 Hirohito Yamaguchi1
Hirohito Yamaguchi1
- 1 Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
- 2 Graduate School of Biomedical Sciences, The University of Texas at Houston, Houston, TX, USA
- 3 Center for Molecular Medicine and Graduate Institute of Cancer Biology, China Medical University, Taichung, Taiwan
The ubiquitin–proteasome system is essential for multiple physiological processes via selective degradation of target proteins and has been shown to plays a critical role in human cancer. Activation of oncogenic factors and inhibition of tumor suppressors have been shown to be essential for cancer development, and protein ubiquitination has been linked to the regulation of oncogenic factors and tumor suppressors. Three kinases, AKT, extracellular signal-regulated kinase, and IκB kinase, we refer to as oncokinases, are activated in multiple human cancers. We and others have identified several key downstream targets that are commonly regulated by these oncokinases, some of which are regulated directly or indirectly via ubiquitin-mediated proteasome degradation, including FOXO3, β-catenin, myeloid cell leukemia-1, and Snail. In this review, we summarize these findings from our and other groups and discuss potential future studies and applications in the clinic.
Ubiquitin–Proteasome System and Cancer
In order to maintain cellular homeostasis, the amount of proteins in cells is selectively controlled not only in protein synthesis but also in protein degradation. The ubiquitin–proteasome pathway is essential for multiple physiological systems via selective degradation of target proteins (Hershko and Ciechanover, 1998). The proteins designated for proteasome-mediated degradation are conjugated with polypeptide of ubiquitin, which are then targeted to 26S proteasome complex (Hochstrasser, 1995). Ubiquitination of target protein is regulated through multi-enzyme processes in an ATP-dependent manner. First, the E1 protein, ubiquitin-activating enzyme, activates ubiquitin, which is then transferred to the E2 protein. The E3 protein is an ubiquitin protein ligase that determines the substrate specificity. The RING finger-containing E3 ligase binds to its substrate and the ubiquitinated E2 protein and then directly transfers the ubiquitin from the E2 protein to the substrate (Lipkowitz and Weissman, 2011). On the other hand, the HECT domain-containing E3 ligase can also receive ubiquitin from the E2 protein first through an active-site cysteine of its HECT domain then interacts with its substrate to catalyze the conjugation of the activated ubiquitin to the substrate (Kee and Huibregtse, 2007). Since the specificity of the target proteins for proteasome-mediated degradation is dependent on the interaction between the E3 ligases and their targets, the E3 ubiquitin ligases are critical for regulating the expression levels of key short-lived proteins.
Cancer is a genetic disease that is caused by multiple genetic mutations. In cancer cells, oncogenic drivers that are frequently mutated or overexpressed activate the signaling pathways to promote cell proliferation, growth, and survival while tumor suppressors that are commonly inactivated by mutation or deletion inhibit these pathways. It has been demonstrated that the expression levels of some key oncoproteins and tumor suppressors are under the control of ubiquitin–proteasome system with some E3 ligases that function as oncogenic factors or tumor suppressors. For example, MDM2 and Skp2 ubiquitinate and inhibit tumor suppressors via proteasomal degradation, and thereby function as oncogenic factors (Marine and Lozano, 2010; Wang et al., 2011). In contrast, other E3 ligases such as the anaphase promoting complex/cyclosome (APC/C) and F-box and WD repeat domain-containing 7 (FBW7) serve as tumor suppressors by downregulating oncogenic factors (Crusio et al., 2010; Wasch et al., 2010).
Accumulating evidence indicates that ubiquitin–proteasome pathways are potential drug targets for cancer therapy. For example, bortezomib is a specific proteasome inhibitor that is currently used for the treatment of multiple myeloma. Moreover, the inhibitors for specific E3 ligases have been also considered as potential anti-cancer drugs. MDM2 is the primary ubiquitin ligase for tumor suppressor protein, p53, which induces apoptosis or senescence in response to oncogenic stress or DNA damage. The p53 pathway is frequently inactivated in human cancer cells, and the small molecules that block the interaction between p53 and MDM2 to inhibit p53 degradation have been tested in clinical trials (Brown et al., 2009). Therefore, dissecting the specific ubiquitin–proteasome signaling facilitating cancer progression may contribute to the development of novel drug targets.
Three Survival Kinases and Their Signaling Pathways
Protein kinases and phosphatases catalyze the protein phosphorylation and dephosphorylation, respectively, which are essential for maintaining signal transduction. When cells receive extracellular signaling and stress, the signals are primarily transduced to the nucleus via protein phosphorylation, resulting in the alteration of gene expression. For example, epidermal growth factor (EGF) stimulates cell proliferation by binding to its receptor, EGF receptor (EGFR), and activating it. EGFR is a receptor tyrosine kinase that phosphorylates and activates multiple downstream targets and promotes cell growth and survival. EGFR, considered as an oncogene, is frequently overexpressed or mutated in multiple human cancers and promotes tumor progression, metastasis, and drug resistance (Nicholson et al., 2001; Hynes and Lane, 2005; Quatrale et al., 2011). Indeed, many oncoproteins include protein kinases, and these oncogenic kinases phosphorylate downstream targets to promote tumor growth, metastasis, and/or angiogenesis.
The serine/threonine kinase AKT is one of the major downstream kinases activated by growth factor signaling such as EGFR, platelet-derived growth factor receptor (PDGFR), and insulin-like growth factor receptor (IGFR). AKT has three isoforms, AKT1, AKT2, and AKT3, and their activities are frequently elevated in multiple human cancers, which contribute to cancer cell survival and growth (Altomare and Testa, 2005). AKT is activated by phosphatidylinositol-3 kinase (PI3K) that converts phosphatidylinositol (3,4)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3) in plasma membrane through lipid phosphorylation (Altomare and Testa, 2005; Yuan and Cantley, 2008). AKT and phosphoinositide dependent kinase 1 (PDK1) are then recruited to the plasma membrane where AKT is directly phosphorylated and activated by PDK1 (Chan et al., 1999). Activated AKT phosphorylates various substrates involved in cell metabolism (GSK3, TSC2), survival (Bad, FOXO), and cell cycles (p21, p27, MDM2; Cross et al., 1995; Datta et al., 1997; Brunet et al., 1999; Zhou et al., 2001a,b; Liang et al., 2002; Manning et al., 2002), and then inhibits apoptosis and promotes cell growth. Therefore, the PI3K–AKT pathway is a potential drug target, and several PI3K or AKT inhibitors have been actively tested in numerous clinical trials (Garcia-Echeverria and Sellers, 2008; Wong et al., 2010; Chappell et al., 2011).
Extracellular signal-regulated kinase (ERK) is another critical downstream kinase in growth factor signaling and plays an essential role in cancer cell proliferation (Robinson and Cobb, 1997; Sebolt-Leopold, 2000). The Ras–Raf–MEK–ERK pathway is a well-characterized signaling pathway and is commonly activated in multiple human cancers. Studies have shown that Ras and Raf are frequently mutated in various human cancer types and responsible for cancer progression (Adjei, 2001; Davies et al., 2002). Therefore, this signaling pathway has been considered a drug target for cancer therapy, and a variety of inhibitors have been developed, including MEK or Raf inhibitors (Davies et al., 2007; Bollag et al., 2010; Hatzivassiliou et al., 2010; Maurer et al., 2011).
IκB kinases (IKKs) are the primary regulator of NF-κB, which plays a key role in immune response, cell proliferation, and survival (Baldwin, 2001; Luo et al., 2005). IKKs are activated in response to various cytokines and inflammatory stimuli such as tumor necrosis factor (TNF)-α, interleukin-1, and lipopolysaccharide. The IKK family includes IKKα, IKKβ, IKKγ, IKKε, and TANK-binding kinase 1 (TBK1). Interestingly, IKKγ has no kinase activity and functions as an adaptor protein for the canonical IKK complex (Hacker and Karin, 2006). IKKs are involved in two distinct pathways for NF-κB activation: the canonical and non-canonical pathways. IKKβ plays a dominant role in the canonical pathway and IKKα in the non-canonical pathway (Perkins, 2007; Israel, 2010). In the canonical pathway, IKKα, IKKβ, and IKKγ form a kinase complex that phosphorylates IκBα, an inhibitor protein of NF-κB, and induces the ubiquitination and subsequent proteasome-dependent degradation of IκBα. In the non-canonical pathway, IKKα forms a homodimer and phosphorylates p100, and generates p52 by partial processing of p100, resulting in the activation of p52/RelB. In contrast to IKKα and IKKβ, IKKε and TBK1 play a role in the induction of interferon signaling in response to viral infection (Shen and Hahn, 2011). Although NF-κB has been known to be involved in the progression of various cancers, increasing evidence suggests that IKKs also play vital roles in cancer independently of NF-κB (Lee and Hung, 2008; Baud and Karin, 2009; Shen and Hahn, 2011). For example, IKKα phosphorylates both estrogen receptor α (ERα) and co-activator SRC3 and enhances ERα transcriptional activity while IKKβ phosphorylates a tumor suppressor, tuberous sclerosis 1 (TSC1), and inhibits its function (Park et al., 2005; Lee et al., 2007). IKKα also phosphorylates CBP and the phosphorylated CBP preferentially interacts with NF-κB rather than p53, resulting in NF-κB activation as well as p53 inactivation (Huang et al., 2007). Recently, we found that IKKα phosphorylates and inhibits FOXA2. Inactivation of FOXA2 results in the decrease of NUMB expression, and subsequent NOTCH activation (Liu et al., 2012). Considering that inhibition of NF-κB may affect inflammatory responses, IKKs may be the potential alternative drug targets for cancer.
The above-mentioned three survival kinases play critical roles in cancer cell survival, metabolism, proliferation, and growth. Interestingly, these kinases have many common targets that they directly or indirectly regulate. Moreover, we and others have identified several common targets of these three kinases that are regulated by the ubiquitin–proteasome system. We will discuss these proteins one by one in the later next section of the review.
Forkhead Box O
The forkhead box O (FOXO) family proteins are critical transcription factors that are involved in the regulation of cell proliferation, cell death, cell metabolism, and DNA repair (Tran et al., 2003; Arden, 2008). FOXO family includes FOXO1, FOXO3, FOXO4, and FOXO6, and is conserved from C. elegans to mammals (Burgering, 2008; Calnan and Brunet, 2008). FOXO family proteins directly activate multiple gene expression involved in cell cycles, apoptosis, metabolism, and DNA damage repair, such as p27kip, Bim, FasL, MnSOD, GADD45 (Dijkers et al., 2000; Kops et al., 2002; Tran et al., 2002; Ciechomska et al., 2003; Sunters et al., 2003). Moreover, it has been shown that FOXO proteins are dysregulated in multiple human cancers such as breast, prostate, leukemia, and glioblastoma (Hu et al., 2004; Seoane et al., 2004; Cornforth et al., 2008; Jagani et al., 2008). Conditional knockout mice of FOXO1, 3, and 4 develop thymic lymphomas and hemangiomas (Paik et al., 2007; Tothova et al., 2007). Therefore, FOXO is believed to function as a potential tumor suppressor.
It has been shown that IKK, AKT, and ERK directly phosphorylate FOXO and induce FOXO ubiquitination and degradation. Among the three kinases, AKT was first identified as a FOXO kinase that phosphorylates FOXO3 at T32, S253, and S315. Phosphorylated FOXO3 protein is excluded from entering the nucleus and binds to 14-3-3 in the cytoplasm (Brunet et al., 1999). In addition to FOXO3, FOXO1, and FOXO4 are also phoshorylated by AKT (Tzivion et al., 2011). AKT-phosphorylated FOXO1 and FOXO3 then undergo degradation in a proteasome-dependent manner (Plas and Thompson, 2003). Specifically, FOXO1 phosphorylated by AKT translocates to the cytosol where it is ubiquitinated by Skp2 and subjected to proteasome-dependent degradation (Huang et al., 2005). We found that IKKβ directly phosphorylates FOXO3 at S644 and induces its ubiquitination and degradation (Hu et al., 2004). Recently, E3 ligase β-transducing repeat-containing protein (β-TrCP) is reported to interact with FOXO3 and induces ubiquitination and degradation in an IKKβ-mediated-phosphorylation-dependent manner (Tsai et al., 2010; Su et al., 2011). Furthermore, we and others demonstrated that ERK phosphorylates FOXO3 and FOXO1, respectively (Asada et al., 2007; Yang et al., 2008). We also showed that FOXO3 is phosphorylated by ERK at S294, S344, and S425, which then undergoes MDM2-mediated ubiquitination, followed by proteasome-dependent degradation (Yang et al., 2008). MDM2-mediated ubiquitination and degradation is also observed with FOXO1, which is dependent on the AKT-mediated phosphorylation (Fu et al., 2009). Interestingly, MDM2 induces mono-ubiquitination of FOXO4, which promotes nuclear localization of FOXO4, and subsequent polyubiquitination by Skp2 and degradation (Brenkman et al., 2008). Taken together, the ubiquitin–proteasome system plays an essential role in regulating FOXO transcription factors by AKT, ERK, and IKK, and MDM2, SKP2, and βTrCP are E3 ligases for FOXO ubiquitination (Figure 1A).
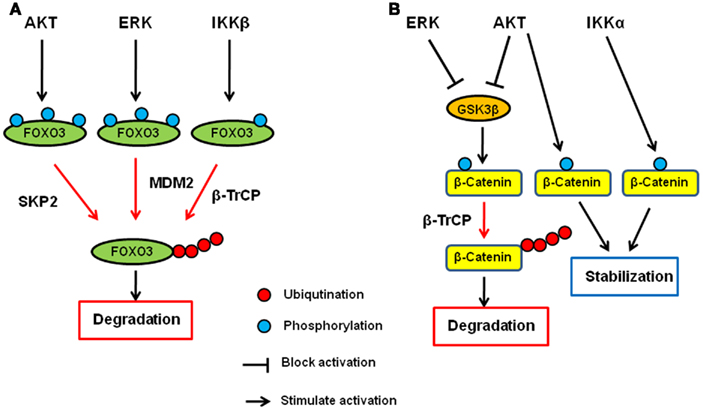
Figure 1. Regulation of FOXO3 and β-catenin by AKT, ERK, and IKK signaling pathway. (A) AKT, ERK, and IKK phosphorylates FOXO3 at different sites and induces its ubiquitination and subsequent degradation via SKP2, MDM2, and β-TrCP, respectively. (B) AKT and ERK phosphorylates GSK3β and inhibit it. GSK3β phosphorylates catenin and induces its ubiquitination and subsequent degradation via β-TrCP. Both AKT and IKKα phosphorylate β-catenin and stabilize it.
β-Catenin
β-Catenin is the key protein in both cadherin junction and Wnt pathway and plays an important role in development and adult homeostasis as well as tumorigenesis (Cadigan, 2008; Stepniak et al., 2009). In the Wnt signaling pathway, β-catenin functions as a transcription co-factor and is involved in the transactivation of several oncogenic proteins such as c-Myc, CyclinD1, and matrix metalloproteases (He et al., 1998; Lin et al., 2000; Mosimann et al., 2009). Glycogen synthase kinase-3β (GSK3β) and casein kinase 1 (CK1) are the major protein kinases regulating the β-catenin stability. In the absence of Wnt ligand, β-catenin forms a complex with Axin, APC, GSK3β, and CK1 and is phosphorylated by these kinases. Once phosphorylated, β-catenin undergoes β-TrCP-mediated ubiquitination and subsequent degradation. When Wnt binds to its receptor, Frizzles, and co-receptor, LRP5/6, the receptor complex recruits Axin–GSK3 complex to cell membrane, releasing β-catenin from the complex for its translocation to the nucleus where it activates gene transcription with T-cell factor (TCF) and lymphocyte enhancer factor (LEF).
AKT has been shown to directly phosphorylate GSK3β and inhibits it (Cross et al., 1995), and therefore, AKT seems to indirectly inhibit β-catenin degradation and inactivation through inhibition of GSK3β (Monick et al., 2001). In addition to the indirect mechanism, AKT directly mediates the β-catenin stability by phosphorylating β-catenin at S552. Once phosphorylated by AKT, β-catenin binds to 14-3-3ζ and is stabilized (Tian et al., 2004; Fang et al., 2007). In addition to AKT, we also demonstrated that ERK upregulates β-catenin via inhibition of GSK3β (Ding et al., 2005). Activated ERK directly interacts with GSK3β and phosphorylates it at T43. After the phosphorylation by ERK, GSK3 is primed for subsequent phosphorylation by p90RSK at S9, which inactivates it. Furthermore, IKK has been shown to be directly involved in the β-catenin regulation, and IKKα but not IKKβ phosphorylates β-catenin and prevent its ubiquitination and degradation (Lamberti et al., 2001; Albanese et al., 2003; Carayol and Wang, 2006). Collectively, β-catenin is another common target of AKT, ERK, and IKK, demonstrating that the ubiquitin–proteasome system plays a critical role in these signaling pathways (Figure 1B).
Myeloid Cell Leukemia-1
Myeloid cell leukemia-1 (Mcl-1) is a member of anti-apoptotic Bcl-2 family proteins which are the central regulators of apoptosis signaling pathway (Inuzuka et al., 2011a). The Bcl-2 family consist of pro-apoptotic and anti-apoptotic proteins that regulate the release of apoptogenic proteins such as cytochrome c and Smac from mitochondria (Youle and Strasser, 2008). Pro-apoptotic Bcl-2 family proteins include the BH3-only proteins such as Bim, Bid, Bik, and Bad and multi-BH domain like Bax and Bak. Bax and Bak are able to form channels on the surface of mitochondria from which cytochrome c and Smac are released. Anti-apoptotic Bcl-2 family proteins, which include Bcl-2, Bcl-XL, and Mcl-1, prevent apoptotic cell death by inhibiting the activation and channel formation of Bax and Bak. In contrast, the BH3-only proteins directly bind to anti-apoptotic Bcl-2 family proteins and inhibit their functions. Thus, the balance of anti-apoptotic and pro-apoptotic proteins determines cell survival and death, and the expression of anti-apoptotic Bcl-2 family protein is the critical for cell survival. Among the anti-apoptotic Bcl-2 family proteins, Mcl-1 has a relatively short protein half-life (Nijhawan et al., 2003; Adams and Cooper, 2007) but its expression is enhanced in various cancer types (Placzek et al., 2010). Thus, Mcl-1 is an important survival factor in human cancer.
The primary kinase that regulates Mcl-1 stability is GSK3β. We and another group reported that GSK3 interacts with and phosphorylates Mcl-1 at S155, S159, and T163 (Maurer et al., 2006; Ding et al., 2007). In addition, phosphorylated Mcl-1 is ubiquitinated and undergoes proteasome-dependent degradation. So far, three possible ubiquitin ligases have been identified for Mcl-1. Mule is the BH3 domain-containing E3 ligase that was the first identified as the Mcl-1 ubiquitin ligase by biochemical purification (Zhong et al., 2005). Later, we found that β-TrCP can ubiquitinate and induce Mcl-1 degradation in GSK3β-mediated-phosphorylation-dependent manner (Ding et al., 2007). Recently, a tumor suppressor protein, FBW7, is shown to function as a Mcl-1 ubiquitin ligase (Inuzuka et al., 2011b; Wertz et al., 2011). FBW7-induced ubiquitination of Mcl-1 is also dependent the phosphorylation induced by GSK3β. Although both β-TrCP and FBW7 recognize the same GSK3β-mediated phosphorylation sites in Mcl-1, the exact roles of these two ubiquitin ligases under various apoptotic stresses are unknown.
AKT and ERK have been shown to upregulate Mcl-1 transcription (Wang et al., 1999; Booy et al., 2011). In addition, both PI3K/AKT and MEK/ERK pathways have also been shown to enhance Mcl-1 protein stability (Derouet et al., 2004). Because AKT inhibits GSK3β as described above, AKT increases Mcl-1 stability, at least in part, by inhibiting GSK3β. Indeed, the inhibition of PI3K induces S159 phosphorylation of Mcl-1 and subsequent ubiquitination and degradation of Mcl-1, which are blocked by GSK3 inhibition (Maurer et al., 2006). In addition to GSK3β, it has been show that ERK phosphorylates Mcl-1 at T92 and T163 (Domina et al., 2004; Ding et al., 2008). In particular, we showed that ERK-phoshorylated Mcl-1 can interact with Pin1 (Ding et al., 2008). Pin1 is a peptidyl–prolyl cis/trans isomerase that binds to specific pS/T-P motifs and then isomerizes its substrates, resulting in their conformational changes. Interestingly, Pin1 stabilizes Mcl-1 protein after the phosphorylation by ERK, and the expression of Mcl-1 correlates with Pin1 in multiple human cancer cell lines (Ding et al., 2008). Thus, ERK and Pin1 cooperatively regulate Mcl-1 stability. Regarding IKK, there is no evidence that IKK is directly involved in Mcl-1 or GSK3β phosphorylation and/or ubiquitination. However, it has been shown that NF-κB is required for EGF-induced Mcl-1 induction, suggesting that the IKK–NF-κB pathway plays a role in Mcl-1 expression or stability (Henson et al., 2003). Thus, Mcl-1 is a critical apoptosis regulator that is controlled by the three kinases at the post-translational as well as transcriptional level (Figure 2A).
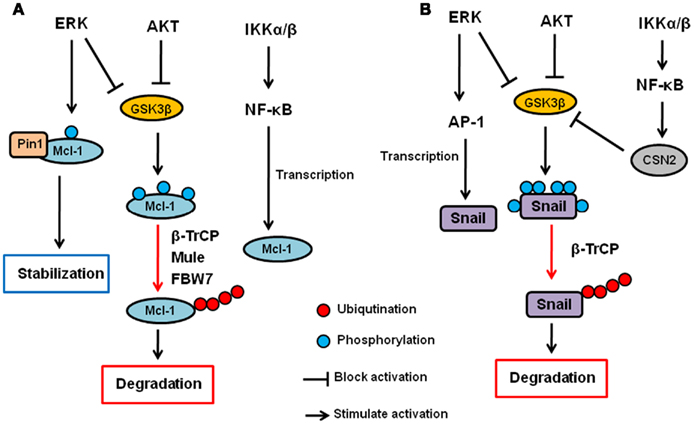
Figure 2. Regulation of Mcl-1 and Snail by AKT, ERK, and IKK signaling pathway. (A) AKT and ERK phosphorylates GSK3β and inhibit it. GSK3β phosphorylates Mcl-1 and induces its ubiquitination and subsequent degradation via β-TrCP, Mule, and/or FBW7. ERK also directly phosphorylates Mcl-1, and phosphorylated Mcl-1 interacts with Pin1, resulting in its stabilization. The IKK–NF-κB pathway also contributes to Mcl-1 upregulation but the mechanisms are uncertain. (B) AKT and ERK phosphorylates GSK3β and inhibit it. GSK3β phosphorylates Snail and induces its nuclear exclusion, ubiquitination and subsequent degradation via β-TrCP. ERK also upregulates Snail transcription via AP-1 transcription factor. The IKK–NF-κB pathway inhibits Snail via upregulation of CSN2, which interferes with the GSK3–Snail interaction and ubiquitination of Snail.
Snail
Epithelial–mesenchymal transition (EMT) is an important physiological process that converts epithelial cells to mesenchymal cells which plays an essential role in embryonic development and tissue repair (Nieto, 2009). In addition, accumulating evidence shows that EMT is also critical for cancer metastasis. Epithelial cells lose cell–cell contacts and gain migratory properties during EMT. During cancer progression, cancer cells undergo EMT, resulting in increased motility, invasiveness, and aggressive behavior (Kalluri and Weinberg, 2009). Snail is a zinc-finger transcription repressor that is one of the EMT regulators. Snail family contains Snail1 (Snail), Snail2 (Slug), and Snail3 (Smuc; de Herreros et al., 2010). Like other EMT regulators, Snail suppresses E-cadherin expression by binding to its promoter. In addition to EMT, Snail is also involved in cell death, survival, stem cell, and immune regulation by controlling multiple target genes (Wu and Zhou, 2010). Downregulation of Snail reduces tumor growth and invasiveness in xenograft animal model (Olmeda et al., 2007). Moreover, Snail is overexpressed in multiple human cancers, and expression of Snail is associated with poor cancer prognosis (Peinado et al., 2007). Therefore, Snail is a critical oncogenic factor.
Snail stability is primarily regulated by GSK3β through the PI3K–AKT pathway (Song et al., 2009). We have shown that GSK3β can phosphorylate Snail at six serine residues in which two of them are responsible for Snail stability while the other four are involved in its nuclear localization (Zhou et al., 2004). First, GSK3β phosphorylates Snail at four serine residues to allow its export from the nucleus. Then, GSK3β phosphorylates the other two sites, resulting in β-TrCP-mediated ubiquitination and degradation of Snail. IKK–NF-κB pathway has also been shown to regulate Snail stability. Specifically, NF-κB induces COP9 signalosome 2 (CSN2), which disrupts the interaction between GSK3 and Snail, resulting in the inhibition of Snail ubiquitination and subsequent degradation (Wu et al., 2009). So far, there is no evidence to show that MEK–ERK signaling pathway is directly involved in Snail stability. However, ERK does play a role in c-Myc-induced EMT via the inhibition of GSK3β, and there by stabilizing Snail (Cho et al., 2010). Furthermore, ERK upregulates Snail gene transcription though activation of AP-1 transcription factor (Hudson et al., 2007; Li et al., 2010). Independently of kinases, wild type p53 but not mutant p53 has been shown to interact with and induce Snail and Slug ubiquitination and degradation (Wang et al., 2009; Lim et al., 2010). MDM2 functions as an ubiquitin ligase for p53-induced Snail ubiquitination. Moreover, F-box and leucine-rich repeat protein 14 (FBXL14) has been shown to interact with and induce Snail ubiquitination and degradation (Vinas-Castells et al., 2010). So far, no other kinases have been reported to be involved in MDM2- or FBXL-mediated Snail ubiquitination. Thus, the three kinases (AKT, ERK, and IKK) seem to be involved in EMT, at least in part, by regulating Snail expression through ubiquitination (Figure 2B).
Conclusion and Future Prospect
Recent advances in signal transduction studies have identified many key oncogenic kinases and their substrates in cancer progression, and the signaling pathways associated with these kinases have been recognized as promising drug targets. Indeed, several kinase inhibitors have been developed and used in clinic that show high efficacy and low toxicity (Sharma and Settleman, 2010). However, several clinical studies have emerged showing that some patients exhibit little or no response to these targeted drugs, and those who originally responded the drugs eventually developed resistance. Although the detailed mechanisms underlying drug resistance are not fully understood, there is evidence to support that alternative pathways are being activated in resistant cells to compensate for the survival signal blocked by targeting agents. For example, EGFR tyrosine kinase inhibitors (TKIs) are effective drugs for EGFR mutant lung cancer, but c-MET amplification or K-Ras mutation causes the resistance to TKIs by bypassing the inhibition of survival signaling (Bean et al., 2007; Linardou et al., 2008). We also showed that cancer cells with high AKT signaling pathway exhibit resistance to ERK inhibitors by inhibiting FOXO3 (Yang et al., 2010). Therefore, in order to develop effective personalized cancer therapy, it would be essential that we understand the cross-talk among the multiple oncogenic signaling pathways.
In this review, we introduced AKT, ERK, and IKK as the key survival kinases for cancer progression and survival. We also mentioned that these three pathways have several common targets that are critical for cancer cell proliferation, survival, and EMT. Because there are many other pathways that are activated in human cancers, we believe that they likely also contribute to the same targets we described here. These three kinase-signaling pathways may have other common targets that are critical for cancer progression. We also introduce some examples of the signaling pathways that are controlled via the ubiquitin–proteasome system, which seems to play an essential role in signaling pathway like phosphorylation. As we described above, inhibition of protein ubiquitination has been demonstrated to be potential drug targets for cancer therapy. Moreover, blockade of specific ubiquitination may exhibit less toxicity because inhibition of upstream molecules in key oncogenic signaling pathways may affect numerous signaling pathways and induce unfavorable side effects. Clearly, further studies for signaling pathways in cancer including post-translational modifications are required for the development of effective personalized cancer therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by NIH PO1 (CA099031), RO1 (CA109311), the National Breast Cancer Foundation Inc., Center for Biological Pathways, and Sister Institution Fund of China Medical University and Hospital, and MD Anderson Cancer Center.
References
Adams, K. W., and Cooper, G. M. (2007). Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J. Biol. Chem. 282, 6192–6200.
Adjei, A. A. (2001). Blocking oncogenic Ras signaling for cancer therapy. J. Natl. Cancer Inst. 93, 1062–1074.
Albanese, C., Wu, K., D’Amico, M., Jarrett, C., Joyce, D., Hughes, J., Hulit, J., Sakamaki, T., Fu, M., Ben-Ze’Ev, A., Bromberg, J. F., Lamberti, C., Verma, U., Gaynor, R. B., Byers, S. W., and Pestell, R. G. (2003). IKKalpha regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol. Biol. Cell 14, 585–599.
Altomare, D. A., and Testa, J. R. (2005). Perturbations of the AKT signaling pathway in human cancer. Oncogene 24, 7455–7464.
Arden, K. C. (2008). FOXO animal models reveal a variety of diverse roles for FOXO transcription factors. Oncogene 27, 2345–2350.
Asada, S., Daitoku, H., Matsuzaki, H., Saito, T., Sudo, T., Mukai, H., Iwashita, S., Kako, K., Kishi, T., Kasuya, Y., and Fukamizu, A. (2007). Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell. Signal. 19, 519–527.
Baldwin, A. S. Jr. (2001). Series introduction: the transcription factor NF-kappaB and human disease. J. Clin. Invest. 107, 3–6.
Baud, V., and Karin, M. (2009). Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 8, 33–40.
Bean, J., Brennan, C., Shih, J. Y., Riely, G., Viale, A., Wang, L., Chitale, D., Motoi, N., Szoke, J., Broderick, S., Balak, M., Chang, W. C., Yu, C. J., Gazdar, A., Pass, H., Rusch, V., Gerald, W., Huang, S. F., Yang, P. C., Miller, V., Ladanyi, M., Yang, C. H., and Pao, W. (2007). MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. U.S.A. 104, 20932–20937.
Bollag, G., Hirth, P., Tsai, J., Zhang, J., Ibrahim, P. N., Cho, H., Spevak, W., Zhang, C., Zhang, Y., Habets, G., Burton, E. A., Wong, B., Tsang, G., West, B. L., Powell, B., Shellooe, R., Marimuthu, A., Nguyen, H., Zhang, K. Y., Artis, D. R., Schlessinger, J., Su, F., Higgins, B., Iyer, R., D’Andrea, K., Koehler, A., Stumm, M., Lin, P. S., Lee, R. J., Grippo, J., Puzanov, I., Kim, K. B., Ribas, A., Mcarthur, G. A., Sosman, J. A., Chapman, P. B., Flaherty, K. T., Xu, X., Nathanson, K. L., and Nolop, K. (2010). Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599.
Booy, E. P., Henson, E. S., and Gibson, S. B. (2011). Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene 30, 2367–2378.
Brenkman, A. B., De Keizer, P. L., van den Broek, N. J., Jochemsen, A. G., and Burgering, B. M. (2008). Mdm2 induces mono-ubiquitination of FOXO4. PLoS ONE 3, e2819.
Brown, C. J., Lain, S., Verma, C. S., Fersht, A. R., and Lane, D. P. (2009). Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer 9, 862–873.
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., Anderson, M. J., Arden, K. C., Blenis, J., and Greenberg, M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868.
Carayol, N., and Wang, C. Y. (2006). IKKalpha stabilizes cytosolic beta-catenin by inhibiting both canonical and non-canonical degradation pathways. Cell. Signal. 18, 1941–1946.
Chan, T. O., Rittenhouse, S. E., and Tsichlis, P. N. (1999). AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu. Rev. Biochem. 68, 965–1014.
Chappell, W. H., Steelman, L. S., Long, J. M., Kempf, R. C., Abrams, S. L., Franklin, R. A., Basecke, J., Stivala, F., Donia, M., Fagone, P., Malaponte, G., Mazzarino, M. C., Nicoletti, F., Libra, M., Maksimovic-Ivanic, D., Mijatovic, S., Montalto, G., Cervello, M., Laidler, P., Milella, M., Tafuri, A., Bonati, A., Evangelisti, C., Cocco, L., Martelli, A. M., and Mccubrey, J. A. (2011). Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget 2, 135–164.
Cho, K. B., Cho, M. K., Lee, W. Y., and Kang, K. W. (2010). Overexpression of c-myc induces epithelial mesenchymal transition in mammary epithelial cells. Cancer Lett. 293, 230–239.
Ciechomska, I., Pyrzynska, B., Kazmierczak, P., and Kaminska, B. (2003). Inhibition of Akt kinase signalling and activation of Forkhead are indispensable for upregulation of FasL expression in apoptosis of glioma cells. Oncogene 22, 7617–7627.
Cornforth, A. N., Davis, J. S., Khanifar, E., Nastiuk, K. L., and Krolewski, J. J. (2008). FOXO3a mediates the androgen-dependent regulation of FLIP and contributes to TRAIL-induced apoptosis of LNCaP cells. Oncogene 27, 4422–4433.
Cross, D. A., Alessi, D. R., Cohen, P., Andjelkovich, M., and Hemmings, B. A. (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789.
Crusio, K. M., King, B., Reavie, L. B., and Aifantis, I. (2010). The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene 29, 4865–4873.
Datta, S. R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y., and Greenberg, M. E. (1997). Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91, 231–241.
Davies, B. R., Logie, A., Mckay, J. S., Martin, P., Steele, S., Jenkins, R., Cockerill, M., Cartlidge, S., and Smith, P. D. (2007). AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol. Cancer Ther. 6, 2209–2219.
Davies, H., Bignell, G. R., Cox, C., Stephens, P., Edkins, S., Clegg, S., Teague, J., Woffendin, H., Garnett, M. J., Bottomley, W., Davis, N., Dicks, E., Ewing, R., Floyd, Y., Gray, K., Hall, S., Hawes, R., Hughes, J., Kosmidou, V., Menzies, A., Mould, C., Parker, A., Stevens, C., Watt, S., Hooper, S., Wilson, R., Jayatilake, H., Gusterson, B. A., Cooper, C., Shipley, J., Hargrave, D., Pritchard-Jones, K., Maitland, N., Chenevix-Trench, G., Riggins, G. J., Bigner, D. D., Palmieri, G., Cossu, A., Flanagan, A., Nicholson, A., Ho, J. W., Leung, S. Y., Yuen, S. T., Weber, B. L., Seigler, H. F., Darrow, T. L., Paterson, H., Marais, R., Marshall, C. J., Wooster, R., Stratton, M. R., and Futreal, P. A. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954.
de Herreros, A. G., Peiro, S., Nassour, M., and Savagner, P. (2010). Snail family regulation and epithelial mesenchymal transitions in breast cancer progression. J. Mammary Gland Biol. Neoplasia 15, 135–147.
Derouet, M., Thomas, L., Cross, A., Moots, R. J., and Edwards, S. W. (2004). Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J. Biol. Chem. 279, 26915–26921.
Dijkers, P. F., Medema, R. H., Pals, C., Banerji, L., Thomas, N. S., Lam, E. W., Burgering, B. M., Raaijmakers, J. A., Lammers, J. W., Koenderman, L., and Coffer, P. J. (2000). Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol. Cell. Biol. 20, 9138–9148.
Ding, Q., He, X., Hsu, J. M., Xia, W., Chen, C. T., Li, L. Y., Lee, D. F., Liu, J. C., Zhong, Q., Wang, X., and Hung, M. C. (2007). Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol. 27, 4006–4017.
Ding, Q., Huo, L., Yang, J. Y., Xia, W., Wei, Y., Liao, Y., Chang, C. J., Yang, Y., Lai, C. C., Lee, D. F., Yen, C. J., Chen, Y. J., Hsu, J. M., Kuo, H. P., Lin, C. Y., Tsai, F. J., Li, L. Y., Tsai, C. H., and Hung, M. C. (2008). Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res. 68, 6109–6117.
Ding, Q., Xia, W., Liu, J. C., Yang, J. Y., Lee, D. F., Xia, J., Bartholomeusz, G., Li, Y., Pan, Y., Li, Z., Bargou, R. C., Qin, J., Lai, C. C., Tsai, F. J., Tsai, C. H., and Hung, M. C. (2005). Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 19, 159–170.
Domina, A. M., Vrana, J. A., Gregory, M. A., Hann, S. R., and Craig, R. W. (2004). MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 23, 5301–5315.
Fang, D., Hawke, D., Zheng, Y., Xia, Y., Meisenhelder, J., Nika, H., Mills, G. B., Kobayashi, R., Hunter, T., and Lu, Z. (2007). Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229.
Fu, W., Ma, Q., Chen, L., Li, P., Zhang, M., Ramamoorthy, S., Nawaz, Z., Shimojima, T., Wang, H., Yang, Y., Shen, Z., Zhang, Y., Zhang, X., Nicosia, S. V., Pledger, J. W., Chen, J., and Bai, W. (2009). MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J. Biol. Chem. 284, 13987–14000.
Garcia-Echeverria, C., and Sellers, W. R. (2008). Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene 27, 5511–5526.
Hacker, H., and Karin, M. (2006). Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13.
Hatzivassiliou, G., Song, K., Yen, I., Brandhuber, B. J., Anderson, D. J., Alvarado, R., Ludlam, M. J., Stokoe, D., Gloor, S. L., Vigers, G., Morales, T., Aliagas, I., Liu, B., Sideris, S., Hoeflich, K. P., Jaiswal, B. S., Seshagiri, S., Koeppen, H., Belvin, M., Friedman, L. S., and Malek, S. (2010). RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435.
He, T. C., Sparks, A. B., Rago, C., Hermeking, H., Zawel, L., Da Costa, L. T., Morin, P. J., Vogelstein, B., and Kinzler, K. W. (1998). Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512.
Henson, E. S., Gibson, E. M., Villanueva, J., Bristow, N. A., Haney, N., and Gibson, S. B. (2003). Increased expression of Mcl-1 is responsible for the blockage of TRAIL-induced apoptosis mediated by EGF/ErbB1 signaling pathway. J. Cell. Biochem. 89, 1177–1192.
Hochstrasser, M. (1995). Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 7, 215–223.
Hu, M. C., Lee, D. F., Xia, W., Golfman, L. S., Ou-Yang, F., Yang, J. Y., Zou, Y., Bao, S., Hanada, N., Saso, H., Kobayashi, R., and Hung, M. C. (2004). IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 117, 225–237.
Huang, H., Regan, K. M., Wang, F., Wang, D., Smith, D. I., Van Deursen, J. M., and Tindall, D. J. (2005). Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc. Natl. Acad. Sci. U.S.A. 102, 1649–1654.
Huang, W. C., Ju, T. K., Hung, M. C., and Chen, C. C. (2007). Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol. Cell 26, 75–87.
Hudson, L. G., Choi, C., Newkirk, K. M., Parkhani, J., Cooper, K. L., Lu, P., and Kusewitt, D. F. (2007). Ultraviolet radiation stimulates expression of Snail family transcription factors in keratinocytes. Mol. Carcinog. 46, 257–268.
Hynes, N. E., and Lane, H. A. (2005). ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 5, 341–354.
Inuzuka, H., Fukushima, H., Shaik, S., Liu, P., Lau, A. W., and Wei, W. (2011a). Mcl-1 ubiquitination and destruction. Oncotarget 2, 239–244.
Inuzuka, H., Shaik, S., Onoyama, I., Gao, D., Tseng, A., Maser, R. S., Zhai, B., Wan, L., Gutierrez, A., Lau, A. W., Xiao, Y., Christie, A. L., Aster, J., Settleman, J., Gygi, S. P., Kung, A. L., Look, T., Nakayama, K. I., Depinho, R. A., and Wei, W. (2011b). SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 471, 104–109.
Israel, A. (2010). The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2, a000158.
Jagani, Z., Singh, A., and Khosravi-Far, R. (2008). FoxO tumor suppressors and BCR-ABL-induced leukemia: a matter of evasion of apoptosis. Biochim. Biophys. Acta 1785, 63–84.
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428.
Kee, Y., and Huibregtse, J. M. (2007). Regulation of catalytic activities of HECT ubiquitin ligases. Biochem. Biophys. Res. Commun. 354, 329–333.
Kops, G. J., Dansen, T. B., Polderman, P. E., Saarloos, I., Wirtz, K. W., Coffer, P. J., Huang, T. T., Bos, J. L., Medema, R. H., and Burgering, B. M. (2002). Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321.
Lamberti, C., Lin, K. M., Yamamoto, Y., Verma, U., Verma, I. M., Byers, S., and Gaynor, R. B. (2001). Regulation of beta-catenin function by the IkappaB kinases. J. Biol. Chem. 276, 42276–42286.
Lee, D. F., and Hung, M. C. (2008). Advances in targeting IKK and IKK-related kinases for cancer therapy. Clin. Cancer Res. 14, 5656–5662.
Lee, D. F., Kuo, H. P., Chen, C. T., Hsu, J. M., Chou, C. K., Wei, Y., Sun, H. L., Li, L. Y., Ping, B., Huang, W. C., He, X., Hung, J. Y., Lai, C. C., Ding, Q., Su, J. L., Yang, J. Y., Sahin, A. A., Hortobagyi, G. N., Tsai, F. J., Tsai, C. H., and Hung, M. C. (2007). IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130, 440–455.
Li, Y., Liu, Y., Xu, Y., Voorhees, J. J., and Fisher, G. J. (2010). UV irradiation induces Snail expression by AP-1 dependent mechanism in human skin keratinocytes. J. Dermatol. Sci. 60, 105–113.
Liang, J., Zubovitz, J., Petrocelli, T., Kotchetkov, R., Connor, M. K., Han, K., Lee, J. H., Ciarallo, S., Catzavelos, C., Beniston, R., Franssen, E., and Slingerland, J. M. (2002). PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 8, 1153–1160.
Lim, S. O., Kim, H., and Jung, G. (2010). p53 inhibits tumor cell invasion via the degradation of snail protein in hepatocellular carcinoma. FEBS Lett. 584, 2231–2236.
Lin, S. Y., Xia, W., Wang, J. C., Kwong, K. Y., Spohn, B., Wen, Y., Pestell, R. G., and Hung, M. C. (2000). Beta-catenin, a novel prognostic marker for breast cancer: its roles in cyclin D1 expression and cancer progression. Proc. Natl. Acad. Sci. U.S.A. 97, 4262–4266.
Linardou, H., Dahabreh, I. J., Kanaloupiti, D., Siannis, F., Bafaloukos, D., Kosmidis, P., Papadimitriou, C. A., and Murray, S. (2008). Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 9, 962–972.
Lipkowitz, S., and Weissman, A. M. (2011). RINGs of good and evil: RING finger ubiquitin ligases at the crossroads of tumour suppression and oncogenesis. Nat. Rev. Cancer 11, 629–643.
Liu, M., Lee, D. F., Chen, C. T., Yen, C. J., Li, L. Y., Lee, H. J., Chang, C. J., Chang, W. C., Hsu, J. M., Kuo, H. P., Xia, W., Wei, Y., Chiu, P. C., Chou, C. K., Du, Y., Dhar, D., Karin, M., Chen, C. H., and Hung, M. C. (2012). IKKa activation of NOTCH links tumorigenesis via FOXA2 suppression. Mol. Cell 45, 171–184.
Luo, J. L., Kamata, H., and Karin, M. (2005). IKK/NF-kappaB signaling: balancing life and death – a new approach to cancer therapy. J. Clin. Invest. 115, 2625–2632.
Manning, B. D., Tee, A. R., Logsdon, M. N., Blenis, J., and Cantley, L. C. (2002). Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162.
Marine, J. C., and Lozano, G. (2010). Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ. 17, 93–102.
Maurer, G., Tarkowski, B., and Baccarini, M. (2011). Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 30, 3477–3488.
Maurer, U., Charvet, C., Wagman, A. S., Dejardin, E., and Green, D. R. (2006). Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol. Cell 21, 749–760.
Monick, M. M., Carter, A. B., Robeff, P. K., Flaherty, D. M., Peterson, M. W., and Hunninghake, G. W. (2001). Lipopolysaccharide activates Akt in human alveolar macrophages resulting in nuclear accumulation and transcriptional activity of beta-catenin. J. Immunol. 166, 4713–4720.
Mosimann, C., Hausmann, G., and Basler, K. (2009). Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 10, 276–286.
Nicholson, R. I., Gee, J. M. W., and Harper, M. E. (2001). EGFR and cancer prognosis. Eur. J. Cancer 37, 9–15.
Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease: old views and new perspectives. Int. J. Dev. Biol. 53, 1541–1547.
Nijhawan, D., Fang, M., Traer, E., Zhong, Q., Gao, W., Du, F., and Wang, X. (2003). Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 17, 1475–1486.
Olmeda, D., Jorda, M., Peinado, H., Fabra, A., and Cano, A. (2007). Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 26, 1862–1874.
Paik, J. H., Kollipara, R., Chu, G., Ji, H., Xiao, Y., Ding, Z., Miao, L., Tothova, Z., Horner, J. W., Carrasco, D. R., Jiang, S., Gilliland, D. G., Chin, L., Wong, W. H., Castrillon, D. H., and Depinho, R. A. (2007). FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128, 309–323.
Park, K. J., Krishnan, V., O’Malley, B. W., Yamamoto, Y., and Gaynor, R. B. (2005). Formation of an IKKalpha-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol. Cell 18, 71–82.
Peinado, H., Olmeda, D., and Cano, A. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428.
Perkins, N. D. (2007). Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat. Rev. Mol. Cell Biol. 8, 49–62.
Placzek, W. J., Wei, J., Kitada, S., Zhai, D., Reed, J. C., and Pellecchia, M. (2010). A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 1, e40.
Plas, D. R., and Thompson, C. B. (2003). Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J. Biol. Chem. 278, 12361–12366.
Quatrale, A. E., Porcelli, L., Silvestris, N., Colucci, G., Angelo, A., and Azzariti, A. (2011). EGFR tyrosine kinases inhibitors in cancer treatment: in vitro and in vivo evidence. Front. Biosci. 16, 1962–1972.
Robinson, M. J., and Cobb, M. H. (1997). Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 9, 180–186.
Sebolt-Leopold, J. S. (2000). Development of anticancer drugs targeting the MAP kinase pathway. Oncogene 19, 6594–6599.
Seoane, J., Le, H. V., Shen, L., Anderson, S. A., and Massague, J. (2004). Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117, 211–223.
Sharma, S. V., and Settleman, J. (2010). Exploiting the balance between life and death: targeted cancer therapy and “oncogenic shock.” Biochem. Pharmacol. 80, 666–673.
Shen, R. R., and Hahn, W. C. (2011). Emerging roles for the non-canonical IKKs in cancer. Oncogene 30, 631–641.
Song, L. B., Li, J., Liao, W. T., Feng, Y., Yu, C. P., Hu, L. J., Kong, Q. L., Xu, L. H., Zhang, X., Liu, W. L., Li, M. Z., Zhang, L., Kang, T. B., Fu, L. W., Huang, W. L., Xia, Y. F., Tsao, S. W., Li, M., Band, V., Band, H., Shi, Q. H., Zeng, Y. X., and Zeng, M. S. (2009). The polycomb group protein Bmi-1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J. Clin. Invest. 119, 3626–3636.
Stepniak, E., Radice, G. L., and Vasioukhin, V. (2009). Adhesive and signaling functions of cadherins and catenins in vertebrate development. Cold Spring Harb. Perspect. Biol. 1, a002949.
Su, J. L., Cheng, X., Yamaguchi, H., Chang, Y. W., Hou, C. F., Lee, D. F., Ko, H. W., Hua, K. T., Wang, Y. N., Hsiao, M., Chen, P. B., Hsu, J. M., Bast, R. C. Jr., Hortobagyi, G. N., and Hung, M. C. (2011). FOXO3a-dependent mechanism of E1A-induced chemosensitization. Cancer Res. 71, 6878–6887.
Sunters, A., Fernandez De Mattos, S., Stahl, M., Brosens, J. J., Zoumpoulidou, G., Saunders, C. A., Coffer, P. J., Medema, R. H., Coombes, R. C., and Lam, E. W. (2003). FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J. Biol. Chem. 278, 49795–49805.
Tian, Q., Feetham, M. C., Tao, W. A., He, X. C., Li, L., Aebersold, R., and Hood, L. (2004). Proteomic analysis identifies that 14-3-3zeta interacts with beta-catenin and facilitates its activation by Akt. Proc. Natl. Acad. Sci. U.S.A. 101, 15370–15375.
Tothova, Z., Kollipara, R., Huntly, B. J., Lee, B. H., Castrillon, D. H., Cullen, D. E., Mcdowell, E. P., Lazo-Kallanian, S., Williams, I. R., Sears, C., Armstrong, S. A., Passegue, E., Depinho, R. A., and Gilliland, D. G. (2007). FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339.
Tran, H., Brunet, A., Grenier, J. M., Datta, S. R., Fornace, A. J. Jr., Distefano, P. S., Chiang, L. W., and Greenberg, M. E. (2002). DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 296, 530–534.
Tran, H., Brunet, A., Griffith, E. C., and Greenberg, M. E. (2003). The many forks in FOXO’s road. Sci. STKE 2003, RE5.
Tsai, W. B., Chung, Y. M., Zou, Y., Park, S. H., Xu, Z., Nakayama, K., Lin, S. H., and Hu, M. C. (2010). Inhibition of FOXO3 tumor suppressor function by betaTrCP1 through ubiquitin-mediated degradation in a tumor mouse model. PLoS ONE 5, e11171.
Tzivion, G., Dobson, M., and Ramakrishnan, G. (2011). FoxO transcription factors; regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813, 1938–1945.
Vinas-Castells, R., Beltran, M., Valls, G., Gomez, I., Garcia, J. M., Montserrat-Sentis, B., Baulida, J., Bonilla, F., De Herreros, A. G., and Diaz, V. M. (2010). The hypoxia-controlled FBXL14 ubiquitin ligase targets SNAIL1 for proteasome degradation. J. Biol. Chem. 285, 3794–3805.
Wang, J. M., Chao, J. R., Chen, W., Kuo, M. L., Yen, J. J., and Yang-Yen, H. F. (1999). The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol. Cell. Biol. 19, 6195–6206.
Wang, S. P., Wang, W. L., Chang, Y. L., Wu, C. T., Chao, Y. C., Kao, S. H., Yuan, A., Lin, C. W., Yang, S. C., Chan, W. K., Li, K. C., Hong, T. M., and Yang, P. C. (2009). p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol. 11, 694–704.
Wang, Z., Gao, D., Fukushima, H., Inuzuka, H., Liu, P., Wan, L., Sarkar, F. H., and Wei, W. (2011). Skp2: a novel potential therapeutic target for prostate cancer. Biochim. Biophys. Acta 1825, 11–17.
Wasch, R., Robbins, J. A., and Cross, F. R. (2010). The emerging role of APC/CCdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene 29, 1–10.
Wertz, I. E., Kusam, S., Lam, C., Okamoto, T., Sandoval, W., Anderson, D. J., Helgason, E., Ernst, J. A., Eby, M., Liu, J., Belmont, L. D., Kaminker, J. S., O’Rourke, K. M., Pujara, K., Kohli, P. B., Johnson, A. R., Chiu, M. L., Lill, J. R., Jackson, P. K., Fairbrother, W. J., Seshagiri, S., Ludlam, M. J., Leong, K. G., Dueber, E. C., Maecker, H., Huang, D. C., and Dixit, V. M. (2011). Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 471, 110–114.
Wong, K. K., Engelman, J. A., and Cantley, L. C. (2010). Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 20, 87–90.
Wu, Y., Deng, J., Rychahou, P. G., Qiu, S., Evers, B. M., and Zhou, B. P. (2009). Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 15, 416–428.
Yang, J. Y., Chang, C. J., Xia, W., Wang, Y., Wong, K. K., Engelman, J. A., Du, Y., Andreeff, M., Hortobagyi, G. N., and Hung, M. C. (2010). Activation of FOXO3a is sufficient to reverse mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor chemoresistance in human cancer. Cancer Res. 70, 4709–4718.
Yang, J. Y., Zong, C. S., Xia, W., Yamaguchi, H., Ding, Q., Xie, X., Lang, J. Y., Lai, C. C., Chang, C. J., Huang, W. C., Huang, H., Kuo, H. P., Lee, D. F., Li, L. Y., Lien, H. C., Cheng, X., Chang, K. J., Hsiao, C. D., Tsai, F. J., Tsai, C. H., Sahin, A. A., Muller, W. J., Mills, G. B., Yu, D., Hortobagyi, G. N., and Hung, M. C. (2008). ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 10, 138–148.
Youle, R. J., and Strasser, A. (2008). The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59.
Yuan, T. L., and Cantley, L. C. (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene 27, 5497–5510.
Zhong, Q., Gao, W., Du, F., and Wang, X. (2005). Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 121, 1085–1095.
Zhou, B. P., Deng, J., Xia, W., Xu, J., Li, Y. M., Gunduz, M., and Hung, M. C. (2004). Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6, 931–940.
Zhou, B. P., Liao, Y., Xia, W., Spohn, B., Lee, M. H., and Hung, M. C. (2001a). Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat. Cell Biol. 3, 245–252.
Keywords: AKT, ERK, IKK, FOXO3, β-catenin, Mcl-1, snail
Citation: Yamaguchi H, Hsu JL and Hung M-C (2012) Regulation of ubiquitination-mediated protein degradation by survival kinases in cancer. Front. Oncol. 2:15. doi: 10.3389/fonc.2012.00015
Received: 06 December 2011; Paper pending published: 18 December 2011;
Accepted: 30 January 2012; Published online: 20 February 2012.
Edited by:
Hui-Kuan Lin, The University of Texas MD Anderson Cancer Center, USAReviewed by:
Shao-Chun Wang, University of Cincinnati, USABinhua (Peter) Zhou, University of Kentucky College of Medicine, USA
Copyright: © 2012 Yamaguchi, Hsu and Hung. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Mien-Chie Hung, Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center, Unit 108, 1515 Holcombe Boulevard, Houston, TX 77030, USA. e-mail:bWh1bmdAbWRhbmRlcnNvbi5vcmc=