGFP–Aurora-A variants, which were treated with 100 ng/ml nocodazole for 6 h to enrich them at M phase. Cells, in Hepes 10 mM, were incubated on ice for 30 min and then the microtubules (MT) were allowed to grow by re-warming the cells at 37°C. At different time points post re-warming, cells were fixed with 4% paraformaldehyde and cold methanol. MT and centrosomes were detected using anti α-tubulin from Sigma (DM1A; 1/1000) and α-CEP135 (kindly provided by Dr. Anthony Hyman Max Plank Institute of Molecular Cell Biology and Genetics, Dresden, Germany; Bird and Hyman, 2008; 1/500).
For the determination of chromosome numbers, metaphase preparations were made using 0.075 M KCl hypotonic treatment and methanol/acetic acid fixation from mouse fibroblasts after a 2 h treatment with colcemid (0.01 μg/ml). Slides were stained with 5% Giemsa in pH 6.8 buffer for 10 min. Chromosome numbers were scored in a minimum of 20 metaphases in each type analyzed.
Protein Extraction and Analysis: Immunoprecipitation, Detection of SUMOylation in vivo, and in vitro Kinase Assays
For immunodetection in protein lysates, cells were washed twice with ice-cold PBS and lysed in RIPA lysis buffer (37 mM NaCl, 0.5% NP-40, 0.1% SDS, 1% TX-100, 20 mM Tris–HCl pH 7.4, 2 mM EDTA, 10% glycerol 1 mM PMSF) supplemented with protease and phosphatase inhibitory cocktails (Sigma). Additionally, N-ethylmaleimide, an inhibitor of SUMO-proteases, was added at 10 mM final concentration. After 30 min on ice, samples were cleared by centrifugation. Proteins were separated on SDS-PAGE, transferred to nitrocellulose membranes (BioRad), probed using specific antibody and detected using fluorescent donkey (Rockland) or goat (Invitrogen) anti rabbit or anti mouse secondary antibodies followed detection using the Odyssey Infrared Imaging System (Li-Cor Biosciences). Primary antibodies used were against Aurora-A (AbCam mouse monoclonal, clone 35C1), V5 tag (Invitrogen, mouse monoclonal #46-0705), GST (SIGMA, mouse monoclonal, #G1160), SUMO1 (BioMol and Active Motif), UBC9 (BD Biosciences, mouse monoclonal, #610749, and Santa Cruz Biotechnology, rabbit polyclonal, #SC-10759), and α-tubulin (Sigma, mouse monoclonal, clone DM1A). Secondary antibodies for immunodetection were coupled to Alexa dies (488, 594, 647) from Molecular Probes (Invitrogen).
For immunoprecipitation, proteins were extracted in the following lysis buffer: Hepes 20 mM pH 7.5, β-glycerophosphate 80 mM, EDTA 20 mM, SDS 0.25%, MgCl2 15 mM, NEM 26 mM, 1× protease inhibitor cocktail Set III, AEBSF 1 mM. Seventy microliters of protein-G Dynabeads (Invitrogen) were washed with 500 μl of acetate buffer 0.5 M pH 5.5 and incubated for 2 h at 4°C with the adequate antibody, and then washed twice with 500 μl PBS. Beads were then incubated with 300 μg of proteins for 2 h at 4°C. The beads were washed once in 500 μl of NaCl 0.5% and five times with 500 μl TBST. Bound proteins were eluted in 10 μl of 2X-Laemmli sample buffer and the proteins were separated on a 12.5% SDS polyacrylamide gel and then immunoblotted. The western blot analyses were performed with the polyclonal UBC9 antibody (1/1000) or the monoclonal Aurora-A antibody (1/200).
For purification of histidine-tagged SUMO1 conjugates under denaturing conditions, we co-transfected HeLa cells with histidine-tagged SUMO1, V5-tagged Aurora-A variants, and Flag-UBC9. Cell lysates and Ni–NTA pull-downs were performed as described (Tatham et al., 2009). V5-tagged Aurora-A was detected by immunoblotting in both inputs and Ni–NTA purified fractions using monoclonal anti-V5 at 1/1000, whereas vinculin was immunodetected using mouse monoclonal anti-vinculin antibody only in inputs at 1/1000. Fluorescent signal from the secondary antibodies was used to quantify V5–Aurora-A signal in each case.
Immunofluorescence, Live Cell Analysis, and FRAP
Cells were cultured on glass coverslips in standard conditions, and fixed, depending on the antibody, with either −20°C methanol for 6 min or with 4% PFA plus TritonX-100 0.15% for 10 min at room temperature. Then, the cells were washed in PBS and preincubated with PBS-3% BSA for 1 h at room temperature to saturate the non-specific binding sites and incubated in PBS plus 1.5% BSA with the primary antibodies for 2 h at room temperature. After washing, the coverslips were incubated with the matching secondary antibodies for 1 h at room temperature and then mounted in Vectashield containing 1 μg/ml DAPI. Images were obtained using a confocal ultra-spectral microscope (Leica TCS-SP5), a Leica-DMRXA microscope or an Olympus IX70 microscope controlled by a Delta Vision SoftWorx (Applied Precision, Issaquah, WA, USA). Image stacks were deconvolved, quick-projected, and saved as tiff images to be processed using Adobe Photoshop.
Fluorescence recovery after photobleaching (FRAP) analysis was performed using a Leica SP5 confocal microscope (Leica, Inc.) fitted with a 63 objective lens. The live cell chamber was maintained at 37°C with 5% CO2. A single Z-section was imaged before and at set time intervals following the bleach. The bleach was performed by setting the laser wavelength to 488 nm for EGFP at maximum power for 75 iterations. The bleached area contained either the centrosomes (cells at interphase) or both the spindle MT and poles (mitotic cells). The fluorescence intensities were determined using LAS AF software (Leica Inc.) in the region of interest (ROI) corresponding to the bleach area. After completion of the photobleach, the signal intensity within the ROI was collected at set time intervals. Intensity values were normalized using the following equation; It = (Xt − Y)/(Z − Y), where I is the normalized intensity at time t, X is the actual intensity at time t, Y is the intensity immediately following the photobleach (when t is equal to zero), and Z is the intensity at the final time point. This sets the initial post-bleach intensity (at t = 0 s) to zero and the final intensity to 1 arbitrary units. Regression analysis was performed using the GraphPad Prism software by calculating the least squares fit using the standard first order logarithmic equation. The t1/2 value was the time at which the normalized intensity reaches 0.5 arbitrary units.
Live images were acquired using DeltaVision RT imaging system [Applied Precision, LLC; IX70/71 (Olympus)] equipped with a charge-coupled device camera (CoolSNAP HQ; Roper Scientific, Inc.). One micrometer serial Z sections were performed for each cell and the images were taken in 10 min intervals using a 60X PlanApo N 1.42 N.A. objective. The datasets were deconvolved using SoftWoRx (Applied Prescision, LLC) software.
For the quantitative analysis of immunofluorescence signals, acquired images were processed and analyzed using Adobe Photoshop CS 8.0 and ImageJ 1.43u. Exogenous EGFP–Aurora-A signal in mitotic cells was determined as follows: first, integrated intensity at MTs and poles was calculated; second, integrated intensity at the rest of the cell was scored; finally, the ratio between the two values was used as the EGFP–Aurora-A signal enrichment at the spindle. The same approach was followed for the determination of endogenous α-tubulin signal in the MT re-growth experiments. Distance between CEP135 signals of mitotic cells was determined using ImageJ. In all the cases external background was eliminated using Adobe Photoshop.
Statistical Analyses
Data are expressed as mean ± SE. Comparisons between the different Aurora-A forms both in transient and stable experiments were performed using unpaired, two-tail, t-test analysis. Statistical analyses were performed using Prism 5 for Windows (GraphPad Software, Inc.). p Values < 0.05 were considered to be statistically significant.
Results
Aurora-A Interacts with the SUMO-Conjugating Enzyme UBC9
To identify new Aurora-A interactors, we performed a two-hybrid screen using full-length Aurora-A as bait. Out of 20 positive clones isolated, we identified one encoding UBC9, a SUMO-conjugating E2 enzyme. To test the specificity of this interaction, the full-length UBC9 cDNA was subcloned into the pGADGH vector and used as bait with pGBT9–Aurora-A. Only cells co-transfected with pGADGH–UBC9 and pGBT9–Aurora-A grew on selective medium. Pull-down assays were undertaken using recombinant proteins for both molecules [Aurora-A-(His)6 and GST–UBC9]. In the first round of experiments, recombinant GST–UBC9 or GST was incubated with Aurora-A(His)6 and then loaded onto glutathione-Sepharose beads. As shown in Figure 1A (left panel), Aurora-A(His)6 was found to bind only to the GST–UBC9 containing sepharose. Binding was also observed in an in vitro histidine pull-down assay (Figure 1A; right panel). In this case recombinant GST–UBC9 associated with recombinant Aurora-A(His)6 bound to the column.
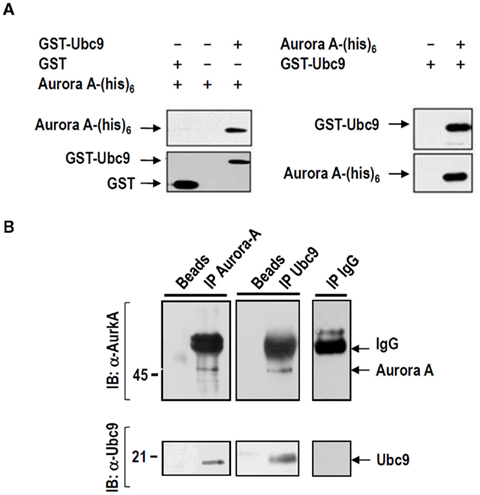
Figure 1. UBC9 is a new partner of Aurora-A. (A) UBC9 and Aurora-A interact in vitro. Recombinant GST–UBC9 protein previously incubated with or without Aurora-A(His)6 was incubated with nickel–agarose beads (Ni–NTA–agarose), whereas Aurora-A (His)6 recombinant protein was incubated in the presence of GST–UBC9 or GST alone and loaded onto glutathione-sepharose. The presence of Aurora-A and UBC9 was tested by blotting using anti-Aurora-A and anti-UBC9 antibodies. (B) Aurora-A and UBC9 interact in vivo. Anti-AurkA antibodies were able to immunoprecipitate UBC9 (left panels) and anti-UBC9 antibodies immunoprecipitated Aurora-A proteins (middle panels) from HeLa cell protein extracts. The same reactions performed with beads only (left lines) or control IgG (right panels) were unable to immunoprecipitate either UBC9 or Aurora-A.
We next asked whether endogenous Aurora-A and UBC9 interact in mammalian cells. Using extracts from asynchronous HeLa cells, we performed immunoprecipitation experiments with antibodies against Aurora-A or UBC9. Consistent with the results obtained with the recombinant proteins, the anti-UBC9 antibody detected endogenous UBC9 in the anti-Aurora-A pull-down (Figure 1B; left panel). In addition, Aurora-A protein was detected by western blot in the UBC9 immunoprecipitates (Figure 1B; middle panel). Finally, associated proteins were not detected in immunoprecipitations performed without the Aurora-A or UBC9 antibody or in those performed using IgG (Figure 1B; “beads” lines and right panel).
Aurora-A is a Novel Target for SUMOylation
To better understand the Aurora-A–UBC9 relationship, we first studied the localization of Aurora-A and UBC9 across the cell cycle in HeLa cells by immunostaining (Figure 2A). As previously described, Aurora-A was concentrated on duplicated centrosomes and, in mitotic cells, it was found to decorate spindle poles, the midzone, and the cytokinesis bridge (Figure 2A). Interestingly, UBC9 immunodetection revealed an overlapping signal at centrosomes at the end of interphase, and at the spindle, midzone, and cytokinesis bridge during mitosis (Figure 2A).
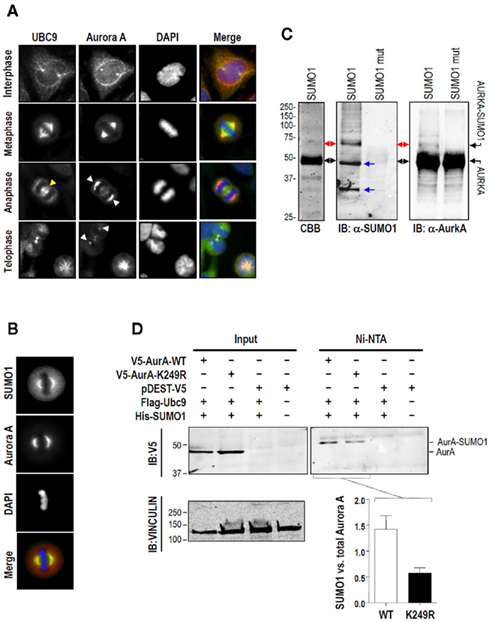
Figure 2. Aurora-A co-localizes with UBC9 and SUMO1 at the spindle and is SUMOylated both in vitro and in vivo. (A) Aurora-A and UBC9 mainly localize at interphase centrosomes and onto the mitotic spindle. HeLa cells were fixed and processed for immunodetection of Aurora-A (red) and UBC9 (green) and observed under a microscope at the indicated phases of the cell cycle. The Aurora-A and UBC9 signals were coincident on the centrosomes and the nuclear membrane in interphase, at the spindle in metaphase and on the cytokinesis bridge at the end of telophase. Note that whereas Aurora-A is still present at the spindle during anaphase and telophase (white arrowheads), this is not the case for UBC9. By contrast, UBC9, and not Aurora-A, is clearly visible at the midzone during anaphase (yellow arrowheads). (B) Co-localization of Aurora-A and SUMO1. Endogenous SUMO1 (red) and Aurora-A (green) were detected by immunofluorescence in HeLa cells. Both proteins co-localize at the spindle (poles and microtubules) during metaphase. (C) Recombinant Aurora-A protein was used as substrate in an in vitro SUMOylation assay in the presence of SUMO1 or SUMO1 mut (a non-conjugatable SUMO1 mutant). Proteins were separated by reducing 12% SDS-PAGE and visualized with Coomassie Blue (left panel). Reaction products were used to detect both Aurora-A and SUMO1 in western blot (right panels). Black arrows indicate unSUMOylated Aurora-A, red arrows indicate SUMOylated Aurora-A, and blue arrows mark other proteins included in the reaction that are also modified by SUMO1. (D) HeLa cells were co-transfected with the indicated expression vectors. Forty-eight hours later proteins were isolated and SUMO1 were conjugates purified under denaturing conditions. V5-tagged Aurora-A was detected by immunoblotting in both the inputs and Ni-NTA purified fractions, whereas vinculin was immunodetected only in the inputs. The V5–Aurora-A signal in the Ni–NTA fractions was quantified after normalization with the inputs signals for V5–Aurora-A.
Because UBC9 was able to bind Aurora-A both in vitro and in vivo, and both proteins co-localized in the same sub-cellular compartments, we hypothesized that this mitotic kinase could be modified by the addition of SUMO residues. We decided to specifically study SUMO1 because it localizes to the mitotic spindle and the spindle midzone whereas SUMO2/3 are found at centromeres and chromatin during prometaphase and metaphase (Azuma et al., 2003; Ayaydin and Dasso, 2004; Zhang et al., 2008b). As shown in Figure 2B, immunodetection revealed the co-localization of SUMO1 with Aurora-A during metaphase, both at centrosomes and the mitotic spindle.
Given the fact that Aurora-A, UBC9, and SUMO1 co-localize onto the same structures, (centrosomes and the mitotic spindle) during the last stages of interphase and during mitosis, we hypothesized that Aurora-A could be a target of SUMOylation. To test our hypothesis we first performed an in vitro SUMOylation assay using bacterially expressed and purified Aurora-A protein. Recombinant Aurora-A protein was incubated with an E1 activating enzyme, the E2 conjugation enzyme UBC9, and SUMO1 or SUMO1 mut (a non-conjugatable form of SUMO1) in the presence of ATP. As shown in Figure 2C, a higher-molecular-weight Aurora-A band was detected by western blot using anti-Aurora-A. This band was also immunoreactive with an antibody against SUMO1 and was not detected when the control (SUMO1 mut) was added to the reaction instead of wild type SUMO1. Additional non-specific bands observed with SUMO1 antibody were also present when other recombinant proteins were studied (Figure A1 in Appendix).
To further determine whether SUMO1 can also modify Aurora-A in vivo, we co-expressed histidine-tagged SUMO1 and V5-tagged Aurora-A constructs in HeLa cells. SUMO1 conjugates were affinity purified under denaturing conditions with Ni–NTA beads following previously described procedure (Tatham et al., 2009). As depicted in Figure 2D, and consistent with the results of the in vitro SUMOylation assay, a V5 positive band was detected in cells co expressing V5–Aurora-A and histidine-tagged-SUMO1.
To identify Aurora-A residues capable of SUMO conjugation, we took advantage of two protein motif finders: the eukaryotic linear motif resource (ELM) and the SUMO motif finder (SUMOFI; see Materials and Methods). Both predictive tools revealed the same hit sequence “IKPE” centered on lysine 258 for human Aurora-A (lysine 249 in mouse). To study the role of this lysine, we generated an Aurora-A mutant that cannot be modified by SUMO at this specific residue; lysine 249 was replaced by the non-SUMOylable residue arginine (K249R). Using denaturing conditions to detect SUMO1 conjugates, the same SUMO band that was observed for wild type Aurora-A was detected upon V5 immunodetection in cells over-expressing the K249R mutant (Figure 2D). Importantly, wild type V5–Aurora-A was SUMOylated at a higher level than the K249R mutant. Therefore, this experiment demonstrated that Aurora-A is SUMOylated at lysine 249 and that other residues may also be modified by SUMO binding.
Expressing a Mutant Form of Aurora-A (K249R) Induces Mitotic Defects
As mentioned above, lysine 249/258 (mouse/human) is a SUMO site conserved in both mouse and human Aurora-A. Subsequent analysis of Aurora-A proteins from other species showed that this putative site for SUMO binding is highly conserved (Figure 3A). Moreover, this SUMO consensus sequence is also conserved in the other Aurora kinase members (Figure 3A; bottom) and it has been shown to participate in the SUMO modification of Aurora-B and -C (Fernandez-Miranda et al., 2010).
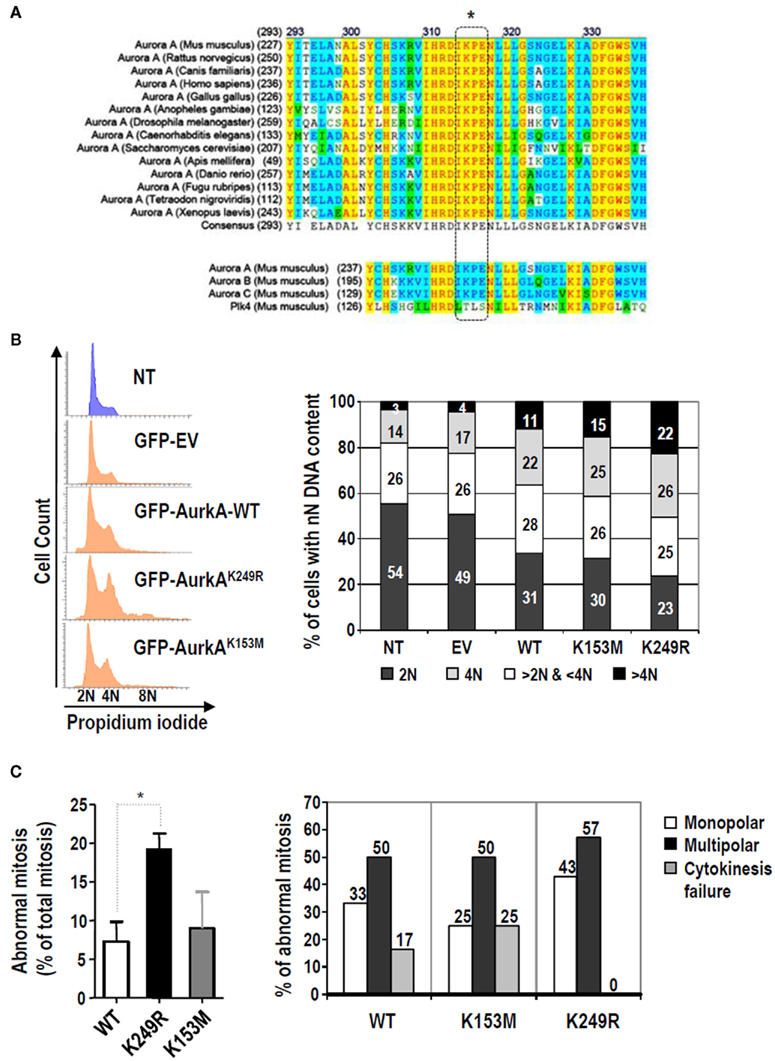
Figure 3. Lack of SUMOylation of Aurora-A alters cell cycle regulation and increases the ratio of mitotic aberrations. (A) Multiple sequence alignment of Aurora-A proteins from different species using ClustalW softwarehttp://www.ch.embnet.org. The SUMO modification motif (dashed box) is highly conserved in all these Aurora-A proteins and in the other two Aurora family members, but it is not present in Polo-like kinase 4 (Plk4), a close mitotic relative of these mitotic kinases. (B) HeLa cells were transiently transfected with an empty vector expressing EGFP (EV) or with any of the three different EGFP-tagger Aurora-A isoforms (WT, K153, K249R). Forty-eight hours later, cell cycle profiles of transfected cells (EGFP-positive cells) and the non-transfected cells (NT; EGFP-negative) were obtained. The histograms and bar-graphs are representative of one of three independent experiments. (C) Evaluation of aberrant mitoses induced by Aurora-AK249R by video-microscopy using EGFP-tagged, Aurora-A stable cell lines. The left panel shows the percentage of abnormal mitoses. Three different stable clones were used for each Aurora-A variant. A total of 118, 81, and 46 mitoses were analyzed for the wild type, K153M and K249R variants, respectively. The bars show the average (±SE) of the percentage of abnormal mitosis found for each clone. The right panel shows the distribution of mitotic aberrations found for each stable Aurora-A type. Only the three main abnormal mitotic phenotypes observed, monopolar spindles, multipolar spindles, and cytokinesis failure, were quantified. The bars show data obtained for the three different clones of each variant and pooled into a single value.
To study the importance of this residue in cell function, we ectopically express its non-SUMOylable form in cell cultures. Asynchronously growing HeLa cells were transfected with EGFP-tagged expression plasmids for wild type mouse Aurora-A (Aurora-Awt), kinase-dead Aurora-A (Aurora-AK153M), or the K249R Aurora-A mutant (Aurora-AK249R). Forty-eight hours after transfection, cell cycle profiles were determined for EGFP-positive (transfected) and EGFP-negative (untransfected) cells (Figure 3B). As previously reported (Anand et al., 2003; Jiang et al., 2003), over-expressing Aurora-A induced a slight accumulation of the 4 N population relative to cells transfected with an empty vector (Figure 3B). A similar accumulation of 4 N cells (25–26%) was found in cells transfected with the kinase-dead or SUMO-deficient Aurora-A mutants. Wild type and kinase-dead Aurora-A forms also induced an increase in the number of cells with DNA content higher than 4 N (11.4% and 15.0%) when compared with the controls (4.4% in untransfected cells and 3.6%, in cells transfected with the empty vector). Interestingly, expressing the Aurora-AK249R mutant induced a significantly higher accumulation (22.3%) of cells with DNA content greater than 4 N. This effect was not a consequence of different expression levels because all Aurora-A isoforms displayed similar levels of expression in these assays (data not shown).
To further investigate the cellular defects induced by the K249R mutation, we generated HeLa cell lines that stably express the wild type, K153M, and K249R isoforms of EGFP-tagged Aurora-A. Three different clones expressing each Aurora-A variant were analyzed under a microscope during mitosis as described in Section “Materials and Methods.” The cells expressing EGFP–Aurora-AK249R showed the highest percentage of mitotic abnormalities (19.28 ± 1.36) compared with EGFP–Aurora-AWT cells (7.29 ± 2.51) or EGFP–Aurora-AK153M cells (8.98 ± 4.70; Figure 3C; left graph). Detailed analysis of the abnormal mitosis occurring in these cells allowed us to identify a specific pattern among the EGFP–Aurora-AK249R expressing cells (Supp videos). Specifically, HeLa cells stably expressing EGFP–Aurora-AK249R showed no cytokinesis phenotypes and more multipolar than monopolar mitoses than did stable EGFP–Aurora-AWT and EGFP–Aurora-AK153M clones (Figure 3C; right graph).
Aurora-AK249R Shows Altered Localization Patterns and Dynamics at the Spindle
During mitosis Aurora-A mainly localizes at spindle poles and along spindle MT. Aurora-A plays a critical role on spindle formation by promoting MT nucleation and stabilization (Sardon et al., 2008). To test whether Aurora-A localization requires SUMOylation at K249, we used HeLa cell lines stably expressing EGFP-tagged Aurora-A variants. As expected, the kinase-dead isoform of Aurora-A (Aurora-AK153M) displayed reduced mitotic spindle localization relative to the wild type form (Figure 4A). Similarly, Aurora-AK249R displayed significantly reduced localization to spindle poles and MT (Figure 4A), indicating that SUMOylation (or some other modifications) at lysine 249 likely plays a role in the proper localization of Aurora-A at the spindle during mitosis.
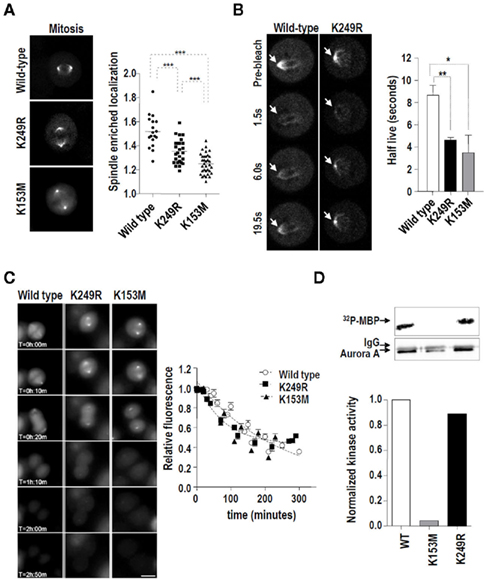
Figure 4. Lack of SUMOylation at K249 alters the dynamics of Aurora-A at the spindle during mitosis without affecting its kinase activity or stability. (A) EGFP-tagged Aurora-A was visualized in HeLa cells stably expressing the wild type, K153M, or K249R variants. EGFP signals were quantified at the spindle (poles and microtubules) and in the rest of the cell. The graph shows the enrichment of EGFP–Aurora-A signal at the spindle [established as the (spindle EGFP signal)/(remaining cellular EGFP signal) ratio] in the stable cell lines. Pictures show representative metaphases for each EGFP-tagged Aurora-A variant. From three different clones of each variant, a total of 18, 26, and 34 mitoses were detected for the wild type, K249R, and K153M variants, respectively ***p < 0.001. (B) The same HeLa cells were used in a series of fluorescence recovery after photobleaching (FRAP) experiments. Representative images of the recovery of Aurora-AWT and Aurora-AK249R at the centrosomes during mitosis are shown. Arrows indicate the spindles (poles and microtubules) that were measured. The graph shows the half-lives obtained using regression analysis to integrate data from 10 cells each. The half-lives of the EGFP–Aurora-A variants at mitotic centrosomes were compared using an unpaired t-test (*p < 0.05, **p < 0.01). (C) The expression of EGFP–Aurora-A variants in HeLa stable clones was measured using time-lapse microscopy. EGFP signals were quantified for 3 h every 10 min from the first frame in which metaphase was detected. Three clones from each different Aurora-A form were analyzed. Five cells from each clone were followed in this experiment. Representative frames and the quantification of the three different variants are shown. (D) In vitro kinase activity toward recombinant MBP of V5-tagged Aurora-A proteins. HEK293 cells were transiently transfected with the indicated V5–Aurora-A expression vectors and the resulting exogenously expressed proteins were immunoprecipitated with α-V5 antibody. The 32P-MBP signal was quantified considering the total amount of V5–Aurora-A protein in each case.
To further characterize the defective localization of Aurora-AK249R, we analyzed the dynamics of this mutant at spindle poles and MTs. FRAP was used to compare the relative mobility of EGFP–Aurora-A variants during interphase and mitosis. Similar recovery rates were observed for wild type and Aurora-AK249R at centrosomes during interphase (Figure A3 in Appendix). During mitosis, wild type EGFP–Aurora-A was found to move rapidly in and out of the spindle during mitosis (Figure 4B; left panels) with a recovery half-life (t1/2) of 8.67 ± 0.9 as previously described (Stenoien et al., 2003). Both the Aurora-AK249R and the Aurora-AK153M mutants displayed shorter t1/2 recovery times (4.60 ± 0.2 and 3.49 ± 1.5 s, respectively; graph in Figure 4B), indicating a more dynamic rate than that of wild type Aurora-A.
Various mechanisms might be responsible for the abnormal dynamics and localization shown by Aurora-AK249R. We first explored the possibility that the mutation at lysine 249 could be affecting the stability of Aurora-A during mitosis. HeLa cell lines stably expressing EGFP–Aurora-A variants (wild type, K249R, and K153M) were followed for 3 h after metaphase by video-microscopy. Quantification of the EGFP signal revealed that all three Aurora-A isoforms showed a similar pattern of expression/degradation from mitosis to the beginning of G1 (Figure 4C). Because Aurora-A localization requires its kinase activity (Sardon et al., 2008), we also tested whether the K249R mutation affects Aurora-A kinase activity. To evaluate the kinase activity of the Aurora-A variants, HeLa cells were transfected with V5-tagged expression vectors for the different Aurora-A variants (WT, K153M, and K249R). Then, the exogenous proteins were immunoprecipitated and tested for their activity toward the substrate MBP. The kinase-dead isoform K135M was not able to efficiently phosphorylate this substrate, the K249R isoform was able to phosphorylate MBP at the same level as the wild type protein (Figure 4D), suggesting no intrinsic deficiencies in kinase activity in this isoform. Thus, we conclude that the abnormal localization and dynamics of the Aurora-AK249R protein is a consequence of neither decreased intrinsic kinase activity nor increased degradation.
Ectopic Expression of Aurora-AK249R Affects Spindle Formation during Mitosis
The major phenotypes resulting from the expression of the Aurora-AK249R form are the formation of monopolar and multipolar spindles (43 and 57% out of the abnormal mitoses vs. 33 and 50% in the case of Aurora-AWT; Figure 3C). As both abnormalities might be due to defects on spindle formation and Aurora-A function has been shown to be crucial to the nucleating capacity of centrosomes during mitosis (Gergely, 2002; Barr and Gergely, 2007), we analyzed microtubule nucleation in HeLa cells stably expressing wild type and variants versions of EGFP-tagged Aurora-A. MTs were depolymerized by incubating the cells on ice for 40 min. Then, the cells were warmed to 37°C and α-tubulin was monitored by immunofluorescence 3 and 10 min later to quantify MT re-growth (Figure 5A). Interestingly, EGFP–Aurora-AK249R expressing cells were not able to re-polymerize MTs as efficiently as cells expressing either EGFP–Aurora-AWT or EGFP–Aurora-AK153M (Figure 5B). This difference between α-tubulin signal detected in Aurora-AWT and Aurora-AK249R cells was significant both 3 and 10 ml after the cells were re-warmed. To analyze the formation of spindles in the different stable cell lines, CEP135 was detected to visualize spindle poles (Figure 5A) and the spindle length was quantified (Figure 5C). HeLa cells stably expressing EGFP–Aurora-AK249R showed shorter spindles than HeLa cells stably expressing either EGFP–Aurora-AWT, EGFP–Aurora-AK153M, or only EGFP. These differences, which were significant at both 3 and 10 min post re-warming, were more pronounced 10 min after the MT were allowed to re-grow (Figure 5C). To gain further support for these results, we studied these EGFP-tagged AurkA stable cell lines by video-microscopy, while they were growing asynchronously. The spindle length of bipolar metaphase cells was established by calculating the distance between poles that were localized by EGFP–AurkA visualization. As in the MT-re-growth experiment, EGFP–Aurora-AK249R cells showed shorter spindles when compared with EGFP–Aurora-AK153M or EGFP–Aurora-AWT cells (Figure 5D). We conclude that over-expression of the K249R Aurora-A protein, but not the wild type or kinase-dead forms, results in a weaker microtubule network and is associated with a defective spindle formation.
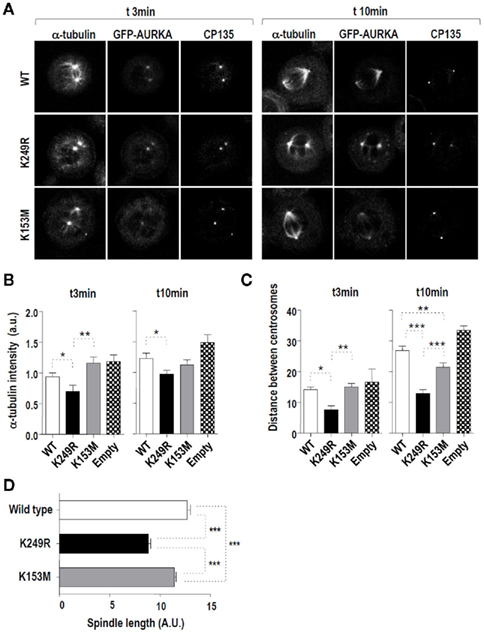
Figure 5. Lack of SUMOylation of Aurora-A affects MT nucleation and spindle formation during mitosis. (A) Analysis of spindle formation in cells stably expressing Aurora-Awt, Aurora-AK249R, or Aurora-AK153M. The cells were incubated with nocodazole for 6 h to enrich the culture for mitotic cells, and MTs were depolymerized by incubating the cells on ice for 40 min. Then, MTs were allowed to grow by re-warming the cells at 37°C, and the cells analyzed for recruitment of endogenous α-tubulin to the spindle and centrosome localization of CEP135 detection. Representative images taken at 3 and 10 min post re-warming. Quantification and statistical analysis of the results of this experiment are shown in (B,C). (B) Graphs show the quantification of α-tubulin signal obtained at the spindle, as described in Section “Materials and Methods,” from each EGFP-Aurora-A cell line. Two different clones of the control (empty) and Aurora-A variants (WT, K153M, and K249R) were analyzed. A total of 36 cells were studied for each type. For the two time point analyzed (3 and 10 min post re-warming) α-tubulin signal was lower among the Aurora-AK249R cells than in the Aurora-AWT ones. (C) Graphs display spindle length in the same HeLa cells that were studied in (B). Spindle length was scored as the distance between the two major CEP135 signals in each mitotic cell. At 3 and 10 min after re-warming, the distance between spindle poles was significantly shorter in Aurora-AK249R expressing cells when compared with wild type and kinase-dead cell lines. (D) Spindle length was also analyzed in the same EGFP–Aurora-A cell lines growing asynchronously. Spindle length was determined as the distance between the two major EGFP–Aurora-A signals detected at metaphase, which should be associated with spindle poles. Three clones from each Aurora-A variant were studied. A total of 28, 42, and 42 mitosis were analyzed for the wild type, K249R, and K153M variants, respectively. Again, the spindle length observed in the Aurora-AK249R cells was shorter than that in the stable cell lines expressing wild type or kinase-dead Aurora-A variants. In all the cases data from a minimum of 20 EGFP-positive cells were compared using unpaired t-tests. Significant differences are indicated as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Aurora-AK249R Displays Altered Tumorigenic and Apoptotic Properties
Ectopic over-expression of Aurora-A allows Rat1 fibroblasts to grow in soft agar (Bischoff et al., 1998) and cooperates with H-Ras in the transformation of mouse fibroblasts (Tatsuka et al., 2005). To examine if lysine 249 has any effect on the role of Aurora-A in inducing the malignant phenotype, we performed a series of focus-formation experiments co-transfecting an oncogenic form of H-Ras with Aurora-A variants. As previously reported (Tatsuka et al., 2005), the wild type form of Aurora-A potentiates H-RasV12-induced transformation (Figure 6A). Importantly, co-transfection of H-RasV12 with the Aurora-AK249R variant significantly increased the number of foci when compared with the wild type form (1.7 fold induction). This increased malignant phenotype was not due to different levels of expression of the exogenous Aurora-A variants used in this experiment (Figure A4 in Appendix). Given the importance of Aurora-A in tumorigenesis, we wanted to ascertain the cause of this difference between the over-expression of Aurora-AK249R and Aurora-AWT. Because Aurora-AK249R cells did not show significant differences when compared with Aurora-AWT expressing cells in colony formation assays (data not shown), we discarded differential proliferation efficiency as the cause of the more aggressive malignant phenotype. We then explored the possibility that Aurora-AK249R may induce high levels of genomic instability as also suggested by the increase in mitotic abnormalities observed upon Aurora-AK249R expression (Figure 3). The number of chromosomes per cell was determined in metaphase cells from foci obtained upon co-transfection of H-RasV12 and Aurora-A. The control mouse fibroblasts used in this experiment had a diploid/tetraploid, mixed karyotype showing an average chromosome number of 48.04 ± 2.38 chromosomes (Figure 6B). Foci obtained upon H-RasV12 transfection significantly differed from those transfected with the empty vector (average chromosome number of 77.75 ± 2.37; p < 0.001). Foci from H-RasV12/Aurora-AWT co-transfected cells showed chromosome numbers (83.71 ± 3.22) significantly higher than those observed in the control cells (p < 0.0001), but they showed no statistical differences when compared with the foci transfected only with H-Ras V12 (p = 0.155). Interestingly, co-transfection with the K249R form of Aurora-A induced the most dramatic aneuploid phenotype. Foci from H-RasV12/Aurora-AK249R co-transfected cells showed an average chromosome number of 97.96 ± 5.07, which significantly differed from the average chromosome numbers found among control cells (p < 0.0001), foci transfected with H-Ras V12 (p = 0.0003) and foci co-transfected with H-Ras V12 and Aurora-AWT (p = 0.0143). These results suggested that the over-expression of Aurora-AK249R cooperates with H-RasV12 in the transformation of mouse fibroblasts by inducing significantly high levels of aneuploidy.
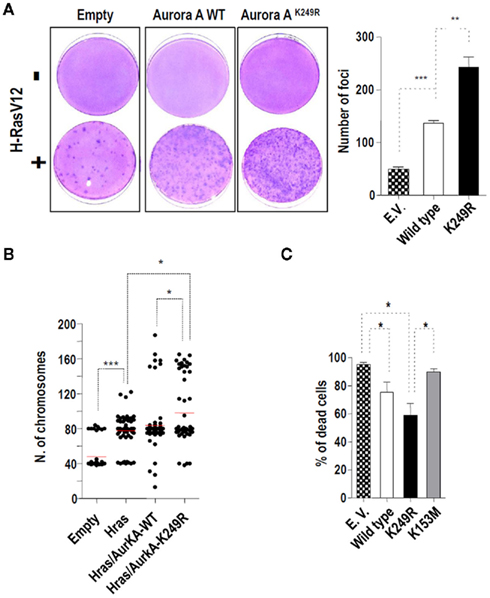
Figure 6. Aurora-A SUMOylation and tumorigenesis. (A) Effects of Aurora-AK249R over-expression on H-RAS-induced transformation in NIH-3T3 cells. Control and Aurora-A expression vectors were co-transfected with an activated G12V–H-RAS vector. Forty-eight hours, post-transfection, the cells were split one to three and kept in culture with 5% calf serum. Two weeks later, dishes were fixed with ethanol and stained with crystal violet. Representative images are shown for each point. A bar graph shows the quantification of the number of foci. (B) Lack of SUMOylation of Aurora-A at lysine 249 increases aneuploidy in H-RAS-transformed mouse fibroblasts. Chromosomes were counted in a minimum of 20 metaphase cells from foci obtained upon transfection with the indicated expression plasmids. Three different foci were analyzed per each transfection type. (C) Lack of SUMOylation increases the taxol chemoresistance induced by Aurora-A over-expression in HeLa cells. HeLa cells stably expressing a control empty vector or EGFP-tagged Aurora-A variants were treated with 150 nM taxol for 48 h. At the end of the treatment, the cells were incubated with propidium iodide (0.02 mg/ml) to determine the number of dead cells (positive for PI). A bar graph shows the quantification for three different clones of each type in one of the two experiments carried out. In all the previous graphs, significant differences are indicated as follows: *p < 0.05, **p < 0.01, and ***p < 0.001.
Finally, we explored the effect of expressing the Aurora-A mutants on the reported resistance to taxol induced by Aurora-A over-expression (Anand et al., 2003). We treated three HeLa clones stably expressing three different EGFP–Aurora-A variants at different levels (Figure A2 in Appendix) with taxol (150 nM) for 48 h. The percentage of apoptotic cells was then measured using propidium iodide (PI) staining and flow cytometry to quantify the PI positive (dead) population. As shown in Figure 6C, stable over-expression of Aurora-A induces a significant resistance to taxol-induced apoptosis, as previously reported for the transient over-expression of wild type Aurora-A in HeLa cells (Anand et al., 2003). Over-expression of the K249R mutant is also associated with a significant protection from the Taxol-induced death (Figure 6C). Interestingly, the protective phenotype is stronger, although not significant (p = 0.203), in Aurora-AK249R over-expressing cells than in those over-expressing Aurora-AWT. On the other hand, the kinase-dead-mutant did not induce the resistance phenotype because the cells stably expressing Aurora-AK153M did not differ from the control cells stably transfected with an empty vector. The percentage of dead cells was significantly different in the Aurora-AK153M and Aurora-AK249R clones suggesting that the kinase activity of the protein is necessary for the K249R Aurora-A form to induce the resistance to taxol-induced apoptosis.
In summary, we conclude that the lysine to arginine mutation at residue 249 of mouse Aurora-A is associated with a high rate of mitotic abnormalities that in turn induces segregation errors that result in severe aneuploidy and cooperate with H-RAS in the induction of malignancy. Furthermore, our data suggest that the over-expression of K249R Aurora-A may protect cells from apoptotic responses to antimitotic poisons such as Taxol.
Discussion
SUMOylation is a reversible process involved in the regulation of multiple protein functions. This post-translational modification affects the sub-cellular localization, activity, and/or stability of an increasing number of substrates. Increasing evidence in recent years has associated SUMOylation with the regulation of multiple cellular processes including meiosis and mitosis (De Carvalho and Colaiacovo, 2006; Dasso, 2008). SUMO peptide modifications are involved in the nucleocytoplasmic shuttling of centrin-2 (Klein and Nigg, 2009), the cell cycle-dependent regulation of Borealin (Klein et al., 2009), the association of CENP-E with kinetochores (Zhang et al., 2008b), mitotic spindle asymmetry and the spindle-assembly checkpoint in yeast via Kar9 (Leisner et al., 2008), and the resolution of sister centromeres by topoisomerase II alpha (Dawlaty et al., 2008). Here, we report that Aurora-A kinase interacts with UBC9, the only E2 conjugating enzyme of the SUMOylation pathway. We observed the interaction of these two proteins both in the two-hybrid system and in biochemical assays in mammalian cells. SUMOylation seems to affect only a small fraction of Aurora-A (Figures 2C,D) as also reported for Aurora-B (Fernandez-Miranda et al., 2010) and many other targets (Geiss-Friedlander and Melchior, 2007).
Aurora-A contains a highly conserved SUMO target site that is also present in other Aurora family members. Indeed, we and others previously observed that modification by SUMO plays a critical role in the regulation of Aurora-B function (Fernandez-Miranda et al., 2010; Ban et al., 2011). Aurora-B can bind SUMO peptides in vitro and the expression of a SUMO-null form of Aurora-B results in abnormal chromosome segregation and failed cytokinesis. We previously proposed that SUMOylation of Aurora-B modulates its function by mediating the extraction of chromosome passenger complexes from chromosome arms during prometaphase (Fernandez-Miranda et al., 2010). Here, we show that the expression of an Aurora-A that cannot be SUMOylated at lysine 249 induces a significant increase in the number of mitotic abnormalities found among proliferating HeLa cells. These Aurora-AK249R-induced mitotic aberrations are specifically enriched in multipolar spindles suggesting that spindle regulation is defective. Aurora-A, UBC9, and SUMO1 all decorate both centrosomes and the mitotic spindle, which favor the hypothesis that these proteins specifically interact in these sub-cellular structures regulating spindle function. Moreover, Aurora-AK249R expressing cells are characterized by an abnormal α-tubulin expression pattern in MT-re-growth experiments and by defective formation of the spindle. The role of Aurora-A in centrosome function and spindle assembly has been widely established in Drosophila, C. elegans, Xenopus egg extracts, and human cells (Hannak et al., 2001; Giet et al., 2002; Andrews et al., 2003). A recent study performed in Xenopus egg extract has demonstrated that Aurora-A is necessary for the generation of a proper spindle through its role at the centrosome of recruiting factors involved in microtubule nucleation and stabilization (Sardon et al., 2008).
However, the identity of the Aurora-A partner or substrate involved in the abnormal spindle formation induced upon over-expression of the Aurora-AK249R mutant remain unknown. MCAK and ch-TOG (also known as CKAP5) emerge as two main candidates. MCAK, a negative regulator of MT plus end growth is regulated at the centrosomes by Aurora-A phosphorylation to control Ran-dependent spindle formation (Zhang et al., 2008a). On the other hand, Aurora-A phosphorylates and localizes TACC3 to spindle poles (Kinoshita et al., 2005; Leroy et al., 2007), which is necessary for ch-TOG localization to spindle MTs (Gergely et al., 2003). Moreover, both MCAK and ch-TOG abnormally accumulate at the spindle of Aurora-A depleted cells (De Luca et al., 2008). All these preliminary results indicate that Aurora-A, MCAK, and ch-TOG are part of a common pathway that controls spindle formation and integrity. How the SUMO modification of Aurora-A is involved in the regulation of this pathway is still an open question. One attractive hypothesis could be that the SUMO modification allows Aurora-A to anchor more firmly to spindle MTs by interacting with the SUMO interacting motifs (SIMs) that are predicted to be present in the ch-TOG sequence (unpublished data). Although we believe that the possibility that ch-TOG acts as a docking platform for SUMOylated Aurora-A deserves further investigation, other possible mechanistic explanations for the abnormal phenotype induced by Aurora-AK249R should not be ruled out. For instance, it is also possible that the SUMOylation of Aurora-A modulates its kinase activity. Indeed, some of the phenotypes induced by the Aurora-AK249R mutant were coincident with the ones induced by the kinase-dead Aurora-A variant; e.g., the altered localization and dynamics at the spindle during mitosis (Figures 4A,B). However, similarly to what has been reported for Aurora-B (Fernandez-Miranda et al., 2010), the Aurora-AK249R mutant does not display reduced kinase activity (Figure 4D) and differs from the kinase-dead in many other phenotypes induced by their over-expression (MT nucleation and spindle length among others). It is also possible that the K249R mutation is affecting Aurora-A function through a mechanism other than SUMO modification. For instance, this mutation could affect the structure of the protein or modification by other post-translational regulators such as ubiquitination. However, the predicted structures for both wild type Aurora-A and K249R do not differ in any of the major catalytic or regulatory domains of this kinase (unpublished results). In addition, the Ubc9–Aurora-A relationship and the actual SUMOylation of Aurora-A described here together with the fact that this residue is highly conserved and regulated by SUMO in other Aurora family members (Fernandez-Miranda et al., 2010) favor the idea that SUMOylation, rather than ubiquitination, is the post-translational modification affecting lysine 249. All these considerations suggest that the functional defects induced by the expression of the Aurora-AK249R protein are likely a consequence of the lack of SUMO modification of this specific residue, which in turn alters the localization of Aurora-A.
Given the fact that SUMOylation is involved in a variety of cellular processes, links between this post-translational modification and cancer and metastasis can be postulated. In fact, the SUMO pathway has been described to be associated with tumorigenesis at multiple levels (Katayama et al., 2003; Seeler et al., 2007). Increased levels of UBC9, the SUMO E3 protein PIAS3, the SUMO E1 enzyme, and the SUMO protease SENP1 are found in several human cancers and, in some cases, they are associated with lower survival rates (Mcdoniels-Silvers et al., 2002; Lee and Thorgeirsson, 2004; Wang and Banerjee, 2004; Jacques et al., 2005; Mo et al., 2005; Cheng et al., 2006). The association of both pro-SUMOylation and pro-deSUMOylation molecules with cancer indicates a complex relationship between this post-translational modification network and tumorigenesis. Indeed, SUMOylation can regulate the activities of both tumor suppressor proteins, including p53, pRB, p63, and p73, and oncogenes such as murine double minute 2 (Mdm2), PML, c-myc, and Wnt-1 (Seeler et al., 2007). Our results further support a role of SUMOylation in the acquisition of malignant properties through the modification of the putative oncogene Aurora-A. First, blocking SUMOylation by mutating lysine 249 is associated with higher susceptibility to cellular transformation by RAS oncogenes. Second, lack of SUMOylation of Aurora-A at lysine 249 results in decreased apoptosis in the presence of mitotic poisons.
Taken together, our data suggest that Aurora-A is modified by SUMO residues through its interaction with UBC9. Conjugation of SUMO residues at lysine 249 may help to control the localization of this kinase at the spindle. This modification in turn modulates the biological function of Aurora-A in a signaling pathway that leads to proper MT nucleation and spindle formation during mitosis. This new regulatory mechanism may have relevant implications in both tumor development and the cellular responses to mitotic poisons currently used in cancer therapy.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Susana Temiño for her technical assistance. Gonzalo Fernández-Miranda was supported by a fellowship from the Comunidad de Madrid. We also want to thank Gulnahar Mortuza and Pilar Redondo (from the Macromolecular Crystallography Group at the CNIO) for the production of the AURKA–K258R recombinant protein. Cristina Aguirre-Portolés was supported by a fellowship from the Spanish Ministry of Science and Innovation. Ignacio Pérez de Castro was supported by a Ramón y Cajal contract. This work was funded by grants from the Association for International Cancer Research (AICR #08-0188), Fundación Ramón Areces, Spanish Ministry of Science and Innovation (MICINN; SAF2010-19710 to Ignacio Pérez de Castro; and SAF2009-07973 to Marcos Malumbres) and by the Canceropole Grand Ouest, the Institut Federatif de Recherches (IFR-Université Rennes), and the COFECUB program. Part of the microscopy work was performed on the IFR140 GFAS fluorescence microscopy platform in Rennes. The Cell Division and Cancer Group of the CNIO is supported by the OncoCycle Programme (S-BIO-0283-2006) from the Comunidad de Madrid, the OncoBIO Consolider-Ingenio 2010 Programme (CSD2007-00017; MICINN), and the MitoSys project (HEALTH-F5-2010-241548; European Union Seventh Framework Program).
Footnotes
References
Anand, S., Penrhyn-Lowe, S., and Venkitaraman, A. R. (2003). AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell 3, 51–62.
Andrews, P. D., Knatko, E., Moore, W. J., and Swedlow, J. R. (2003). Mitotic mechanics: the auroras come into view. Curr. Opin. Cell Biol. 15, 672–683.
Arlot-Bonnemains, Y., Klotzbucher, A., Giet, R., Uzbekov, R., Bihan, R., and Prigent, C. (2001). Identification of a functional destruction box in the Xenopus laevis aurora-A kinase pEg2. FEBS Lett. 508, 149–152.
Ayaydin, F., and Dasso, M. (2004). Distinct in vivo dynamics of vertebrate SUMO paralogues. Mol. Biol. Cell 15, 5208–5218.
Azuma, Y., Arnaoutov, A., and Dasso, M. (2003). SUMO-2/3 regulates topoisomerase II in mitosis. J. Cell Biol. 163, 477–487.
Ban, R., Nishida, T., and Urano, T. (2011). Mitotic kinase Aurora-B is regulated by SUMO-2/3 conjugation/deconjugation during mitosis. Genes Cells 16, 652–669.
Barr, A. R., and Gergely, F. (2007). Aurora-A: the maker and breaker of spindle poles. J. Cell. Sci. 120, 2987–2996.
Bird, A. W., and Hyman, A. A. (2008). Building a spindle of the correct length in human cells requires the interaction between TPX2 and Aurora A. J. Cell Biol. 182, 289–300.
Bischoff, J. R., Anderson, L., Zhu, Y., Mossie, K., Ng, L., Souza, B., Schryver, B., Flanagan, P., Clairvoyant, F., Ginther, C., Chan, C. S., Novotny, M., Slamon, D. J., and Plowman, G. D. (1998). A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 17, 3052–3065.
Breeden, L., and Nasmyth, K. (1985). Regulation of the yeast HO gene. Cold Spring Harb. Symp. Quant. Biol. 50, 643–650.
Carmena, M., and Earnshaw, W. C. (2003). The cellular geography of aurora kinases. Nat. Rev. Mol. Cell Biol. 4, 842–854.
Castro, A., Arlot-Bonnemains, Y., Vigneron, S., Labbe, J. C., Prigent, C., and Lorca, T. (2002). APC/Fizzy-Related targets Aurora-A kinase for proteolysis. EMBO Rep. 3, 457–462.
Cheng, J., Bawa, T., Lee, P., Gong, L., and Yeh, E. T. (2006). Role of desumoylation in the development of prostate cancer. Neoplasia 8, 667–676.
Dawlaty, M. M., Malureanu, L., Jeganathan, K. B., Kao, E., Sustmann, C., Tahk, S., Shuai, K., Grosschedl, R., and Van Deursen, J. M. (2008). Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 133, 103–115.
De Carvalho, C. E., and Colaiacovo, M. P. (2006). SUMO-mediated regulation of synaptonemal complex formation during meiosis. Genes Dev. 20, 1986–1992.
De Luca, M., Brunetto, L., Asteriti, I. A., Giubettini, M., Lavia, P., and Guarguaglini, G. (2008). Aurora-A and ch-TOG act in a common pathway in control of spindle pole integrity. Oncogene 27, 6539–6549.
Ewart-Toland, A., Briassouli, P., De Koning, J. P., Mao, J. H., Yuan, J., Chan, F., Maccarthy-Morrogh, L., Ponder, B. A., Nagase, H., Burn, J., Ball, S., Almeida, M., Linardopoulos, S., and Balmain, A. (2003). Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nat. Genet. 34, 403–412.
Eyers, P. A., Erikson, E., Chen, L. G., and Maller, J. L. (2003). A novel mechanism for activation of the protein kinase Aurora A. Curr. Biol. 13, 691–697.
Fernandez-Miranda, G., Perez De Castro, I., Carmena, M., Aguirre-Portoles, C., Ruchaud, S., Fant, X., Montoya, G., Earnshaw, W. C., and Malumbres, M. (2010). SUMOylation modulates the function of Aurora-B kinase. J. Cell. Sci. 123, 2823–2833.
Geiss-Friedlander, R., and Melchior, F. (2007). Concepts in sumoylation: a decade on. Nat. Rev. Mol. Cell Biol. 8, 947–956.
Gergely, F., Draviam, V. M., and Raff, J. W. (2003). The ch-TOG/XMAP215 protein is essential for spindle pole organization in human somatic cells. Genes Dev. 17, 336–341.
Giet, R., Mclean, D., Descamps, S., Lee, M. J., Raff, J. W., Prigent, C., and Glover, D. M. (2002). Drosophila Aurora A kinase is required to localize D-TACC to centrosomes and to regulate astral microtubules. J. Cell Biol. 156, 437–451.
Gritsko, T. M., Coppola, D., Paciga, J. E., Yang, L., Sun, M., Shelley, S. A., Fiorica, J. V., Nicosia, S. V., and Cheng, J. Q. (2003). Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin. Cancer Res. 9, 1420–1426.
Hannak, E., Kirkham, M., Hyman, A. A., and Oegema, K. (2001). Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J. Cell Biol. 155, 1109–1116.
Jacques, C., Baris, O., Prunier-Mirebeau, D., Savagner, F., Rodien, P., Rohmer, V., Franc, B., Guyetant, S., Malthiery, Y., and Reynier, P. (2005). Two-step differential expression analysis reveals a new set of genes involved in thyroid oncocytic tumors. J. Clin. Endocrinol. Metab. 90, 2314–2320.
Jiang, Y., Zhang, Y., Lees, E., and Seghezzi, W. (2003). AuroraA overexpression overrides the mitotic spindle checkpoint triggered by nocodazole, a microtubule destabilizer. Oncogene 22, 8293–8301.
Katayama, H., Brinkley, W. R., and Sen, S. (2003). The Aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev. 22, 451–464.
Kinoshita, K., Noetzel, T. L., Pelletier, L., Mechtler, K., Drechsel, D. N., Schwager, A., Lee, M., Raff, J. W., and Hyman, A. A. (2005). Aurora A phosphorylation of TACC3/maskin is required for centrosome-dependent microtubule assembly in mitosis. J. Cell Biol. 170, 1047–1055.
Kitajima, S., Kudo, Y., Ogawa, I., Tatsuka, M., Kawai, H., Pagano, M., and Takata, T. (2007). Constitutive phosphorylation of aurora-a on ser51 induces its stabilization and consequent overexpression in cancer. PLoS ONE 2, e944. doi:10.1371/journal.pone.0000944
Klein, U. R., Haindl, M., Nigg, E. A., and Muller, S. (2009). RanBP2 and SENP3 function in a mitotic SUMO2/3 conjugation-deconjugation cycle on Borealin. Mol. Biol. Cell 20, 410–418.
Klein, U. R., and Nigg, E. A. (2009). SUMO-dependent regulation of centrin-2. J. Cell Sci. 122, 3312–3321.
Lee, J. S., and Thorgeirsson, S. S. (2004). Genome-scale profiling of gene expression in hepatocellular carcinoma: classification, survival prediction, and identification of therapeutic targets. Gastroenterology 127, S51–S55.
Leisner, C., Kammerer, D., Denoth, A., Britschi, M., Barral, Y., and Liakopoulos, D. (2008). Regulation of mitotic spindle asymmetry by SUMO and the spindle-assembly checkpoint in yeast. Curr. Biol. 18, 1249–1255.
Lens, S. M., Voest, E. E., and Medema, R. H. (2010). Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 10, 825–841.
Leroy, P. J., Hunter, J. J., Hoar, K. M., Burke, K. E., Shinde, V., Ruan, J., Bowman, D., Galvin, K., and Ecsedy, J. A. (2007). Localization of human TACC3 to mitotic spindles is mediated by phosphorylation on Ser558 by Aurora A: a novel pharmacodynamic method for measuring Aurora A activity. Cancer Res. 67, 5362–5370.
Li, D., Zhu, J., Firozi, P. F., Abbruzzese, J. L., Evans, D. B., Cleary, K., Friess, H., and Sen, S. (2003). Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin. Cancer Res. 9, 991–997.
Littlepage, L. E., Wu, H., Andresson, T., Deanehan, J. K., Amundadottir, L. T., and Ruderman, J. V. (2002). Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc. Natl. Acad. Sci. U.S.A. 99, 15440–15445.
Marumoto, T., Zhang, D., and Saya, H. (2005). Aurora-A – a guardian of poles. Nat. Rev. Cancer 5, 42–50.
Mcdoniels-Silvers, A. L., Nimri, C. F., Stoner, G. D., Lubet, R. A., and You, M. (2002). Differential gene expression in human lung adenocarcinomas and squamous cell carcinomas. Clin. Cancer Res. 8, 1127–1138.
Meraldi, P., Honda, R., and Nigg, E. A. (2002). Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J. 21, 483–492.
Miyoshi, Y., Iwao, K., Egawa, C., and Noguchi, S. (2001). Association of centrosomal kinase STK15/BTAK mRNA expression with chromosomal instability in human breast cancers. Int. J. Cancer 92, 370–373.
Mo, Y. Y., Yu, Y., Theodosiou, E., Ee, P. L., and Beck, W. T. (2005). A role for Ubc9 in tumorigenesis. Oncogene 24, 2677–2683.
Moreno-Bueno, G., Sanchez-Estevez, C., Cassia, R., Rodriguez-Perales, S., Diaz-Uriarte, R., Dominguez, O., Hardisson, D., Andujar, M., Prat, J., Matias-Guiu, X., Cigudosa, J. C., and Palacios, J. (2003). Differential gene expression profile in endometrioid and nonendometrioid endometrial carcinoma: STK15 is frequently overexpressed and amplified in nonendometrioid carcinomas. Cancer Res. 63, 5697–5702.
Perez De Castro, I., De Carcer, G., Montoya, G., and Malumbres, M. (2008). Emerging cancer therapeutic opportunities by inhibiting mitotic kinases. Curr. Opin. Pharmacol. 8, 375–383.
Roghi, C., Giet, R., Uzbekov, R., Morin, N., Chartrain, I., Le Guellec, R., Couturier, A., Doree, M., Philippe, M., and Prigent, C. (1998). The Xenopus protein kinase pEg2 associates with the centrosome in a cell cycle-dependent manner, binds to the spindle microtubules and is involved in bipolar mitotic spindle assembly. J. Cell. Sci. 111(Pt 5), 557–572.
Royce, M. E., Xia, W., Sahin, A. A., Katayama, H., Johnston, D. A., Hortobagyi, G., Sen, S., and Hung, M. C. (2004). STK15/Aurora-A expression in primary breast tumors is correlated with nuclear grade but not with prognosis. Cancer 100, 12–19.
Sakakura, C., Hagiwara, A., Yasuoka, R., Fujita, Y., Nakanishi, M., Masuda, K., Shimomura, K., Nakamura, Y., Inazawa, J., Abe, T., and Yamagishi, H. (2001). Tumour-amplified kinase BTAK is amplified and overexpressed in gastric cancers with possible involvement in aneuploid formation. Br. J. Cancer 84, 824–831.
Sardon, T., Peset, I., Petrova, B., and Vernos, I. (2008). Dissecting the role of Aurora A during spindle assembly. EMBO J. 27, 2567–2579.
Seeler, J. S., Bischof, O., Nacerddine, K., and Dejean, A. (2007). SUMO, the three Rs and cancer. Curr. Top. Microbiol. Immunol. 313, 49–71.
Sen, S., Zhou, H., and White, R. A. (1997). A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene 14, 2195–2200.
Sen, S., Zhou, H., Zhang, R. D., Yoon, D. S., Vakar-Lopez, F., Ito, S., Jiang, F., Johnston, D., Grossman, H. B., Ruifrok, A. C., Katz, R. L., Brinkley, W., and Czerniak, B. (2002). Amplification/overexpression of a mitotic kinase gene in human bladder cancer. J. Natl. Cancer Inst. 94, 1320–1329.
Smith, D. B., and Johnson, K. S. (1988). Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67, 31–40.
Stenoien, D. L., Sen, S., Mancini, M. A., and Brinkley, B. R. (2003). Dynamic association of a tumor amplified kinase, Aurora-A, with the centrosome and mitotic spindle. Cell Motil. Cytoskeleton 55, 134–146.
Taguchi, S., Honda, K., Sugiura, K., Yamaguchi, A., Furukawa, K., and Urano, T. (2002). Degradation of human Aurora-A protein kinase is mediated by hCdh1. FEBS Lett. 519, 59–65.
Tatham, M. H., Rodriguez, M. S., Xirodimas, D. P., and Hay, R. T. (2009). Detection of protein SUMOylation in vivo. Nat. Protoc. 4, 1363–1371.
Tatsuka, M., Sato, S., Kitajima, S., Suto, S., Kawai, H., Miyauchi, M., Ogawa, I., Maeda, M., Ota, T., and Takata, T. (2005). Overexpression of Aurora-A potentiates HRAS-mediated oncogenic transformation and is implicated in oral carcinogenesis. Oncogene 24, 1122–1127.
Wang, L., and Banerjee, S. (2004). Differential PIAS3 expression in human malignancy. Oncol. Rep. 11, 1319–1324.
Zhang, X., Ems-Mcclung, S. C., and Walczak, C. E. (2008a). Aurora A phosphorylates MCAK to control ran-dependent spindle bipolarity. Mol. Biol. Cell 19, 2752–2765.
Zhang, X. D., Goeres, J., Zhang, H., Yen, T. J., Porter, A. C., and Matunis, M. J. (2008b). SUMO-2/3 modification and binding regulate the association of CENP-E with kinetochores and progression through mitosis. Mol. Cell 29, 729–741.
Zhou, H., Kuang, J., Zhong, L., Kuo, W. L., Gray, J. W., Sahin, A., Brinkley, B. R., and Sen, S. (1998). Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 20, 189–193.
Appendix
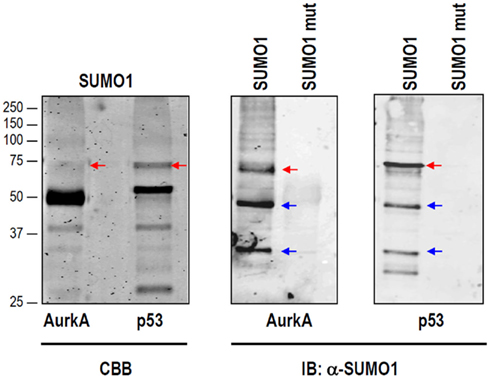
Figure A1. Non-specific SUMO1-reactive bands are detected upon in vitro SUMOylation. In vitro SUMO1 reactions were performed for Aurora-A and p53 as in Figure 2C. In both cases, the resulting bands were visualized by Coomassie (CBB) staining (left panel). Immunoblotting using an anti-SUMO1 antibody detects a number of common bands (blue arrows) that do not correspond to the expected SUMOylated bands (red arrows).
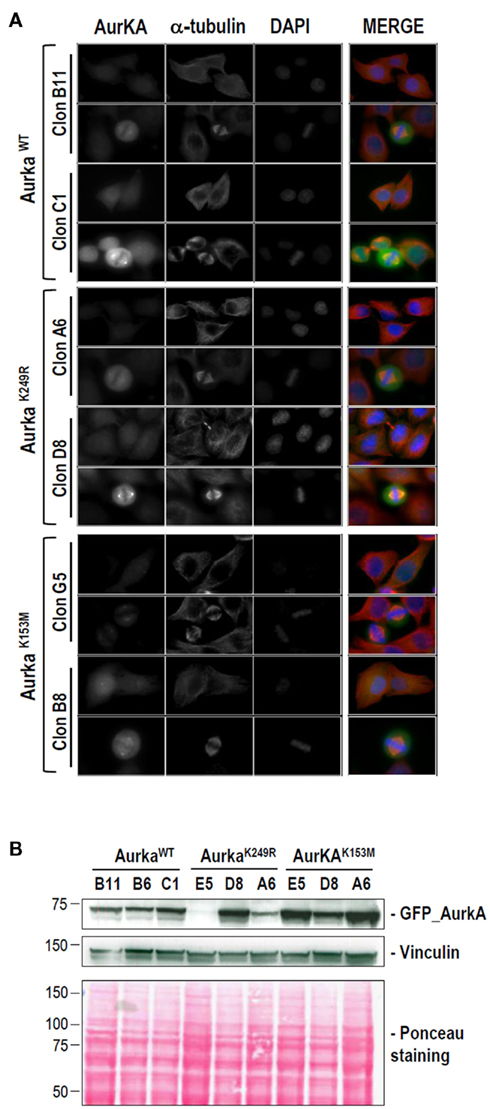
Figure A2. Stable HeLa cell lines used in this work for the analysis of K249. EGFP-tagged expression vectors were used to transfect wild-type, K249R, and K153M Aurora-A into HeLa cells. G418-resistant, EGFP-positive colonies were expanded and Aurora-A exogenous expression was established by western blotting and immunofluorescence. (A) Representative clones for the expression of high and low levels of EGFP–Aurora-A (green) variants. DNA (blue) and α-tubulin (red) are also shown. Mouse monoclonal anti-α-tubulin (clone DM1A) from SIGMA was used at 1/1000. (B) Detection of EGFP–Aurora-A proteins in stable cell lines by immunoblotting. Three different clones for each Aurora-A variant are shown. Vinculin detection and Ponceau staining are shown as loading controls. The antibodies and dilutions used were as follows: anti-EGFP (Roche, mouse monoclonal; clones 7.1 plus 13.1) at 1/1000 and anti-vinculin (SIGMA mouse monoclonal; clone hVIN-1) at 1/1000.
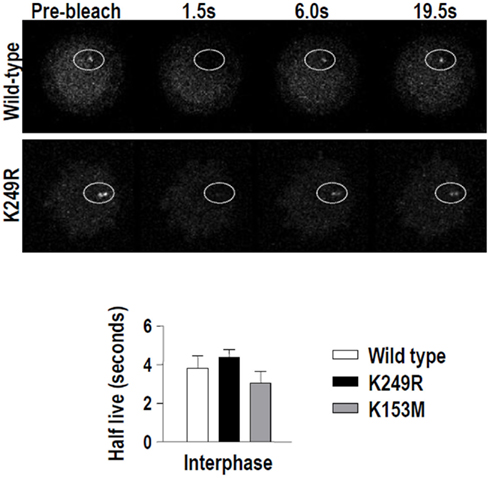
Figure A3. Lack of SUMOylation at K249 does not alter Aurora-A dynamics at centrosomes during interphase. HeLa cells stably expressing EGP–AurkA variants were used in a series of fluorescence recovery after photobleaching (FRAP) experiments. The upper panels show representative images of Aurora-AWT and Aurora-AK249R recovery at centrosomes during interphase. A graph shows the half-lives obtained using regression analysis to integrate data from 10 cells each. The half-lives of EGFP–Aurora-A variants at interphase centrosomes were not different when compared using a unpaired t-test.
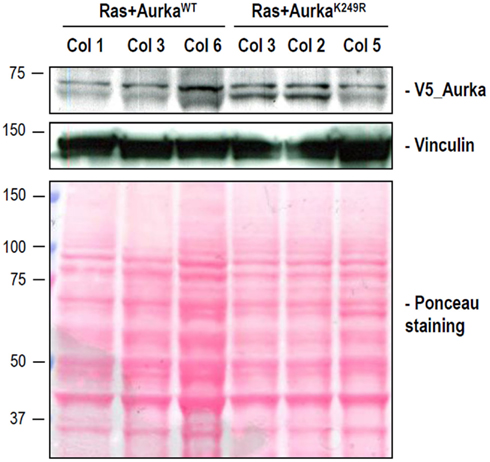
Figure A4. Foci induced upon Ras plus Aurora-A transfection show similar expression levels of exogenous Aurora-A. Detection of V5-Aurora-A proteins in stable cell lines by immunoblotting is shown for three different foci of the indicated types. Anti-V5 tag (Invitrogen, mouse monoclonal #46-0705) was used to detect exogenous Aurora-A fused to V5. Vinculin detection and Ponceau staining are shown as loading controls.
Keywords: mitosis, kinases, Aurora-A, SUMO, spindle, cancer
Citation: Pérez de Castro I, Aguirre-Portolés C, Martin B, Fernández-Miranda G, Klotzbucher A, Kubbutat MHG, Megías D, Arlot-Bonnemains Y and Malumbres M (2011) A SUMOylation motif in Aurora-A: implications for spindle dynamics and oncogenesis. Front. Oncol. 1:50. doi: 10.3389/fonc.2011.00050
Received: 22 June 2011;
Accepted: 22 November 2011;
Published online: 14 December 2011.
Edited by:
Hugues De Thé, University of Paris, FranceReviewed by:
Stephen Tait, St. Jude Children’s Research Hospital, USAGuillaume Bossis, CNRS, France
Mar Carmena, The University of Edinburgh, UK
Susanne Lens, University Medical Center Utrecht, Netherlands
Copyright: © 2011 Pérez de Castro, Aguirre-Portolés, Martin, Fernández-Miranda, Klotzbucher, Kubbutat, Megías, Arlot-Bonnemains and Malumbres. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ignacio Pérez de Castro and Marcos Malumbres, Centro Nacional de Investigaciones Oncológicas, Melchor Fernández Almagro 3, E-28029 Madrid, Spain. e-mail:bW1tQGNuaW8uZXM=;aXBlcmV6QGNuaW8uZXM=
†Yannick Arlot-Bonnemains and Marcos Malumbres have contributed equally to this work.