
- Cleveland Clinic Taussig Cancer Center, Cleveland Clinic Lerner College of Medicine of CWRU, Pepper Pike, OH, USA
Angiogenesis in general and the vascular endothelial growth factor (VEGF) signaling axis in particular is a validated target in renal cell carcinoma (RCC). Clear-cell carcinoma of the kidney is now recognized as a malignancy that is sensitive to inhibitors of the VEGF pathway. Treatment options for patients with metastatic renal cell carcinoma have evolved in dramatic fashion over the past 6 years, and a new paradigm has developed. The cytokines interferon-α and interleukin-2 were previously utilized for therapy, but since December 2005, six new agents have been approved in the United States for the treatment of advanced RCC. Two are tyrosine kinase inhibitors (TKI’s) including sunitinib and recently pazopanib, and the multikinase inhibitor sorafenib. The current review examines the evolving data with the next generation of TKI’s, axitinib and tivozanib being developed for the treatment of advanced RCC. These agents were synthesized to provide increased target specificity and enhanced target inhibition. The preclinical and clinical data are examined, an overview of the development of these TKI’s is provided, and discussion plus speculation concerning their potential roles as RCC therapy is provided.
Treatment of patients with metastatic renal cell carcinoma (mRCC) has changed dramatically during the past 5 years, and a new paradigm has evolved. Interferon-α (INF-α) and interleukin-2 (IL-2) were previously utilized (McDermott, 2009) but since December 2005, a series of targeted agents have been approved in the United States for the treatment of mRCC, including the three multi-targeted tyrosine kinase inhibitors (TKI’s) sunitinib, sorafenib, and pazopanib (Bukowski, 2009). The first generation of TKI’s included sunitinib, which has emerged as the standard of care for treatment-naïve mRCC patients. The recent approval of pazopanib, a second generation agent with increased specificity for vascular endothelial growth factor (VEGF) receptors (VEGFR), provided another option for frontline therapy. This review will focus on several third generation TKI’s under investigation, axitinib and tivozanib, and explore their current development as therapy for mRCC. The number of agents approved within a very short time, raises a series of questions, such as, do we need another tyrosine kinase inhibitor for this patient population, if yes, what should its characteristics and clinical activity be, and finally, how should these novel agent be studied in the future?
Introduction
Clear-cell renal cell carcinoma (RCC) is the commonest subtype of renal epithelial cancer, and accounts for over 85% of sporadic tumors (Choueiri et al., 2003). The von Hippel–Lindau (VHL) tumor suppressor gene located on chromosome 3p25, is silenced in the majority of clear-cell renal tumors (Kaelin, 2007). This gene encodes the VHL protein (p-VHL) that targets the various hypoxia-inducible factors (HIFs) for proteolysis. HIF-α is a regulator of the hypoxic stress response, and is a primary target of p-VHL. In hypoxic conditions or when VHL is mutated or inactivated, p-VHL does not bind HIF-α, and levels of this transcription factor are increased (Bukowski, 2009). The result is increased transcription of a variety of hypoxia-inducible genes such as VEGF, platelet-derived growth factor (PDGF), transforming growth factor-α (TGF-α), epidermal growth factor receptor (EGFR), and erythropoietin (Kaelin, 2007). VEGF plays a prominent role in angiogenesis, binds to the VEGFR on endothelial cells, and promotes proliferation and migration of these cells (Cohen and McGovern, 2005). This makes VEGF or VEGFR attractive targets for treatment of mRCC, a tumor characterized by VEGF overproduction and increased vascularity. In view of this, the development of drugs for RCC therapy has focused on these targets. TKI’s such as sunitinib and pazopanib are now approved for treatment of these patients.
Preclinical Investigations
The TKI’s are multi-targeted kinase inhibitors which inhibit signaling in a variety of receptors such asVEGFR-1,-2 and -3, platelet-derived growth factor receptors (PDGFR)-α and -β, c-RET, macrophage colony-stimulating factor 1 (CSF-1R), FMS-like tyrosine kinase 3 receptor (FLT3), and c-KIT (Motzer et al., 2007). Recently, a series of small molecular third generation TKI’s including axitinib (Hu-Lowe et al., 2008) and tivozanib (Nakamura et al., 2006) have been developed. These agents have more restricted target spectrums, as well as higher affinity for VEGFR-1, -2, and -3 when compared with sunitinib and pazopanib. The in vitro kinase specificity of these agents is summarized in Tables 1 and 2. The activity of these agents has been assessed against various tyrosine kinases using either cell-free kinase assays employing recombinant enzymes, or inhibition of ligand-induced phosphorylation of growth factor receptors in a variety of cellular based assays. It is not clear which assay best reflects the in vivo inhibitory activity or efficacy of these TKI’s, but their utility is in demonstrating the relative kinase specificity of the various drugs. Additionally, comparing the various IC50 values is problematic, since the different assay procedures and methods were utilized.

Table 1. Receptor tyrosine kinase inhibitory activity of selected TKI’s – cell-free kinase assay (IC50).
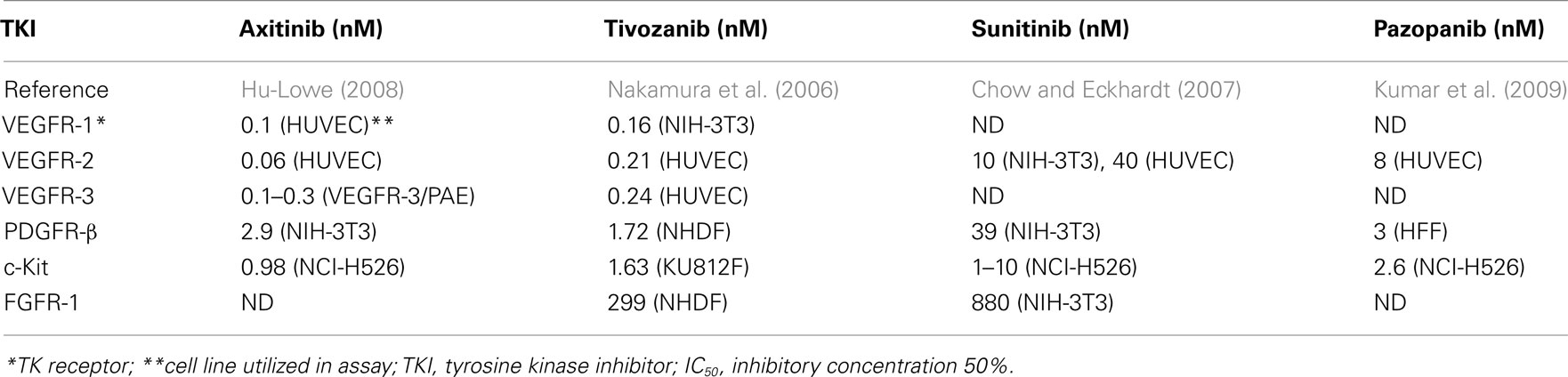
Table 2. Receptor tyrosine kinase inhibitory activity of selected TKI’s – inhibition ligand dependent in proliferation cell-based assay (IC50).
Figure 1 illustrates the molecular structures and characteristics of these two drugs. Axitinib (formerly known as AG-013736) is a substituted indazole derivative, and was developed using structure-based drug design technology (Kania, 2009). The resulting agent was a highly potent and selective inhibitor of the various kinase domains of VEGFRs, distinguishing it from previous multi-targeted TKIs. In vitro, axitinib inhibits the autophosphorylation of VEGFR-1, 2, and 3 in picomolar quantities (IC50), and has approximately a 10-fold lower potency for inhibition of PDGFR-α and -β, and the stem-cell factor receptor KIT. Axitinib produces in vivo inhibition of angiogenesis in various preclinical models (Hu-Lowe et al., 2008). In various xenograft mouse models, its antitumor activity was associated with decreased tumor vascularization and blood flow, and a reduction in tumor size, related to its plasma concentrations.
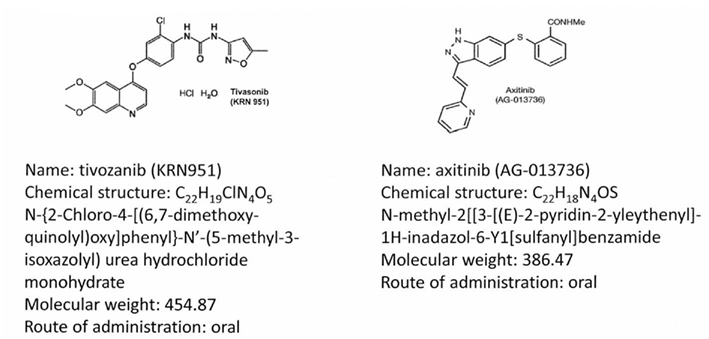
Figure 1. Molecular structures and chemical data for two third generation tyrosine kinase inhibitors, axitinib (Hu-Lowe et al., 2008), and tivozanib (Gupta and Fishman, 2011).
Tivozanib (formerly AV-951, KRN-951) is also a highly potent and selective VEGFR tyrosine kinase inhibitor of VEGFR-1, -2, and -3, which inhibits angiogenesis and vascular permeability in tumor tissues (Nakamura et al., 2006). It has antitumor effects in human breast, colon, liver, lung, ovarian, pancreas, prostate, brain, and RCC xenograft models (Nakamura et al., 2006; Taguchi et al., 2008). Studies utilizing dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) analysis have demonstrated the antitumor activity of this agent is associated with a significant reduction vascular permeability in tumors (Nakamura et al., 2006). Both of these agents have subsequently been developed for treatment of advanced RCC.
Phase 1 Studies
Axitinib
A phase 1 trial of axitinib (Rugo et al., 2005) was conducted in 36 patients with various solid tumors (see Table 3). Dose levels ranged from 5 to 30 mg orally twice daily. Dose-limiting toxicities included primarily hypertension, with hemoptysis, and stomatitis. These were seen at the higher dose levels. The hypertension observed was easily managed with antihypertensive medications. Stomatitis was mild to moderate, and managed by dose reduction or drug holidays. Two patients with adenocarcinoma of the lung receiving 20 mg BID developed fatal hemoptysis. Pharmacokinetic studies demonstrated rapid absorption, with maximum plasma concentrations found within 2–6 h after dosing. The dose recommended for subsequent trials was 5 mg, twice daily, administered while fasting. Three confirmed partial responses were noted, one in a patient with adenoid cystic carcinoma, and two patients with mRCC. Other evidence of clinical activity was noted in three additional patients, all of whom had reductions in tumor size that did however, not meet RECIST criteria for a partial response. This phase 1 trial demonstrated the potential clinical activity of axitinib in advanced and refractory malignancies, and importantly, the toxicities observed were essentially similar to those reported with other antiangiogenic agents.
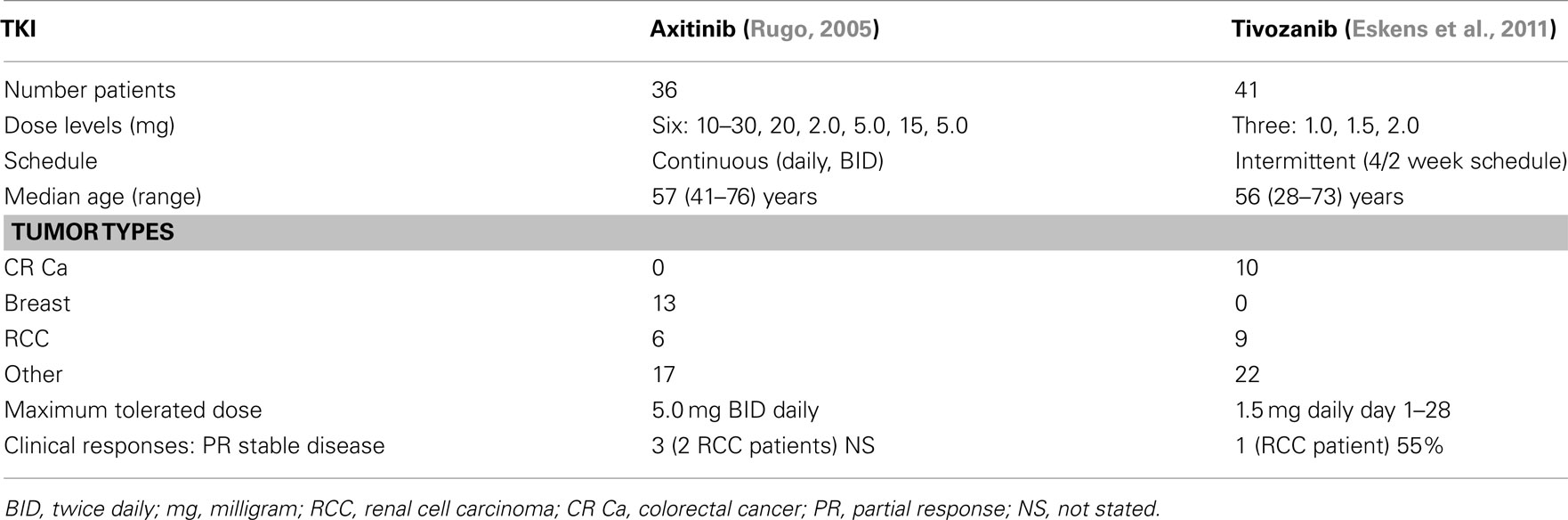
Table 3. Phase 1 trials third generation tyrosine kinase inhibitors (TKI’s) – axitinib and tivozanib.
This study also demonstrated rapid absorption of axitinib when administered with food. Plasma concentrations demonstrated a terminal half-life from 2 to 5 h. Nine patients were investigated for the effects of food on drug levels. The rate and extent of absorption appeared increased during fasting, (higher Cmax and AUC0–24, and a shorter Tmax), with peak concentrations seen within 1–2 h. The investigators noted a 49% (median) increase in plasma exposure compared to non-fasting patients.
The likely hood of drug interactions was also considered, since in vitro studies with human liver cells demonstrated axitinib was metabolized by conversion to glucuronide metabolites as well as oxidation by the CYP3A4 and CYP1A2 isozymes, both P450 inducible enzymes. Potential drug interactions were investigated in a subsequent study which assessed the effects of rifampin on axitinib pharmacokinetics in normal volunteers (Pithavala et al., 2010). In this trial, axitinib exposure was decreased when concomitantly administered with rifampin. Concomitant treatment with axitinib and potent inducers of CYP3A4 therefore require dose adjustment for optimal clinical efficacy. The similarity of axitinib pharmacokinetics in Caucasian and Japanese subjects also supported a single starting dose and regimen for these patient populations.
Tivozanib
A phase 1 and pharmacologic study of tivozanib (Eskens et al., 2011) was conducted in 41 advanced solid tumor patients, including nine individuals with RCC (see Table 3). Tivozanib was administered at dose levels of 1.0, 1.5, and 2.0 mg/day for 28 days followed by a 14-day break. This intermittent schedule resembling the one utilized for sunitinib in mRCC patients (Motzer et al., 2007). The initial starting dose was 2.0 mg/day, however, a series of adverse events (AE’s) were noted which included grade 3 proteinuria, hypertension, and ataxia. Lower dose levels were then explored. Ten patients received 1.0 mg/day, and no dose-limiting toxicity was noted. The MTD was therefore reported as1.5 mg/day for 28 days of therapy every 6 weeks. The dose-limiting toxicity described was similar to other oral VEGFR inhibitors and included hypertension, fatigue, and transaminase elevations. Clinical activity was noted in 2/9 patients with metastatic RCC. One confirmed and one unconfirmed partial response were reported. Additionally, 35% of patients had tumor shrinkage that did not meet the criteria for a partial response, and 55% of patients had stable disease. DCE-MRI analysis was also conducted in eight patients, and variable decreases in tumor vascularization were noted over time. In one patient with mRCC, decreased tumor vascularization was accompanied by some reduction in tumor size.
Pharmacokinetic studies demonstrated slow absorption of tivozanib after single or multiple doses, with maximal plasma levels observed between 2 and 24 h. Significant inter-patient variability was found, and the possibility of enterohepatic recirculation was suggested. The mean half-life of tivozanib was 112 h (range 31–233 h).
Phase 2 Trials: Renal Cell Carcinoma
Axitinib
Three single arm phase 2 trials of axitinib were conducted in previously treated patients with metastatic RCC. These included either cytokine refractory or treatment refractory individuals. The results of these studies are summarized in Tables 4– 6. Axitinib was administered at a starting dose of 5.0 mg twice daily (BID) orally with food, and was escalated in a step-wise fashion to 7 mg BID, then to 10 mg BID, in the absence of toxicity greater than grade 2 for a continuous 2 week period, and absence of hypertension (defined as two blood pressure measurements of >150/90 mmHg taken in the clinic >1 h apart).
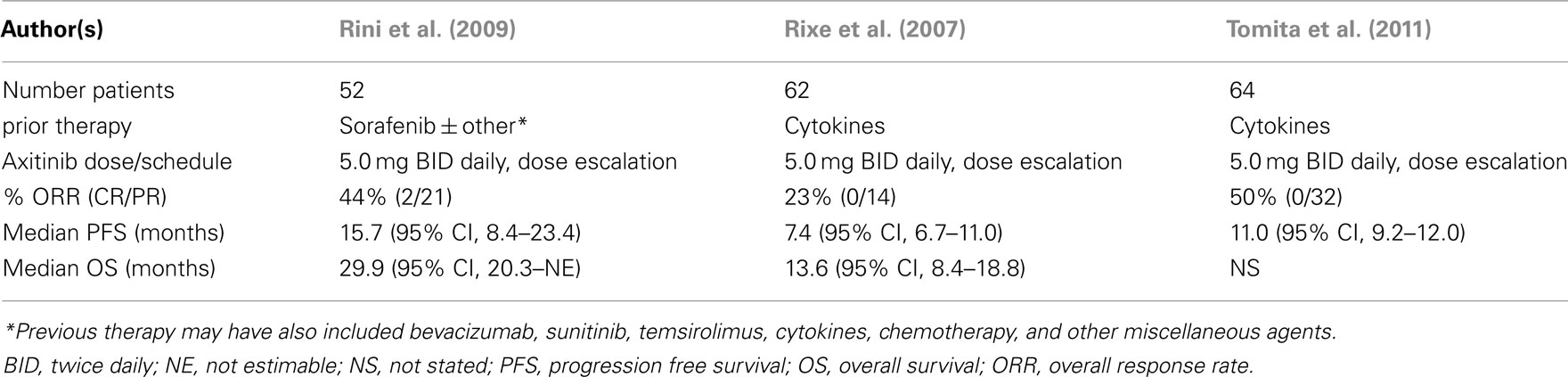
Table 4. Axitinib phase 2 trials – refractory metastatic RCC.
The first study accrued 52 cytokine refractory patients (Rixe et al., 2007). The results of this trial were remarkable, with an overall response rate (ORR) of 44% (2/23 CR’s), a stable disease rate (>8 weeks) of 42%, a median progression free survival (PFS) of 15.7 months, and median overall survival (OS) of 29.9 months. 53.2% of patients had their initial axitinib dose escalated to >5.0 mg BID. A second phase 2 trial was conducted in 62 patients with refractory mRCC who had previously failed therapy with sorafenib, but also may have received prior chemotherapy, cytokines, and/or other targeted therapies such as sunitinib, bevacizumab, and temsirolimus. In this trial, axitinib dose escalation was reported in 11.5% of patients, who received >5.0 mg BID. The ORR was reported as 22.6% (95% CI, 12.9–35.0), with stable disease in 18%. The median PFS was 7.6 months, and median OS 13.6 months. The ORR in the 33 patients receiving >5.0 mg BID was 21%. The investigators suggested higher doses may have been associated with a higher frequency of tumor regression. A third phase 2 study was conducted in Japan, and included cytokine refractory patients. Sixty-four individuals received axitinib (starting dose: 5 mg BID) with dose escalation in 8%. An ORR of 50% was reported with a median PFS of 11.0 months. In summary, these studies provided evidence of efficacy in refractory RCC patients, with median PFS values similar or in excess of those reported in the phase 3 trials of either sunitinib or pazopanib (Motzer et al., 2007; Sternberg et al., 2010). With axitinib monotherapy, the most common AE’s reported included hypertension, fatigue, and gastrointestinal toxicity (Table 5). The majority of AE’s were ≤grade 2, and demonstrate that the continuous administration of this axitinib dose appears to have acceptable toxicity, and is compatible with chronic therapy.
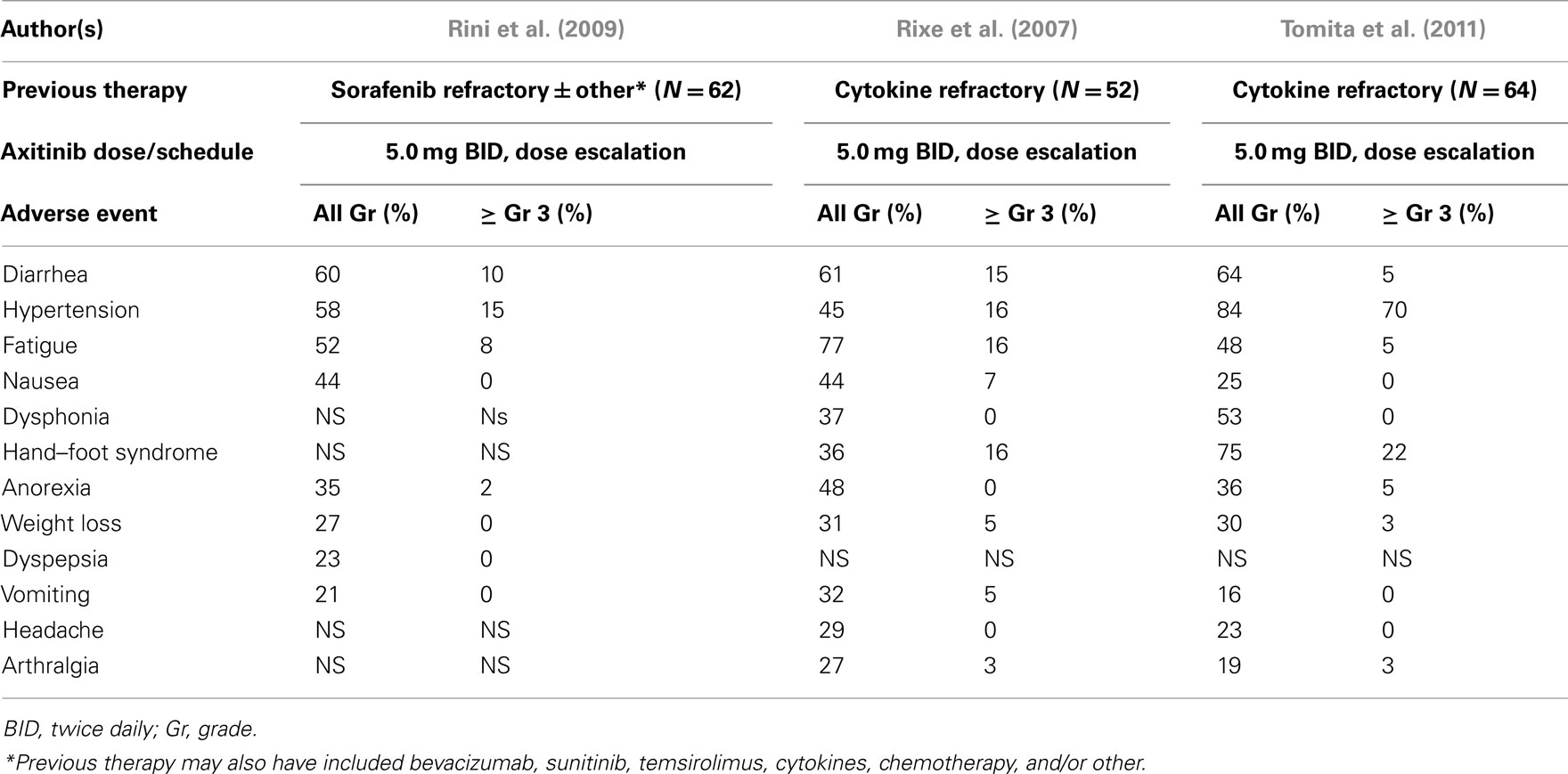
Table 5. Phase 2 axitinib trials – adverse events.
An interesting association between diastolic blood pressure and clinical outcome has been suggested by retrospective reviews of these trials. Hypertension is common following administration of a third generation TKI such as axitinib. It is generally easily managed with standard antihypertensive agents. In multiple tumor types, across, the occurrence of diastolic hypertension ≥90 mmHg during axitinib treatment was associated with increased efficacy, including OS (Rini et al., 2011a). A pooled analysis of data from the axitinib phase 2 renal cancer trials (N = 114) explored the relationship between pharmacokinetics, diastolic hypertension, and clinical efficacy (Rixe et al., 2009). Both diastolic hypertension ≥90 mmHg and AUC appear to be independent predictors of clinical efficacy, with clinical responses also associated with increasing axitinib plasma exposure. These observations are of interest, and are being examined in a prospective manner. The possibility that hypertension is not merely related to higher axitinib drug levels, but itself is a biomarker of efficacy exists.
The laboratory abnormalities reported in these studies are summarized in Table 6. In the two trials conducted in the United States and Western Europe, minimal hematologic toxicity was reported, with treatment-related proteinuria recorded in 2/17 patients by Rixe et al. (2007). In contrast, proteinuria was reported in a much higher frequency by Tomita et al. (2011) in the Japanese population studied, with 28% developing levels ≥2.0 g/24 h requiring treatment interruption and/or discontinuation.
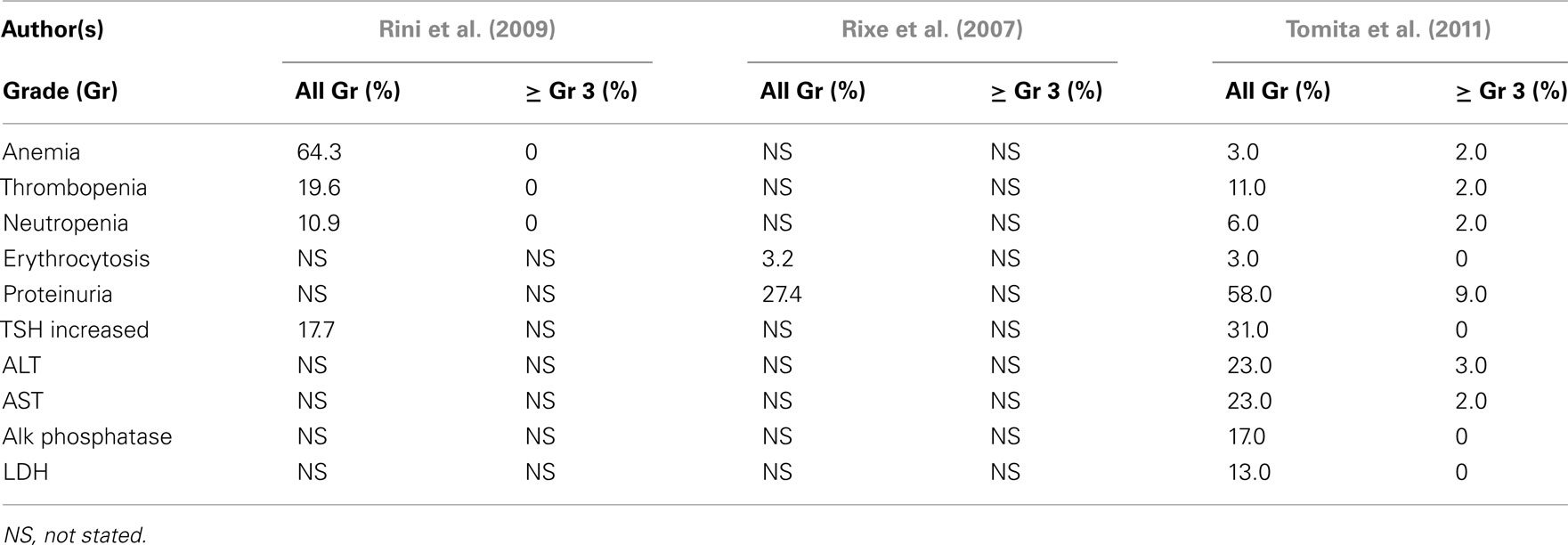
Table 6. Phase 2 axitinib trials – laboratory abnormalities.
Tivozanib
A preliminary report of the single phase 2 trial of tivozanib in mRCC patients is available. The study utilized a random discontinuation design (Bhargava et al., 2010), with 272 patients predominantly from Eastern Europe treated. Tivozanib was administered at a dose of 1.5 mg daily for 3 weeks, followed by a 1-week rest (3/1 week schedule). This open-label study phase involved 16 weeks of initial therapy, at which time patients were randomized dependent upon the degree of tumor regression noted: (1) Group A: ≥25% tumor regression – continue open-label tivozanib; (2) Group B: <25% change from baseline – randomized in a double blind fashion to continue tivozanib or placebo for 12 weeks; (3) Group C: ≥25% tumor growth – stop therapy. The patient population included untreated (53.7%) as well as refractory individuals, as well as patients with clear cell (83.1%) and non-clear cell (16.9%) histology. This design therefore was somewhat different that the phase 2 RTD pazopanib trial (Hutson et al., 2010), and not only included a significant proportion of treatment-naïve patients, but also patients with non-clear cell histology.
In the RTD portion of this trial, 111 patients with SD were randomized to either tivozanib or a placebo (Nosov et al., 2010). The median PFS was longer for patients continuing tivozanib (n = 58; 12.1 months) compared to the placebo group (n = 53; 6.3 months). A significantly greater proportion of patients remained progression free after 12 weeks of on the tivozanib arm (p = 0.003). In the placebo group, 26/29 patients with PD on placebo, 26 crossed back to open-label tivozanib. Of this group, 24/26 had either SD or a response.
The data for all patients entered as well as the various patient subsets is summarized in Table 7.The median treatment duration was reported as 8.5 months (range 0.03–23.8 months). Of interest, the median PFS for all patients was reported as 11.8 months. It is not clear how the RTD design impacted these data. The various subset analyses demonstrate increased efficacy for the patients with clear cell histology and prior nephrectomy. The reported toxicity profile of tivozanib is summarized in Table 8. The AE’s reported are similar to those seen with other VEGFR inhibitors, including hypertension in 50% of individuals. Tivozanib associated hepatic toxicity was modest.
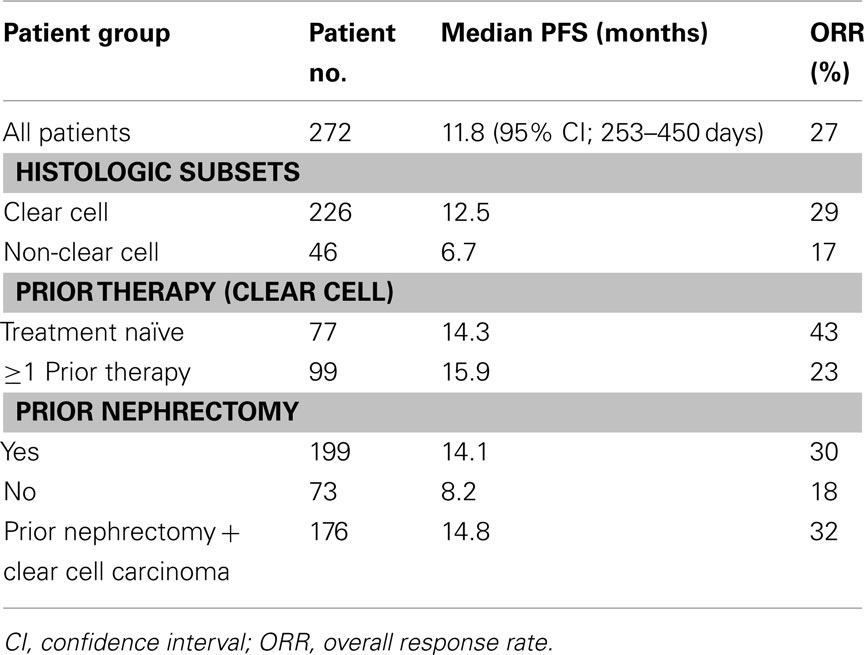
Table 7. Phase 2 tivozanib randomized discontinuation trial (Bhargava et al., 2010; Bhargava and Robinson, 2011).
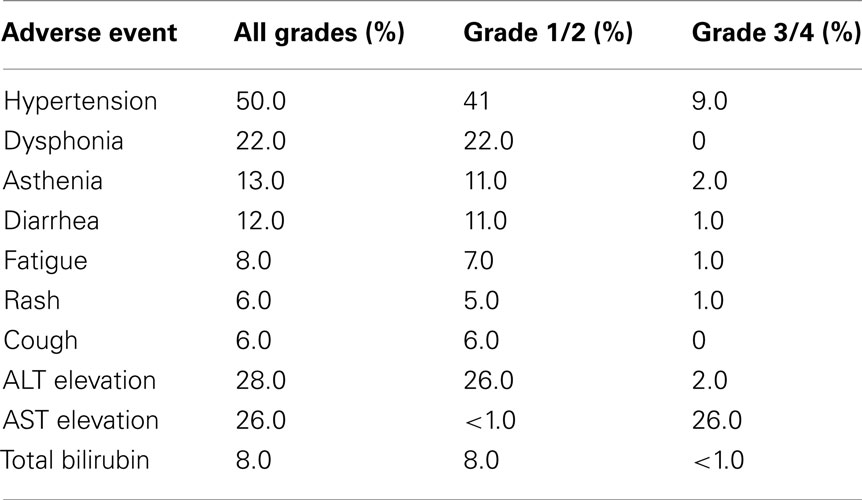
Table 8. Phase 2 trial tivozanib – adverse events and laboratory abnormalities (Bhargava et al., 2010; Bhargava and Robinson, 2011).
In summary, the results of this trial demonstrate tivozanib has significant efficacy in RCC with an acceptable AE profile. The study design was not optimal, and the RTD approach was probably unnecessary for an agent expected to be active in RCC. It is also unclear how limiting patient entry to predominantly Eastern Europe affected the evaluation of efficacy and toxicity. Additional studies to further delineate the clinical activity of tivozanib are now required.
Phase 3 Trials: Metastatic RCC
Phase 3 trials examining either axitinib or tivozanib in patients with mRCC have been conducted. The overall study designs were similar, and utilized a common active comparator arm, sorafenib. Patient populations were however different, and included treatment refractory patients in the phase 3 axitinib trial, in contrast to the inclusion of cytokine refractory and treatment-naïve patients in the phase 3 tivozanib trial (see Figure 2). The primary endpoint in each trial was PFS determined by independent review.
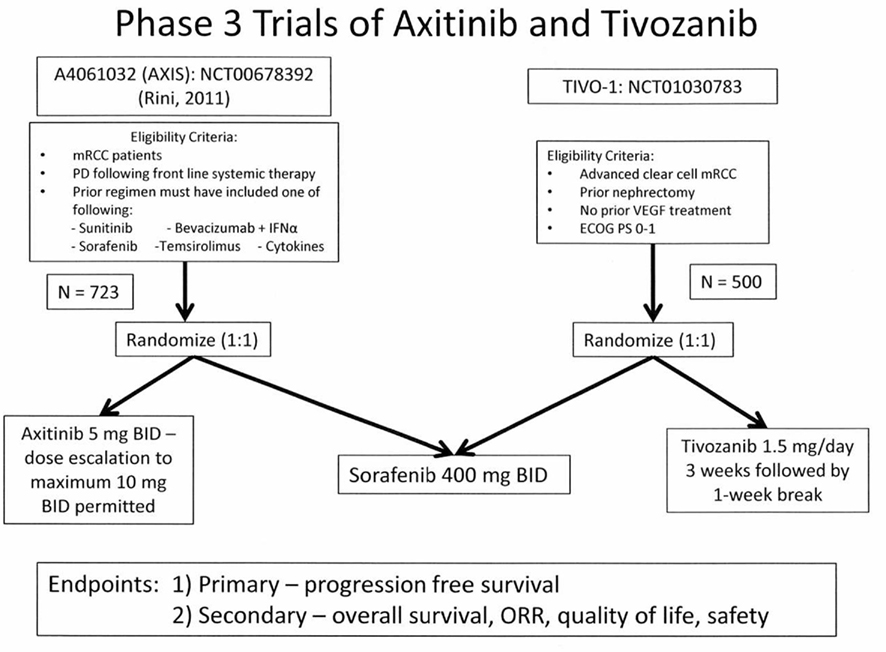
Figure 2. Phase 3 pivotal trial (AXIS and TIVO-1) designs for studies of axitinib or tivozanib vs sorafenib.
Phase 3 Axitinib Trial (AXIS)
The pivotal Phase 3 mRCC Study (Rini et al., 2011b) investigating axitinib was a randomized comparative trial vs the approved TKI, sorafenib (Figure 2). Patients with advanced mRCC after failure of one prior systemic first-line regimen were eligible. Prior therapy may have included one or more of the following agents: sunitinib, bevacizumab, temsirolimus, or cytokine(s). Patients also had histologically or cytologically confirmed RCC with a component of clear-cell subtype, with measurable disease per RECIST criteria (version 1.0). Patients were randomized in a 1:1 ratio to axitinib (5 mg BID administered orally with food), or sorafenib (400 mg BID administered orally without food). In patients receiving axitinib, who tolerated therapy, dose escalation to 7 mg BID and then to a maximum of 10 mg BID was permitted. The trial design investigated whether treatment could increase the median PFS from 5 months (sorafenib cohort), to 7 months for axitinib therapy. A sample size of 650 patients with 409 events (progressive disease or death), based on 90% power (one-sided log-rank test, p value 0.025) was estimated.
A total of 723 patients were randomized to either axitinib (n = 361) or sorafenib (n = 362). The median PFS was 6.7 months for axitinib treated patients, compared to 4.7 months for sorafenib group (hazard ratio 0.665; 95% CI 0.544, 0.812; one-sided p < 0.0001). When the four prespecified subgroups of patients were examined (Table 9), the majority of individuals had previously received either sunitinib (54%) or cytokines (35%). The major difference in PFS was noted in the cytokine refractory cohort, and in the sunitinib refractory group, the difference in PFS was <1.5 months (4.8 vs 3.4 months). The groups previously treated with bevacizumab or temsirolimus were of limited size.
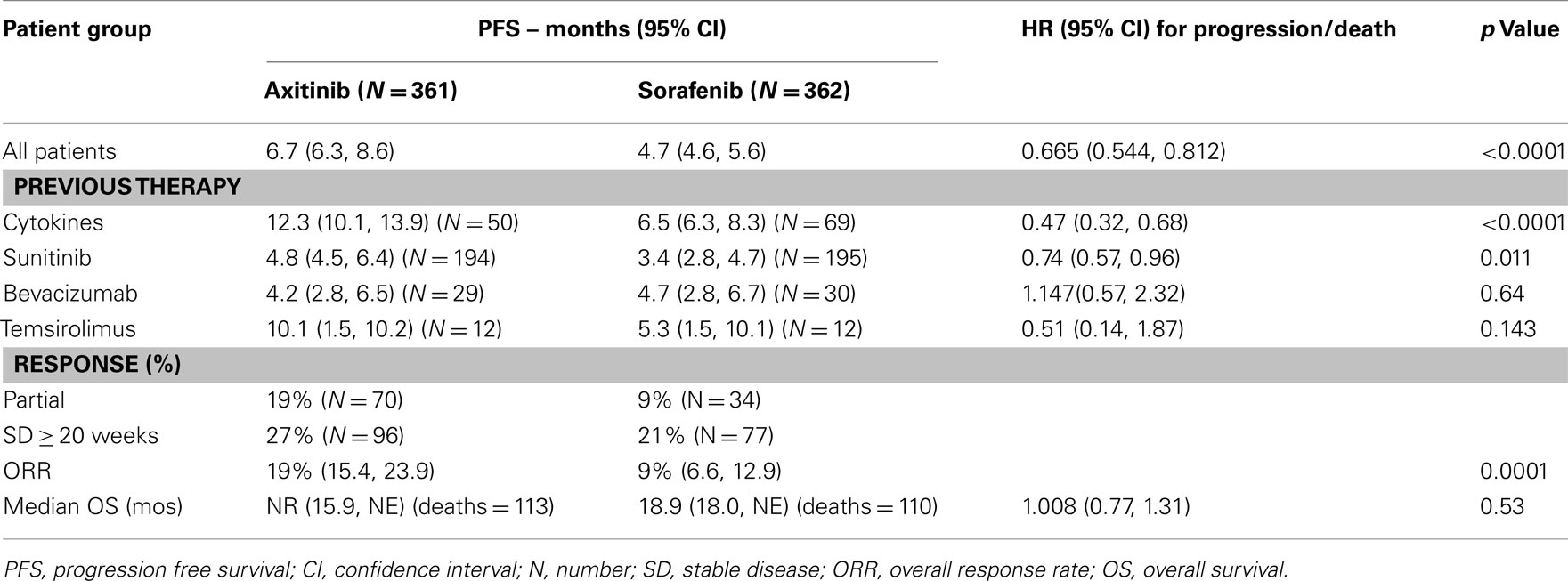
Table 9. Phase 3 trial axitinib versus sorafenib – efficacy (FDA, 2011; Rini et al., 2011b).
Secondary endpoints in this study included ORR and survival. Partial responses assessed by an independent review committee, were more frequent in axitinib treated patients. The median response durations reported were 11.0 (95% CI; 7.4, not estimable) months for the axitinib group, and 10.6 (95% CI; 8.8, 11.5) months for sorafenib treated patients. The survival data were described as immature (Rini et al., 2011b), however, an interim analysis presented at the December 7, 2011 meeting of the FDA ODAC committee demonstrated overlapping survival curves (FDA, 2011), with the median OS demonstrating no differences. The final survival analysis however, is pending. Potential issues confounding the survival data include crossover from sorafenib to axitinib, however this was not permitted. A second issue involved continuation of treatment following disease progression. Fifty of one hundred eighty-seven (26.7%) axitinib patients compared to 74/214 (34.6%) sorafenib treated patients continued the respective TKI ≥ 28 days following PD.
The axitinib AE profile appeared similar to other small molecule VEGF pathway inhibitors. Selected AE’s reported in the AXIS study are summarized in Table 10. These include common AEs such as diarrhea, nausea, hypertension, fatigue, and cutaneous toxicity, as well as uncommon events such as arterial and venous thrombosis, gastrointestinal perforation, bleeding, hypothyroidism, proteinuria, and the reversible posterior leukoencephalopathy syndrome. In general, higher rates of gastrointestinal events, fatigue, asthenia, hypertension, hypothyroidism (19 vs 9%), and dysphonia (32 vs 14%) were seen in patients receiving axitinib compared to sorafenib. In contrast, axitinib treated patients had lower rates of dermatologic AEs. Hepatic toxicity produced by axitinib appears less compared to other to VEGF pathway small molecule inhibitors, with a lower degree of liver enzyme elevation. In the current trial, treatment was discontinued because of toxicity in 14/359 (4%) patients receiving axitinib and 29/355 (8%) sorafenib patients. Diarrhea and hypertension accounted for the majority of dose modifications in the axitinib cohort, while sorafenib patients more often required dose modifications for palmar–plantar erythrodysesthesia and diarrhea.
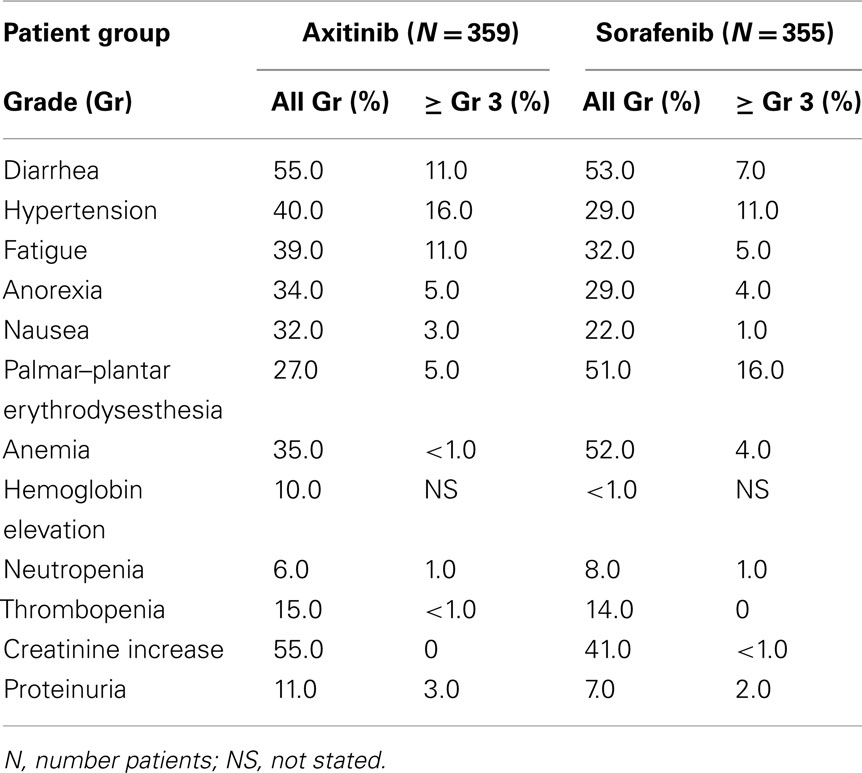
Table 10. Phase 3 trial axitinib versus sorafenib (AXIS) – selected AE’s and laboratory abnormalities (Rini et al., 2011b).
Patient questionnaires (FKSI and FSKI-DRS) were completed by over 90% of subjects. The estimated means in the FKSI-15 and FKSI-DRS were similar between the two treatment arms. Overall, axitinib treatment resulted in patient reported outcomes comparable to sorafenib in second-line mRCC patients (Cella et al., 2011). A composite time to deterioration endpoint (death, progression, worsening of FSKI), showed a 25% risk reduction for axitinib vs sorafenib (p = 0.0001; Rini et al., 2011b).
An application for axitinib approval as therapy for a broad mRCC indication has been submitted to the FDA, and is currently under review. Several additional studies investigating the efficacy of axitinib in mRCC patients are in progress. A phase 2 double blind randomized trial in treatment-naïve patients investigating the effects of dose escalation on safety and efficacy is underway (NCT00835978, 2011). A second phase 3 trial in 447 treatment naïve, or sunitinib and/or cytokine refractory patients comparing the efficacy of axitinib and sorafenib is also in progress (NCT00920816, 2011). These studies will provide additional information on the safety and efficacy of axitinib, prospective data on the efficacy of dose escalation, and additional information on outcomes in the refractory and treatment-naïve patient subsets.
Phase 3 Tivozanib Trial (TIVO-1)
Figure 2 also illustrates the design of the pivotal TIVO-1 trial in mRCC patients (NCT01030783, 2011). As in the AXIS trial, an active comparator arm employing sorafenib was utilized. The patients eligible however, include individuals who have not received prior VEGF therapy, therefore essentially comprising a treatment naïve as well as a cytokine refractory group. Additionally, prior nephrectomy and an ECOG performance status of 0–1 are required. The primary endpoint is PFS, with secondary endpoints of OS, ORR, and quality of life. A total of 500 patients have been randomized (1:1) to receive either sorafenib or tivozanib. This study is ongoing, accrual has been completed, and final data analysis is pending.
Summary and Conclusion
The currently available targeted agents provide significant clinical benefit for patients with mRCC, however, these agents also produce significant off-target toxicities that may limit their effectiveness. The development of additional VEGFR TKIs with improved potency and selectivity therefore may provide more effective and better tolerated treatment options. Additionally, the possibility that combinations may be easier to develop utilizing the more selective third generation TKI’s exists.
1. Do we need another TKI for metastatic RCC? The currently available therapeutic options have clearly improved patient outcomes as measured by the surrogate PFS. Improvement of OS has been more difficult to demonstrate, possibly because of study design and the inclusion of crossover treatment options in many of the trials. The third generation of TKI’s now under study, are still utilizing PFS as the primary endpoint, however, the study designs involve the active comparator sorafenib. Examination of the two pivotal trials which compare either axitinib or tivozanib to sorafenib suggest the following:
a. Studies in the treatment-naïve population to determine if the increased in vitro potency and selectivity for VEGFR will translate into increased efficacy are required. Comparisons with existing TKI’s such as sunitinib or pazopanib are necessary to provide this type of evidence. If increased effectiveness cannot be demonstrated, than the issue of tolerability and toxicity should be critically addressed. Again, this will require comparative studies with current standards of care. The currently completed studies have not addressed these issues. The TIVO-1 trial will provide information on the treatment-naïve patient population, but this is a subset of the study population, with sorafenib, an agent never demonstrated in a phase 3 trial to improve outcomes for mRCC patients, utilized as the comparator. Similarly, data on the efficacy of axitinib in the treatment-naïve patient subset will be available, but the comparator remains sorafenib.
b. Studies in refractory patient populations to determine if this third generation of TKI’s can provide clinical benefit for patients when utilized as sequential therapy following either VEGF inhibitor therapy or cytokines. From the available data, it is clear these two patient populations are very different clinically in their responses to second-line agents.
(1) Cytokine refractory or intolerant mRCC: this patient population has become increasingly uncommon as the utilization of TKI’s for treatment-naïve patients has been adopted. In recent phase 3 trials, this is clearly demonstrated with 46% of patients in the phase 3 pazopanib trial and 35% of patients in the phase 3 axitinib trial classified as cytokine refractory. Additionally, the frequency of initial cytokine therapy will vary from country to country, and in the United States, initial cytokine therapy is increasingly less common. Therefore, the relevance of including this group as a subset of patients in future clinical trials has been questioned. In the TIVO-1 trial, this patient group will also be prominently represented.
(2) TKI refractory mRCC: this patient group is the most frequent refractory population encountered in the United States. The RECORD 1 trial investigating everolimus vs a placebo clearly demonstrated an oral mTOR inhibitor can improve PFS compared to supportive care alone (Motzer et al., 2008). This agent is therefore a reasonable comparator in TKI refractory patient studies. In the AXIS trial, 54% of patients had received sunitinib as frontline therapy, representing the majority of individuals in this trial. In Record 1, 211 patients had received one prior TKI, including 124 who had only received sunitinib. In this trial, patients may also have received other agents including cytokines or bevacizumab, therefore the patients subsets in these two studies are potentially different. The number of patients and their outcome in the RECORD 1 trial receiving only sunitinib as frontline therapy are not available. These two trials in the TKI refractory patient subset are contrasted in Table 11. An analysis of the RECORD 1 data has demonstrated that patients receiving prior sunitinib therapy had a poorer prognosis compared to those receiving sorafenib. The timing of therapy is however not directly addressed by this analysis. The results of these two trials does permit even indirect comparisons of which agent is best utilized in the second line setting. Both study results demonstrate modest effects on PFS in the refractory disease setting, and as described, the characteristics of each treatment population are different. Direct prospective comparisons in a well-defined population are therefore necessary. In the absence of such a trial, sequential use of agents with different mechanisms of action seems a reasonable alternative.
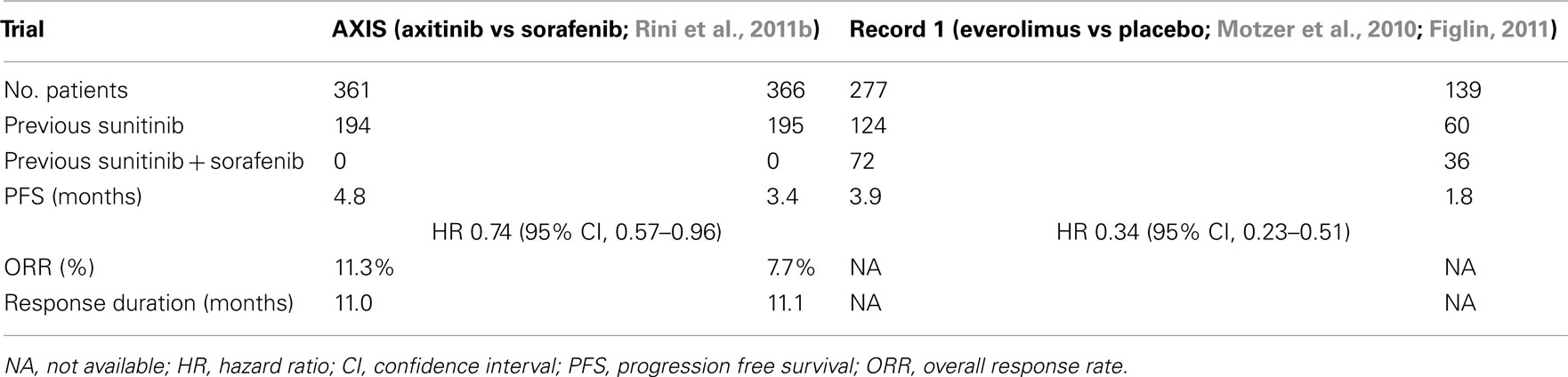
Table 11. AXIS and RECORD 1 trials – results in sunitinib refractory patients.
In summary, in view of the lack of prospective comparative data in frontline patients, limited information on the mechanisms of resistance and progression in RCC patients receiving TKI’s, and prospective data in which comparisons are limited, treatment decisions, and recommendations should be based on available level 1 evidence. The third generation of TKI’s appears potentially interesting with efficacy similar or better than existing agents, and acceptable AE profiles. Demonstration of improved or less toxicity requires randomized studies utilizing appropriate comparisons and patient populations. The possibility that the third generation of TKI’s can be utilized in combination approaches in view of their apparent improved safety profiles is an important consideration. Finally, the lack of Cooperative group interest or participation in such trials is a major concern limiting the further development of the knowledge base delineating therapy for advanced RCC.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Bhargava, P., Esteves, B., Al-Adhami, M., Nosov, D. A., Lipatov, O. N., Lyulko, A. A., Anischenko, A. A., Chacko, R. T., Doval, D. C., and Slichenmyer, W. (2010). Activity of tivozanib (AV-951) in patients with renal cell carcinoma (RCC): subgroup analysis from a phase II randomized discontinuation trial (RDT). J. Clin. Oncol. 28(Suppl.), :15s.
Bhargava, P., and Robinson, M. O. (2011). Development of second-generation VEGFR tyrosine kinase inhibitors: current status. Curr. Oncol. Rep. 13, 103–111.
Bukowski, R. M. (2009). Tyrosine kinase inhibitors in advanced renal cell carcinoma: the evolving treatment paradigm. Clin. Genitourin. Cancer 7, 9–10.
Cella, D., Escudier, B., Rini, B. I., Chen, C., Bhattacharyya, H., Tarazi, J. C., Rosbrook, B., Kim, S., and Motzer, R. J. (2011). Patient-reported outcomes (PROs) in a phase III AXIS trial of axitinib versus sorafenib as second-line therapy for metastatic renal cell carcinoma (mRCC). J. Clin. Oncol. 29(Suppl.), abstr. :4504.
Choueiri, T. K., Hutson, T. E., and Bukowski, R. M. (2003). Evolving role of pegylated interferons in metastatic renal cell carcinoma. Expert Rev. Anticancer Ther. 3, 823–829.
Chow, L. Q., and Eckhardt, S. G. (2007). Sunitinib: from rational design to clinical efficacy. J. Clin. Oncol. 25, 884–896.
Eskens, F., de Jonge, M., Bhargava, P., Isoe, T., Cotreau, M. M., Esteves, B., Hayashi, K., Burger, H., Thomeer, M., van Doorn, L., and Verweij, J. (2011). Biologic and clinical activity of tivozanib (AV-951, KRN-951), a selective inhibitor of VEGF receptor-1, -2, and -3 tyrosine kinases, in a 4-week-on, 2-week-off schedule in patients with advanced solid tumors. Clin. Cancer Res. 17, 1–8.
FDA. (2011). “FDA Briefing Document,” in Oncologic Drugs Advisory Committee Meeting, December 7, NDA 202324 Axitinib (Inlyta®) (New York: Pfizer, Inc.), 1–27.
Figlin, R. A., Calvo, E., Motzer, R. J., Hutson, T. E., Oudard, S., Porta, C., Grunwald, V., Ravaud, A., and Kpamegan, E. (2011). Everolimus in metastatic renal cell carcinoma (mRCC): subgroup analysis of patients (pts) with one versus two prior vascular endothelial growth factor receptor tyrosine kinase inhibitor (VEGFR-TKI) therapies enrolled in the phase III RECORD-1 study. J. Clin. Oncol. 29(Suppl.), abstr. :304.
Gupta, S., and Fishman, M. (2011). Progress and contrasts of the development of tivozanib for therapy of kidney cancer. Expert Opin. Pharmacother. 12, 2915–2922.
Hu-Lowe, D. D., Zou, H. Y., Grazzini, M. L., Hallin, M. E., Wickman, G. R., Amundson, K., Chen, J. H., Rewolinski, D. A., Yamazaki, S., Wu, E. Y., McTigue, M. A., Murray, B. W., Kania, R. S., O’Connor, P., Shalinsky, D. R., and Bender, S. L. (2008). Nonclinical antiangiogenesis and antitumoractivities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 14, 7272–7283.
Hutson, T. E., Davis, I. D., Michiels, J. P. H., De Souza, P. L., Rottey, S., Hong, B., Epstein, R. J., Baker, K. L., McCann, L., Crofts, T., Pandite, L., and Figlin, R. A. (2010). Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 28, 475–480.
Kaelin, W. G. Jr. (2007). The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin. Cancer Res. 13, 680s–684s.
Kania, R. S. (2009). “Structure-based design and characterization of axitinib,” in Kinase Inhibitor Drugs, eds L. Rongshi and J. A. Stafford (Hoboken: John Wiley & Sons Inc.), 167–201.
Kumar, R., Crouthamel, M. C., Rominger, D. H., Gontarek, R. R., Tummino, P. J., Levin, R. A., and King, A. G. (2009). Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br. J. Cancer 101, 1717–1723.
McDermott, D. F. (2009). Immunotherapy of metastatic renal cell carcinoma. Cancer 115(Suppl.), 2298–2305.
Motzer, R. J., Escudier, B., Oudard, S., Hutson, T. E., Porta, C., Bracarda, S., Grunwald, V., Thompson, J. A., Figlin, R. A., Hollaender, N., Kay, A., Ravaud, A., RECORD-1 Study Group. (2010). Phase 3 trial of everolimus for metastatic renal cell carcinoma final results and analysis of prognostic factors. Cancer 116, 4256–4265.
Motzer, R. J., Escudier, B., Oudard, S., Hutson, T. E., Porta, C., Bracarda, S., Grünwald, V., Thompson, J. A., Figlin, R. A., Hollaender, N., Urbanowitz, G., Berg, W. J., Kay, A., Lebwohl, D., Ravaud, A., RECORD-1 Study Group. (2008). Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 372, 449–456.
Motzer, R. J., Hutson, T. E., Tomczak, P., Michaelson, M. D., Bukowski, R. M., Rixe, O., Oudard, S., Negrier, S., Szczylik, C., Kim, S. T., Chen, I., Baum, C. M., and Figlin, R. A. (2007). Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 356, 115–124.
Nakamura, K., Taguchi, E., Miura, T., Yamamoto, A., Takahashi, K., Bichat, F., Guilbaud, N., Hasegawa, K., Kubo, K., Fujiwara, Y., Suzuki, R., Kubo, K., Shibuya, M., and Isoe, T. (2006). KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res. 66, 9134–9142.
NCT00835978. (2011). Randomized, Double-Blind Phase 2 Study of Axitinib (AG-013736) With or Without Dose Titration in Patients with Metastatic Renal Cell Carcinoma. Available at: clinicaltrial.gov
NCT00920816. (2011). Axitinib (AG-013736) for the Treatment of Metastatic Renal Cell Cancer. Available at: clinicaltrial.gov
NCT01030783. (2011). A Study to Compare Tivozanib (AV-951) to Sorafenib in Subjects with Advanced Renal Cell Carcinoma. Available at: clinicaltrial.gov
Nosov, D. A., Bhargava, P., Esteves, B., Al-Adhami, M., Lipatov, O., Lyulko, A. A., Anischenko, A. A., Chacko, R., Doval, D. C., and Slichenmyer, W. (2010). Phase 2 randomized discontinuation trial (RDT) of tivozanib in patients with renal cell carcinoma (RCC): results in patients randomized to tivozanib vs. placebo. Ann. Oncol. 21(Suppl. 8), viii271–viii303.
Pithavala, Y. K., Tortorici, M., Toh, M., Garrett, M., Hee, B., Kuruganti, U., Ni, G., and Klameruset, K. J. (2010). Effect of rifampin on the pharmacokinetics of axitinib (AG-013736) in Japanese and Caucasian healthy volunteers. Cancer Chemother. Pharmacol. 65, 563–570.
Rini, B. I., Schiller, J. H., Fruehauf, J. P., Cohen, E. E. W., Tarazi, J. C., Rosbrook, B., Bair, A. H., Ricart, A. D., Olszanski, A. J., Letrent, K. J., Kim, S., and Rixe, O. (2011a). Diastolic blood pressure as a biomarker of axitinib efficacy in solid tumors. Clin. Cancer Res. 17, 3841–3849.
Rini, B., Escudier, B., Tomczak, P., Kaprin, A., Szczylik, C., Hutson, T. E., Michaelson, M. D., Gorbunova, V. A., Gore, M. E., Rusakov, I. G., Negrier, S., Ou, Y. C., Castellano, D., Lim, H. O., Uemura, H., Tarazi, J., Cella, D., Chen, C., Rosbrook, B., Kim, S., and Motzer, R. J. (2011b). Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 378, 1931–1939.
Rini, B. I., Wilding, G., Hudes, G., Stadler, W. M., Kim, S., Tarazi, J., Rosbrook, B., Trask, P. C., Wood, L., and Dutcher, J. P. (2009). Phase II study of axitinib in sorafenib-refractory metastatic renal cell carcinoma. J. Clin. Oncol. 27, 4462–4468.
Rixe, O., Bukowski, R. M., Michaelson, M. D., Wilding, G., Hudes, G. R., Bolte, O., Motzer, R. J., Bycott, P., Liau, K. F., Freddo, J., Trask, P. C., Kim, S., and Rini, B. I. (2007). Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 8, 975–984.
Rixe, O., Dutcher, J., Motzer, R. J., Wilding, G., Stadler, W. M., Garrett, M., Pithavala, Y., Kim, S., Tarazi, J., and Rini, B. I. (2009). Diastolic blood pressure (dBP) and pharmacokinetics (PK) as predictors of axitinib efficacy in metastatic renal cell cancer (mRCC). J. Clin. Oncol. 27(Suppl.), abstr. :5045.
Rugo, H. S., Herbst, R. S., Liu, G., Park, J. W., Kies, M. S., Steinfeldt, H. M., Pithavala, Y. K., Reich, S. D., Freddo, J. L., and George Wilding, G. (2005). Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J. Clin. Oncol. 23, 5474–5483.
Sternberg, C. N., Davis, I. D., Mardiak, J., Szczylik, C., Lee, E., Wagstaff, J., Barrios, C. H., Salman, P., Gladkov, O. A., Kavina, A., Zarba, J. J., Chen, M., McCann, L., Pandite, L., Roychowdhury, D. F., and Hawkins, R. E. (2010). Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J. Clin. Oncol. 28, 1061–1068.
Taguchi, E., Nakamura, K., Miura, T., Shibuya, M., and Isoe, T. (2008). Anti-tumor activity and tumor vessel normalization by the vascular endothelial growth factor receptor tyrosine kinase inhibitor KRN951 in a rat peritoneal disseminated tumor model. Cancer Sci. 99, 623–630.
Tomita, Y., Uemura, H., Fujimoto, H., Kanayama, H., Shinohara, N., Nakazawa, H., Imai, K., Umeyama, Y., Ozono, S., Naito, S., Akaza, H., Japan Axitinib Phase II Study Group. (2011). Key predictive factors of axitinib (AG-013736)-induced proteinuria and efficacy: a phase II study in Japanese patients with cytokine-refractory metastatic renal cell carcinoma. Eur. J. Cancer 47, 2592–2602.
Keywords: renal cell carcinoma, tyrosine kinase inhibitors
Citation: Bukowski RM (2012) Third generation tyrosine kinase inhibitors and their development in advanced renal cell carcinoma. Front. Oncol. 2:13. doi: 10.3389/fonc.2012.00013
Received: 15 December 2011;
Accepted: 27 January 2012;
Published online: 15 February 2012.
Edited by:
Ganesh V. Raj, UT Southwestern Medical Center, USAReviewed by:
Scott T. Tagawa, Weill Cornell Medical College, USATanya Dorff, University of Southern California Keck School of Medicine, USA
Copyright: © 2012 Bukowski. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Ronald M. Bukowski, Cleveland Clinic Taussig Cancer Center, Cleveland Clinic Lerner College of Medicine of CWRU, Cleveland Clinic Foundation, 28099 Gates Mills Blvd., Pepper Pike, 44124 OH, USA. e-mail:YnVrb3c0NjRAc2JjZ2xvYmFsLm5ldA==