 Howard Donninger1 Thibaut Barnoud1 Nick Nelson1 Suzanna Kassler1 Jennifer Clark1 Timothy D. Cummins2 David W. Powell2 Sarah Nyante3 Robert C. Millikan3
Howard Donninger1 Thibaut Barnoud1 Nick Nelson1 Suzanna Kassler1 Jennifer Clark1 Timothy D. Cummins2 David W. Powell2 Sarah Nyante3 Robert C. Millikan3 Geoffrey J. Clark1*
Geoffrey J. Clark1*- 1 Molecular Targets Program, Department of Medicine, James Graham Brown Cancer Center, University of Louisville, Louisville, KY, USA
- 2 Department of Biochemistry and Molecular Biology, University of Louisville, Louisville, KY, USA
- 3 The University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
RASSF1A is one of the most frequently inactivated tumor suppressors yet identified in human cancer. It is pro-apoptotic and appears to function as a scaffolding protein that interacts with a variety of other tumor suppressors to modulate their function. It can also complex with the Ras oncoprotein and may serve to integrate pro-growth and pro-death signaling pathways. A SNP has been identified that is present in approximately 29% of European populations [rs2073498, A(133)S]. Several studies have now presented evidence that this SNP is associated with an enhanced risk of developing breast cancer. We have used a proteomics based approach to identify multiple differences in the pattern of protein/protein interactions mediated by the wild type compared to the SNP variant protein. We have also identified a significant difference in biological activity between wild type and SNP variant protein. However, we have found only a very modest association of the SNP with breast cancer predisposition.
Introduction
RASSF1A is a tumor suppressor that suffers epigenetic inactivation in over 50% of human tumors (Dammann et al., 2005; Donninger et al., 2007; van der and Adams, 2007). Knockout RASSF1A mice demonstrate an enhanced predisposition to develop spontaneous tumors (Dammann et al., 2000) and this effect is amplified in a p53 null mouse background (Tommasi et al., 2011). RASSF1A is pro-apoptotic and can induce cell cycle arrest in G2 and in G1 (Shivakumar et al., 2002; Vos et al., 2004, 2006). RASSF1A has no enzymatic activity, but appears to act by modulating microtubule polymerization and by scaffolding other tumor suppressors. Several tumor suppressor pathways are now known to be directly modulated by RASSF1A including MOAP-1/Bax (Baksh et al., 2005; Vos et al., 2006), the hippo pathway (Guo et al., 2007), and p53 (Song et al., 2008).
Key RASSF1A Interactions
RASSF1A and K-Ras
RASSF1A contains a Ras association domain and a cysteine rich domain (CRD). By analogy to the classic Ras effector Raf, both of these domains have the potential to bind directly to Ras oncoproteins (Drugan et al., 1996). RASSF1A can be detected in an endogenous complex with Ras (Calvisi et al., 2006), can form a complex with exogenously expressed activated K-Ras and serve as pro-apoptotic K-Ras effector (Vos et al., 2006; Donninger et al., 2007). There remains some controversy over the physiological nature of the interaction between Ras and RASSF1A. This may have arisen because only the K-Ras specific isoform interacts with RASSF1A, and the K-Ras protein must be post-translationally modified to support the interaction (Donninger et al., 2007). Moreover, our experience working with the Ras effector Raf-1 showed us that inclusion of EDTA or strong detergents in the binding buffer can destroy the interaction between Ras and CRD structures (data not shown).
RASSF1A and microtubules
Early work showed that RASSF1A forms a complex with the microtubule network in cells and co-immunoprecipitates with most tubulin isoforms. Moreover, the interaction with RASSF1A appears to promote a robust, Taxol-like increase in the polymerization of microtubules (Liu et al., 2003; Dallol et al., 2004; Vos et al., 2004). Yeast two-hybrid studies have found that RASSF1A can directly bind a series of microtubule associating proteins (MAPS) including MAP1a/MAP1b, C19ORF5 (Dallol et al., 2004), and MAP4 (Vos et al., 2004). Thus, it seems likely that the association with microtubules is indirect. Interestingly, certain tumor derived mutants of RASSF1A appear to lose the ability to associate with specific tubulin isoforms (El-Kalla et al., 2010). Moreover, deletion mutants of RASSF1A that lose the ability to complex with microtubules are defective for the induction of cell cycle arrest (Vos et al., 2004). These data suggest that the interaction of RASSF1A with microtubules may be unusually complex and essential for its tumor suppressing activity.
RASSF1A and MOAP-1/Bax
MOAP-1 is a BH3 (Bcl-2 homology3) domain containing protein that binds and activates the pro-apoptotic effector Bax (Tan et al., 2005). RASSF1A was found to bind directly to MOAP-1 and thereby stimulate Bax apoptotic activity (Baksh et al., 2005; Vos et al., 2006). As MOAP-1 can associate with TNF-R1 and TRAIL-R1/DR4 death receptors, this links RASSF1A into TNF alpha/TRAIL death signaling pathways (Baksh et al., 2005). The interaction of RASSF1A and MOAP-1 is enhanced by the presence of activated K-Ras. Thus RASSF1A can act as a connector between Ras and Bax. Cells knocked out for RASSF1A exhibit reduced Bax activation and apoptosis when transfected with activated K-Ras (Baksh et al., 2005; Vos et al., 2006).
RASSF1A and the hippo pathway
RASSF1A directly binds and activates the pro-apoptotic MST1 and MST2 kinases (Praskova et al., 2004; Avruch et al., 2006). These kinases drive the hippo pathway by phosphorylating and kinases LATs1 and LATs2 (Harvey and Tapon, 2007). These kinases then phosphorylate the transcription factors YAP and TAZ, which in turn activate the p73 tumor suppressor (Matallanas et al., 2007). The full role of RASSF1A in this pathway may be more complex as it also directly binds a protein called Salvador. Salvador may serve as an adaptor to promote the phosphorylation of LATs by MST (Guo et al., 2007). However, Salvador also appears to be a tumor suppressor in its own right, with significant functions independent of the classic hippo pathway (Donninger et al., 2011).
In summary, we now know that RASSF1A can complex with multiple known tumor suppressors and modulate their activity. This gives RASSF1A the potential to serve as a tumor suppressor node, integrating the activity of multiple tumor suppressors and connecting K-Ras to their action.
The Rs2073498A SNP
Several SNPs have been identified in the RASSF1A gene. The best characterized (rs2073498) is a C–A variation resulting in the presence of a Serine instead of an Alanine at amino acid 133 in the RASSF1A protein. This A(133)S variant protein has been associated with a reduced ability to regulate the cell cycle at G1/S (Shivakumar et al., 2002). Structurally, the A133S variant falls in a rather an intriguing position in the RASSF1A protein, within the minimal microtubule association domain (residues 120–185; Vos et al., 2004). Although the A(133)S variant retains an overtly similar association with microtubules as the wild type protein, it is defective for the interaction with gamma and alpha (but not beta) tubulin (El-Kalla et al., 2010). Moreover, the change of Alanine133 to a Serine destroys a consensus phosphorylation site for the ATM kinase, an essential component of the DNA damage response (Hamilton et al., 2009). In fact it generates a new consensus phosphorylation site for several other kinases, including Casein kinase II. Thus, the biology and the regulation of the A(133)S variant may be significantly different to that of the wild type protein.
The Rs2073498 SNP and Cancer Predisposition
In 2005, an analysis was performed to determine if SNPs detected in RASSF1A might have any association with the development of cancer (Schagdarsurengin et al., 2005). A striking result was obtained showing that the rs2073498 SNP was present in approximately 2% of non-cancer patient controls but almost 20% of breast cancer patients. A subsequent study (Gao et al., 2008) showed a much more modest increased association of the SNP with breast cancer development but linked it to a predisposition to early onset of disease in BRCA1 mutant positive patients. The presence of the SNP in the normal control population was found to be dramatically higher in this study (~18%). Moreover, the dbSNP (Sherry et al., 2001) gives a frequency of ~29% for the SNP in normal European populations. A third study (Bergqvist et al., 2010) has now been performed and has been unable to confirm a link between the SNP and breast cancer predisposition. However, this study did not address age of onset issues. This study also found a much higher frequency of the SNP in their non-cancer controls. Thus, there remains a controversy regarding the importance of the SNP for the development of breast cancer.
Here we have sought to address the role of the rs2073498 SNP in RASSF1A function and in breast cancer predisposition. We have compared the biological characteristics of the wild type and SNP derived proteins and identified differences in protein/protein interactions as well as in the effects of the proteins on cellular adhesion. We have also screened a larger cohort of breast cancer patients for the presence of the SNP. However, we found only a very modest increase in breast cancer amongst carriers of the SNP.
Results
Frequency of the Rs2073498 SNP in Breast Cancer
We have analyzed the largest breast cancer population group to date for the presence of the rs2073498 C/A SNP in RASSF1A. This group consisted of a primarily Caucasian, Non-African American (NAA) group of 1118 normal, and 1230 breast cancer patients as well as an African American (AA) group of 658 normal and 742 breast cancer patients (Table 1). We find that 20% of the normal NAA population are Heterozygous, and 2% are Homozygous, for the allele. Curiously, only ~6.1% of 658 normal AA women carried the allele. This shows that there is a dramatic racial disparity in the presence of the allele. We observed a very modest 4.5% (odds ratio 1.066) increase in frequency for the presence of the SNP in NAA populations with breast cancer. This is not considered statistically significant (P-value 0.6). No increase was observed in the much smaller AA sample. In fact, the SNP was less present in the AA breast cancer patients.
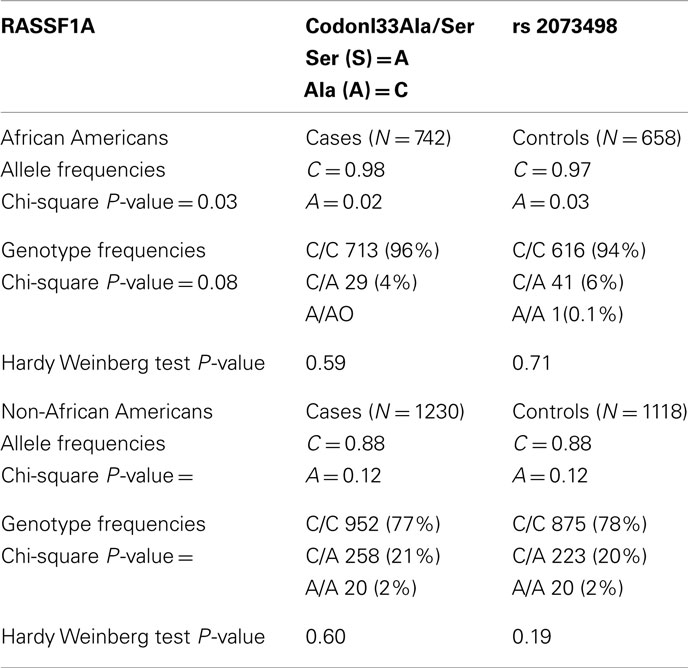
Table 1. Frequency of the A(133)S SNP in normal and breast cancer patient populations of Non-African American and African American women.
When breaking up the NAA breast cancers by sub-type, the SNP was present in 22% of normal control samples and 26.3% of Luminal A breast cancer patients (P-value = 0.094, odds ratio 1.4), no differences were detected in Luminal B or Triple negative patients. Moreover, no significant (P = 0.85) differences in age of onset were observed (Table 2). However, data was not available to consider the role of the BRCA1 mutation status in the samples.

Table 2. Average age of onset of breast cancer in wt vs. rs2073498 SNP patients.
We have also performed a series of experiments in an attempt to determine if there are significant differences in the biological activity of between the wild type RASSF1A protein and the A(133)S variant produced by the SNP allele that might support a role in cancer predisposition.
The RASSF1A A(133)S Variant Alters Cellular Adhesion
In order to examine any potential differences in biological activity between the wild type RASSF1A and the A(133)S variant, we transfected H1299 lung carcinoma cells (RASSF1A negative) with expression constructs to generate stable cell lines expressing approximately equal amounts of each protein (Figure 1). We found little obvious difference in the rate of cell growth between the matched pair of cells. However, we detected a significant increase in the degree of adhesion in the A(133)S variant compared to the wild type RASSF1A expressing cells (Figure1). Whereas transformed cells often exhibit reduced adhesion which correlates with enhanced motility, here we found that the variant RASSF1A expressing cells demonstrate stronger adhesion to the substratum than the wild type RASSF1A expressing cells. However, there are multiple examples where enhanced adhesion has been associated with enhanced transformation (Kenny et al., 2008) or survival (Tsujii and DuBois, 1995).
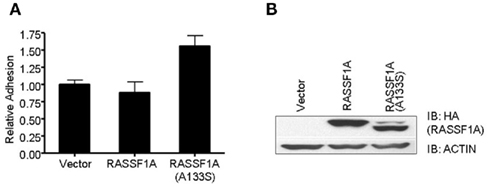
Figure 1. The A(133)S SNP induces enhanced adhesion. (A) H1299 cells stably transfected with HA tagged RASSF1A expression vector were assayed for the strength of their adhesion to culture dishes. The difference between the wild type and the A(133)S variant transfected cells was statistically significant (P = 0.03). (B) Levels of RASSF1A expression in the stable cell lines was confirmed by western analysis using an HA antibody.
The RASSF1A A(133)S Variant Exhibits Differential Binding to a Variety of Potential RASSF1A Targets
In an attempt to identify proteins which bind differentially between the wild type and the SNP derived protein, we performed a proteomic analysis of immunoprecipitations of wild type and A(133)S RASSF1A transfected into 293-T cells. Problems in interpreting this type of experiment can arise because of the degree of background. However, here we were focusing on differences between the two forms of the protein rather than the total composition of the pull down. We identified a number of known RASSF1A interacting proteins that bound to the same degree, such as MST1 and C19ORF5 (Table 3, denoted in blue), but we also identified a small group of proteins that appeared to exhibit differential association (Table 3). Some proteins appeared to gain association with the SNP derived variant, and some appeared to lose association.
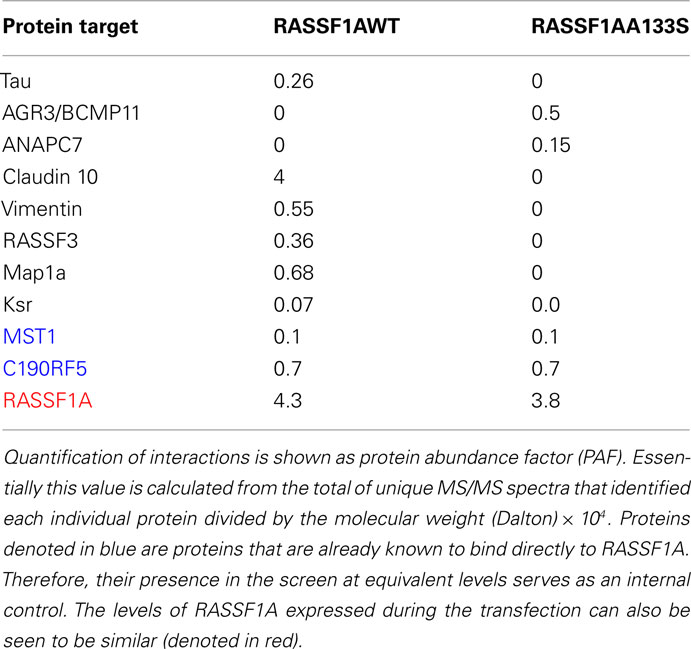
Table 3. Summary of proteins identified as demonstrating differential interactions with the wild type and A(133)S variant of RASSF1A.
We have begun to validate these results by Western blot. Our initial progress suggests that these results may be accurate as MAP1a appeared to show differential binding when we performed co-transfection/co-immunoprecipitation experiments (Figure 2). This is a specific defect, as MAP4 showed equivalent binding. Further validation is proceeding.
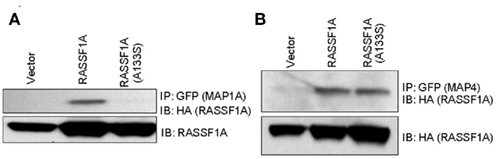
Figure 2. Differential binding of the A(133)s RASSF1A variant to MAPs. GFP–MAP1a (A) and GFP–MAP4 (B) were co-transfected into 293-T cells with HA tagged RASSF1A constructs. Equally fluorescent transfected dishes were lysed, immunoprecipitated with GFP and Western blotted with HA and GFP.
Discussion
RASSF1A is an important tumor suppressor with the potential to impact many different biological processes that are critical to the development of cancer. The A(133)S SNP derived protein has now been shown to exhibit a number of subtle defects in biological function. These include differential binding to various interaction partners, differential effects on the cell cycle and cellular adhesion. We have also recently observed a differential interaction with components of the DNA repair machinery and found that the A(133)S variant form of RASSF1A can act as a dominant negative in certain aspects of DNA repair (manuscript submitted). Thus it would be perfectly reasonable to find that the SNP is associated with an enhanced risk of cancer development.
However, dramatic early results in breast cancer patients have not yet been reproduced. We performed a larger study to determine if we could do so. We were unable to confirm results showing a strong association between presence of the SNP and breast cancer development. We found only a much more modest association and only with the luminal A sub-type. Thus, the association of the SNP with breast cancer appears to be more subtle than originally anticipated and may be dependent upon a background of BRCA1 deficiency (Gao et al., 2008). Indeed, the most curious feature of the initial breast cancer study is not the high frequency of the SNP in the cancer patients, but rather the very low frequency reported for the non-cancer patient controls. Unlike the other studies, most of these controls were suffering from some non-cancer, malady of the breast, mostly cystic diseases. Perhaps the low frequency of the SNP in these patients is telling us that the SNP is protective against cystic diseases.
The strongest correlation with cancer susceptibility described so far for the rs2073498 SNP is with lung cancer in Japanese populations (Kanzaki et al., 2006). In Japan the SNP is present at ~11% in the normal population and lung cancer patients were almost twice as likely to carry the SNP (Kanzaki et al., 2006). This tissue is likely to be exposed to mutagenic insults at a higher frequency than breast cancer tissue. As we have recently observed reduced DNA repair activity in lung tumor cells expressing the A(133)S variant derived from the SNP (manuscript submitted), this tissue may be more sensitive to the cancer predisposition effect. Thus, a larger study in lung cancer could prove very interesting.
Our proteomics screen has identified several intriguing potential differences in the protein complexes formed by the wild type and A(133)S variant. Most of the differences identified involved proteins with known functions in cancer. These include proteins that appear to lose association with the SNP derived variant and those that appear to gain it. Claudin 10 is a tight junction protein with multiple functions (Gunzel et al., 2009) that has been implicated in modulating invasion (Ip et al., 2007). Vimentin over-expression in cancer has been documented and a potential role in metastasis identified (Satelli and Li, 2011). Vimentin has been implicated in adhesion (Ivaska et al., 2007) and so the differential association might explain the differential adhesion promoted by the two forms of RASSF1A (Figure 1).
The A(133)S variant of RASSF1A has previously been shown to be defective for the interaction with gamma and alpha tubulin, although the interaction with beta tubulin was retained (El-Kalla et al., 2010). This implies that it is defective for the interaction with MAPs which are already known to bind directly to RASSF1A (Dallol et al., 2004). Potential differences in the interaction with the MAPs Tau and MAP1a were identified in the screen. The defective interaction with MAP1a was confirmed by co-immunoprecipitation studies.
Ksr is thought to act as a scaffold facilitating the activation of the Raf oncoprotein by Ras. Ksr can complex with a second Raf scaffold called CNK (Claperon and Therrien, 2007). CNK has been reported to be able to bind RASSF1A and to mediate some of the apoptotic effects of RASSF1A (Rabizadeh et al., 2004).
RASSF3 remains the most obscure member of the RASSF family (Tommasi et al., 2002). In our hands it is pro-apoptotic but we have been unable to detect any loss of mRNA or protein expression in human tumor cells (data not shown). It has, however, been implicated in mediating resistance to transformation by deregulated Her/Neu in breast cancer (Jacquemart et al., 2009).
ANAPC7 is a sub-unit of the anaphase promoting complex (APC). The APC acts to regulate the levels of a variety of key components of the cell cycle. RASSF1A has previously been implicated in modulating APC activity via binding cdc20 (Song and Lim, 2004). However, this interaction remains controversial (Liu et al., 2007).
AGR3/BCMP11 remains a relatively obscure protein but has recently been associated with differentiation and survival in ovarian cancer (King et al., 2011). It has also been identified as an over-expressed protein in breast cancer (Adam et al., 2003). Further validation, including endogenous co-immunoprecipitations, will be required to validate the data set.
An important point that may be worth considering is that when tumors lose expression of RASSF1A by promoter methylation, a smaller isoform of RASSF1 called RASSF1C usually remains (Richter et al., 2009). In these cells, the SNP will manifest at a protein level as RASSF1C A(63)S. It is possible that it is this protein, not RASSF1A A(133)S that may be driving any cancer susceptibility phenotype. Indeed, some evidence has been presented that RASSF1C can serve as an oncogene (Reeves et al., 2010).
A further consideration is that the SNP shows a dramatic difference in its occurrence between different racial populations. According to the dbSNP (Sherry et al., 2001), the frequency of the SNP is almost 15 times higher in European populations than in African populations. Our studies based on populations in the USA found an approximately fourfold higher frequency in NAAs than in AAs. Therefore, to determine if the SNP predisposes toward cancer, perhaps we should focus on cancers that are more frequent in the NAA populations such as ovarian (Ness et al., 2000). This observation also means that if study populations are not controlled for race, then any epidemiology results obtained may be seriously flawed.
Materials and Methods
Plasmids and DNA
RASSF1A plasmids have been described previously (Dallol et al., 2004; Vos et al., 2006). GFP–MAP1a was a kind gift of Chien et al., 2005; Chung Shan Medical University, Taichung, Taiwan).
Tissue Culture and Western Analysis
293-T cells were cultured and transfected as described previously Vos and Clark (2005). After 24 h, transfected cells were lysed as described previously Vos and Clark (2005) and immunoprecipitated with GFP-trap beads (Allele Biotech, Belgium) according to the manufacturer’s instructions. The immunoprecipitates were Western blotted with anti-GFP (Santa Cruz Bio, Santa Cruz, CA, USA) and anti-HA antibodies (Sigma, St. Louis, MO, USA).
Adhesion Assays
H1299 cells (ATCC, Manassas, VA, USA) were stably transfected with RASSF1A wild type or mutant cloned in the vector pZIP-neoHA (Fiordalisi et al., 2001). Cells were selected in G418 and adhesion assays were performed as described in (Humphries, 2001). Essentially, cells were plated at 5 × 104 cells per well in BSA-coated 96-well plates and allowed to adhere for 45 min at 37°C. The medium was removed and the adhering cells fixed and stained with crystal violet. The dye was solubilized in 10% acetic acid and absorbance at 570 nm was used as a measure of adhesion.
Proteomics
Ten 10 cm dishes of 293-T cells were transfected with 5 μg of HA tagged RASSF1A expression plasmid. Cells were lysed and immunoprecipitated with HA conjugated beads after 24 h. Samples were washed and subjected to trypsin digestion. Trypsin-digested samples were analyzed by 2D-LC-MS/MS. Briefly, protein samples were digested with modified trypsin (Promega, Madison, WI, USA) overnight at 37°C as previously described (Cummins et al., 2010). Resulting peptides were loaded onto an analytical 2D capillary chromatography column packed with strong cation exchange (SCX) and C18 reversed-phase (RP) resin (Phenomenex, Torrance, CA, USA). This biphasic column was attached to an analytical RP chromatography column with an integrated, laser pulled emitter tip. Peptides were eluted from SCX with seven step gradients of 5, 10, 15, 30, 50, 70, and 100% of 500 mM ammonium acetate. Following each SCX elution step, peptides were ionized and eluted into a linear ion trap mass spectrometer according to the following linear HPLC gradient: 20 min: 0% B, 80 min: 40% B, 90 min: 60% B at a flow rate of 200 nl/min (mobile phase A: 5% acetonitrile/0.1% formic acid and mobile phase B: 80% acetonitrile/0.1% formic acid). MS/MS spectra were acquired with a LTQ ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Protein identification was performed with Sequest Sorcerer (Sage-N Research, San Jose, CA, USA), which was set up to search a FASTA formatted human protein database (Human RefSeq) with a fragment ion mass tolerance of 1.00 Da and a parent ion tolerance of 1.2 Da. High-probability peptide and protein assignments from were made using Peptide and Protein Prophet algorithms (Nesvizhskii et al., 2003). Abundance of each identified protein was determined by normalizing the number of unique spectral counts matching to the protein by its predicted molecular weight. This value has been termed a protein abundance factor (Powell et al., 2004). Comparative analysis was performed with an in-house, web-based program (McAfee et al., 2006).
Genomics
Experiments were performed as described previously (Nyante et al., 2011). Essentially, the Carolina Breast Cancer Study (CBCS) is an IRB approved, comprehensive, interdisciplinary investigation into the causes of invasive and in-situ breast cancer among AA and white women in North Carolina. Enrollment was conducted in two phases, Phase 1 (1993–1996) and Phase 2 (1996–2001), and the total number of enrolled participants is 4333. Phase 1 and Phase 2 of the study includes 2311 cases (894 AA, 1417 NAA, primarily Caucasian) and 2022 controls (788 AA, 1234 NAA). Peripheral blood lymphocytes were collected and DNA extracted for 90% of participants. Genotyping was completed using the Illumina Golden Gate platform: 96% of samples had call rates greater than 95; 90% of SNPs were successfully genotyped; control DNA samples had concordance rates of 99.9%. SAS was used to calculate chi-squared tests and Hardy Weinberg tests.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by NIH AND USAMRMC grants 1R01CA153132-01 and PR-081350 (GJC), NIH/NIDD K01-DK076743-01 and COBRE-RR-02-007 (DWP), Program of Research Excellence (SPORE) in Breast Cancer (NIH/NCI P50-CA58223 (RCM).
References
Adam, P. J., Boyd, R., Tyson, K. L., Fletcher, G. C., Stamps, A., Hudson, L., Poyser, H. R., Redpath, N., Griffiths, M., Steers, G., Harris, A. L., Patel, S., Berry, J., Loader, J. A., Townsend, R. R., Daviet, L., Legrain, P., Parekh, R., and Terrett, J. A. (2003). Comprehensive proteomic analysis of breast cancer cell membranes reveals unique proteins with potential roles in clinical cancer. J. Biol. Chem. 278, 6482–6489.
Avruch, J., Praskova, M., Ortiz-Vega, S., Liu, M., and Zhang, X. F. (2006). Nore1 and RASSF1 regulation of cell proliferation and of the MST1/2 kinases. Methods Enzymol. 407, 290–310.
Baksh, S., Tommasi, S., Fenton, S., Yu, V. C., Martins, L. M., Pfeifer, G. P., Latif, F., Downward, J., and Neel, B. G. (2005). The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol. Cell 18, 637–650.
Bergqvist, J., Latif, A., Roberts, S. A., Hadfield, K. D., Lalloo, F., Howell, A., Evans, D. G., and Newman, W. G. (2010). RASSF1A polymorphism in familial breast cancer. Fam. Cancer 9, 263–265.
Calvisi, D. F., Ladu, S., Gorden, A., Farina, M., Conner, E. A., Lee, J. S., Factor, V. M., and Thorgeirsson, S. S. (2006). Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 130, 1117–1128.
Chien, C. L., Lu, K. S., Lin, Y. S., Hsieh, C. J., and Hirokawa, N. (2005). The functional cooperation of MAP1A heavy chain and light chain 2 in the binding of microtubules. Exp. Cell Res. 308, 446–458.
Claperon, A., and Therrien, M. (2007). KSR and CNK: two scaffolds regulating RAS-mediated RAF activation. Oncogene 26, 3143–3158.
Cummins, T. D., Barati, M. T., Coventry, S. C., Salyer, S. A., Klein, J. B., and Powell, D. W. (2010). Quantitative mass spectrometry of diabetic kidney tubules identifies GRAP as a novel regulator of TGF-beta signaling. Biochim. Biophys. Acta 1804, 653–661.
Dallol, A., Agathanggelou, A., Fenton, S. L., hmed-Choudhury, J., Hesson, L., Vos, M. D., Clark, G. J., Downward, J., Maher, E. R., and Latif, F. (2004). RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer Res. 64, 4112–4116.
Dammann, R., Li, C., Yoon, J. H., Chin, P. L., Bates, S., and Pfeifer, G. P. (2000). Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 25, 315–319.
Dammann, R., Schagdarsurengin, U., Seidel, C., Strunnikova, M., Rastetter, M., Baier, K., and Pfeifer, G. P. (2005). The tumor suppressor RASSF1A in human carcinogenesis: an update. Histol. Histopathol. 20, 645–663.
Donninger, H., Allen, N., Henson, A., Pogue, J., Williams, A., Gordon, L., Kassler, S., Dunwell, T., Latif, F., and Clark, G. J. (2011). Salvador protein is a tumor suppressor effector of RASSF1A with hippo pathway-independent functions. J. Biol. Chem. 286, 18483–18491.
Donninger, H., Vos, M. D., and Clark, G. J. (2007). The RASSF1A tumor suppressor. J. Cell Sci. 120, 3163–3172.
Drugan, J. K., Khosravi-Far, R., White, M. A., Der, C. J., Sung, Y. J., Hwang, Y. W., and Campbell, S. L. (1996). Ras interaction with two distinct binding domains in Raf-1 may be required for Ras transformation. J. Biol. Chem. 271, 233–237.
El-Kalla, M., Onyskiw, C., and Baksh, S. (2010). Functional importance of RASSF1A microtubule localization and polymorphisms. Oncogene 29, 5729–5740.
Fiordalisi, J. J., Johnson, R. L., Ulku, A. S., Der, C. J., and Cox, A. D. (2001). Mammalian expression vectors for Ras family proteins: generation and use of expression constructs to analyze Ras family function. Methods Enzymol. 332, 3–36.
Gao, B., Xie, X. J., Huang, C., Shames, D. S., Chen, T. T., Lewis, C. M., Bian, A., Zhang, B., Olopade, O. I., Garber, J. E., Euhus, D. M., Tomlinson, G. E., and Minna, J. D. (2008). RASSF1A polymorphism A133S is associated with early onset breast cancer in BRCA1/2 mutation carriers. Cancer Res. 68, 22–25.
Gunzel, D., Stuiver, M., Kausalya, P. J., Haisch, L., Krug, S. M., Rosenthal, R., Meij, I. C., Hunziker, W., Fromm, M., and Müller, D. (2009). Claudin-10 exists in six alternatively spliced isoforms that exhibit distinct localization and function. J. Cell Sci. 122, 1507–1517.
Guo, C., Tommasi, S., Liu, L., Yee, J. K., Dammann, R., and Pfeifer, G. P. (2007). RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 17, 700–705.
Hamilton, G., Yee, K. S., Scrace, S., and O’neill, E. (2009). ATM regulates a RASSF1A-dependent DNA damage response. Curr. Biol. 19, 2020–2025.
Harvey, K., and Tapon, N. (2007). The Salvador-Warts-Hippo pathway – an emerging tumour-suppressor network. Nat. Rev. Cancer 7, 182–191.
Ip, Y. C., Cheung, S. T., Lee, Y. T., Ho, J. C., and Fan, S. T. (2007). Inhibition of hepatocellular carcinoma invasion by suppression of claudin-10 in HLE cells. Mol. Cancer Ther. 6, 2858–2867.
Ivaska, J., Pallari, H. M., Nevo, J., and Eriksson, J. E. (2007). Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 313, 2050–2062.
Jacquemart, I. C., Springs, A. E., and Chen, W. Y. (2009). Rassf3 is responsible in part for resistance to mammary tumor development in neu transgenic mice. Int. J. Oncol. 34, 517–528.
Kanzaki, H., Hanafusa, H., Yamamoto, H., Yasuda, Y., Imai, K., Yano, M., Aoe, M., Shimizu, N., Nakachi, K., Ouchida, M., and Shimizu, K. (2006). Single nucleotide polymorphism at codon 133 of the RASSF1 gene is preferentially associated with human lung adenocarcinoma risk. Cancer Lett. 238, 128–134.
Kenny, H. A., Kaur, S., Coussens, L. M., and Lengyel, E. (2008). The initial steps of ovarian cancer cell metastasis are mediated by MMP-2 cleavage of vitronectin and fibronectin. J. Clin. Invest. 118, 1367–1379.
King, E. R., Tung, C. S., Tsang, Y. T., Zu, Z., Lok, G. T., Deavers, M. T., Malpica, A., Wolf, J. K., Lu, K. H., Birrer, M. J., Mok, S. C., Gershenson, D. M., and Wong, K. K. (2011). The anterior gradient homolog 3 (AGR3) gene is associated with differentiation and survival in ovarian cancer. Am. J. Surg. Pathol. 35, 904–912.
Liu, L., Baier, K., Dammann, R., and Pfeifer, G. P. (2007). The tumor suppressor RASSF1A does not interact with Cdc20, an activator of the anaphase-promoting complex. Cell Cycle 6, 1663–1665.
Liu, L., Tommasi, S., Lee, D. H., Dammann, R., and Pfeifer, G. P. (2003). Control of microtubule stability by the RASSF1A tumor suppressor. Oncogene 22, 8125–8136.
Matallanas, D., Romano, D., Yee, K., Meissl, K., Kucerova, L., Piazzolla, D., Baccarini, M., Vass, J. K., Kolch, W., and O’neill, E. (2007). RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 27, 962–975.
McAfee, K. J., Duncan, D. T., Assink, M., and Link, A. J. (2006). Analyzing proteomes and protein function using graphical comparative analysis of tandem mass spectrometry results. Mol. Cell. Proteomics 5, 1497–1513.
Ness, R. B., Grisso, J. A., Klapper, J., and Vergona, R. (2000). Racial differences in ovarian cancer risk. J. Natl. Med. Assoc. 92, 176–182.
Nesvizhskii, A. I., Keller, A., Kolker, E., and Aebersold, R. (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658.
Nyante, S. J., Gammon, M. D., Kaufman, J. S., Bensen, J. T., Lin, D. Y., Barnholtz-Sloan, J. S., Hu, Y., He, Q., Luo, J., and Millikan, R. C. (2011). Common genetic variation in adiponectin, leptin, and leptin receptor and association with breast cancer subtypes. Breast Cancer Res. Treat. 129, 593–606.
Powell, D. W., Weaver, C. M., Jennings, J. L., McAfee, K. J., He, Y., Weil, P. A., and Link, A. J. (2004). Cluster analysis of mass spectrometry data reveals a novel component of SAGA. Mol. Cell Biol. 24, 7249–7259.
Praskova, M., Khoklatchev, A., Ortiz-Vega, S., and Avruch, J. (2004). Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 381, 453–462.
Rabizadeh, S., Xavier, R. J., Ishiguro, K., Bernabeortiz, J., Lopez-Ilasaca, M., Khokhlatchev, A., Mollahan, P., Pfeifer, G. P., Avruch, J., and Seed, B. (2004). The scaffold protein CNK1 interacts with the tumor suppressor RASSF1A and augments RASSF1A-induced cell death. J. Biol. Chem. 2.79, 29247–29254.
Reeves, M. E., Baldwin, S. W., Baldwin, M. L., Chen, S. T., Moretz, J. M., Aragon, R. J., Li, X., Strong, D. D., Mohan, S., and Amaar, Y. G. (2010). Ras-association domain family 1C protein promotes breast cancer cell migration and attenuates apoptosis. BMC Cancer 10, 562. doi:10.1186/1471-2407-10-562
Richter, A. M., Pfeifer, G. P., and Dammann, R. H. (2009). The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta 1796, 114–128.
Satelli, A., and Li, S. (2011). Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 68, 3033–3046.
Schagdarsurengin, U., Seidel, C., Ulbrich, E. J., Kolbl, H., Dittmer, J., and Dammann, R. A. (2005). Polymorphism at codon 133 of the tumor suppressor RASSF1A is associated with tumorous alteration of the breast. Int. J. Oncol. 27, 185–191.
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., and Sirotkin, K. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311.
Shivakumar, L., Minna, J., Sakamaki, T., Pestell, R., and White, M. A. (2002). The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Mol. Cell Biol. 22, 4309–4318.
Song, M. S., and Lim, D. S. (2004). Control of APC-Cdc20 by the tumor suppressor RASSF1A. Cell Cycle 3, 574–576.
Song, M. S., Song, S. J., Kim, S. Y., Oh, H. J., and Lim, D. S. (2008). The tumour suppressor RASSF1A promotes MDM2 self-ubiquitination by disrupting the MDM2-DAXX-HAUSP complex. EMBO J. 27, 1863–1874.
Tan, K. O., Fu, N. Y., Sukumaran, S. K., Chan, S. L., Kang, J. H., Poon, K. L., Chen, B. S., and Yu, V. C. (2005). MAP-1 is a mitochondrial effector of Bax. Proc. Natl. Acad. Sci. U.S.A. 102, 14623–14628.
Tommasi, S., Besaratinia, A., Wilczynski, S. P., and Pfeifer, G. P. (2011). Loss of Rassf1a enhances p53-mediated tumor predisposition and accelerates progression to aneuploidy. Oncogene 30, 690–700.
Tommasi, S., Dammann, R., Jin, S. G., Zhang, X. X., Avruch, J., and Pfeifer, G. P. (2002). RASSF3 and NORE1: identification and cloning of two human homologues of the putative tumor suppressor gene RASSF1. Oncogene 21, 2713–2720.
Tsujii, M., and DuBois, R. N. (1995). Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell 83, 493–501.
van der, W. L., and Adams, D. J. (2007). The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 1776, 58–85.
Vos, M. D., and Clark, G. J. (2005). RASSF family proteins and Ras transformation. Methods Enzymol. 407, 311–322.
Vos, M. D., Dallol, A., Eckfeld, K., Allen, N. P., Donninger, H., Hesson, L. B., Calvisi, D., Latif, F., and Clark, G. J. (2006). The RASSF1A tumor suppressor activates Bax via MOAP-1. J. Biol. Chem. 281, 4557–4563.
Keywords: RASSF1A, SNP, breast cancer, Ras
Citation: Donninger H, Barnoud T, Nelson N, Kassler S, Clark J, Cummins TD, Powell DW, Nyante S, Millikan RC and Clark GJ (2011) RASSF1A and the rs2073498 cancer associated SNP. Front. Oncol. 1:54. doi: 10.3389/fonc.2011.00054
Received: 26 September 2011; Accepted: 06 December 2011;
Published online: 28 December 2011.
Edited by:
Sven Bilke, National Cancer Institute/National Institutes of Health, USACopyright: © 2011 Donninger, Barnoud, Nelson, Kassler, Clark, Cummins, Powell, Nyante, Millikan and Clark. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Geoffrey J. Clark, Department of Biochemistry and Molecular Biology, University of Louisville, Louisville, KY 40202, USA. e-mail:Z2pjbGFyMDFAbG91aXN2aWxsZS5lZHU=